 ,2, 王兴春,1
,2, 王兴春,1Cas9 protein variant VQR recognizes NGAC protospacer adjacent motif in rice
Gaowei Xin1,2, Xixun Hu2, Kejian Wang,2, Xingchun Wang,1通讯作者:
编委: 高彩霞
收稿日期:2018-05-10修回日期:2018-06-20网络出版日期:2018-12-20
| 基金资助: |
Received:2018-05-10Revised:2018-06-20Online:2018-12-20
| Fund supported: |
作者简介 About authors
辛高伟,硕士研究生,专业方向:生物化学与分子生物学E-mail:592950985@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (606KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
辛高伟, 胡熙璕, 王克剑, 王兴春. Cas9蛋白变体VQR高效识别水稻NGAC前间区序列邻近基序[J]. 遗传, 2018, 40(12): 1112-1119 doi:10.16288/j.yczz.18-126
Gaowei Xin, Xixun Hu, Kejian Wang, Xingchun Wang.
基因组编辑技术是一种在基因组水平上对目标基因序列进行碱基删除、插入或替换的操作技术。该技术的原理是利用人工构建序列特异性核酸酶(sequence-specific nucleases, SSNs)在特定的基因组位置切断DNA,切断的DNA在细胞内DNA修复系统修复过程中产生突变,从而实现定点改造基因组的目的[1]。目前,常用的SSNs主要包括锌指核酸酶(zinc-finger nucleases, ZFNs)技术[2]、类转录激活因子效应物核酸酶(Transcription activator-like effector nucleases, TALENs)技术[3]和成簇的规律间隔的短回文重复序列/相关蛋白(clustered regularly interspaced short palindromic repeats/CRISPR-associated 9, CRISPR/ Cas9)技术[4]。与ZFN和TALEN技术相比,CRISPR/ Cas9技术具有载体构建简单、基因编辑效率高、成本低等特点[5,6],目前被广泛应用于基因功能研究和动植物精准分子育种等领域[7,8,9,10,11]。
2013年,在细菌天然免疫系统中发现的CRISPR/ Cas9系统作为第三代基因组编辑技术迅猛发展起 来[12,13]。该基因编辑系统通过单链向导RNA (single guide RNA, sgRNA)与基因组上的靶位点进行碱基互补配对完成识别过程,进而与核酸酶Cas9形成的sgRNA-Cas9复合体在特定靶位点造成DNA双链断裂,并借助于生物体内的同源或非同源重组修复途径完成基因组编辑。其中,靶位点的选择需符合不同系统的PAM要求[14]。目前,广泛使用的酿脓链球菌(Streptococcus pyogenes) SpCas9在植物中所识别的PAM主要是NGG[4],极大地限制了SpCas9的基因组编辑范围。为了扩展CRISPR/Cas9在基因组中的编辑范围,人们从不同微生物中鉴定了识别不同PAM的同源蛋白,比如嗜热性链球菌(Streptococcus thermophiles) CRISPR3 Cas9识别NGGNG PAM[15],嗜热性链球菌(Streptococcus thermophiles) CRISPR1 Cas9识别NNAGAAW PAM[16],脑膜炎奈瑟菌(Neisseria meningitides) NmeCas9识别NNNNGATT PAM[17]。此外,通过改造SpCas9的方法也可以改变其识别的靶位点。SpCas9变体VQR (D1135V/R1335Q/T1337R)和VRER (D1135V/G1218R/R1335E/T1337R)分别可以识别NGA PAM和NGCG PAM[18];而变体xCas9可以识别NG、GAA和GAT 3种PAM[19]。类似地,本课题组对VQR和VRER变体在水稻中的研究表明,VQR和VRER在水稻中也分别识别NGA PAM和NGCG PAM[20]。生物信息学分析表明,水稻基因组中VQR、VRER和SpCas9可编辑位点数目分别为45176043、7972298和38923028[20]。在水稻中VQR的基因编辑范围大于VRER和SpCas9,其中NGAG、NGAC、NGAT和NGAA 4种类型PAM靶位点的个数分别为9784476、9784158、12807613和12799796,NGAC所占比例为22%[20]。然而,VQR变体能够高效识别NGAA、NGAT和NGAG 3种PAM,NGAC是否能够被编辑仍然未知[20,21]。
