 ,1,2,3
,1,2,3Applications of metabonomics in animal genetics and breeding
Meng Zhou1,2,3, Junhong Jing1,2,3, Ruihan Mao1,2,3, Jing Guo1,2,3, Zhipeng Wang,1,2,3通讯作者:
编委: 李明洲
收稿日期:2018-08-8修回日期:2018-10-17网络出版日期:2019-02-25
| 基金资助: |
Editorial board:
Received:2018-08-8Revised:2018-10-17Online:2019-02-25
| Fund supported: |
作者简介 About authors
周萌,硕士研究生,专业方向:动物分子数量遗传学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (592KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
周萌, 景军红, 毛瑞涵, 郭静, 王志鹏. 代谢组学在家养动物遗传育种中的应用[J]. 遗传, 2019, 41(2): 111-124 doi:10.16288/j.yczz.18-226
Meng Zhou, Junhong Jing, Ruihan Mao, Jing Guo, Zhipeng Wang.
代谢组学(metabonomics)是对生物体内所有代谢分子进行定量分析,寻找代谢分子与生理、病理变化的相对关系的研究,是继基因组学和蛋白质组学之后新近发展起来的研究某一时刻细胞内所有代谢分子集合的一门学科[1,2]。与其他组学相比,代谢组学具有以下特点:基因和蛋白质表达的微小变化在代谢分子上得到的放大使代谢组学检测更加容易;代谢组学技术需要相对完整的代谢分子信息库,但其远没有全基因组测序及大量表达序列标签的数据库复杂;代谢分子种类远小于基因和蛋白质的数量,其物质的分子结构也更为简单[3]。
随着高通量测序技术和生物信息学的发展,代谢组学也已成为生物学研究的重要领域,已广泛应用在人类生物医学研究中,并取得了丰硕的成果,特别是在疾病的生物标志物研究、疾病机理研究等方面都展现出巨大的应用潜力与优势[4,5,6,7,8]。但是,在畜牧生产和研究中代谢组学的应用相对较少。根据已发表的相关文献,其应用主要集中在评估胴体品质和牛奶质量、预测饲料效率或剩余饲料摄入量(residual feed intake, RFI)、生殖生理学研究、营养生理学研究、疾病检测的生物标记开发和药物使用检测等非遗传学研究方面。而利用代谢组学开展动物遗传学研究长期被忽视,直到最近以精确、高通量为特点的动物表型组学以及生理基因组学、生理遗传学研究的开展,科研人员在解析动物重要经济性状遗传机制的研究中才更多地使用了代谢组学的技术和方法[9]。
动物基因组上的变异不一定直接对动物表型产生影响,而相关代谢分子的变化可能是该变异的最终结果。换言之,基因组上的变异需通过相关代谢分子的转化而最终传递到表型。这可能是科研人员在将表型和遗传变异直接进行关联分析时,遗漏一些变异位点的原因之一,利用代谢组学数据则可以筛查出这些遗漏的位点[10]。随着代谢组学的快速发展,与其他组学数据的整合研究将有助于解析动物生命活动许多复杂的调控机制。
本文比较了代谢组学检测技术的优缺点,概述了利用代谢组学技术在主要家养动物体液中所检测到的代谢分子的分布情况,综述了关键代谢标志物在遗传育种领域中的应用,以期为进一步利用代谢组学技术开展动物重要经济性状的遗传基础研究提供参考。
1 代谢组学的分类及检测平台
1.1 代谢组学的分类
代谢组学的研究对象大都是相对分子质量1000以内的小分子物质,到目前为止,有114 100个代谢分子被收录到人类代谢组学数据库[11]。根据不同的理化属性可以将代谢组学所包含的物质主要分为氨基酸类(amino acid)、肽类(peptide)、碳水化合物类(carbohydrate)、能量类(energy)、脂类(lipid)、核苷酸(nucleotide)、维生素和辅助因子(cofactors and vitamins)及外源化合物类(xenobiotics);根据代谢分子的产生来源则可以分为内源性代谢分子和外源性代谢分子。根据代谢组学研究策略的不同,代谢组学研究可分为非靶标代谢组学和靶标代谢组学[9],这两种研究策略相辅相成,从发现到验证,从“无假设”到“假设驱动”,共同组成了代谢组学研究[12]。代谢组学包含物质及研究策略的分类特点见表1。Table 1
表1
表1 代谢组学包含物质及研究策略的分类特点
Table 1
| 分类类别 | 种类 | 特点 |
|---|---|---|
| 理化属性 | 氨基酸类 | 主要包括参与甘氨酸、丝氨酸和苏氨酸代谢,丙氨酸和天门冬氨酸代谢,谷氨酸代谢,组氨酸代谢,赖氨酸代谢,苯丙氨酸和酪氨酸代谢,色氨酸代谢,缬氨酸、亮氨酸和异亮氨酸代谢,半胱氨酸、蛋氨酸、牛磺酸代谢,精氨酸类和脯氨酸类代谢,以及尿素循环等代谢过程的小分子 |
| 肽类 | 主要包括参与二肽和γ-谷氨酰等相关代谢过程的小分子 | |
| 碳水化合物类 | 主要包括参与果糖、甘露糖、半乳糖、淀粉和蔗糖、核苷酸糖、戊糖等物质代谢,以及糖酵解、糖异生和丙酮酸代谢过程的小分子 | |
| 能量类 | 主要包括参与三羧酸循环和氧化磷酸化等相关代谢过程的小分子 | |
| 脂类 | 主要包括参与必需脂肪酸、中链脂肪酸、长链脂肪酸、肉碱、胆汁酸、甘油酯、溶血磷脂、单酰甘油、鞘脂类和固醇/甾体等相关代谢过程的小分子 | |
| 核苷酸 | 主要包括参与嘌呤或次黄嘌呤或肌苷、嘧啶或胸腺嘧啶、缬氨酸、亮氨酸和异亮氨酸等相关代谢过程的小分子 | |
| 维生素和辅助因子 | 主要包括参与抗坏血酸盐和醛酸盐、血红蛋白与卟啉、泛酸和辅酶A和生育酚等相关代谢过程的小分子 | |
| 外源化合物 | 主要包括外源化学分子、药物等小分子物质 | |
| 产生来源 | 内源性代谢分子 | 直接由机体产生,分为初级代谢分子(磷酸糖、氨基酸、核苷酸、有机酸等)和次级代谢分子(由初级代谢分子产生,如激素、脂类、植物素等) |
| 外源性代谢分子 | 来源于体外的化学混合物,部分物质参与体内代谢;主要有药物分子、药物代谢分子等 | |
| 研究策略 | 靶标代谢组学 | 从定量角度研究样品中特定代谢分子的生物学意义,是一种“假设驱动”的代谢组学研究 策略[12] |
| 非靶标代谢组学 (或发现代谢组学) | 主要检测样品中代谢分子的相对含量。尽可能多的同时测量样本中包含的所有代谢分子,是从定性和半定量角度分析代谢产物的生物学意义 |
新窗口打开|下载CSV
1.2 代谢组学检测平台
目前用于开展高通量代谢组学研究最常见的仪器平台主要包括核磁共振仪(nuclear magnetic resonance, NMR)、质谱仪(mass spectrometer, MS)、色谱-质谱联用仪(chromatograph-mass spectrometer, C-MS)。Goldansaz等[13]详细介绍了上述检测技术平台的特性(图1)及其应用。NMR是最早应用于代谢组学研究的高通量分析技术,其优势在于对样品检测前的预处理较为简单,且对样品无破坏性;其缺点在于检测灵敏度较低,只能检测μmol/L~mmol/L浓度的代谢分子。质谱技术具有高灵敏度和专属性等优势,能够检测nmol/L~pmol/L浓度的代谢分子。近年来,代谢组学研究普遍采用的方法是色谱-质谱联用技术,主要包括气相色谱-质谱(gas chromatography- mass spectrometry, GC-MS)和液相色谱-质谱(high performance liquid chromatography-mass spectrometry, LC-MS)联用技术。该技术的优势在于检测覆盖率达到前所未有的高度,可以检测到更多的小分子代谢产物,包括糖、糖醇、氨基酸、有机酸、脂肪酸和芳胺,以及包括大量次级代谢分子在内的数百种化学性质不同的化合物[14]。由于GC-MS需要对挥发性较低的代谢分子进行衍生化预处理,样品制备较为繁琐,甚至会引起样品的变化,因此与LC-MS相比,GC-MS没有被普遍使用。图1
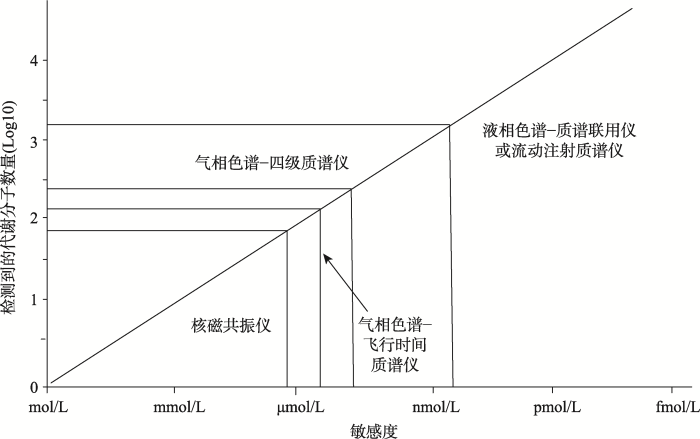 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1不同代谢组学检测平台的敏感度
Fig. 1Relative sensitivity of metabolomics platforms
根据文献[13]修改绘制。
2 代谢组学技术检测家养动物体液中关键代谢分子
在家养动物代谢组学研究中,主要是利用核磁技术和质谱技术测定血浆、血清、乳汁、尿液、粪便、瘤胃、肌肉、脂肪等样品的代谢分子。其中对血浆、血清、乳汁中的代谢组学研究最多,所检测到的代谢分子也最多。基于目前报道的检测结果,Goldansaz等[13]将在不同物种、不同样品中通过代谢组学技术所检测到的1070个代谢分子收录在LMDB数据库,该数据库中除了收集每个代谢分子的理化特性以外,重点收录了特定物种的特定样品内该代谢分子的标准化浓度等注释信息。通过检索该数据库,在不同动物的血浆中累计检测到408个代谢分子,在血清中累计检测到351个代谢分子,在乳汁中累计检测到422个代谢分子。上述检测到的代谢分子约70%来自牛和猪的组织或体液,在不同年份猪和牛各种组织或体液中所检测到代谢分子的分布情况见图2。随着敏感度更高的检测设备在动物代谢组学中的应用,动物各种组织或体液中的代谢分子将会被陆续检测出来。图2
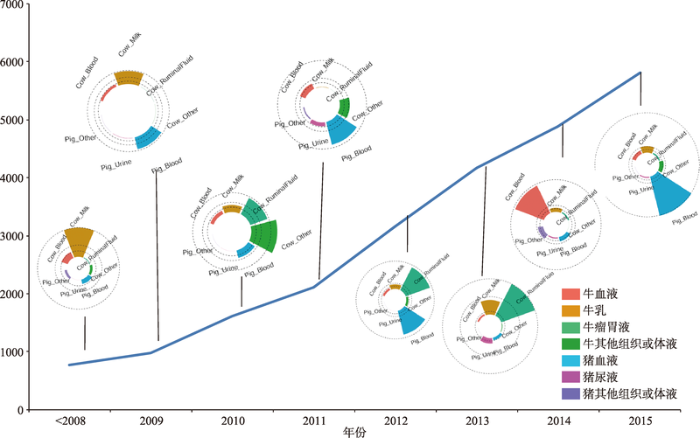 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2在不同年份牛和猪中检测到的代谢分子数
Fig. 2The timeline of metabolites detected in cattle and pig
数据来自于LMDB数据库。X轴表示年份,Y轴表示每年度在牛和猪不同组织或体液上检测到的代谢分子总数;每年度的圈图表示该年度在牛和猪不同组织或体液中检测到的代谢分子的分布情况,其中扇面颜色表示所检测的组织或体液类型,扇面的面积大小代表检测到的代谢分子数。
3 家养动物关键代谢标志物在遗传育种中的应用
如前所述,在家养动物上利用代谢组学的研究手段开展复杂性状遗传学机制的研究较少。目前所开展的相关研究主要集中在代谢分子遗传参数估计、筛选能够鉴别不同品种(系)的生物标记、代谢分子全基因组关联分析研究(genome-wide association studies with metaotypes, mGWAS)以及寻找代谢分子与重要经济性状的关系等方面。为了系统地解析乳品质或肉品质等相关性状的遗传学机制,目前对牛和猪的研究较多。相对而言,在其他家养动物上利用代谢组学开展遗传育种方面的研究较少,其研究多集中于营养生理学研究、疾病检测的生物标记开发和药物或食品添加剂使用效果检测等非遗传学研究。3.1 动物代谢分子遗传参数估计
机体内的代谢水平会随时间而发生变化,更易受到环境和生活习惯的影响。因此,每个生物样品的代谢组学图谱仅仅是刻画了该个体在特定时间的代谢状态。在将代谢分子的表达量作为中间性状开展遗传学分析时,需要充分了解遗传因素解释这些代谢分子表型变异的份额,即需要对代谢分子表达水平开展遗传参数估计工作。如果期望将鉴定的代谢分子应用于动物遗传评估,估计这些代谢分子的遗传参数则更是一项必要的研究工作。在牛、猪和鸡等家养动物群体中,科研人员已经系统地估计了各类代谢分子的遗传参数。对牛的代谢分子估计遗传参数的研究较多。Soyeurt等[15]检测了瑞士褐牛(Brown Swiss)、比利时蓝牛(Dual-Purpose Belgian Blue)、荷斯坦-弗里斯牛(Holstein Friesian)、泽西牛(Jersey)、蒙贝利亚牛(Montbeliarde)、诺曼地牛(Normande)等总计7700个牛乳样品,分析发现牛乳中不同长链脂肪酸代谢分子的遗传力在0.05~0.38之间,它们之间的遗传相关在-0.06~0.84之间。Stoop等[16]估计了1953只荷兰荷斯坦(Holstein)黑白花母牛尿素氮的遗传力,分析发现该代谢分子为低遗传力性状(h2=0.14)。Oikonomou等[17]对初次泌乳奶牛的血清代谢分子开展了遗传参数估计,研究发现葡萄糖的遗传力为0.12~0.39,β-羟基丁酸酯为0.08~0.40,非酯化脂肪酸为0.08~ 0.35。Nogi等[18]对日本黑牛(Japanese Black cattle)进行了研究,单不饱和脂肪酸、饱和脂肪酸和多不饱和脂肪酸遗传力估计值分别为0.68、0.66和0.47。Buitenhuis等[19]对456只丹麦荷斯坦奶牛和436只丹麦泽西牛的牛乳开展了代谢分子的遗传力估计,其中乳清酸和β-羟基丁酸的遗传力均大于0.8。Wittenburg等[20]利用GC-MS测定了1295头奶牛乳中190种代谢分子的遗传力,发现这些代谢分子的广义遗传力为0~0.699,中位数为0.125。Gebreyesus等[21]对650头丹麦荷斯坦奶牛主要乳蛋白的遗传力进行估计,发现不同乳蛋白的遗传力为0.05~0.78。
在对猪的研究中,重点估计了与肉质性状相关的各种长链脂肪酸的遗传力。Ntawubizi等[22]估计猪肉肌内脂肪酸的组成和参与多不饱和脂肪酸代谢的去饱和酶和延长酶活性指标的遗传参数,发现长链多不饱和脂肪酸的遗传力值通常在0.50以上。Ibá?ez-Escriche等[23]对伊比利亚猪(Iberian pig)皮下脂肪组织的不同长链脂肪酸的遗传力进行估计,发现这些脂质代谢分子的遗传力范围为0.06~0.53。
另外,科研人员对特定几项鸡血液生化指标开展了遗传参数估计研究。Dong等[24]以肉鸡高、低脂双向选择品系为实验材料,估计了多项血液生化指标的遗传参数,研究发现总胆汁酸、肌酐、低密度脂蛋白胆固醇的遗传力为0.60~0.85。Zhang等[25]测定了332只广西黄鸡进食和禁食状况下的血液生化指标以及腹部脂肪性状,研究发现,在进食状态下,甘油三酯、总胆固醇等血液生化指标的遗传力较高,从0.26~0.60不等,在禁食状态下,这些血液生化指标的遗传力为0.22~0.59。
3.2 动物代谢分子在品种(系)中的鉴定
在对牛、猪和鸡等家养动物的代谢组学研究中,通过检测动物血清、组织、乳汁等研究材料,筛选出一些代谢分子作为区分品种(系)的生物标记物。目前,已有多篇文献报道了牛、猪、鸡的不同品种(系)间存在显著差异表达的代谢分子,这些代谢分子可以作为品种(系)鉴定的生物标记。例如:Karisa等[26]利用NMR技术检测了纯种安格斯牛(Augus)和杂交牛(58.3%安格斯、30.6%西门塔尔(Simmemal)和11.1%海福特牛(Hareford)等欧系肉牛)的血浆代谢图谱,发现肌酸、肉碱、马尿酸等代谢分子的表达在两个品种间差异显著。D'Alessandro等[27]分析发现可以用甘油-3-磷酸脱氢酶、甘油3-磷酸和甘油代谢分子来区分高脂的卡斯塔纳猪(Casertana)和瘦肉型大白猪(Large White);He等[28]用代谢组学方法比较肥胖型猪(宁乡品系)和瘦肉型杂交猪间的血清代谢图谱,发现宁乡品系猪血清的胰岛素、胰高血糖素、脂质、不饱和脂质、糖蛋白、肌肉肌醇、丙酮酸盐、苏氨酸、酪氨酸和肌酸均显著高于瘦肉型猪,而血清中葡萄糖和尿素则低于瘦肉型猪;Straadt等[29]通过NMR技术,检测了杜洛克/长白/约克夏(Duroc/ Landrace/Yorkshire)、伊比利亚/杜洛克(Iberian/Duroc)、伊比利亚/杜洛克/长白(Iberian/Duroc/Landrace)、曼加利萨/杜洛克(Mangalitza/Duroc)和曼加利萨/长白/约克夏(Mangalitza/Landrace/Yorkshire)5个杂交组合的猪背最长肌的代谢分子,发现氨基酸(丙氨酸、肌肽、异亮氨酸、甲硫氨酸、苯丙氨酸和缬氨酸)、乳酸盐、肌苷酸、肌苷、甘油和含胆碱的化合物等可以区分这些杂交猪;Bovo等[30]利用质谱技术检测了大白猪和杜洛克猪的血浆、血清,共检测到180种代谢分子,其中鞘磷脂在杜洛克猪中表达量较高,乙酰鸟氨酸在大白猪中表达量较高。Ji等[31]利用LC-MS检测来航鸡(Leghorn)、Fayoumi和商业肉鸡的脂肪组织,共检测到92种代谢分子,与商业肉鸡相比,Fayoumi和来航鸡脂肪组织中的肉碱、鸟苷、胞嘧啶、腺苷和磷酸戊糖含量显著增加,来航鸡脂肪组织中的3-磷酸甘油酸和磷酸烯醇丙酮酸含量较高;Baéza等[32]利用H-NMR技术,通过比较饲喂同样日粮、同一日龄的腹脂双向选择系公鸡的血浆,发现谷氨酰胺、组氨酸、甜菜碱等代谢分子的表达在两系间差异显著。3.3 动物代谢分子的全基因组关联分析
基于基因组学研究技术,特别是全基因组SNP标记检测平台,将血清、血液、尿液等体液所检测到的代谢分子表达量作为表型值,开展了代谢分子全基因组关联分析,这是将代谢组学与基因组学耦合在一起的关键,也是目前代谢组学领域以遗传学研究为落脚点,鉴定代谢分子功能与遗传调控方面最主要的研究进展之一。从已有的文献报道可以发现,人类代谢组学mGWAS研究结果极为丰富,且已经筛选到多个与代谢分子表达显著相关的遗传标记或基因,通过进一步的分析发现这些代谢分子可以作为人类疾病的生物标记。家养动物mGWAS的报道相对较少,主要集中在牛和猪上,其目的是通过对某一类代谢分子的全基因组关联分析筛选出影响肉质或乳质的遗传标记或重要候选基因。例如,已筛选出DGAT1、FASN、SCD、ELOVL家族等重要候选基因与牛乳液和肌肉中各种不同链长的脂肪酸含量显著相关;筛选出FASD家族、ELOVL家族等重要候选基因与猪最长肌中脂肪酸含量显著相关。其他研究结果详见表2。在动物群体上开展mGWAS的样本量规模都比人类研究群体的规模小很多,这是由于:一方面,在饲养过程中可以有效的控制饲养环境,使动物所受到的环境因素趋于一致;另一方面,试验群体的遗传背景相似。Table 2
表2
表2 牛、猪、鸡mGWAS研究结果汇总
Table 2
| 物种 | 动物群体 | 样品 | 代谢分子(类别) | 重要候选基因 | 文献 |
|---|---|---|---|---|---|
