 ,1, 赵志虎,2
,1, 赵志虎,2Bioinformatics analysis of Alu components at the level of genome 3D structure
Chao He1,2, Wenlong Shen2, Ping Li2, Yan Zhang2, Jing Zeng1, Zuoming Yin,1, Zhihu Zhao,2通讯作者:
编委: 吴强
收稿日期:2018-10-30修回日期:2019-01-1网络出版日期:2019-01-14
| 基金资助: |
Received:2018-10-30Revised:2019-01-1Online:2019-01-14
| Fund supported: |
作者简介 About authors
何超,硕士,工程师,研究方向:生物信息学E-mail:hechao2010@tsinghua.org.cn。

摘要
关键词:
Abstract
Keywords:
PDF (423KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
何超, 沈文龙, 李平, 张彦, 曾晶, 殷作明, 赵志虎. Alu元件在染色质三维结构层次上的生物信息学分析[J]. 遗传, 2019, 41(3): 254-261 doi:10.16288/j.yczz.18-296
Chao He, Wenlong Shen, Ping Li, Yan Zhang, Jing Zeng, Zuoming Yin, Zhihu Zhao.
短散在元件(short interspersed elements, SINEs)是以散在方式分布于基因组中的较短的重复序列,其单元长度通常在50 bp以下。Alu序列是哺乳动物的短散在元件中所占比例最多的一种重复元件[1]。Alu序列的左右两端各分布着一个不等长的、包含一个RNA聚合酶Ⅲ启动子序列的单体[2],左侧单体比右侧单体短31 bp且只有左侧单体的RNA聚合酶Ⅲ启动子具有活性。研究表明,在Alu元件附近往往富集着长非编码RNA (long non-coding RNAs, lncRNAs),可辅助lncRNAs在细胞核内进行定位[3],而且还能在基因组转录过程中能够充当剪接受体,抑制mRNA的翻译并导致基因组不稳定[4]。此外,还有研究发现Alu元件在基因组中与增强子(enhancer)广泛相互作用[5,6,7]。
染色质在细胞核内的结构与蛋白质的三维高级结构相似,包括基础的核小体、核小体组成的“串珠”结构、螺线管纤维结构、染色质/DNA环结构(chromatin/DNA loop)、拓扑结构域(topological associated domain, TAD)等多层次结构[8,9]。其中,TAD因在不同的细胞类型中相对稳定且保守,被认为是染色质三维结构的基本单元[10]。目前对于染色质三维结构的研究主要基于近距离连接反应的染色体构象捕获技术、基于图谱技术根据多聚物聚合等原理进行计算机建模、基于CRISPR的DNA片段编辑等的遗传学技术,以及基因光谱反射的显微镜技术。迄今为止,对染色质结构的直接观测受到技术的限制,难以直接在高分辨率下观测染色质的动态变化,因此研究者们采用先将染色质内的相互作用固定下来,通过分子生物学手段进行分析,从而推断生理条件下真实的染色质动态结构。目前对染色质三维结构的研究方法主要包括研究点与点之间距离水平的3C技术[11]、研究一点到基因组范围内其他位点距离水平的4C技术[12]、研究已知范围内多点到多点间相互作用的5C技术[13]、全基因组内染色质相互作用的Hi-C技术[14,15]及进一步衍生出的DNase Hi-C[16]、Micro-C[17]、Capture Hi-C[18]、COLA[19]、C-walk[20]等技术[21,22]。本研究通过生物信息学方法,结合Hi-C实验数据,在染色质三维结构层次上对Alu元件在染色质三维结构形成中发挥的作用进行探索,并根据染色质三维折叠数据对Alu元件的相关功能进行分析。
1 材料与方法
1.1 数据集来源
Hi-C实验数据来源于Rao等[19]所公开的数据集,包括GM12878、IMR90和K562 3个细胞系,均可在最高5 kb分辨率下开展研究。Alu元件数据来自UCSC genome browser repeat masker,包含了Alu元件不同子类在基因组上所在位点的信息[23]。参考基因组选取为人类参考基因组hg19版本。1.2 数据预处理
目前已经有较多处理Hi-C数据的比较成熟的软件,如GenomicInteractions[24]、HiCPlotter[25]、HiC-Pro[26]等,其中GenomicInteractions可用于分析特殊位点间相互作用网络,然而这一软件并不能整合多组学数据,也不能够进行后续的功能挖掘等分析[24];HiCPlotter侧重于将Hi-C相互作用热图与相应的包括组蛋白修饰等表观遗传特征的结合,但并未关注染色质上特殊序列间的相互作用[25]。利用HiC-Pro软件对原始数据进行预处理,因Hi-C数据是包含相互作用的两个基因组片段的整合序列,通过将原始测序序列比对到基因组上,得到发生相互作用的两个基因组位点。过滤掉自连接或未连接的无用、冗余相互作用对,将剩余相互作用对进行整合汇总,以便后续分析处理。1.3 Alu元件相互作用频率分布分析
利用开源的R语言及附属软件包对Alu元件不同子类相互作用频率分布进行分析。将不同位点的全部相互作用汇总,设定相互作用上下阈值,超过上限阈值认为其过度连接,低于下线阈值认为其为染色质内随机连接。根据上下阈值进行过滤后,按照相互作用频率强度将所有位点的相互作用分成若干子集。根据Alu元件所在的位置生成随机位点组成随机组,将每一组内的相互作用数据与随机组数据相比较,从而帮助用户判断特殊位点的相互作用在生物学上是否显著,以及是否与染色质三维结构相关。1.4 基于染色质三维结构的Alu元件演化分析
将Alu元件子族参与的相互作用从全局染色质间与染色质内相互作用中提取出来,并根据Alu元件不同子族进行分类。然后根据不同Alu元件子族间的平均相互作用强度,推算三维结构上的距离关系,并与已知的Alu元件进化树相比较,从而判断这些元件之间是否存在关联。2 结果与分析
