 ,2, 邓子新,1, 刘天罡,1
,2, 邓子新,1, 刘天罡,1The third-generation sequencing combined with targeted capture technology for high-resolution HLA typing and MHC region haplotype identification
Jia Chen1, Mingyue Shu1, Jin Li2, Aisi Fu1, Fan Yang3, Zou Wang3, Yirong Li,2, Zixin Deng,1, Tiangang Liu,1通讯作者:
编委: 方向东
收稿日期:2018-11-26修回日期:2019-01-25网络出版日期:2019-04-20
| 基金资助: |
Received:2018-11-26Revised:2019-01-25Online:2019-04-20
| Fund supported: |
作者简介 About authors
陈佳,在读硕士研究生,专业方向:微生物与生化制药E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (843KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
陈佳, 舒明月, 里进, 付爱思, 杨帆, 王邹, 李一荣, 邓子新, 刘天罡. 三代测序与靶向捕获技术联用进行高分辨HLA基因分型及MHC区域单倍体型精细鉴定[J]. 遗传, 2019, 41(4): 337-348 doi:10.16288/j.yczz.18-282
Jia Chen, Mingyue Shu, Jin Li, Aisi Fu, Fan Yang, Zou Wang, Yirong Li, Zixin Deng, Tiangang Liu.
人类白细胞抗原(human leukocyte antigen,HLA)基因位于人体第6号染色体的短臂,受控于人类主要组织相容性复合体(major histocompatibility complex, MHC)的基因簇[1],全长约3.6 Mb,是目前所知人体最复杂的遗传多态性系统[2,3]。
前期研究表明,HLA基因的变异与传染病[4]、药物过敏反应[5]、自身免疫疾病[6]、器官移植反应[7]以及恶性肿瘤[1]等均有关联。此外,近期研究也表明HLA特定等位基因多态性与原发性肝细胞癌的发生相关[8]。因此,准确的HLA分型技术对于组织配型以及研究HLA与疾病相关性具有重要意义。
由于MHC区域存在高度的多态性和广泛的连锁不平衡,因此研究人员对该区域所涉及到的分子机制的研究受到一定的限制。传统基于聚合酶链式反应等分型方法存在分辨率低、无法获得单倍体型结果以及新等位基因信息等诸多问题[9,10]。虽然以往Sanger测序技术被视为是HLA分型的金标准[11],但其因通量低而逐步被各种第二代测序(next-generation sequencing, NGS)平台所取代[9,12,13]。但是,NGS对于个体所具备的两套同源染色体的独特核苷酸信息,即“对单倍体型的判定”(phasing)的解析依然存在困难,而已有文献表明准确的单倍体分型能更好解读基因与表型(包括疾病)之间的关系[14],尤其是对于HLA-DP等较大的HLA基因,NGS因其读长短而很难准确获得单倍体型结果[15]。基于单分子实时测序技术(single molecule real-time, SMRT)的第三代测序仪能产生平均长度在10 kb以上的数据,这不但有利于基因精确分型,还能实现对基因组复杂区域的组装以及对某个基因内及等位基因间差异的细致解析。目前利用该技术进行基因组完成图组装或用于多倍体基因组中单倍体型的解析研究已有所报道[16,17]。
三代测序技术预期能实现对HLA基因及MHC区域更精细的分析,从而准确地确定每个基因甚至整个MHC区域的单倍体分型结果,并且可有效挖掘包括单核苷酸多态性(single nucleotide polymorphhism, SNPs)在内的一系列遗传信息,这将极大地推进各类与人体免疫相关研究的发展。本研究以12位原发性肝细胞癌病人的外周血为供试样本,分析二、三代测序数据用于高分辨率HLA分型的优劣势,同时结合探针捕获与三代测序技术对MHC区域进行靶向分析,探究长读长测序技术对于整个MHC区域精细分析的潜力。
1 材料与方法
1.1 研究对象
本研究收集的12位原发性肝细胞癌(hepaticellular carcinoma, HCC)患者外周血样本均由武汉大学中南医院提供,所有患者均签署了知情同意书。YH标准细胞系由中国国家基因库提供,HeLa细胞采购自美国菌种保藏中心。1.2 HLA基因的测序分型
外周血样本总DNA提取使用Hipure Blood DNA Mini Kit (广州美基生物科技有限公司)完成,YH、HeLa细胞系总DNA的提取采用酚-氯仿提取法。HLA基因扩增使用GENDX NGSgo-AmpX试剂盒以及QIAGEN Long range PCR试剂盒(QIAGEN公司,德国),扩增产物使用Qubit 2.0定量后等摩尔混合,不同样本的HLA扩增子混合物分别采用Illumina Miseq以及PacBio RSII的标准混样建库流程进行文库制备及测序。PacBio RSII原始数据使用SMRT Portal中的RS_ReadsOfInsert方法进行质量过滤,得到环形一致性序列(circular consensus sequences, CCS)的数据,将过滤的质量值(即minimum predicted accuracy参数)分别设为0.80、0.85、0.90、0.95和0.99,分别得到CCS0.80、CCS0.85、CCS0.90、CCS0.95和CCS0.99的高质量数据。HLA分型分析中使用的各种开源或商业分型软件均采用从官方网站或者商业公司处获得的最新版本,其中,NGSengine、HLAssign、HLA-reporter、Omixon、HLAminer、HLA-VBseq 和 OptiType用于二代数据分型,NGSengine和HLAminer用于三代不同数据类型以及不同质量值CCS的数据分型。
1.3 MHC区域捕获测序以及数据分析
使用Roche NimbleGen MHC探针捕获试剂盒对MHC相关区域(包括传统MHC区域和约1.0 Mb的MHC周边区域)进行捕获,实验过程根据三代测序长读长的特点对DNA打断、DNA纯化体系以及DNA杂交时间等操作进行了优化,采用PacBio RSII的标准流程进行文库制备及测序。使用FALCON软件进行数据组装并且使用SMRT Portal中RS_Resequencing标准流程将原始测序数据比对到对应参考基因组MHC参考序列,以计算数据覆盖度并根据原始覆盖度(该流程的默认参数)统计SNPs在MHC区域的分布(未对覆盖度进行筛选,95%以上的覆盖度为100×)。MHC区域单体型分析分别采用了FALCON- Unzip软件以及targeted-phasing-consensus脚本(https://github.com/PacificBiosciences/targeted-phasing-consensus)两套方法,并使用MUMmer软件将得到的单倍体分型结果与其对应的HLA基因分型结果进行比对,评估上述两套方法对MHC区域单倍体分型结果的差异。
2 结果与分析
2.1 基于二代测序数据对HLA基因分型
利用包括全基因组、全外显子组、转录组及HLA基因扩增子数据在内的多种数据类型进行HLA分型的各种学术和商业化软件已被广泛使用(表1)。为评估分型软件的准确性以便从中选择最佳的分型软件用于与后续三代数据分型结果的比较,比较了7种不同的HLA分型软件对基于全长扩增子二代测序数据的分析性能。为了确保分型结果的可靠性,不论是Illumina Miseq还是PacBio RSII均保证了足够的数据量。由于二代数据存在一定偏好,因此其覆盖度会存在不均一的情况,不同基因的覆盖度有一定的差异(图1),但是总体而言,其95%以上的区域覆盖度会大于200×。而三代数据的覆盖度比二代数据更高(图1)。通过比较,7种软件对二代数据的分型结果展现出较大差异。从总体上来看,NGSengine对classⅠ和classⅡ基因的分型结果敏感度都较高,结果与血清学鉴定结果吻合。而其他6种分型软件仅对classⅠ类基因的分型均较为准确,但对classⅡ类基因则不敏感,部分软件甚至不能给出分型结果。例如,HLAssgin和HLAreporter只有2个(2/12)和0个样本(0/12)预测到了DPA1基因分型。而HLAminer和Omixon对classⅠ基因的分型结果的判定与NGSengine/血清学结果相比有很大的差异。由于HCC27样本的HLA基因分型结果经过了血清学结果及MHC区域捕获测序结果的双重验证,故以该样本作为示例(表2),NGSengine对classⅠ和classⅡ基因的分型结果与血清学鉴定结果以及MHC区域捕获测序结果高度吻合。但是HLAssgin和HLAreporter无法预测到DPA1的基因分型,而HLAminer和Omixon对DPB1、DQB1、DRB1等classⅡ基因的分型结果判定错误率较高(表2)。Table 1