| 牛 | 荷斯坦牛 | 牛乳 | 短链、中链脂肪酸 | DGAT1、SCD1、PPARGC1A和FASN | [33] |
| 丹麦奶牛 | 牛乳 | 中链、长链脂肪酸 | ABCG2、PPARGC1A、ACSS2、DGAT1、ACLY、SREBF1、STAT5A、GH、FASN、SCD1和AGPAT6 | [34] | |
| 荷斯坦牛 | 牛乳 | 丙二酸、半乳糖-1-磷酸、顺式-乌头酸酯、肉碱、甘油磷酸胆碱 | APORB | [19] | |
| 荷斯坦牛 | 牛乳 | 维生素B12 | LRP2、MMADHC、CD320、LMBRD1、CUBN、GIF、TCN1、MMAA、MMAB、TCN2、MTRR、AMN、MUT、ABCC1和MTR | [35] | |
| 荷斯坦牛和娟姗牛 | 牛乳 | 中链、长链脂肪酸 | DGAT、SCD和ACSS3 | [36] | |
| 中国荷斯坦牛 | 牛乳 | 中链、长链脂肪酸 | FASN、IGF1、PPARGC1A、ABCG2、SCD、HTR1B、CPM、PRKG1、MINPP1、LIPJ、LIPK、EHHADH、MOGAT1、ECHS1、STAT1、SORBS1、NFKB2、AGPAT3、CHUK、OSBPL8、PRLR、IGF1R、ACSL3、GHR和OXCT1 | [37] | |
| 瑞士褐牛和 荷斯坦牛 | 牛乳 | 葡萄糖 | UEVLD | [38] | |
| β-羟基丁酸酯 | DNAJC30和WBSCR22 | ||||
| 非酯化脂肪酸 | SANI2、UGT2B15和MGC152010 | ||||
| 丹麦荷斯坦牛、 中国荷斯坦牛 | 牛乳 | 中链、长链脂肪酸 | SEMA5B、AGPAT3、DGAT1、SREBF1、FASN、SCD1和GHR | [39] | |
| 荷斯坦牛和娟珊牛 | 牛乳 | 核黄素 | SLC52A3 | [40] | |
| 荷斯坦牛 | 牛乳 | 甘油磷酸胆碱 | APORB | [41] | |
| 瑞士褐牛 荷斯坦牛 | 牛乳 牛乳 | 短链、中链脂肪酸 | LEP、PRL、STAT5A、CCL3、 ACACA、GHR、ADRB2、LPIN1、 STAT1、FABP4和CSN2 | [42,43] | |
| 短链脂肪酸 | LARP1B和PKL4 | ||||
| 荷斯坦牛和娟姗牛 | 牛乳 | 乳糖 | ABCG2、DGAT1、STAT5B、KCNH4、NPFFR2和RNF214 | [44] | |
| 日本黑牛 | 最长肌 | 中链、长链脂肪酸 | FASN和SCD | [45] | |
| 安格斯牛 | 最长肌 | 中链、长链脂肪酸 | FASN、SCD、THRSP、 SREBP1、ACACA、PPARG、FABP4、ACSL1、LEP和LXRA | [46] | |
| 安格斯牛 | 最长肌 | 磷脂 | PFKFB2和CDH2 | [47] | |
| 三酰基甘油 | FASN、THRSP、SCD和INPP4B | ||||
| 内洛尔肉牛 | 最长肌 | 中链、长链脂肪酸 | ELOVL5、ESSRG、PCYT1A、ABC5、ABC6和ABC10 | [48] | |
| 日本黑牛 | 最长肌 | 果糖 | PMP22和HS3ST3B1 | [49] | |
| 中链、长链脂肪酸 | FASN和SCD | ||||
| 日本黑牛 | 最长肌 | 牛磺酸 | SLC6A6、RAB7A、RPN1和CCDC12 | [50] | |
| 肌苷、次黄嘌呤 | NT5E | ||||
| 中国西门塔尔牛 | 最长肌 | 中链脂肪酸 | FASN和ELOVL5 | [51] | |
| 日本黑牛 | 最长肌 | 油酸 | VNN1和LYPLA1 | [52] | |
| 日本黑牛 | 最长肌 | 丙氨酸 | STT3B、SUV420H1、CPT1A、MRPL21和IGHMBP2 | [53] | |
| 安格斯牛 | 最长肌、 皮下脂肪 | 中链、长链脂肪酸 | FASN、SCD、THRSP和GCLC | [54] | |
| 物种 | 动物群体 | 样品 | 代谢分子(类别) | 重要候选基因 | 文献 |
| 牛 | 日本黑牛 | 肌间脂肪 | 油酸 | FMNL1、FASN、HRNBP3和SMURF2 | [55] |
| 内洛尔肉牛 | 肌间脂肪 | 不同链长脂肪酸 | SLITRK6、DHRS7、NUP214、SREBP-SCAP、GNG11、RGS5、WARS2、HMGCS2和PHGDH、HSD3B1、HAO2、GAD1、Sp5、ABCG5、GPC6、GPC4、MGCS2, PHGDH、RAPGEF2、AQP7、RORA、LOXL2、SPAG17和WDR3 | [56] | |
| 韩牛 | 肌间脂肪 | 肉豆蔻酸、油酸 | CCDC57和FASN | [57] | |
| 夏洛莱牛×德国 荷斯坦牛F2群体 | 血清 | 精氨酸 | NCAPG | [58] | |
| 猪 | 伊比利亚猪×长白 | 背最长肌 | 中链、长链脂肪酸 | DECR1、FABP4、FABP5、APOA2、USF1、FAS、MTTP、CYP2U1、PLA2G12A、PLA2、HADH、AACS和ELOVL7 | [59] |
| 伊比利亚猪×长白 | 背最长肌 | 中链、长链脂肪酸 | LDLR, LIPG、ELOVL6, MGST2 KIT、RDH16和NUDT7 | [60] | |
| 杜洛克× 皮特兰杂交猪 | 背最长肌 | 三羧酸循环中间 代谢产物 | PIK3C3、TTLL5、PTPRT、VAPB ANK3、RASGEF1A、SAMD4A、LRGUK、AKT3、ENPP3、CREB3L2、NFE2L3、HLCS、NTNG1、GBP4、PKN2、ZNHIT6、DDAH1和WDR63 | [61] | |
| 杜洛克×二花脸?F2、 杜洛克× (长白× 约克夏)、苏太猪、 二花脸猪和莱芜猪 | 背最长肌 | 不同链长脂肪酸 | FADS2、SREBF1和PLA2G7 | [62] | |
| 杜洛克 | 背最长肌 | 中链、长链脂肪酸 | NDUFC2、FASN、ACO、RANBP9、PSMD1、WNT8B、APBB2、ROBO2、ADGRL2、LIN7A、ZNF37A和TENM2 | [63] | |
| 八马香猪 | 背最长肌 | 长链脂肪酸 | FADS2、FADS1、ABCD2、ELOVL7 ACSBG1、ELOVL7和ACOX2 | [64] | |
| 二花脸猪 | 背最长肌 | 长链脂肪酸 | FASN、ELOVL5、ELOVL6、ELOVL7、ABCD3, ABCA4和FADS2 | ||
| 杜洛克 | 背最长肌 | 不同链长脂肪酸 | GBF1、SCD、CUEDC2、NFKB2、HPS6、ELOVL3、FBXW4、BTRC、TMEM180、ACTR1A、SUFU、TRIM8、ABCC2 PAX2、HPSE2、DNMBP和HOGA1 | [65] | |
| 伊比利亚猪×长白 | 背最长肌、背膘 | 棕榈油酸 | ELOVL6 | [66] | |
| 杜洛克×二花脸F2和 苏太猪 | 背最长肌、腹脂 | 长链脂肪酸 | ADIPOR2、ABCD2、PPARD、HMGA1、ACSBG1、ELOVL7和SCD | [67] | |
| 杜洛克 | 皮下脂肪 | 不同链长脂肪酸 | CPN1、PKD2L1 、PAX2、ENTPD7和SEMAG4 | [68] | |
| 长白猪 | 皮下脂肪 | 棕榈油酸 | ELOVL6 | ||
| 意大利大白猪 | 背膘组织 | 不同链长脂肪酸 | ELOVL6、ACSBG1、IDH3A、SCD、ELOVL3、APBB1IP、ADIPOR2、PNLIPRP1、PNLIPRP2、PNLIP、NLIPRP3、ME3、MTMR3、INPP5J、PLA2G3和PISD | [69] | |
| 公猪 | 颈下脂肪 | 雄甾酮 | SULT2A1、SULT2B1、HSD17B14和CYP2A19 | [70] | |
| 鸡 | Fayoumi鸡 | 血浆 | 葡萄糖 | TPGS2 | [71] |
| 伊朗Urmia鸡× AA鸡构成F2 | 血浆 | 甘油三酯 | DOCK10和AP1S3 | [72] |
新窗口打开|下载CSV
3.4 与动物重要经济性状相关的代谢分子鉴定
代谢组学所反映的小分子物质的产生和代谢能更直接、准确地反映生物体的生理状态和生理表型,是复杂表型的分子展现。相对于表型数据而言,代谢分子更容易进行标准化和批量化的分析检测,所检测得到的数据更为准确。筛选与重要经济性状或疾病相关的代谢分子是代谢组学研究的重要热点。在人类代谢组学研究方面,通过比较对照与处理组之间的代谢组学差异筛选出表征疾病的生物标记,或通过将代谢组学和其他组学整合分析筛选出影响复杂疾病发生、发展的重要代谢分子。
在动物群体中,目前主要是利用研究代谢分子与重要经济性状之间的相关性或差异分析来筛选生物标记物,育种者也期望能够在育种实践中利用这些筛选到的代谢分子作为生物标记物,并由此应用于标记辅助选择中,从而提高选择的准确性。目前,一些代谢分子已被筛选出来作为奶牛产奶性状或疾病的生物标记;尿碳酸酐酶-Ⅵ等代谢分子可以作为猪肾病诊断的生物标记;甘油磷脂等代谢分子可以作为鸡腹水综合征的生物标记。其他相关研究结果详见表3。
Table 3
表3
表3 牛、猪、鸡代谢分子与重要经济性状之间的关系
Table 3
| 物种 | 性状 | 动物群体 | 样品 | 代谢分子 | 文献来源 |
|---|---|---|---|---|---|
| 牛 | 亚临床酮症 | 荷斯坦牛 | 血浆和牛乳 | β-羟基丁酸酯 | [73] |
| 产奶相关性状 | 荷斯坦牛 | 牛乳 | 尿素氮 | [16] | |
| 鱼腥味性状 | 瑞典奶牛 | 牛乳 | 三甲胺 | [74] | |
| 牛生长性状和 体脂性状 | 夏洛来× 德国荷斯坦F2群体 | 肌肉 | 精氨酸和肉碱 | [75] | |
| [58] | |||||
| 乳腺炎 | 荷斯坦牛 | 牛乳 | 乳酸 | [76] | |
| 酮体耐受性 | 荷斯坦牛 | 牛乳 | 甘油磷酸胆碱 | [41,77] | |
| 产奶量和 乳蛋白量 | 荷斯坦牛 | 瘤胃液、牛乳和 血清和尿液 | 参与甘氨酸、丝氨酸、苏氨酸、酪氨酸和 苯丙氨酸代谢通路的代谢分子 | [78] | |
| 牛胰岛素 功能紊乱 | 荷斯坦牛和 西门塔尔牛 | 血清 | 血清磷脂浓度与长链鞘磷脂含量 | [79] | |
| 剩余采食量 | 夏洛来牛 | 血清 | 甘氨酸和肌氨酸 | [80] | |
| 饲料转化率 | 天冬氨酸、肌肽和丝氨酸 | ||||
| 胎盘滞留 | 荷斯坦牛 | 血清 | 乙酰鸟氨酸、赖氨酸、天冬酰胺和亮氨酸 | [81] | |
| 公牛繁殖力 | 荷斯坦牛 | 精液 | 2-羟基戊二酸和果糖 | [82] | |
| 猪 | 瘦肉率、 日采食量 | 大白、长白和 皮特兰 | 血清 | 肌酐、肌酸、胆碱、胆碱磷酸或甘油磷酸胆碱、 谷氨酰胺、乳酸、丙氨酸和异亮氨酸 | [83] |
| 肾脏疾病 | 家猪 | 猪尿液 | 尿碳酸酐酶-VI | [84] | |
| 肉滴水损失 | 杜洛克× 皮特兰杂交猪 | 背最长肌 | 甘氨酸和磷酸甘油酸变位酶2 | [61] | |
| 鸡 | 鱼腥味性状 | 肉鸡 | 鸡蛋 | 三甲胺 | [85] |
| 腹水综合征 | Ross-308 | 血清 | 牛磺脱氧胆酸、胆酸葡萄糖醛酸和甘氨胆酸 | [86] | |
| 鸡胸肉质 | 商品肉鸡 | 肌肉 | 次黄嘌呤、黄嘌呤和尿酸 | [87] | |
| 腹水综合征 | Ross-308 | 血清 | 甘油磷脂 | [88] | |
| 鸡胸肉质 | 商品肉鸡 | 肌肉 | 鹅脚碱、肌肽和肌酸 | [89] | |
| 消化效率 | 高、低消化 效率杂交鸡 | 血清 | 脯氨酸 | [90] | |
| 回肠 | 延胡索酸 | ||||
| 盲肠 | 葡萄糖 | ||||
| 早期胚胎发育 | Ross-308 | 血清 | 谷氨酰胺、苏氨酸和雌酮 | [91] |
新窗口打开|下载CSV
随着家养动物基因组数据的积累,研究者开始整合分析代谢组学与基因组、转录组、表观组、表型组的数据,尝试构建出“遗传标记或基因-代谢分子-表型”的关系网络,从而筛选出相关的生物标记,同时进一步解析相关性状的遗传机制。如Weikard等[75]整合肉牛基因组、代谢分子和生产性能数据,发现“NCAPG基因、GDF8基因-精氨酸、肉碱-牛生长性状和体脂性状”的通路,从而推测精氨酸、肉碱可以作为生长性状和体脂性状的生物标记物;Tetens等[47]通过整合基因组、代谢组学数据,发现“APORB基因-甘油磷酸胆碱-酮体耐受性”的关系;Ha等[38]通过比较产犊前3周、产犊后4周、产犊后13周3个时间点的代谢产物,结合GWAS和基因组富集分析,筛选出参与脂质和类固醇代谢的代谢分子与奶牛哺乳早期代谢适应性性状相关;Widmann等[58]和Weikard等[75]整合表型、代谢组学和基因组学数据推断NCAPG基因上的突变位点,通过调控精氨酸代谢而影响NO通路,从而促进血管平滑肌收缩,最终影响牛的生长;Lundén等[74]发现了“FMO3基因-三甲胺-鱼腥味性状”的关系,Chu等[92]利用此关系在北京油鸡群体中剔除FMO3基因有害等位基因,从而培育出没有鱼腥味性状的北京油鸡。
4 结语与展望
目前,在人和家养动物群体中,代谢组学已广泛应用于遗传学、功能基因组学、疾病预测、药物设计等领域[93]。同时,代谢组学的研究也为动物育种技术开辟了新思路,如利用特定生理状态的标记代谢分子对动物进行选育,或利用代谢组学信息理解动物潜在的遗传差异。随着代谢组学技术的日趋臻熟,该技术将会更为广泛地应用于牛、猪、鸡等家养动物重要经济性状的选育上,而且将会有助于阐明复杂的生物学机制问题。当然,代谢组学研究还有很多亟待解决的问题。例如,生物体代谢产物的变化除受生理刺激和遗传因素影响,与环境因素也密切相关,所以在代谢组学样品收集和实验设计时应该考虑代谢对环境条件的敏感性;其次,目前已知的代谢组学技术平台在仪器的灵敏度、检测的覆盖度上仍存在一定的局限性,检测到的所有代谢分子可能也只代表了基因变异的一小部分,所以应该提高代谢组学检测技术的灵敏性、稳定性、广谱性和特异性;另外,代谢组学研究产生的海量高维数据,需要利用多元统计、生物信息等多种数据挖掘技术进行分析,代谢组学数据的分析是代谢组学研究的重要组成部分之一,但目前相关的研究方法还不够成熟,仍需要进一步的完善。
随着代谢组学数据的积累,将基因组、转录组、表观组、蛋白组与代谢组学、表型组整合分析将成为今后的研究热点,代谢组学信息的使用和发展也将有助于各种组学数据的整合分析。例如通过整合分析,筛选出影响重要经济性状的“基因-蛋白质-代谢分子-表型”网络或通路;通过将代谢组学和表观遗传组整合分析,发现某些代谢分子与染色质活跃区域的关系,从而更全面地解析机体内的代谢分子遗传学机制。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
URLPMID:3446258 [本文引用: 1]

Increasingly sophisticated measurement technologies have allowed the fields of metabolomics and genomics to identify, in parallel, risk factors of disease; predict drug metabolism; and study metabolic and genetic diversity in large human populations. Yet the complementarity of these fields and the utility of studying genes and metabolites together is belied by the frequent separate, parallel applications of genomic and metabolomic analysis. Early attempts at identifying co-variation and interaction between genetic variants and downstream metabolic changes, including metabolic profiling of human Mendelian diseases and quantitative trait locus mapping of individual metabolite concentrations, have recently been extended by new experimental designs that search for a large number of gene-metabolite associations. These approaches, including metabolomic quantitiative trait locus mapping and metabolomic genome-wide association studies, involve the concurrent collection of both genomic and metabolomic data and a subsequent search for statistical associations between genetic polymorphisms and metabolite concentrations across a broad range of genes and metabolites. These new data-fusion techniques will have important consequences in functional genomics, microbial metagenomics and disease modeling, the early results and implications of which are reviewed.