虽然VQR变体大大扩展了CRISPR/Cas9的应用范围,但其编辑效率低于原CRISPR/Cas9系统,这极大地限制了该系统在水稻中的推广应用[20]。在哺乳动物细胞中,将sgRNA中连续4个胸腺嘧啶(T)中的一个碱基T突变为胞嘧啶(C),同时将sgRNA双链特异性延长5 bp可以提高CRISPR/Cas9系统的基因编辑效率[22]。最近,本课题组通过优化sgRNA的结构以及使用水稻内源性强启动子来驱动VQR变体的表达,成功地将CRISPR/Cas9-VQR系统的编辑效率提高到了原有系统的3~7倍[21]。
为了验证改进后的VQR变体是否可以高效编辑NGAC靶位点,本文在水稻中使用改进后的CRISPR/VQR系统,即改造后的sgRNA和Actin1启动子表达VQR来编辑PAM为NGAC的两个靶位点。在获得的57株转基因水稻中,两个靶位点附近分别有27株和44株发生移码突变。进一步分析表明,改进后的VQR系统编辑效率显著高于未改进的VQR系统。这表明改进后的VQR系统可以高效识别NGAC PAM,为今后利用改进后的VQR变体编辑NGAC靶位点提供了理论依据。
1 材料和方法
1.1 材料
水稻品种为日本晴(Oryza sativa L. ssp. japonica),所有材料均种植于浙江省杭州市富阳区中国水稻研究所实验基地,正季常规水肥管理。载体分别为SK-gRNA、pC1300-ACT1-Cas9和pC1300-ACT1-VQR,载体详细信息参见文献[21]。
1.2 基因编辑载体的构建
在正向靶序列5′端加上GGCA,合成引物g++;在反向互补靶序列的5′端加上AAAC,合成引物g--。g++和g--等量混合,100℃ 5 min,室温复性成双链DNA,并与AarⅠ (Thermo Scientific,美国)线性化的SK-gRNA载体进行连接。靶标位点序列及其所对应的引物g++和g--序列详见表1。Table 1
表1
表1 靶位点及构建基因编辑载体的引物
Table 1
| 靶位点 | 靶位点序列(5′→3′) | 引物名称 | 引物序列(5′→3′) |
|---|---|---|---|
| NAL1-Q1 | GCGTTTATCTTGGAGCTGTTGAA | NAL1-Q1-g++ | GGCAGCGTTTATCTTGGAGCTGT |
| NAL1-Q1-g-- | AAACACAGCTCCAAGATAAACGC | ||
| NAL1-Q2 | GCTATTGAGCGGACACTGCAGAT | NAL1-Q2-g++ | GGCAGCTATTGAGCGGACACTGC |
| NAL1-Q2-g-- | AAACGCAGTGTCCGCTCAATAGC | ||
| LPA1-Q | CAGCGGGAGCAGAACCTGCAGAT | LPA1-Q-g++ | GGCACAGCGGGAGCAGAACCTGC |
| LPA1-Q-g-- | AAACGCAGGTTCTGCTCCCGCTG | ||
| NAL1-C | ACCCCATATAATTCCAATTGGAC | NAL1-C-g++ | GGCAACCCCATATAATTCCAATT |
| NAL1-C-g-- | AAACAATTGGAATTATATGGGGT | ||
| GL1-C | CCGCCATGCGCCTCCCCAAGGAC | GL1-C-g++ | GGCACCGCCATGCGCCTCCCCAA |
| GL1-C-g-- | AAACTTGGGGAGGCGCATGGCGG |
新窗口打开|下载CSV
基因编辑的构建采用同尾酶连接策略:首先将5个靶位点NAL1-Q1、NAL1-Q2、LPA1-Q、GL1-C和NAL1-C序列分别组装至中间载体SK-gRNA,所得载体分别命名为SK-gRNA-NAL1-Q1、SK-gRNA- NAL1-Q2、SK-gRNA-LPA1-Q、SK-gRNA-GL1-C和SK-gRNA-NAL1-C。然后将SK-gRNA-NA L1-Q1 (KpnⅠ/SalⅠ)、SK-gRNA-NAL1-Q2 (XhoⅠ/BglⅡ)和SK-gRNA-LPA1-Q (BamHⅠ/NheⅠ)同时组装至pC1300-ACT-VQR (KpnⅠ/XbaⅠ);将SK-gRNA-GL1C (KpnⅠ/SalⅠ)和SK-gRNA-NAL1C (XhoⅠ/NheⅠ)同时组装至pC1300-ACT-VQR (KpnⅠ/XbaⅠ);将SK- gRNA-GL1C (KpnⅠ/SalⅠ)和SK-gRNA-NAL1C(XhoⅠ/NheⅠ)同时组装至pC1300-ACT-Cas9 (KpnⅠ/ XbaⅠ)。
1.3 农杆菌介导的水稻遗传转化
水稻遗传转化采用Hiei等[23]农杆菌介导法进行,所用的农杆菌菌株为EHA105。1.4 靶位点片段的扩增和测序
采用十六烷基三甲基溴化铵(hexadecyl trimethyl ammonium bromide, CTAB)方法提取水稻愈伤组织及植株基因组DNA。利用KOD FX Polymerase (TOYOBO, 日本)扩增靶标位点片段,引物序列详见表2。PCR产物通过Sanger测序,峰图使用DSD (degenerate sequence decoding)方法进行解读[24]。Table 2
表2
表2 靶位点片段扩增所用引物
Table 2
| 引物名称 | 引物序列(5′→3′) |
|---|---|
| NAL1-Q1 | F:TGTCCAGATTTGCTGTGACC |
| R:GGGCGGCATATATGTTTGG | |
