 ,, 杨勇, 张灵敏, 戴宪华,中山大学电子与信息工程学院,广州 510006
,, 杨勇, 张灵敏, 戴宪华,中山大学电子与信息工程学院,广州 510006Application of machine learning in the CRISPR/Cas9 system
Guishan Zhang,, Yong Yang, Lingmin Zhang, Xianhua Dai,School of Electronics and Information Technology, Sun Yat-sen University, Guangzhou 510006, China通讯作者:
编委: 谷峰
收稿日期:2018-05-15修回日期:2018-07-19网络出版日期:2018-09-20
| 基金资助: |
Editorial board:
Received:2018-05-15Revised:2018-07-19Online:2018-09-20
| Fund supported: |
作者简介 About authors
张桂珊,博士研究生,研究方向:生物信息学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (653KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
张桂珊, 杨勇, 张灵敏, 戴宪华. 机器学习方法在CRISPR/Cas9系统中的应用[J]. 遗传, 2018, 40(9): 704-723 doi:10.16288/j.yczz.18-135
Guishan Zhang, Yong Yang, Lingmin Zhang, Xianhua Dai.
CRISPR/Cas9 (clustered regularly interspaced short palindromic repeat/CRISPR-associated protein 9)系统介导的基因组编辑技术[1,2,3]是继锌指核酸酶(zinc-finger nucleases, ZFNs)、类转录激活因子效应物核酸酶(transcription activator-like effector nuclease, TALENs)后出现的第三代“基因组定点编辑技术”,可对特定位置上的DNA序列进行编辑与修改。近年来,CRISPR/Cas9技术凭借成本低廉、容易操作等优点迅速成为基因工程领域的关注热点。目前,CRISPR/Cas9技术主要应用于基因敲除、基因敲入、DNA大片段删除、转录调控、基因检测、基因标记。但是,该技术仍存在许多科学问题有待研究。比如,CRISPR/Cas9是一种单链酶,其自身具有不稳定性,容易引发突变导致脱靶效应[4, 5]。而且,不同靶点Cas9内切酶切割效率存在明显的差异[6,7,8,9,10],靶点位于核小体核心区域,Cas9的活性受到抑制;靶点位于DNA邻接区,核小体结构不影响Cas9的活性[7]。此外,CRISPR/Cas9可能在靶点远处删除大规模DNA[11]。因此,克服脱靶效应和提高基因组编辑效率成为研究人员亟待解决的问题。
机器学习(machine learning, ML)是人工智能(artificial intelligence, AI)的核心,主要研究如何通过计算的方法,利用经验改善系统的性能[12]。机器学习能有效地分析经验数据,为生物信息学提供重要的技术支撑。基于机器学习预测CRISPR/Cas9脱靶效应的主要思路,是利用先验知识(priori knowledge)学习引起脱靶效应的sgRNA与目标基因组序列碱基错配数目的统计学规律,进而评估sgRNA的脱靶效应[13]。此外,机器学习在CRISPR/Cas9系统优化设计高效sgRNA序列[14,15,16]、预测sgRNA活性[17]、设计CRISPR干扰(CRISPR interference, CRISPRi)/ CRISPR激活(CRISPR activation, CRISPRa)高效率的sgRNA[8]、设计全基因组CRISPR/Cas9基因敲除文库[18]、预测CRISPR重复序列的方向[19]、鉴定必需基因[18, 20~24]等方面有着日渐广泛的应用。本文在阐述CRISPR/Cas9系统作用机制的基础上,对现阶段CRISPR/Cas9研究领域所存在的科学问题进行讨论与分析,然后对机器学习应用于CRISPR/Cas9系统设计高效sgRNA、脱靶位点预测、打靶活性评估、基因敲除、高通量功能基因筛选等展开综述,以期为相关领域的研究提供参考。
1 CRISPR/Cas9系统的组成及其作用机制
CRISPR序列最早发现于细菌和古细菌中,几乎所有的古细菌和40%的细菌都具有此类序列[25, 26]。CRISPR序列中含有大量的重复序列和间隔序列(protospacer),其间隔序列长度大致相同,并具有特异性[27]。CRISPR/Cas9是细菌和古细菌在长期演化过程中形成的一种适应性免疫防御系统,用于对抗入侵的病毒及外源DNA。CRISPR/Cas9系统通过将入侵噬菌体和质粒DNA片段整合到CRISPR中,指导相应的CRISPR RNA (crRNA)降解同源序列,同时该系统具有免疫记忆能力[28, 29]。CRISPR/Cas9系统由CRISPR序列元件和Cas9核酸酶组成。Cas9核酸酶在crRNA (CRISPR RNA)和反式激活crRNA (trans-activating crRNA, tracrRNA)的指导下,在具有前间区序列邻近基序(protospacer adjacent motif, PAM)的DNA双链靶点(PAM上游3个碱基处)进行靶向双链切割,形成钝末端DNA双链断裂(double strand breaks, DSBs)[25, 30, 31]。其作用过程大致如图1所示:首先,Cas9蛋白复合物与crRNA和tracrRNA组成功能复合物,在crRNA引导下靶向识别并结合具有PAM位点相匹配的DNA靶序列。然后,Cas9依靠其自身的两个核酸内切酶结构域发挥内切酶的作用,在PAM上游约3个碱基位点处进行切割。在切割过程中,Cas9核酸酶的HNH (His-Asn-His)结构域切割模板链,RuvC (核糖核酸酶H/整合酶超家族的成员)结构域切割非模板链,进而导致目标DNA双链断裂[32]。最后,引发细胞启动自动修复机制。通过非同源末端连接(nonhomologous end-joining, NHEJ),细胞引入插入/缺失(indel)碱基引起靶点位置基因的突变;通过同源重组修复(homology-directed repair, HDR),细胞利用外源DNA提供的“供体模板”与突变靶点重组,实现对基因组的DNA定点编辑[33,34,35]。
图1
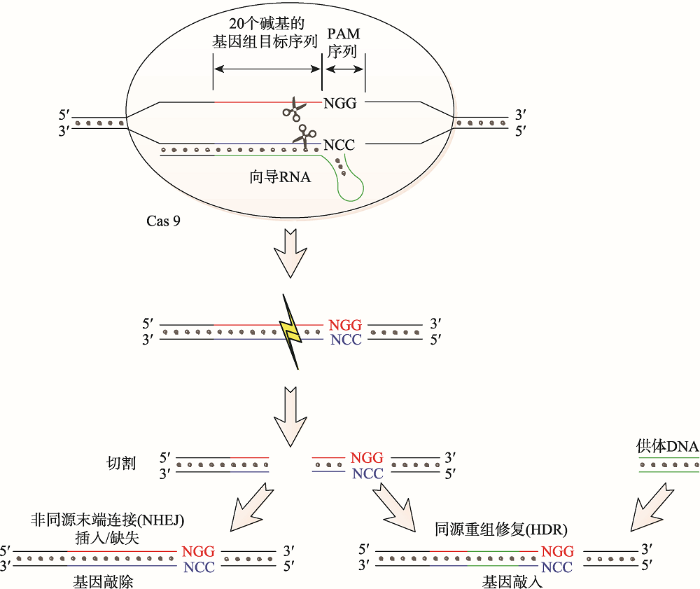 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1CRISPR/Cas9对靶基因的切割示意图
Fig. 1Schema of CRISPR/Cas9-mediated cleavage of target genes
2 CRISPR/Cas9基因组编辑效率与特异性
2.1 PAM序列对CRISPR/Cas9系统基因组编辑效率与特异性的影响