[本文引用: 1]
URLPMID:24740590 [本文引用: 1]
Abstract PURPOSE: Childhood obesity is an increasing problem and is accompanied by metabolic disturbances. Recently, we have identified 14 serum metabolites by a metabolomics approach (FIA-MS/MS), which showed altered concentrations in obese children as compared to normal-weight children. Obese children demonstrated higher concentrations of two acylcarnitines and lower levels of three amino acids, six acyl-alkyl phosphatidylcholines, and three lysophosphatidylcholines. The aim of this study was to analyze whether these alterations normalize in weight loss. METHODS: We analyzed the changes of these 14 metabolites by the same metabolic kit as in our previous study in serum samples of 80 obese children with substantial weight loss (BMI-SDS reduction >0.5) and in 80 obese children with stable weight status all participating in a 1-year lifestyle intervention. RESULTS: In the children without weight change, no significant changes of metabolite concentrations could be observed. In children with substantial weight loss, glutamine, methionine, the lysophosphatidylcholines LPCaC18:1, LPCaC18:2, and LPCa20:4, as well as the acyl-alkyl phosphatidylcholine PCaeC36:2 increased significantly, while the acylcarnitines C12:1 and C16:1, proline, PCaeC34:1, PCaeC34:2, PCaeC34:3, PCaeC36:3, and PCaeC38:2 did not change significantly. CONCLUSIONS: The changes of glutamine, methionine, LPCaC18:1, LPCaC18:2, LPCa20:4, and PCaeC36:2 seem to be related to the changes of dieting or exercise habits in lifestyle intervention or to be a consequence of overweight since they normalized in weight loss. Further studies should substantiate our findings.
[本文引用: 1]
URLPMID:26196272 [本文引用: 1]
Until recently, the study of mycobacterial diseases was trapped in culture-based technology that is more than a century old. The use of nucleic acid amplification is changing this, and powerful new technologies are on the horizon. Metabolomics, which is the study of sets of metabolites of both the bacteria and host, is being used to clarify mechanisms of disease, and can identify changes leading to better diagnosis, treatment, and prognostication of mycobacterial diseases. Metabolomic profiles are arrays of biochemical products of genes in their environment. These complex patterns are biomarkers that can allow a more complete understanding of cell function, dysfunction, and perturbation than genomics or proteomics. Metabolomics could herald sweeping advances in personalized medicine and clinical trial design, but the challenges in metabolomics are also great. Measured metabolite concentrations vary with the timing within a condition, the intrinsic biology, the instruments, and the sample preparation. Metabolism profoundly changes with age, sex, variations in gut microbial flora, and lifestyle. Validation of biomarkers is complicated by measurement accuracy, selectivity, linearity, reproducibility, robustness, and limits of detection. The statistical challenges include analysis, interpretation, and description of the vast amount of data generated. Despite these drawbacks, metabolomics provides great opportunity and the potential to understand and manage mycobacterial diseases.
URLPMID:25693144 [本文引用: 1]
The metabolic profiles of breast cancer cells are different from normal mammary epithelial cells. Breast cancer cells that gain resistance to therapeutic interventions can reprogram their endogenous metabolism in order to adapt and proliferate despite high oxidative stress and hypoxic conditions. Drug resistance in breast cancer, regardless of subgroups, is a major clinical setback. Although recent advances in genomics and proteomics research has given us a glimpse into the heterogeneity that exists even within subgroups, the ability to precisely predict a tumor’s response to therapy remains elusive. Metabolomics as a quantitative, high through put technology offers promise towards devising new strategies to establish predictive, diagnostic and prognostic markers of breast cancer. Along with other “omics” technologies that include genomics, transcriptomics, and proteomics, metabolomics fits into the puzzle of a comprehensive systems biology approach to understand drug resistance in breast cancer. In this review, we highlight the challenges facing successful therapeutic treatment of breast cancer and the innovative approaches that metabolomics offers to better understand drug resistance in cancer.
[本文引用: 2]
URLPMID:23032255Magsci [本文引用: 1]
Many complex disorders are linked to metabolic phenotypes. Revealing genetic influences on metabolic phenotypes is key to a systems-wide understanding of their interactions with environmental and lifestyle factors in their aetiology, and we can now explore the genetics of large panels of metabolic traits by coupling genome-wide association studies and metabolomics. These genome-wide association studies are beginning to unravel the genetic contribution to human metabolic individuality and to demonstrate its relevance for biomedical and pharmaceutical research. Adopting the most appropriate study designs and analytical tools is paramount to further refining the genotype-phenotype map and eventually identifying the part played by genetic influences on metabolic phenotypes. We discuss such design considerations and applications in this Review.
URLPMID:29140435 [本文引用: 1]
Abstract The Human Metabolome Database or HMDB (www.hmdb.ca) is a web-enabled metabolomic database containing comprehensive information about human metabolites along with their biological roles, physiological concentrations, disease associations, chemical reactions, metabolic pathways, and reference spectra. First described in 2007, the HMDB is now considered the standard metabolomic resource for human metabolic studies. Over the past decade the HMDB has continued to grow and evolve in response to emerging needs for metabolomics researchers and continuing changes in web standards. This year's update, HMDB 4.0, represents the most significant upgrade to the database in its history. For instance, the number of fully annotated metabolites has increased by nearly threefold, the number of experimental spectra has grown by almost fourfold and the number of illustrated metabolic pathways has grown by a factor of almost 60. Significant improvements have also been made to the HMDB's chemical taxonomy, chemical ontology, spectral viewing, and spectral/text searching tools. A great deal of brand new data has also been added to HMDB 4.0. This includes large quantities of predicted MS/MS and GC-MS reference spectral data as well as predicted (physiologically feasible) metabolite structures to facilitate novel metabolite identification. Additional information on metabolite-SNP interactions and the influence of drugs on metabolite levels (pharmacometabolomics) has also been added. Many other important improvements in the content, the interface, and the performance of the HMDB website have been made and these should greatly enhance its ease of use and its potential applications in nutrition, biochemistry, clinical chemistry, clinical genetics, medicine, and metabolomics science.
[本文引用: 2]
[本文引用: 2]
URLPMID:5439675 [本文引用: 3]
Abstract Metabolomics uses advanced analytical chemistry techniques to comprehensively measure large numbers of small molecule metabolites in cells, tissues and biofluids. The ability to rapidly detect and quantify hundreds or even thousands of metabolites within a single sample is helping scientists paint a far more complete picture of system-wide metabolism and biology. Metabolomics is also allowing researchers to focus on measuring the end-products of complex, hard-to-decipher genetic, epigenetic and environmental interactions. As a result, metabolomics has become an increasingly popular "omics" approach to assist with the robust phenotypic characterization of humans, crop plants and model organisms. Indeed, metabolomics is now routinely used in biomedical, nutritional and crop research. It is also being increasingly used in livestock research and livestock monitoring. The purpose of this systematic review is to quantitatively and objectively summarize the current status of livestock metabolomics and to identify emerging trends, preferred technologies and important gaps in the field. In conducting this review we also critically assessed the applications of livestock metabolomics in key areas such as animal health assessment, disease diagnosis, bioproduct characterization and biomarker discovery for highly desirable economic traits (i.e., feed efficiency, growth potential and milk production). A secondary goal of this critical review was to compile data on the known composition of the livestock metabolome (for 5 of the most common livestock species namely cattle, sheep, goats, horses and pigs). These data have been made available through an open access, comprehensive livestock metabolome database (LMDB, available at http://www.lmdb.ca). The LMDB should enable livestock researchers and producers to conduct more targeted metabolomic studies and to identify where further metabolome coverage is needed.
URLPMID:24905732Magsci [本文引用: 1]
61Metabolite-GWAS provide insight in the genetics of biochemical conversions.61Pathway analysis using prior knowledge improves interpretation of mGWAS data.61Systems biology will further interpretation of mGWAS data and disease associations.
URLPMID:17699064Magsci [本文引用: 1]
The current cattle selection program for dairy cattle in the Walloon region of Belgium does not consider the relative content of the different fatty acids (FA) in milk. However, interest by the local dairy industry in differentiated milk products is increasing. Therefore, farmers may be interested in selecting their animals based on the fat composition. The aim of this study was to evaluate the feasibility of genetic selection to improve the nutritional quality of bovine milk fat. The heritabilities and correlations among milk yield, fat, protein, and major FA contents in milk were estimated. Heritabilities for FA in milk and fat ranged from 5 to 38%. The genetic correlations estimated among FA reflected the common origin of several groups of FA. Given these results, an index including FA contents with the similar metabolic process of production in the mammary gland could be used, for example, to increase the monounsaturated and conjugated fatty acids in milk. Moreover, the genetic correlations between the percentage of fat and the content of C14:0, C12:0, C16:0, and C18:0 in fat were 0.06, 0.55, 0.60, and 0.84, respectively. This result demonstrates that an increase in fat content is not directly correlated with undesirable changes in FA profile in milk for human health. Based on the obtained genetic parameters, a future selection program to improve the FA composition of milk fat could be initiated.
Magsci [本文引用: 1]
<h2 class="secHeading" id="section_abstract">Abstract</h2><p id="">The aim of this study was to estimate genetic parameters for test-day milk urea nitrogen (MUN) and its relationships with milk production traits. Three test-day morning milk samples were collected from 1,953 Holstein-Friesian heifers located on 398 commercial herds in the Netherlands. Each sample was analyzed for somatic cell count, net energy concentration, MUN, and the percentage of fat, protein, and lactose. Genetic parameters were estimated using an animal model with covariates for days in milk and age at first calving, fixed effects for season of calving and effect of test or proven bull, and random effects for herd-test day, animal, permanent environment, and error. Coefficient of variation for MUN was 33%. Estimated heritability for MUN was 0.14. Phenotypic correlation of MUN with each of the milk production traits was low. The genetic correlation was close to zero for MUN and lactose percentage (−0.09); was moderately positive for MUN and net energy concentration of milk (0.19), fat yield (0.41), protein yield (0.38), lactose yield (0.22), and milk yield (0.24), and percentage of fat (0.18), and percentage of protein (0.27); and was high for MUN and somatic cell score (0.85). Herd-test day explained 58% of the variation in MUN, which suggests that management adjustments at herd-level can reduce MUN. This study shows that it is possible to influence MUN by herd practice and by genetic selection.</p>
URLPMID:18565939Magsci [本文引用: 1]
The objectives of this study were to characterize the changes of body condition score (BCS), energy content (EC), cumulative effective energy balance (CEEB), and blood serum concentrations of glucose, -hydroxybutyrate (BHBA), and nonesterified fatty acids (NEFA) across the first lactation of Holstein cows, and to estimate variance components for these traits. Four hundred ninety-seven cows kept on a commercial farm in Greece that had calved during 2005 and 2006 were used. Body condition score, estimated live weight, and blood metabolic traits were recorded weekly for the first 3 mo of lactation and monthly thereafter until the end of lactation. Body condition score and estimated live weight records were used to calculate EC and CEEB throughout the first lactation. Estimates of fixed curves and genetic parameters for each trait, by week of lactation, were obtained with the use of random regression models. The estimated fixed curves were indicative of changes in the metabolic process and energy balance of the cows. Significant genetic variance existed in all studied traits, and was particularly high during the first weeks of lactation (except for the genetic variance of CEEB, which was not significant at the beginning of lactation). Significant heritability estimates for BCS ranged from 0.34 to 0.79, for EC from 0.19 to 0.87, for CEEB from 0.58 to 0.93, for serum glucose from 0.12 to 0.39, for BHBA from 0.08 to 0.40, and for NEFA from 0.08 to 0.35. Genetic correlations between different weeks of lactation were near unity for adjacent weeks and decreased for weeks further apart, becoming practically zero for measurements taken more than 3 to 4 mo apart, especially with regard to blood metabolic traits. Significant heritability estimates were also obtained for BCS recorded before first calving. Results suggest that genetic evaluation and selection of dairy cows for early-lactation body energy and blood metabolic traits is possible.
