 ,四川农业大学动物科技学院,动物遗传育种研究所,成都 611130
,四川农业大学动物科技学院,动物遗传育种研究所,成都 611130Advances in mammalian three-dimensional genome by using Hi-C technology approach
Chunyou Ning, Mengnan He, Qianzi Tang, Qing Zhu, Mingzhou Li, Diyan Li,Institute of Animal Genetics and Breeding, College of Animal Science and Technology, Sichuan Agricultural University, Chengdu 611130, China通讯作者:
编委: 赵方庆
收稿日期:2018-11-21修回日期:2019-01-23网络出版日期:2019-02-28
| 基金资助: |
Received:2018-11-21Revised:2019-01-23Online:2019-02-28
| Fund supported: |
作者简介 About authors
宁椿游,博士研究生,研究方向:动物遗传育种与繁殖E-mail:ningchunyou@hotmail.com。

摘要
关键词:
Abstract
Keywords:
PDF (868KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
宁椿游, 何梦楠, 唐茜子, 朱庆, 李明洲, 李地艳. 基于Hi-C技术哺乳动物三维基因组研究进展[J]. 遗传, 2019, 41(3): 215-233 doi:10.16288/j.yczz.18-317
Chunyou Ning, Mengnan He, Qianzi Tang, Qing Zhu, Mingzhou Li, Diyan Li.
染色质是遗传物质的载体,其活性和功能由线性的基因组序列、序列之间的相互作用和动态变化的染色质三维空间构象共同决定。早期对基因的表达调控研究大都是基于一维(基因序列)和二维(不同序列的相互作用)的层面,将基因组作为线性分子模型去研究机体或细胞内的各种调控机理。随着更多的一维和二维基因组数据的产生,现有的线性模型不足以揭示这些离散的调控元件、结构变异与基因功能的联系。由此,基于染色质空间构象解释基因表达调控机制的三维基因组学应运而生。
2009年,Lieberman-Aiden等[1]在Science上首次报道了以整个细胞核为研究对象,利用高通量测序技术,结合生物信息学分析方法,研究全基因组范围内DNA序列在空间位置上任意两位点间互作关系的高通量染色体构象捕获(high-througnput chromosome conformation capture, Hi-C)技术。Hi-C及其衍生技术的出现,使得人们能够从技术上突破对于三维基因组学认识的障碍。三维基因组学研究能够解释那些距离目标基因几kb甚至几Mb的调控元件如何调控基因表达[2],其研究重点在于解析细胞核内染色质的不同空间构象及其结构单元,探究不同类型的结构单元如何介导转录调控元件与基因间的互作关系,从而阐明基因功能与转录调控的分子机制。本文通过目前已有的研究,对哺乳动物细胞核内三维基因组结构划分、构象单元作用以及目前Hi-C技术在三维基因组学应用等方面进行了介绍。
1 哺乳动物三维基因组的结构单元
目前,部分哺乳动物和其他真核生物的细胞核内染色质三维折叠组装的基本规律被揭示[3]。在哺乳动物细胞核内,染色质以严密的层级结构折叠组装成高级构象,这些层级结构单元由大到小依次为染色体疆域(chromosome territory, CT)、染色质区室(chromatin compartment A/B)、拓扑关联结构域(topological associated domain, TAD)和染色质环(chromatin loop)[4,5](图1)。其中,染色体疆域是普遍存在的基因组空间结构,不同的染色体在细胞核内占据不同的疆域;染色质区室是由基因组表观状态所决定的较大的结构单元,与染色质活性密切相关;拓扑关联结构域是细胞核内稳定存在的空间结构单元,在局部范围内介导基因的表达调控;染色质环是直接调控基因表达的最精细的结构和功能单元,通常由启动子与远端增强子互作形成,在介导基因的转录激活中发挥着重要作用。图1
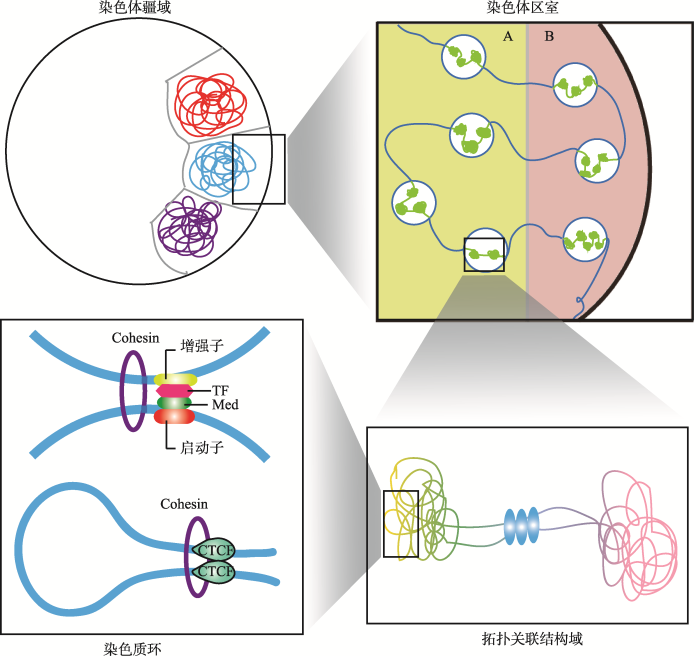 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1哺乳动物细胞核内染色质的层级结构
Fig. 1The hierarchical structure of chromatin in mammalian nuclei
1.1 染色体疆域
早在20世纪,细胞学家Theodore Boveri研究蛔虫(Ascaris lumbricoides)间期核中的染色质时,发现染色质在细胞核中并非随机排列[6]。后来的研究发现,每条染色体在间期中都各自占据了一块特定的不重叠的核区域,即染色体疆域(CT)[7]。不同染色体之间的重叠仅限于CTs的边界[8]。CTs的定位与其基因密度有关,不同基因密度的CTs占据着不同的核位置[9]。在CTs中,每条染色体都被限制在各自具体的核空间中,仅仅一小部分延伸到邻近的核空间中,因此染色体折叠形成CT被认为是内部核运动的屏障[4,10]。CTs的定位还与细胞类型的特异性因素如复制时间和转录活性相关,早期复制位点和活性基因倾向定位于细胞核内部,而晚期复制位点和抑制基因倾向于核边缘[11,12]。Solovei等[13]发现,与白天活动的动物和大多数真核生物相比,夜行哺乳动物的视网膜杆状细胞中的CT位置是倒置的。在夜行性视网膜杆状细胞中,异染色质定位于核中心,常染色质位于核外围。核组织的模型计算发现这种倒置的CT结构形式能够有效地引导光线,从而有助于夜行动物适应夜晚的生活方式。1.2 染色体区室
2009年,Lieberman-Aiden等[1]首次运用Hi-C技术揭示了人淋巴母细胞(GM06990)的三维基因组结构。该研究证实了之前通过3C (chromosome conformation capture)技术和3D-FISH (3D fluorescence in situ hybridization)技术发现的CTs的存在,即那些较小的、基因富集(gene-rich)的染色体在空间上更接近。对同一条染色体内部互作(cis interaction)进行分析发现,染色质间互作强度随着基因组线性距离的增加而降低,且同一条染色体内部互作强度高于不同染色体间的互作。该研究也首次提出了基因组空间结构的另一重要特征,即染色质是由compartment A和B两种基因组间隔区交叉分布构成(图2)。同一种compartment内部具有更高的染色质互作频率,且在同一线性距离上,compartment B之间的互作频率高于compartment A。其中,compartment A为开放(open)染色质区室,多为常染色质,是基因富集区域,GC含量高,基因高表达;而compartment B为封闭(close)染色质区室,多为异染色质区域,通常是基因沙漠(gene-desert)区域,GC含量低,基因表达量相对compartment A低。Compartment A和B的特征与其他基因组和表观特征呈现高度相关,其中compartment A区域有激活的染色质标签H3K36me3,具有更高的染色质可接近性(DNAseⅠ 高度敏感),而compartment B与抑制性组蛋白标签H3K27me3高度相关。因此,compartment A是更加开放的、可接近的、转录激活的染色质区域。图2
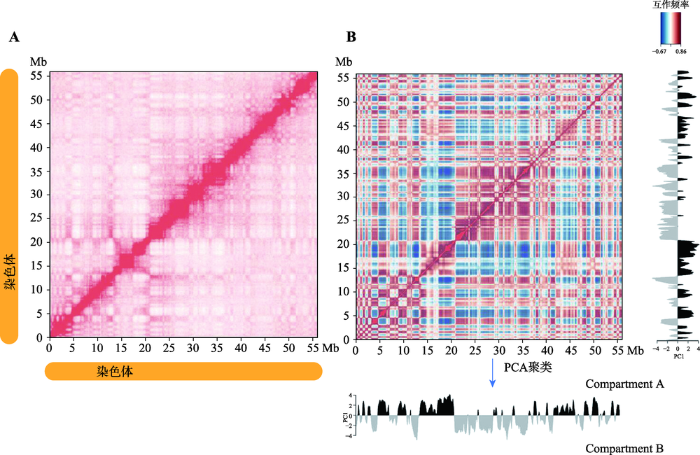 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2Hi-C数据显示每条染色体两种类型的compartments
A:Hi-C原始互作矩阵;B:相关系数矩阵。根据本课题组对猪第18号染色体的Hi-C测序结果(数据未发表)绘制。
Fig. 2Hi-C data reveal two types of compartments in each chromosome
高阶染色质结构是基因表达的重要调节因子。虽然在基因组中已经发现了动态染色质结构,但在哺乳动物发育和谱系规范中染色质动态的完整范围仍有待确定。美国加州大学Bing Ren 教授及其团队通过绘制人类ES细胞核4个ES细胞衍生谱系的全基因组染色质相互作用图谱,揭示了在谱系规范中广泛的染色质重组[14]。在胚胎干细胞分化成4种特定细胞系的过程中,至少有36%的基因组发生了空间可塑性重排(即compartment A/B switch)。这些重排与特定的细胞功能相关,B到A状态改变的基因倾向于更高表达,而A到B状态改变的基因倾向于更低的表达。这说明在一个全局范围内,compartment A/B具有较高可塑性,并且与细胞特异性基因表达相关,并不起决定性的作用。
在更高分辨率的Hi-C互作图谱中,compartment A/B还能被分成更小的subcompartments,即A1、A2和B1、B2、B3,并且每一个subcompartment都与部分特异性的组蛋白修饰模式相关联[15]。在果蝇(Drosophila)细胞中同样存在这5种主要的subcompartments染色质类型(2个激活性,3个抑制性)[16],表明这种相似的compartments染色质结构在后生动物中高度保守。
1.3 拓扑关联结构域
2012年5月,Nature同时报道了美国麻省大学医学院分子遗传学家Job Dekker以及美国加州大学Ludwig癌症研究所Bing Ren教授的研究成果,他们均发现了哺乳动物细胞内染色质折叠的二级结构单元——TAD[17,18]。研究发现,将Hi-C互作图谱的分辨率提高到40 kb或更高时,高度自我相关的染色质区域在互作热图上表现为间隔的三角形,即拓扑关联结构域(TAD) (图3)。其中,Bing Ren教授及其团队研究了小鼠(Mus musculus)的胚胎干细胞(mESCs)、大脑皮层(cortex)以及人的胚胎干细胞(hESCs)和肺成纤维细胞(IMR90)的Hi-C数据,在小鼠胚胎干细胞的Hi-C数据分析中找到了约2200个平均大小为0.88 Mb、约占基因组91%区域的TAD结构,且在这些TAD内部的互作显著高于TAD间的互作[17]。此外,非哺乳动物如果蝇[19]、斑马鱼(Danio rerio)[20]、线虫(Caenorhabditis elegans)[21]以及酵母(Saccharomyce)[22,23]等基因组也具有这种相似的TAD结构,而在拟南芥(Arabidopsis thaliana)的Hi-C结果中并未发现类似TAD样的结构[24,25]。但最近的研究发现,水稻(Oryza sativa L.)中同样存在非典型的TAD结构,并且平均分布在水稻的12条染色体中,表明TAD结构在植物中可能并不保守[26,27]。图3
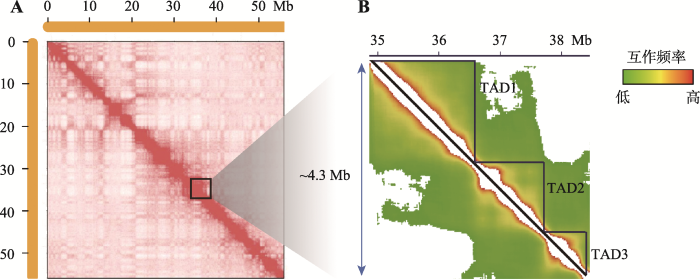 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Hi-C测序数据中鉴定得到的拓扑结构域(TAD)
A:500 kb Hi-C互作矩阵;B:20 kb Hi-C互作矩阵。根据本课题组对猪第18号染色体的Hi-C测序结果(数据未发表)绘制。
Fig. 3Identification of topologically associating domains (TADs) from Hi-C data
越来越多的证据表明,TAD作为基因组折叠的功能单元,在不同的动物细胞中稳定存在[28,29]。首先,TAD在不同细胞间的位置相对稳定,并且其似乎并不与组织特异性的基因表达或组蛋白修饰相关;其次,TAD的定位也具有保守性,在人和小鼠的ES细胞中,共有的TAD边界达到50%~70%[14],并且这种保守性还体现在果蝇等昆虫上[30],表明TAD是动物基因组的固有特性。此外,研究还发现,TAD边界与复制域(replication domain)边界存在着大量的重合,说明TAD是复制时间调节的稳定单位[31]。
TAD作为基因组三维结构单元具有重要特征,其具体形成机制正在被不断揭示。研究显示,TAD边界富集着大量的标记因子,包括H3K4me3和H3K36me3组蛋白修饰位点、转录起始位点(transcription start site, TSS)、看家基因、tRNA、短散在元件(SINE)以及阻遏子CTCF和黏连蛋白复合物(cohesin complex),暗示这些因子在建立TAD的过程中存在着重要作用。在小鼠ES细胞中,分别有75%和33%的TAD边界在CTCF结合位点、看家基因位置的20 kb以内[17];而在基因表达时,CTCF能够与黏连蛋白协同合作,使得线性距离较远的增强子与基因的启动子相结合,激活转录表达,由此说明CTCF的结合和高表达水平的转录活性可能与TAD的形成有关。为了揭示CTCF和cohesin在TAD形成中的作用,Bing Ren教授团队分别对CTCF和cohesin进行了精确敲除,并结合4C(chromosome conformation capture-on-chip)、Hi-C和3D- FISH技术检测了染色质组装的变化及其对基因表达的影响[32]。研究发现,CTCF和cohesin对于TAD的形成具有不同的作用,cohesin主要参与TAD内部的染色质互作,而CTCF主要参与它们之间的空间隔离。CTCF稳定地绑定在染色质上,并且决定了cohesin的定位从而维持边界的稳定。如果没有CTCF,cohesin将不能准确定位,会形成跨越边界的非特异性互作。当cohesin被降解后,所有的loop域(同一条染色体上的两个位点之间具有CTCF和cohesin蛋白绑定的区域)都消失了,但compartment域(具有相似组蛋白修饰的间隔区域)或组蛋白标签并不会受到影响[33]。Loop域的缺失并不会导致广泛的基因异常表达,但确实会显著影响小部分基因的表达活性。Schwarzer等[34]也发现,TAD的形成依赖于cohesin,而compartment域却不受影响。在对染色质结构的进化分析中发现,CTCF和cohesin对于驱动染色质结构的改变也起着直接的作用[35]。也有部分研究发现,将CTCF或cohesin进行功能性敲除或敲低虽然能够引起局部互作的缺失和基因表达紊乱,但compartment或TAD在敲除之后仍然得以保留[36,37]。Barutcu等[38]也发现,敲除或者插入Firre(X染色体中存在15个CTCF位点的保守区域)序列片段都不足以以特定性别或等位基因的方式改变活性X染色体中的TAD边界。这可能是由于在这些实验中对CTCF或cohesin的敲除并 不完全或所用细胞类型的差异所致,或者是除了CTCF/cohesin结合外,可能还存在着其他的机制调控TAD的形成。
最近对果蝇的研究发现,TAD并不是由CTCF和cohesin所定义。通过超高深度Hi-C技术方法,研究人员发现果蝇基因组中TAD的实际数目是现有注释的10倍,而且整个基因组全部为TAD所覆盖,并且果蝇染色质中绝大多数的TAD边界都是由特异性的绝缘子蛋白复合物BEAF-32/CP190或BEAF-32/ Chromator所定义,而不是与人同源的CTCF/cohesin[39]。现有证据表明,BEAF-32是果蝇中特异性结合DNA的绝缘蛋白之一,而CP190/Chromator恰好可与BEAF-32结合并介导远距离相互作用,类似于哺乳动物细胞中的cohesin。这些结果表明,虽然CTCF/cohesin在果蝇中并不参与TAD的形成,但是,与其功能相似但不同源的蛋白复合物起到了哺乳动物细胞中CTCF/cohesin相同的作用。此外,除了CTCF和cohesin,DNA超螺旋在TAD的形成可能也发挥着作用[40],而超螺旋结构域的边界在位置上与TAD边界确实有着部分的重叠区域[41],但这种因素是否真正影响了TAD结构的形成,则需要进一步的实验验证。
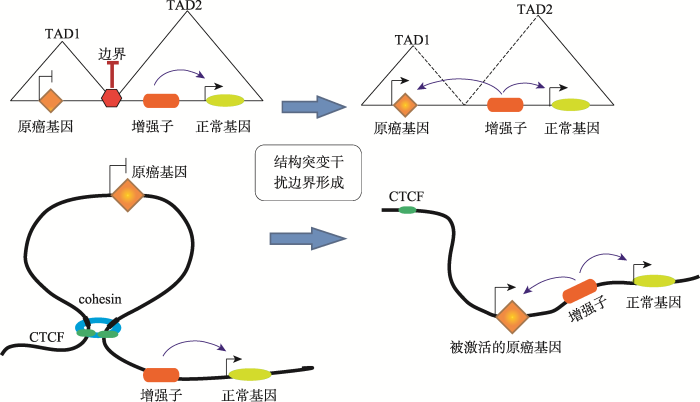
TAD是基因组的基本特性,其结构的完整性是基因调控所必须的。删除TAD的边界片段会使基因调控陷入混乱,原本沉默的基因开始表达,而原本表达的基因被沉默。研究发现,在癌症病人中,TAD边界区域往往与大量的超级增强子的位点相重合,说明其稳定性与癌症的发生密切相关[42]。2015年,Cell首次报道了TAD与遗传学疾病的关联[43]。德国马克斯普朗克分子遗传学研究所和柏林夏洛蒂医科大学的科学家运用最新的基因组编辑技术CRISPR/ Cas成功将调控3种人类罕见疾病(短指症、多趾畸形和并趾)的基因所在的TAD边界破坏,使得小鼠模型产生相应遗传疾病表型。在小鼠的肢体组织和患者的成纤维细胞中,与疾病有关的染色质结构改变使启动子和非编码DNA出现异常互作。在野生型小鼠中,Epha4基因的增强子正常激活其自身表达,而在3种患病的小鼠模型中,由于DNA结构变异,Epha4的增强子分别错误地激活了Pax3、Ihh以及Wnt6基因,使其发生异位表达,从而产生短指、多趾畸形和并趾的疾病表型;进一步研究表明,只有在CTCF相关的TAD边界区域被破坏时, 才会出现这种问题。该研究证实了TAD结构的破坏会导致远距离调控元件的重排,使得增强子会作用于错误的靶基因而引起异位表达,导致致病表型。这项研究证明了TAD功能的重要性,人们可以在此基础上预测人类结构变异的致病性,尤其是在基因组的非编码区域。该研究对基因组变化引起疾病的机制提出了新的见解。
1.4 染色质环
在哺乳动物细胞核内,由于染色质浓缩聚合的性质,导致基因纤维上两个远端位点会产生随机碰撞而以较低的频率相互作用。然而,在某些确定的基因位点间,这种远距离互作的频率却显著高于预测值。在sub-Mb分辨率的Hi-C互作图谱中,这些远距离互作的位点大量存在,它们构成了染色质稳定结构的基础,或直接参与转录等调控过程。因此,由基因位点的远距离互作而介导染色质纤维折叠形成的环状结构,称之为“Chromatin loop”,即染色质环。2014年,美国Broad研究院Aiden教授团队通过超高分辨率的原位Hi-C方法(in situ Hi-C, 1 kb),详细地展示了长达2米的人类基因组在直径约10微米的细胞核内的全部折叠方式[15],获得了人的类淋巴母细胞(GM12878)49亿个染色质互作信息,并首次列出了整个人类基因组上形成的9448个染色质环(loop)。研究发现,这些loop的两端通常连接着已知基因的启动子和增强子,并且这些loop相关的启动子所在基因具有更高的表达水平和更强的细胞特异性。因此,这些loop确实是启动子-增强子的长距离互作所形成,并直接调控基因的表达。随着技术的发展,越来越多的超高分辨率的Hi-C结果都先后揭示出不同细胞间由于长距离互作而形成的loop结构[44,45,46,47]。尽管这些研究对于鉴定不同细胞的长距离互作的算法各有差异,但是都发现了一些loop互作共有的规律特征。首先,这些长距离互作通常发生在同一个TAD或者sub-TAD内部,排除一些特异性的基因区域(如Hox基因)外,基因组中发生极长距离的互作相对较少[48];其次,活性的启动子、增强子以及CTCF结合位点通常与长距离互作密切相关[46,49]。此外,除启动子-增强子互作形成的loop结构外,启动子-启动子以及增强子-增强子的互作也能形成复杂的loop网络结构[49,50]。研究发现,38%的 loop与contact domain具有一致性,即形成loop的锚点通常位于domain的边界区域,65%的loop的出现通常伴随着domain的出现,因此这些domain被称为loop domain。并且,这些临近的loop通常具有传递性(transitivity),即形成相邻两个loop的互作位点L1-L2和L2-L3,往往在L1-L3之间也会有loop的产生,说明这3个位点具有同一个空间位置。进一步研究loop形成机制时发现,大部分的loop (peak)所涉及的两个peak loci具有显著的绝缘蛋白CTCF (86%)和cohesin的两个亚基—RAD21 (86%)和SMC3 (87%)的富集,说明CTCF和cohesin参与loop结构的形成[15]。随后,Aiden教授团队对CTCF和cohesin参与loop结构形成的机制进行研究,并提出了CTCF/cohesin介导的环挤压模型(Loop Extrusion Model)[51]。在这个模型中(图4),cohesin环在NIPBL装载蛋白作用下形成cohesin复合物并结合到染色质上,延DNA序列向相反的方向滑动,挤压染色质形成loop环,直到遇到绑定在CTCF motif序列的阻遏子CTCF蛋白,挤压过程即被终止[51,52,53]。在环挤压过程中,染色体结构维持(stuctural maintenance of chromosomes, SMC)蛋白家族中的SMC1、SMC3以及RAD21参与形成cohesin的亚基结构[54]。此外,部分cohesin环能够在WAPL和PDS5蛋白作用下从挤压过程中释放[54]。研究还发现,由CTCF和cohesin介导形成的loop结构在不同的细胞间具有稳定的保守性,并以此划分TAD以及sub-TAD[15,55,56]。此外,这些CTCF结合位点都具有收敛的CTCF模体序列,因而能够解释在所有的CTCF结合位点中只有一小部分参与domain边界的界定[15,47,57]。
图4
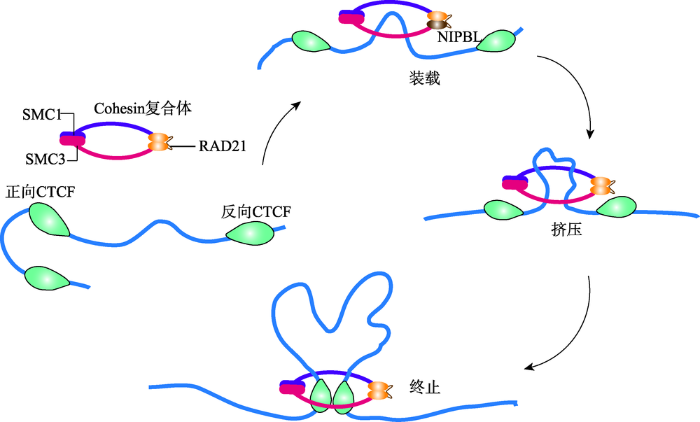 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4CTCF/cohesin介导的环挤压模型
Fig. 4The loop extrusion model mediated by CTCF/cohesin
2 Hi-C技术在三维基因组学中的应用
随着Hi-C技术的不断发展,Single cell Hi-C、In situ Hi-C、Dnase Hi-C等一系列衍生技术相继出现[22,58~61]。这些技术不仅能够用来揭示哺乳动物细胞核内染色质空间构象方式,阐明其折叠规律及其作用机制,在三维基因组学应用方面也发挥着重要作用。因此,Hi-C及其衍生技术能够用来辅助组装基因组,构建哺乳动物全基因组单倍型,比较不同细胞/物种间染色质互作的差异及其介导的基因表达差异,探究机体发育规律以及复杂疾病的发病机制等。2.1 辅助基因组组装