表1
表1 7种HLA分型软件比较
Table 1
| 软件 | 原理 | 分辨率 | 数据类型 | 是否测试 | 参考文献 |
|---|---|---|---|---|---|
| NGSengine | 8-digit | Amplicon | Y | - | |
| HLAminer | 比对/组装 | 4-digit | WGS/WES/RNA-seq/amplicon | Y | [18] |
| ATHLATES | 组装 | 4-digit | WGS/WES/amplicon | N | [19] |
| HLAFOREST | 比对 | 4-digit | RNA-seq | N | [20] |
| OptiType | 比对 | 4-digit | WGS/WES/RNA-seq | Y | [21] |
| Omixon | 比对 | 6-digit | WGS/WES/amplicon | Y | [22] |
| HLAreporter | 比对和组装 | 4-digit | WGS/WES | Y | [23] |
| HLA-VBSeq | 比对 | 8-digit | WGS | Y | [24] |
| HLAssign | 比对 | 6-digit | Sequence capture | Y | [25] |
| SEQ2HLA | 比对 | 4-digit | RNA-seq | N | [26] |
新窗口打开|下载CSV
Table 2
表2
表2 HCC27样本基于二代测序数据的7种软件分型结果比较
Table 2
| 分型结果 | 软件名称 | ||||||
|---|---|---|---|---|---|---|---|
| NGSengine | HLAminer | HLAssign | HLA-reporter | Omixon | HLA-VBseq | Opti Type | |
| HLA-A | A*02:01:01:01 | A*02:01P | A*02:01:01 | A*02:01:01G | A*02:01:01 | A*02:01:01:01 | A*02:01 |
| A*31:01:02:01 | A*31:01P | A*31:01:02 | A*31:01:02G | A*31:01:02 | A*31:01:02 | A*31:01 | |
| HLA-B | B*40:01:02 | B*40:01:02:04 | B*40:01:02 | B*40:01:01G | B*40:01:02 | B*40:01:02 | B*40:01 |
| B*46:01:01 | B*46:01P | B*46:01:01 | B*46:01:01G | B*46:01:01 | B*46:01:01 | B*46:01 | |
| HLA-C | C*01:02:01 | C*01:02P | C*01:02:01 | C*01:02:01G | C*01:02:01 | C*01:02:01 | C*01:02 |
| C*07:02:01:01 | C*15:102 | C*07:02:01 | C*07:02:01G | C*07:02:01 | C*07:02:01:01 | C*07:02 | |
| DPA1 | DPA1*02:02:02 | DPA1*02:01P | - | - | DPA1*02:02:02 | - | - |
| DPA1*04:01 | DPA1*01:03P | - | - | DPA1*04:01 | - | - | |
| DPB1 | DPB1*05:01:01 | DPB1*40:01 | DPB1*05:01:01 | DPB1*05:01:01G | DPB1*135:01 | - | - |
| DPB1*13:01:01 | DPB1*19:01P | DPB1*13:01:01 | DPB1*13:01:01G | DPB1*519:01 | - | - | |
| DQA1 | DQA1*01:03:01:04 | DQA1*01:02P | DQA1*01:03:01 | DQA1*01:03:01G | DQA1*01:03:01 | DQA1*01:03:01:01 | - |
| DQA1*03:02 | DQA1*02:01P | DQA1*03:02 | DQA1*03:01:01G | DQA1*03:02 | DQA1*03:02 | - | |
| DQB1 | DQB1*03:03:02:02 | DQB1*03:05P | - | DQB1*03:03:02G | DQB1*03:03:02 | DQB1*03:03:02:03 | - |
| DQB1*06:01:01 | DQB1*04:32 | - | DQB1*06:01:01G | DQB1*06:01:15 | DQB1*06:01:01 | - | |
| DRB1 | DRB1*08:03:02 | DRB1*07:01P | - | DRB1*08:03:02 | DRB1*08:03:02 | DRB1*08:03:02 | - |
| DRB1*09:01:02 | DRB1*13:02P | - | DRB1*09:01:02 | DRB1*09:01:02 | DRB1*09:21 | - | |
| DRB4 | DRB4*01:03:01:01 | DRB4*01:01P | DRB4*01:03:01 | - | DRB4*01:03:01N | - | - |
| - | - | - | - | DRB4*01:10 | - | - | |
新窗口打开|下载CSV
图1
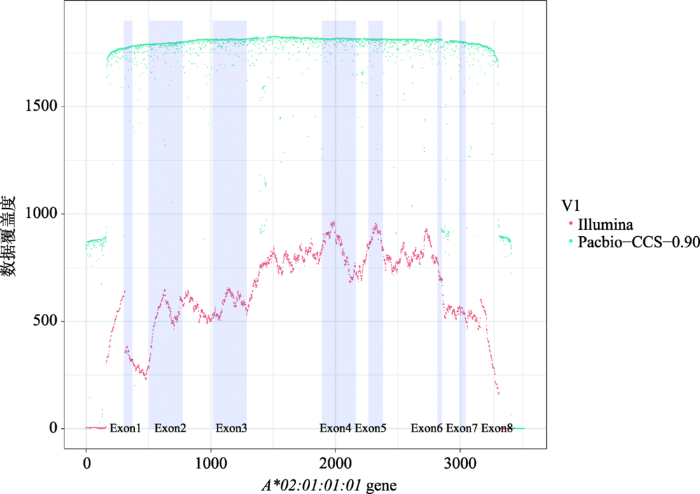 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1HCC27样本HLA-A基因Illimina和PacBio-CCS-0.90数据覆盖度
Fig. 1Comparison of coverage based on Illumina and PacBio-CCS-0.90 data from HLA-A of HCC27
此外,OptiType与NGSengine的分型结果很相似,但是其分辨率只有4位(表2)。有文献表明,位于外显子外部的单核苷酸变异可能在疾病的发病机制中起关键作用[27]。因此,高分辨率的HLA分型软件具有更高的应用价值。在测试的7种分型软件中,NGSengine分型最准确,分辨率最高。后续将以NGSengine产生的结果为参照,评估三代测序数据分析结果。
2.2 基于三代测序数据对HLA基因分型
采用两种已公开能够使用三代测序数据的分型软件——HLAminer和NGSengine对基于三代测序数据的HLA基因进行分型。此外,PacBio三代测序数据分为subreads和环形一致性序列(circular consensus sequences, CCS)两类:subreads是去除接头序列和低质量部分所得到的未经矫正的数据,而CCS是将来自于同一个DNA分子经过环状反复测序产生的多条subreads相互矫正后得到的高准确性数据。理论上,CCS数据的准确性越高越有利于获得准确的分型结果,但是矫正准确性设置越高最终所获得的CCS数据量也会减少,存在不能满足分型最低数据量需求的风险。因此,本研究测试了不同的数据类型和CCS准确性对分型结果的影响。首先,HLAminer软件三代与二代数据的分型结果间存在差异,而NGSengine软件三代与二代数据的分型结果保持一致(表3)。此外,当subreads的数据量足够时,基于subreads数据的分型结果与CCS数据基本一致,但前者往往会多出一个代表错配信息的后缀。以HCC5样本的DRB5基因为例,其基于subreads的分型结果为DRB5*02:023,而CCS对应的分型结果为DRB5*02:02,这表明subreads的结果在外显子区域存在3个错配碱基。从单碱基准确性角度考虑,使用CCS数据对HLA基因分型应该是更好的选择。
Table 3
表3
表3 NGSengine和HLAminer的分型结果分辨率及与Illumina分型结果一致性统计
Table 3
| 软件 | 分型分辨率6位 以上的基因个数 | 分型分辨率8位 以上的基因个数 | 与NGSengine-Illumina分型结果一致的基因个数 | |||
|---|---|---|---|---|---|---|
| CCS0.80 | CCS0.85 | CCS0.90 | CCS0.95 | |||