2.1 Alu元件广泛参与染色质三维折叠
首先使用bowtie2软件对数据集进行序列比对,通过比对后得到染色质相互作用序列对。将全部相互作用序列对进行统计,得到不同位点间相互作用频率,根据上下阈值进行过滤,然后按照相互作用频率大小分成若干子集,将每一组内的相互作用数据与随机组数据相比较,从而判断Alu元件的相互作用在生物学上是否显著,以及是否与染色质三维结构相关。为了判断差异是否真实存在,根据相应Alu元件的位置信息,生成了与之对应的随机对照组进行比较[27]。经计算发现,在不同细胞系中Alu元件所参与的相互作用比例结果显示均具有较高的一致性,Pearson相关系数均在0.997以上,说明Alu元件参与相互作用的比例在不同细胞系中是相对一致的,这些结果提示在不同细胞系中,Alu元件可能对染 色质三维结构的构建、传递和维持发挥着重要的作用。
2.2 Alu元件参与高强度染色质相互作用
根据UCSC上Repeat Masker数据集中Alu的位点信息,对其在不同强度的染色质相互作用中所占的比例进行了分析,因Hi-C实验数据是若干条序列,其中每条序列为染色质上两段相互靠近的不同位点连接形成,通过序列双端比对即可获取全基因组范围内的染色质相互作用水平[28],将全部相互作用根据上下阈值进行过滤,按照相互作用频率大小分成若干子集,根据Alu元件的位点信息,将Alu元件参与的相互作用从全局相互作用中提取出来,进一步计算其在不同子集中所占的比例。结果发现,Alu元件在高强度的相互作用中所占比例随着强度增加而逐渐增高,这表明Alu在染色质三维结构构建过程中发挥了重要的作用(图1)。之前的研究也表明,Alu富集的区域会使序列有更高的GC含量,从而也会有更多的CpG岛与甲基化水平,这些使序列和转录调控变得更加灵活,也就更有可能富集着较多的染色质相互作用。同时Alu序列经常在细胞中发挥增强子的作用,这也可能是其广泛参与高强度染色质相互作用的原因[29]。
图1
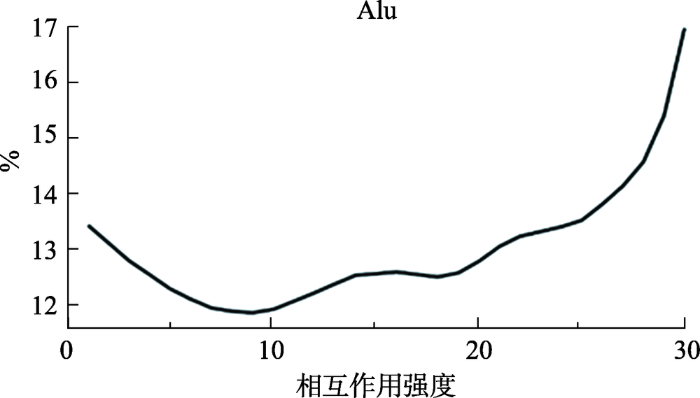 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1Alu元件参与的染色质相互作用分布
Fig. 1Distribution of chromatin interactions of Alu elements
2.3 从染色质三维结构的角度分析Alu元件的进化过程
本研究从染色质三维结构层面上对Alu元件家族中不同子类之间的分布进行了分析与验证。通过计算不同子类之间的相互作用强度,即染色质三维结构层次上的平均距离,结果见图2。颜色越深表明两种元件在染色质三维结构层面上平均距离越近,可以看出AluJ与AluS子家族呈现出一个局部的热区,而AluY子家族则与其余子家族间相互作用强度都较低。图2
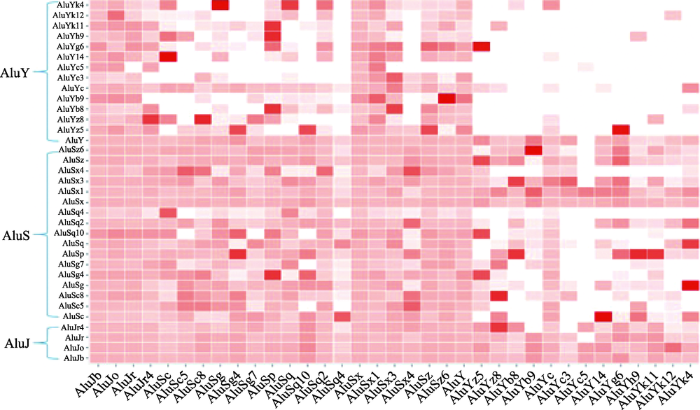 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2Alu元件子族间相互作用强度分布
横纵坐标代表相应的Alu元件子类。
Fig. 2Intensity distribution of the Alu sub-families
为排除细胞类型特异性因素的影响,对GM12878、K562和IMR90 3个细胞系中Alu家族所有二级子类所参与的相互作用强度进行了一致性检验。结果发现,3个细胞系的分析结果均具有相对较高的一致性,尤其是GM12878与K562,Pearson相关系数可以达到0.827,说明不同Alu子类参与的相互作用强度水平并不是细胞类型特异的,而是普遍存在且相对一致的。
从上面的研究中可以发现,Alu的3个子家族AluJ、AluS和AluY在相互作用强度上存在着差异,因此对这3个子家族的相互作用强度进行了进一步的观察,并根据三者之间相互作用强度绘制了热图(图3),结果发现AluJ和AluS都是与自身相互作用最强,与相邻的子家族次强,与距离最远的子家族相互作用强度最弱,也就是说,AluJ与AluS、AluS与AluY在染色质三维结构上距离较近,而AluJ与AluY距离较远。这一发现与之前文献中报道的仅仅根据一维核酸序列分析得到的Alu家族进化树(图4)相一致[30,31],AluY自身作用强度较弱,这也许与其较近年代才产生有关,相对于AluJ (21 575个)、AluS (16 536个)而言,AluY的数量较少,只有1731个,分布也较为分散,相互作用强度较弱。因此推测,在进一步的进化过程中,当Alu继续衍生出新的子家族,AluY数量在人类基因组中逐渐增多时,其参与的相互作用的强度也会随之增加。根据相互作用强度做了层次聚类,发现与上面的推测也基本一致(图5)。
图3
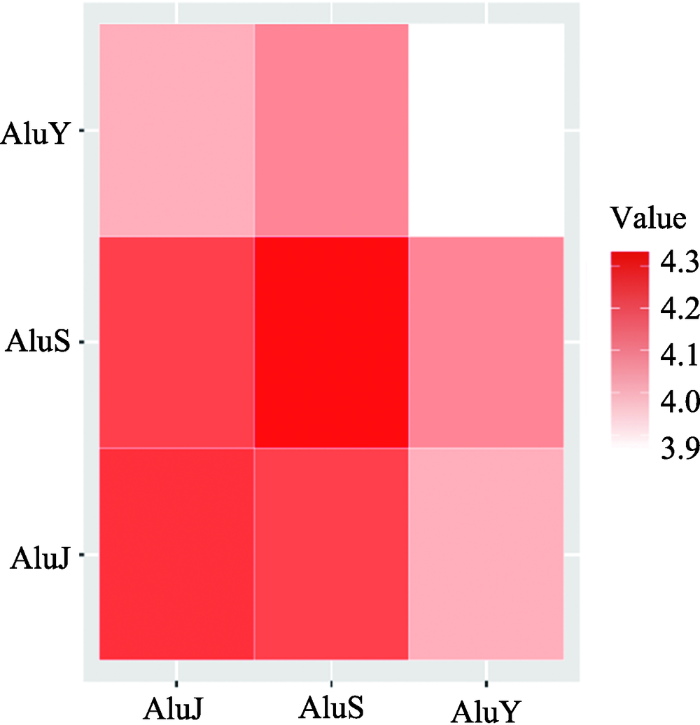 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Alu元件3个子族间相互作用强度分布
Fig. 3Intensity distribution of interactions between the three sub-families of Alu elements
图4
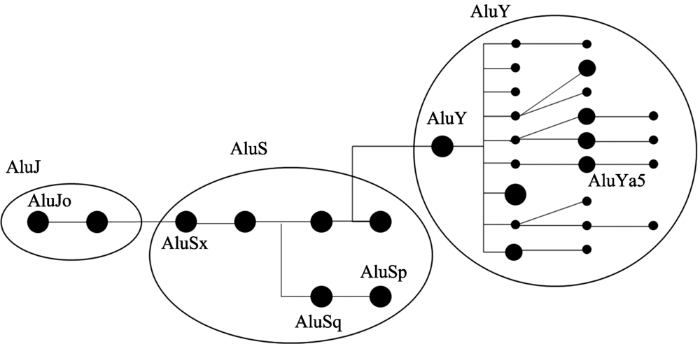 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4Alu元件进化树
Fig. 4The evolutionary tree of Alu elements
图5
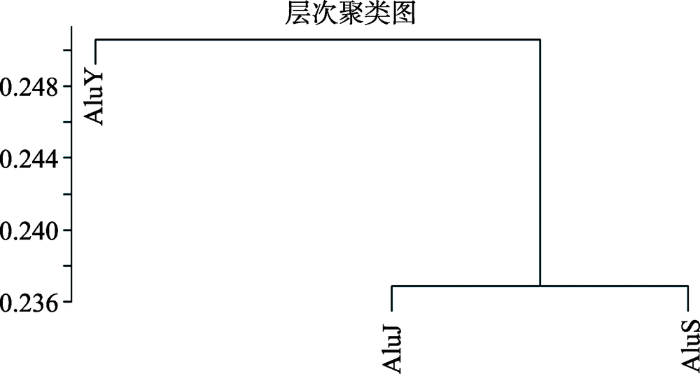 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5Alu元件相互作用强度层次聚类图
Fig. 5Hierarchical clustering diagram of Alu element interaction intensity