Hi-C技术用于辅助基因组组装是目前提高基因组组装质量的一种必要手段,具体是指在已经完成基本组装的基因组草图(Draft genome)序列(Scaffolds/ Contigs)和染色体数目已知的前提下,利用Hi-C测序数据将Draft genome序列进行不同染色体的群组划分,并确定各序列在染色体上的顺序和方向,使基因组组装水平提升到染色体水平。其主要原理是染色体内互作强度高于染色体间的互作,同一染色体上近距离互作强于远距离互作[62]。Hi-C辅助基因组组装主要分为3步:(1) Cluster:将contigs或scaffolds聚类到不同的染色体组;(2) Order:在每个染色体组中按顺序排列contigs或scaffolds;(3) Orient:为每一个排好顺序的相邻的contigs或scaffolds确定方向。自2013年Burton等[62]首次利用Hi-C技术辅助组装了人、小鼠及果蝇的基因组后,近年来,研究者相继对拟南芥[63]、山羊(Capra hircus)[64]、藜麦(Chenopodium quinoa)[65]、埃及伊蚊(Aedes aegypti)[66]、大麦(Hordeum vulgare L.)[67]以及甘蔗(Saccharum L.)[68]等动植物的基因组进行Hi-C辅助组装,为进行更深入的基因组学研究奠定了基础。2.2 构建全基因组单倍型
单倍型是存在于染色单体内具有统计学关联性的一类单核苷酸多态性(single nucleotide polymorphisms, SNPs),这些进行共同遗传的多个基因座上等位基因的组合信息对人类遗传、疾病风险预测以及农业动植物经济性状连锁标记等方面研究具有重要价值[69]。相比于传统的单倍型分析技术对于DNA片段分析长度的限制,Hi-C技术能够使其在全基因范围内进行单倍型组装,且检测效率以及分析的准确性都较高。早2013年,Bing Ren教授团队首次利用Hi-C技术对人细胞进行了全基因组单倍型组装,构建了准确率达98%的人的单倍型群体[70]。此后,越来越多的研究报道了Hi-C技术用于构建基因组单倍型[14,71,72]。另外,研究者还开发了直接针对Hi-C测序数据的单倍型分析工具HapCUT2[73]。这些研究结果都说明Hi-C技术具有革命性的优势,能够广泛用于哺乳动物群体的单倍型构建。2.3 基因的表达调控
Hi-C技术除了可以进行辅助组装基因组分析外,还可对基因的表达调控以及基因功能进行研究。染色质互作的形成和功能对于细胞的命运决定和分化等过程至关重要,在基因特异性表达调控中发挥重要作用。之前的研究表明,染色质环(chromatin loop)的两端通常连接着基因的启动子和增强子,线性距离较远的增强子能够通过loop结构被募集到已知基因的启动子区域,从而激活基因的转录。Mifsud等[74]通过高分辨捕获Hi-C (Capture Hi-C, CHi-C)技术,构建了两种人类血细胞(GM12878和CD34+)中超过 22 000个长距离的启动子互作图谱,鉴定了超过11 600 000个两种细胞类型共有的互作,它们跨越启动子和远端位点之间的数百个碱基;研究还发现,与疾病相关的SNPs位点明显富集在基因的互作区域,暗示着远距离突变可能会破坏相关基因的表达调控而导致疾病的发生。Rubin等[75]利用CHi-C联合ChIP-seq技术,在全基因组范围内研究了分离培养的人原代角质细胞分化过程中增强子和启动子的互作模式,确认了两种类型的启动子-增强子互作:获得型(gained)互作,在分化过程中增强,并与enhancer获得H3K27ac活化标记一致;稳定型(stable)互作,在未分化细胞中已预先建立,enhancer有H3K27ac的标记,并与黏连蛋白cohesin相关。但这两种互作均未在多能性细胞中检测到,表明这种谱系特异的染色质构象在组织的前体细胞中形成,并且在终末分化中重塑。Bonev等[76]对小鼠神经细胞分化过程中的染色质结构进行了超高分辨率的解析,发现基因的转录活动与染色质的绝缘以及远距离互作相关,但dCas9介导的激活不足以重新形成TAD边界;此外,在所有的细胞类型中,长距离互作主要发生在外显子富集的gene body与激活基因间,且在神经细胞分化过程中,活性TADs之间的互作变得不明显,而非活动TADs之间的互作则越来越强,说明由分化引起的基因转录激活使得TADs的构象发生变化。X染色体失活(X-chromosome inactivation, XCI)会引起X染色体结构重塑,转变成沉默的异染色质[77]。在雌性哺乳动物发育中,X染色体失活由两条X染色体中一条的非编码RNA Xist发生上调引起[78,79,80]。Giorgetti等[81]利用Hi-C技术解析了小鼠失活X染色体的结构特征以及基因表达情况:在小鼠神经前体细胞(NPCs)和胚胎干细胞中,失活的X染色体结构重塑中Xist和含有DXZ4边界发挥着重要作用,并且在失活的X染色体中,除了“逃脱”沉默的基因附近,其他位置失去了有活性和失活的compartment A/B以及TADs。2.4 机体与细胞发育
2015年,Battulin等[82]对小鼠精细胞和胚胎成纤维细胞进行Hi-C结果的比较,在1 Mb分辨率下,精细胞的compartment A/B与胚胎成纤维细胞具有高度相似性,这与之前报道的小鼠胚胎干细胞的三维基因组结构相一致[17]。而当研究人员将分辨率提高到40 kb时,彼此之间的TAD边界出现差异,且在特定的基因座位点上,两种细胞染色质的互作差异显著。与成纤维细胞相比,精子细胞的间期细胞核小10倍左右,其基因组高度浓缩的包装形式导致了精子细胞染色质远距离互作的富集,由此说明配子细胞的染色质构象与体细胞存在差异。目前的研究认为,染色质不同层级的构象(如compartment A/B和TAD)在体细胞中是稳定存在的保守结构单元,但这种构象是与生俱来还是从配子转变为合子的早期胚胎发育时期形成的,值得人们关注。之前由于细胞数量和实验手段的限制,染色体三维结构在哺乳动物早期胚胎发育过程中的动态变化鲜为人知。近年来,随着单细胞Hi-C (single-cell Hi-C)技术的运用,研究者不仅能从普通Hi-C大量群体细胞中获得平均数据评估染色质折叠和潜在的互作,还能利用单细胞Hi-C技术分辨单个染色体的构象模型,精确调控细胞的状态和功能[58,83]。2017年,Nature和Cell“背靠背”发表研究论文,研究者们都发现哺乳动物染色体三维结构在着床前胚胎发育过程中的动态重组过程[84,85]。研究结果显示,精子保留经典的染色质高级结构,包括TADs和compartments;相反,处于MⅡ期的卵子染色体呈现出一种均一性结构,缺乏TADs和compartments结构。染色体三维结构在受精后首先呈现出一种极其松散的状态,两套亲本基因组在空间上部分分离且染色体compartments不同,差异持续到8细胞期。在随后的胚胎早期发育过程中,染色质高级结构逐步以亲本特异的方式建立和成熟,并且不完全依赖于合子基因组的转录激活。Flyamer等[86]也发现,受精完成后,父源和母源染色质需要在受精卵中进行空间排布重组,并且此过程中父源和母源染色质的重组方式不同。此外,Kaaij等[87]研究发现,在斑马鱼的早期胚胎发育过程中,因为缺乏合子的转录活动,其基因组高度结构化;当合子基因组被激活后,斑马鱼染色体失去结构特征,并且这些特征在随后的发育过程中被重新建立。染色质重塑是调控基因时序性表达的重要环节,往往发生在衰老细胞中,并且衰老细胞核中会形成衰老相关异染色质聚集(senescence-associated heterochromatic foci, SAHF)。Chandra等[88]利用Hi-C技术对衰老细胞和正常ES细胞的染色质空间构象进行探究,发现与正常细胞相比,在衰老细胞的异染色质中存在大量依赖于序列和核纤层蛋白的局部互作缺失,且衰老细胞中出现特有的异染色质聚集,这可能是SAHF形成的中间产物。此外,另一项研究也发现,当衰老发生时,染色质重塑是由于CTCF簇的形成,导致loop的重组,并且HMGB2蛋白参与此过程[89]。
2.5 在疾病研究中的应用
许多复杂疾病的发生往往与其组织细胞的三维基因组构象改变密切相关。Won等[90]通过Hi-C技术构建了人大脑皮质的高分辨率3D图谱,分析鉴定了数百个在人类谱系中已知的启动子-增强子互作基因,并且将染色质互作与GWAS研究中确定的精神分裂症相关非编码变异相结合,突出了多个候选精神分裂症风险基因和相关通路,其中一个远端的精神分裂症变异位点能够调控风险基因FOXG1的表达,支持其作为精神分裂症易感基因的潜在作用。心力衰竭主要是由心肌细胞的生物化学变化引起,已有研究表明这一复杂的细胞功能障碍是基因表达改变的结果,受转录因子和染色质重塑酶的影响[91,92,93,94]。Rosa-Garrido等[95]利用5 kb分辨率的全基因组捕获Hi-C与DNA测序结合,对心脏特异性敲除CTCF的小鼠心肌细胞进行了研究,发现CTCF元件缺失和心肌压力超负荷能大幅减少loop结构,重塑loop内部互作,导致功能元件与启动子区域的互作明显减弱,从而扰乱基因的转录调控。Anene Nzelu等[96]也发现,患病小鼠与正常小鼠心室肌细胞的染色质构象单元compartments A/B的变化与基因的表达变化模式相关,通过对H3K27ac标记的富集区域进行分析,确定了细胞特异性基因表达的调控元件,并通过CRISPR敲除Nppb和Nppa基因座上游的一个调控区域,导致与其互作基因的表达下调。Loviglio等[97]通过Hi-C、FISH以及4C-seq技术确认了染色体16p11.2上两个拷贝数变异(copy number variants,CNV)倾向的区域:16p11.2远端BP2-BP3间220 kb区域和16p11.2近端BP4-BP5间600 kb区域影响染色质的成环作用,并影响成环区域间所含基因的协调表达和调控,提示染色质互作异常与孤独症谱系障碍(autism spectrum disorders)、肥胖/体重不足及巨头/小头畸形的表型相关,并且确认了在基因组其他区域类似表型相关的顺式及反式染色体互作,表明染色体互作图谱可以揭示功能和临床诊断相关的疾病易感基因。此外,也有研究报道利用Hi-C技术对类风湿关节炎、Crohn氏病等自身免疫疾病的发病机制进行探究,发现启动子互作区域的异常与这些疾病的调控机制密切相关[44]。与正常细胞相比,癌细胞由于遗传以及表观遗传的改变,使得基因表达紊乱[98,99,100,101];并且癌症是一种以细胞核的主要形态变化为特征的疾病[102,103]。因此,染色质空间构象的变化与癌症的发生密切相关(图5)。Barutcu等[104]通过Hi-C技术对乳腺上皮细胞(MCF-10A)和乳腺癌细胞系(MCF-7)进行分析,发现与MCF-10A细胞相比,MCF-7细胞中小而基因富集的16~22号染色体间的互作频率更低,且两种细胞的染色体内部互作也区别明显;此外,MCF-10A细胞在端粒区以及亚端粒区的互作要强于MCF-7细胞。Taberlay等[105]利用Hi-C技术对前列腺癌中包括拷贝数变异、远距离染色质互作重塑以及非典型基因表达的染色体三维结构破坏进行了研究,发现癌细胞保留了将其基因组分割成Mb级别的TAD的能力,但由于附加的TAD边界的建立,这些TAD比正常细胞中的TAD小,且很大一部分新的与癌症相关的特异性TAD边界发生在CNV变化的区域。此外,前列腺癌患者17p13.1的一个常见缺失导致了单个TAD分为两个明显更小的TAD,而TAD的改变伴随着TAD内新的肿瘤特异性染色质互作的形成,并在启动子、增强子以及绝缘子等调控元件上富集,引发基因表达的改变。此外,研究者还利用Hi-C技术探究了神经母细胞瘤[106]、胶质瘤[107]以及急性T淋巴细胞白血病[108]等恶性肿瘤的发病机制,旨在从三维基因组角度解析染色体空间构象重组对于癌症发生的重要作用。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5癌症中染色质三维结构的破坏导致基因异常表达
Fig. 5Disruption of 3D chromosomal structure in cancer cells leads to abnormal gene expression
癌症的发生往往伴随着基因组变异。癌症基因组的测序提供的第一个直接信息是体细胞基因组变异率如何在正常细胞与癌细胞之间变化[109,110,111,112,113,114,115]。研究发现,癌症基因组中的突变率与染色质折叠密切相关,在Mb尺度下,异染色质相关的组蛋白修饰标志物H3K9me3的单一特征水平可占到变异率的40%以上[116]。Litchfield等[117]联合利用Hi-C与GWAS对睾丸生殖细胞肿瘤(testicular germ cell tumors, TGCTs)中的SNPs进行鉴定,确认了19个新的风险位点,并结合之前已发现的25个位点[118,119],将总共44个风险位点与候选的致病基因进行互作网络分析,发现TGCTs的易感基础是发育调控因子的大范围紊乱导致的。Romanel等[120]利用Hi-C技术对引起前列腺癌的早发性体细胞变异的非编码多态性调节元件7p14.3与其调控基因的结构进行了分析,表明7p14.3位点的多态性可能通过雄性激素依赖的DNA损失修复功能影响前列腺癌的致病倾向。此外,还有研究利用捕获Hi-C技术对乳腺癌[121,122]、结直肠癌[123]等癌症的风险位点进行了鉴定,揭示出基因座中的重要远距离染色质互作参与癌症发生的致病机制。
Hi-C技术还能通过检测癌症病人原发性肿瘤样本组织中平衡和非平衡的染色质重塑,包括易位和反转,以及获得性CNV,预测癌症的发生及预后,这将极大降低癌症的检测成本[124]。此外,Hi-C技术还用于鉴定经福尔马林固定的石蜡包埋肿瘤样本的结构变异,这种与Hi-C技术类似的高通量构象捕获技术被称为“Fix-C”技术,能够识别未被其他方法检测到的新结构变异,这种方法能够在癌症进展期间从FFPE样本中详细解析全基因组重塑事件,为患者护理的目标分子诊断提供信息[125]。
越来越多的研究表明,染色体的三维空间构象紊乱正在成为癌症等疾病发生过程中一种新的致病机制,以Hi-C技术为代表的三维基因组技术将极大地促进复杂疾病以及癌症的相关研究,通过解析病发细胞中基因与其调控元件之间互作的改变,找到新的致病位点,并为开发新的靶向治疗药物提供线索。
3 结语与展望
随着时代的进步以及技术的革新,基因组学的发展取得了长足的进步,从“人类基因组计划”(HGP)[126]到“人类基因组百科全书计划”(ENCODE)[127]的顺利完成,科研人员可以深入分析、解读和注释基因组序列信息和功能。但是,基因组DNA并不是在染色体上呈线性排列,其三维空间构象对DNA复制、基因转录调控、染色质浓缩和分离等基本生物学过程都有着不可或缺的重要作用。Hi-C技术的提出以及其大规模的运用,使得人们可以从空间层面去揭示这些不同调控元件的互作关系,认识染色质构象对基因表达调控的机制及作用。2015年,科学家开始正式实施一个全新的全球合作项目——“4D核体计划”[128],计划用5年或者更长的时间从空间(三维)和时间(四维)角度来研究细胞核结构形成原理,探索细胞核结构对基因表达、细胞功能,以及对发育和疾病发生、发展的影响。因此,Hi-C技术以及三维基因组的全面发展,必然会为全面解读人类基因组信息、攻克复杂疾病和促进人类医学进步提供有力的支持。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 2]
URLPMID:26967279 [本文引用: 1]

Proper expression of genes requires communication with their regulatory elements that can be located elsewhere along the chromosome. The physics of chromatin fibers imposes a range of constraints on such communication. The molecular and biophysical mechanisms by which chromosomal communication is established, or prevented, have become a topic of intense study, and important roles for the spatial organization of chromosomes are being discovered. Here we present a view of the interphase 3D genome characterized by extensive physical compartmentalization and insulation on the one hand and facilitated long-range interactions on the other. We propose the existence of topological machines dedicated to set up and to exploit a 3D genome organization to both promote and censor communication along and between chromosomes.
URLPMID:23414654 [本文引用: 1]
We review recent developments in mapping chromosomal contacts and compare emerging insights on chromosomal contact domain organization in Drosophila and mammalian cells. Potential scenarios leading to the observation of Hi-C domains and their association with the epigenomic context of the chromosomal elements involved are discussed. We argue that even though the mechanisms and precise physical structure underlying chromosomal domain demarcation are yet to be fully resolved, the implications to genome regulation and genome evolution are profound. Specifically, we hypothesize that domains are facilitating genomic compartmentalization that support the implementation of complex, modular, and tissue specific transcriptional program in metazoa.
URLPMID:23473598 [本文引用: 2]
Mammalian genomes encode genetic information in their linear sequence, but appropriate expression of their genes requires chromosomes to fold into complex three-dimensional structures. Transcriptional control involves the establishment of physical connections among genes and regulatory elements, both along and between chromosomes. Recent technological innovations in probing the folding of chromosomes are providing new insights into the spatial organization of genomes and its role in gene regulation. It is emerging that folding of large complex chromosomes involves a hierarchy of structures, from chromatin loops that connect genes and enhancers to larger chromosomal domains and nuclear compartments. The larger these structures are along this hierarchy, the more stable they are within cells, while becoming more stochastic between cells. Here, we review the experimental and theoretical data on this hierarchy of structures and propose a key role for the recently discovered topologically associating domains.
URLPMID:4517094 [本文引用: 1]
In humans, nearly two meters of genomic material must be folded to fit inside each micrometer-scale cell nucleus while remaining accessible for gene transcription, DNA replication, and DNA repair. This fact highlights the need for mechanisms governing genome organization during any activity and to maintain the physical organization of chromosomes at all times. Insight into the functions and three-dimensional structures of genomes comes mostly from the application of visual techniques such as fluorescence in situ hybridization (FISH) and molecular approaches including chromosome conformation capture (3C) technologies. Recent developments in both types of approaches now offer the possibility of exploring the folded state of an entire genome and maybe even the identification of how complex molecular machines govern its shape. In this review, we present key methodologies used to study genome organization and discuss what they reveal about chromosome conformation as it relates to transcription regulation across genomic scales in mammals.