| NGSengine | 103/114 | 49/114 | 99/114 | 99/114 | 99/114 | 99/114 |
| HLAminer | 4/114 | 2/114 | 28/114 | 30/114 | 36/114 | 43/114 |
新窗口打开|下载CSV
随着CCS数据准确性的不断提高,其数据量会显著下降(图2)。以HCC27样本为例,数据量下降最大的两个断层分别在subreads到CCS0.80,以及CCS0.95到CCS0.99两处,其数据量分别降低了78.86%和89.84%。虽然CCS0.99准确性最高,但其reads数量从subreads的3000条降至50条,无法满足后续的分型需求。另一方面,数据量远多于CCS数据的subreads并没有获得最好的覆盖度,除了DQB1和DRB1,其他基因的subreads覆盖度甚至还低于CCS0.80 (图2),这可能是由于subreads错误率较高导致大量结果被过滤。因此,考虑到准确性和数据量之间的平衡,本研究最终选用CCS0.90的数据用于最终分型结果的比较。
图2
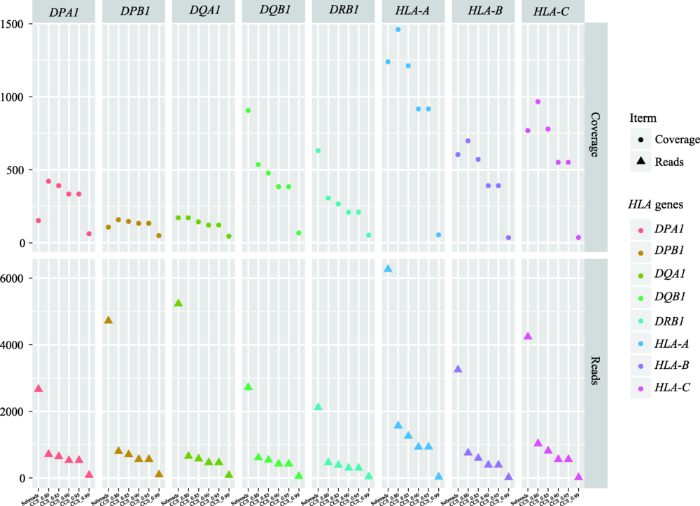 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2HCC27样本在基于不同数据类型和CCS准确性下各HLA基因的覆盖度和数据量
Fig. 2Coverage and reads of HLA genes in HCC27 based on different data types and CCS accuracy
虽然基于二代和三代CCS0.90数据的分型结果一致(表3),但是两种数据的基因覆盖度却存在较大差异(图1)。Illumina数据存在一定偏好,在某些区段上会出现明显的“断层”现象,尤其在基因两端区域。相比之下,CCS0.90数据的覆盖度更均匀,对于一些复杂的或全新的HLA基因具有更强的分型能力和更低的错误风险。
2.3 基于三代测序数据对HLA基因进行单倍体分型
Phasing regions是用以评估单倍体分型效果的重要指标,主要代表目的基因中能准确分型单倍体区域的数目。分析结果显示基于二、三代数据进行HLA基因单倍体分型的结果间存在一定的差异(表4)。基于三代数据,有92.79% (103/111)的基因可以得到一条完整的单体型结果,而这一比例在NGS数据里仅占75.65% (87/115)。与此同时,同一个单体型被定相到3个以上区域的比例中,NGS占比13.91% (16/115),而三代测序数据对应的占比仅为3.6% (4/111) (表4)。因此,三代测序更有利于提高单倍体分型的准确性,减少不确定性。Table 4
表4
表4 二代(Illumina)和三代(PacBio)测序数据中所有HLA基因phasing regions个数的差异
Table 4
| 测序平台 | 单倍体分型区域数目 | ||
|---|---|---|---|
| 1 | 2 | >3 | |
| Illumina | 87 | 12 | 16 |
| PacBio | 103 | 4 | 4 |
新窗口打开|下载CSV
2.4 利用PacBio数据组装MHC区域以及SNP在MHC区域上的分布
MHC捕获探针设计区域大小为4 970 458 bp (Chr.6:28 477 797~33 448 354 bp),使用FALCON软件对YH标准细胞系进行组装,得到的最佳组装结果为:MHC组装大小为4.46 Mb,Contig N50为85 kb,Contig总数154个。将组装的Contig比对到YH基因组的参考序列上(图3),可以完整覆盖其MHC的参考序列。数据总覆盖度97.86% (4 864 179/4 970 458 bp),跟以往使用二代数据结果获得的覆盖度为97.29%的结果相比有所提升[28]。图3
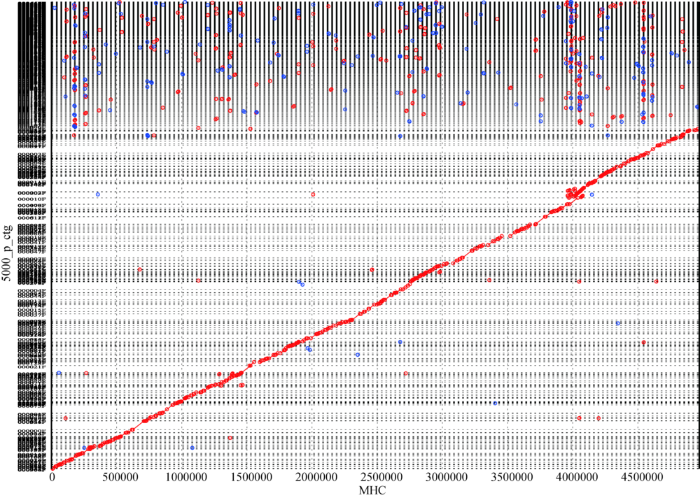 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3YH细胞系MHC区域FALCOM组装序列与人类MHC的参考序列的比对结果
Fig. 3Alignment of the FALCOM assembly sequence of the MHC region of the YH cell line with the reference sequence of human MHC
随后,使用根据YH细胞系组装优化参数对HCC27样本MHC区域进行组装,得到的最佳组装效果为:MHC组装大小为4.79 Mb,Contig N50为90 kb,Contig总数223个。数据覆盖度99.8% (4 960 480 bp/ 4 970 458)。0×以下的覆盖度比例为0.21% (10 363/ 4 970 458 bp),30×以下的覆盖度比例为2.23% (110 974/4 970 458 bp),意味着有97.77%的序列覆盖度达到30×以上,组装效果进一步提升。
此外,本研究统计了HCC27样本、YH和HeLa细胞系MHC区域SNP的分布情况(图4),发现HLA基因区域的SNP频率显著升高,该结果与之前报道的结果一致[28],这也是HLA基因多态性高的重要表现。
图4
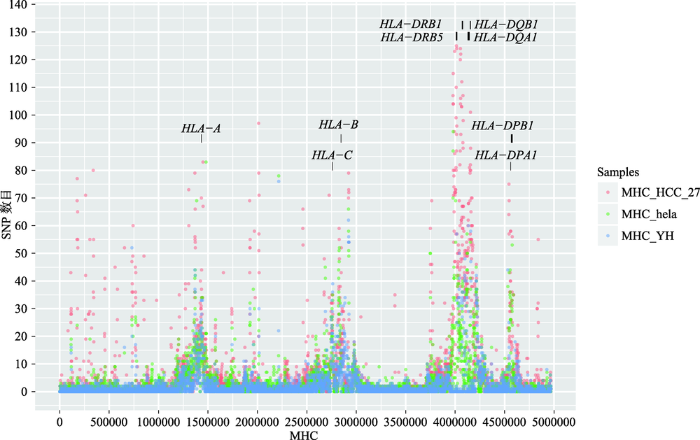 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4HCC27、YH和HeLa标准细胞系SNP在MHC区域的分布图
Fig. 4Distribution of SNPs in the MHC region of HCC27,YH and HeLa standard cell lines
2.5 关于HCC27样本MHC区域的单倍体分型
FALCON+FALCON-Unzip以及targeted-phasing- consensus采用了两种不同的单倍体分型原理。前者是基于数据的从头组装,是无参考序列的单倍体型分析方法。后者的分析思路是基于数据比对,将原始的测序数据比对到参考序列上,然后根据比对的结果得到两条单倍体型结果,即consensus0和consensus1。两种方法所得到结果与本文2.2部分的结果基本一致(表5)。为进一步验证上述HCC27样本HLA基因单倍体分型结果的准确性,对其产生的两条单倍体型序列中所含有HLA-A序列与各外显子区域内HLA-A基因进行比对(图5,A和B),两者在单碱基水平上均保持一致。这表明基于长读长的三代测序数据,两种方法均可以对MHC区域上各HLA基因进行较为准确的单倍体型分析。此外,基于捕获的测序结果,还可以获得扩增测序难以得到的内含子信息。Table 5
表5
表5 HCC27基于FALCON+FALCON-Unzip以及Targeted-phasing-consensus的单倍体分型结果
Table 5
| FALCON+FALCON-Unzip | Targeted-phasing-consensus | ||
|---|---|---|---|
| contig_Tags | 单倍体分型结果 | consensus_Tags | 单倍体分型结果 |
| 000009F|arrow | A*02:01:01:01_gen | consensus1 | A*02:01:01:01_gen |
| 000009F_001|arrow | A*02:01:01:01_gen | ||
| 000009F_001|arrow | A*31:01:02:01_gen | consensus0 | A*31:01:02:01_gen |
| 000018F|arrow | B*46:01:01_gen | consensus1 | B*46:01:01_gen |
| 000018F_002|arrow | B*46:01:01_gen | ||
| 000018F_002|arrow | B*40:01:02:01_gen | consensus0 | B*40:01:02:01_gen |
| 000018F|arrow | C*01:02:01_gen | consensus1 | C*01:02:01_gen |
| 000018F_002|arrow | C*01:02:01_gen | ||
| 000018F_002|arrow | C*07:02:01:01_gen | consensus0 | C*07:02:01:01_gen |
| 000027F_001|arrow | DPA1*02:02:02_gen | consensus0 | DPA1*02:02:02_gen |
| 000027F|arrow | DPA1*04:01_nuc | consensus1 | DPA1*04:01_nuc |
| 000027F_001|arrow | DPB1*05:01:01_nuc | consensus0 | DPB1*05:01:01_nuc |