CRISPR/Cas9切割位点的编辑效率与特异性依赖于PAM序列[36]。在CRISPR/Cas9系统中,PAM序列是位于靶点DNA链3'端的3个碱基(通常为NGG,N = A,C,G或T),PAM在该系统中发挥着关键作用。研究表明,PAM序列有助于Cas9结合与切割目标DNA序列[37],Cas9利用PAM快速识别潜在的靶点。若目标DNA序列3°端不存在PAM,即使目标序列与sgRNA序列完全匹配,Cas9也无法切割该序列靶点[37]。CRISPR/Cas9系统“经典PAM序列”(canonical PAM)为NGG,“非经典PAM序列”(non-canonical PAM),如NAG、NCG和NGA也能够激发Cas9的活性[17, 38~40]。PAM类型影响CRISPR/ Cas9的目标DNA序列编辑效率[38]。Zhang等[38]发现,PAM影响的基因组编辑效率大小依次为NGG > NGA > NAG,Doench等[22]发现NGG (97%) > NAG (26%) > NCG (11%) > NGA (7%)。CRISPR/Cas9系统近PAM端1~12位碱基的序列称为“种子序列”(seed sequence)[5, 36, 39, 41],近PAM端1~5位碱基序列是更精确的种子区域,该子区域被称为“核心区域”(core region)[42, 43]。与远PAM端碱基相比,近PAM端的碱基对CRISPR/Cas9的特异性影响更大。Jinek等[36]发现Cas9对种子序列区域sgRNA与目的DNA碱基错配的耐受能力较差,而对非核心区的碱基错配具有较好耐受能力。Hsu等[39]研究发现,在近PAM端 (PAM序列5°端第1至10~12位碱基),sgRNA与目标DNA序列碱基配对数目影响Cas9的特异性。近PAM端1~12位碱基区域,sgRNA与目标DNA序列碱基错配导致靶点切割效率降低(消失),远PAM端序列错配也可能剪切该位点[1]。Zhang等[39]发现,紧邻PAM上游8~14个碱基序列是影响CRISPR/Cas9特异性的关键因素。近PAM端两个碱基或3个碱基突变,无论是相邻的还是分散的,对CRISPR/Cas9特异性影响较大。而且,错配碱基数目大于3时,CRISPR/Cas9切割基本消失。Mali等[7]发现,在非种子序列区域,Cas9 可以容忍sgRNA与目标DNA序列存在1~3个碱基错配;而在种子序列区域,错配两个碱基导致Cas9活性降低。此外,Cas9对sgRNA与目标DNA序列错配碱基数目的耐受能力与反应条件有关。当sgRNA和Cas9浓度较高时,sgRNA与目的DNA错配碱基数目不能大于5个碱基[4]。PAM如何影响该系统基因组编辑效率的作用机制将有待进一步研究。
2.2 sgRNA对CRISPR/Cas9系统基因组编辑效率与特异性的影响
CRISPR/Cas9系统靶点识别特异性主要依赖sgRNA,因此,设计与选择sgRNA是CRISPR/Cas9技术成功的关键。sgRNA的编辑效率与特异性受诸多因素的影响:(1) sgRNA上20个碱基的向导序列(guide RNA, gRNA)对Cas9靶向性和特异性有着重要的作用[22, 24],近PAM端gRNA与目的DNA碱基配对数目决定Cas9的特异性。(2)考虑微同源特征(microhomology feature)能够提高sgRNA的活性[44]。Doench等[45]分析了影响sgRNA效率活性的序列特征,发现在第20号位置的sgRNA偏好C而排斥G,在sgRNA的中间区域,存在A的sgRNA活性较高。此外,富含G且仅含少数A的sgRNA稳定性和活性较高[46]。然而,过高或过低的GC含量均导致sgRNA的编辑活性降低[45, 47]。(3) sgRNA序列长度影响CRISPR/Cas9的剪切特异性。适当改变sgRNA的长度,在识别序列前增加两个G[48]或截断5°端2~3个碱基[48, 49],能够减少脱靶效应[22, 39]。(4)优化sgRNA的结构可以提高CRISPR/Cas9的编辑效率。利用double-nikase策略,突变Cas9 (D10A),设计成对sgRNA从而提高CRISPR/Cas9的编辑效率。而且,两条sgRNA的剪切位点之间的距离越近,编辑效率越高[50, 51]。(5)结合表观遗传修饰特征可以提高sgRNA的编辑效率。如蛋白质紊乱状态(protein disorder status)影响sgRNA与目的DNA相结合[24],研究表明,结合染色质的易接近性(脱氧核糖核酸酶超敏位点,DNase I hypersensitive sites)可以提高gRNA的活性[14]。目前,利用计算方法设计编辑效率高的sgRNA主要考虑sgRNA序列特征及其二级结构特征,将所有可能的sgRNA按分数排序,选择具有较高切割效率的sgRNA[52]。自2013年以来,已有多款在线或单机版sgRNA设计和脱靶效应评估软件,包括CRISPR DESIGN[39]、sgRNAcas9[53]、Protospacer[54]、Cas9 Design[55]、CCTop[27]和CFD[22]等。
2.3 CRISPR/Cas9系统存在潜在的脱靶效应
CRISPR/Cas9系统靶向生物基因组存在潜在的脱靶效应[17, 39, 48, 49, 56, 57]。Cas9核酸酶对sgRNA与目标DNA序列碱基匹配具有一定的容错能力[39]。sgRNA除了正常切割靶点DNA双链,也可能与靶点同源性较高的非靶点DNA序列局部匹配(partial match),激活Cas9切割非目标序列,产生脱靶效 应[39]。Hsu等[39]发现脱靶效应主要取决于sgRNA与靶DNA序列错配碱基的数目、错配碱基所在位置、碱基错配类型(转换、颠换)等。存在非经典PAM序列且sgRNA与靶DNA序列碱基错配数目较大时,CRISPR/Cas9 脱靶可能发生在非目的位点,甚至脱靶位点的编辑效率高于目标靶点[52]。目前,已有诸多研究策略旨在降低CRISPR/ Cas9系统的脱靶效应,主要包括:(1)选择合适的靶点。合理地避开GC含量较高的靶点可以降低脱靶效应[1]。(2)适当改变sgRNA的长度。(3)控制sgRNA与靶DNA序列碱基错配数目。(4)合理控制Cas9与sgRNA表达的剂量与持续时间。通过滴定控制Cas9和sgRNA的表达剂量,可以有效地降低脱靶效应[39]。当Cas9与sgRNA的复合物含量较低时,脱靶效应较低[5]。(5)利用全基因组无偏鉴定方法分析脱靶效应,包括基于寡核苷酸整合成双链断裂(GUIDE- Seq)[56, 58],高通量全基因组易位测序(HTGTS)[59]等。
3 机器学习在CRISPR/Cas9系统的应用
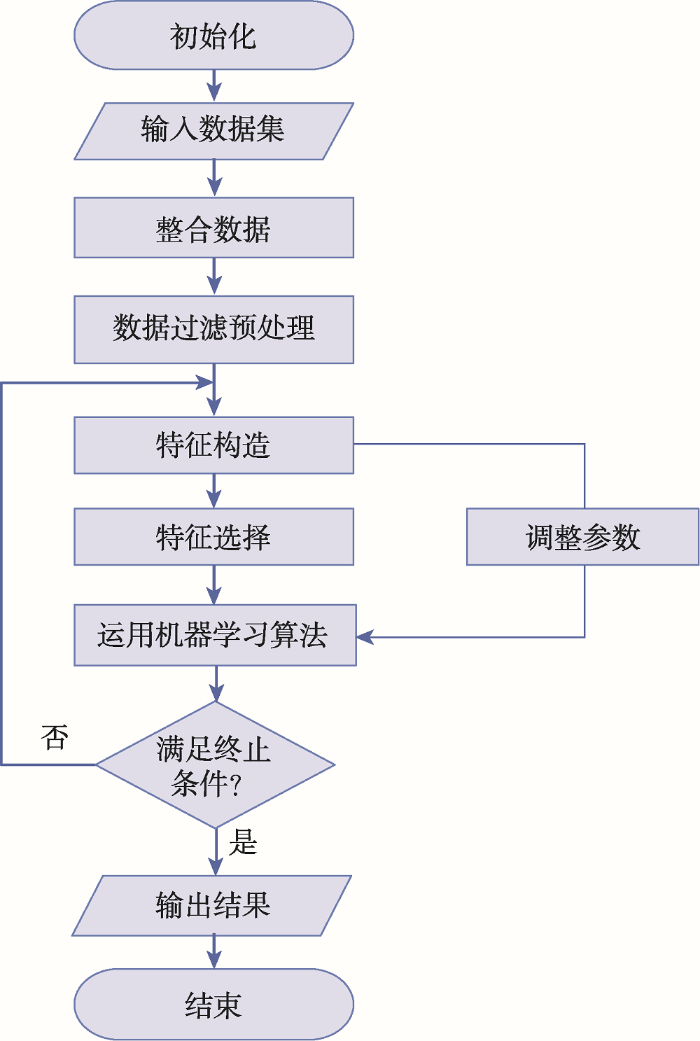
自2014年以来,机器学习已逐步应用于优化设计高效sgRNA序列、预测sgRNA的活性、脱靶位点预测、基因敲除、高通量功能基因筛选等。运用机器学习优化CRISPR/Cas9系统主要分6个步骤(图2):第一,整合不同实验平台、数据库的数据;第二,数据预处理(数据清洗),剔除冗余信息;第三,构造特征,由sgRNA与靶序列碱基配对情况、sgRNA序列本身的特征与二级结构特征、染色质状态等构造特征集;第四,特征选择,运用特征选择算法选取与目标相关的特征;第五,训练模型,通过交叉验证判断模型是否存在过拟合(overfitting)/欠拟合(underfitting),调整参数,进一步优化模型;第六,模型分析,通过实验测试进行误差分析,评估所得模型的有效性、泛化性。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2机器学习优化CRISPR/Cas9系统流程图
Fig. 2Schematic flow of the machine learning method for optimizing the CRISPR/Cas9 system
3.1 数据整合