[本文引用: 1]
URL [本文引用: 1]
The motion of subchromosomal foci and of whole chromosome territories in live human cell nuclei was investigated in four-dimensional space-time images. Visualization of subchromosomal foci was achieved by incorporating Cy3-dUTP into the nuclear DNA of two different cell types after microinjection. A subsequent segregation of the labeled cell nuclei led to the presence of only a few labeled chromosome territories on a background of nonlabeled chromatin (Zink et al.,1998. Hum. Genet. 102:241-251). This procedure yielded many distinct signals in a given cell nucleus. Motion analysis in four-dimensional space-time images was performed using single-particle tracking and a statistical approach to the detection of a possible directional motion of foci relative to the center of mass of a chromosome territory. The accuracy of the analysis was tested using simulated data sets that closely mirrored the experimental setup and using microparticles of known size. Application of the analysis tools to experimental data showed that mutual diffusion-like movements between foci located on different chromosomes were more pronounced than inside the territories. In the time range observed, movements of individual foci could best be described by a random diffusion process. The statistical test for joint directed motion of several foci inside chromosome territories revealed that foci occasionally switched from random to directional motion inside the territories.
URL [本文引用: 1]
URLPMID:11283701 [本文引用: 1]
Abstract The expression of genes is regulated at many levels. Perhaps the area in which least is known is how nuclear organization influences gene expression. Studies of higher-order chromatin arrangements and their dynamic interactions with other nuclear components have been boosted by recent technical advances. The emerging view is that chromosomes are compartmentalized into discrete territories. The location of a gene within a chromosome territory seems to influence its access to the machinery responsible for specific nuclear functions, such as transcription and splicing. This view is consistent with a topological model for gene regulation.
URLPMID:2515109 [本文引用: 1]
During decondense, but fluorescent in situ hybridization experiments reveal the existence of distinct territories occupied by individual inside the nuclei of most eukaryotic . We use computer simulations to show that the existence and stability of territories is a kinetic effect that can be explained without invoking an underlying nuclear scaffold or -mediated interactions between DNA sequences. In particular, we show that the experimentally observed territory shapes and spatial distances between marked sites for , , and budding can be reproduced by a parameter-free minimal model of decondensing . Our results suggest that the observed structure and dynamics are due to generic polymer effects: confined Brownian motion conserving the local topological state of long chain molecules and segregation of mutually unentangled chains due to topological constraints.
URLPMID:18951171 [本文引用: 1]
Abstract Fluorescence in situ hybridization (FISH) of specific DNA probes has become a widely used technique mostly for chromosome analysis and for studies of the chromosomal location of specific DNA segments in metaphase preparations as well as in interphase nuclei. FISH on 3D-preserved nuclei (3D-FISH) in combination with 3D-microscopy and image reconstruction is an efficient tool to analyze the spatial arrangement of targeted DNA sequences in the nucleus. Recent developments of a "new generation" of confocal microscopes that allow the distinct visualization of at least five different fluorochromes within one experiment opened the way for multicolor 3D-FISH experiments. Thus, numerous differently labeled nuclear targets can be delineated simultaneously and their spatial interrelationships can be analyzed on the level of individual nuclei.In this chapter, we provide protocols for the preparation of complex DNA-probe sets suitable for 3D-FISH with up to six different fluorochromes, for 3D-FISH on cultured mammalian cells (growing in suspension or adherently) as well as on tissue sections, and for 3D immuno-FISH.In comparison with FISH on metaphase chromosomes and conventional interphase cytogenetics, FISH on 3D-preserved nuclei requires special demands with regard to probe quality, fixation, and pretreatment steps of cells in order to achieve the two goals, namely the best possible preservation of the nuclear structure and at the same time an efficient probe accessibility.
URLPMID:18854147 [本文引用: 1]
There is no doubt that genomes are organized nonrandomly in the nucleus of higher eukaryotes. But what is the functional relevance of this nonrandomness? In this Essay, we explore the biological meaning of spatial gene positioning by examining the functional link between the activity of a gene and its radial position in the nucleus.
URLPMID:19379699 [本文引用: 1]
We show that the nuclear architecture of rod photoreceptor cells differs fundamentally in nocturnal and diurnal mammals. The rods of diurnal retinas possess the conventional architecture found in nearly all eukaryotic cells, with most heterochromatin situated at the nuclear periphery and euchromatin residing toward the nuclear interior. The rods of nocturnal retinas have a unique inverted pattern, where heterochromatin localizes in the nuclear center, whereas euchromatin, as well as nascent transcripts and splicing machinery, line the nuclear border. The inverted pattern forms by remodeling of the conventional one during terminal differentiation of rods. The inverted rod nuclei act as collecting lenses, and computer simulations indicate that columns of such nuclei channel light efficiently toward the light-sensing rod outer segments. Comparison of the two patterns suggests that the conventional architecture prevails in eukaryotic nuclei because it results in more flexible chromosome arrangements, facilitating positional regulation of nuclear functions.
URL [本文引用: 3]
URLPMID:25497547 [本文引用: 5]
Using 3D genome sequencing, we find 6510,000 DNA loops across the human genome. Loop anchors typically occur at domain boundaries and bind CTCF in a convergent orientation, with the asymmetric motifs “facing” one another. On the inactive X chromosome, large loops are anchored at CTCF-binding repeats. Loops are conserved across cell types and species.
URLPMID:20888037 [本文引用: 1]
Abstract Chromatin is important for the regulation of transcription and other functions, yet the diversity of chromatin composition and the distribution along chromosomes are still poorly characterized. By integrative analysis of genome-wide binding maps of 53 broadly selected chromatin components in Drosophila cells, we show that the genome is segmented into five principal chromatin types that are defined by unique yet overlapping combinations of proteins and form domains that can extend over > 100 kb. We identify a repressive chromatin type that covers about half of the genome and lacks classic heterochromatin markers. Furthermore, transcriptionally active euchromatin consists of two types that differ in molecular organization and H3K36 methylation and regulate distinct classes of genes. Finally, we provide evidence that the different chromatin types help to target DNA-binding factors to specific genomic regions. These results provide a global view of chromatin diversity and domain organization in a metazoan cell. Copyright 2010 Elsevier Inc. All rights reserved.
URLPMID:22495300 [本文引用: 4]
The spatial organization of the genome is intimately linked to its biological function, yet our understanding of higher order genomic structure is coarse, fragmented and incomplete. In the nucleus of eukaryotic cells, interphase chromosomes occupy distinct chromosome territories, and numerous models have been proposed for how chromosomes fold within chromosome territories. These models, however, provide only few mechanistic details about the relationship between higher order chromatin structure and genome function. Recent advances in genomic technologies have led to rapid advances in the study of three-dimensional genome organization. In particular, Hi-C has been introduced as a method for identifying higher order chromatin interactions genome wide. Here we investigate the three-dimensional organization of the human and mouse genomes in embryonic stem cells and terminally differentiated cell types at unprecedented resolution. We identify large, megabase-sized local chromatin interaction domains, which we term 'topological domains', as a pervasive structural feature of the genome organization. These domains correlate with regions of the genome that constrain the spread of heterochromatin. The domains are stable across different cell types and highly conserved across species, indicating that topological domains are an inherent property of mammalian genomes. Finally, we find that the boundaries of topological domains are enriched for the insulator binding protein CTCF, housekeeping genes, transfer RNAs and short interspersed element (SINE) retrotransposons, indicating that these factors may have a role in establishing the topological domain structure of the genome.