| 000027F|arrow | DPB1*13:01:01_nuc | consensus1 | DPB1*13:01:01_nuc |
| 000017F|arrow | DQA1*01:03:01:04_gen | consensus0 | DQA1*01:03:01:04_gen |
| 000017F|arrow | DQA1*03:02_gen | consensus1 | DQA1*03:02_gen |
| 000017F|arrow | DQB1*03:03:02:02_gen | consensus1 | DQB1*03:03:02:02_gen |
新窗口打开|下载CSV
图5
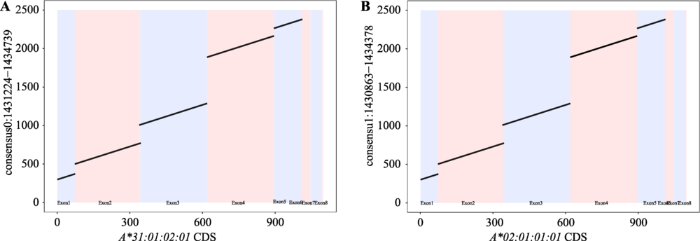 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5HCC27样本consensus0和consensus1与HLA-A基因的比对结果
A:HCC27样本consensus0与A*31:01:02:01的序列比对结果;B:HCC27样本consensus1与A*02:01:01:01的序列比对结果。
Fig. 5Sequence alignment of consensus0 and consensus1 and HLA-A gene
另一方面,FALCON+FALCON-Unzip会出现同一基因被比对到不同contig上的情况(表5),表明该组装方法对于MHC这类的多态性极高的区域可能会存在一定的单倍体分型错误。而基于比对方法得到的consensus序列,由于捕获测序长度限制,也可能导致在进行单倍体分型时,同一个位点的SNP信息难以准确定位到不同的consensus序列,从而导致模棱两可的结果。
因此,本研究整合了上述两种单倍体分型方法的所有信息以及各HLA基因扩增子的分型信息,对HCC27样本MHC区域的单倍体上的HLA基因的分布进行了校正和预测(图6),两套HLA基因分别被定位到consensus0和consensus1两个单倍体型上。其中,没有用虚线标注的,即DPA1和DPB1等位基因,还通过从头组装的方法(FALCON+FALCON- Unzip结果)验证了这两个等位基因的确位于一条contig上。基于此,可以大致了解来自双亲的两套HLA等位基因、以及基因间其他功能原件在MHC上的确切位置与连锁关系,这对更深入研究基因与表型(包括疾病)之间的关系具有重要意义。
图6
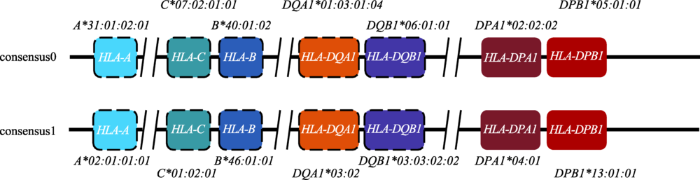 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6HCC27 MHC区域的单倍体分型结果预测
consensus0和consensus1表示利用targeted-phasing-consensus脚本得到的两个单倍体型,不同颜色表示不同的等位HLA基因。未用虚线标注的,表明其结果可通过从头组装的结果(FALCON+FALCON-Unzip结果)得到验证,虚线边框标注的表示通过从头组装结果无法得到验证信息。
Fig. 6Proposed phasing results of MHC region of HCC27
3 讨论
本研究评估了7种可使用二代测序数据和2种可使用三代数据的HLA分型软件。结果显示,二代数据的分型结果差异很大,其主要归因于各软件最适合输入数据类型、分析原理、数据库的差异(表1)。而不论是基于二代还是三代数据,NGSengine均能产生准确,分辨率高的分型结果,这说明分型实验设计与分析流程匹配的重要性。CCS数据有助于提高分型结果的单碱基准确性,基于CCS分型结果其外显子错配信息会大幅减少,需要进行单碱基级别分析的研究可采用CCS0.90的数据进行分析。不同于二代数据需要通过多条数据组装/计算等分析手段对基因上各个位点的相位进行单倍体分型。单条三代测序数据可跨越较长的区域,基因分型与单倍体分型过程不涉及数据组装,减少了因组装而导致的错误。虽然在本研究中二、三代的分型结果基本一致,但是三代的覆盖均一度和单倍体分型结果均优于二代(图1,表4),可以大幅提高单倍体分型的准确性,减少模棱两可的分型结果,更适用于HLA基因的分型与单体型分析。YH、HeLa和HCC样本的MHC区域捕获和三代测序的结果表明,三代数据的组装结果优于以往文献使用二代测序的结果,且结果准确性可以达到单碱基水平。此外,FALCON+FALCON-Unzip软件由于是基于三代测序原始数据的无参考基因组单倍体分型方式,可能出现同一基因被比对到不同Contig上的情况(表5),从而导致组装出错。而targeted- phasing-consensus方法虽然是基于参考基因组序列的单倍体分型方法,但由于受到捕获产物测序数据长度的限制,同一个位点的SNP位点难以准确定位到不同的consensus序列,同样可能导致模棱两可的结果。因此,本研究将上述两种基于组装和比对的单倍体分型方法所得到的单倍体型信息以及本文2.2部分的HLA基因扩增子分型结果进行整合,对HCC27样本MHC区域的单倍体上的HLA基因的分布进行了校正和预测,通过从头组装的方法对预测结果进行验证,发现DPA1和DPB1等位基因的确位于同一contig上。基于该方法,可以从整体上了解来自于双亲的两套HLA等位基因、以及基因间其他功能原件在MHC上的位置与连锁关系,这将有助于对MHC这类结构复杂的基因区域进行系统研究并极大的推进各类相关疾病的相关性分析。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLMagsci [本文引用: 2]

鼻咽癌是一种多因素影响的复杂性疾病, 其发病具有显著的地理分布差异。Epstein-Barr(EB)病毒感染与鼻咽癌发病密切相关已得到公认, 但环境因素及遗传因素在鼻咽癌的发病中也具有重要作用。在鼻咽癌的遗传相关因素中, 位于6号染色体上具有高度多态性的人类白细胞抗原(Human leukocyte antigen, <em>HLA</em>)与鼻咽癌发病风险相关在多个研究组中被报道。随着DNA测序技术的发展, 高分辨基因分型技术的应用, <em>HLA</em>新等位基因数目呈指数级的上升, 更多的<em>HLA</em>全基因序列被研究者所报道。近年来, 等位基因关联性分析、微卫星连锁不平衡分析及全基因组关联性分析的研究结果均证实了6号染色体<em>HLA</em>区域与鼻咽癌具有显著关联。为了进一步探讨遗传相关性因子<em>HLA</em>在鼻咽癌发生发展中的作用, 文章着重综述了<em>HLA</em>与鼻咽癌相关性研究的最新进展, 为鼻咽癌<em>HLA</em>相关性研究提供新的思路。
URLMagsci [本文引用: 2]
鼻咽癌是一种多因素影响的复杂性疾病, 其发病具有显著的地理分布差异。Epstein-Barr(EB)病毒感染与鼻咽癌发病密切相关已得到公认, 但环境因素及遗传因素在鼻咽癌的发病中也具有重要作用。在鼻咽癌的遗传相关因素中, 位于6号染色体上具有高度多态性的人类白细胞抗原(Human leukocyte antigen, <em>HLA</em>)与鼻咽癌发病风险相关在多个研究组中被报道。随着DNA测序技术的发展, 高分辨基因分型技术的应用, <em>HLA</em>新等位基因数目呈指数级的上升, 更多的<em>HLA</em>全基因序列被研究者所报道。近年来, 等位基因关联性分析、微卫星连锁不平衡分析及全基因组关联性分析的研究结果均证实了6号染色体<em>HLA</em>区域与鼻咽癌具有显著关联。为了进一步探讨遗传相关性因子<em>HLA</em>在鼻咽癌发生发展中的作用, 文章着重综述了<em>HLA</em>与鼻咽癌相关性研究的最新进展, 为鼻咽癌<em>HLA</em>相关性研究提供新的思路。
URLMagsci [本文引用: 1]
人类遗传多样性表现为世界各种族、民族和个体间存在的基因组差异, 是探讨人类进化与迁徙、环境与遗传背景相互作用、疾病与健康影响因素的主要资源和工具。中国具有世界1/5的人口, 有56个民族和丰富的遗传多样性资源。经过几十年的努力, 中国人类遗传多样性研究已积累了丰富的资料, 部分成果已经达到国际先进水平。文章重点论述了近年来形态学标记、生化及免疫学标记、DNA遗传标记在我国人类遗传多样性研究中的应用, 线粒体DNA、Y染色体DNA和HLA标记在中国不同民族源流和相互关系、东亚现代人起源和迁移等研究中的应用, 以及我国在中华民族遗传资源保存和利用、疾病易感基因和环境适应相关基因的鉴定及全基因组关联分析和第二代测序技术的应用、中国人基因组结构研究等方面取得的重要成果和进展。
URLMagsci [本文引用: 1]