自2014年以来,可用于研究机器学习在CRISPR/Cas9系统中的应用的开源数据集与在线资源逐年增加,Hart等对此进行了详细的汇总。例如,FC_RES数据集包含17种基因,共含有4380条sgRNA序列,包括6种大鼠基因(Cd5, Cd28, H2-K, Cd45, Thy1, Cd43)、11种人类基因(CD13, CD15, CD33, CCDC101, MED12, TADA2B, TADA1, HPRT, CUL3, NF1, NF2)。FC数据集包含人类和大鼠9种基因共1841条sgRNA序列,RES数据集包含8种基因,共2549条sgRNA序列[60]。机器学习已应用FC_RES数据集预测sgRNA的脱靶效应[13]和打靶活性[60],优化了sgRNA编辑效率[24],且进一步结合表达策略预测sgRNA的编辑效率[61]等。表1总结了常用的机器学习方法应用于CRISPR/Cas9系统研究所采用的数据集。Table 1
表1
表1 优化CRISPR/Cas9系统的机器学习方法常用数据集
Table 1
| 工具名 | 年份 | Cas类型 | 数据集|数据集来源 | 数据集URL | 参考文献 |
|---|---|---|---|---|---|
| Elevation | 2018 | Cas9 | FC, “Sch?nig”, “Concordet”, “Eschstruth”, “Shkumatava”| U2OS, HEK293 | https://www.ncbi.nlm.nih.gov/sra?term=SRP117146%5BAccession%5D https://github.com/maximilianh/crisporWebsite | [13] |
| DeepCpf1 | 2018 | Cpf1 | HT 1-2, HT 2, HT 3, HEK-lenti, HEK-plasmid, HCT-plasmid| HEK293T cells | http://www.rgenome.net/cpf1-database | [14] |
| CRISTA | 2017 | Cas9 | GUIDE-seq data, BLESS data, HTGTS data| HEK293, U2OS | http://crista.tau.ac.il/ | [52] |
| CRISPRpred | 2017 | Cas9 | FC, RES | http://research.microsoft.com/en-us/projects/azimuth/ | [60] |
| sgRNA Designer (Rule Set 2) | 2016 | Cas9 | FC, RES| A375, HL60, KBM7, mouse ESC JM8 | https://www.nature.com/articles/nbt.3437#supplementary-information | [22] |
| predictSGRNA | 2017 | Cas9 | ribosomal genes, non-ribosomal genes, essential genes| HL-60, KBM-7, mouse ESC JM8 | http://genome.cshlp.org/content/25/8/1147/suppl/DC1 http://www.sciencemag.org/content/343/6166/80/suppl/DC1 http://www.nature.com/nbt/journal/v32/n3/full/nbt.2800.html#supplementary information | [23] |
| Big Papi | 2017 | Cas9 | A375, 293T, MOLM13 | https://github.com/mhegde | [16] |
| — | 2017 | Cas9 | FC, RES, UniRef100 | http://research.microsoft.com/en-us/projets/azimuth | [24] |
| sgRNA Scorer 2.0 | 2017 | Cas9 Cpf1 | 293T | https://pubs.acs.org/doi/abs/%2010.1021%2Facssynbio.6b00343 | [62] |
| 工具名 | 年份 | Cas类型 | 数据集|数据集来源 | 数据集URL | 参考文献 |
| CRISPR-DO | 2016 | Cas9 | full human (GRCh37/hg19, GRCh38/hg38) mouse (NCBI37/mm9 GRCm38/mm10) zebrafish (danRer7), fly (dm6), worm (ce10)| HL60, 293T, KBM7 | https://www.ncbi.nlm.nih.gov/pubmed/?term=CRISPR-DO+for+genome-wide+CRISPR+design+and+optimization | [63] |
| CRISPR multitargeter | 2015 | Cas9 | BioMart, zebrafish ohnologs | https://github.com/SergeyPry/CRISPR_MultiTargeter | [64] |
| CRISPRscan | 2015 | Cas9 | One-cell-stage zebrafish embryos, germ | https://www.nature.com/articles/nmeth.3543 | [46] |
| WU-CRISPR | 2015 | Cas9 | FC| 293T, K562, A549, HepG2, SKNAS, U2OS, PGP1-iPS, HEK293 | https://www.ncbi.nlm.nih.gov/sra/ | [6] |
| CRISPR (SSC) | 2015 | Cas9 | HL60, KBM7, ABL1, BCR, 293T, LNCaP- abl, mouse ESC JM8| A promoter-level mammalian expression atlas; Determinants of nucleosome organization in primary human cells; An integrated encyclopedia of DNA elements in the human genome | http://fantom.gsc.riken.jp/5/datafiles/phase1.3/extra/TSS_classifier/ https://www.encodeproject.org/files/ENCFF000VNN https://www.encodepro ject.org/files/EN-CFF000TLU | [65] |
| CRISPRko | 2014 | Cas9 | FC| A375, EL4, AML, MOLM13, NB4, TF1 | https://www.nature.com/articles/nbt.3026#supplementary-information | [45] |
| — | 2014 | Cas9 | HL60, 293T, KBM7, DH5α | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3972032/#SD1 | [47] |