基于上述结果,推断Alu元件在染色质相互作用的强度与进化上的关系是存在着一定的正相关性,也就是说一维序列进化距离上比较接近的Alu元件在染色质的三维结构上也会彼此相互靠近。至于是因为它们进化上首先随机跳跃扩展到基因组中的不同区域,但是由于一维序列比较类似因而结合类似的、共同的调控因子从而导致所在区域染色质三维结构相互聚集,还是因为原本在染色质三维构象上,原始的序列位点就比较接近,在进一步的演化过程中,新出现的元件优先扩展到三维距离上更近的区域最终使得所有这些同族的Alu子族在三维分布上也比较接近,还需要后续更多的实验验证。
3 讨论
3.1 Alu元件在染色质三维构象的动态折叠中发挥了重要的作用
细胞核是真核生物独有的一种细胞器,具有高度不均一、严格区室化、亚结构相对独立等特点,可以从外至内分为核被膜、核纤层、核基质、染色质及核仁层结构,其中核纤层、核基质、核仁3者相互连接,构成细胞核内的骨架及支撑结构,染色质围绕这一骨架以复杂而精致的三维形态高度折叠,这一折叠能够维持着相对稳定且高度动态变化的结构,在DNA复制、重排、重组、修复以及基因转录调控等方面发挥重要的功能[16,32,33]。随着技术的发展,人们逐渐发现染色质与蛋白质的多级折叠结构相似,可以分成若干个不同的层级。DNA双螺旋链通过缠绕形成核小体,核小体通过连接DNA形成核小体“串珠”结构,进一步转曲折叠形成螺线管显微结构。共定位、共表达的染色质位点又在染色质内相互靠近,形成染色质或DNA环结构。染色质环又进一步根据相互作用折叠成拓扑结构域(TAD)[28,34]。经研究发现同一TAD内染色质会存在较强的相互作用,TAD间的则相对较弱,且TAD的边界在不同物种、不同类型的细胞系中会相对稳定,因此TAD被认为是染色质三维构象的结构和功能基本单元。近期Bing等[35]整合比较了21种人类不同类型细胞的染色质相互作用,进一步证实TAD边界在不同类型细胞相对保守,且发现了局部相互作用高度富集的染色质活跃区域(frequently interacting regions, FIREs)与超级增强子部分重叠,表明染色质三维折叠与细胞生命活动紧密相关。
在哺乳动物基因组中,一半以上都是各类重复元件序列。重复元件根据结构和复制形式,可分为串联重复序列(tandem repeat)和转座元件(transposable elements)。转座元件又可分为长散在重复元件(long interspersed nuclear elements, LINE)与短散在重复元件(short interspersed nuclear elements, SINE)等[36]。短散在重复元件又根据结构等可分为Alu元件与Mir元件等不同子类。其中Alu元件在哺乳动物尤其是灵长类动物基因组中广泛存在且比例较高,然而对其功能上的研究却一直进展缓慢,早起甚至被认为是垃圾序列[1,37]。最近的一些研究才发现Alu元件在基因组的结构及基因表达调控等方面发挥了一些功能[38,39]。
本研究发现Alu元件在染色质三维构象的动态折叠过程中发挥了重要的作用,在不同种类的细胞系中均参与了较多高强度的染色质间相互作用。且目前多有研究发现,Alu元件与染色质内的增强子有强烈的相互作用。这些都表明Alu元件可能在染色质三维结构的构建、传递和维持中发挥着作用。染色质在细胞核内维持着动态平衡、永不缠绕的结构状态,有着相对稳定的结构,而这样的结构需要一定的网络节点加以固定,因此认为Alu元件在其中可能就发挥着高强度的相互作用网络中心固定节点的作用,从而维持着染色质动态结构的稳定。
3.2 染色质三维构象与Alu元件的演化存在关联
Alu元件在短散在重复序列中广泛分布,且种类繁多[40],目前已有较多根据序列层面分析Alu家族演化过程的研究,但尚未有人从其在染色质三维结构上的分布来分析其演化过程。本研究基于染色质三维结构的数据,对Alu元件的演化历程进行了分析,证明了一维序列演化关系上相对靠近的子类,在染色质三维结构中的相互作用强度也会较高,并用层次聚类的方式得到了基于染色质相互作用强度的进化树。这些工作表明,Alu元件在染色质上相互作用的强度与进化上的关系存在着一定的正相关,可目前的工作尚无法对这一现象的原因进行证实。一方面有可能是因为在进化过程中,Alu元件初代子族随机跳跃扩展到基因组内,某些调控因子对这些初代子族进行调控,使得这些子族所在的染色质位点相互靠拢,对染色质的三维构象进行改变;另一方面有可能在进化过程中,Alu元件的初代子族本身就分布在空间距离比较接近的染色质位点,随着基因组的演化,新产生的Alu元件子族也逐渐跳跃扩展到三维距离相近的染色质位点上,从而使得相同的Alu子族在染色质三维分布上比较靠近。两方面共同作用,从而使得Alu元件的演化与染色质的三维构象紧密相关。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
.
URLPMID:12764185 [本文引用: 2]

CiteSeerX - Scientific documents that cite the following paper: Genomics. Not junk after all
URL [本文引用: 1]
Alu家族是灵长类动物特有的且是最重要的短散在元件(short interspersed elements,SINEs),经过6千5百万年的进化,Alu序列在基因组中约有120万份拷贝,占基因组的10%以上。Alu家族在基因组中有很多功能,如介导重组、基因插入和删除、甲基化和A-to-I的编辑作用、调控转录和翻译、选择性剪接等等。Alu家族的变异与疾病和进化存在密切关系。
URL [本文引用: 1]
Alu家族是灵长类动物特有的且是最重要的短散在元件(short interspersed elements,SINEs),经过6千5百万年的进化,Alu序列在基因组中约有120万份拷贝,占基因组的10%以上。Alu家族在基因组中有很多功能,如介导重组、基因插入和删除、甲基化和A-to-I的编辑作用、调控转录和翻译、选择性剪接等等。Alu家族的变异与疾病和进化存在密切关系。
.