人类遗传多样性表现为世界各种族、民族和个体间存在的基因组差异, 是探讨人类进化与迁徙、环境与遗传背景相互作用、疾病与健康影响因素的主要资源和工具。中国具有世界1/5的人口, 有56个民族和丰富的遗传多样性资源。经过几十年的努力, 中国人类遗传多样性研究已积累了丰富的资料, 部分成果已经达到国际先进水平。文章重点论述了近年来形态学标记、生化及免疫学标记、DNA遗传标记在我国人类遗传多样性研究中的应用, 线粒体DNA、Y染色体DNA和HLA标记在中国不同民族源流和相互关系、东亚现代人起源和迁移等研究中的应用, 以及我国在中华民族遗传资源保存和利用、疾病易感基因和环境适应相关基因的鉴定及全基因组关联分析和第二代测序技术的应用、中国人基因组结构研究等方面取得的重要成果和进展。
URLMagsci [本文引用: 1]
文章利用20个中国汉族个体样本建立了稳定精确的<em>HLA-A、-B</em>基因全长序列的克隆测序方法, 获得<em>HLA-A </em>10个等位基因4.2 kb序列, <em>HLA-B </em>6个等位基因3.7 kb序列, 序列涵盖了两个基因的所有外显子、所有内含子、5′启动子区以及3′非翻译区(3′UTR)。<em>A</em>*1153是文章发现的一个新等位基因, <em>B</em>*151101的内含子序列、5个<em>HLA-A</em>以及2个<em>HLA-B</em>等位基因的5′启动子序列和3′UTR序列为国际上首次报道, 其他等位基因均延伸了IMGT/HLA数据库中释放的全长序列。文章首次在中国汉族个体中测定了IMGT/HLA数据库中没有覆盖的HLA-A、-B基因的上游5′启动子以及下游3′UTR区域的多态性模式。<em>HLA-A</em>基因5′启动子延伸区域共发现26个SNPs和一处3 bp(AAA/-)的插入/缺失, 3′UTR延伸区域共发现14个SNPs; <em>HLA-B</em>基因5′启动子延伸区域共发现5个SNPs和一处1 bp(T/-)的插入/缺失, 3′UTR延伸区域共发现8个SNPs。通过对两个基因的5′启动子、外显子以及3′UTR的系统发育树分析, 发现两个基因调控区与外显子的进化关系有所不同, <em>HLA-A</em>基因除<em>A</em>*24020101外, 其他等位基因两端调控区与外显子连锁比较紧密, <em>HLA-B</em>基因两端调控区与外显子之间则发生了较为频繁的重组事件。
URLMagsci [本文引用: 1]
文章利用20个中国汉族个体样本建立了稳定精确的<em>HLA-A、-B</em>基因全长序列的克隆测序方法, 获得<em>HLA-A </em>10个等位基因4.2 kb序列, <em>HLA-B </em>6个等位基因3.7 kb序列, 序列涵盖了两个基因的所有外显子、所有内含子、5′启动子区以及3′非翻译区(3′UTR)。<em>A</em>*1153是文章发现的一个新等位基因, <em>B</em>*151101的内含子序列、5个<em>HLA-A</em>以及2个<em>HLA-B</em>等位基因的5′启动子序列和3′UTR序列为国际上首次报道, 其他等位基因均延伸了IMGT/HLA数据库中释放的全长序列。文章首次在中国汉族个体中测定了IMGT/HLA数据库中没有覆盖的HLA-A、-B基因的上游5′启动子以及下游3′UTR区域的多态性模式。<em>HLA-A</em>基因5′启动子延伸区域共发现26个SNPs和一处3 bp(AAA/-)的插入/缺失, 3′UTR延伸区域共发现14个SNPs; <em>HLA-B</em>基因5′启动子延伸区域共发现5个SNPs和一处1 bp(T/-)的插入/缺失, 3′UTR延伸区域共发现8个SNPs。通过对两个基因的5′启动子、外显子以及3′UTR的系统发育树分析, 发现两个基因调控区与外显子的进化关系有所不同, <em>HLA-A</em>基因除<em>A</em>*24020101外, 其他等位基因两端调控区与外显子连锁比较紧密, <em>HLA-B</em>基因两端调控区与外显子之间则发生了较为频繁的重组事件。
URLPMID:24010680 [本文引用: 1]
Genetic polymorphisms within the MHC encoding region have the strongest impact on disease progression of any in the genome and provide important clues to the mechanisms of immune control. Few analyses have been undertaken of alleles associated with rapid disease progression. *07:02 is an class I molecule that is prevalent in most populations worldwide and that has previously been consistently linked to accelerated disease progression in B-clade . This study investigates the observation that *07:02 is not associated with a high viral setpoint in C-clade . We examine the hypothesis that this clade-specific difference in association with disease outcome may be related to distinct targeting of CD8(+) T epitopes. We observed that C-clade-infected individuals with *07:02 target a broader range of epitopes, and to higher magnitudes, than do individuals infected with B-clade . In particular, a novel -(Gag22-30, RPGGKKHYM) epitope is targeted in >50% of *07:02-positive C-clade-infected individuals but clade-specific differences in this epitope result in nonimmunogenicity in B-clade . Only the C-clade -"GL9" (Gag355-363, GPSHKARVL) epitope-specific CD8(+) T response out of 16 studied was associated with a low viral setpoint. Although this epitope was also targeted in B-clade , the escape mutant S357S is present at higher frequency in B-clade than in C-clade (70% versus 43% in *07:02-negative subjects). These data support earlier studies suggesting that increased breadth of the -specific CD8(+) T response may contribute to improved immune control irrespective of the particular molecules expressed.
URLPMID:11888582 [本文引用: 1]