| SgRNA Scorer 1.0 | 2015 | Cas9 | DNase-seq (GSM1008573), H3K4-trimethylation (GSM945288) |293T, K562, A549, HepG2, SKNAS, U2OS, PGP1-iPS, HEK293 | http://arep.med.harvard.edu/CasFinder/ | [66] |
| CRoatan | 2017 | Cas9 | FC| A375, K562 | http://dx.doi.org/10.1016/j.molcel.2017.06.030 | [61] |
| TKOv3 | 2017 | Cas9 | essential genes, nonessential genes| CEG2, KBM7, HL60, RPE1, DLD1, GBM, HAP1, HCT116, RPE1dTP53 | http://tko.ccbr.utoronto.ca/ | [18] |
| BAGEL | 2016 | Cas9 | essential genes, nonessential genes| GBM, HCT116, HeLa, RPE1 | http://tko.ccbr.utoronto.ca/ | [20] |
| CRISPRiaDesign | 2016 | Cas9 | FANTOM Consortium、ENCODE; Consortium (accession no. ENCFF000VNN); ENCODE Consortium (accession no. ENCFF000TLU)|; K562, HEK293T; A promoter-level mammalian expression atlas; Determinants of nucleosome, organization in primary human cells | http://fantom.gsc.riken.jp/5/datafiles/phase1.3/extra/TSS_classifier/ https://www.encodeproject.org/files/ENCFF000VNN https://www.encodeproject.org/files/ENCFF000TLU | [8] |
| CRISPRstrand | 2014 | Cas | REPEATSLange,; REPEATSKunin; REPEATSShah | http://www.ncbi.nlm.nih.gov/ | [19] |
| H1/H2 library | 2018 | Cas9 | K562, Raji, Jiyoye, KBM7 | https://doi.org/10.1093/bioinformatics/bty450 | [67] |
新窗口打开|下载CSV
3.2 数据预处理
由于计算机只能处理数字信号,利用计算的方法分析CRISPR/Cas9系统,需要将DNA/RNA符号序列数值化。选择合适的映射机制,在保留生物学信息的前提下,将DNA/RNA符号序列转换成数值序列。由于不同实验平台数据的格式不一致,故应选择适当的变换方法进行数据预处理分析。3.2.1 数据变换
当数据存在偏移时,先对原始数据进行变换(如对数变换、Box-Cox变换[68]),使该数据服从正态分布。Abadi等[52]将Kleinsteiver[5],Frock[48],Ran[69]和Slaymaker[70]的数据统一整理成Tsai[71]的格式。Abadi等[52]做切割效率预测分析,为了强化学习过程,仅保留序列比对分数大于14.75的位点(95%的切割位点序列匹配个数均值为16.7)。 Doench等[22]在评估基因敲除时,将sgRNA分数进行尺度变换成[0,1],1表示成功敲除基因。
3.2.2 单字母编码法
单字母编码法(one-letter code)[24] 对4种核苷酸(A、C、G、T)进行编码,将长度为N的sgRNA序列编码成一个4维向量,依据碱基存在性将向量的每一个分量设置为0或1。如sgRNA中“G”编码为“0010”。单字母编码法将第[i,i+1](i=1,2,…,N-1)个所有可能的成对核苷酸(AA, AC, …, TG, TT)分别编码成一个16维的向量。如“CG”编码成“0000001000000000”。长度为30个碱基的sgRNA编码成584 (30×4+29×16)个单核苷酸和双核苷酸对序列。
3.2.3 独热编码法
独热编码(one-hot coding)通过有效地增加额外的列,分别用0和1表示分类值的有或无。Doench等[22]采用独热编码,对于一阶特征,将长度为30 bp的sgRNA第一个位置可能出现的A/C/G/T转换成4个二进制变量,分别代表一种可能的核苷酸。二阶特征(所有相邻的两个核苷酸作为一个特征:AA/AT/AG等)共4×4=16个,独热编码将每个二阶特征变编码成一个16位二进制变量。
3.3 特征构造
机器学习应用于CRISPR/Cas9系统sgRNA设计、sgRNA活性预测、脱靶效应评估、基因敲除、高通量功能基因筛选等,特征选择的目标大致如下:第一,提高算法预测的准确性;第二,构造计算低复杂度的机器学习预测模型;第三,更好地理解和解析模型。运用机器学习分析CRISPR/Cas9系统主要考虑以下特征:(1)序列的特征,如序列本身的特征(序列中GC含量、ACGT各碱基数)、位置依赖的特征(sgRNA中与位置有关的单核苷酸、二核苷酸、3个连续的核苷酸),以及sgRNA与目标DNA序列碱基错配类型[6, 8, 13, 14, 16, 19, 22~24, 42, 43, 45~47, 52, 60, 61, 63~66]。(2)热力学特征,包括局部配对概率、sgRNA的最小自由能量(minimum free energy)[23, 52, 60, 64],DNA焓(DNA enthalpy)[52]。DNA焓与染色质的状态有关,反映基因组位点或附近双螺旋的结合亲和力。DNA焓在预测Cas9效率方面可能有着重要的作用。研究表明,DNA焓影响转录因子与其他DNA结合蛋白的结合[52]。但是,其对Cas9蛋白的亲和力的贡献还不明确。(3)核染色质的状态,如超敏位点[13, 14, 43, 52, 64]。(4)氨基酸的切割位置(amino acid cut position)和肽百分比(percent peptide)[24, 60]。(5)核小体占位(nuclosome occupancy)[7, 8, 33, 34, 72]。(6)微同源特征(microhomology feature)[22]。(7)蛋白质紊乱状态[24]。(8)靶位点与PAM序列的距离[62, 64]。(9)切割位点核苷酸的偏好性[42, 63]。(10)外显子类型[47]。(11) sgRNA靶向必需基因与非必需基因的概率分布[18, 20]。(12)序列折叠成二级结构的倾向性[5]。常用优化CRISPR/Cas9系统的机器学习算法及其在该系统的应用与所构建的特征见表2。Table 2
表2
表2 常用优化CRISPR/Cas9系统的机器学习方法总结
Table 2
| 工具名 | 年份 | 机器学习方法 | 机器学习方法在 设计该工具的应用 | 特征构造与选择 | 参考文献 |
|---|---|---|---|---|---|
| Elevation | 2018 | 朴素贝叶斯; 梯度提升回归树; L1正则线性回归 | 预测sgRNA的脱靶效应 | sgRNA与目的DNA碱基错配位置; sgRNA与目的DNA碱基错配类型; 与位置相关的sgRNA与目的DNA; 碱基错配类型; 突变类型(转换、颠换); 染色质的易接近性 | [13] |
| DeepCpf1 | 2018 | 卷积神经网络 | 预测CRISPR-Cpf1 sgRNA的编辑效率 | sgRNA序列特征; 染色质的易接近性 | [14] |