URLPMID:21036930Magsci [本文引用: 1]
Fatty acid composition and carcass traits of 2,275 Japanese Black steers and heifers were analyzed to estimate the heritabilities and genetic correlations using the REML procedure. Slices of LM at the 6th to 7th rib section were minced and homogenized, and total lipids were extracted for the analysis by a gas chromatograph. Oleic acid accounted for the majority (51.3%), followed by palmitic (26.4%) and stearic (10.8%) acids. Heritabilities of carcass traits were moderate to high, ranging from 0.34 to 0.61, and heritabilities of individual fatty acids varied largely from 0.00 to 0.78. Those of MUFA, SFA, and PUFA were estimated to be 0.68, 0.66, and 0.47, respectively. Predicted breeding values for MUFA in 99 sires ranged from -3.0 to 5.4%. Genetic correlations of fatty acid compositions with carcass traits were generally weak (-0.28 to 0.39). Low but positive genetic correlations were obtained between beef marbling, on which emphasis of selection has been placed, and oleic acid (0.19) or MUFA (0.23). The results indicated the possibility not only for genetic improvement in fat quality traits but also simultaneous improvements with carcass traits by appropriate selection program.
URLPMID:23497994Magsci [本文引用: 1]
Small components and metabolites in milk are significant for the utilization of milk, not only in dairy food production but also as disease predictors in dairy cattle. This study focused on estimation of genetic parameters and detection of quantitative trait loci for metabolites in bovine milk. For this purpose, milk samples were collected in mid lactation from 371 Danish Holstein cows in first to third parity. A total of 31 metabolites were detected and identified in bovine milk by using 1H nuclear magnetic resonance (NMR) spectroscopy. Cows were genotyped using a bovine high-density single nucleotide polymorphism (SNP) chip. Based on the SNP data, a genomic relationship matrix was calculated and used as a random factor in a model together with 2 fixed factors (herd and lactation stage) to estimate the heritability and breeding value for individual metabolites in the milk. Heritability was in the range of 0 for lactic acid to >0.8 for orotic acid and hydroxybutyrate. A single SNP association analysis revealed 7 genome-wide significant quantitative trait loci [malonate: Bos taurus autosome (BTA)2 and BTA7; galactose-1-phosphate: BTA2; cis-aconitate: BTA11; urea: BTA12; carnitine: BTA25; and glycerophosphocholine: BTA25]. These results demonstrate that selection for metabolites in bovine milk may be possible.
URLPMID:23403187Magsci [本文引用: 1]
The composition of milk is crucial to evaluate milk performance and quality measures. Milk components partly contribute to breeding scores, and they can be assessed to judge metabolic and energy status of the cow as well as to serve as predictive markers for diseases. In addition to the milk composition measures (e.g., fat, protein, lactose) traditionally recorded during milk performance test via infrared spectroscopy, novel techniques, such as gas chromatography-mass spectrometry, allow for a further analysis of milk into its metabolic components. Gas chromatography-mass spectrometry is suitable for measuring several hundred metabolites with high throughput, and thus it is applicable to study sources of genetic and nongenetic variation of milk metabolites in dairy cows. Heritability and mode of inheritance of metabolite measurements were studied in a linear mixed model approach including expected (pedigree) and realized (genomic) relationship between animals. The genetic variability of 190 milk metabolite intensities was analyzed from 1,295 cows held on 18 farms in Mecklenburg-Western Pomerania, Germany. Besides extensive pedigree information, genotypic data comprising 37,180 single nucleotide polymorphism markers were available. Goodness of fit and significance of genetic variance components based on likelihood ratio tests were investigated with a full model, including marker- and pedigree-based genetic effects. Broad-sense heritability varied from zero to 0.699, with a median of 0.125. Significant additive genetic variance was observed for highly heritable metabolites, but dominance variance was not significantly present. As some metabolites are particularly favorable for human nutrition, for instance, future research should address the identification of locus-specific genetic effects and investigate metabolites as the molecular basis of traditional milk performance test traits.
URLPMID:26805988 [本文引用: 1]
Genetic parameters were estimated for the major milk proteins using bivariate and multi-trait models based on genomic relationships between animals. The analyses included, apart from total protein percentage, αS1-casein (CN), αS2-CN, β-CN, κ-CN, α-lactalbumin, and β-lactoglobulin, as well as the posttranslational sub-forms of glycosylated κ-CN and αS1-CN-8P (phosphorylated). Standard errors of the estimates were used to compare the models. In total, 650 Danish Holstein cows across 4 parities and days in milk ranging from 9 to 481 d were selected from 21 herds. The multi-trait model generally resulted in lower standard errors of heritability estimates, suggesting that genetic parameters can be estimated with high accuracy using multi-trait analyses with genomic relationships for scarcely recorded traits. The heritability estimates from the multi-trait model ranged from low (0.05 for β-CN) to high (0.78 for κ-CN). Genetic correlations between the milk proteins and the total milk protein percentage were generally low, suggesting the possibility to alter protein composition through selective breeding with little effect on total milk protein percentage.
URLPMID:20042548 [本文引用: 1]
The aim of this study was to estimate genetic parameters for pork intramuscular fatty acid (FA) composition and indices for desaturase and elongase activities involved in n-3 and n-6 PUFA metabolism. The LM of 437 slaughter pigs was analyzed for FA composition (expressed as g/100 g of FA). Indices for enzyme activities were calculated from product to precursor FA ratios. Genetic parameters were estimated with single- and multi-trait animal models. The total FA content, reflecting the intramuscular fat content, was either included or not in the model. Results from the models without total FA content showed relatively high heritability estimates, generally above 0.50, for the proportion of the most important MUFA and PUFA, compared with much smaller values for the SFA. When total FA content was included in the models, heritability values decreased (P < 0.001) for most individual FA and for all sums of FA groups, except for C18:0, C18:3n-6, and C18:3n-3. Heritability estimates for the ratios C20:4n-6/C18:2n-6 and C22:6n-3/C18:3n-3, reflecting the overall conversion in the n-6 and n-3 PUFA pathway, respectively, were 0.29 and 0.35, respectively, with total FA content in the model and increased to 0.38 and 0.49, respectively, if total FA content was not in the model. Heritabilities for other more specific indices were of the same order. Genetic correlations between PUFA proportions and indices for enzyme activities with ADG were mostly negative, whereas the correlations with carcass lean meat percentage were mostly positive. It was concluded that there is meaningful genetic variation for long-chain PUFA metabolism that is only partly dependent on the carcass and muscle fat content. This may allow selection for improved FA composition of pork.
URLPMID:26812309 [本文引用: 1]
Abstract The aim of this study was to estimate the genetic and environmental parameters and crossbreeding effects on fatty acid and fat traits in the Iberian pig. Our final goal is to explore target selection traits and define crossbreeding strategies. The phenotypes were obtained under intensive management from 470 animals in a diallelic experiment involving Retinto, Torbiscal, and Entrepelado lines. The data set was composed of backfat thickness at the fourth rib (BFT), intramuscular fat (IMF) in the longissimus thoracis (LT), and the fatty acid profile for IMF and subcutaneous fat (SCF) traits. Data were analyzed through a Bayesian bivariate animal model by using a reparameterization of Dickerson's model. The results obtained showed an important genetic determinism for all traits analyzed with heritability ranging from 0.09 to 0.67. The common environment litter effect also had an important effect on IMF (0.34) and its fatty acid composition (0.06-0.53) at slaughter. The additive genetic correlation between BFT and IMF (additive genetic correlation [] = 0.31) suggested that it would be possible to improve lean growth independent of the IMF with an appropriate selection index. Furthermore, the high additive genetic correlation ( = 0.68) found between MUFA tissues would seem to indicate that either the LT or SCF could be used as the reference tissue for MUFA selection. The relevance of the crossbreeding parameters varied according to the traits analyzed. Backfat thickness at the fourth rib and the fatty acid profile of the IMF showed relevant differences between crosses, mostly due to line additive genetic effects associated with the Retinto line. On the contrary, those for IMF crosses were probably mainly attributable to heterosis effects. Particularly, heterosis effects were relevant for the Retinto and Entrepelado crosses (approximately 16% of the trait), which could be valuable for a crossbreeding system involving these lines.
[本文引用: 1]
URLPMID:28905661 [本文引用: 1]
(2017). Genetic parameters for the prediction of abdominal fat traits using blood biochemical indicators in broilers. British Poultry Science. Ahead of Print.
URL [本文引用: 1]
61This study identified metabolites associated with variation in production traits in beef cattle.61Plasma metabolites found significant could be used as markers for selection for these traits.61We identified metabolites accounting for >74% of the variation in feed efficiency.61The accuracy of prediction using all the metabolites associated with feed efficiency was 95%.61Significant and validated metabolites can be developed to rapid and accurate tests for selection.
URLPMID:21920477Magsci [本文引用: 1]
Higher levels of glycolytic enzymes and lactate accumulation were related to slow pH drop in Casertana pigs, albeit not to rapid pH lowering in LW counterparts.On the other hand, the individuation of pyruvate kinaseM1 and tropomyosin levels in LW were related to water holding capacity and Minolta values at 24h after slaughter.Bioinformatic analyses strengthened the correlation between over-expression of structural proteins in LW and more accentuated growth aptitude in this breed. Conversely, enzymes taking part into branching glycolytic reactions, such as glycerol 3-phosphate and creatine kinase M, were related to accentuated lipogenesis and slower albeit prolonged glycolytic rate in Casertana, respectively.Breed-specific differences at the protein level were not only related to growth performances and fat accumulation tendency Graphical abstractView high quality image (97K)
URLPMID:21429726Magsci [本文引用: 1]
Childhood obesity has become a prevalent risk to health of children and teenagers. To develop biomarkers in serum for altered lipid metabolism, genetically obese (Ningxiang strain) and lean (Duroc×Landrace×Large Yorkshire strain) growing pigs were used as models to identify potential differences in the serum metabonome between the two strains of pigs after consuming the same diet for 46 days. At the end of the study, pigs were euthanized for analysis of the serum metabonome and determination of body composition. Obese pigs had higher fat mass (42.3±8.8% vs. 21.9±4.5%) and lower muscle mass (35.4±4.5% vs. 58.9±2.5%) than lean pigs ( P<.01). Serum concentrations of insulin and glucagon were higher ( P<.02) in obese than in lean pigs. With the use of an NMR-based metabonomic technology, orthogonal projection to latent structure with discriminant analysis showed that serum HDL, VLDL, lipids, unsaturated lipids, glycoprotein, myo-inositol, pyruvate, threonine, tyrosine and creatine were higher in obese than in lean pigs ( P<.05), while serum glucose and urea were lower in obese pigs ( P<.05). In addition, changes in gut microbiota-related metabolites, including trimethylamine- N-oxide and choline, were observed in sera of obese pigs relatively to lean pigs ( P<.05). These novel findings indicate that obese pigs have distinct metabolism, including lipogenesis, lipid oxidation, energy utilization and partition, protein and amino acid metabolism, and fermentation of gastrointestinal microbes, compared with lean pigs. The obese Ningxiang pig may be a useful model for childhood obesity research.
URLPMID:24200563Magsci [本文引用: 1]
61Crossbreeds differed in their meat metabolite profile.61A strong effect of crossbreed on the meat content of choline was observed.61Carnosine was the meat metabolite showing highest correlation to sensory attributes.
[本文引用: 1]
URLPMID:24550212Magsci [本文引用: 1]
Domestic broiler rapidly accumulate fat and are naturally hyperglycemic and resistant, making them an attractive model for studies of . We previously demonstrated that short-term (5 h) fasting rapidly upregulates pathways of in broiler and proposed that activation of these pathways may promote leanness. The objective of the current study was to characterize adipose tissue from relatively lean and fatty lines of and determine if heritable leanness in is associated with activation of some of the same pathways induced by fasting. We compared adipose and metabolite profiles in white adipose tissue of lean Leghorn and Fayoumi breeds to those of fattier commercial broiler . Both and expression of genes involved in were upregulated in lean compared with broilers. Although there were strong similarities between the lean lines compared with broilers, distinct expression signatures were also found between Fayoumi and Leghorn, including differences in adipogenic genes. Similarities between genetically lean and fasted suggest that in white adipose tissue is adaptively coupled to and plays a role in heritable differences in fatness. Unique signatures of leanness in Fayoumi and Leghorn lines highlight distinct pathways that may provide insight into the basis for leanness in . Collectively, our results provide a number of future directions through which to fully exploit as unique models for the study of and adipose .
URLPMID:25568361 [本文引用: 1]
Excessive deposition of body fat is detrimental to production efficiency. The aim of this study was to provide plasma indicators of chickens' ability to store fat. From 3 to 9 wk of age, chickens from 2 experimental lines exhibiting a 2.5-fold difference in abdominal fat content and fed experimental diets with contrasted feed energy sources were compared. The diets contained 80 vs. 20 g of lipids and 379 vs. 514 g of starch per kg of feed, respectively, but had the same ME and total protein contents. Cellulose was used to dilute energy in the high-fat diet. At 9 wk of age, the body composition was analyzed and blood samples were collected. A metabolome-wide approach based on proton nuclear magnetic resonance spectroscopy was associated with conventional measurements of plasma parameters. A metabolomics approach showed that betaine, glutamine, and histidine were the most discriminating metabolites between groups. Betaine, uric acid, triglycerides, and phospholipids were positively correlated (r > 0.3; P < 0.05) and glutamine, histidine, triiodothyronine, homocysteine, and hydroxybutyrate were negatively correlated (r < -0.3; P < 0.05) with relative weight of abdominal fat and/or fat situated at the top of external face of the thigh. The combination of plasma free fatty acids, total cholesterol, phospholipid, hydroxybutyrate, glutamine, and methionine levels accounted for 74% of the variability of the relative weight of abdominal fat. On the other hand, the combination of plasma triglyceride and homocysteine levels accounted for 37% of the variability of fat situated at the top of external face of the thigh. The variations in plasma levels of betaine, homocysteine, uric acid, glutamine, and histidine suggest the implication of methyl donors in the control of hepatic lipid synthesis and illustrate the interplay between AA, glucose, and lipid metabolisms in growing chickens.
URLPMID:19700730Magsci
A genome-wide scan was performed to identify quantitative trait loci (QTL) for short- and medium-chain fatty acids (expressed in wt/wt %). Milk samples were available from 1,905 cows from 398 commercial herds in the Netherlands, and milk-fat composition was measured by gas chromatography. DNA was available from 7 of the paternal half-sib families: 849 cows and their 7 sires. A genetic map was constructed comprising 1,341 SNP and 2,829 cM, with an average information content of 0.83. Multimarker interval mapping was used in an across-family regression on corrected phenotypes for the 7 half-sib families. Four QTL were found: on gene. Quantitative trait loci that affect specific fatty acids might increase the understanding of physiological processes regarding fat synthesis and the position of the causal genes.
URLPMID:21569316
Background Identifying genomic regions, and preferably individual genes, responsible for genetic variation in milk fat composition of bovine milk will enhance the understanding of biological pathways involved in fatty acid synthesis and may point to opportunities for changing milk fat composition via selective breeding. An association study of 50,000 single nucleotide polymorphisms (SNPs) was performed for even-chain saturated fatty acids (C4:0-C18:0), even-chain monounsaturated fatty acids (C10:1-C18:1), and the polyunsaturated C18:2cis9,trans11 (CLA) to identify genomic regions associated with individual fatty acids in bovine milk. Results The two-step single SNP association analysis found a total of 54 regions on 29 chromosomes that were significantly associated with one or more fatty acids. Bos taurus autosomes (BTA) 14, 19, and 26 showed highly significant associations with seven to ten traits, explaining a relatively large percentage of the total additive genetic variation. Many additional regions were significantly associated with the fatty acids. Some of the regions harbor genes that are known to be involved in fat synthesis or were previously identified as underlying quantitative trait loci for fat yield or content, such as ABCG2 and PPARGC1A on BTA 6; ACSS2 on BTA 13; DGAT1 on BTA 14; ACLY, SREBF1, STAT5A, GH, and FASN on BTA 19; SCD1 on BTA26; and AGPAT6 on BTA 27. Conclusions Medium chain and unsaturated fatty acids are strongly influenced by polymorphisms in DGAT1 and SCD1. Other regions also showed significant associations with the fatty acids studied. These additional regions explain a relatively small percentage of the total additive genetic variance, but they are relevant to the total genetic merit of an individual and in unraveling the genetic background of milk fat composition. Regions identified in this study can be fine mapped to find causal mutations. The results also create opportunities for changing milk fat composition through breeding by selecting individuals based on their genetic merit for milk fat composition.