| NAL1-Q2 | F:TCATATTAATGCCGGATTGC |
| R:CAAGTTCCAGACGGTCGAG | |
| LPA1-Q | F:TCCTTGCAGTTATGGCACTG |
| R:TTGAGGTGGGCCTTGTAGTC | |
| NAL1-C | F:AGCTCTGGTCACACAACTGG |
| R:CCCCAACAGCTGAGGTAACG | |
| GL1-C | F:GTCGCTGTTGCTGAGACGTA |
| R:CCAGCGAGTACTGCACAAGA |
新窗口打开|下载CSV
2 结果与分析
2.1 改进的CRISPR/VQR系统可以高效编辑NAL1和LPA1基因
前期研究发现,在NAL1和LPA1(LOOSE PLANT ARCHITECTURE 1)基因外显子上存在3个相对低效的VQR靶位点NAL1-Q1、NAL1-Q2和LPA1-Q,其PAM分别为NGAA、NGAT、NGAG[20]。在未改进的VQR系统下,这3个位点的基因编辑效率分别为0%、13.30%和2.10%[20]。为了进一步研究改进后VQR系统的编辑效率,本研究构建了同时靶向这3个位点的基因编辑载体(图1A),并利用农杆菌介导法将其转入水稻,获得41株转基因水稻。图1
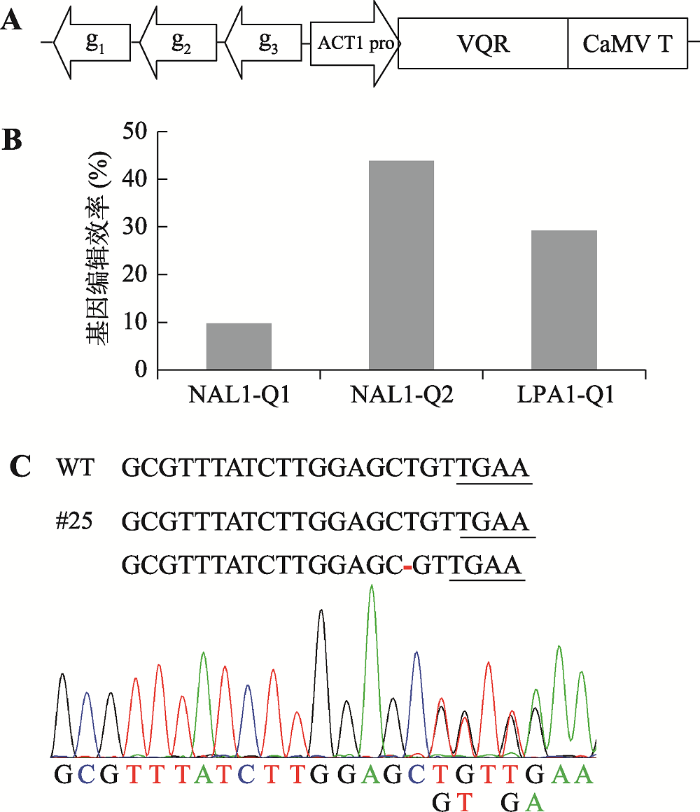 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1NAL1-Q1、NAL1-Q2和LPA1-Q位点基因编辑
A:NAL1-Q1、NAL1-Q2和LPA1-Q靶位点基因编辑载体示意图。g1、g2和g3分别表示gRNA-NAL1-Q1、gRNA-NAL1-Q2和gRNA-LPA1-Q,ACT1 pro表示Actin1启动子,CaMV T表示CaMV终止子。B:NAL1-Q1、NAL1-Q2和LPA1-Q位点基因编辑效率。C:#25转基因植株靶位点NAL1-Q1的测序结果。红色短线表示碱基缺失,下划线的序列为PAM。
Fig. 1Gene editing of NAL1-Q1, NAL1-Q2 and LPA1-Q
经Sanger测序检测,NAL1-Q1、NAL1-Q2和LPA1-Q靶位点的基因突变率分别为9.75%、43.90%和29.26% (图1B),表明改进后的VQR系统可以大幅提高这3个位点的编辑效率。值得一提的是,在原系统无法编辑的NAL1-Q1位点上,改进后的VQR系统也造成了一定水平的突变(图1C)。
2.2 改进的CRISPR/VQR系统高效识别NGAC PAM序列
在水稻中,NAL1通过影响细胞分裂调控叶片宽度,该基因的突变导致叶片变窄[25];GL1是蜡质合成中的一个基因,GL1突变破坏叶表皮蜡质形成叶片高度亲水表型[26]。为了进一步研究改进后的CRISPR/VQR系统能否对NGAC PAM靶位点进行识别和编辑,本研究选择水稻中GL1和NAL1基因的靶位点GL1-C和NAL1-C进行基因编辑(图 2A)。利用同尾酶连接策略,构建了GL1-C和NAL1-C共敲除载体(图 2B),然后通过农杆菌转染法将其转入水稻中,最终获得57株转基因水稻。靶位点PCR扩增及测序结果显示,NAL1-C和GL1-C靶位点的基因突变植株分别为27株和44株,突变率分别为47.36%和77.19%;NAL1-C和GL1-C靶位点的双基因突变植株为26株,突变率为45.61% (图2C)。此外,也检测出基因功能缺失型突变体(双等位突变及纯合突变)。为了验证改进VQR系统的编辑效果,本研究进一步分析了基因编辑水稻的表型。如图2D所示,野生型水稻叶片较宽,疏水性较强,水滴呈球形;而双基因纯合突变叶片较窄,亲水性较强,水滴扩散开。这些结果表明在改进后的系统中,VQR可以高效地识别NGAC PAM序列。图2
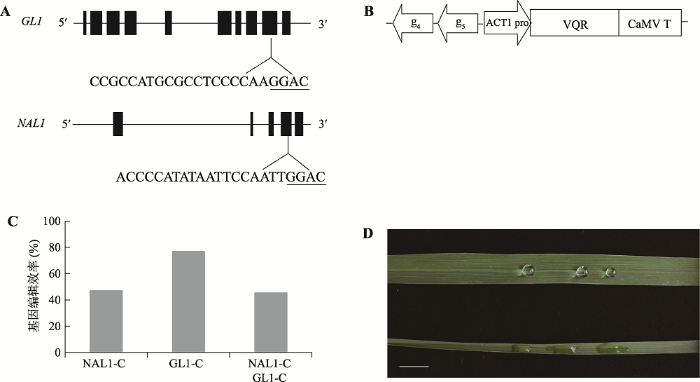 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2GL1-C和NAL1-C位点基因编辑
A:GL1和NAL1基因编辑靶位点示意图。下划线序列表示PAM。B:GL1-C和NAL1-C靶位点基因编辑载体示意图。g4和g5分别表示gRNA-GL1-C和gRNA-NAL1-C,ACT1 pro表示Actin1启动子,CaMV T表示CaMV终止子。C:NAL1-C靶位点和GL1-C靶位点及NAL1-C/GL1-C双靶位点的基因编辑效率。D:GL1和NAL1基因共敲除水稻叶片。上面为野生型,下面为基因编辑的突变体,标尺为1 cm。
Fig. 2Gene editing of GL1-C and NAL1-C
2.3 突变类型分析
本研究进一步对CRISPR/VQR系统造成的突变进行分类,发现共有4种突变类型,分别为杂合突变、双等位突变、嵌合体突变和纯合突变。其中,以杂合突变和双等位突变为主,其次是嵌合体突变和纯合突变(表3)。双等位突变多数情况意味着基因功能丧失,而转基因T1代则能产生一半的纯合突变体,在研究中具备重要的价值。Table 3
表3
表3 靶位点突变类型
Table 3
| 靶位点 | 突变率(%) | 杂合突变 | 双等位突变 | 嵌合体 | 纯合突变 | ||||