URLPMID:29466324 [本文引用: 1]
Abstract Long noncoding RNAs (lncRNAs) are emerging as key parts of multiple cellular pathways, but their modes of action and how these are dictated by sequence remain unclear. lncRNAs tend to be enriched in the nuclear fraction, whereas most mRNAs are overtly cytoplasmic, although several studies have found that hundreds of mRNAs in various cell types are retained in the nucleus. It is thus conceivable that some mechanisms that promote nuclear enrichment are shared between lncRNAs and mRNAs. Here, to identify elements in lncRNAs and mRNAs that can force nuclear localization, we screened libraries of short fragments tiled across nuclear RNAs, which were cloned into the untranslated regions of an efficiently exported mRNA. The screen identified a short sequence derived from Alu elements and bound by HNRNPK that increased nuclear accumulation. Binding of HNRNPK to C-rich motifs outside Alu elements is also associated with nuclear enrichment in both lncRNAs and mRNAs, and this mechanism is conserved across species. Our results thus identify a pathway for regulation of RNA accumulation and subcellular localization that has been co-opted to regulate the fate of transcripts with integrated Alu elements.
.
URLPMID:28355180 [本文引用: 1]
Transposable elements are viewed as elfish genetic elements , yet they contribute to gene regulation and genome evolution in diverse ways. More than half of the human genome consists of transposable elements. Alu elements belong to the short interspersed nuclear element (SINE) family of repetitive elements, and with over 1 illion insertions they make up more than 10% of the human genome. Despite their abundance and the potential evolutionary advantages they confer, Alu elements can be mutagenic to the host as they can act as splice acceptors, inhibit translation of mRNAs and cause genomic instability. Alu elements are the main targets of the RNA-editing enzyme ADAR and the formation of Alu exons is suppressed by the nuclear ribonucleoprotein HNRNPC, but the broad effect of massive secondary structures formed by inverted-repeat Alu elements on RNA processing in the nucleus remains unknown. Here we show that DHX9, an abundant nuclear RNA helicase, binds specifically to inverted-repeat Alu elements that are transcribed as parts of genes. Loss of DHX9 leads to an increase in the number of circular-RNA-producing genes and amount of circular RNAs, translational repression of reporters containing inverted-repeat Alu elements, and transcriptional rewiring (the creation of mostly nonsensical novel connections between exons) of susceptible loci. Biochemical purifications of DHX9 identify the interferon-inducible isoform of ADAR (p150), but not the constitutively expressed ADAR isoform (p110), as an RNA-independent interaction partner. Co-depletion of ADAR and DHX9 augments the double-stranded RNA accumulation defects, leading to increased circular RNA production, revealing a functional link between these two enzymes. Our work uncovers an evolutionarily conserved function of DHX9. We propose that it acts as a nuclear RNA resolvase that neutralizes the immediate threat posed by transposon insertions and allows these elements to evolve as tools for the post-transcriptional regulation of gene expression.
.
URLPMID:24703844 [本文引用: 1]
正Prof.Han and colleagues from CAS-MPG Partner Institute for Computational Biology published their research result entitled"Evolution of Alu elements toward enhancers"in Cell Reports(2014,7(2):376—385).Of the human genome 10%is comprised of more than one million Alu repetitive elements,whose functions remain unclear.In this work,Prof.Han's team showed that Alus are more conserved when loca-
.
URLPMID:28586325 [本文引用: 1]
Abstract Two new studies show that RNA-binding proteins can mediate distinct and beneficial effects to cells by binding to the extensive double-stranded RNA (dsRNA) structures of inverted-repeat Alu elements (IRAlus). One study reports stress-induced export of the 110-kDa isoform of the adenosine deaminase acting on RNA 1 protein (ADAR1p110) to the cytoplasm, where it binds IRAlus so as to protect many mRNAs encoding anti-apoptotic proteins from degradation. The other study demonstrates that binding of the nuclear helicase DHX9 to IRAlus embedded within RNAs minimizes defects in RNA processing. 2017 Nature America, Inc., part of Springer Nature. All rights reserved.
.
URLPMID:29698748 [本文引用: 1]
Abstract Alu elements occupy 10% of the human genome. However, although they contribute to genomic and transcriptomic diversity, their function is still not fully understood. We hypothesized that intronic Alu elements may contribute to alternative splicing. We therefore examined their effect on splicing using minigene constructs including exon 9-exon 11 inclusive of ACAT1 with truncated introns 9 and 10. These constructs contained a suboptimal splice acceptor site for intron 9. Insertion of AluY-partial AluSz6-AluSx, originally located in ACAT1 intron 5, in an antisense direction within intron 9 had a negative effect on exon 10 inclusion. This effect was additive with that of an exonic splicing enhancer mutation in exon 10, and was canceled by the substitution of G for C at the first nucleotide of exon 10 which optimized the splice acceptor site of intron 9. A sense AluY-partial AluSz6-AluSx insertion had no effect on exon 10 inclusion, and one antisense AluSx insertion had a similar effect to antisense AluY-partial AluSz6-AluSx insertion. The shorter the distance between the antisense Alu element and exon 10, the greater the negative effect on exon 10 inclusion. This distance effect was more evident for suboptimal than optimal splice sites. Based on our data, we propose that intronic antisense Alu elements contribute to alternative splicing and transcriptomic diversity in some genes, especially when splice acceptor sites are suboptimal.
.
URLPMID:22495300 [本文引用: 1]
The spatial organization of the genome is intimately linked to its biological function, yet our understanding of higher order genomic structure is coarse, fragmented and incomplete. In the nucleus of eukaryotic cells, interphase chromosomes occupy distinct chromosome territories, and numerous models have been proposed for how chromosomes fold within chromosome territories. These models, however, provide only few mechanistic details about the relationship between higher order chromatin structure and genome function. Recent advances in genomic technologies have led to rapid advances in the study of three-dimensional genome organization. In particular, Hi-C has been introduced as a method for identifying higher order chromatin interactions genome wide. Here we investigate the three-dimensional organization of the human and mouse genomes in embryonic stem cells and terminally differentiated cell types at unprecedented resolution. We identify large, megabase-sized local chromatin interaction domains, which we term 'topological domains', as a pervasive structural feature of the genome organization. These domains correlate with regions of the genome that constrain the spread of heterochromatin. The domains are stable across different cell types and highly conserved across species, indicating that topological domains are an inherent property of mammalian genomes. Finally, we find that the boundaries of topological domains are enriched for the insulator binding protein CTCF, housekeeping genes, transfer RNAs and short interspersed element (SINE) retrotransposons, indicating that these factors may have a role in establishing the topological domain structure of the genome.