URLPMID:23626813
Vitamin B-12 (also called cobalamin) is essential for human health and current intake levels of vitamin B-12 are considered to be too low. Natural enrichment of the vitamin B-12 content in milk, an important dietary source of vitamin B-12, may help to increase vitamin B-12 intake. Natural enrichment of the milk vitamin B-12 content could be achieved through genetic selection, provided there is genetic variation between cows with respect to the vitamin B-12 content in their milk. A substantial amount of genetic variation in vitamin B-12 content was detected among raw milk samples of 544 first-lactation Dutch Holstein Friesian cows. The presence of genetic variation between animals in vitamin B-12 content in milk indicates that the genotype of the cow affects the amount of vitamin B-12 that ends up in her milk and, consequently, that the average milk vitamin B-12 content of the cow population can be increased by genetic selection. A genome-wide association study revealed significant association between 68 SNP and vitamin B-12 content in raw milk of 487 first-lactation Dutch Holstein Friesian cows. This knowledge facilitates genetic selection for milk vitamin B-12 content. It also contributes to the understanding of the biological mechanism responsible for the observed genetic variation in vitamin B-12 content in milk. None of the 68 significantly associated SNP were in or near known candidate genes involved in transport of vitamin B-12 through the gastrointestinal tract, uptake by ileum epithelial cells, export from ileal cells, transport through the blood, uptake from the blood, intracellular processing, or reabsorption by the kidneys. Probably, associations relate to genes involved in alternative pathways of well-studied processes or to genes involved in less well-studied processes such as ruminal production of vitamin B-12 or secretion of vitamin B-12 by the mammary gland
URLPMID:4032272
Detecting genes associated with milk fat composition could provide valuable insights into the complex genetic networks of genes underling variation in fatty acids synthesis and point towards opportunities for changing milk fat composition via selective breeding. In this study, we conducted a genome-wide association study (GWAS) for 22 milk fatty acids in 784 Chinese Holstein cows with the PLINK software. Genotypes were obtained with the Illumina BovineSNP50 Bead chip and a total of 40,604 informative, high-quality single nucleotide polymorphisms (SNPs) were used. Totally, 83 genome-wide significant SNPs and 314 suggestive significant SNPs associated with 18 milk fatty acid traits were detected. Chromosome regions that affect milk fatty acid traits were mainly observed on BTA1, 2, 5, 6, 7, 9, 13, 14, 18, 19, 20, 21, 23, 26 and 27. Of these, 146 SNPs were associated with more than one milk fatty acid trait; most of studied fatty acid traits were significant associated with multiple SNPs, especially C18:0 (105 SNPs), C18 index (93 SNPs), and C14 index (84 SNPs); Several SNPs are close to or within the DGAT1, SCD1 and FASN genes which are well-known to affect milk composition traits of dairy cattle. Combined with the previously reported QTL regions and the biological functions of the genes, 20 novel promising candidates for C10:0, C12:0, C14:0, C14:1, C14 index, C18:0, C18:1n9c, C18 index, SFA, UFA and SFA/UFA were found, which composed of HTR1B, CPM, PRKG1, MINPP1, LIPJ, LIPK, EHHADH, MOGAT1, ECHS1, STAT1, SORBS1, NFKB2, AGPAT3, CHUK, OSBPL8, PRLR, IGF1R, ACSL3, GHR and OXCT1. Our findings provide a groundwork for unraveling the key genes and causal mutations affecting milk fatty acid traits in dairy cattle.
URLPMID:25789767 [本文引用: 1]
The metabolic adaptation of dairy cows during the transition period has been studied intensively in the last decades. However, until now, only few studies have paid attention to the genetic aspects of this process. Here, we present the results of a gene-based mapping and pathway analysis with the measurements of three key metabolites, (1) non-esterified fatty acids (NEFA), (2) beta-hydroxybutyrate (BHBA) and (3) glucose, characterizing the metabolic adaptability of dairy cows before and after calving. In contrast to the conventional single-marker approach, we identify 99 significant and biologically sensible genes associated with at least one of the considered phenotypes and thus giving evidence for a genetic basis of the metabolic adaptability. Moreover, our results strongly suggest three pathways involved in the metabolism of steroids and lipids are potential candidates for the adaptive regulation of dairy cows in their early lactation. From our perspective, a closer investigation of our findings will lead to a step forward in understanding the variability in the metabolic adaptability of dairy cows in their early lactation.
URLPMID:26364108
The identification of causal genes or genomic regions associated with fatty acids (FA) will enhance our understanding of the pathways underlying FA synthesis and provide opportunities for changing milk fat composition through a genetic approach. The linkage disequilibrium between adjacent markers is highly consistent between the Chinese and Danish Holstein populations, such that a joint genome-wide association study (GWAS) can be performed. In this study, a joint GWAS was performed for 16 milk FA traits based on data of 784 Chinese and 371 Danish Holstein cows genotyped by a high-density bovine single nucleotide polymorphism (SNP) array. A total of 486,464 SNP markers on 29 bovine autosomes were used. Bonferroni corrections were applied to adjust the significance thresholds for multiple testing at the genome- and chromosome-wide levels. According to the analysis of either the Chinese or Danish data individually, the total numbers of overlapping SNP that were significant at the chromosome level were 94 for C14:1, 208 for the C14 index, and 1 for C18:0. Joint analysis using the combined data of the 2 populations detected greater numbers of significant SNP compared with either of the individual populations alone for 7 and 10 traits at the genome- and chromosome-wide significance levels, respectively. Greater numbers of significant SNP were detected for C18:0 and the C18 index in the Chinese population compared with the joint analysis. Sixty-five significant SNP across all traits had significantly different effects in the 2 populations. Ten FA were influenced by a quantitative trait loci (QTL) region includingDGAT1. Both C14:1 and the C14 index were influenced by a QTL region includingSCD1in the combined population. Other QTL regions also showed significant associations with the studied FA. A large region (14.9-24.9 Mbp) in BTA26 significantly influenced C14:1 and the C14 index in both populations, mostly likely due to the SNP inSCD1. A QTL region (69.97-73.69 Mbp) on BTA9 showed a significantly different effect on C18:0 between the 2 populations. Detection of these important SNP and the corresponding QTL regions will be helpful for follow-up studies to identify causal mutations and their interaction with environments for milk FA in dairy cattle.
URLPMID:25771056
Riboflavin (vitamin B2) is an essential water-soluble vitamin; elderly people and adolescents in particular can have poor riboflavin status. In Western diets, milk and dairy products are primary sources of riboflavin, but little is known about the natural variation within and among bovine breeds, and how genetic and environmental factors can affect the riboflavin content in milk. As a part of the Danish-Swedish Milk Genomics Initiative, the aim of the study was to quantify milk riboflavin content using reverse-phase HPLC in 2 major Danish dairy breeds. The results showed substantial interbreed differences in milk riboflavin content. Milk from Danish Jersey cows contained significantly higher levels of riboflavin (1.93mg/L of milk) than milk from Danish Holstein cows (1.40mg/L of milk). Furthermore, genetic analyses revealed high heritabilities in both breeds (0.52 for Danish Holstein and 0.31 for Danish Jersey). A genomic association study found 35 significant single nucleotide polymorphisms (false discovery rate<0.10) to be associated with riboflavin content in milk in Jersey cows (all on BTA14 and BTA17), and 511 significant single nucleotide polymorphisms in Holstein cows spread over 25 different autosomes with BTA13 and BTA14 having the most promising quantitative trait loci. The best candidate gene found within the identified quantitative trait loci wasSLC52A3, a riboflavin transporter gene, which was among the significant markers on BTA13 in Holstein cows.
URLPMID:25670729
Essentially all high-yielding dairy cows experience a negative energy balance during early lactation leading to increased lipomobilization, which is a normal physiological response. However, a severe energy deficit may lead to high levels of ketone bodies and, subsequently, to subclinical or clinical ketosis. It has previously been reported that the ratio of glycerophosphocholine to phosphocholine in milk is a prognostic biomarker for the risk of ketosis in dairy cattle. It was hypothesized that this ratio reflects the ability to break down blood phosphatidylcholine as a fatty acid resource. In the current study, 248 animals from a previous study were genotyped with Illumina BovineSNP50 BeadChip, and genome-wide association studies were carried out for the milk levels of phosphocholine, glycerophosphocholine, and the ratio of both metabolites. It was demonstrated that the latter two traits are heritable with h2 = 0.43 and h2 = 0.34, respectively. A major quantitative trait locus was identified on cattle chromosome 25. The APOBR gene, coding for the apolipoprotein B receptor, is located within this region and was analyzed as a candidate gene. The analysis revealed highly significant associations of polymorphisms within the gene with glycerophosphocholine as well as the metabolite ratio. These findings support the hypothesis that differences in the ability to take up blood phosphatidylcholine from low-density lipoproteins play an important role in early lactation metabolic stability of dairy cows and indicate APOBR to contain a causative variant.
URLPMID:26995140
Association analyses between candidate genes and bovine milk fatty acids can improve our understanding of genetic variation in milk fatty acid profiles and reveal potential opportunities to tailor milk fat composition through selection strategies. In this work, we investigated the association of 51 single nucleotide polymorphisms (SNP) selected from 37 candidate genes using a functional and positional approach, with 47 fatty acids, 9 fatty acid groups, and 59-desaturation indices in milk samples from Brown Swiss cows. Individual milk samples were collected from 1,158 Italian Brown Swiss cows, and gas chromatography was used to obtain detailed milk fatty acid compositions. A GoldenGate assay system (Illumina, San Diego, CA) was used to perform genotype 96 selected SNP located in 54 genes across 22 chromosomes. In total, 51 polymorphic SNP in 37 candidate genes were retained for the association analysis. A Bayesian linear animal model was used to estimate the contribution of each SNP. A total of 129 tests indicated relevant additive effects between a given SNP and a single fatty acid trait; 38 SNP belonging to 30 genes were relevant for a total of 57 fatty acid traits. Most of the studied fatty acid traits (~81%) were relevantly associated with multiple SNP. Relevantly associated SNP were mainly found in genes related to fat metabolism, linked to or contained in previously identified quantitative trait loci for fat yield or content, or associated with genes previously identified in association analyses with milk fatty acid profiles in other cow breeds. The most representative candidate genes wereLEP,PRL,STAT5A,CCL3,ACACA,GHR,ADRB2,LPIN1,STAT1,FABP4, andCSN2. In particular, relevant associations with SNP located on bovine chromosome 19 (BTA19) were found. Two candidate genes on BTA19 (CCL3andACACA)were relevantly associated with de novo short- and medium-chain fatty acids, likely explaining the high heritability values found for these fatty acids (with the exception of C6:0). Two additional genes on BTA19 (CCL2andGH1) showed associations with saturated and branched-chain fatty acids. Our findings provide basic information on genes and SNP affecting the milk fatty acid composition of dairy cows. These results may support the possibility of using genetic selection to modify milk fatty acid profiles to promote beneficial health-related effects.
URLPMID:28865853
The aim of this study was to fine-map a genomic region associated with milk fatty acids (FA) on Bos taurus autosome (BTA) 17. This genomic region has been discovered with 50,000 (50k) single nucleotide polymorphisms (SNP) imputed to 777,000 (777k) SNP. In this study, high-density genotypes were imputed to whole-genome sequences level to identify candidate gene(s) associated with milk FA composition on BTA17. Phenotypes and genotypes were available for 1,640 cows sampled in winter, and for 1,581 cows sampled in summer. Phenotypes consisted of gas chromatography measurements in winter and in summer milk samples of 6 individual FA and the indicator of de novo synthesis, C6:0-C14:0. Genotypes consisted of imputed 777k SNP, and 89 sequenced ancestors of the population of genotyped cows. In addition, 450 whole-genome sequences from the 1,000 Bull Genome Consortium were available. Using 495 Holstein-Friesian sequences as a reference population, the 777k SNP genotypes of the cows were imputed to sequence level. We then applied single-variant analyses with an animal model, and identified thousands of significant associations with C6:0, C8:0, C10:0, C12:0, C14:0, and C6:0-C14:0. For C8:0 in summer milk samples, the genomic region located between 29 and 34 Mbp on BTA17 revealed a total of 646 significant associations. The most significant associations [-log10(P-value) = 7.82] were 8 SNP in perfect linkage disequilibrium. After fitting one of these 8 SNP as a fixed effect in the model, and re-running the single-variant analyses, no further significant associations were found for any of the 6 FA or C6:0-C14:0. These findings suggest that one polymorphism underlying this QTL on BTA17 influences multiple de novo synthesized milk FA. Thirteen genes in the QTL region were identified and analyzed carefully. Six out of the 8 SNP that showed the strongest associations were located in the La ribonucleoprotein domain family, member 1B (LARP1B) gene, and we suggest LARP1B as a primary candidate gene. Another gene of interest for this QTL region might be PKL4. None of these suggested candidate genes have previously been associated with milk fat synthesis or milk FA composition.
URL
Lactose provides an easily-digested energy source for neonates, and is the primary carbohydrate in milk in most species. Bovine lactose is also a key component of many human food products. However, compared to analyses of other milk components, the genetic control of lactose has been little studied. Here we present the first GWAS focussed on analysis of milk lactose traits. Using a discovery population of 12,000 taurine dairy cattle, we detail 27 QTL for lactose concentration and yield, and subsequently validate the effects of 26 of these loci in a distinct population of 18,000 cows. We next present data implicating causative genes and variants for these QTL. Fine mapping of these regions using imputed, whole genome sequence-resolution genotypes reveals protein-coding candidate causative variants affecting theABCG2,DGAT1,STAT5B,KCNH4,NPFFR2andRNF214genes. Eleven of the remaining QTL appear to be driven by regulatory effects, suggested by the presence of co-locating, co-segregating eQTL discovered using mammary RNA sequence data from a population of 357 lactating cows. Pathway analysis of genes representing all lactose-associated loci shows significant enrichment of genes located in the endoplasmic reticulum, with functions related to ion channel activity mediated through theLRRC8C,P2RX4,KCNJ2andANKHgenes. A number of the validated QTL are also found to be associated with additional milk volume, fat and protein phenotypes. Overall, these findings highlight novel candidate genes and variants involved in milk lactose regulation, whose impacts on membrane transport mechanisms reinforce the key osmo-regulatory roles of lactose in milk. The online version of this article (doi:10.1186/s12864-017-4320-3) contains supplementary material, which is available to authorized users.
URLPMID:23607548Magsci
Fatty acid composition is one of the important traits in beef. The aim of this study was to identify candidate genomic regions for fatty acid composition by genome-wide association study with 5065K single nucleotide polymorphism (SNP) array in Japanese Black cattle. A total of 461 individuals and 4065657 SNPs were used in this study. We applied genome-wide rapid association using mixed model and regression (GRAMMAR) and genomic control approaches to estimate the associations between genotypes and fatty acid composition. In addition, two SNPs in fatty acid synthase (FASN) (T1952A) and stearoyl-CoA desaturase (SCD) (V293A) genes were also genotyped. Association analysis revealed that 30 significant SNPs for several fatty acids (C14:0, C14:1, C16:1 and C18:1) were located in the BTA1961FASN gene located within this region but the FASN mutation had no significant effect on any traits. We also detected one significant SNP for C18:1 on BTA23 and two SNPs for C16:0 on BTA25. The region around 1765Mb on BTA26 harbored two significant SNPs for C14:1 and SNP in SCD in this region showed the strongest association with C14:1. This study demonstrated novel candidate regions in BTA19, 23 and 25 for fatty acid composition.
URLPMID:24156620
As consumers continue to request food products that have health advantages, it will be important for the livestock industry to supply a product that meet these demands. One such nutrient is fatty acids, which have been implicated as playing a role in cardiovascular disease. Therefore, the objective of this study was to determine the extent to which molecular markers could account for variation in fatty acid composition of skeletal muscle and identify genomic regions that harbor genetic variation. Subsets of markers on the Illumina 54K bovine SNPchip were able to account for up to 57% of the variance observed in fatty acid composition. In addition, these markers could be used to calculate a direct genomic breeding values (DGV) for a given fatty acids with an accuracy (measured as simple correlations between DGV and phenotype) ranging from -0.06 to 0.57. Furthermore, 57 1-Mb regions were identified that were associated with at least one fatty acid with a posterior probability of inclusion greater than 0.90. 1-Mb regions on BTA19, BTA26 and BTA29, which harbored fatty acid synthase, Sterol-CoA desaturase and thyroid hormone responsive candidate genes, respectively, explained a high percentage of genetic variance in more than one fatty acid. It was also observed that the correlation between DGV for different fatty acids at a given 1-Mb window ranged from almost 1 to -1. Further investigations are needed to identify the causal variants harbored within the identified 1-Mb windows. For the first time, Angus breeders have a tool whereby they could select for altered fatty acid composition. Furthermore, these reported results could improve our understanding of the biology of fatty acid metabolism and deposition.