|---|---|---|---|---|---|---|---|---|---|
| 植株数 | 所占比例(%) | 植株数 | 所占比例(%) | 植株数 | 所占比例(%) | 植株数 | 所占比例(%) | ||
| NAL1-Q1 | 9.75 | 3 | 75.00 | 0 | 0 | 1 | 25.00 | 0 | 0 |
| NAL1-Q2 | 43.90 | 15 | 83.33 | 3 | 16.66 | 0 | 0 | 0 | 0 |
| LPA1-Q | 29.26 | 6 | 50.00 | 5 | 41.66 | 1 | 8.33 | 0 | 0 |
| NAL1-C | 47.36 | 19 | 70.37 | 6 | 22.22 | 1 | 3.70 | 1 | 3.70 |
| GL1-C | 77.19 | 19 | 43.18 | 12 | 27.27 | 11 | 25.00 | 2 | 4.54 |
新窗口打开|下载CSV
2.4 SpCas9低效识别NGAC PAM
前人研究表明SpCas9可以切割NGA PAM类型的靶位点[18],但切割效率尚不清楚。为此,本研究检测了SpCas9对NAL1-C和GL1-C靶位点的切割能力。结果表明,NAL1-C和GL1-C靶位点在SpCas9切割下,转基因愈伤组织的基因突变率分别为10.42%和12.50% (表4)。与转基因植株测序结果相比,通常愈伤组织中敲除效率要略高一些。然而,SpCas9在NAL1-C和GL1-C位点的表现却远逊于VQR在转基因苗中的结果(图2C),这表明VQR对NGAC PAM的编辑能力要高于SpCas9。Table 4
表4
表4 SpCas9对NAL1-C和GL1-C靶位点的编辑效率
Table 4
| 靶点名称 | 检测愈伤组织总数 | 突变愈伤组织个数 | 突变率(%) |
|---|---|---|---|
| NAL1-C | 48 | 5 | 10.42 |
| GL1-C | 48 | 6 | 12.50 |
新窗口打开|下载CSV
3 讨 论
目前,广泛应用的CRISPR/Cas9基因编辑系统仅能识别NGG PAM,极大地限制了其靶位点的选择范围。SpCas9的变体VQR能够高效识别NGAA、NGAT和NGAG3种PAM,对于CRISPR/Cas9系统是一个强大补充。本研究表明,在水稻中利用改进后的CRISPR/VQR也可以高效编辑NGAC PAM,并产生丰富的突变类型,从而为水稻NGAC PAM位点的编辑提供了理论依据。在人类细胞中,基于增强型绿色荧光蛋白(enhanced green fluorescent protein, EGFP)报告基因破坏实验的结果表明,VQR对包含NGAN PAM位点的切割效率为:NGAG>NGAT=NGAA>NGAC,这表明VQR对包含NGAC PAM的位点有切割能力[18]。在水稻中,由于NGAC的位点尚未被验证,22%的NGA位点是否能够被VQR编辑仍然不清楚[20,21]。在原始的CRISPR/VQR系统中,VQR无法切割NGAC PAM位点[20]。在改进的CRISPR/VQR系统中,sgRNA特异性延长5 bp,增强了sgRNA的稳定性;将多聚胸腺嘧啶(poly T)的第4个碱基T突变为C,消除了转录终止信号,从而使sgRNA转录水平得到提高;强内源启动子表达VQR使其蛋白表达水平得到提高,因此基因编辑效率得到显著提高[22]。本研究利用改进后的CRISPR/VQR系统也高效地编辑了水稻中NAL1-Q1、NAL1-Q2和LPA1-Q等3个位点(图1),而原始CRISPR/VQR系统对这3个位点的编辑效率极低甚至无法编辑。在此基础上,本研究进一步验证了改进后的CRISPR/VQR系统对NGAC PAM识别和编辑的情况。结果表明,改进后的CRISPR/VQR系统可以高效识别并编辑NAL1和GL1基因的NGAC PAM (图2),从而为水稻中含有NGAC PAM位点基因的编辑提供了理论依据。此外,我们推测原始的CRISPR/VQR系统也应该能够识别NGAC PAM位点,但由于编辑效率极低,在转基因株系较少的情况可能无法获得基因编辑植株。对于相同靶位点NAL1-C和GL1-C,SpCas9的基因编辑效率远低于VQR (图2,表4),因此VQR在实际应用中仍具有不可被SpCas9替代的优势。从突变类型来看,VQR主要产生杂合突变和双等位突变(表3)。其中双等位突变与纯合体一样,同为基因功能缺失型突变,并且其自交一代理论上可以产生出约1/2的纯合体,在育种或基因研究方面均具备巨大应用价值。
CRISPR/Cas9基因编辑系统的脱靶问题会引起基因组非靶向位点的突变,导致研究结果的不确定性。本课题组前期的研究表明,在利用改进后的CRISPR/VQR编辑NGAG和NGAT PAM位点时也存在一定的脱靶效率[21]。由于脱靶效率主要与错配碱基距离PAM位点的远近关,因此在利用改进后的CRISPR/VQR系统编辑NGAC位点时也存在类似的脱靶问题。
综上所述,本文利用改进的CRISPR/VQR基因编辑系统成功地编辑了水稻NGAC PAM序列,并产生丰富的突变型,为水稻及其他植物含有NGAC PAM基因的编辑提供了借鉴。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
.