| CRISTA | 2017 | 回归模型; 随机森林 | 评估sgRNA切割 基因组位点的倾向性 | PAM序列类型,sgRNA序列核苷酸 组成,GC含量; 染色质的结构,编码区基因表达水平; sgRNA的二级结构、热力学特征; sgRNA与目标DNA序列错配; DNA凸起/RNA凸起的数量 | [52] |
| CRISPRpred | 2017 | 支持向量机(SVM); 逻辑回归; 随机森林 | 预测sgRNA的 在靶活性 | 单核苷酸、双核苷酸、三个连续的 核苷酸在sgRNA中的位置; 最小自由能、局部配对概率、sgRNA的热量; sgRNA序列GC含量、AT含量、A/C/G/T数; 氨基酸的切割位置,肽所占百分比 | [60] |
| sgRNA Designer (Rule Set 2) | 2016 | 线性回归; L1/L2正则逻辑回归; 支持向量机; 随机森林; 梯度上升回归树 | 预测sgRNA的 在靶活性 | 二核苷酸特征; 与位置有关的单核苷酸和双核苷酸; sgRNA中GC含量; 位置独立的核苷酸数; sgRNA靶点在基因中的位置; 微同源特征 | [22] |
| predictSGRNA | 2017 | 逻辑回归;随机森林 | 设计高编辑效率 sgRNA | 位置依赖的单核苷酸; 位置独立的单核苷酸; 单核苷酸与二核苷酸的频率; sgRNA与目的DNA比对得分; 热力学特征及二级结构、理化性质; 由PseKNC模型生成伪k-元组核苷酸特征 | [23] |
| Big Papi | 2017 | 梯度上升回归树 | 优化设计sgRNA 文库 | 位置独立的单核苷酸、二核苷酸; 位置依赖的单核苷酸、二核苷酸; 热力学特性(解链温度); 3’PAM序列最接近的胸腺嘧啶(T) | [16] |
| — | 2017 | 最小冗余最大相关性; 支持向量机 | 优化sgRNA 编辑效率 | 单个、成对的核苷酸(SNTs, PNTs); sgRNA与目的DNA序列保守性; 氨基酸切割位置,编码肽的氨基酸组成; 靶蛋白序列的紊乱状态 | [24] |
| sgRNA Scorer 2.0 | 2017 | 支持向量机 | 预测sgRNA编辑效率 | 靶点与PAM序列的距离 | [62] |
| CRISPR-DO | 2016 | LASSO回归; 弹性网络线性回归 | 预测sgRNA的 编辑效率 | sgRNA序列特征; 切割位点处胞嘧啶的偏好性 | [63] |
| CRISPR multitargeter | 2015 | 逻辑回归 | 预测sgRNA的 编辑效率 | GC百分比,与位置有关的单核苷酸、 相邻的二核苷酸; sgRNA中G,C的含量,G/C比值; 局部染色质结构 | [64] |
| 工具名 | 年份 | 机器学习方法 | 机器学习方法在 设计该工具的应用 | 特征构造与选择 | 参考文献 |
| CRISPRscan | 2015 | 逻辑回归 | 预测sgRNA的 编辑效率 | 位置依赖的单核苷酸、二核苷酸; GC含量 | [46] |
| WU-CRISPR | 2015 | 支持向量机 | 预测sgRNA的编辑 效率与编辑特异性 | 位置独立的单核苷酸、二核苷酸; 位置依赖的单核苷酸、二核苷酸; RNA的二级结构(折叠自由能、 核苷酸的易接近性) ; 位置依赖的sgRNA核苷酸的易接近性; sgRNA重复碱基的分布 | [6] |
| CRISPR (SSC) | 2015 | LASSO回归 | 预测全基因组功能 基因筛选sgRNA 的编辑效率 | sgRNA序列特征; 切割位点处胞嘧啶的偏好性 | [65] |
| CRISPRko | 2014 | 支持向量机; 逻辑回归 | 预测sgRNA的 编辑效率 | 与位置有关的单核苷酸、相邻的二核苷酸; sgRNA中G,C的含量,G/C比值; 局部染色质结构 | [45] |
| — | 2014 | 支持向量机 | 预测sgRNA的 编辑效率 | 序列特征,GC含量; sgRNA链,外显子类型 | [47] |
| SgRNA Scorer 1.0 | 2015 | 支持向量机 | 分析不同活性(高/低) sgRNA之间的关系 研究sgRNA特异性 与活性的关系 | 位置依赖的序列特征 | [66] |
| CRoatan | 2017 | 随机森林; 线性回归 | 结合表达策略预测 sgRNA的效能 预测同源引导修复 Cas9切割引起的 DSBs的可能性 | 序列长度,GC含量; 双链断裂与对应位点的距离 | [61] |
| TKOv3 | 2017 | 贝叶斯分析 | 鉴定必需基因 设计全基因组 CRISPR/Cas9 基因文库 | 评估利用倍数变化分析sgRNA靶向; 必需基因与非必需基因的分布情况; 评估概率分布(贝叶斯因子BF) | [18] |
| BAGEL | 2016 | 贝叶斯分析 | 混合文库筛选 鉴定必需基因 | 评估利用倍数变化分析sgRNA靶向; 必需基因与非必需基因的分布情况; 评估概率分布(贝叶斯因子BF) | [20] |
| CRISPRiaDesign | 2016 | 弹性网络线性回归; 支持向量回归模型 | 鉴定CRISPRa/i 高效率的sgRNA | 序列特征,位置依赖的序列特征; 染色质的状态,核小体占位率 | [8] |
| CRISPRstrand | 2014 | 基于随机梯度下降 的支持向量机 | 预测CRISPR 重复序列的方向 | ATTGAAAN重复出现次数; CRISPR 序列核苷酸的组成; 序列特定位置的突变; 序列折叠成二级结构的倾向性 | [19] |
| H1/H2 library | 2018 | 弹性网络回归算法 | 筛选sgRNA异常值 | sgRNA序列特征 | [67] |
新窗口打开|下载CSV
3.4 特征选择
利用机器学习优化CRISPR/Cas9系统,特征集往往较大,特征之间可能存在相关性。训练特征过多导致训练模型所需时间较长,容易引起“维度灾难”,导致模型复杂,泛化性差[73]。机器学习预测模型与特征选择息息相关,选择不同的特征训练将得到不同的模型[74]。特征选择(feature selection)[75]旨在从特征集中选取一个代表全局特征的子集。特征选择主要分为4个部分(图3):(1)产生过程;(2)评价函数;(3)停止准则;(4)验证过程。首先,从特征集中选取一个特征子集。利用评价函数对该子集进行计分并与停止准则进行比较,若满足停止条件,程序结束;否则继续产生下一组特征子集,重复上述步骤。最后,验证特征子集的有效性。评价函数主要包括三种方法:筛选器(filter)、封装器(wrapper)及嵌入法(embedded)。筛选器对每一维的特征赋予权重,依据权重排序,如信息增益、相关系数。封装器将选择子集视为一个搜索寻优问题,生成不同的特征组合分别进行评价与比较。嵌入法在模型给定的情况下,学习提高模型准确性的属性。正则化对权重进行约束,岭回归(ridge regression)在线性回归中加入了正则项。停止准则通常是一个与评价函数有关的阈值,当评价函数值达到该阈值则停止搜索。验证过程主要验证已选特征子集的有效性。
图3
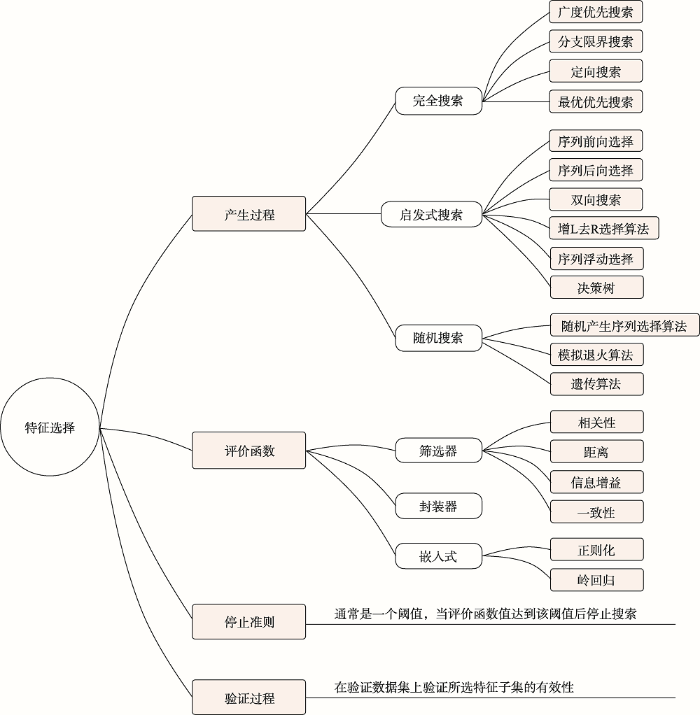 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3特征选择方法
Fig. 3Feature selection methods
3.5 评估方法