URLPMID:4913692 [本文引用: 1]
The fatty acid profile of beef is a complex trait that can benefit from gene-interaction network analysis to understand relationships among loci that contribute to phenotypic variation. Phenotypic measures of fatty acid profile from triacylglycerol and phospholipid fractions of longissimus muscle, pedigree information, and Illumina 54 k bovine SNP genotypes were utilized to derive an annotated gene network associated with fatty acid composition in 1,833 Angus beef cattle. The Bayes-B statistical model was utilized to perform a genome wide association study to estimate associations between 54 k SNP genotypes and 39 individual fatty acid phenotypes within each fraction. Posterior means of the effects were estimated for each of the 54 k SNP and for the collective effects of all the SNP in every 1-Mb genomic window in terms of the proportion of genetic variance explained by the window. Windows that explained the largest proportions of genetic variance for individual lipids were found in the triacylglycerol fraction. There was almost no overlap in the genomic regions explaining variance between the triacylglycerol and phospholipid fractions. Partial correlations were used to identify correlated regions of the genome for the set of largest 1 Mb windows that explained up to 35% genetic variation in either fatty acid fraction. SNP were allocated to windows based on the bovine UMD3.1 assembly. Gene network clusters were generated utilizing a partial correlation and information theory algorithm. Results were used in conjunction with network scoring and visualization software to analyze correlated SNP across 39 fatty acid phenotypes to identify SNP of significance. Significant pathways implicated in fatty acid metabolism through GO term enrichment analysis included homeostasis of number of cells, homeostatic process, coenzyme/cofactor activity, and immunoglobulin. These results suggest different metabolic pathways regulate the development of different types of lipids found in bovine muscle tissues. Network analysis using partial correlations and annotation of significant SNPs can yield information about the genetic architecture of complex traits.
URLPMID:4784275
Saturated fatty acids can be detrimental to human health and have received considerable attention in recent years. Several studies using taurine breeds showed the existence of genetic variability and thus the possibility of genetic improvement of the fatty acid profile in beef. This study identified the regions of the genome associated with saturated, mono- and polyunsaturated fatty acids, and n-6 to n-3 ratios in theLongissimus thoracisof Nellore finished in feedlot, using the single-step method. The results showed that 115 windows explain more than 1% of the additive genetic variance for the 22 studied fatty acids. Thirty-one genomic regions that explain more than 1% of the additive genetic variance were observed for total saturated fatty acids, C12:0, C14:0, C16:0 and C18:0. Nineteen genomic regions, distributed in sixteen different chromosomes accounted for more than 1% of the additive genetic variance for the monounsaturated fatty acids, such as the sum of monounsaturated fatty acids, C14:1 cis-9, C18:1 trans-11, C18:1 cis-9, and C18:1 trans-9. Forty genomic regions explained more than 1% of the additive variance for the polyunsaturated fatty acids group, which are related to the total polyunsaturated fatty acids, C20:4 n-6, C18:2 cis-9 cis12 n-6, C18:3 n-3, C18:3 n-6, C22:6 n-3 and C20:3 n-6 cis-8 cis-11 cis-14. Twenty-one genomic regions accounted for more than 1% of the genetic variance for the group of omega-3, omega-6 and the n-6:n-3 ratio. The identification of such regions and the respective candidate genes, such asELOVL5,ESSRG,PCYT1Aand genes of theABCgroup (ABC5,ABC6andABC10), should contribute to form a genetic basis of the fatty acid profile of Nellore (Bos indicus) beef, contributing to better selection of the traits associated with improving human health. The online version of this article (doi:10.1186/s12864-016-2511-y) contains supplementary material, which is available to authorized users.
URLPMID:27112906
Abstract We performed a genome-wide association study (GWAS) and candidate gene analysis to: (i) evaluate the effectiveness of the GWAS in our small population by performing GWAS for carcass weight (CW) and fatty acid composition; (ii) detect novel candidate regions affecting non-CW carcass traits, chemical composition and sugar; and (iii) evaluate the association of the candidate genes previously detected in CW and fatty acid composition with other economically important traits. A total of 574 Japanese Black cattle and 40-657 Single nucleotide polymorphisms were used. In addition, candidate gene analyses were performed to evaluate the association of three CW-related genes and two fatty acid-related genes with carcass traits, fatty acid composition, chemical composition and sugar. The significant regions with the candidate genes were detected for CW and fatty acid composition, and these results showed that a significant region would be detectable despite the small sample size. The novel candidate regions were detected on BTA23 for crude protein and on BTA19 for fructose. CW-related genes associated with the rib-eye area and fatty acid composition were identified, and fatty acid-related genes had no relationship with other traits. Moreover, the favorable allele of CW-related genes had an unfavorable effect on fatty acid composition.
URLPMID:28615065
Fatty acid composition of muscle is an important trait contributing to meat quality. Recently, genome-wide association study (GWAS) has been extensively used to explore the molecular mechanism underlying important traits in cattle. In this study, we performed GWAS using high density SNP array to analyze the association between SNPs and fatty acids and evaluated the accuracy of genomic prediction for fatty acids in Chinese Simmental cattle. Using the BayesB method, we identified 35 and 7 regions in Chinese Simmental cattle that displayed significant associations with individual fatty acids and fatty acid groups, respectively. We further obtained several candidate genes which may be involved in fatty acid biosynthesis including elongation of very long chain fatty acids protein 5 (ELOVL5), fatty acid synthase (FASN), caspase 2 (CASP2) and thyroglobulin (TG). Specifically, we obtained strong evidence of association signals for one SNP located at 51.3Mb forFASNusing Genome-wide Rapid Association Mixed Model and Regression-Genomic Control (GRAMMAR-GC) approaches. Also, region-based association test identified multiple SNPs withinFASNandELOVL5for C14:0. In addition, our result revealed that the effectiveness of genomic prediction for fatty acid composition using BayesB was slightly superior over GBLUP in Chinese Simmental cattle. We identified several significantly associated regions and loci which can be considered as potential candidate markers for genomics-assisted breeding programs. Using multiple methods, our results revealed thatFASNandELOVL5are associated with fatty acids with strong evidence. Our finding also suggested that it is feasible to perform genomic selection for fatty acids in Chinese Simmental cattle. The online version of this article (doi:10.1186/s12864-017-3847-7) contains supplementary material, which is available to authorized users.
URL
URLPMID:29770529
URLPMID:4654876
Background Identification of genetic variants that are associated with fatty acid composition in beef will enhance our understanding of host genetic influence on the trait and also allow for more effective improvement of beef fatty acid profiles through genomic selection and marker-assisted diet management. In this study, 81 and 83 fatty acid traits were measured in subcutaneous adipose (SQ) and longissimus lumborum muscle (LL), respectively, from 1366 purebred and crossbred beef steers and heifers that were genotyped on the Illumina BovineSNP50 Beadchip. The objective was to conduct genome-wide association studies (GWAS) for the fatty acid traits and to evaluate the accuracy of genomic prediction for fatty acid composition using genomic best linear unbiased prediction (GBLUP) and Bayesian methods. Results In total, 302 and 360 significant SNPs spanning all autosomal chromosomes were identified to be associated with fatty acid composition in SQ and LL tissues, respectively. Proportions of total genetic variance explained by individual significant SNPs ranged from 0.03 to 11.06% in SQ, and from 0.005 to 24.28% in the LL muscle. Markers with relatively large effects were located near fatty acid synthase (FASN), stearoyl-CoA desaturase (SCD), and thyroid hormone responsive (THRSP) genes. For the majority of the fatty acid traits studied, the accuracy of genomic prediction was relatively low (<0.40). Relatively high accuracies (>???=???0.50) were achieved for 10:0, 12:0, 14:0, 15:0, 16:0, 9c-14:1, 12c-16:1, 13c-18:1, and health index (HI) in LL, and for 12:0, 14:0, 15:0, 10t,12c-18:2, and 11t,13c???+???11c,13t-18:2 in SQ. The Bayesian method performed similarly as GBLUP for most of the traits but substantially better for traits that were affected by SNPs of large effects as identified by GWAS. Conclusions Fatty acid composition in beef is influenced by a few host genes with major effects and many genes of smaller effects. With the current training population size and marker density, genomic prediction has the potential to predict the breeding values of fatty acid composition in beef cattle at a moderate to relatively high accuracy for fatty acids that have moderate to high heritability.
URLPMID:20590532Magsci
Summary Fatty acid composition, especially oleic acid (C18:1), plays an important role in the eating quality of meat in Japanese Black cattle. Therefore, the objective of this study was to identify loci associated with C18:1 in the intramuscular fat of the trapezius muscles in Japanese Black cattle using the Illumina BovineSNP50 BeadChip whole genome single nucleotide polymorphism (SNP) assay. We also evaluated the relationship between C18:1 and three fatty acid synthesis genes, fatty acid synthase ( FASN ), stearoyl-CoA desaturase and sterol regulatory element-binding protein-1 . In this experiment, we applied a mixed model and Genomic Control approach using selective genotyping to perform a genome-wide association study. A total of 160 animals (80 animals with higher values and 80 animals with lower values), selected from 3356 animals based on corrected phenotype, were genotyped using the Illumina BovineSNP50 BeadChip and three fatty acid synthesis genes, and the quality of these SNPs was assessed. In this study, a total of 38-955 SNPs, which included SNPs in the three fatty acid synthesis genes, were used, and the estimated inflation factor was 1.06. In the studied population, a total of 32 SNPs, including the FASN gene, had significant effects, and in particular 30 SNPs of all significant SNPs were located between 49 and 55-Mbp on chromosome 19. This study is one of the first genome-wide association studies for fatty acid composition in a cattle population using the recently released Illumina BovineSNP50 BeadChip.
URLPMID:4230646
Background Meat from Bos taurus and Bos indicus breeds are an important source of nutrients for humans and intramuscular fat (IMF) influences its flavor, nutritional value and impacts human health. Human consumption of fat that contains high levels of monounsaturated fatty acids (MUFA) can reduce the concentration of undesirable cholesterol (LDL) in circulating blood. Different feeding practices and genetic variation within and between breeds influences the amount of IMF and fatty acid (FA) composition in meat. However, it is difficult and costly to determine fatty acid composition, which has precluded beef cattle breeding programs from selecting for a healthier fatty acid profile. In this study, we employed a high-density single nucleotide polymorphism (SNP) chip to genotype 386 Nellore steers, a Bos indicus breed and, a Bayesian approach to identify genomic regions and putative candidate genes that could be involved with deposition and composition of IMF. Results Twenty-three genomic regions (1-Mb SNP windows) associated with IMF deposition and FA composition that each explain?????????1% of the genetic variance were identified on chromosomes 2, 3, 6, 7, 8, 9, 10, 11, 12, 17, 26 and 27. Many of these regions were not previously detected in other breeds. The genes present in these regions were identified and some can help explain the genetic basis of deposition and composition of fat in cattle. Conclusions The genomic regions and genes identified contribute to a better understanding of the genetic control of fatty acid deposition and can lead to DNA-based selection strategies to improve meat quality for human consumption.
URL
URLPMID:3840609 [本文引用: 1]
Background Systems biology enables the identification of gene networks that modulate complex traits. Comprehensive metabolomic analyses provide innovative phenotypes that are intermediate between the initiator of genetic variability, the genome, and raw phenotypes that are influenced by a large number of environmental effects. The present study combines two concepts, systems biology and metabolic analyses, in an approach without prior functional hypothesis in order to dissect genes and molecular pathways that modulate differential growth at the onset of puberty in male cattle. Furthermore, this integrative strategy was applied to specifically explore distinctive gene interactions of non-SMC condensin I complex, subunit G ( NCAPG ) and myostatin ( GDF8 ), known modulators of pre- and postnatal growth that are only partially understood for their molecular pathways affecting differential body weight. Results Our study successfully established gene networks and interacting partners affecting growth at the onset of puberty in cattle. We demonstrated the biological relevance of the created networks by comparison to randomly created networks. Our data showed that GnRH (Gonadotropin-releasing hormone) signaling is associated with divergent growth at the onset of puberty and revealed two highly connected hubs, BTC and DGKH , within the network. Both genes are known to directly interact with the GnRH signaling pathway. Furthermore, a gene interaction network for NCAPG containing 14 densely connected genes revealed novel information concerning the functional role of NCAPG in divergent growth. Conclusions Merging both concepts, systems biology and metabolomic analyses, successfully yielded new insights into gene networks and interacting partners affecting growth at the onset of puberty in cattle. Genetic modulation in GnRH signaling was identified as key modifier of differential cattle growth at the onset of puberty. In addition, the benefit of our innovative concept without prior functional hypothesis was demonstrated by data suggesting that NCAPG might contribute to vascular smooth muscle contraction by indirect effects on the NO pathway via modulation of arginine metabolism. Our study shows for the first time in cattle that integration of genetic, physiological and metabolomics data in a systems biology approach will enable (or contribute to) an improved understanding of metabolic and gene networks and genotype-phenotype relationships.
URLPMID:22785162Magsci
The lipid content and fatty acid (FA) profile have an important impact in human health as well as in the technological transformation and nutritional and organoleptic quality of meat. A genome-wide association study (GWAS) on 144 backcross pigs (25% Iberian 75% Landrace) was performed for 32 traits associated with intramuscular FA composition and indices of FA metabolism. The GWAS was carried out using Qxpak 5.0 and the genotyping information obtained from the Porcine SNP60K BeadChip (Illumina Inc., San Diego, CA). Signals of significant association considering a false- discovery rate (q-value < 0.05) were observed in 15 of the 32 analyzed traits, and a total of 813 trait-associated SNP (TAS), distributed in 43 chromosomal intervals on almost all autosomes, were annotated. According to the clustering analysis based on functional classification, several of the annotated genes are related to FA composition and lipid metabolism. Some interesting positional concordances among TAS and previously reported QTL for FA compositions and/or other lipid traits were also found. These common genomic regions for different traits suggest pleiotropic effects for FA composition and were found primarily on SSC4, SSC8, and SSC16. These results contribute to our understanding of the complex genetic basis of FA composition and FA metabolism.
URLPMID:24295214
Porcine fatty acid composition is a key factor for quality and nutritive value of pork. Several QTLs for fatty acid composition have been reported in diverse fat tissues. The results obtained so far seem to point out different genetic control of fatty acid composition conditional on the fat deposits. Those studies have been conducted using simple approaches and most of them focused on one single tissue. The first objective of the present study was to identify tissue-specific and tissue-consistent QTLs for fatty acid composition in backfat and intramuscular fat, combining linkage mapping and GWAS approaches and conducted under single and multitrait models. A second aim was to identify powerful candidate genes for these tissue-consistent QTLs, using microarray gene expression data and following a targeted genetical genomics approach. The single model analyses, linkage and GWAS, revealed over 30 and 20 chromosomal regions, 24 of them identified here for the first time, specifically associated to the content of diverse fatty acids in BF and IMF, respectively. The analyses with multitrait models allowed identifying for the first time with a formal statistical approach seven different regions with pleiotropic effects on particular fatty acids in both fat deposits. The most relevant were found on SSC8 for C16:0 and C16:1(n-7) fatty acids, detected by both linkage and GWAS approaches. Other detected pleiotropic regions included one on SSC1 for C16:0, two on SSC4 for C16:0 and C18:2, one on SSC11 for C20:3 and the last one on SSC17 for C16:0. Finally, a targeted eQTL scan focused on regions showing tissue-consistent effects was conducted with Longissimus and fat gene expression data. Some powerful candidate genes and regions were identified such as the PBX1, RGS4, TRIB3 and a transcription regulatory element close to ELOVL6 gene to be further studied. Complementary genome scans have confirmed several chromosome regions previously associated to fatty acid composition in backfat and intramuscular fat, but even more, to identify new ones. Although most of the detected regions were tissue-specific, supporting the hypothesis that the major part of genes affecting fatty acid composition differs among tissues, seven chromosomal regions showed tissue-consistent effects. Additional gene expression analyses have revealed powerful target regions to carry the mutation responsible for the pleiotropic effects.
URLPMID:5037705
The aim of this study was to integrate multi omics data to characterize underlying functional pathways and candidate genes for drip loss in pigs. The consideration of different omics levels allows elucidating the black box of phenotype expression. Metabolite and protein profiling was applied inMusculus longissimus dorsisamples of 97 Duroc × Pietrain pigs. In total, 126 and 35 annotated metabolites and proteins were quantified, respectively. In addition, all animals were genotyped with the porcine 60 k Illumina beadchip. An enrichment analysis resulted in 10 pathways, amongst others, sphingolipid metabolism and glycolysis/gluconeogenesis, with significant influence on drip loss. Drip loss and 22 metabolic components were analyzed as intermediate phenotypes within a genome-wide association study (GWAS). We detected significantly associated genetic markers and candidate genes for drip loss and for most of the metabolic components. On chromosome 18, a region with promising candidate genes was identified based on SNPs associated with drip loss, the protein “phosphoglycerate mutase 2” and the metabolite glycine. We hypothesize that association studies based on intermediate phenotypes are able to provide comprehensive insights in the genetic variation of genes directly involved in the metabolism of performance traits. In this way, the analyses contribute to identify reliable candidate genes.