URL [本文引用: 1]

基因组编辑技术已经在多个模式植物、动物以及其他生物中得到成功应用。基因组编辑是利用序列特异核酸酶(Sequence-specific nucleases,SSNs)在基因组特定位点产生DNA双链断裂(Double-strand breaks,DSBs),从而激活细胞自身修复机制——非同源末端连接(Non-homologous end joining,NHEJ)或同源重组(Homologous recombination,HR),实现基因敲除、染色体重组以及基因定点插入或替换等。锌指核酸酶(Zinc finger nuclease,ZFN)、TALEN(Transcription activator-like effector nuclease)和CRISPR/Cas9(Clustered regularly interspaced short palindromic repeats/CRISPR-associated 9)系统是最主要的3类SSNs。ZFN和TALEN是利用蛋白与DNA结合方式靶向特定的基因组位点,而最新的CIRISPR/Cas9系统则是利用更简单的核苷酸互补配对方式结合在基因组靶位点,其构建简单、效率更高效,因而促进了基因组编辑在植物中的广泛应用。利用基因组编辑技术除了实现植物基因定点突变外,还可以将SSNs的DNA结合域与其他功能蛋白融合,实现基因的靶向激活、抑制和表观调控等衍生技术。本文从基因组编辑技术的原理与优势、SSNs组成及构建方法、基因组编辑及衍生技术在植物中应用、优化SSNs突变效率和减少脱靶突变方法等方面进行了系统介绍,并对未来需要迫切解决的一些问题进行了分析和展望。
URL [本文引用: 1]
基因组编辑技术已经在多个模式植物、动物以及其他生物中得到成功应用。基因组编辑是利用序列特异核酸酶(Sequence-specific nucleases,SSNs)在基因组特定位点产生DNA双链断裂(Double-strand breaks,DSBs),从而激活细胞自身修复机制——非同源末端连接(Non-homologous end joining,NHEJ)或同源重组(Homologous recombination,HR),实现基因敲除、染色体重组以及基因定点插入或替换等。锌指核酸酶(Zinc finger nuclease,ZFN)、TALEN(Transcription activator-like effector nuclease)和CRISPR/Cas9(Clustered regularly interspaced short palindromic repeats/CRISPR-associated 9)系统是最主要的3类SSNs。ZFN和TALEN是利用蛋白与DNA结合方式靶向特定的基因组位点,而最新的CIRISPR/Cas9系统则是利用更简单的核苷酸互补配对方式结合在基因组靶位点,其构建简单、效率更高效,因而促进了基因组编辑在植物中的广泛应用。利用基因组编辑技术除了实现植物基因定点突变外,还可以将SSNs的DNA结合域与其他功能蛋白融合,实现基因的靶向激活、抑制和表观调控等衍生技术。本文从基因组编辑技术的原理与优势、SSNs组成及构建方法、基因组编辑及衍生技术在植物中应用、优化SSNs突变效率和减少脱靶突变方法等方面进行了系统介绍,并对未来需要迫切解决的一些问题进行了分析和展望。
.
URLPMID:8577732 [本文引用: 1]
A long-term goal in the field of restriction-modification enzymes has been to generate restriction endonucleases with novel sequence specificities by mutating or engineering existing enzymes. This will avoid the increasingly arduous task of extensive screening of bacteria and other microorganisms for new enzymes. Here, we report the deliberate creation of novel site-specific endonucleases by linking two different zinc finger proteins to the cleavage domain of Fok I endonuclease. Both fusion proteins are active and under optimal conditions cleave DNA in a sequence-specific manner. Thus, the modular structure of Fok I endonuclease and the zinc finger motifs makes it possible to create ``artificial'' nucleases that will cut DNA near a predetermined site. This opens the way to generate many new enzymes with tailor-made sequence specificities desirable for various applications.
.
URLPMID:19933107 [本文引用: 1]
TAL effectors are important virulence factors of bacterial plant pathogenic Xanthomonas, which infect a wide variety of plants including valuable crops like pepper, rice, and citrus. TAL proteins are translocated via the bacterial type III secretion system into host cells and induce transcription of plant genes by binding to target gene promoters. Members of the TAL effector family differ mainly in their central domain of tandemly arranged repeats of typically 34 amino acids each with hypervariable di-amino acids at positions 12 and 13. We recently showed that target DNA-recognition specificity of TAL effectors is encoded in a modular and clearly predictable mode. The repeats of TAL effectors feature a surprising one repeat-to-one-bp correlation with different repeat types exhibiting a different DNA base pair specificity. Accordingly, we predicted DNA specificities of TAL effectors and generated artificial TAL proteins with novel DNA recognition specificities. We describe here novel artificial TALs and discuss implications for the DNA recognition specificity. The unique TAL-DNA binding domain allows design of proteins with potentially any given DNA recognition specificity enabling many uses for biotechnology.
.
URLPMID:24906146 [本文引用: 2]
Derived from a microbial defense system, Cas9 can be guided to specific locations within complex genomes by a short RNA. The development, applications, and future directions of the CRISPR-Cas9 system for genome engineering are discussed here.
URLPMID:25430965 [本文引用: 1]
The article focuses on the use of site-specific nucleases such as Transcription Activator-Like Effector Nucleases (TALENs) and Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) for carrying out genome modification in non-human primates (NHPs). It states that p53 tumor suppressor gene, injected via a zygote injection using a CRISPR system, was used to produce biallelic mutant monkeys. It states that addition of desired template DNA to the CRISPR system can modify nucleotide.
.
URL [本文引用: 1]
.