URLPMID:22495304 [本文引用: 1]
Abstract In eukaryotes transcriptional regulation often involves multiple long-range elements and is influenced by the genomic environment. A prime example of this concerns the mouse X-inactivation centre (Xic), which orchestrates the initiation of X-chromosome inactivation (XCI) by controlling the expression of the non-protein-coding Xist transcript. The extent of Xic sequences required for the proper regulation of Xist remains unknown. Here we use chromosome conformation capture carbon-copy (5C) and super-resolution microscopy to analyse the spatial organization of a 4.5-megabases (Mb) region including Xist. We discover a series of discrete 200-kilobase to 1 Mb topologically associating domains (TADs), present both before and after cell differentiation and on the active and inactive X. TADs align with, but do not rely on, several domain-wide features of the epigenome, such as H3K27me3 or H3K9me2 blocks and lamina-associated domains. TADs also align with coordinately regulated gene clusters. Disruption of a TAD boundary causes ectopic chromosomal contacts and long-range transcriptional misregulation. The Xist/Tsix sense/antisense unit illustrates how TADs enable the spatial segregation of oppositely regulated chromosomal neighbourhoods, with the respective promoters of Xist and Tsix lying in adjacent TADs, each containing their known positive regulators. We identify a novel distal regulatory region of Tsix within its TAD, which produces a long intervening RNA, Linx. In addition to uncovering a new principle of cis-regulatory architecture of mammalian chromosomes, our study sets the stage for the full genetic dissection of the X-inactivation centre.
URLPMID:22265598 [本文引用: 1]
A genome-wide chromosomal contact map of the Drosophila genome at kilobase resolution shows that the genome is partitioned into physical domains and that contact between domains is dependent upon their epigenetic profile.
URLPMID:26034287 [本文引用: 1]
Abstract Increasing evidence in the last years indicates that the vast amount of regulatory information contained in mammalian genomes is organized in precise 3D chromatin structures. However, the impact of this spatial chromatin organization on gene expression and its degree of evolutionary conservation is still poorly understood. The Six homeobox genes are essential developmental regulators organized in gene clusters conserved during evolution. Here, we reveal that the Six clusters share a deeply evolutionarily conserved 3D chromatin organization that predates the Cambrian explosion. This chromatin architecture generates two largely independent regulatory landscapes (RLs) contained in two adjacent topological associating domains (TADs). By disrupting the conserved TAD border in one of the zebrafish Six clusters, we demonstrate that this border is critical for preventing competition between promoters and enhancers located in separated RLs, thereby generating different expression patterns in genes located in close genomic proximity. Moreover, evolutionary comparison of Six-associated TAD borders reveals the presence of CCCTC-binding factor (CTCF) sites with diverging orientations in all studied deuterostomes. Genome-wide examination of mammalian HiC data reveals that this conserved CTCF configuration is a general signature of TAD borders, underscoring that common organizational principles underlie TAD compartmentalization in deuterostome evolution.
URLPMID:26030525 [本文引用: 1]
The three-dimensional organization of a genome plays a critical role in regulating gene expression, yet little is known about the machinery and mechanisms that determine higher-order chromosome structure. Here we perform genome-wide chromosome conformation capture analysis, fluorescent in situ hybridization (FISH), and RNA-seq to obtain comprehensive three-dimensional (3D) maps of the Caenorhabditis elegans genome and to dissect X chromosome dosage compensation, which balances gene expression between XX hermaphrodites and XO males. The dosage compensation complex (DCC), a condensin complex, binds to both hermaphrodite X chromosomes via sequence-specific recruitment elements on X (rex sites) to reduce chromosome-wide gene expression by half. Most DCC condensin subunits also act in other condensin complexes to control the compaction and resolution of all mitotic and meiotic chromosomes. By comparing chromosome structure in wild-type and DCC-defective embryos, we show that the DCC remodels hermaphrodite X chromosomes into a sex-specific spatial conformation distinct from autosomes. Dosage-compensated X chromosomes consist of self-interacting domains ( approximately 1 Mb) resembling mammalian topologically associating domains (TADs). TADs on X chromosomes have stronger boundaries and more regular spacing than on autosomes. Many TAD boundaries on X chromosomes coincide with the highest-affinity rex sites and become diminished or lost in DCC-defective mutants, thereby converting the topology of X to a conformation resembling autosomes. rex sites engage in DCC-dependent long-range interactions, with the most frequent interactions occurring between rex sites at DCC-dependent TAD boundaries. These results imply that the DCC reshapes the topology of X chromosomes by forming new TAD boundaries and reinforcing weak boundaries through interactions between its highest-affinity binding sites. As this model predicts, deletion of an endogenous rex site at a DCC-dependent TAD boundary using CRISPR/Cas9 greatly diminished the boundary. Thus, the DCC imposes a distinct higher-order structure onto X chromosomes while regulating gene expression chromosome-wide.
URLPMID:26119342 [本文引用: 2]
Mononucleosome resolution mapping of chromosome folding in yeast reveals self-associating domains similar to those found in other organisms. But they are far shorter, with domain size being scaled by gene number rather than linear distance.
URLPMID:26096785 [本文引用: 1]
Advanced techniques including the chromosome conformation capture (3C) methodology and its derivatives are complementing microscopy approaches to study genome organization, and are revealing new details of three-dimensional (3D) genome architecture at increasing resolution. The fission yeast Schizosaccharomyces pombe ( S. pombe ) comprises a small genome featuring organizational elements of more complex eukaryotic systems, including conserved heterochromatin assembly machinery. Here we review key insights into genome organization revealed in this model system through a variety of techniques. We discuss the predominant role of Rabl-like configuration for interphase chromosome organization and the dynamic changes that occur during mitosis and meiosis. High resolution Hi-C studies have also revealed the presence of locally crumpled chromatin regions called lobules along chromosome arms, and implicated a critical role for pericentromeric heterochromatin in imposing fundamental constraints on the genome to maintain chromosome territoriality and stability. These findings have shed new light on the connections between genome organization and function. It is likely that insights gained from the S. pombe system will also broadly apply to higher eukaryotes. .overlined { text-decoration: overline; } .struck { text-decoration:line-through; } .underlined { text-decoration:underline; } .doubleUnderlined { text-decoration:underline;border-bottom:1px solid #000; }
[本文引用: 1]
URLPMID:4347903 [本文引用: 1]
Using Hi-C analysis, Feng et02al. show extensive interactions among regions of Arabidopsis chromatin. These interactions correlate with epigenetic marks, and mutants of epigenetic pathways affect interaction patterns.
URL [本文引用: 1]
URLPMID:29175436 [本文引用: 1]
Abstract The spatial organization of the genome plays an important role in the regulation of gene expression. However, the core structural features of animal genomes, such as topologically associated domains (TADs) and chromatin loops, are not prominent in the extremely compact Arabidopsis genome. In this study, we examine the chromatin architecture, as well as their DNA methylation, histone modifications, accessible chromatin, and gene expression, of maize, tomato, sorghum, foxtail millet, and rice with genome sizes ranging from 0.4 to 2.4 Gb. We found that these plant genomes can be divided into mammalian-like A/B compartments. At higher resolution, the chromosomes of these plants can be further partitioned to local A/B compartments that reflect their euchromatin, heterochromatin, and polycomb status. Chromatins in all these plants are organized into domains that are not conserved across species. They show similarity to the Drosophila compartment domains, and are clustered into active, polycomb, repressive, and intermediate types based on their transcriptional activities and epigenetic signatures, with domain border overlaps with the local A/B compartment junctions. In the large maize and tomato genomes, we observed extensive chromatin loops. However, unlike the mammalian chromatin loops that are enriched at the TAD border, plant chromatin loops are often formed between gene islands outside the repressive domains and are closely associated with active compartments. Our study indicates that plants have complex and unique 3D chromatin architectures, which require further study to elucidate their biological functions.
URLPMID:26348399 [本文引用: 1]
Recent studies have shown that chromosomes in a range of organisms are compartmentalized in different types of chromatin domains. In mammals, chromosomes form compartments that are composed of smaller Topologically Associating Domains (TADs). TADs are thought to represent functional domains of gene regulation but much is still unknown about the mechanisms of their formation and how they exert their regulatory effect on embedded genes. Further, similar domains have been detected in other organisms, including flies, worms, fungi and bacteria. Although in all these cases these domains appear similar as detected by 3C-based methods, their biology appears to be quite distinct with differences in the protein complexes involved in their formation and differences in their internal organization. Here we outline our current understanding of such domains in different organisms and their roles in gene regulation.
URLPMID:27259200 [本文引用: 1]
We review recent literature related to chromatin domains, a prominent feature of the chromosome organization in animal cells. We discuss general properties, biological functions, and potential mechanisms of such structural partitions of chromosomes.
URLPMID:29503869 [本文引用: 1]
Abstract Deciphering the rules of genome folding in the cell nucleus is essential to understand its functions. Recent chromosome conformation capture (Hi-C) studies have revealed that the genome is partitioned into topologically associating domains (TADs), which demarcate functional epigenetic domains defined by combinations of specific chromatin marks. However, whether TADs are true physical units in each cell nucleus or whether they reflect statistical frequencies of measured interactions within cell populations is unclear. Using a combination of Hi-C, three-dimensional (3D) fluorescent in situ hybridization, super-resolution microscopy, and polymer modeling, we provide an integrative view of chromatin folding in Drosophila . We observed that repressed TADs form a succession of discrete nanocompartments, interspersed by less condensed active regions. Single-cell analysis revealed a consistent TAD-based physical compartmentalization of the chromatin fiber, with some degree of heterogeneity in intra-TAD conformations and in cis and trans inter-TAD contact events. These results indicate that TADs are fundamental 3D genome units that engage in dynamic higher-order inter-TAD connections. This domain-based architecture is likely to play a major role in regulatory transactions during DNA-dependent processes.
URLPMID:25409831 [本文引用: 1]
Eukaryotic chromosomes replicate in a temporal order known as the replication-timing program. In mammals, replication timing is cell-type-specific with at least half the genome switching replication timing during development, primarily in units of 400-800 kilobases ('replication domains'), whose positions are preserved in different cell types, conserved between species, and appear to confine long-range effects of chromosome rearrangements. Early and late replication correlate, respectively, with open and closed three-dimensional chromatin compartments identified by high-resolution chromosome conformation capture (Hi-C), and, to a lesser extent, late replication correlates with lamina-associated domains (LADs). Recent Hi-C mapping has unveiled substructure within chromatin compartments called topologically associating domains (TADs) that are largely conserved in their positions between cell types and are similar in size to replication domains. However, TADs can be further sub-stratified into smaller domains, challenging the significance of structures at any particular scale. Moreover, attempts to reconcile TADs and LADs to replication-timing data have not revealed a common, underlying domain structure. Here we localize boundaries of replication domains to the early-replicating border of replication-timing transitions and map their positions in 18 human and 13 mouse cell types. We demonstrate that, collectively, replication domain boundaries share a near one-to-one correlation with TAD boundaries, whereas within a cell type, adjacent TADs that replicate at similar times obscure replication domain boundaries, largely accounting for the previously reported lack of alignment. Moreover, cell-type-specific replication timing of TADs partitions the genome into two large-scale sub-nuclear compartments revealing that replication-timing transitions are indistinguishable from late-replicating regions in chromatin composition and lamina association and accounting for the reduced correlation of replication timing to LADs and heterochromatin. Our results reconcile cell-type-specific sub-nuclear compartmentalization and replication timing with developmentally stable structural domains and offer a unified model for large-scale chromosome structure and function.
URLPMID:24335803 [本文引用: 1]
Abstract Recent studies of genome-wide chromatin interactions have revealed that the human genome is partitioned into many self-associating topological domains. The boundary sequences between domains are enriched for binding sites of CTCC-binding factor (CTCF) and the cohesin complex, implicating these two factors in the establishment or maintenance of topological domains. To determine the role of cohesin and CTCF in higher-order chromatin architecture in human cells, we depleted the cohesin complex or CTCF and examined the consequences of loss of these factors on higher-order chromatin organization, as well as the transcriptome. We observed a general loss of local chromatin interactions upon disruption of cohesin, but the topological domains remain intact. However, we found that depletion of CTCF not only reduced intradomain interactions but also increased interdomain interactions. Furthermore, distinct groups of genes become misregulated upon depletion of cohesin and CTCF. Taken together, these observations suggest that CTCF and cohesin contribute differentially to chromatin organization and gene regulation.
URLPMID:28985562 [本文引用: 1]
The human genome folds to create thousands of intervals, called “contact domains,” that exhibit enhanced contact frequency within themselves. “Loop domains” form because of tethering between two loci—almost always bound by CTCF and cohesin—lying on the same chromosome. “Compartment domains” form when genomic intervals with similar histone marks co-segregate. Here, we explore the effects of degrading cohesin. All loop domains are eliminated, but neither compartment domains nor histone marks are affected. Loss of loop domains does not lead to widespread ectopic gene activation but does affect a significant minority of active genes. In particular, cohesin loss causes superenhancers to co-localize, forming hundreds of links within and across chromosomes and affecting the regulation of nearby genes. We then restore cohesin and monitor the re-formation of each loop. Although re-formation rates vary greatly, many megabase-sized loops recovered in under an hour, consistent with a model where loop extrusion is rapid.
URLPMID:29117157 [本文引用: 1]
Tackling the challenges of genomics and studies of the immune system should help to create much-needed diagnostics and treatments.
URLPMID:25732821 [本文引用: 1]
To explore the mechanisms underlying the evolution of chromosomal domain structures, Vietri Rudan et al. compare four mammalian species and reveal a direct link between insulator site divergence and the evolution of chromatin domain structure. Their data point to a direct role for CTCF/cohesin in driving structural change in the genome.
URL [本文引用: 1]
Chromosome conformation capture approaches have shown that interphase chromatin is partitioned into spatially segregated Mb-sized compartments and sub-Mb-sized topological domains. This compartmentalization is thought to facilitate the matching of genes and regulatory elements, but its precise function and mechanistic basis remain unknown. Cohesin controls chromosome topology to enable DNA repair and chromosome segregation in cycling cells. In addition, cohesin associates with active enhancers and promoters and with CTCF to form long-range interactions important for gene regulation. Although these findings suggest an important role for cohesin in genome organization, this role has not been assessed on a global scale. Unexpectedly, we find that architectural compartments are maintained in noncycling mouse thymocytes after genetic depletion of cohesin in vivo. Cohesin was, however, required for specific long-range interactions within compartments where cohesin-regulated genes reside. Cohesin depletion diminished interactions between cohesin-bound sites, whereas alternative interactions between chromatin features associated with transcriptional activation and repression became more prominent, with corresponding changes in gene expression. Our findings indicate that cohesin-mediated long-range interactions facilitate discrete gene expression states within preexisting chromosomal compartments.
URLPMID:24185899 [本文引用: 1]
To ensure proper gene regulation within constrained nuclear space, chromosomes facilitate access to transcribed regions, while compactly packaging all other information. Recent studies revealed that chromosomes are organized into megabase-scale domains that demarcate active and inactive genetic elements, suggesting that compartmentalization is important for genome function. Here, we show that very specific long-range interactions are anchored by cohesin/CTCF sites, but not cohesin-only or CTCF-only sites, to form a hierarchy of chromosomal loops. These loops demarcate topological domains and form intricate internal structures within them. Post-mitotic nuclei deficient for functional cohesin exhibit global architectural changes associated with loss of cohesin/CTCF contacts and relaxation of topological domains. Transcriptional analysis shows that this cohesin-dependent perturbation of domain organization leads to widespread gene deregulation of both cohesin-bound and non-bound genes. Our data thereby support a role for cohesin in the global organization of domain structure and suggest that domains function to stabilize the transcriptional programmes within them.
URL [本文引用: 1]
The binding of the transcriptional regulator CTCF to the genome has been implicated in the formation of topologically associated domains (TADs). However, the general mechanisms of folding the genome into TADs are not fully understood. Here we test the effects of deleting a CTCF-rich locus on TAD boundary formation. Using genome-wide chromosome conformation capture (Hi-C), we focus on one TAD boundary on chromosome X harboring ~6515 CTCF binding sites and located at the long non-coding RNA (lncRNA) locusFirre. Specifically, this TAD boundary is invariant across evolution, tissues, and temporal dynamics of X-chromosome inactivation. We demonstrate that neither the deletion of this locus nor the ectopic insertion ofFirrecDNA or its ectopic expression are sufficient to alter TADs in a sex-specific or allele-specific manner. In contrast,Firre’sdeletion disrupts the chromatin super-loop formation of the inactive X-chromosome. Collectively, our findings suggest that apart from CTCF binding, additional mechanisms may play roles in establishing TAD boundary formation. Although CTCF binding has been implicated in the formation of topologically associated domains (TADs) the mechanisms folding the genome into TADs are not fully understood. Here the authors investigate the TAD boundary on lncRNA locusFirre, which has ~6515 CTCF binding sites, and its organization.