The use of abacavir a potent HIV—1 nucleosideanalogue reverse—transcriptase inhibitor is complicated by a potentially life-threatening hypersensitivity syndrome in about 5% of cases. Genetic factors influencing the immune response to abacavir might confer susceptibility. We aimed to find associations between MHC alleles and abacavir hypersensitivity in HIV—1-positive individuals treated with abacavir. MHC region typing was done in the first 200 Western Australian HIV Cohort Study participants exposed to abacavir. Definite abacavir hypersensitivity was identified in 18 cases, and was excluded in 167 individuals with more than 6 weeks' exposure to the drug (abacavir tolerant). 15 individuals experienced some symptoms but did not meet criteria for abacavir hypersensitivity. p values were corrected for comparisons of multiple HLA alleles (p c) by multiplication of the raw p value by the estimated number of HLA alleles present within the loci examined. HLA-B *5701 was present in 14 (78%) of the 18 patients with abacavir hypersensitivity, and in four (2%) of the 167 abacavir tolerant patients (odds ratio 117 [95% CI 29–481], p c<0·0001), and the HLA-DR7 and HLA-DQ3 combination was found in 13 (72%) of hypersensitive and five (3%) of tolerant patients (73 [20–268], p c<0·0001). HLA-B *5701, HLA-DR7, and HLA-DQ3 were present in combination in 13 (72%) hypersensitive patients and none of the tolerant patients (822 [43–1564675], p c<0·0001). Other MHC markers also present on the 57·1 ancestral haplotype to which the three markers above belong confirmed the presence of haplotype-specific linkage disequilibrium, and mapped potential susceptibility loci to a region bounded by C4A6 and HLA-C. Within the entire abacavir-exposed cohort (n=200), presence of HLA-B *5701, HLA-DR7, and HLA-DQ3 had a positive predictive value for hypersensitivity of 100%, and a negative predictive value of 97%. Genetic susceptibility to abacavir hypersensitivity is carried on the 57·1 ancestral haplotype. In our population, withholding abacavir in those with HLA-B *5701, HLA-DR7, and HLA-DQ3 should reduce the prevalence of hypersensitivity from 9% to 2·5% without inappropriately denying abacavir to any patient.
URLPMID:1556698 [本文引用: 1]
Abstract In order to ascertain whether the HLA-DP locus plays a role in the genetic predisposition for systemic lupus erythematosus (SLE), 42 patients with SLE were typed for 17 different DPBeta haplotypes by locus specific amplification followed by allele specific oligonucleotide hybridization. Sera from the same patients were assayed for the presence of autoantibodies to Sm, RNP, Ro, La and of anticardiolipin antibodies (aCL). DPB1*0301 and DPB1*1401 were increased in patients compared with 107 healthy controls, mainly in those anti-Sm/RNP positive and aCL positive. Remarkably, DPB1*1401 and DPB1*0301 have a nearly identical nucleotide sequence, suggesting that an epitope shared by their membrane products is of major importance for the DP related susceptibility to SLE.
URL [本文引用: 1]
URLPMID:11318984 [本文引用: 1]
Abstract: Background/Aims: Recent reports of an association between human leucocyte antigens (HLA) and persistence of hepatitis B virus infection, and the familial clustering of hepatocellular carcinoma raise the question of genetic susceptibility. Previous studies have been limited to serological phenotyping of HLA B and DR antigens. The aim of this study was to use molecular genotyping to investigate HLA class II as a risk factor for the development of hepatocellular carcinoma in Hong Kong Chinese. Methods: We determined HLA DRB1 , DQA1 , DQB1 and DPB1 alleles in 123 hepatitis B surface antigen positive patients (84 with hepatocellular carcinoma and 39 without) and 124 matched controls. Results: The alleles DRB1*1501 (36% of HCC patients versus 19% of controls, odds ratio=2.44), DQA1*0102 (42% versus 26%, odds ratio=2.07), and DPB1*0501 (80% versus 63%, odds ratio=2.35) were significantly more common in patients with hepatocellular carcinoma, and DQA1*03 (36% versus 56%, odds ratio=0.53), DQB1*0302 (4.% versus 13%, odds ratio=0.25) and DPB1*0201 (14% versus 29%, odds ratio=0.4) were found at significantly lower frequencies. Conclusions: Although none of these associations was significant after correction for multiple testing, this report suggests that further investigations are warranted.