URLPMID:23451779 [本文引用: 1]
Abstract Recent advances in genome engineering provide newfound control over a plant's genetic material. It is now possible for most bench scientists to alter DNA in living plant cells in a variety of ways, including introducing specific nucleotide substitutions in a gene that change a protein's amino acid sequence, deleting genes or chromosomal segments, and inserting foreign DNA at precise genomic locations. Such targeted DNA sequence modifications are enabled by sequence-specific nucleases that create double-strand breaks in the genomic loci to be altered. The repair of the breaks, through either homologous recombination or nonhomologous end joining, can be controlled to achieve the desired sequence modification. Genome engineering promises to advance basic plant research by linking DNA sequences to biological function. Further, genome engineering will enable plants' biosynthetic capacity to be harnessed to produce the many agricultural products required by an expanding world population.
.
URLPMID:25038773 [本文引用: 1]
Sequence-specific nucleases have been applied to engineer targeted modifications in polyploid genomes, but simultaneous modification of multiple homoeoalleles has not been reported. Here we use transcription activator-like effector nuclease (TALEN) and clustered, regularly interspaced, short palindromic repeats (CRISPR)-Cas9 (refs. 4,5) technologies in hexaploid bread wheat to introduce targeted mutations in the three homoeoalleles that encode MILDEW-RESISTANCE LOCUS (MLO) proteins. Genetic redundancy has prevented evaluation of whether mutation of all three MLO alleles in bread wheat might confer resistance to powdery mildew, a trait not found in natural populations. We show that TALEN-induced mutation of all three TaMLO homoeologs in the same plant confers heritable broad-spectrum resistance to powdery mildew. We further use CRISPR-Cas9 technology to generate transgenic wheat plants that carry mutations in the TaMLO-A1 allele. We also demonstrate the feasibility of engineering targeted DNA insertion in bread wheat through nonhomologous end joining of the double-strand breaks caused by TALENs. Our findings provide a methodological framework to improve polyploid crops.
.
URLPMID:28349304 [本文引用: 1]
The clustered regularly interspaced short palindromic repeats (CRISPR)-associated endonuclease 9 (CRISPR/Cas9) system has emerged as a promising technology for specific genome editing in many species. Here we constructed one vector targeting eight agronomic genes in rice using the CRISPR/Cas9 multiplex genome editing system. By subsequent genetic transformation and DNA sequencing, we found that the eight target genes have high mutation efficiencies in the T 0 generation. Both heterozygous and homozygous mutations of all editing genes were obtained in T 0 plants. In addition, homozygous sextuple, septuple, and octuple mutants were identified. As the abundant genotypes in T 0 transgenic plants, various phenotypes related to the editing genes were observed. The findings demonstrate the potential of the CRISPR/Cas9 system for rapid introduction of genetic diversity during crop breeding.
URL [本文引用: 1]
基因组编辑技术的飞速发展,尤其是近年来CRISPR/Cas9基因组编辑体系的出现,使得研究人员能高效地在细胞系和动物模型中对基因组进行精确编辑。基于基因组编辑技术的各种实验研究平台被相继开发,包括通过在细胞系中引入疾病相关突变位点建立疾病模型,通过高通量筛选寻找导致肿瘤耐药性的突变基因,通过体内原位靶向致病基因并修改突变进行基因治疗等。这些基因组编辑技术研究平台极大推动了精准医学研究领域的发展。本文对基因组编辑技术在精准医学领域的基础研究、转化应用、目前存在的问题以及未来发展的方向进行了讨论。
URL [本文引用: 1]
基因组编辑技术的飞速发展,尤其是近年来CRISPR/Cas9基因组编辑体系的出现,使得研究人员能高效地在细胞系和动物模型中对基因组进行精确编辑。基于基因组编辑技术的各种实验研究平台被相继开发,包括通过在细胞系中引入疾病相关突变位点建立疾病模型,通过高通量筛选寻找导致肿瘤耐药性的突变基因,通过体内原位靶向致病基因并修改突变进行基因治疗等。这些基因组编辑技术研究平台极大推动了精准医学研究领域的发展。本文对基因组编辑技术在精准医学领域的基础研究、转化应用、目前存在的问题以及未来发展的方向进行了讨论。
[本文引用: 1]
[本文引用: 1]
.
URLPMID:23929338 [本文引用: 1]
The article offers information on genome modification of crop plants using a CRISPR-Cas system. It states that genome editing technologies using zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) can also generate genome modifications. Photographs related to genome editing in rice and wheat using an engineered type II CRISPR-Cas system are also presented.
.
URL [本文引用: 1]
.
URLPMID:28375731 [本文引用: 1]
Abstract Many bacterial clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated (Cas) systems employ the dual RNA-guided DNA endonuclease Cas9 to defend against invading phages and conjugative plasmids by introducing site-specific double-stranded breaks in target DNA. Target recognition strictly requires the presence of a short protospacer adjacent motif (PAM) flanking the target site, and subsequent R-loop formation and strand scission are driven by complementary base pairing between the guide RNA and target DNA, Cas9-DNA interactions, and associated conformational changes. The use of CRISPR-Cas9 as an RNA-programmable DNA targeting and editing platform is simplified by a synthetic single-guide RNA (sgRNA) mimicking the natural dual trans-activating CRISPR RNA (tracrRNA)-CRISPR RNA (crRNA) structure. This review aims to provide an in-depth mechanistic and structural understanding of Cas9-mediated RNA-guided DNA targeting and cleavage. Molecular insights from biochemical and structural studies provide a framework for rational engineering aimed at altering catalytic function, guide RNA specificity, and PAM requirements and reducing off-target activity for the development of Cas9-based therapies against genetic diseases.