URLPMID:29335463 [本文引用: 1]
Topologically associating domains (TADs) are fundamental elements of the eukaryotic genomic structure. However, recent studies suggest that the insulating complexes, CTCF/cohesin, present at TAD borders in mammals are absent from those inDrosophila melanogaster, raising the possibility that border elements are not conserved among metazoans. Using in situ Hi-C with sub-kb resolution, here we show that theD.melanogastergenome is almost completely partitioned into >4000 TADs, nearly sevenfold more than previously identified. The overwhelming majority of these TADs are demarcated by the insulator complexes, BEAF-32/CP190, or BEAF-32/Chromator, indicating that these proteins may play an analogous role in flies as that of CTCF/cohesin in mammals. Moreover, extended regions previously thought to be unstructured are shown to consist of small contiguous TADs, a property also observed in mammals upon re-examination. Altogether, our work demonstrates that fundamental features associated with the higher-order folding of the genome are conserved from insects to mammals. Topologically associating domain (TAD) boundaries in flies seem to be different from those in mammals. Here, the authors use Hi-C with sub-kb resolution to identify about 4000 TADs in flies, most demarcated by the insulator complexes BEAF-32/CP190 or BEAF-32/Chromator like CTCF/cohesin in mammals.
URLPMID:3950722 [本文引用: 1]
Understanding the structure of interphase chromosomes is essential to elucidate regulatory mechanisms of gene expression. During recent years, high-throughput DNA sequencing expanded the power of chromosome conformation capture (3C) methods that provide information about reciprocal spatial proximity of chromosomal loci. Since 2012, it is known that entire chromatin in interphase chromosomes is organized into regions with strongly increased frequency of internal contacts. These regions, with the average size of 1 Mb, were named topological domains. More recent studies demonstrated presence of unconstrained supercoiling in interphase chromosomes. Using Brownian dynamics simulations, we show here that by including supercoiling into models of topological domains one can reproduce and thus provide possible explanations of several experimentally observed characteristics of interphase chromosomes, such as their complex contact maps.
URLPMID:23416946 [本文引用: 1]
DNA supercoiling is an inherent consequence of twisting DNA and is critical for regulating gene expression and DNA replication. However, DNA supercoiling at a genomic scale in human cells is uncharacterized. To map supercoiling, we used biotinylated trimethylpsoralen as a DNA structure probe to show that the human genome is organized into supercoiling domains. Domains are formed and remodeled by RNA polymerase and topoisomerase activities and are flanked by GC-AT boundaries and CTCF insulator protein-binding sites. Underwound domains are transcriptionally active and enriched in topoisomerase I, 'open' chromatin fibers and DNase I sites, but they are depleted of topoisomerase II. Furthermore, DNA supercoiling affects additional levels of chromatin compaction as underwound domains are cytologically decondensed, topologically constrained and decompacted by transcription of short RNAs. We suggest that supercoiling domains create a topological environment that facilitates gene activation, providing an evolutionary purpose for clustering genes along chromosomes.
URLPMID:29416042 [本文引用: 1]
Abstract The metazoan genome is compartmentalized in areas of highly interacting chromatin known as topologically associating domains (TADs). TADs are demarcated by boundaries mostly conserved across cell types and even across species. However, a genome-wide characterization of TAD boundary strength in mammals is still lacking. In this study, we first use fused two-dimensional lasso as a machine learning method to improve Hi-C contact matrix reproducibility, and, subsequently, we categorize TAD boundaries based on their insulation score. We demonstrate that higher TAD boundary insulation scores are associated with elevated CTCF levels and that they may differ across cell types. Intriguingly, we observe that super-enhancers are preferentially insulated by strong boundaries. Furthermore, we demonstrate that strong TAD boundaries and super-enhancer elements are frequently co-duplicated in cancer patients. Taken together, our findings suggest that super-enhancers insulated by strong TAD boundaries may be exploited, as a functional unit, by cancer cells to promote oncogenesis.
URLPMID:4791538 [本文引用: 1]
Disease-associated structural variants, when affecting CTCF-associated boundary elements, cause pathogenicity by disrupting the structure of topologically associated chromatin domains leading to ectopic promoter interactions and altered gene expression.
URLPMID:27863249 [本文引用: 2]
As part of the IHEC consortium, this study deploys a Hi-C promoter capture approach in 17 primary blood cell types to match collaborating regulatory regions and identify genes regulated by noncoding disease-associated variants. Explore the Cell Press IHEC webportal athttp://www.cell.com/consortium/IHEC.
URLPMID:26971819 [本文引用: 1]
Krijger et al. report that the reprogramming of four somatic cell types with highly distinct 3D genomes results in pluripotent cells with largely identical, ESC-like, genome conformations carrying founder-dependent topological hallmarks. The latter are not remnants of somatic chromosome topologies but are acquired during reprogramming in a cell-of-origin-dependent manner.
URLPMID:3838900 [本文引用: 2]
A large number of cis-regulatory sequences have been annotated in the human genome(1,2), but defining their target genes remains a challenge(3). One strategy is to identify the long-range looping interactions at these elements with the use of chromosome conformation capture (3C)-based techniques(4). However, previous studies lack either the resolution or coverage to permit a whole-genome, unbiased view of chromatin interactions. Here we report a comprehensive chromatin interaction map generated in human fibroblasts using a genome-wide 3C analysis method (Hi-C)(5). We determined over one million long-range chromatin interactions at 5-10-kb resolution, and uncovered general principles of chromatin organization at different types of genomic features. We also characterized the dynamics of promoter-enhancer contacts after TNF-alpha signalling in these cells. Unexpectedly, we found that TNF-alpha-responsive enhancers are already in contact with their target promoters before signalling. Such pre-existing chromatin looping, which also exists in other cell types with different extracellular signalling, is a strong predictor of gene induction. Our observations suggest that the three-dimensional chromatin landscape, once established in a particular cell type, is relatively stable and could influence the selection or activation of target genes by a ubiquitous transcription activator in a cell-specific manner.
URLPMID:4734140 [本文引用: 2]
Advanced ChIA-PET shows that CTCF/cohesin and RNA polymerase II arrange spatial organization for coordinated transcription. Haplotype variants exhibit allelic effects on chromatin topology and transcription that link disease susceptibility.
URLPMID:26637943 [本文引用: 1]
Stunnenberg and colleagues used CHi-C to identify Extremely Long-Range Promoter-Promoter Interactions (ELRIs) in mESCs. Their analysis points to a spatiotemporal mechanism for repressingHoxand other developmentally important genes during the transition from the 2i ground-state to the primed serum state regulated by PRC2.
URLPMID:22955621 [本文引用: 2]
Abstract The vast non-coding portion of the human genome is full of functional elements and disease-causing regulatory variants. The principles defining the relationships between these elements and distal target genes remain unknown. Promoters and distal elements can engage in looping interactions that have been implicated in gene regulation. Here we have applied chromosome conformation capture carbon copy (5C) to interrogate comprehensively interactions between transcription start sites (TSSs) and distal elements in 1% of the human genome representing the ENCODE pilot project regions. 5C maps were generated for GM12878, K562 and HeLa-S3 cells and results were integrated with data from the ENCODE consortium. In each cell line we discovered >1,000 long-range interactions between promoters and distal sites that include elements resembling enhancers, promoters and CTCF-bound sites. We observed significant correlations between gene expression, promoter-enhancer interactions and the presence of enhancer RNAs. Long-range interactions show marked asymmetry with a bias for interactions with elements located 090804120 kilobases upstream of the TSS. Long-range interactions are often not blocked by sites bound by CTCF and cohesin, indicating that many of these sites do not demarcate physically insulated gene domains. Furthermore, only 0908047% of looping interactions are with the nearest gene, indicating that genomic proximity is not a simple predictor for long-range interactions. Finally, promoters and distal elements are engaged in multiple long-range interactions to form complex networks. Our results start to place genes and regulatory elements in three-dimensional context, revealing their functional relationships.
URLPMID:20 [本文引用: 1]
In multicellular organisms, transcription regulation is one of the central mechanisms modelling lineage differentiation and cell-fate determination(1). Transcription requires dynamic chromatin configurations between promoters and their corresponding distal regulatory elements(2). It is believed that their communication occurs within large discrete foci of aggregated RNA polymerases termed transcription factories in three-dimensional nuclear space(3). However, the dynamic nature of chromatin connectivity has not been characterized at the genome-wide level. Here, through a chromatin interaction analysis with paired-end tagging approach(3-5) using an antibody that primarily recognizes the pre-initiation complexes of RNA polymerase II6, we explore the transcriptional interactomes of three mouse cells of progressive lineage commitment, including pluripotent embryonic stem cells(7), neural stem cells(8) and neurosphere stem/progenitor cells(9). Our global chromatin connectivity maps reveal approximately 40,000 long-range interactions, suggest precise enhancer-promoter associations and delineate cell-type-specific chromatin structures. Analysis of the complex regulatory repertoire shows that there are extensive colocalizations among promoters and distal-acting enhancers. Most of the enhancers associate with promoters located beyond their nearest active genes, indicating that the linear juxtaposition is not the only guiding principle driving enhancer target selection. Although promoter-enhancer interactions exhibit high cell-type specificity, promoters involved in interactions are found to be generally common and mostly active among different cells. Chromatin connectivity networks reveal that the pivotal genes of reprogramming functions are transcribed within physical proximity to each other in embryonic stem cells, linking chromatin architecture to coordinated gene expression. Our study sets the stage for the full-scale dissection of spatial and temporal genome structures and their roles in orchestrating development.
URLPMID:26499245 [本文引用: 2]
We recently used in situ Hi-C to create kilobase-resolution 3D maps of mammalian genomes. Here, we combine these maps with new Hi-C, microscopy, and genome-editing experiments to study the physical structure of fibers, domains, and loops. We find that the observed contact domains are inconsistent with the equilibrium state for an ordinary condensed polymer. Combining Hi-C data and novel mathematical theorems, we show that contact domains are also not consistent with a fractal globule. Instead, we use physical simulations to study two models of genome folding. In one, intermonomer attraction during polymer condensation leads to of an anisotropic "tension globule." In the other, () and act together to extrude unknotted loops during . Both models are consistent with the observed contact domains and with the observation that contact domains tend to form inside loops. However, the extrusion model explains a far wider array of observations, such as why loops tend not to overlap and why the -motifs at pairs of loop anchors lie in the convergent orientation. Finally, we perform 13 genome-editing experiments examining the effect of altering -sites on folding. The convergent rule correctly predicts the affected loops in every case. Moreover, the extrusion model accurately predicts in silico the 3D maps resulting from each experiment using only the location of -sites in the WT. Thus, we show that it is possible to disrupt, restore, and move loops and domains using targeted mutations as small as a single base pair.
URLPMID:4889513 [本文引用: 1]
Topologically associating domains (TADs) are fundamental building blocks of human interphase chromosomes. Fudenberg et al. propose that TADs emerge as a consequence of loop extrusion limited by boundary elements. The authors use polymer simulations and genomic analyses to identify molecular roles for the architectural proteins cohesin and CTCF.
URLPMID:4745123 [本文引用: 1]
The architectural protein CTCF plays a complex role in decoding the functional output of the genome. Guo et al. now show that the orientation of a CTCF site restricts its choice of interacting partner, thus creating a code that predicts the three-dimensional organization of the genome. We propose a DNA extrusion model to account for orientation-specific loop formation.
URLPMID:27134029 [本文引用: 2]
SMC dimeric complexes combine with a subunit of the kleisin family and form ring complexes entrapping DNA. In eukaryotes, cohesins, condensins, and SMC5-6 act on sister chromatid cohesion, chromosome condensation, and DNA repair. Cohesin complexes are dynamic as a result of two opposing activities: a DNA-loading activity mediated by the Scc2-Scc4 complex and a releasing activity elicited by the Pds5-Wapl-Scc3 complex. Crystal structures of these proteins are providing insight into their function. Pds5 and Scc3 have distinct positive functions in cohesion maintenance after S phase. The concept emerging is that different arrangements of these complexes exist in different stages of the cell cycle. SMCs are found in both open V-shaped and closed rod-shaped forms, which likely represent functional states of the complexes. Their formation depends on interactions with DNA and ATP hydrolysis.
URLPMID:25303531 [本文引用: 1]
In embryonic stem cells, both super-enhancer-driven cell identity genes and repressed lineage-specifying genes occur in insulated neighborhoods. The integrity of these structural neighborhoods is important for proper expression of nearby genes.
URLPMID:26686465 [本文引用: 1]
Ji et al. map the chromosome organizational structures that underlie gene regulation in human naive and primed pluripotent cells. Their framework of cohesin-associated CTCF loops, and the cohesin-associated enhancer-promoter loops within them, provides a reference map for future interrogation of regulatory interactions.
URL [本文引用: 1]
哺乳动物中原钙粘蛋白(Protocadherin,Pcdh)基因簇包含50多个串联排列的基因,这些基因形成3个紧密相连的基因簇(Pcdh?、Pcdh?和Pcdh?),所编码的原钙粘蛋白质群在神经元多样性(Neuronal diversity)和单细胞特异性(Single cell identity)以及神经突触信号转导中发挥重要作用。前期的工作已证实转录因子CTCF(CCCTC-binding factor)与CTCF结合位点(CTCF-binding site,CBS)的方向性结合能够决定增强子和启动子环化的方向以及其远距离交互作用的特异性,并进一步在Pcdh基因座(Locus)形成两个(Pcdh?和Pcdh?)染色质拓扑结构域(CTCF/cohesin-mediated chromatin domain,CCD),而且染色质拓扑结构域对于控制基因表达调控至关重要。本文通过生物信息学方法对比人类和小鼠序列,发现Pcdh??染色质拓扑结构域调控区域中的DNase I超敏位点(DNase I hypersensitive sites,HSs)较为保守。染色质免疫沉淀及大规模测序实验(Chromatin immunoprecipitation and massive parallel sequencing,Ch IP-Seq)揭示CBS位点在Pcdh??调控区域中成簇分布并且具有相同的方向。凝胶电泳迁移实验(Electrophoresis mobility shift assay,EMSA)确定Pcdh??调控区域内具体的42 bp CBS位点并且发现一个CTCF峰包含两个CBS位点。在全基因组范围内,运用计算生物学方法分析CTCF和增强子、启动子等调控元件的关系,发现CBS位点在调控元件附近有较多分布,推测CTCF通过介导增强子和启动子的特异性交互作用,在细胞核三维基因组内形成活性转录枢纽调控基因精准表达。
URL [本文引用: 1]
哺乳动物中原钙粘蛋白(Protocadherin,Pcdh)基因簇包含50多个串联排列的基因,这些基因形成3个紧密相连的基因簇(Pcdh?、Pcdh?和Pcdh?),所编码的原钙粘蛋白质群在神经元多样性(Neuronal diversity)和单细胞特异性(Single cell identity)以及神经突触信号转导中发挥重要作用。前期的工作已证实转录因子CTCF(CCCTC-binding factor)与CTCF结合位点(CTCF-binding site,CBS)的方向性结合能够决定增强子和启动子环化的方向以及其远距离交互作用的特异性,并进一步在Pcdh基因座(Locus)形成两个(Pcdh?和Pcdh?)染色质拓扑结构域(CTCF/cohesin-mediated chromatin domain,CCD),而且染色质拓扑结构域对于控制基因表达调控至关重要。本文通过生物信息学方法对比人类和小鼠序列,发现Pcdh??染色质拓扑结构域调控区域中的DNase I超敏位点(DNase I hypersensitive sites,HSs)较为保守。染色质免疫沉淀及大规模测序实验(Chromatin immunoprecipitation and massive parallel sequencing,Ch IP-Seq)揭示CBS位点在Pcdh??调控区域中成簇分布并且具有相同的方向。凝胶电泳迁移实验(Electrophoresis mobility shift assay,EMSA)确定Pcdh??调控区域内具体的42 bp CBS位点并且发现一个CTCF峰包含两个CBS位点。在全基因组范围内,运用计算生物学方法分析CTCF和增强子、启动子等调控元件的关系,发现CBS位点在调控元件附近有较多分布,推测CTCF通过介导增强子和启动子的特异性交互作用,在细胞核三维基因组内形成活性转录枢纽调控基因精准表达。
URLPMID:24067610 [本文引用: 2]
Large-scale chromosome structure and spatial nuclear arrangement have been linked to control of gene expression and DNA replication and repair. Genomic techniques based on chromosome conformation capture (3C) assess contacts for millions of loci simultaneously, but do so by averaging chromosome conformations from millions of nuclei. Here we introduce single-cell Hi-C, combined with genome-wide statistical analysis and structural modelling of single-copy X chromosomes, to show that individual chromosomes maintain domain organization at the megabase scale, but show variable cell-to-cell chromosome structures at larger scales. Despite this structural stochasticity, localization of active gene domains to boundaries of chromosome territories is a hallmark of chromosomal conformation. Single-cell Hi-C data bridge current gaps between genomics and microscopy studies of chromosomes, demonstrating how modular organization underlies dynamic chromosome structure, and how this structure is probabilistically linked with genome activity patterns.