URLPMID:20603174 [本文引用: 2]
Human leukocyte antigen (HLA) typing has been a challenge for more than 50 years. Current methods (Sanger sequencing, sequence-specific primers [SSP], sequence-specific oligonucleotide probes [SSOP]) continue to generate ambiguities that are time-consuming and expensive to resolve. However, next-generation sequencing (NGS) overcomes ambiguity through the combination of clonal amplification, which provides on-phase sequence and a high level of parallelism, whereby millions of sequencing reads are produced enabling an expansion of the HLA regions sequenced. We explored HLA typing using NGS through a three-step process. First, HLA-A, -B, -C, -DRB1, and -DQB1 were amplified with long-range PCR. Subsequently, amplicons were sequenced using the 454 GS-FLX platform. Finally, sequencing data were analyzed with Assign-NG software. In a single experiment, four individual samples and two mixtures were sequenced producing >75 Mb of sequence from >300,000 individual sequence reads (average length, 244 b). The reads were aligned and covered 100%of the regions amplified. Allele assignment was 100%concordant with the known HLA alleles of our samples. Our results suggest this method can be a useful tool for complete genomic characterization of new HLA alleles and for completion of sequence for existing, partially sequenced alleles. NGS can provide complete, unambiguous, high-resolution HLA typing; however, further evaluation is needed to explore the feasibility of its routine use.
URLPMID:22861646 [本文引用: 1]
Current human leukocyte antigen (HLA) DNA typing methods such as the sequence-based typing (SBT) and sequence-specific oligonucleotide (SSO) methods generally yield ambiguous typing results because of oligonucleotide probe design limitations or phase ambiguity for HLA allele assignment. Here we describe the development and application of the super high-resolution single-molecule sequence-based typing (SS-SBT) of HLA loci at the 8-digit level using next generation sequencing (NGS). NGS which can determine an HLA allele sequence derived from a single DNA molecule is expected to solve the phase ambiguity problem. Eight classical HLA loci-specific polymerase chain reaction (PCR) primers were designed to amplify the entire gene sequences from the enhancer-promoter region to the 3 untranslated region. Phase ambiguities of HLA-A, -B, -C, -DRB1 and -DQB1 were completely resolved and unequivocally assigned without ambiguity to single HLA alleles. Therefore, the SS-SBT method described here is a superior and effective HLA DNA typing method to efficiently detect new HLA alleles and null alleles without ambiguity.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:26027777 [本文引用: 1]
Clinical immunogenetics laboratories performing routine sequencing of human leukocyte antigen (HLA) genes in support of hematopoietic cell transplantation are motivated to upgrade to next-generation sequencing (NGS) technology by its potential for cost savings as well as testing accuracy and flexibility. While NGS machines are available and simple to operate, there are few systems available that provide comprehensive sample preparation and data analysis workflows to complete the process. We report on the development and testing of the Integrated Genotyping System (IGS), which has been designed to specifically address the challenges associated with the adoption of NGS in clinical laboratories. To validate the system for a variety of sample DNA sources, we have tested 336 DNA specimens from whole blood, dried blood spots, buccal swabs, and lymphoblastoid cell lines. HLA class I and class II genotypes were derived from amplicon sequencing of HLA-A, -B, -C for exons 1 7 and HLA-DPA1, -DPB1, -DQA1, -DQB1, -DRB1, -DRB3, -DRB4, -DRB5 for exons 1 4. Additionally, to demonstrate the extensibility of the IGS to other genetic loci, KIR haplotyping of 93 samples was carried out in parallel with HLA typing using a workflow based on the HLA system. These results are discussed with respect to their applications in the clinical setting and consequent potential for advancing precision medicine.
URLPMID:23644548 [本文引用: 1]
We present a hierarchical genome-assembly process (HGAP) for high-quality de novo microbial genome assemblies using only a single, long-insert shotgun DNA library in conjunction with Single Molecule, Real-Time (SMRT) DNA sequencing. Our method uses the longest reads as seeds to recruit all other reads for construction of highly accurate preassembled reads through a directed acyclic graph-based consensus procedure, which we follow with assembly using off-the-shelf long-read assemblers. In contrast to hybrid approaches, HGAP does not require highly accurate raw reads for error correction. We demonstrate efficient genome assembly for several microorganisms using as few as three SMRT Cell zero-mode waveguide arrays of sequencing and for BACs using just one SMRT Cell. Long repeat regions can be successfully resolved with this workflow. We also describe a consensus algorithm that incorporates SMRT sequencing primary quality values to produce de novo genome sequence exceeding 99.999% accuracy.
URLPMID:4162275 [本文引用: 1]
While the identification of individual SNPs has been readily available for some time, the ability to accurately phase SNPs and structural variation across a haplotype has been a challenge. With individual reads of up to 30kb in length, SMRT Sequencing technology allows the identification of combinations of mutations such as microdeletions, insertions, and substitutions without any predetermined reference sequence. Long amplicon analysis is a novel protocol that identifies and reports the abundance of differing clusters of sequencing reads within a single library. Graphs generated via hierarchical clustering of individual sequencing reads are used to generate Markov models representing the consensus sequence of individual clusters found to be significantly different. Long amplicon analysis is capable of differentiating between underlying sequences that are 99.9% similar, such as haplotypes and pseudogenes. This protocol allowed for the identification of structural variation in the MUC5AC gene sequence, despite the presence of a gap in the current genome assembly. Long amplicon analysis allows for the elucidation of complex regions otherwise missed by other sequencing technologies, which may contribute to the diagnosis and understanding of otherwise mysterious diseases.
URLPMID:23228053
The human leukocyte antigen (HLA) is key to many aspects of human physiology and medicine. All current sequence-based HLA typing methodologies are targeted approaches requiring the amplification of specific HLA gene segments. Whole genome, exome and transcriptome shotgun sequencing can generate prodigious data but due to the complexity of HLA loci these data have not been immediately informative regarding HLA genotype. We describe HLAminer, a computational method for identifying HLA alleles directly from shotgun sequence datasets (http://www.bcgsc.ca/platform/bioinfo/software/hlaminer webcite). This approach circumvents the additional time and cost of generating HLA-specific data and capitalizes on the increasing accessibility and affordability of massively parallel sequencing.
URLPMID:23748956
Human leukocyte antigen (HLA) typing at the allelic level can in theory be achieved using whole exome sequencing (exome-seq) data with no added cost but has been hindered by its computational challenge. We developed ATHLATES, a program that applies assembly, allele identification and allelic pair inference to short read sequences, and applied it to data from Illumina platforms. In 15 data sets with adequate coverage for HLA-A, -B, -C, -DRB1 and -DQB1 genes, ATHLATES correctly reported 74 out of 75 allelic pairs with an overall concordance rate of 99% compared with conventional typing. This novel approach should be broadly applicable to research and clinical laboratories.
URLPMID:23840783
Correctly matching the HLA haplotypes of donor and recipient is essential to the success of allogenic hematopoietic stem cell transplantation. Current HLA typing methods rely on targeted testing of recognized antigens or sequences. Despite advances in Next Generation Sequencing, general high throughput transcriptome sequencing is currently underutilized for HLA haplotyping due to the central difficulty in aligning sequences within this highly variable region. Here we present the method, HLAforest, that can accurately predict HLA haplotype by hierarchically weighting reads and using an iterative, greedy, top down pruning technique. HLAforest correctly predicts >99% of allele group level (2 digit) haplotypes and 93% of peptide-level (4 digit) haplotypes of the most diverse HLA genes in simulations with read lengths and error rates modeling currently available sequencing technology. The method is very robust to sequencing error and can predict 99% of allele-group level haplotypes with substitution rates as high as 8.8%. When applied to data generated from a trio of cell lines, HLAforest corroborated PCR-based HLA haplotyping methods and accurately predicted 16/18 (89%) major class I genes for a daughter-father-mother trio at the peptide level. Major class II genes were predicted with 100% concordance between the daughter-father-mother trio. In fifty HapMap samples with paired end reads just 37 nucleotides long, HLAforest predicted 96.5% of allele group level HLA haplotypes correctly and 83% of peptide level haplotypes correctly. In sixteen RNAseq samples with limited coverage across HLA genes, HLAforest predicted 97.7% of allele group level haplotypes and 85% of peptide level haplotypes correctly.