URLPMID:27097669
Fatty acid composition profiles are important indicators of meat quality and tasting flavor. Metabolic indices of fatty acids are more authentic to reflect meat nutrition and public acceptance. To investigate the genetic mechanism of fatty acid metabolic indices in pork, we conducted genome-wide association studies (GWAS) for 33 fatty acid metabolic traits in five pig populations. We identified a total of 865 single nucleotide polymorphisms (SNPs), corresponding to 11 genome-wide significant loci on nine chromosomes and 12 suggestive loci on nine chromosomes. Our findings not only confirmed seven previously reported QTL with stronger association strength, but also revealed four novel population-specific loci, showing that investigations on intermediate phenotypes like the metabolic traits of fatty acids can increase the statistical power of GWAS for end-point phenotypes. We proposed a list of candidate genes at the identified loci, including three novel genes (FADS2,SREBF1andPLA2G7). Further, we constructed the functional networks involving these candidate genes and deduced the potential fatty acid metabolic pathway. These findings advance our understanding of the genetic basis of fatty acid composition in pigs. The results from European hybrid commercial pigs can be immediately transited into breeding practice for beneficial fatty acid composition.
URLPMID:28402008
react-text: 380 The leptin receptor (LEPR) gene is considered a candidate gene for fatness traits. It is located on SSC 6 in a region in which quantitative trait loci (QTLs) for backfat thickness (BF), fat area ratios, and serum leptin concentration (LEPC) have previously been detected in a Duroc purebred population. The objectives of the present study were to identify porcine LEPR polymorphisms and examine... /react-text react-text: 381 /react-text [Show full abstract]
URLPMID:28940847
Abstract Chinese indigenous pigs display marked genetic and phenotypic differences compared with western commercial pigs. In this study, we tested the association between 660K SNPs and longissimus muscle fatty acid composition traits in Chinese Erhualian (n0002=0002331) and Bamaxiang (n0002=0002315) pigs based on a customized 1.4 million SNP array. We identified a total of 64 significant associations for 20 fatty acid composition traits at the p-value threshold of 100020103000210 -6 among which 42 associations in low linkage disequilibrium (r 20002 <0002.2) with previously reported loci were considered novel. We substantially improved the strength and precision of the associations at four previously detected loci near FADS2, ELOVL7, ELOVL6 and FASN genes, facilitating follow-up candidate gene studies. Moreover, we also identified loci near ABCD2, ACSBG1, ELOVL5, HPGDS, DAGT2, ACAD10 and ACSL1 genes with function relevant to metabolism of fatty acids. In this study, valuable genetic variants and candidate genes associated with fatty acid composition traits were identified in Erhualian and Bamaxiang pigs. Some identified loci could be used to improve pork nutrition in pig breeding practice. Using the SNP array with higher marker density and less ascertainment bias improved QTL detection power and precision in Chinese indigenous pigs. 0008 2017 Blackwell Verlag GmbH.
URL
URLPMID:23341976
APOA2 is a protein implicated in triglyceride, fatty acid and glucose metabolism. In pigs, the APOA2 gene is located on pig chromosome 4 (SSC4) in a QTL region affecting fatty acid composition, fatness and growth traits. In this study, we evaluated APOA2 as a candidate gene for meat quality traits in an Iberian Landrace backcross population. The APOA2:c.131T>A polymorphism, located in exon 3... [Show full abstract]
URLPMID:3676363
Abstract Fatty acid composition is an important phenotypic trait in pigs as it affects nutritional, technical and sensory quality of pork. Here, we reported a genome-wide association study (GWAS) for fatty acid composition in the longissimus muscle and abdominal fat tissues of 591 White Duroc×Erhualian F2 animals and in muscle samples of 282 Chinese Sutai pigs. A total of 46 loci surpassing the suggestive significance level were identified on 15 pig chromosomes (SSC) for 12 fatty acids, revealing the complex genetic architecture of fatty acid composition in pigs. Of the 46 loci, 15 on SSC5, 7, 14 and 16 reached the genome-wide significance level. The two most significant SNPs were ss131535508 (P66=662.48×10(-25)) at 41.39 Mb on SSC16 for C20∶0 in abdominal fat and ss478935891 (P66=663.29×10(-13)) at 121.31 Mb on SSC14 for muscle C18∶0. A meta-analysis of GWAS identified 4 novel loci and enhanced the association strength at 6 loci compared to those evidenced in a single population, suggesting the presence of common underlying variants. The longissimus muscle and abdominal fat showed consistent association profiles at most of the identified loci and distinct association signals at several loci. All loci have specific effects on fatty acid composition, except for two loci on SSC4 and SSC7 affecting multiple fatness traits. Several promising candidate genes were found in the neighboring regions of the lead SNPs at the genome-wide significant loci, such as SCD for C18∶0 and C16∶1 on SSC14 and ELOVL7 for C20∶0 on SSC16. The findings provide insights into the molecular basis of fatty acid composition in pigs, and would benefit the final identification of the underlying mutations.
URLPMID:28494783
Fatty acid composition contributes importantly to meat quality and is essential to the nutritional value of the meat. Identification of genetic factors underlying levels of fatty acids can be used to breed for pigs with healthier meat. The aim of this study was to conduct genome-wide association studies (GWAS) to identify QTL regions affecting fatty acid composition in backfat from the pig breeds Duroc and Landrace. Using data from the Axiom porcine 660K array, we performed GWAS on 454 Duroc and 659 Landrace boars for fatty acid phenotypes measured by near-infrared spectroscopy (NIRS) technology (C16:0, C16:1n-7, C18:0, C18:1n-9, C18:2n-6, C18:3n-3, total saturated fatty acids, monounsaturated fatty acids and polyunsaturated fatty acids). Two QTL regions on SSC4 and SSC14 were identified in Duroc for thede novosynthesized fatty acids traits, whereas one QTL on SSC8 was detected in Landrace for C16:1n-7. The QTL region on SSC14 has been reported in previous studies and a putative causative mutation has been suggested in the promoter region of theSCDgene. Whole genome re-sequencing data was used for genotype imputation and to fine map the SSC14 QTL region in Norwegian Duroc. This effort confirms the location of the QTL on this chromosome as well as suggesting other putative candidate genes in the region. The most significant single nucleotide polymorphisms (SNPs) located on SSC14 explain between 55 and 76% of the genetic variance and between 27 and 54% of the phenotypic variance for thede novosynthesized fatty acid traits in Norwegian Duroc. For the QTL region on SSC8 in Landrace, the most significant SNP explained 19% of the genetic variance and 5% of the phenotypic variance for C16:1n-7. This study confirms a major QTL affecting fatty acid composition on SSC14 in Duroc, which can be used in genetic selection to increase the level of fatty acid desaturation. The SSC14 QTL was not segregating in the Landrace population, but another QTL on SSC8 affecting C16:1n-7 was identified and might be used to increase the level of desaturation in meat products from this breed. The online version of this article (doi:10.1186/s12864-017-3752-0) contains supplementary material, which is available to authorized users.
URLPMID:29580300
Abstract Dietary fatty acid (FA) composition has an impact on human health. There is an increasing request from consumers for healthier food and pork industry must respond to it without worsening performance and the technological properties of pork products. The inclusion of genetic markers for carcass FA composition in pig selection schemes could be a useful tool to reach the right balance between unsaturated and saturated FAs to satisfy market demands. With the aim of finding genomic regions associated with porcine backfat FA composition, a genome-wide association study was performed on 798 Italian Large White pigs genotyped using Illumina PorcineSNP60 k. The strongest associations with backfat contents of palmitic, palmitoleic, oleic, medium-chain and long-chain FAs were found for the Sus scrofa chromosome (SSC) 8 region located at 119 to 122 Mb, where the gene ELOVL FA elongase 6 is mapped. Palmitic, palmitoleic, stearic and oleic acid contents were also found associated with SSC14, in particular with the genomic region at 121 to 124 Mb, where stearoyl-CoA desaturase 9 gene lies. On the other hand, the genomic regions associated with backfat contents of arachidic, arachidonic, n-6 and n-3 FAs showed to harbour mainly genes involved in dietary lipids and carbohydrates digestion, absorption and utilisation. To our knowledge, this is the first study performed in Large White pigs identifying markers and genomic regions associated with backfat FA composition. The results validate in Large White some associations previously detected in other pig breeds and indicate the involvement of distinct metabolic pathways in the deposition pattern of essential and non-essential FAs.
URLPMID:25149343Magsci
Androstenone is one of the compounds causing boar taint of pork and is highly heritable (approximately 0.6). Recently, indirect genetic effects (IGE; also known as associative effects or social genetic effects) were found for androstenone, meaning that pen mates (boars) affect each other's androstenone level genetically. Similar to estimating variance components with a direct-indirect animal model, direct and indirect genetic SNP effects can be estimated for androstenone. This study aims to detect SNP with significant direct genetic effects and IGE on androstenone. The dataset consisted of 1,282 noncastrated boars (993 boars genotyped) from 184 groups of pen members. After quality control, 46,421 SNP were included in the analysis. One model for single-SNP regression was fitted, where both the direct SNP effect of the individual itself and the indirect SNP effects of its pen mates were included. None of the SNP (direct or indirect) were found genomewide significant. One QTL on SSC6 was chromosome-wide significant for the direct effect. A single SNP on SSC9 and 2 regions and a single SNP on SSC14 were found for the indirect effect. A backwards elimination method and haplotype analysis were used to quantify the variance explained by the SNP. The backwards elimination method identified 4 independent regions affecting androstenone. The QTL on SSC6 explained 2.1 and 2.6% of the phenotypic variance using the backwards elimination method or the haplotype analysis. The QTL on SSC14 explained 3.4 and 2.7% of the phenotypic variance using the backwards elimination method or the haplotype analysis. The single association on SSC9 explained 2.2% of the phenotypic variance. All significant QTL together explained 7 to 8% of phenotypic variance and 40 to 44% of the total genetic variance available for response to selection. Besides the newly discovered QTL and the confirmation of known QTL, this study also presents a methodology to model SNP for IGE.
URLPMID:27076351
Heat stress in poultry results in considerable economic losses and is a concern for both animal health and welfare. Physiological changes occur during periods of heat stress, including changes in blood chemistry components. A highly advanced intercross line, created from a broiler (heat susceptible) by Fayoumi (heat resistant) cross, was exposed to daily heat cycles for seven days starting at 2202days of age. Blood components measured pre-heat treatment and on the seventh day of heat treatment included pH, pCO2, pO2, base excess, HCO3, TCO2, K, Na, ionized Ca, hematocrit, hemoglobin, sO2, and glucose. A genome-wide association study (GWAS) for these traits and their calculated changes was conducted to identify quantitative trait loci (QTL) using a 60002K SNP panel. There were significant increases in pH, base excess, HCO3, TCO2, ionized Ca, hematocrit, hemoglobin, and sO2, and significant decreases in pCO2and glucose after 702days of heat treatment. Heritabilities ranged from 0.01-0.21 for pre-heat measurements, 0.01-0.23 for measurements taken during heat, and 0.00-0.10 for the calculated change due to heat treatment. All blood components were highly correlated within measurement days, but not correlated between measurement days. The GWAS revealed 61 QTL for all traits, located on GGA (Gallus galluschromosome) 1, 3, 6, 9, 10, 12–14, 17, 18, 21–28, and Z. A functional analysis of the genes in these QTL regions identified the Angiopoietin pathway as significant. The QTL that co-localized for three or more traits were on GGA10, 22, 26, 28, and Z and revealed candidate genes for birds’ response to heat stress. The results of this study contribute to our knowledge of levels and heritabilities of several blood components of chickens under thermoneutral and heat stress conditions. Most components responded to heat treatment. Mapped QTL may serve as markers for genomic selection to enhance heat tolerance in poultry. The Angiopoietin pathway is likely involved in the response to heat stress in chickens. Several candidate genes were identified, giving additional insight into potential mechanisms of physiologic response to high ambient temperatures. The online version of this article (doi:10.1186/s12864-016-2601-x) contains supplementary material, which is available to authorized users.
URL
Evidence indicates that high62grade serous ovarian carcinoma arises from the fallopian tube, rather than ovarian surface epithelium. This is termed the ‘tubal origin’ theory. The aim of the present study was to compare the immunophenotype and gene expression profiling among high62grade serous ovarian carcinoma (HGSOC), fallopian tube epithelium (FTE) and ovarian surface epithelium (OSE) based on tubal origin theory, and identify the differential genes associated with ovarian carcinogenesis. A total of 61 cases of fresh tissue samples including 21 cases of HGSOC, 20 cases of OSE, and 20 cases of FTE were obtained following surgical resection. Immunostaining was performed to detect the expression of PAX8, which has been considered as a potential immunophenotype marker of Müllerian origin. Illumina BeadChip was applied for gene expression profiling. Reverse transcription62quantitative polymerase chain reaction (RT62qPCR) was performed to confirm the differential expression of candidate genes between HGSOC and FTE. The results of the present study demonstrated that PAX8 was highly expressed in HGSOC (19/21, 90.4%) and FTE (20/20, 100%), but not in OSE (3/20, 14.3%). A dendrogram generated by cluster analysis indicated a higher similarity of gene expression profile between HGSOC and FTE than OSE. A total of 2,412 differentially expressed genes were identified (absolute fold change >2) between HGSOC and FTE, including 822 upregulated genes in cancer and 1,590 downregulated genes. S100 calcium binding protein P, Ras62interacting protein 1, Wnt family member 5A, tumor62associated calcium signal transducer 2, Dickkopf Wnt signaling pathway inhibitor 3 and tumor suppressor candidate 3 genes were identified as candidate markers, of which the differential gene expression in HGSOC and FTE was confirmed by RT62qPCR (P<0.05). The results indicate the presence of a greater similarity in the immunophenotype and gene expression profile of HGSOC and FTE, when compared with OSE, which was consistent with the tubal origin theory of HGSOC.
URLPMID:10714863
The objective of this study was to evaluate eight cowside ketone tests when used with milk for detection of subclinical ketosis. A total of 469 dairy cows in the first week of lactation were studied. Twelve percent of these cows had subclinical ketosis, defined as >1400 mol of hydroxybutyrate/L of blood serum. The Pink test liquid and the Ketolac test strip were highly sensitive for subclinical ketosis when used with milk. The Uriscan and Rapignost test strips were poorly sensitive; the Ketostix, Ketur-Test, and Medi-Test-Keton test strips and the Acetonreagenz test tablet were insensitive for subclinical ketosis when used with milk. Pink and Ketolac milk ketone tests are potentially useful tools for use in a routine monitoring program to detect subclinical ketosis in early postpartal dairy cows.
URLPMID:12466292 [本文引用: 1]
Fish-odor syndrome or Trimethylaminuria (OMIM #602079) in humans is an inborn error of metabolism associated with a characteristic fishy body odor due to elevated levels of trimethylamine (TMA) in body fluids. It is caused by loss-of-function mutations in FMO3 encoding flavin-containing mono-oxygenase 3. A fishy off-flavor is occasionally observed in cow's milk and it has been established recently that this phenotype is due to elevated TMA levels. Here, we report that fishy off-flavor in cow's milk is caused by a nonsense mutation (R238X) in the bovine FMO3 ortholog. RT-PCR analysis indicated that the mutant transcript is present in a very low amount. The mutation was found to be surprisingly common (q = 0.155) in one breed of cattle. [The sequence data described in this paper have been submitted to GenBank with accession nos. AF488417-AF488422. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: K. Sandberg and I. Hansson.]
URLPMID:20647382 [本文引用: 2]
Identifying trait-associated genetic variation offers new prospects to reveal novel physiological pathways modulating complex traits. Taking advantage of a unique animal model, we identified the I442M mutation in the non-SMC condensin I complex, subunit G (NCAPG) gene and the Q204X mutation in the growth differentiation factor 8 (GDF8) gene as substantial modulators of pre- and/or postnatal growth in cattle. In a combined metabolomic and genotype association approach, which is the first respective study in livestock, we surveyed the specific physiological background of the effects of both loci on body-mass gain and lipid deposition. Our data provided confirming evidence from two historically and geographically distant cattle populations that the onset of puberty is the key interval of divergent growth. The locus-specific metabolic patterns obtained from monitoring 201 plasma metabolites at puberty mirror the particular NCAPG I442M and GDF8 Q204X effects and represent biosignatures of divergent physiological pathways potentially modulating effects on proportional and disproportional growth, respectively. While the NCAPG I442M mutation affected the arginine metabolism, the 204X allele in the GDF8 gene predominantly raised the carnitine level and had concordant effects on glycerophosphatidylcholines and sphingomyelins. Our study provides a conclusive link between the well-described growth-regulating functions of arginine metabolism and the previously unknown specific physiological role of the NCAPG protein in mammalian metabolism. Owing to the confirmed effect of the NCAPG/LCORL locus on human height in genome-wide association studies, the results obtained for bovine NCAPG might add valuable, comparative information on the physiological background of genetically determined divergent mammalian growth.