URLPMID:2238196 [本文引用: 1]
Clustered regularly interspaced short palindromic repeats (CRISPR) are hypervariable loci widely distributed in prokaryotes that provide acquired immunity against foreign genetic elements. Here, we characterize a novel Streptococcus thermophilus locus, CRISPR3, and experimentally demonstrate its ability to integrate novel spacers in response to bacteriophage. Also, we analyze CRISPR diversity and activity across three distinct CRISPR loci in several S. thermophilus strains. We show that both CRISPR repeats and cas genes are locus specific and functionally coupled. A total of 124 strains were studied, and 109 unique spacer arrangements were observed across the three CRISPR loci. Overall, 3,626 spacers were analyzed, including 2,829 for CRISPR1 (782 unique), 173 for CRISPR2 (16 unique), and 624 for CRISPR3 (154 unique). Sequence analysis of the spacers revealed homology and identity to phage sequences (77%), plasmid sequences (16%), and S. thermophilus chromosomal sequences (7%). Polymorphisms were observed for the CRISPR repeats, CRISPR spacers, cas genes, CRISPR motif, locus architecture, and specific sequence content. Interestingly, CRISPR loci evolved both via polarized addition of novel spacers after exposure to foreign genetic elements and via internal deletion of spacers. We hypothesize that the level of diversity is correlated with relative CRISPR activity and propose that the activity is highest for CRISPR1, followed by CRISPR3, while CRISPR2 may be degenerate. Globally, the dynamic nature of CRISPR loci might prove valuable for typing and comparative analyses of strains and microbial populations. Also, CRISPRs provide critical insights into the relationships between prokaryotes and their environments, notably the coevolution of host and viral genomes.
URLPMID:22382282 [本文引用: 1]
Abstract Clustered regularly interspaced short palindromic repeats (CRISPR) and their associated genes are linked to a mechanism of acquired resistance against bacteriophages. Bacteria can integrate short stretches of phage-derived sequences (spacers) within CRISPR loci to become phage resistant. In this study, we further characterized the efficiency of CRISPR1 as a phage resistance mechanism in Streptococcus thermophilus. First, we show that CRISPR1 is distinct from previously known phage defense systems and is effective against the two main groups of S. thermophilus phages. Analyses of 30 bacteriophage-insensitive mutants of S. thermophilus indicate that the addition of one new spacer in CRISPR1 is the most frequent outcome of a phage challenge and that the iterative addition of spacers increases the overall phage resistance of the host. The added new spacers have a size of between 29 to 31 nucleotides, with 30 being by far the most frequent. Comparative analysis of 39 newly acquired spacers with the complete genomic sequences of the wild-type phages 2972, 858, and DT1 demonstrated that the newly added spacer must be identical to a region (named proto-spacer) in the phage genome to confer a phage resistance phenotype. Moreover, we found a CRISPR1-specific sequence (NNAGAAW) located downstream of the proto-spacer region that is important for the phage resistance phenotype. Finally, we show through the analyses of 20 mutant phages that virulent phages are rapidly evolving through single nucleotide mutations as well as deletions, in response to CRISPR1.
URLPMID:3694421 [本文引用: 1]
61Unlike previously described CRISPRs, each Neisseria repeat carries its own promoter61Pre-crRNA processing is dispensable for CRISPR interference in Neisseria spp.61CRISPR interference blocks natural transformation in the pathogen N.02meningitidis61Neisseria CRISPR/Cas systems are the most streamlined observed to date
.
URLPMID:4540238 [本文引用: 3]
Although CRISPR-Cas9 nucleases are widely used for genome editing1,2, the range of sequences that Cas9 can recognize is constrained by the need for a specific protospacer adjacent motif (PAM)3 6. As a result, it can often be difficult to target double-stranded breaks (DSBs) with the precision that is necessary for various genome editing applications. The ability to engineer Cas9 derivatives with purposefully altered PAM specificities would address this limitation. Here we show that the commonly usedStreptococcus pyogenesCas9 (SpCas9) can be modified to recognize alternative PAM sequences using structural information, bacterial selection-based directed evolution, and combinatorial design. These altered PAM specificity variants enable robust editing of endogenous gene sites in zebrafish and human cells not currently targetable by wild-type SpCas9, and their genome-wide specificities are comparable to wild-type SpCas9 as judged by GUIDE-Seq analysis7. In addition, we identified and characterized another SpCas9 variant that exhibits improved specificity in human cells, possessing better discrimination against off-target sites with non-canonical NAG and NGA PAMs and/or mismatched spacers. We also found that two smaller-size Cas9 orthologues,Streptococcus thermophilusCas9 (St1Cas9) andStaphylococcus aureusCas9 (SaCas9), function efficiently in the bacterial selection systems and in human cells, suggesting that our engineering strategies could be extended to Cas9s from other species. Our findings provide broadly useful SpCas9 variants and, more importantly, establish the feasibility of engineering a wide range of Cas9s with altered and improved PAM specificities.
.
URLPMID:29512652 [本文引用: 1]
Programmable DNA nucleases have provided scientists with the unprecedented ability to probe, regulate, and manipulate the human genome. Zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeat-Cas9 system (CRISPR-Cas9) represent a powerful array of tools that can bind to and cleave a specified DNA... [Show full abstract]
.
URLPMID:26995294 [本文引用: 9]
.
URLPMID:28605576 [本文引用: 5]
Abstract Clustered regularly interspaced short palindromic repeats-associated protein 9 (CRISPR-Cas9) is a revolutionary technology that enables efficient genomic modification in many organisms. Currently, the wide use of Streptococcus pyogenes Cas9 (SpCas9) primarily recognises sites harbouring a canonical NGG protospacer adjacent motif (PAM). The newly developed VQR (D1135V/R1335Q/T1337R) variant of Cas9 has been shown to cleave sites containing NGA PAM in rice, which greatly expanded the range of genome editing. However, the low editing efficiency of the VQR variant remains, which limits its wide application in genome editing. In this study, by modifying the single guide RNA (sgRNA) structure and using strong endogenous promoters, we significantly increased the editing efficiency of the VQR variant. The modified CRISPR-Cas9-VQR system provides a robust toolbox for multiplex genome editing at sites containing non-canonical NGA PAM. This article is protected by copyright. All rights reserved. This article is protected by copyright. All rights reserved.