URL [本文引用: 1]
在细胞分裂间期,每条染色质都占据着特定的染色质领域(chromosome territory,CT)。每个CT领域内进一步分成不同的拓扑学相关区域(topological associated domain,TAD),每个TAD又由若干子TAD(sub-TAD)构成。不同的TAD相互聚集,形成基因活跃表达和不表达的A、B两种组份或区室(compartment)。然而,目前对于染色质折叠方式及维持机制的研究尚无定论。核基质附着区(matrix attachment regions,MARs)是在不同物种基因组中广泛存在的一类富含AT序列的与核基质结合的DNA元件,能够通过与CTCF、SATB1等调控蛋白质相互作用,对远距离的基因表达进行调控。本研究以染色质三维结构为背景,通过整合染色质三维结构及组蛋白修饰等组学数据,对MARs元件与染色质三维结构的关系进行研究,对MARs元件参与形成的相互作用网络的结构及功能进行探索。结果发现,MARs元件与染色质三维结构高度相关,而且在高强度相互作用中占据较大的比例,提示MARs元件在染色质折叠方面发挥作用。此外,通过拓扑结构聚类分析还首次揭示,MARs元件分为不同类型,包括维持染色质领域及空间构象等的结构单元部分,以及调控基因表达等的功能单元部分。这表明,MARs元件在基因组三维高级结构的建立、维持以及功能等方面发挥重要作用。
URL [本文引用: 1]
在细胞分裂间期,每条染色质都占据着特定的染色质领域(chromosome territory,CT)。每个CT领域内进一步分成不同的拓扑学相关区域(topological associated domain,TAD),每个TAD又由若干子TAD(sub-TAD)构成。不同的TAD相互聚集,形成基因活跃表达和不表达的A、B两种组份或区室(compartment)。然而,目前对于染色质折叠方式及维持机制的研究尚无定论。核基质附着区(matrix attachment regions,MARs)是在不同物种基因组中广泛存在的一类富含AT序列的与核基质结合的DNA元件,能够通过与CTCF、SATB1等调控蛋白质相互作用,对远距离的基因表达进行调控。本研究以染色质三维结构为背景,通过整合染色质三维结构及组蛋白修饰等组学数据,对MARs元件与染色质三维结构的关系进行研究,对MARs元件参与形成的相互作用网络的结构及功能进行探索。结果发现,MARs元件与染色质三维结构高度相关,而且在高强度相互作用中占据较大的比例,提示MARs元件在染色质折叠方面发挥作用。此外,通过拓扑结构聚类分析还首次揭示,MARs元件分为不同类型,包括维持染色质领域及空间构象等的结构单元部分,以及调控基因表达等的功能单元部分。这表明,MARs元件在基因组三维高级结构的建立、维持以及功能等方面发挥重要作用。
.
URLPMID:25409831 [本文引用: 1]
Eukaryotic chromosomes replicate in a temporal order known as the replication-timing program. In mammals, replication timing is cell-type-specific with at least half the genome switching replication timing during development, primarily in units of 400-800 kilobases ('replication domains'), whose positions are preserved in different cell types, conserved between species, and appear to confine long-range effects of chromosome rearrangements. Early and late replication correlate, respectively, with open and closed three-dimensional chromatin compartments identified by high-resolution chromosome conformation capture (Hi-C), and, to a lesser extent, late replication correlates with lamina-associated domains (LADs). Recent Hi-C mapping has unveiled substructure within chromatin compartments called topologically associating domains (TADs) that are largely conserved in their positions between cell types and are similar in size to replication domains. However, TADs can be further sub-stratified into smaller domains, challenging the significance of structures at any particular scale. Moreover, attempts to reconcile TADs and LADs to replication-timing data have not revealed a common, underlying domain structure. Here we localize boundaries of replication domains to the early-replicating border of replication-timing transitions and map their positions in 18 human and 13 mouse cell types. We demonstrate that, collectively, replication domain boundaries share a near one-to-one correlation with TAD boundaries, whereas within a cell type, adjacent TADs that replicate at similar times obscure replication domain boundaries, largely accounting for the previously reported lack of alignment. Moreover, cell-type-specific replication timing of TADs partitions the genome into two large-scale sub-nuclear compartments revealing that replication-timing transitions are indistinguishable from late-replicating regions in chromatin composition and lamina association and accounting for the reduced correlation of replication timing to LADs and heterochromatin. Our results reconcile cell-type-specific sub-nuclear compartmentalization and replication timing with developmentally stable structural domains and offer a unified model for large-scale chromosome structure and function.
.
URL [本文引用: 1]
.
URLPMID:17033624 [本文引用: 1]
Accumulating evidence converges on the possibility that chromosomes interact with each other to regulate transcription in trans. To systematically explore the epigenetic dimension of such interactions, we devised a strategy termed circular chromosome conformation capture (4C). This approach involves a circularization step that enables high-throughput screening of physical interactions between chromosomes without a preconceived idea of the interacting partners. Here we identify 114 unique sequences from all autosomes, several of which interact primarily with the maternally inherited H19 imprinting control region. Imprinted domains were strongly overrepresented in the library of 4C sequences, further highlighting the epigenetic nature of these interactions. Moreover, we found that the direct interaction between differentially methylated regions was linked to epigenetic regulation of transcription in trans. Finally, the patterns of interactions specific to the maternal H19 imprinting control region underwent reprogramming during in vitro maturation of embryonic stem cells. These observations shed new light on development, cancer epigenetics and the evolution of imprinting.
.
URL [本文引用: 1]
.
URLPMID:2858594 [本文引用: 1]
We describe Hi-C, a method that probes the three-dimensional architecture of whole genomes by coupling proximity-based ligation with massively parallel sequencing. We constructed spatial proximity maps of the human genome with Hi-C at a resolution of 1 megabase. These maps confirm the presence of chromosome territories and the spatial proximity of small, gene-rich chromosomes. We identified an additional level of genome organization that is characterized by the spatial segregation of open and closed chromatin to form two genome-wide compartments. At the megabase scale, the chromatin conformation is consistent with a fractal globule, a knot-free, polymer conformation that enables maximally dense packing while preserving the ability to easily fold and unfold any genomic locus. The fractal globule is distinct from the more commonly used globular equilibrium model. Our results demonstrate the power of Hi-C to map the dynamic conformations of whole genomes.
URL [本文引用: 1]
Hi-C(highest-throughput chromosome conformation capture)技术是近年出现的一种研究染色质相互作用的关键技术。该技术步骤多、耗时长,涉及的试剂耗材繁杂,目前常规流程还有较多可以改进优化的步骤。本研究以GM12878细胞为材料,通过优化常规Hi-C实验中的交联、酶切、限制性内切酶的失活、末端生物素标记、原位连接等关键步骤,建立了稳健的Hi-C流程,制备了相应的Hi-C文库。文库经初步的Sanger测序等质量控制以后,两个生物学重复文库进行了高通量测序。测序结果利用生物信息学处理发现:原始测序数据中可比对率和配对率分别达到90%和72%左右。此外,去除自连片段(self-circular ligation)和dangling-ends片段以后,可获得超过96%的有效相互作用对,其中染色体内相互作用数据达到60%。进一步的染色体相互作用热图分析可见清晰拓扑学相关结构域TADs(topologically associated domains),与已发表的文献报道一致。而两次生物学重复之间的相关性分析则表明bin coverage和all bin pairs的相关性都极强。上述结果表明通过对常规Hi-C流程关键步骤的优化,本研究建立了高效、稳健、可靠的Hi-C流程,对Hi-C技术的进一步使用与推广具有重要的参考价值。
URL [本文引用: 1]
Hi-C(highest-throughput chromosome conformation capture)技术是近年出现的一种研究染色质相互作用的关键技术。该技术步骤多、耗时长,涉及的试剂耗材繁杂,目前常规流程还有较多可以改进优化的步骤。本研究以GM12878细胞为材料,通过优化常规Hi-C实验中的交联、酶切、限制性内切酶的失活、末端生物素标记、原位连接等关键步骤,建立了稳健的Hi-C流程,制备了相应的Hi-C文库。文库经初步的Sanger测序等质量控制以后,两个生物学重复文库进行了高通量测序。测序结果利用生物信息学处理发现:原始测序数据中可比对率和配对率分别达到90%和72%左右。此外,去除自连片段(self-circular ligation)和dangling-ends片段以后,可获得超过96%的有效相互作用对,其中染色体内相互作用数据达到60%。进一步的染色体相互作用热图分析可见清晰拓扑学相关结构域TADs(topologically associated domains),与已发表的文献报道一致。而两次生物学重复之间的相关性分析则表明bin coverage和all bin pairs的相关性都极强。上述结果表明通过对常规Hi-C流程关键步骤的优化,本研究建立了高效、稳健、可靠的Hi-C流程,对Hi-C技术的进一步使用与推广具有重要的参考价值。
.