URLPMID:23438684
In the field of dairy cattle research, it is of great interest to improve the detection and prevention of diseases (e.g., mastitis and ketosis) and monitor specific traits related to the state of health and management. During the standard milk performance test, traditional milk traits are monitored, and quality and quantity are screened. In addition to the standard test, it is also now possible to analyze milk metabolites in a high-throughput manner and to consider them in connection with milk traits to identify functionally important metabolites that can also serve as biomarker candidates. We present a study in which 190 milk metabolites and 14 milk traits of 1,305 Holstein cows on 18 commercial farms were investigated to characterize interrelations of milk metabolites between each other, to milk traits from the milk standard performance test, and to influencing factors such as farm and sire effect (half-sib structure). The effect of influencing factors (e.g., farm) varied among metabolites and traditional milk traits. The investigations of associations between metabolites and milk traits revealed groups of metabolites that show, for example, positive correlations to protein and casein, and negative correlations to lactose and pH. On the other hand, groups of metabolites jointly associated with the investigated milk traits can be identified and functionally discussed. To enable a multivariate investigation, 2 machine learning methods were applied to detect important metabolites that are highly correlated with the investigated traditional milk traits. For somatic cell score, uracil, lactic acid, and 9 other important metabolites were detected. Lactic acid has already been proposed as a biomarker candidate for mastitis in the recent literature. In conclusion, we found sets of metabolites eligible to predict milk traits, enabling the analysis of milk traits from a metabolic perspective and discussion of the possible functional background for some of the detected associations.
URLPMID:22098372Magsci
Abstract Ketosis is a common metabolic disease in dairy cows. Diagnostic markers for ketosis such as acetone and beta-hydroxybutyric acid (BHBA) are known, but disease prediction remains an unsolved challenge. Milk is a steadily available biofluid and routinely collected on a daily basis. This high availability makes milk superior to blood or urine samples for diagnostic purposes. In this contribution, we show that high milk glycerophosphocholine (GPC) levels and high ratios of GPC to phosphocholine (PC) allow for the reliable selection of healthy and metabolically stable cows for breeding purposes. Throughout lactation, high GPC values are connected with a low ketosis incidence. During the first month of lactation, molar GPC/PC ratios equal or greater than 2.5 indicate a very low risk for developing ketosis. This threshold was validated for different breeds (Holstein-Friesian, Brown Swiss, and Simmental Fleckvieh) and for animals in different lactations, with observed odds ratios between 1.5 and 2.38. In contrast to acetone and BHBA, these measures are independent of the acute disease status. A possible explanation for the predictive effect is that GPC and PC are measures for the ability to break down phospholipids as a fatty acid source to meet the enhanced energy requirements of early lactation.
URLPMID:25599412
Abstract The fundamental understanding of the mechanisms regulating milk protein synthesis is limited. This study aimed to elucidate the metabolic mechanisms of milk production affected by forage quality through studying metabolites from four biofluids (rumen fluid, milk, serum, and urine) collected from 16 lactating cows fed alfalfa hay (AH, high-quality, n = 8) and corn stover (CS, low-quality, n = 8) using gas chromatography-time-of-flight/mass spectrometry. The cows fed AH exhibited higher milk yield (P 1 and P < 0.05) between the AH- and CS-fed animals. These metabolites were involved in glycine, serine, and threonine metabolism; tyrosine metabolism; and phenylalanine metabolism. Further integrated key metabolic pathway analysis showed that the AH-fed cows may have more comprehensive amino acid metabolisms, suggesting that these metabolite-associated pathways may serve as biomarkers for higher milk yield and better milk protein quality.
URLPMID:4934687
A decrease in insulin sensitivity enhances adipose tissue lipolysis helping early lactation cows counteracting their energy deficit. However, excessive lipolysis poses serious health risks for cows, and its underlying mechanisms are not clearly understood. The present study used targeted ESI-LC-MS/MS-based metabolomics and indirect insulin sensitivity measurements to evaluate metabolic alterations in the serum of dairy cows of various parities experiencing variable lipolysis early postpartum. Thirty (12 primiparous and 18 multiparous) cows of Holstein Friesian and Simmental breeds, fed the same diet and kept under the same management conditions, were sampled at d 21 postpartum and classified as low (n = 10), medium (n = 8), and high (n = 12) lipolysis groups, based on serum concentration of nonesterified fatty acids. Overall, excessive lipolysis in the high group came along with impaired estimated insulin sensitivity and characteristic shifts in acylcarnitine, sphingomyelin, phosphatidylcholine and lysophospholipid metabolome profiles compared to the low group. From the detected phosphatidylcholines mainly those with diacyl-residues showed differences among lipolysis groups. Furthermore, more than half of the detected sphingomyelins were increased in cows experiencing high lipomobilization. Additionally, strong differences in serum acylcarnitines were noticed among lipolysis groups. The study suggests an altered serum phospholipidome in dairy cows associated with an increase in certain long-chain sphingomyelins and the progression of disturbed insulin function. In conclusion, the present study revealed 37 key metabolites as part of alterations in the synthesis or breakdown of sphingolipids and phospholipids associated with lowered estimated insulin sensitivity and excessive lipolysis in early-lactating cows.
URL
The efficiency with which ruminants convert feed to desirable products is difficult to measure under normal commercial settings. We explored the use of potential biological markers from easily obtainable samples, that is, blood, hair, and feces, to characterize potential causes of divergent efficiency when considered as residual feed intake (RFI) or feed conversion efficiency (FCE). A total of 54 Charolais bulls, 20 in period 1 and 34 in period 2, were examined for individual dry matter intake (DMI) and growth. Bulls were offered a diet of 70:30 wrapped grass silage to concentrate for 99 d. At the conclusion of the test period, blood samples were collected for the determination of vitamins B-2 and B-6, and plasma used for the determination of metabolites, natural isotopic N-15 abundance (N-15 NIA, expressed as delta N-15 parts per thousand) and fractionation (Delta N-15(plasma) (proteins-diet) and Delta C-13(plasma) (proteins-diet)) and near-infrared spectroscopy (NIRS). Feces were analyzed by NIRS. Bulls were slaughtered at 15-17 months of age and carcass characteristics determined. Bulls were ranked according to RFI with extremes (SD +/- 0.5; n = 31) classified as either efficient (Neg-RFI) or inefficient (Pos-RFI). Extreme bulls were then classified for FCE (high vs low FCE), changing the groups. Pos-RFI bulls consumed 14% more feed than Neg-RFI bulls for the same level of weight gain. Low FCE bulls tended to eat more, but had lower weight gains than high FCE bulls. No differences were detected in carcass conformation, fat scores, hot carcass weight, or dressing percentage. Yet, heart and bladder weights were heavier in Pos-RFI, and rumen weight tended to be heavier in Pos-RFI bulls. RFI did not affect bulk N-15 or BC fractionation. A negative correlation was observed between FCE and Delta N-15(plasma) (proteins-diet*) Inefficient bulls (Pos-RFI) had higher delta N-15 in glycine compared to Neg-RFI bulls. Similarly, metabolomic analysis showed a tendency for concentrations of glycine and sarcosine to be elevated in Pos-RFI bulls, whereas aspartic acid and carnosine tended to be elevated, and serine tended to be lower in High FCE. Among vitamins, only flavin adenine dinucleotide concentration was higher in the blood of bulls with High FCE. These results suggest that the two feed efficiency metrics differ in the underlying mechanisms of metabolism, where RFI is driven by differences in the energetic requirements of visceral organs and the extent of AA catabolism.
URLPMID:29032783
Abstract A targeted quantitative metabolomics approach was used to study temporal changes of serum metabolites in cows that normally released their fetal membranes and those that retained the placenta. We identified and measured serum concentrations of 128 metabolites including amino acids, acylcarnitines, biogenic amines, glycerophospholipids, sphingolipids and hexose at -8 and -4 weeks before parturition, during the week of retained placenta (RP) diagnosis, and at +4 and +8 weeks after parturition. In addition, we aimed at identifying metabolite signatures of pre-RP in the serum that might be used as predictive biomarkers for risk of developing RP in dairy cows. Results revealed major alterations in the metabolite fingerprints of pre-RP cows starting as early as -8 weeks before parturition and continuing as far as +8 weeks after calving. Biomarker candidates found in this study are mainly biomarkers of inflammation which might not be specific to RP. Therefore, the relevance of serum Lys, Orn, acetylornithine, lysophophatidylcholine LysoPC a C28:0, Asp, Leu and Ile as potential serum biomarkers for prediction of risk of RP in dairy cows will have to be tested in the future. In addition, lower concentrations of LysoPCs, Trp, and higher kynurenine in the serum during prepartum and the week of occurrence of RP suggest involvement of inflammation in the pathobiology of RP.
URL
URLPMID:23100586Magsci
Predicting phenotypes is a statistical and biotechnical challenge, both in medicine (predicting an illness) and animal breeding (predicting the carcass economical value on a young living animal). High-throughput fine phenotyping is possible using metabolomics, which describes the global metabolic status of an individual, and is the closest to the terminal phenotype. The purpose of this work was to quantify the prediction power of metabolomic profiles for commonly used production phenotypes from a single blood sample from the growing pig. Several statistical approaches were investigated and compared on the basis of cross validation: raw data vs. signal preprocessing (wavelet transformation), with a single-feature selection method. The best results in terms of prediction accuracy were obtained when data were preprocessed using wavelet transformations on the Daubechies basis. The phenotypes related to meat quality were not well predicted because the blood sample was taken some time prior to slaughter, and slaughter is known to have a strong influence on these traits. In contrast, phenotypes of potential economic interest (e.g., lean meat percentage and ADFI) were well predicted (R(2) = 0.7; P < 0.0001) using metabolomic data.
URLPMID:25087569Magsci
This study investigated whether carbonic anhydrase (CA)-VI has utility as a biomarker in swine kidney disease. Serum chemistry, histopathology, immunohistochemical staining and enzyme-linked immunosorbent assay (ELISA) analyses were performed. In the kidney of normal healthy pigs, CA-VI was localized in the epithelial cells of the renal distal straight tubules. CA-VI levels were 1665±653565ng/g wet tissue and 5065±656665ng/mL in normal pig kidney and urine, respectively, and 13665±6517365ng/mL in the urine of pigs with kidney disease. CA-VI urinary concentration was not correlated with urinary urea nitrogen (UUN), urinary creatinine (Cre), or urinary albumin levels in pigs with kidney disease. However, UUN and Cre levels were positively correlated in the urine of pigs with kidney disease. These data suggest that urinary CA-VI may represent a biomarker for kidney disease in pigs, particularly for disorders affecting distal straight tubules.
URL
1. Fishy taints in eggs from hens fed on rapeseed meal, which have been reported to occur in brown090006shelled eggs of some hens with Rhode Island Red ancestry, occur also in white090006shelled eggs of some hens of a strain of Brown Leghorns.
URLPMID:24700147Magsci
Ascites is a major problem for both human health and animal production, due to its association with high rates of morbidity and mortality, low efficiency of nutrient utilization, and permanent adverse effects on performance. Although it is one of the three major metabolic diseases in poultry production, the underlying mechanisms are largely unknown. In this study, six ascites syndrome (AS) chickens and six normal chickens were obtained from each group (108 chickens) at 21 and 35 days. A liver metabolomics method based on ultra-performance liquid chromatography/quadruple time-of-flight mass spectrometry (UPLC/Q-TOF/MS) was used to explore the metabolic pattern of low molecular mass metabolites in chickens with low-temperature-induced AS. Coupled with blood biochemistry and histopathology results, the significant difference in metabolic profiling between the AS group and the control group, as determined through pattern recognition analysis, indicated changes in global tissue metabolites. The results showed that a primary bile acid synthesis disorder and inflammation had occurred by 21 days and that lysophospholipid metabolism was disrupted by 35 days with the continuation of low temperatures. Several metabolites, including taurodeoxycholic acid, cholic acid glucuronide, glycocholic acid, LysoPC(15-:-0) and taurocholic acid, were identified as the potential and proper biomarkers of AS. These biochemical changes in tissue metabolites are related to perturbations of lipid metabolism, which may be helpful to further understand the AS mechanisms. This work shows that the metabolomics is a valuable tool for studying metabolic diseases.
URLPMID:4838225
This study was conducted to characterize metabolic features of the breast muscle (pectoralis major) in chickens affected with the Wooden Breast myopathy. Live birds from two purebred chicken lines and one crossbred commercial broiler population were clinically examined by manual palpation of the breast muscle (pectoralis major) at 47-48 days of age. Metabolite abundance was determined by gas chromatography/mass spectrometry (GC/MS) and liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) using breast muscle tissue samples from 16 affected and 16 unaffected chickens. Muscle glycogen content was also quantified in breast muscle tissue samples from affected and unaffected chickens. In total, levels of 140 biochemicals were significantly different (FDR < 0.1 and fold-change A/U > 1.3 or < 0.77) between affected and unaffected chickens. Glycogen content measurements were considerably lower (1.7-fold) in samples taken from Wooden Breast affected birds when compared with samples from unaffected birds. Affected tissues exhibited biomarkers related to increased oxidative stress, elevated protein levels, muscle degradation, and altered glucose utilization. Affected muscle also showed elevated levels of hypoxanthine, xanthine, and urate molecules, the generation of which can contribute to altered redox homeostasis. In conclusion, our findings show that Wooden Breast affected tissues possess a unique metabolic signature. This unique profile may identify candidate biomarkers for diagnostic utilization and provide mechanistic insight into altered biochemical processes contributing to tissue hardening associated with the Wooden Breast myopathy in commercial chickens.
URLPMID:28539642
To select metabolic biomarkers and differentially expressed genes (DEGs) associated with resistant-ascites syndrome (resistant-AS), we used innovative techniques such as metabolomics and transcriptomics to comparatively examine resistant-AS chickens and AS controls. Metabolomic evaluation of chicken serum using ultra-performance liquid chromatography-quadruple time-of-flight high-sensitivity mass spectrometry (UPLC-QTOF/HSMS) showed significantly altered lysoPC(18:1), PE(18:3/16:0), PC(20:1/18:3), DG(24:1/22:6/0:0), PS(18:2/18:0), PI(16:0/16:0), PS(18:0/18:1), PS(14:1/14:0), dihydroxyacetone, ursodeoxycholic acid, tryptophan, L-valine, cycloserine, hypoxanthine, and 4-O-Methylmelleolide concentrations on day 21 and LysoPC(18:0), LysoPE(20:1/0:0), LysoPC(16:0), LysoPE(16:0/0:0), hypoxanthine, dihydroxyacetone, 4-O-Methylmelleolide, LysoPC(18:2), and PC(14:1/22:1) concentrations on day 35, between the susceptible and resistant groups. Compared to the susceptible group, transcriptomic analysis of liver samples using RNA-seq revealed 413 DEGs on day 21 and 214 DEGs on day 35 in the resistant group. Additional evaluations using gene ontology (GO) indicate that significant enrichment occurred in the oxygen transportation, defensive reactions, and protein modifications of the decreased DEGs as well as in the cell morphological formation, neural development, and transforming growth factor (TGF)-beta signalling of the increased DEGs on day 21. Oxygen transportation was also significantly enriched for downregulated DEGs on day 35. The combinatory evaluation of the metabolome and the transcriptome suggests the possible involvement of glycerophospholipid metabolism in the development of resistant-AS in broilers.
URLPMID:27664620
Increased incidences of pectoralis muscle dystrophy are observed in commercial chicken products, but the muscle physiological causes for the condition remain to be identified. In the present study a high-resolution magic angle spinning (HR-MAS) proton (1H) NMR spectroscopic examination of intact pectoralis muscle samples (n=77) were conducted to explore metabolite perturbations associated with the muscle dystrophy condition for the very first time. Both in chicken with an age of 21 and 31days, respectively, pectoralis muscle dystrophy was associated with a significantly lower content of anserine (p=0.034), carnosine (p=0.019) and creatine (p=0.049). These findings must be considered intriguing as they corroborate that characteristic muscle di-peptides composed of alanine and histidine derivatives such as anserine are extremely important in homeostasis of contractile muscles as a results of their role as buffering, anti-oxidative, and anti-glycation capacities.
URLPMID:29806083
Background: Chicken embryos are widely used as a model for studies of obesity; however, no detailed information is available about the dynamic changes of proteins during the regulation of adipose biology and metabolism. Thus, the present study used an isobaric tags for relative and absolute quantitation (iTRAQ)-based proteomic approach to identify the changes in protein abundance at different... [Show full abstract]
URLPMID:24235218 [本文引用: 1]
In this study, using a newly developed TaqMan-based real-time PCR method for the T329S mutation in the flavin-containing monooxygenase 3 (FMO3) gene, a marker-assisted selective breeding program against the unfavorable T allele was implemented in the Beijing You chicken breeding stock from 2010 to 2012. A total of 2,359 breeder candidate chickens were detected. After 1-generation culling in both males and females and 2-generation culling only in males, genotyping results in 2013 showed that there still remained a low unfavorable allele frequency of 0.022 in this population. The results indicated that to ensure a complete eradication of the defective tainting mutation in FMO3 out of the Beijing You population, more strict breeding and management schemes should be carried out in the future.
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}