.
URLPMID:4699467 [本文引用: 2]
Single-guide RNA (sgRNA) is one of the two key components of the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 genome-editing system. The current commonly used sgRNA structure has a shortened duplex compared with the native bacterial CRISPR RNA (crRNA) ransactivating crRNA (tracrRNA) duplex and contains a continuous sequence of thymines, which is the pause signal for RNA polymerase III and thus could potentially reduce transcription efficiency. Here, we systematically investigate the effect of these two elements on knockout efficiency and showed that modifying the sgRNA structure by extending the duplex length and mutating the fourth thymine of the continuous sequence of thymines to cytosine or guanine significantly, and sometimes dramatically, improves knockout efficiency in cells. In addition, the optimized sgRNA structure also significantly increases the efficiency of more challenging genome-editing procedures, such as gene deletion, which is important for inducing a loss of function in non-coding genes. By a systematic investigation of sgRNA structure we find that extending the duplex by approximately 5 bp combined with mutating the continuous sequence of thymines at position 4 to cytosine or guanine significantly increases gene knockout efficiency in CRISPR-Cas9-based genome editing experiments. The online version of this article (doi:10.1186/s13059-015-0846-3) contains supplementary material, which is available to authorized users.
URLPMID:7920717 [本文引用: 1]
A large number of morphologically normal, fertile, transgenic rice plants were obtained by co-cultivation of rice tissues with Agrobacterium tumefaciens . The efficiency of transformation was similar to that obtained by the methods used routinely for transformation of dicotyledons with the bacterium. Stable integration, expression and inheritance of transgenes were demonstrated by molecular and genetic analysis of transformants in the R 0 , R 1 and R 2 generations. Sequence analysis revealed that the boundaries of the T-DNA in transgenic rice plants were essentially identical to those in transgenic dicotyledons. Calli induced from scutella were very good starting materials. A strain of A. tumefaciens that carried a so-called 'super-binary' vector gave especially high frequencies of transformation of various cultivars of japonica rice that included Koshihikari, which normally shows poor responses in tissue culture.
.
URLPMID:25747846 [本文引用: 1]
Dear Editor,The recent development of sequence-specific nuclease systems,i.e.,TALENs and CRISPR/Cas9,has made genomic targeting easier in many organisms including plants (Li et al.,2012;Cong et al.,2013;Joung and Sander,2013;Li,et al.,2013;Shan et al.,2013;Liang et al.,2014;Zhang et al.,2014).Mutations induced by CRISPR/Cas9 usually occur around the cleavage sites at three bases upstream of the protospacer-adjacent motif (PAM),producing insertion and deletion of nucleotides.For diploid organisms,such targeted mutations may happen in one or both homologous chromosomes.
URLPMID:25658704 [本文引用: 1]
Leaf morphology is closely associated with cell division. In rice, mutations in Narrow leaf 1 (NAL1) show narrow leaf phenotypes. Previous studies have shown that NAL1 plays a role in regulating vein patterning and increasing grain yield in indica cultivars, but its role in leaf growth and development remains unknown. In this report, we characterized two allelic mutants of NARROW LEAF1 (NAL1), nal1-2 and nal1-3, both of which showed a 50% reduction in leaf width and length, as well as a dwarf culm. Longitudinal and transverse histological analyses of leaves and internodes revealed that cell division was suppressed in the anticlinal orientation but enhanced in the periclinal orientation in the mutants, while cell size remained unaltered. In addition to defects in cell proliferation, the mutants showed abnormal midrib in leaves. Map-based cloning revealed that nal1-2 is a null allelic mutant of NAL1 since both the whole promoter and a 404-bp fragment in the first exon of NAL1 were deleted, and that a 6-bp fragment was deleted in the mutant nal1-3. We demonstrated that NAL1 functions in the regulation of cell division as early as during leaf primordia initiation. The altered transcript level of G1- and S-phase-specific genes suggested that NAL1 affects cell cycle regulation. Heterogeneous expression of NAL1 in fission yeast (Schizosaccharomyces pombe) further supported that NAL1 affects cell division. These results suggest that NAL1 controls leaf width and plant height through its effects on cell division.
URLPMID:21511810 [本文引用: 1]
Cuticular wax forms a hydrophobic barrier on aerial plant organs; it plays an important role in protecting a plant from damage caused by many forms of environmental stress. In the present study, we characterized a rice leaf wax-deficient mutant osgl1-1 derived from a spontaneous mutation, which exhibited a wax-deficient and highly hydrophilic leaf phenotype. We cloned the OsGL1-1 gene by the map-based cloning method and performed a complementation test to confirm the function of the candidate gene. Molecular studies revealed that OsGL1-1 was a member of the OsGL1 family, and contained regions that were homologous to some regions in sterol desaturases and short-chain dehydrogenases/reductases. Compared to the wild-type, the osgl1-1 mutant showed decreased cuticular wax deposition, thinner cuticular membrane, decreased chlorophyll leaching, increased rate of water loss, and enhanced sensitivity to drought. OsGL1-1 is expressed ubiquitously in rice. The transient expression of OsGL1-1 reen fluorescent protein fusion protein indicated that OsGL1-1 is localized in the cytoplasm, plasma membrane, and nucleus.

{kind=link}
{kind=link}
{kind=link}
{kind=link}