URLPMID:25437436 [本文引用: 2]
High-throughput methods based on conformation capture have greatly advanced our understanding of the three-dimensional (3D) organization of genomes but are limited in resolution by their reliance on restriction enzymes. Here we describe a method called Hi-C for comprehensively mapping global contacts. Hi-C uses for fragmentation, leading to greatly improved efficiency and resolution over that of Hi-C. Coupling this method with DNA-capture technology provides a high-throughput approach for targeted mapping of fine-scale architecture. We applied targeted Hi-C to characterize the 3D organization of 998 large intergenic noncoding (lincRNA) promoters in two cell lines. Our results revealed that expression of lincRNAs is tightly controlled by complex mechanisms involving both super-enhancers and the Polycomb repressive complex. Our results provide the first glimpse of the cell type-specific 3D organization of lincRNA genes.
.
URLPMID:26119342 [本文引用: 1]
Mononucleosome resolution mapping of chromosome folding in yeast reveals self-associating domains similar to those found in other organisms. But they are far shorter, with domain size being scaled by gene number rather than linear distance.
.
URLPMID:25122612 [本文引用: 1]
Genome-wide association studies have identified more than 70 common variants that are associated with breast cancer risk. Most of these variants map to non-protein-coding regions and several map to gene deserts, regions of several hundred kilobases lacking protein-coding genes. We hypothesized that gene deserts harbor long-range regulatory elements that can physically interact with target genes to influence their expression. To test this, we developed Capture Hi-C (CHi-C), which, by incorporating a sequence capture step into a Hi-C protocol, allows high-resolution analysis of targeted regions of the genome. We used CHi-C to investigate long-range interactions at three breast cancer gene deserts mapping to 2q35, 8q24.21, and 9q31.2. We identified interaction peaks between putative regulatory elements ("bait fragments") within the captured regions and "targets" that included both protein-coding genes and long noncoding (Inc) RNAs over distances of 6.6 kb to 2.6 Mb. Target protein-coding genes were IGFBP5, KLF4, NSMCE2, and MYC; and target IncRNAs included DIRC3, PVT1, and CCDC26. For one gene desert, we were able to define two SNPs (rs12613955 and rs4442975) that were highly correlated with the published risk variant and that mapped within the bait end of an interaction peak. In vivo ChIP-qPCR data show that one of these, rs4442975, affects the binding of FOXA1 and implicate this SNP as a putative functional variant.
.
URLPMID:25497547 [本文引用: 2]
Using 3D genome sequencing, we find 6510,000 DNA loops across the human genome. Loop anchors typically occur at domain boundaries and bind CTCF in a convergent orientation, with the asymmetric motifs “facing” one another. On the inactive X chromosome, large loops are anchored at CTCF-binding repeats. Loops are conserved across cell types and species.
.
URLPMID:27919068 [本文引用: 1]
Chromosomes are folded into highly compacted structures to accommodate physical constraints within nuclei and to regulate access to genomic information. Recently, global mapping of pairwise contacts showed that loops anchoring topological domains (TADs) are highly conserved between cell types and species. Whether pairwise loops synergize to form higher-order structures is still unclear. Here we develop a conformation capture assay to study higher-order organization using chromosomal walks (C-walks) that link multiple genomic loci together into proximity chains in human and mouse cells. This approach captures chromosomal structure at varying scales. Inter-chromosomal contacts constitute only 7-10% of the pairs and are restricted by interfacing TADs. About half of the C-walks stay within one chromosome, and almost half of those are restricted to intra-TAD spaces. C-walks that couple 2-4 TADs indicate stochastic associations between transcriptionally active, early replicating loci. Targeted analysis of thousands of 3-walks anchored at highly expressed genes support pairwise, rather than hub-like, chromosomal topology at active loci. Polycomb-repressed Hox domains are shown by the same approach to enrich for synergistic hubs. Together, the data indicate that chromosomal territories, TADs, and intra-TAD loops are primarily driven by nested, possibly dynamic, pairwise contacts.
.
URLPMID:27340173 [本文引用: 1]
The relevance of three-dimensional (3D) genome organization for transcriptional regulation and thereby for cellular fate at large is nowwidely accepted. Our understanding of the fascinating architecture underlying this function is based on microscopy studies as well as the chromosome conformation capture (3C) methods, which entered the stage at the beginning of the millennium. The first decade of 3C methods rendered unprecedented insights into genome topology. Here, we provide an update of developments and discoveries made over the more recent years. As we discuss, established and newly developed experimental and computational methodsenabled identification of novel, functionally important chromosome structures. Regulatory and architectural chromatin loops throughout thegenomeare being cataloged and compared between cell types, revealing tissue invariant and developmentally dynamic loops. Architectural proteins shaping thegenome were disclosed, and their mode of action is being uncovered. We explain how more detailed insights into the 3D genome increase our understanding of transcriptional regulation in development and misregulation in disease. Finally, to help researchers in choosing the approach best tailored for their specific research question, we explain the differences and commonalities between the various 3C-derived methods.
.
URLPMID:5617335 [本文引用: 1]
The spatial organization of the genome and its dynamics contribute to gene expression and cellular function in normal development as well as in disease. Although we are increasingly well equipped to determine a genome9s sequence and linear chromatin composition, studying the three-dimensional organization of the genome with high spatial and temporal resolution remains challenging. The 4D Nucleome Network aims to develop and apply approaches to map the structure and dynamics of the human and mouse genomes in space and time with the long term goal of gaining deeper mechanistic understanding of how the nucleus is organized. The project will develop and benchmark experimental and computational approaches for measuring genome conformation and nuclear organization, and investigate how these contribute to gene regulation and other genome functions. Further efforts will be directed at applying validated experimental approaches combined with biophysical modeling to generate integrated maps and quantitative models of spatial genome organization in different biological states, both in cell populations and in single cells.
.
URLPMID:18428725 [本文引用: 1]
The RepeatMasker program is used for identifying repetitive elements in nucleotide sequences for further detailed analyses. Users can run RepeatMasker remotely via a Web site, or, for larger input sequences, the program and its dependent programs may be downloaded and run locally on Unix/Linux computers. The protocols in this chapter detail how to use RepeatMasker both remotely and locally to extract repetitive elements data and mask these repetitive elements in nucleotide sequences.