URLPMID:27685100
Abstract With the advent of massively parallel sequencing, considerable work has gone into adapting chromosome conformation capture (3C) techniques to study chromosomal architecture at a genome-wide scale. We recently demonstrated that the inactive murine X chromosome adopts a bipartite structure using a novel 3C protocol, termed in situ DNase Hi-C. Like traditional Hi-C protocols, in situ DNase Hi-C requires that chromatin be chemically cross-linked, digested, end-repaired, and proximity-ligated with a biotinylated bridge adaptor. The resulting ligation products are optionally sheared, affinity-purified via streptavidin bead immobilization, and subjected to traditional next-generation library preparation for Illumina paired-end sequencing. Importantly, in situ DNase Hi-C obviates the dependence on a restriction enzyme to digest chromatin, instead relying on the endonuclease DNase I. Libraries generated by in situ DNase Hi-C have a higher effective resolution than traditional Hi-C libraries, which makes them valuable in cases in which high sequencing depth is allowed for, or when hybrid capture technologies are expected to be used. The protocol described here, which involves 4 d of bench work, is optimized for the study of mammalian cells, but it can be broadly applicable to any cell or tissue of interest, given experimental parameter optimization.
URLPMID:28435001
Chromosome conformation capture-based methods such as Hi-C have become mainstream techniques for the study of the 3D organization of genomes. These methods convert chromatin interactions reflecting topological chromatin structures into digital information (counts of pair-wise interactions). Here, we describe an updated protocol for Hi-C (Hi-C 2.0) that integrates recent improvements into a single protocol for efficient and high-resolution capture of chromatin interactions. This protocol combines chromatin digestion and frequently cutting enzymes to obtain kilobase (Kb) resolution. It also includes steps to reduce random ligation and the generation of uninformative molecules, such as unligated ends, to improve the amount of valid intra-chromosomal read pairs. This protocol allows for obtaining information on conformational structures such as compartment and topologically associating domains, as well as high-resolution conformational features such as DNA loops.
URLPMID:22198700 [本文引用: 1]
We describe tethered conformation capture (TCC), a method for genome-wide mapping of chromatin interactions. By performing ligations on solid substrates rather than in solution, TCC substantially enhances the signal-to-noise ratio, thereby facilitating a detailed analysis of interactions within and between chromosomes. We identified a group of regions in each chromosome in human cells that account for the majority of interchromosomal interactions. These regions are marked by high transcriptional activity, suggesting that their interactions are mediated by transcriptional machinery. Each of these regions interacts with numerous other such regions throughout the genome in an indiscriminate fashion, partly driven by the accessibility of the partners. As a different combination of interactions is likely present in different cells, we developed a computational method to translate the TCC data into physical chromatin contacts in a population of three-dimensional genome structures. Statistical analysis of the resulting population demonstrates that the indiscriminate properties of interchromosomal interactions are consistent with the well-known architectural features of the human genome.
URLPMID:4117202 [本文引用: 2]
Genomes assembled de novo from short reads are highly fragmented relative to the finished chromosomes of Homo sapiens and key model organisms generated by the Human Genome Project. To address this problem, we need scalable, cost-effective methods to obtain assemblies with chromosome-scale contiguity. Here we show that genome-wide chromatin interaction data sets, such as those generated by Hi-C, are a rich source of long-range information for assigning, ordering and orienting genomic sequences to chromosomes, including across centromeres. To exploit this finding, we developed an algorithm that uses Hi-C data for ultra-long-range scaffolding of de novo genome assemblies. We demonstrate the approach by combining shotgun fragment and short jump mate-pair sequences with Hi-C data to generate chromosome-scale de novo assemblies of the human, mouse and Drosophila genomes, achieving-for the human genome-98% accuracy in assigning scaffolds to chromosome groups and 99% accuracy in ordering and orienting scaffolds within chromosome groups. Hi-C data can also be used to validate chromosomal translocations in cancer genomes.
URLPMID:25667002 [本文引用: 1]
URLPMID:28263316 [本文引用: 1]
Abstract The decrease in sequencing cost and increased sophistication of assembly algorithms for short-read platforms has resulted in a sharp increase in the number of species with genome assemblies. However, these assemblies are highly fragmented, with many gaps, ambiguities, and errors, impeding downstream applications. We demonstrate current state of the art for de novo assembly using the domestic goat (Capra hircus) based on long reads for contig formation, short reads for consensus validation, and scaffolding by optical and chromatin interaction mapping. These combined technologies produced what is, to our knowledge, the most continuous de novo mammalian assembly to date, with chromosome-length scaffolds and only 649 gaps. Our assembly represents a 400-fold improvement in continuity due to properly assembled gaps, compared to the previously published C. hircus assembly, and better resolves repetitive structures longer than 1 kb, representing the largest repeat family and immune gene complex yet produced for an individual of a ruminant species.
URLPMID:28178233 [本文引用: 1]
Abstract Chenopodium quinoa (quinoa) is a highly nutritious grain identified as an important crop to improve world food security. Unfortunately, few resources are available to facilitate its genetic improvement. Here we report the assembly of a high-quality, chromosome-scale reference genome sequence for quinoa, which was produced using single-molecule real-time sequencing in combination with optical, chromosome-contact and genetic maps. We also report the sequencing of two diploids from the ancestral gene pools of quinoa, which enables the identification of sub-genomes in quinoa, and reduced-coverage genome sequences for 22 other samples of the allotetraploid goosefoot complex. The genome sequence facilitated the identification of the transcription factor likely to control the production of anti-nutritional triterpenoid saponins found in quinoa seeds, including a mutation that appears to cause alternative splicing and a premature stop codon in sweet quinoa strains. These genomic resources are an important first step towards the genetic improvement of quinoa.
URLPMID:28336562 [本文引用: 1]
The Zika outbreak, spread by the Aedes aegypti mosquito, highlights the need to create high-quality assemblies of large genomes in a rapid and cost-effective way. Here we combine Hi-C data with existing draft assemblies to generate chromosome-length scaffolds. We validate this method by assembling a human genome, de novo, from short reads alone (67 coverage). We then combine our method with draft sequences to create genome assemblies of the mosquito disease vectors Ae. aegypti and Culex quinquefasciatus, each consisting of three scaffolds corresponding to the three chromosomes in each species. These assemblies indicate that almost all genomic rearrangements among these species occur within, rather than between, chromosome arms. The genome assembly procedure we describe is fast, inexpensive, and accurate, and can be applied to many species.
URLPMID:28447635 [本文引用: 1]
Abstract Cereal grasses of the Triticeae tribe have been the major food source in temperate regions since the dawn of agriculture. Their large genomes are characterized by a high content of repetitive elements and large pericentromeric regions that are virtually devoid of meiotic recombination. Here we present a high-quality reference genome assembly for barley (Hordeum vulgare L.). We use chromosome conformation capture mapping to derive the linear order of sequences across the pericentromeric space and to investigate the spatial organization of chromatin in the nucleus at megabase resolution. The composition of genes and repetitive elements differs between distal and proximal regions. Gene family analyses reveal lineage-specific duplications of genes involved in the transport of nutrients to developing seeds and the mobilization of carbohydrates in grains. We demonstrate the importance of the barley reference sequence for breeding by inspecting the genomic partitioning of sequence variation in modern elite germplasm, highlighting regions vulnerable to genetic erosion.
[本文引用: 1]
URLPMID:15660514 [本文引用: 1]
Advances in genotyping and sequencing technologies, coupled with the of sophisticated statistical methods, have afforded investigators novel opportunities to define the role of sequence variation in the of common diseases. At the forefront of these investigations is the use of dense maps of single-nucleotide polymorphisms (SNPs) and the haplotypes derived from these polymorphisms. Here we review basic concepts of high-density genetic maps of SNPs and haplotypes and how they are typically generated and used in genetic research. We also provide useful examples and tools available for researchers interested in incorporating haplotypes into their studies. Finally, we discuss the latest concepts for the analysis of haplotypes related to disease, including haplotype blocks, the International HapMap Project, and the future directions of these resources.
URLPMID:4180835 [本文引用: 1]
Rapid advances in high-throughput sequencing facilitate variant discovery and genotyping, but linking variants into a single haplotype remains challenging. Here we demonstrate HaploSeq, an approach for assembling chromosome-scale haplotypes by exploiting the existence of 'chromosome territories'. We use proximity ligation and sequencing to show that alleles on homologous chromosomes occupy distinct territories, and therefore this experimental protocol preferentially recovers physically linked DNA variants on a homolog. Computational analysis of such data sets allows for accurate (similar to 99.5%) reconstruction of chromosome-spanning haplotypes for similar to 95% of alleles in hybrid mouse cells with 30x sequencing coverage. To resolve haplotypes for a human genome, which has a low density of variants, we coupled HaploSeq with local conditional phasing to obtain haplotypes for similar to 81% of alleles with similar to 98% accuracy from just 17x sequencing. Whereas methods based on proximity ligation were originally designed to investigate spatial organization of genomes, our results lend support for their use as a general tool for haplotyping.
URLPMID:24316648 [本文引用: 1]
Genome sequencing is improved by information on long-range chromatin interactions.
URL [本文引用: 1]
Abstract Motivation: Complex interactions among alleles often drive differences in inherited properties including disease predisposition. Isolating the effects of these interactions requires phasing information that is difficult to measure or infer. Furthermore, prevalent sequencing technologies used in the essential first step of determining a haplotype limit the range of that step to the span of reads, namely hundreds of bases. With the advent of pseudo-long read technologies, observable partial haplotypes can span several orders of magnitude more. Yet, measuring whole-genome-single-individual haplotypes remains a challenge. A different view of whole genome measurement addresses the 3D structure of the genome ith great development of Hi-C techniques in recent years. A shortcoming of current Hi-C, however, is the difficulty in inferring information that is specific to each of a pair of homologous chromosomes. Results: In this work, we develop a robust algorithmic framework that takes two measurement derived datasets: raw Hi-C and partial short-range haplotypes, and constructs the full-genome haplotype as well as phased diploid Hi-C maps. By analyzing both data sets together we thus bridge important gaps in both technologies rom short to long haplotypes and from un-phased to phased Hi-C. We demonstrate that our method can recover ground truth haplotypes with high accuracy, using measured biological data as well as simulated data. We analyze the impact of noise, Hi-C sequencing depth and measured haplotype lengths on performance. Finally, we use the inferred 3D structure of a human genome to point at transcription factor targets nuclear co-localization. Availability and Implementation: The implementation available at https://github.com/YakhiniGroup/SpectraPh . Contact:zohar.yakhini@gmail.com Supplementary information:Supplementary data are available at Bioinformatics online.
URLPMID:27940952 [本文引用: 1]
Abstract Many tools have been developed for haplotype assembly - the reconstruction of individual haplotypes using reads mapped to a reference genome sequence. Due to increasing interest in obtaining haplotype-resolved human genomes, a range of new sequencing protocols and technologies have been developed to enable the reconstruction of whole-genome haplotypes. However, existing computational methods designed to handle specific technologies do not scale well on data from different protocols. We describe a new algorithm, HapCUT2, that extends our previous method (HapCUT) to handle multiple sequencing technologies. Using simulations and whole-genome sequencing (WGS) data from multiple different data types -- dilution pool sequencing, linked-read sequencing, single molecule real-time (SMRT) sequencing, and proximity ligation (Hi-C) sequencing -- we show that HapCUT2 rapidly assembles haplotypes with best-in-class accuracy for all data types. In particular, HapCUT2 scales well for high sequencing coverage and rapidly assembled haplotypes for two long-read WGS datasets on which other methods struggled. Further, HapCUT2 directly models Hi-C specific error modalities resulting in significant improvements in error rates compared to HapCUT, the only other method that could assemble haplotypes from Hi-C data. Using HapCUT2, haplotype assembly from a 90x coverage whole-genome Hi-C dataset yielded high-resolution haplotypes (78.6% of variants phased in a single block) with high pairwise phasing accuracy (~98% across chromosomes). Our results demonstrate that HapCUT2 is a robust tool for haplotype assembly applicable to data from diverse sequencing technologies. Published by Cold Spring Harbor Laboratory Press.
URL [本文引用: 1]
URLPMID:28805829 [本文引用: 1]
Abstract Chromosome conformation is an important feature of metazoan gene regulation; however, enhancer-promoter contact remodeling during cellular differentiation remains poorly understood. To address this, genome-wide promoter capture Hi-C (CHi-C) was performed during epidermal differentiation. Two classes of enhancer-promoter contacts associated with differentiation-induced genes were identified. The first class ('gained') increased in contact strength during differentiation in concert with enhancer acquisition of the H3K27ac activation mark. The second class ('stable') were pre-established in undifferentiated cells, with enhancers constitutively marked by H3K27ac. The stable class was associated with the canonical conformation regulator cohesin, whereas the gained class was not, implying distinct mechanisms of contact formation and regulation. Analysis of stable enhancers identified a new, essential role for a constitutively expressed, lineage-restricted ETS-family transcription factor, EHF, in epidermal differentiation. Furthermore, neither class of contacts was observed in pluripotent cells, suggesting that lineage-specific chromatin structure is established in tissue progenitor cells and is further remodeled in terminal differentiation.
URLPMID:29053968 [本文引用: 1]
Chromosome conformation capture technologies have revealed important insights into genome folding. Yet, how spatial genome architecture is related to gene expression and cell fate remains unclear. We comprehensively mapped 3D chromatin organization during mouse neural differentiationin02vitroandin02vivo, generating the highest-resolution Hi-C maps available to date. We found that transcription is correlated with chromatin insulation and long-range interactions, but dCas9-mediated activation is insufficient for creating TAD boundariesde novo. Additionally, we discovered long-range contacts between gene bodies of exon-rich, active genes in all cell types. During neural differentiation, contacts between active TADs become less pronounced while inactive TADs interact more strongly. An extensive Polycomb network in stem cells is disrupted, while02dynamic interactions between neural transcription factors appearin02vivo. Finally, cell type-specific enhancer-promoter contacts are established concomitant to gene expression. This work shows that multiple factors influence the dynamics of chromatin interactions in development. Graphical Abstract 61Ultra-deep Hi-C during mouse neural differentiation, bothin02vitroandin02vivo61Transcription is correlated with, but not sufficient for, local chromatin insulation61Polycomb network is disrupted, while novel contacts between neural TF sites appear61Dynamic contacts among exon-rich gene bodies, enhancer-promoters, and TF sites Ultra-deep Hi-C during mouse neural differentiation, bothin02vitroandin02vivo Transcription is correlated with, but not sufficient for, local chromatin insulation Polycomb network is disrupted, while novel contacts between neural TF sites appear Dynamic contacts among exon-rich gene bodies, enhancer-promoters, and TF sites An ultrahigh resolution Hi-C map of mouse neural differentiation yields insights into the multiple factors that influence the dynamics of chromatin interactions during development.
URLPMID:20550932 [本文引用: 1]
78 LINE and SINE repeats help to nucleate heterochromatin during X inactivation 78 Young LINEs are active on the X chromosome during its inactivation 78 Young LINE expression on the inactive X requires functional Xist RNA 78 LINE activity may facilitate X inactivation of genes in escape-prone regions
URL [本文引用: 1]
URLPMID:11780141 [本文引用: 1]
The gene Xist initiates the chromosomal silencing process of X inactivation in mammals. Its product, a noncoding RNA, is expressed from and specifically associates with the inactive X chromosome in female cells. Here we use an inducible Xist expression system in mouse embryonic stem cells that recapitulates long-range chromosomal silencing to elucidate which Xist RNA sequences are necessary for chromosomal association and silencing. We show that chromosomal association and spreading of Xist RNA can be functionally separated from silencing by specific mutations. Silencing requires a conserved repeat sequence located at the 5' end of Xist. Deletion of this element results in Xist RNA that still associates with chromatin and spreads over the chromosome but does not effect transcriptional repression. Association of Xist RNA with chromatin is mediated by functionally redundant sequences that act cooperatively and are dispersed throughout the remainder of Xist but show little or no homology.
URLPMID:25843628 [本文引用: 1]
Development of a general method for identifying RNA-protein interactions in02vivo reveals 81 endogenous proteins that associate with Xist RNA in two waves to control mammalian dosage compensation.
URLPMID:5443622 [本文引用: 1]
X-chromosome inactivation (XCI) involves major reorganization of the X chromosome as it becomes silent and heterochromatic. During female mammalian development, XCI is triggered by upregulation of the non-coding Xist RNA from one of the two X chromosomes. Xist coats the chromosome in cis and induces silencing of almost all genes via its A-repeat region, although some genes (constitutive escapees) avoid silencing in most cell types, and others (facultative escapees) escape XCI only in specific contexts. A role for Xist in organizing the inactive X (Xi) chromosome has been proposed. Recent chromosome conformation capture approaches have revealed global loss of local structure on the Xi chromosome and formation of large mega-domains, separated by a region containing the DXZ4 macrosatellite. However, the molecular architecture of the Xi chromosome, in both the silent and expressed regions, remains unclear. Here we investigate the structure, chromatin accessibility and expression status of the mouse Xi chromosome in highly polymorphic clonal neural progenitors (NPCs) and embryonic stem cells. We demonstrate a crucial role for Xist and the DXZ4-containing boundary in shaping Xi chromosome structure using allele-specific genome-wide chromosome conformation capture (Hi-C) analysis, an assay for transposase-accessible chromatin with high throughput sequencing (ATAC eq) and RNA sequencing. Deletion of the boundary disrupts mega-domain formation, and induction of Xist RNA initiates formation of the boundary and the loss of DNA accessibility. We also show that in NPCs, the Xi chromosome lacks active/inactive compartments and topologically associating domains (TADs), except around genes that escape XCI. Escapee gene clusters display TAD-like structures and retain DNA accessibility at promoter-proximal and CTCF-binding sites. Furthermore, altered patterns of facultative escape genes in different neural progenitor clones are associated with the presence of different TAD-like structures after XCI. These findings suggest a key role for transcription and CTCF in the formation of TADs in the context of the Xi chromosome in neural progenitors.