模型和参数确定之后,通过实验测试评估学习器的泛化性能。一般地,通过学习训练数据,比较模型对输入数据集的预测值与实际值的差异验证模型。常用的模型验证方法是交叉验证(cross validation),数据的每个子集既是训练集又是验证集。如三折交叉验证(three-fold cross-validation)[42]、五折交叉验证(five-fold cross-validation)[13, 20]、十折交叉验证(ten-fold cross-validation)[13, 23, 24, 60, 62]、二十折交叉验证(twenty-fold cross-validation)[13]、嵌套交叉验证(nested cross-validation)[13, 22]、留一法(leave-one-out, LOO)[52, 60]等。三折交叉验证将数据分成3组,每一轮依次训练其中的两组数据,由训练所得参数预测第3组数据,评估模型的准确性。LOO验证法每次仅测试一个样本,其他样本用于训练模型。LOO不受随机样本划分方式的影响,评估效果比较准确。然而,数据集较大的情况下,LOO训练模型计算成本高。3.6 性能度量
评估优化CRISPR/Cas9系统的机器学习算法学习器的泛化性能,需要可行的实验评估方法与衡量模型泛化能力的评价体系。通常,真正例率(true positive rate, TPR)表示正确预测的正样本比例,假正例率(false positive rate, FPR)表示错误预测的负样本 比例。$\text{TPR}=\frac{\text{TP}}{\text{TP}+\text{FN}},\ \text{FPR}=\frac{\text{TN}}{\text{TN}+\text{FP}}$,
其中,TP (true positive)为真阳性,表示正确预测的正样本数;TN (true negative)为真阴性,表示正确预测的负样本数;FP (false positive)为假阳性,表示错误预测的负样本数;FN (false negative)为假阴性,表示错误预测的正样本数。准确度(accuracy, ACC)和马修相关系数(Matthew correlation coefficient, MCC)衡量所有分类器的预测性能:
$\begin{align} & \text{ACC}=\frac{\text{TP}+\text{TN}}{\text{TP}+\text{TN}+\text{FP}+\text{FN}},\ \\ & \text{MCC}=\frac{\text{TP}\times \text{TN}-\text{FP}\times \text{FN}}{\sqrt{(\text{TP}+\text{FP})(\text{TP}+\text{FN})(\text{TN}+\text{FP})(\text{TN}+\text{FN})}} \\ \end{align}$
当正样本量与负样本量差别较大时,MCC能够更公平地反映预测能力。此外,以“真正例率”为纵轴,“假正例率”为横轴的受试者工作特征曲线(receiver operating characteristic, ROC)可作为机器学习算法预测性能的衡量标准[76],ROC到达坐标左上角表示所得模型具有较好的性能。
4 常用优化CRISPR/Cas9系统的机器学习算法
机器学习在DNA编码序列中学习影响CRISPR/ Cas9系统的模型特征,为提升该系统基因组编辑效率、最大限度降低脱靶效应提供有力的计算工具。近年来,机器学习已被应用于预测sgRNA的脱靶效应[13]、优化设计高效sgRNA [10, 14, 64]、预测sgRNA活性[17]、设计全基因组CRISPR/Cas9基因敲除文 库[18]、鉴定用于CRISPRi/a技术的高效率sgRNA[8]、鉴定必需基因[18, 20]等。下面简述几种基于机器学习优化CRISPR/Cas9系统的计算方法。4.1 优化设计高效sgRNA
设计高效sgRNA对提高CRISPR/Cas9系统靶点识别特异性有着重要的作用。CRISPR/Cas9系统可以容忍sgRNA与靶DNA序列存在一定数量范围的碱基错配,因此,靶点可能存在多个候选的sgRNA。sgRNA序列特征与其二级结构特征影响靶点切割活性,设计sgRNA的指导思想是,由序列的基本特征预测候选sgRNA的脱靶效应(打靶效率),依据候选sgRNA排序结果,从中选取得分较高的sgRNA。应用机器学习设计sgRNA主要考虑可能优化该系统靶点切割效率的特征,通过训练模型、调整参数、误差分析,从而确定影响sgRNA打靶效率的特征。Chen等[24]基于最小冗余最大相关性(maximal-relevance minimal-redundancy, mRMR)、增量特征选择算法(incremental feature selection, IFS)、支持向量机(support vector machine, SVM),设计选择影响CRISPR/ Cas9系统基因组编辑效率与特异性关键因素的算法,有效地剔除冗余特征。CRISTA方法[52]基于随机森林(random forest)与回归模型(regression model),考虑了DNA凸起 (DNA bulge)/RNA凸起(RNA bulge)对sgRNA编辑效率的影响。DNA凸起(RNA凸起)指脱靶位点DNA序列长度比sgRNA多(少)若干个碱基,通过形成凸起完成碱基的正确配对。CRISTA方法考虑DNA凸起,结合基因组核苷酸含量、sgRNA热力学特征、sgRNA与靶DNA序列元素相似性等,评估sgRNA切割基因组位点的倾向性。该作者改进Needleman- Wunch双序列比对算法。在sgRNA与目标DNA序列比对中,最多容忍3个单碱基的间隙(gap),这有利于高效预测潜在的脱靶位点。由于最多允许3个单碱基的间隙,故将长度为20 bp的靶DNA序列缩短(延伸) 3个碱基。随机选择其中7个长度为17~ 23 bp的靶DNA序列与相应sgRNA序列做序列比对,选取最高分数的DNA序列作为靶序列。最后,计算sgRNA对靶DNA序列切割倾向性与序列比对分数的最大均方皮尔逊相关系数(maximal averaged squared Pearson correlation coefficient),确定最优模型参数。研究表明,DNA凸起/RNA凸起是CRISPR/Cas9系统的组成部分(integral part)[52]。在该作者测试的数据集中,凸起约占20%。大多数凸起sgRNA对目标DNA序列的切割效率较低,少数可达到中等切割 效率。
4.2 预测sgRNA的编辑特异性
CRISPRpred方法[60]基于支持向量机预测sgRNA对目标DNA序列的打靶活性。利用随机森林算法计算平均下降基尼系数(Gini index),选取所有影响sgRNA编辑特异性的特征。基尼系数表示模型的不纯度。基尼系数越小,不纯度越低,特征越好。决策树选择基尼系数增益值最大的特征,作为节点的分裂条件[77]。CRISPRpred方法利用十折交叉验证,计算均方根误差(root-mean-square error, RMSE)验证特征之间的相关性。通过设置参数(学习率、随机森林树的数量)验证模型防止过拟合。利用已选特征的不同组合分析FC-RES数据集。该作者利用留一法交叉验证计算评估矩阵,由ROC曲线、PR (precision- recall)曲线评估模型的性能。研究表明,考虑氨基酸的切割位置、肽百分比有利于估计sgRNA的编辑特异性。Kuan等[23]根据寡核苷酸设计(oligonucleotide design)[78]与核小体占用模型[79]的先验知识,基于机器学习评估影响sgRNA对靶DNA序列切割效率的特征,提出设计高编辑效率的sgRNA的计算方法。