.
URLPMID:4650858 [本文引用: 2]
Precise quantitative and spatiotemporal control of gene expression is necessary to ensure proper cellular differentiation and the maintenance of homeostasis. The relationship between gene expression and the spatial organisation of chromatin is highly complex, interdependent and not completely understood. The development of experimental techniques to interrogate both the higher-order structure of chromatin and the interactions between regulatory elements has recently lead to important insights on how gene expression is controlled. The ability to gain these and future insights is critically dependent on computational tools for the analysis and visualisation of data produced by these techniques. We have developed GenomicInteractions, a freely available R/Bioconductor package designed for processing, analysis and visualisation of data generated from various types of chromosome conformation capture experiments. The package allows the easy annotation and summarisation of large genome-wide datasets at both the level of individual interactions and sets of genomic features, and provides several different methods for interrogating and visualising this type of data. We demonstrate this package utility by showing example analyses performed on interaction datasets generated using Hi-C and ChIA-PET.
.
URLPMID:26392354 [本文引用: 2]
Metazoan genomic material is folded into stable non-randomly arranged chromosomal structures that are tightly associated with transcriptional regulation and DNA replication. Various factors including...
.
URLPMID:26619908 [本文引用: 1]
HiC-Pro is an optimized and flexible pipeline for processing Hi-C data from raw reads to normalized contact maps. HiC-Pro maps reads, detects valid ligation products, performs quality controls and generates intra- and inter-chromosomal contact maps. It includes a fast implementation of the iterative correction method and is based on a memory-efficient data format for Hi-C contact maps. In addition, HiC-Pro can use phased genotype data to build allele-specific contact maps. We applied HiC-Pro to different Hi-C datasets, demonstrating its ability to easily process large data in a reasonable time. Source code and documentation are available athttp://github.com/nservant/HiC-Pro. The online version of this article (doi:10.1186/s13059-015-0831-x) contains supplementary material, which is available to authorized users.
.
URLPMID:25448293 [本文引用: 1]
Over the last decade, development and application of a set of molecular genomic approaches based on the chromosome conformation capture method (3C), combined with increasingly powerful imaging approaches, have enabled high resolution and genome-wide analysis of the spatial organization of chromosomes. The aim of this paper is to provide guidelines for analyzing and interpreting data obtained with genome-wide 3C methods such as Hi-C and 3C-seq that rely on deep sequencing to detect and quantify pairwise chromatin interactions.
.
URLPMID:29626919 [本文引用: 2]
4D nucleome research aims to understand the impact of nuclear organization in space and time on nuclear functions, such as gene expression patterns, chromatin replication, and the maintenance of genome integrity. In this review we describe evidence that the origin of 4D genome compartmentalization can be traced back to the prokaryotic world. In cell nuclei of animals and plants chromosomes occupy distinct territories, built up from ~1 Mb chromatin domains, which in turn are composed of smaller chromatin subdomains and also form larger chromatin domain clusters. Microscopic evidence for this higher order chromatin landscape was strengthened by chromosome conformation capture studies, in particular Hi-C. This approach demonstrated ~1 Mb sized, topologically associating domains in mammalian cell nuclei separated by boundaries. Mutations, which destroy boundaries, can result in developmental disorders and cancer. Nucleosomes appeared first as tetramers in the Archaea kingdom and later evolved to octamers built up each from two H2A, two H2B, two H3, and two H4 proteins. Notably, nucleosomes were lost during the evolution of the Dinoflagellata phylum. Dinoflagellate chromosomes remain condensed during the entire cell cycle, but their chromosome architecture differs radically from the architecture of other eukaryotes. In summary, the conservation of fundamental features of higher order chromatin arrangements throughout the evolution of metazoan animals suggests the existence of conserved, but still unknown mechanism(s) controlling this architecture. Notwithstanding this conservation, a comparison of metazoans and protists also demonstrates species-specific structural and functional features of nuclear organization.
.
URLPMID:26609133 [本文引用: 1]
The potential roles of the numerous repetitive elements found in the genomes of multi-cellular organisms remain speculative. Several studies have suggested a role in stabilizing specific 3D genomic contacts. To test this hypothesis, we exploited inter-chromosomal contacts frequencies obtained from Hi-C experiments and show that the folding of the human, mouse andDrosophilagenomes is associated with a significant co-localization of several specific repetitive elements, notably many elements of the SINE family. These repeats tend to be the oldest ones and are enriched in transcription factor binding sites. We propose that the co-localization of these repetitive elements may explain the global conservation of genome folding observed between homologous regions of the human and mouse genome. Taken together, these results support a contribution of specific repetitive elements in maintaining and/or reshaping genome architecture over evolutionary times.
.
URLPMID:8576965 [本文引用: 1]
Using Kimura's distance measure we have calculated the average age of all major Alu subfamilies based on the most recent available data. We conclude that AluJ sequences are some 26 Myr older than previously thought. Furthermore, the origin of the FLA (Free Left Arm) Alu family can be traced back to the very beginning of the mammalian radiation. One new minor subfamily is reported and discussed in the context of sequence diversity in major Alu subfamilies.
.
URLPMID:15520288 [本文引用: 1]
Alu repeats are the most abundant family of repeats in the human genome, with over 1 million copies comprising 10% of the genome. They have been implicated in human genetic disease and in the enrichment of gene-rich segmental duplications in the human genome, and they form a rich fossil record of primate and human history. Alu repeat elements are believed to have arisen from the replication of a small number of source elements, whose evolution over time gives rise to the 31 Alu subfamilies currently reported in Repbase Update. We apply a novel method to identify and statistically validate 213 Alu subfamilies. We build an evolutionary tree of these subfamilies and conclude that the history of Alu evolution is more complex than previous studies had indicated.
URL [本文引用: 1]
CTCF是脊椎动物关键的绝缘子蛋白,在细胞生命过程中发挥重要作用,敲除CTCF基因会导致小鼠胚胎死亡。为进一步探讨CTCF的功能,本文利用CRISPR/Cas9介导的同源重组,在内源性CTCF表达框上游敲入一个有丝分裂期降解结构域(Mitosis-special degradation domain,MD),该结构域可以带动CTCF融合蛋白在M期降解。作为对照,将MD结构域的第42位的精氨酸突变为丙氨酸,形成无降解活性的MD*,可使MD*-CTCF融合蛋白始终稳定存在。将嘌呤霉素与融合蛋白同时表达,即可利用抗生素筛选,高效地筛选到纯合克隆。利用蛋白印迹技术和免疫荧光检测3种细胞在不同细胞周期的CTCF蛋白变化情况,发现MD-CTCF细胞系CTCF蛋白含量约为野生型细胞的10%,MD*-CTCF细胞系的CTCF含量与野生型没有显著差别;通过流式细胞术观测降解CTCF对细胞的影响,发现MD-CTCF细胞系G1期明显延长。总之,利用CRISPR/Cas9技术在CTCF表达框上游高效地插入MD,首个CTCF特异性降解的人类细胞系获得成功构建。
URL [本文引用: 1]
CTCF是脊椎动物关键的绝缘子蛋白,在细胞生命过程中发挥重要作用,敲除CTCF基因会导致小鼠胚胎死亡。为进一步探讨CTCF的功能,本文利用CRISPR/Cas9介导的同源重组,在内源性CTCF表达框上游敲入一个有丝分裂期降解结构域(Mitosis-special degradation domain,MD),该结构域可以带动CTCF融合蛋白在M期降解。作为对照,将MD结构域的第42位的精氨酸突变为丙氨酸,形成无降解活性的MD*,可使MD*-CTCF融合蛋白始终稳定存在。将嘌呤霉素与融合蛋白同时表达,即可利用抗生素筛选,高效地筛选到纯合克隆。利用蛋白印迹技术和免疫荧光检测3种细胞在不同细胞周期的CTCF蛋白变化情况,发现MD-CTCF细胞系CTCF蛋白含量约为野生型细胞的10%,MD*-CTCF细胞系的CTCF含量与野生型没有显著差别;通过流式细胞术观测降解CTCF对细胞的影响,发现MD-CTCF细胞系G1期明显延长。总之,利用CRISPR/Cas9技术在CTCF表达框上游高效地插入MD,首个CTCF特异性降解的人类细胞系获得成功构建。
.