URLPMID:25143287
Motivation: The human leukocyte antigen (HLA) gene cluster plays a crucial role in adaptive immunity and is thus relevant in many biomedical applications. While next-generation sequencing data are often available for a patient, deducing the HLA genotype is difficult because of substantial sequence similarity within the cluster and exceptionally high variability of the loci. Established approaches, therefore, rely on specific HLA enrichment and sequencing techniques, coming at an additional cost and extra turnaround time. Result: We present OptiType, a novel HLA genotyping algorithm based on integer linear programming, capable of producing accurate predictions from NGS data not specifically enriched for the HLA cluster. We also present a comprehensive benchmark dataset consisting of RNA, exome and whole-genome sequencing data. OptiType significantly outperformed previously published in silico approaches with an overall accuracy of 97% enabling its use in a broad range of applications.
URLPMID:3819389
Specific HLA genotypes are known to be linked to either resistance or susceptibility to certain diseases or sensitivity to certain drugs. In addition, high accuracy HLA typing is crucial for organ and bone marrow transplantation. The most widespread high resolution HLA typing method used to date is Sanger sequencing based typing (SBT), and next generation sequencing (NGS) based HLA typing is just starting to be adopted as a higher throughput, lower cost alternative. By HLA typing the HapMap subset of the public 1000 Genomes paired Illumina data, we demonstrate that HLA-A, B and C typing is possible from exome sequencing samples with higher than 90% accuracy. The older 1000 Genomes whole genome sequencing read sets are less reliable and generally unsuitable for the purpose of HLA typing. We also propose using coverage % (the extent of exons covered) as a quality check (QC) measure to increase reliability.
URLPMID:4407542
Human leukocyte antigen (HLA) typing from next generation sequencing (NGS) data has the potential for widespread applications. Here we introduce a novel tool (HLAreporter) for HLA typing from NGS data based on read-mapping using a comprehensive reference panel containing all known HLA alleles, followed by de novo assembly of the gene-specific short reads. Accurate HLA typing at high-digit resolution was achieved when it was tested on publicly available NGS data, outperforming other newly developed tools such as HLAminer and PHLAT. HLAreporter can be downloaded from http://paed.hku.hk/genome/.
URLPMID:4331721
Abstract Background Human leucocyte antigen (HLA) genes play an important role in determining the outcome of organ transplantation and are linked to many human diseases. Because of the diversity and polymorphisms of HLA loci, HLA typing at high resolution is challenging even with whole-genome sequencing data. Results We have developed a computational tool, HLA-VBSeq, to estimate the most probable HLA alleles at full (8-digit) resolution from whole-genome sequence data. HLA-VBSeq simultaneously optimizes read alignments to HLA allele sequences and abundance of reads on HLA alleles by variational Bayesian inference. We show the effectiveness of the proposed method over other methods through the analysis of predicting HLA types for HLA class I (HLA-A, -B and -C) and class II (HLA-DQA1,-DQB1 and -DRB1) loci from the simulation data of various depth of coverage, and real sequencing data of human trio samples. Conclusions HLA-VBSeq is an efficient and accurate HLA typing method using high-throughput sequencing data without the need of primer design for HLA loci. Moreover, it does not assume any prior knowledge about HLA allele frequencies, and hence HLA-VBSeq is broadly applicable to human samples obtained from a genetically diverse population.
URLPMID:4477639
The human leukocyte antigen (HLA) complex contains the most polymorphic genes in the human genome. The classical HLA class I and II genes define the specificity of adaptive immune responses. Genetic variation at the HLA genes is associated with susceptibility to autoimmune and infectious diseases and plays a major role in transplantation medicine and immunology. Currently, the HLA genes are characterized using Sanger- or next-generation sequencing (NGS) of a limited amplicon repertoire or labeled oligonucleotides for allele-specific sequences. High-quality NGS-based methods are in proprietary use and not publicly available. Here, we introduce the first highly automated open-kit/open-source HLA-typing method for NGS. The method employs in-solution targeted capturing of the classical class I (HLA-A, HLA-B, HLA-C) and class II HLA genes (HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, HLA-DPB1). The calling algorithm allows for highly confident allele-calling to three-field resolution (cDNA nucleotide variants). The method was validated on 357 commercially available DNA samples with known HLA alleles obtained by classical typing. Our results showed on average an accurate allele call rate of 0.99 in a fully automated manner, identifying also errors in the reference data. Finally, our method provides the flexibility to add further enrichment target regions.
URLPMID:23259685
We present a method, seq2HLA, for obtaining an individual's human leukocyte antigen (HLA) class I and II type and expression using standard next generation sequencing RNA-Seq data. RNA-Seq reads are mapped against a reference database of HLA alleles, and HLA type, confidence score and locus-specific expression level are determined. We successfully applied seq2HLA to 50 individuals included in the HapMap project, yielding 100% specificity and 94% sensitivity at a P-value of 0.1 for two-digit HLA types. We determined HLA type and expression for previously un-typed Illumina Body Map tissues and a cohort of Korean patients with lung cancer. Because the algorithm uses standard RNA-Seq reads and requires no change to laboratory protocols, it can be used for both existing datasets and future studies, thus adding a new dimension for HLA typing and biomarker studies.
URLPMID:26311539 [本文引用: 1]
In the past decade, the development of next-generation sequencing (NGS) has paved the way for whole-genome analysis in individuals. Research on the human leukocyte antigen (HLA), an extensively studied molecule involved in immunity, has benefitted from NGS technologies. The HLA region, a 3.6-Mb segment of the human genome at 6p21, has been associated with more than 100 different diseases, primarily autoimmune diseases. Recently, the HLA region has received much attention because severe adverse effects of various drugs are associated with particular HLA alleles. Owing to the complex nature of the HLA genes, classical direct sequencing methods cannot comprehensively elucidate the genomic makeup of HLA genes. Thus far, several high-throughput HLA-typing methods using NGS have been developed. In HLA research, NGS facilitates complete HLA sequencing and is expected to improve our understanding of the mechanisms through which HLA genes are modulated, including transcription, regulation of gene expression and epigenetics. Most importantly, NGS may also permit the analysis of HLA-omics. In this review, we summarize the impact of NGS on HLA research, with a focus on the potential for clinical applications.
URLPMID:3722289 [本文引用: 2]
The major histocompatibility complex (MHC) is one of the most variable and gene-dense regions of the human genome. Most studies of the MHC, and associated regions, focus on minor variants and HLA typing, many of which have been demonstrated to be associated with human disease susceptibility and metabolic pathways. However, the detection of variants in the MHC region, and diagnostic HLA typing, still lacks a coherent, standardized, cost effective and high coverage protocol of clinical quality and reliability. In this paper, we presented such a method for the accurate detection of minor variants and HLA types in the human MHC region, using high-throughput, high-coverage sequencing of target regions. A probe set was designed to template upon the 8 annotated human MHC haplotypes, and to encompass the 5 megabases (Mb) of the extended MHC region. We deployed our probes upon three, genetically diverse human samples for probe set evaluation, and sequencing data show that 97% of the MHC region, and over 99% of the genes in MHC region, are covered with sufficient depth and good evenness. 98% of genotypes called by this capture sequencing prove consistent with established HapMap genotypes. We have concurrently developed a one-step pipeline for calling any HLA type referenced in the IMGT/HLA database from this target capture sequencing data, which shows over 96% typing accuracy when deployed at 4 digital resolution. This cost-effective and highly accurate approach for variant detection and HLA typing in the MHC region may lend further insight into immune-mediated diseases studies, and may find clinical utility in transplantation medicine research. This one-step pipeline is released for general evaluation and use by the scientific community.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}