该作者考虑可能影响sgRNA靶向目标DNA序列切割效率的特征:(1)位置依赖的单核苷酸(position- dependent mono-nucleotide, PD Mono),如sgRNA第一个符号为A记为“A_1”;(2)位置独立的单核苷酸(position-dependent dinucleotide, PD Dinuc)特征,如sgRNA序列A的数目;(3)单核苷酸与二核苷酸的频率;(4) sgRNA与目标DNA序列的比对得分;(5) 热力学特征及二级结构、理化性质(physiochemical properties);(6)由PseKNC模型生成伪k-元组核苷酸特征。结合逻辑回归、随机森林分析每个特征对sgRNA编辑效率的贡献。研究发现,T和TT二核苷酸的频率与sgRNA的编辑效率呈强负相关。TT二核苷酸在移位方面灵活性较小,在丰度较高的区域sgRNA的编辑效率较低。结合位置依赖的二核苷酸特征优化ROC曲线下的面积(area under ROC curve, AUC)的性能预测sgRNA的活性。染色质可达性(chromatin accessibility)已被证明是dCas9-sgRNA基因组结合的主要决定因素[43]。与染色质重塑、染色质可达性相关的表观遗传标记包括DNase I 超敏感位点、转录因子结合、DNA甲基化和组蛋白修饰。综合考虑核苷酸组成、染色质结构特征与sgRNA表达载体依赖性水平的特征有利于预测sgRNA的特异性。
4.3 评估sgRNA的脱靶效应
Elevation方法[13]基于CFD[22]分三步评估给定sgRNA的脱靶效应。首先,利用已知数据训练第一层机器学习模型。根据目标DNA序列与sgRNA至多存在N个碱基不匹配的原则,筛选sgRNA在基因组所有可能的靶点。接着,为每个潜在的靶点评分,量化sgRNA与靶序列的脱靶活性。最后,评估其脱靶活性,计算每个sgRNA的综合得分。该方法运用二层回归模型,第一层模型(单个碱基不匹配)考虑位置依赖的sgRNA与目标DNA序列碱基不匹配的位置、不匹配核苷酸类型、转换突变、颠换突变等,计算基尼系数分析特征的重要性;第二层模型(多错配组合)计算sgRNA与目标DNA序列碱基不匹配得分与第一层单个碱基不匹配得分之和。Listgarten等[13]利用美国哈佛医学院和马萨诸塞州总医院的公开数据集对第二层模型进行训练,将第一层模型加以细化,推广至碱基错配数目大于1的靶标区域。基于机器学习方法计算基因组可能发生脱靶的各个区域的概率计算脱靶分值。Elevation算法计算sgRNA两类脱靶分值:该sgRNA在特定靶标区域内的脱靶分值及其在所有可能的靶区域内对应的脱靶分值。4.4 CRISPR遗传筛选
CRISPR/Cas9为功能性基因筛选(functional genetic screening)提供高效简便的技术支持。利用CRISPR可以通过诱导基因突变进行功能缺失型(loss of function)筛选,也可以激活转录进行功能获得型(gain of function)筛选。CRISPR基因筛选可以作用于基因编码区(coding region),也可以靶向基因非编码区(non-coding region)[4]。基于CRISPR/Cas9功能基因筛选技术在细胞生存必需基因鉴定[18, 20]、潜在治疗靶标筛选[6]、肿瘤转移[80]等方面已取得重要的研究进展。近年来,CRISPR/Cas9在基因扰动(genetic perturbation)领域已被应用于基因编辑、基因的上调和下调表达,主要包括基因敲除(gene knockout, KO)、基因敲入(gene knockin, KI)、CRISPR干扰/CRISPR激活[12, 81, 82]。CRISPRi将Cas9突变失活(dead Cas9, dCas9),使其无法切割DNA双链,再与转录抑制因子(transcription repression factor)作用,从而实现在sgRNA的指导下抑制特定基因的表达[83]。CRISPRa利用dCas9与转录激活因子(transcriptional activator)作用,上调靶基因的表达水平,用于功能获得型筛选[84]。运用CRISPR/Cas9系统进行基因扰动,sgRNA的特异性对靶基因筛选至关重要。合理构建包含靶点信息的sgRNA文库,可以有效地提高靶基因筛选的效率,实现最大限度地降低脱靶效应[85]。CRISPR筛选(CRISPR screen)常用混合文库(pooled-library),将合成混合寡链核苷酸库装入逆转录病毒(如慢病毒)载体侵染宿主细胞,再将文库序列整合至基因组中表达,检测细胞生长表型[36]。混合文库通常采用定向筛选,包括正向筛选(positive screening)与逆向筛选(negative screening)。正向筛选通过施加一个强的选择压力,经过文库扰动,野生型基因致死,获得抗性的克隆细胞存活,从而鉴定可产生抗性的基因[86]。逆向筛选用于鉴定细胞必需基因[87]。
机器学习在CRISPR遗传筛选领域有着巨大的潜能。机器学习已逐渐应用于设计高编辑效率sgRNA文库[67]、鉴定必需基因[18, 20]、结合基因表达策略预测sgRNA的编辑效率[61]、鉴定CRISPRa/i高效率的sgRNA[8, 19]等。
4.4.1 设计全基因组sgRNA文库
基因筛选sgRNA文库构建以全基因组基因为靶点,针对每个靶点设计效率较高的一组sgRNA。设计全基因组sgRNA文库仍存在以下问题: (1) sgRNA异常值、sgRNA与靶向相同基因的其他sgRNA活性存在较大的差异;(2) CRISPR/Cas9系统中,间隔区(spacer)长度可能不同[77, 88],仅对单个向导(靶序列)分析其最佳间隔区长度;(3)间 隔区长度与信噪比(signal-to-nosie ratio, SNR)的关系尚不明 确[67]。基于机器学习算法设计全基因组sgRNA文库的指导思想是,考虑影响sgRNA编辑效率的特征,利用机器学习算法进行训练并评分[85, 89],根据得分选取高效的sgRNA构建大规模文库。
Chen等[67]基于MAGeCK-VISPR模型[90]识别sgRNA异常值(sgRNA outlier),利用弹性网络回归算法(elastic-net regression)[42]提取sgRNA序列特征,根据sgRNA是否为异常值分别赋值为1或0。由于不同sgRNA的切割活性与修复效率、局部染色质结构与潜在的脱靶效应不同。在基因筛选中,不同sgRNA靶向同一个基因,将产生不同的细胞生长表型与选择水平[39, 91, 92]。该作者分析已有文库发现,在非种子区域G较高的sgRNA具有较强的脱靶活性,导致较强的异常表型。将sgRNA靶向多个非必需基因作为阴性对照,能够降低假阳性。比较不同长度(18,19,20个碱基) sgRNA的切割效率及信噪比,得到长度为19个碱基的sgRNA具有最高的切割效率,提示其在基因敲除效应中更稳定(潜在的脱靶效应小)。基于此,该作者提出一种全新的全基因组CRISPR/Cas9筛选文库(H1/H2),与Brunello文库[22]、TKO文库[93]、Ong文库[94]比较,该文库sgRNA异常值比例最小。比较GeCKOv2与TKO文库,H1/H2、Brunello与Ong文库对已知必需基因的识别性能更优。
4.4.2 鉴定必需基因