URLPMID:4434584 [本文引用: 1]
Abstract BACKGROUND: The three-dimensional organization of the genome is tightly connected to its biological function. The Hi-C approach was recently introduced as a method that can be used to identify higher-order chromatin interactions genome-wide. The aim of this study was to determine genome-wide chromatin interaction frequencies using the Hi-C approach in mouse sperm cells and embryonic fibroblasts. RESULTS: The obtained data demonstrate that the three-dimensional genome organizations of sperm and fibroblast cells show a high degree of similarity both with each other and with the previously described mouse embryonic stem cells. Both A- and B-compartments and topologically associated domains are present in spermatozoa and fibroblasts. Nevertheless, sperm cells and fibroblasts exhibit statistically significant differences between each other in the contact probabilities of defined loci. Tight packaging of the sperm genome results in an enrichment of long-range contacts compared with the fibroblasts. However, only 30% of the differences in the number of contacts are based on differences in the densities of their genome packages; the main source of the differences is the gain or loss of contacts that are specific for defined genome regions. We find that the dependence of the contact probability on genomic distance for sperm is close to the dependence predicted for the fractal globular folding of chromatin. CONCLUSIONS: Overall, we can conclude that the three-dimensional structure of the genome is passed through generations without being dramatically changed in sperm cells.
URLPMID:28289288 [本文引用: 1]
The folding of genomic DNA from the beads-on-a-string-like structure of nucleosomes into higher-order assemblies is crucially linked to nuclear processes. Here we calculate 3D structures of entire mammalian genomes using data from a new chromosome conformation capture procedure that allows us to first image and then process single cells. The technique enables genome folding to be examined at a scale of less than 100 b, and chromosome structures to be validated. The structures of individual topological-associated domains and loops vary substantially from cell to cell. By contrast, A and B compartments, lamina-associated domains and active enhancers and promoters are organized in a consistent way on a genome-wide basis in every cell, suggesting that they could drive chromosome and genome folding. By studying genes regulated by pluripotency factor and nucleosome remodelling deacetylase (NuRD), we illustrate how the determination of single-cell genome structure provides a new approach for investigating biological processes.
URLPMID:28703188 [本文引用: 1]
Abstract In mammals, chromatin organization undergoes drastic reprogramming after fertilization. However, the three-dimensional structure of chromatin and its reprogramming in preimplantation development remain poorly understood. Here, by developing a low-input Hi-C (genome-wide chromosome conformation capture) approach, we examined the reprogramming of chromatin organization during early development in mice. We found that oocytes in metaphase II show homogeneous chromatin folding that lacks detectable topologically associating domains (TADs) and chromatin compartments. Strikingly, chromatin shows greatly diminished higher-order structure after fertilization. Unexpectedly, the subsequent establishment of chromatin organization is a prolonged process that extends through preimplantation development, as characterized by slow consolidation of TADs and segregation of chromatin compartments. The two sets of parental chromosomes are spatially separated from each other and display distinct compartmentalization in zygotes. Such allele separation and allelic compartmentalization can be found as late as the 8-cell stage. Finally, we show that chromatin compaction in preimplantation embryos can partially proceed in the absence of zygotic transcription and is a multi-level hierarchical process. Taken together, our data suggest that chromatin may exist in a markedly relaxed state after fertilization, followed by progressive maturation of higher-order chromatin architecture during early development.
URLPMID:28709003 [本文引用: 1]
Abstract High-order chromatin structure plays important roles in gene expression regulation. Knowledge of the dynamics of 3D chromatin structures during mammalian embryo development remains limited. We report the 3D chromatin architecture of mouse gametes and early embryos using an optimized Hi-C method with low-cell samples. We find that mature oocytes at the metaphase II stage do not have topologically associated domains (TADs). In sperm, extra-long-range interactions (>4 Mb) and interchromosomal interactions occur frequently. The high-order structures of both the paternal and maternal genomes in zygotes and two-cell embryos are obscure but are gradually re-established through development. The establishment of the TAD structure requires DNA replication but not zygotic genome activation. Furthermore, unmethylated CpGs are enriched in A compartment, and methylation levels are decreased to a greater extent in A compartment than in B compartment in embryos. In summary, the global reprogramming of chromatin architecture occurs during early mammalian development. Copyright 2017 Elsevier Inc. All rights reserved.
URLPMID:28355183 [本文引用: 1]
Chromatin is reprogrammed after fertilization to produce a totipotent zygote with the potential to generate a new organism1. The maternal genome inherited through the oocyte and the paternal genome provided by sperm coexist as separate haploid nuclei in the zygote. How these two epigenetically distinct genomes are spatially organized is poorly understood. Existing chromosome conformation capture-based methods2–5are inapplicable to oocytes and zygotes due to a paucity of material. To study the 3D chromatin organization in rare cell types, we developed a single-nucleus Hi-C (snHi-C) protocol that provides >10-fold more contacts per cell than the previous method2. Here we show that chromatin architecture is uniquely reorganized during the mouse oocyte-to-zygote transition and is distinct in paternal and maternal nuclei within single-cell zygotes. Features of genomic organization including compartments, topologically associating domains (TADs) and loops are present in individual oocytes when averaged over the genome; each feature at a locus is variable between cells. At the sub-megabase level, we observe stochastic clusters of contacts that violate TAD boundaries but average into TADs. Strikingly, we found that TADs and loops but not compartments are present in zygotic maternal chromatin, suggesting that these are generated by different mechanisms. Our results demonstrate that the global chromatin organization of zygote nuclei is fundamentally different from other interphase cells. An understanding of this zygotic chromatin “ground state” has the potential to provide insights into reprogramming to totipotency.
URLPMID:29972771 [本文引用: 1]
Abstract The spatial organization of chromosomes is critical in establishing gene expression programs. We generated in situ Hi-C maps throughout zebrafish development to gain insight into higher-order chromatin organization and dynamics. Zebrafish chromosomes segregate in active and inactive chromatin (A/B compartments), which are further organized into topologically associating domains (TADs). Zebrafish A/B compartments and TADs have genomic features similar to those of their mammalian counterparts, including evolutionary conservation and enrichment of CTCF binding sites at TAD borders. At the earliest time point, when there is no zygotic transcription, the genome is highly structured. After zygotic genome activation (ZGA), the genome loses structural features, which are re-established throughout early development. Despite the absence of structural features, we see clustering of super-enhancers in the 3D genome. Our results provide insight into vertebrate genome organization and demonstrate that the developing zebrafish embryo is a powerful model system to study the dynamics of nuclear organization.
URLPMID:4542308 [本文引用: 1]
Chandra, Ewels, et al. map changes in genome organization in cellular senescence using Hi-C. Contrary to the believed increase in heterochromatin in senescence-associated heterochromatic foci formation, they describe a loss of local interactions in heterochromatic regions. This is in agreement with changes observed in progeria cells.
URL [本文引用: 1]
Ageing-relevant processes, like cellular senescence, are characterized by complex events giving rise to heterogeneous cell populations. However, the early molec
URLPMID:27760116 [本文引用: 1]
Three-dimensional physical interactions within chromosomes dynamically regulate gene expression in a tissue-specific manner. However, the 3D organization of chromosomes during human brain development and its role in regulating gene networks dysregulated in neurodevelopmental disorders, such as autism or schizophrenia, are unknown. Here we generate high-resolution 3D maps of chromatin contacts during human corticogenesis, permitting large-scale annotation of previously uncharacterized regulatory relationships relevant to the evolution of human cognition and disease. Our analyses identify hundreds of genes that physically interact with enhancers gained on the human lineage, many of which are under purifying selection and associated with human cognitive function. We integrate chromatin contacts with non-coding variants identified in schizophrenia genome-wide association studies (GWAS), highlighting multiple candidate schizophrenia risk genes and pathways, including transcription factors involved in neurogenesis, and cholinergic signalling molecules, several of which are supported by independent expression quantitative trait loci and gene expression analyses. Genome editing in human neural progenitors suggests that one of these distal schizophrenia GWAS loci regulates FOXG1 expression, supporting its potential role as a schizophrenia risk gene. This work provides a framework for understanding the effect of non-coding regulatory elements on human brain development and the evolution of cognition, and highlights novel mechanisms underlying neuropsychiatric disorders.
URL [本文引用: 1]
URLPMID:15171251 [本文引用: 1]
Abstract Diverse aetiological factors, including myocardial infarction, hypertension and contractile abnormalities, trigger a cardiac remodelling process in which the heart becomes abnormally enlarged with a consequent decline in cardiac function and eventual heart failure. Pathological cardiac hypertrophy is accompanied by the activation of a fetal cardiac gene programme, which contributes to maladaptive changes in contractility and calcium handling. Traditional treatment for heart failure involves administration of drugs that antagonize early signalling events at or near the cell membrane (e.g. cell surface receptor or ion channels). Given the complexity and redundant nature of the signalling networks that drive cardiac pathogenesis, a potentially more efficacious therapeutic strategy for disrupting the disease process would be to target common downstream elements in pathological signalling cascades. We have shown that class II histone deacetylases (HDACs) suppress cardiac hypertrophy, and mice lacking class II HDACs are sensitized to hypertrophic signals. Paradoxically, HDAC inhibitors also block cardiac hypertrophy and fetal gene activation. Based on these findings, we propose that distinct HDACs play positive or negative roles in the control of cardiac growth by regulating opposing sets of target genes via their interactions with different sets of transcription factors.
URL [本文引用: 1]
URLPMID:27022893 [本文引用: 1]
Abstract Epigenetic regulatory mechanisms play key roles in cardiac development, differentiation, homeostasis, response to stress and injury and disease. Human heart failure epigenetic regulatory mechanisms have not been deciphered to date. This 2-part review distills the rapidly evolving research focused on human heart failure epigenetic regulatory mechanisms. Part I focuses on epigenetic regulatory mechanisms involving RNA, specifically the role of short, intermediate and long noncoding RNAs and endogenous competing RNA regulatory networks. Part II focuses on the epigenetic regulatory mechanisms involving DNA, including DNA methylation, histone modifications, and chromatin conformational changes. Part II concludes with 2 examples of well-studied integrated epigenetic regulatory mechanisms, the structural and functional roles of the mediator complex in regulating transcription, and the epigenetic networked "cross-talk" regulating atrial natriuretic peptide and brain natriuretic peptide promoter activation.
URLPMID:28802249 [本文引用: 1]
Supplemental Digital Content is available in the text. Cardiovascular disease is associated with epigenomic changes in the heart; however, the endogenous structure of cardiac myocyte chromatin has never been determined. To investigate the mechanisms of epigenomic function in the heart, genome-wide chromatin conformation capture (Hi-C) and DNA sequencing were performed in adult cardiac myocytes following development of pressure overload nduced hypertrophy. Mice with cardiac-specific deletion of CTCF (a ubiquitous chromatin structural protein) were generated to explore the role of this protein in chromatin structure and cardiac phenotype. Transcriptome analyses by RNA-seq were conducted as a functional readout of the epigenomic structural changes. Depletion of CTCF was sufficient to induce heart failure in mice, and human patients with heart failure receiving mechanical unloading via left ventricular assist devices show increased CTCF abundance. Chromatin structural analyses revealed interactions within the cardiac myocyte genome at 5-kb resolution, enabling examination of intra- and interchromosomal events, and providing a resource for future cardiac epigenomic investigations. Pressure overload or CTCF depletion selectively altered boundary strength between topologically associating domains and A/B compartmentalization, measurements of genome accessibility. Heart failure involved decreased stability of chromatin interactions around disease-causing genes. In addition, pressure overload or CTCF depletion remodeled long-range interactions of cardiac enhancers, resulting in a significant decrease in local chromatin interactions around these functional elements. These findings provide a high-resolution chromatin architecture resource for cardiac epigenomic investigations and demonstrate that global structural remodeling of chromatin underpins heart failure. The newly identified principles of endogenous chromatin structure have key implications for epigenetic therapy.
[本文引用: 1]
URLPMID:27240531 [本文引用: 1]
Copy number variants (CNVs) are major contributors to genomic imbalance disorders. Phenotyping of 137 unrelated deletion and reciprocal duplication carriers of the distal 16p11.2 22065kb BP2-BP3 interval showed that these rearrangements are associated with autism spectrum disorders and mirror phenotypes of obesity/underweight and macrocephaly/microcephaly. Such phenotypes were previously associated with rearrangements of the non-overlapping proximal 16p11.2 60065kb BP4-BP5 interval. These two CNV-prone regions at 16p11.2 are reciprocally engaged in complex chromatin looping, as successfully confirmed by 4C-seq, fluorescencein situhybridization and Hi-C, as well as coordinated expression and regulation of encompassed genes. We observed that genes differentially expressed in 16p11.2 BP4-BP5 CNV carriers are concomitantly modified in their chromatin interactions, suggesting that disruption of chromatin interplays could participate in the observed phenotypes. We also identifiedcis- andtrans-acting chromatin contacts to other genomic regions previously associated with analogous phenotypes. For example, we uncovered that individuals with reciprocal rearrangements of thetrans-contacted 2p15 locus similarly display mirror phenotypes on head circumference and weight. Our results indicate that chromosomal contacts’ maps could uncover functionally and clinically related genes.
URLPMID:3465532 [本文引用: 1]
We analyzed primary breast cancers by genomic DNA copy number arrays, DNA methylation, exome sequencing, mRNA arrays, microRNA sequencing and reverse phase protein arrays. Our ability to integrate information across platforms provided key insights into previously-defined gene expression subtypes and demonstrated the existence of four main breast cancer classes when combining data from five platforms, each of which shows significant molecular heterogeneity. Somatic mutations in only three genes (TP53,PIK3CAandGATA3) occurred at > 10% incidence across all breast cancers; however, there were numerous subtype-associated and novel gene mutations including the enrichment of specific mutations inGATA3,PIK3CAandMAP3K1with the Luminal A subtype. We identified two novel protein expression-defined subgroups, possibly contributed by stromal/microenvironmental elements, and integrated analyses identified specific signaling pathways dominant in each molecular subtype including a HER2/p-HER2/HER1/p-HER1 signature within the HER2-Enriched expression subtype. Comparison of Basal-like breast tumors with high-grade Serous Ovarian tumors showed many molecular commonalities, suggesting a related etiology and similar therapeutic opportunities. The biologic finding of the four main breast cancer subtypes caused by different subsets of genetic and epigenetic abnormalities raises the hypothesis that much of the clinically observable plasticity and heterogeneity occurs within, and not across, these major biologic subtypes of breast cancer.
URLPMID:22722201 [本文引用: 1]
Abstract All cancers carry somatic mutations in their genomes. A subset, known as driver mutations, confer clonal selective advantage on cancer cells and are causally implicated in oncogenesis, and the remainder are passenger mutations. The driver mutations and mutational processes operative in breast cancer have not yet been comprehensively explored. Here we examine the genomes of 100 tumours for somatic copy number changes and mutations in the coding exons of protein-coding genes. The number of somatic mutations varied markedly between individual tumours. We found strong correlations between mutation number, age at which cancer was diagnosed and cancer histological grade, and observed multiple mutational signatures, including one present in about ten per cent of tumours characterized by numerous mutations of cytosine at TpC dinucleotides. Driver mutations were identified in several new cancer genes including AKT2, ARID1B, CASP8, CDKN1B, MAP3K1, MAP3K13, NCOR1, SMARCD1 and TBX3. Among the 100 tumours, we found driver mutations in at least 40 cancer genes and 73 different combinations of mutated cancer genes. The results highlight the substantial genetic diversity underlying this common disease.
URLPMID:3927368 [本文引用: 1]
The Cancer Genome Atlas (TCGA) has used the latest sequencing and analysis methods to identify somatic variants across thousands of tumours. Here we present data and analytical results for point mutations and small insertions/deletions from 3,281 tumours across 12 tumour types as part of the TCGA Pan-Cancer effort. We illustrate the distributions of mutation frequencies, types and contexts across tumour types, and establish their links to tissues of origin, environmental/carcinogen influences, and DNA repair defects. Using the integrated data sets, we identified 127 significantly mutated genes from well-known (for example, mitogen-activated protein kinase, phosphatidylinositol-3-OH kinase, Wnt/ -catenin and receptor tyrosine kinase signalling pathways, and cell cycle control) and emerging (for example, histone, histone modification, splicing, metabolism and proteolysis) cellular processes in cancer. The average number of mutations in these significantly mutated genes varies across tumour types; most tumours have two to six, indicating that the number of driver mutations required during oncogenesis is relatively small. Mutations in transcriptional factors/regulators show tissue specificity, whereas histone modifiers are often mutated across several cancer types. Clinical association analysis identifies genes having a significant effect on survival, and investigations of mutations with respect to clonal/subclonal architecture delineate their temporal orders during tumorigenesis. Taken together, these results lay the groundwork for developing new diagnostics and individualizing cancer treatment.