URLPMID:28273065 [本文引用: 1]
The organization of the genome in the nucleus and the interactions of genes with their regulatory elements are key features of transcriptional control and their disruption can cause disease. Here we report a genome-wide method, genome architecture mapping (GAM), for measuring chromatin contacts and other features of three-dimensional chromatin topology on the basis of sequencing DNA from a large collection of thin nuclear sections. We apply GAM to mouse embryonic stem cells and identify enrichment for specific interactions between active genes and enhancers across very large genomic distances using a mathematical model termed SLICE (statistical inference of co-segregation). GAM also reveals an abundance of three-way contacts across the genome, especially between regions that are highly transcribed or contain super-enhancers, providing a level of insight into genome architecture that, owing to the technical limitations of current technologies, has previously remained unattainable. Furthermore, GAM highlights a role for gene-expression-specific contacts in organizing the genome in mammalian nuclei.
.
[本文引用: 1]
.
URLPMID:5478386 [本文引用: 1]
Schmitt et al. analyze Hi-C maps in 21 human cell lines and primary tissues and uncover a class of genome organizational features termed FIREs. FIREs are local interaction hotspots, highly tissue-specific, and correspond to active enhancers. We discuss the implications of our findings for the study of gene regulation and disease. Explore the Cell Press IHEC web portal athttp://www.cell.com/consortium/IHEC.
.
URLPMID:27867194 [本文引用: 1]
Transposable elements (TEs) are a prolific source of tightly regulated, biochemically active non-coding elements, such as transcription factor-binding sites and non-coding RNAs. Many recent studies reinvigorate the idea that these elements are pervasively co-opted for the regulation of host genes. We argue that the inherent genetic properties of TEs and the conflicting relationships with their hosts facilitate their recruitment for regulatory functions in diverse genomes. We review recent findings supporting the long-standing hypothesis that the waves of TE invasions endured by organisms for eons have catalysed the evolution of gene-regulatory networks. We also discuss the challenges of dissecting and interpreting the phenotypic effect of regulatory activities encoded by TEs in health and disease.
.
URLPMID:53242214 [本文引用: 1]
Instead of testing predefined hypotheses, the goal of exploratory data analysis (EDA) is to find what data can tell us. Following this strategy, we re-analyzed a large body of genomic data to study the complex gene regulation in mouse pre-implantation development (PD). Starting with a single-cell RNA-seq dataset consisting of 259 mouse embryonic cells derived from zygote to blastocyst stages, we reconstructed the temporal and spatial gene expression pattern during PD. The dynamics of gene expression can be partially explained by the enrichment of transposable elements in gene promoters and the similarity of expression profiles with those of corresponding transposons. Long Terminal Repeats (LTRs) are associated with transient, strong induction of many nearby genes at the 2-4 cell stages, probably by providing binding sites for Obox and other homeobox factors. B1 and B2 SINEs (Short Interspersed Nuclear Elements) are correlated with the upregulation of thousands of nearby genes during zygotic genome activation. Such enhancer-like effects are also found for human Alu and bovine tRNA SINEs. SINEs also seem to be predictive of gene expression in embryonic stem cells (ESCs), raising the possibility that they may also be involved in regulating pluripotency. We also identified many potential transcription factors underlying PD and discussed the evolutionary necessity of transposons in enhancing genetic diversity, especially for species with longer generation time. Together with other recent studies, our results provide further evidence that many transposable elements may play a role in establishing the expression landscape in early embryos. It also demonstrates that exploratory bioinformatics investigation can pinpoint developmental pathways for further study, and serve as a strategy to generate novel insights from big genomic data. The online version of this article (doi:10.1186/s12864-017-3566-0) contains supplementary material, which is available to authorized users.
.
URLPMID:25917896 [本文引用: 1]
Eukaryotic genomes contain millions of copies of repetitive elements (RE). Although the euchromatic parts of most genomes are clearly annotated, the repetitive/heterochromatic parts are poorly defined. It is estimated that between 50 and 70% of the human genome is composed of REs. Despite this, we know surprisingly little about the physiological relevance, molecular regulation and the composition of these regions. This primarily reflects the difficulty that REs pose for PCR-based assays, and their poor map-ability in next generation sequencing experiments. Here we first summarize the nature and classification of REs and then examine how this has been used in the recent years to broaden our understanding of mechanisms that keep the repetitive regions of our genomes silent and stable.
.
URLPMID:26861146 [本文引用: 1]
Transposable elements (TEs) have no longer been totally considered as “junk DNA” for quite a time since the continual discoveries of their multifunctional roles in eukaryote genomes. As one of the most important and abundant TEs that still active in human genome,Alu, a SINE family, has demonstrated its indispensable regulatory functions at sequence level, but its spatial roles are still unclear. Technologies based on 3C (chromosome conformation capture) have revealed the mysterious three-dimensional structure of chromatin, and make it possible to study the distal chromatin interaction in the genome. To find the role TE playing in distal regulation in human genome, we compiled the new released Hi-C data, TE annotation, histone marker annotations, and the genome-wide methylation data to operate correlation analysis, and found that the density ofAluelements showed a strong positive correlation with the level of chromatin interactions (hESC:r=020.9,P< 2.2 × 1016; IMR90 fibroblasts:r=020.94,P< 2.2 × 1016) and also have a significant positive correlation with some remote functional DNA elements like enhancers and promoters (Enhancer: hESC:r=020.997,P=022.3 × 10614; IMR90:r=020.934,P=022 × 10612; Promoter: hESC:r=020.995,P=023.8 × 10614; IMR90:r=020.996,P=023.2 × 10614). Further investigation involving GC content and methylation status showed the GC content ofAlucovered sequences shared a similar pattern with that of the overall sequence, suggesting thatAluelements also function as the GC nucleotide and CpG site provider. In all, our results suggest that theAluelements may act as an alternative parameter to evaluate the Hi-C data, which is confirmed by the correlation analysis ofAluelements and histone markers. Moreover, the GC-richAlusequence can bring high GC content and methylation flexibility to the regions with more distal chromatin contact, regulating the transcription of tissue-specific genes. The online version of this article (doi:10.1007/s13238-015-0240-7) contains supplementary material, which is available to authorized users.
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}