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:4587679 [本文引用: 1]
Higher-order chromatin structure is often perturbed in cancer and other pathological states. Although several genetic and epigenetic differences have been charted between normal and breast cancer tissues, changes in higher-order chromatin organization during tumorigenesis have not been fully explored. To probe the differences in higher-order chromatin structure between mammary epithelial and breast cancer cells, we performed Hi-C analysis on MCF-10A mammary epithelial and MCF-7 breast cancer cell lines. Our studies reveal that the small, gene-rich chromosomes chr16 through chr22 in the MCF-7 breast cancer genome display decreased interaction frequency with each other compared to the inter-chromosomal interaction frequency in the MCF-10A epithelial cells. Interestingly, this finding is associated with a higher occurrence of open compartments on chr16 22 in MCF-7 cells. Pathway analysis of the MCF-7 up-regulated genes located in altered compartment regions on chr16 22 reveals pathways related to repression of WNT signaling. There are also differences in intra-chromosomal interactions between the cell lines; telomeric and sub-telomeric regions in the MCF-10A cells display more frequent interactions than are observed in the MCF-7 cells. We show evidence of an intricate relationship between chromosomal organization and gene expression between epithelial and breast cancer cells. Importantly, this work provides a genome-wide view of higher-order chromatin dynamics and a resource for studying higher-order chromatin interactions in two cell lines commonly used to study the progression of breast cancer. The online version of this article (doi:10.1186/s13059-015-0768-0) contains supplementary material, which is available to authorized users.
URLPMID:27053337 [本文引用: 1]
A three-dimensional chromatin state underpins the structural and functional basis of the genome by bringing regulatory elements and genes into close spatial proximity to ensure proper, cell-type-specific gene expression profiles. Here, we performed Hi-C chromosome conformation capture sequencing to investigate how three-dimensional chromatin organization is disrupted in the context of copy-number variation, long-range epigenetic remodeling, and atypical gene expression programs in prostate cancer. We find that cancer cells retain the ability to segment their genomes into megabase-sized topologically associated domains (TADs); however, these domains are generally smaller due to establishment of additional domain boundaries. Interestingly, a large proportion of the new cancer-specific domain boundaries occur at regions that display copy-number variation. Notably, a common deletion on 17p13.1 in prostate cancer spanning the TP53 tumor suppressor locus results in bifurcation of a single TAD into two distinct smaller TADs. Change in domain structure is also accompanied by novel cancer-specific chromatin interactions within the TADs that are enriched at regulatory elements such as enhancers, promoters, and insulators, and associated with alterations in gene expression. We also show that differential chromatin interactions across regulatory regions occur within long-range epigenetically activated or silenced regions of concordant gene activation or repression in prostate cancer. Finally, we present a novel visualization tool that enables integrated exploration of Hi-C interaction data, the transcriptome, and epigenome. This study provides new insights into the relationship between long-range epigenetic and genomic dysregulation and changes in higher-order chromatin interactions in cancer.
URL [本文引用: 1]
N6-methyladenosine (m6A), the most prevalent modification of mammalian RNA, has received increasing attention. Although m6A has been shown to be associated with biological activities, such as spermatogenesis modulation, cell spermatogenesis and pluripotency, Drosophila sex determination, and the control of T cell homeostasis and response to heat shock, little is known about its roles in cancer... [Show full abstract]
URLPMID:4831574 [本文引用: 1]
Gain-of-functionIDHmutations are initiating events that define major clinical and prognostic classes of gliomas1,2. Mutant IDH protein produces a novel onco-metabolite, 2-hydroxyglutarate (2-HG), that interferes with iron-dependent hydroxylases, including the TET family of 5 -methylcytosine hydroxylases3 7. TET enzymes catalyze a key step in the removal of DNA methylation8,9.IDHmutant gliomas thus manifest a CpG island methylator phenotype (G-CIMP)10,11, though the functional significance of this altered epigenetic state remains unclear. Here we show thatIDHmutant gliomas exhibit hyper-methylation at CTCF binding sites, compromising binding of this methylation-sensitive insulator protein. Reduced CTCF binding is associated with loss of insulation between topological domains and aberrant gene activation. We specifically demonstrate that loss of CTCF at a domain boundary permits a constitutive enhancer to aberrantly interact with the receptor tyrosine kinase genePDGFRA, a prominent glioma oncogene. Treatment ofIDHmutant gliomaspheres with demethylating agent partially restores insulator function and down-regulatesPDGFRA. Conversely, CRISPR-mediated disruption of the CTCF motif inIDHwildtype gliomaspheres up-regulatesPDGFRAand increases proliferation. Our study suggests thatIDHmutations promote gliomagenesis by disrupting chromosomal topology and allowing aberrant regulatory interactions that induce oncogene expression.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:20016488 [本文引用: 1]
Nature is the international weekly journal of science: a magazine style journal that publishes full-length research papers in all disciplines of science, as well as News and Views, reviews, news, features, commentaries, web focuses and more, covering all branches of science and how science impacts upon all aspects of society and life.
[本文引用: 1]
URL [本文引用: 1]
URLPMID:3322590 [本文引用: 1]
Chronic lymphocytic leukaemia (CLL), the most frequent leukaemia in adults in Western countries, is a heterogeneous disease with variable clinical presentation and evolution. Two major molecular subtypes can be distinguished, characterized respectively by a high or low number of somatic hypermutations in the variable region of immunoglobulin genes. The molecular changes leading to the pathogenesis of the disease are still poorly understood. Here we performed whole-genome sequencing of four cases of CLL and identified 46 somatic mutations that potentially affect gene function. Further analysis of these mutations in 363 patients with CLL identified four genes that are recurrently mutated: notch 1 (NOTCH1), exportin 1 (XPO1), myeloid differentiation primary response gene 88 (MYD88) and kelch-like 6 (KLHL6). Mutations in MYD88 and KLHL6 are predominant in cases of CLL with mutated immunoglobulin genes, whereas NOTCH1 and XPO1 mutations are mainly detected in patients with unmutated immunoglobulins. The patterns of somatic mutation, supported by functional and clinical analyses, strongly indicate that the recurrent NOTCH1, MYD88 and XPO1 mutations are oncogenic changes that contribute to the clinical evolution of the disease. To our knowledge, this is the first comprehensive analysis of CLL combining whole-genome sequencing with clinical characteristics and clinical outcomes. It highlights the usefulness of this approach for the identification of clinically relevant mutations in cancer.
URLPMID:20505728 [本文引用: 1]
Lung cancer is the leading cause of cancer-related mortality worldwide, with non-small-cell lung carcinomas in smokers being the predominant form of the disease. Although previous studies have identified important common somatic mutations in lung cancers, they have primarily focused on a limited set of genes and have thus provided a constrained view of the mutational spectrum. Recent cancer sequencing efforts have used next-generation sequencing technologies to provide a genome-wide view of mutations in leukaemia, breast cancer and cancer cell lines. Here we present the complete sequences of a primary lung tumour (60 coverage) and adjacent normal tissue (46 ). Comparing the two genomes, we identify a wide variety of somatic variations, including >50,000 high-confidence single nucleotide variants. We validated 530 somatic single nucleotide variants in this tumour, including one in the KRAS proto-oncogene and 391 others in coding regions, as well as 43 large-scale structural variations. These constitute a large set of new somatic mutations and yield an estimated 17.7 per megabase genome-wide somatic mutation rate. Notably, we observe a distinct pattern of selection against mutations within expressed genes compared to non-expressed genes and in promoter regions up to 5kilobases upstream of all protein-coding genes. Furthermore, we observe a higher rate of amino acid-changing mutations in kinase genes. We present a comprehensive view of somatic alterations in a single lung tumour, and provide the first evidence, to our knowledge, of distinct selective pressures present within the tumour environment.
URLPMID:21953857 [本文引用: 1]
Abstract Recently, the genome sequences from several cancers have been published, along with the genome from a noncancer tissue from the same individual, allowing the identification of new somatic mutations in the cancer. We show that there is significant variation in the density of mutations at the 1-Mb scale within three cancer genomes and that the density of mutations is correlated between them. We also demonstrate that the density of mutations is correlated to that in the germline, as measured by the divergence between humans and chimpanzees and humans and macaques. We show that the density of mutations is correlated to the guanine and cytosine (GC) conent, replication time, distance to telomere and centromere, gene density, and nucleosome occupancy in the cancer genomes. However, overall, all factors explain less than 40% of the variance in mutation density and each factor explains very little of the variance. We find that genes associated with cancer occupy regions of the genome with significantly lower mutation rates than the average. Finally, we show that the density of mutations varies at a 10-Mb and a chromosomal scale, but that the variation at these scales is weak. Hum Mutat 33:136 143, 2012. 2011 Wiley Periodicals, Inc.
URLPMID:22820252 [本文引用: 1]
Cancer genome sequencing provides the first direct information on how mutation rates vary across the human genome in somatic cells. Testing diverse genetic and epigenetic features, here we show that mutation rates in cancer genomes are strikingly related to chromatin organization. Indeed, at the megabase scale, a single feature—levels of the heterochromatin-associated histone modification H3K9me3—can account for more than 40% of mutation-rate variation, and a combination of features can account for more than 55%. The strong association between mutation rates and chromatin organization is upheld in samples from different tissues and for different mutation types. This suggests that the arrangement of the genome into heterochromatin- and euchromatin-like domains is a dominant influence on regional mutation-rate variation in human somatic cells.
URLPMID:28604728 [本文引用: 1]
Genome-wide association studies (GWAS) have transformed understanding of susceptibility to testicular germ cell tumors (TGCTs), but much of the heritability remains unexplained. Here we report a new GWAS, a meta-analysis with previous GWAS and a replication series, totaling 7,319 TGCT cases and 23,082 controls. We identify 19 new TGCT risk loci, roughly doubling the number of known TGCT risk loci to 44. By performing in situ Hi-C in TGCT cells, we provide evidence for a network of physical interactions among all 44 TGCT risk SNPs and candidate causal genes. Our findings implicate widespread disruption of developmental transcriptional regulators as a basis of TGCT susceptibility, consistent with failed primordial germ cell differentiation as an initiating step in oncogenesis. Defective microtubule assembly and dysregulation of KIT-MAPK signaling also feature as recurrently disrupted pathways. Our findings support a polygenic model of risk and provide insight into the biological basis of TGCT.
URLPMID:2871592 [本文引用: 1]
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human traits and on other model organisms, including mouse, fly, nematode and yeast. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
URLPMID:23666239 [本文引用: 1]
We conducted a meta-analysis to identify new susceptibility loci for testicular germ cell tumor (TGCT). In the discovery phase, we analyzed 931 affected individuals and 1,975 controls from 3 genome-wide association studies (GWAS). We conducted replication in 6 independent sample sets comprising 3,211 affected individuals and 7,591 controls. In the combined analysis, risk of TGCT was significantly associated with markers at four previously unreported loci: 4q22.2 in HPGDS (per-allele odds ratio (OR) = 1.19, 95% confidence interval (CI) = 1.12-1.26; P = 1.11 × 10(-8)), 7p22.3 in MAD1L1 (OR = 1.21, 95% CI = 1.14-1.29; P = 5.59 × 10(-9)), 16q22.3 in RFWD3 (OR = 1.26, 95% CI = 1.18-1.34; P = 5.15 × 10(-12)) and 17q22 (rs9905704: OR = 1.27, 95% CI = 1.18-1.33; P = 4.32 × 10(-13) and rs7221274: OR = 1.20, 95% CI = 1.12-1.28; P = 4.04 × 10(-9)), a locus that includes TEX14, RAD51C and PPM1E. These new TGCT susceptibility loci contain biologically plausible genes encoding proteins important for male germ cell development, chromosomal segregation and the DNA damage response.
URLPMID:28663546 [本文引用: 1]
Prostate cancer is a highly heritable molecularly and clinically heterogeneous disease. To discover germline events involved in prostate cancer predisposition, we develop a computational approach to nominate heritable facilitators of somatic genomic events in the context of the androgen receptor signaling. Here, we use a ranking score and benign prostate transcriptomes to identify a non-coding polymorphic regulatory element at 7p14.3 that associates with DNA repair and hormone-regulated transcript levels and with an early recurrent prostate cancer-specific somatic mutation in the Speckle-Type POZ protein (SPOP) gene. The locus shows allele-specific activity that is concomitantly modulated by androgen receptor and by CCAAT/enhancer-binding protein (C/EBP) beta (CEBPB). Deletion of this locus via CRISPR-Cas9 leads to deregulation of the genes predicted to interact with the 7p14.3 locus by Hi-C chromosome conformation capture data. This study suggests that a polymorphism at 7p14.3 may predispose to SPOP mutant prostate cancer subclass through a hormone-dependent DNA damage response. Prostate cancer is a heterogeneous disease, and many cases show somatic mutations of SPOP. Here, the authors show that a non-coding polymorphic regulatory element at 7p14.3 may predispose to SPOP mutant prostate cancer subclass through a hormone dependent DNA damage response.
URLPMID:25122612 [本文引用: 1]
Genome-wide association studies have identified more than 70 common variants that are associated with breast cancer risk. Most of these variants map to non-protein-coding regions and several map to gene deserts, regions of several hundred kilobases lacking protein-coding genes. We hypothesized that gene deserts harbor long-range regulatory elements that can physically interact with target genes to influence their expression. To test this, we developed Capture Hi-C (CHi-C), which, by incorporating a sequence capture step into a Hi-C protocol, allows high-resolution analysis of targeted regions of the genome. We used CHi-C to investigate long-range interactions at three breast cancer gene deserts mapping to 2q35, 8q24.21, and 9q31.2. We identified interaction peaks between putative regulatory elements ("bait fragments") within the captured regions and "targets" that included both protein-coding genes and long noncoding (Inc) RNAs over distances of 6.6 kb to 2.6 Mb. Target protein-coding genes were IGFBP5, KLF4, NSMCE2, and MYC; and target IncRNAs included DIRC3, PVT1, and CCDC26. For one gene desert, we were able to define two SNPs (rs12613955 and rs4442975) that were highly correlated with the published risk variant and that mapped within the bait end of an interaction peak. In vivo ChIP-qPCR data show that one of these, rs4442975, affects the binding of FOXA1 and implicate this SNP as a putative functional variant.
URLPMID:29531215 [本文引用: 1]
Genome-wide association studies (GWAS) have identified approximately 100 breast cancer risk loci. Translating these findings into a greater understanding of the mechanisms that influence disease risk requires identification of the genes or non-coding RNAs that mediate these associations. Here, we use Capture Hi-C (CHi-C) to annotate 63 loci; we identify 110 putative target genes at 33 loci. To assess the support for these target genes in other data sources we test for associations between levels of expression and SNP genotype (eQTLs), disease-specific survival (DSS), and compare them with somatically mutated cancer genes. 22 putative target genes are eQTLs, 32 are associated with DSS and 14 are somatically mutated in breast, or other, cancers. Identifying the target genes at GWAS risk loci will lead to a greater understanding of the mechanisms that influence breast cancer risk and prognosis.
URL [本文引用: 1]
URLPMID:5488307 [本文引用: 1]
Chromosomal rearrangements occur constitutionally in the general population and somatically in the majority of cancers. Detection of balanced rearrangements, such as reciprocal translocations and inversions, is troublesome, which is particularly detrimental in oncology where rearrangements play diagnostic and prognostic roles. Here we describe the use of Hi-C as a tool for detection of both balanced and unbalanced chromosomal rearrangements in primary human tumour samples, with the potential to define chromosome breakpoints to bp resolution. In addition, we show copy number profiles can also be obtained from the same data, all at a significantly lower cost than standard sequencing approaches. The online version of this article (doi:10.1186/s13059-017-1253-8) contains supplementary material, which is available to authorized users.
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:5617335 [本文引用: 1]
The spatial organization of the genome and its dynamics contribute to gene expression and cellular function in normal development as well as in disease. Although we are increasingly well equipped to determine a genome9s sequence and linear chromatin composition, studying the three-dimensional organization of the genome with high spatial and temporal resolution remains challenging. The 4D Nucleome Network aims to develop and apply approaches to map the structure and dynamics of the human and mouse genomes in space and time with the long term goal of gaining deeper mechanistic understanding of how the nucleus is organized. The project will develop and benchmark experimental and computational approaches for measuring genome conformation and nuclear organization, and investigate how these contribute to gene regulation and other genome functions. Further efforts will be directed at applying validated experimental approaches combined with biophysical modeling to generate integrated maps and quantitative models of spatial genome organization in different biological states, both in cell populations and in single cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}