BAGEL (Bayesian analysis of gene EssentiaLity)方法[20]基于贝叶斯分析基因敲除筛选,识别混合文库筛选中的必需基因。BAGEL首先估计所有靶向必需基因、靶向非必需基因训练集的sgRNA的表达倍数变化的分布(distribution of fold changes)。其次,使用核密度估计(kernel density estimation)sgRNA靶向参考必需基因与非必需基因的似然函数,计算贝叶斯因子(Bayes factor, BF)。Hart等[20]将必需基因、非必需基因数据集作为测试集,计算PR曲线评估筛选性能。研究表明,利用BAGEL分析基因敲除筛选,能够灵敏、准确地鉴定适应性基因(fitness gene),且大大降低计算时间。运用BAGEL鉴定人类细胞系混合库基因敲除筛选2000个适应性基因,错误发现率(false discovery rate, FDR)为5%。而且,BAGEL对不同平台文库筛选具有高灵敏性与特异性。
CRISPR/Cas9为哺乳动物细胞高通量功能基因筛选提供技术支持。人类蛋白编码基因与病毒载体表达的CRISPR sgRNA混合文库已应用于人类多种癌症细胞与永生细胞系基因敲除[18]。研究表明,CRISPR筛选比混合shRNA筛选更敏感[95],然而,CRISPR文库设计和实验方案中存在显著的偏差。Hart等[18]报道了利用CRISPR/Cas9系统评估与设计全基因组遗传筛选。该作者使用来自3个研究组不同基因组规模的sgRNA文库分析人类细胞系17个基因敲除筛选,使用BAGEL算法[20]鉴定必需基因,将已定义的人类核心必需基因由360个扩展至684。根据扩展的核心必需基因参考基(CEG2)以及来自6个CRISPR敲除筛选的经验数据,设计优化序列的sgRNA文库(Toronto KnockOut version 3.0, TKOv3)[18]。结合CEG2参考基的优化TKOv3文库,为评估人类细胞系基因敲除筛选提供一个高效的平台[18]。
4.4.3 鉴定CRISPRi/a高效率的sgRNA
CRISPRi/a主要靶向基因启动子区,设计sgRNA所考虑的特征与CRISPR基因敲除(CRISPR KO)系统稍有不同。与CRISPR/Cas9 基因敲除类似,CRISPRi/a系统序列间隔区也具有嘌呤偏好性。与CRISPR/Cas9系统相比,CRISPRi/a具有独特的效应区(effector domain),而且,该结构域在基因扰动中发挥关键作用[42, 96]。由于缺乏足够的数据来源,仅有少数工具用于CRISPRi/a系统设计sgRNA (如CRISPR-ERA)[97]。序列特征分析将有助于提高对sgRNA编辑效率的预测性能[85]。
Horlbeck等[98]报道了核小体能够直接阻断CRISPR/Cas9接近目标DNA。CRISPRi/a需要持续的dCas9与DNA相结合,考虑核小体占位有利于预测CRISPRi/a高编辑效率的sgRNA[8]。Horlbeck等[8]基于机器学习结合染色质、sgRNA序列等特征,设计CRISPRiaDesign工具用于预测CRISPRi/a高效率的sgRNA。CRISPRi的活性与靶点与转录起始点的距离之间具有周期性与不对称性[98]。基于此,该作者首先运用支持向量回归(support vector regression, SVR) 拟合sgRNA位置特征,从而预测靶位点特征的连续函数。接着,利用弹性网络线性回归对30个CRISPRi筛选的数据进行训练分析,结合靶位点距离转录起始点、核小体占位、位置依赖的二核苷酸、位置依赖的单核苷酸、sgRNA二级结构、靶位点染色质的易接近性、sgRNA的长度等特征,预测sgRNA的编辑活性分数。最后,利用该算法设计人类(hCRISPRi-v2)和小鼠基因组CRISPRi/a文库(version 2)。K562细胞必需基因CRISPRi筛选实验表明,大多数sgRNA具有较高的活性。PR曲线分析显示,采用一个紧凑的sgRNA 基因文库检测超过90%的必需基因假阳性最小。CRISPRi/a作为功能缺失型筛选与功能获得型筛选研究的主要工具,能够为识别Cas9靶点提供一个通用的工具。
4.4.4 预测sgRNA的活性
Croatan方法[61]结合随机森林与sgRNA的表达策略优化CRISPR/Cas9系统基因敲除效率。研究表明,利用多个RNA聚合酶Ⅲ可以促进sgRNA的独立性,优化基因的表达策略[99]。Cpf1可以靶向独立表达sgRNA的crRNA阵列的细胞的多个靶点[100]。上述策略有助于研究细胞中单基因敲除的位点,但是,Cas9同时靶向目标序列多个位点可能导致更大的功能性后果(functional consequence)。结合选择算法与基因表达策略将有利于选择高效能的sgRNA。Erard等[61]基于随机森林设计Croatan算法用于预测sgRNA切割效率,结合基因表达策略,可用于人类细胞系单个基因敲除、多个基因敲除。Croatan结合核苷酸组合、移码突变(frameshift mutation, FSM)可能性与靶点是否在编码区特征,训练Doench等[43]与Chari等[66]的数据集选择高编辑效率的sgRNA识别目标靶点。研究表明,靶序列的保守性(target conservation)与靶序列侧翼同源序列(target-flanking homologous sequence)影响sgRNA的切割效率。而 且,Cas9作用于多个靶位点将增强功能影响。当两个独立的sgRNA同时引导Cas9靶向目标基因,编辑效率将显著提高。对于每个靶位点,依据预测分数较高的两个sgRNA设计文库。基于此,该作者设计与建立全基因组CRISPR阵列文库(arrayed CRISPR library),该文库可用多重或阵列格式进行单个(合并)遗传筛选。
5 存在的问题与潜在的解决方案
CRISPR/Cas9系统基因组编辑效率和特异性受诸多因素的影响。自2014年起,已有多种基于机器学习的计算方法用于优化该系统。然而,这些方法对该系统基因组编辑效率和脱靶效应的研究尚不够深入,研究结果并不一致。这反映在不同计算方法数据整合、特征选择、sgRNA的编辑活性的评价标准不一致。机器学习优化CRISPR/Cas9系统依赖先验知识,需要大量学习已知实验数据。不同sgRNA设计和脱靶效应评估软件的操作平台、参数设置与评估指标不同,输出数据格式也不一致。如Digenome-Seq[101, 102]未提供活体实验的基因编辑切割频率的数据。此外,由于算法原理不同,运用不同的计算的方法得到的研究结论不完全一致。如针对影响sgRNA切割效率的特征这一问题,Haeussler等[103]发现凸起极少发生,其切割效率忽略不计。而Abadi 等[52]测试的数据集中,凸起约占20%。而且大多数凸起sgRNA对靶DNA序列的切割效率较低,少数凸起sgRNA达到中等切割效率水平。运用机器学习预测CRISPR/Cas9系统基因组编辑效率主要分析序列的特征。有些计算方法为了降低计算成本,忽略一些重要的特征。如Elevation方法[13]第二层整合分数模型,考虑sgRNA与目标DNA序列碱基错配类型的组合容易产生组合爆炸。为了简化模型,该方法忽略插入/缺失,主要考虑sgRNA与目标DNA序列碱基不匹配特征。另外,与表观遗传修饰有关的特征可能影响CRISPR/Cas9系统的性能。然而,大多数计算方法并未考虑这方面的特征。
单碱基编辑技术(base editor)[104, 105]是基于CRISPR系统的新型靶基因定点修饰技术,它不需要产生DNA双链断裂及DNA模板,能够对基因组特定碱基进行高效的替换。单碱基编辑PAM序列主要为NGG,能够进行修饰的序列较少[106]。目前,单碱基编辑技术的作用机制有待深入探究。David[106]等发现xCas9酶在单碱基编辑领域得到更广泛的PAM,而且脱靶效应远低于spCas9,但其作用机制尚不明确。运用机器学习学习先验知识,可以帮助科研工作者寻找影响单碱基编辑效率的关键特征,从而提高单碱基编辑效率,降低脱靶效应。运用机器学习优化单碱基编辑技术将为实现基因的功能缺失突变、基因的表达调控方面、植物基因组功能解析和作物遗传改良及新品种培育提供了重要技术支撑[104, 106, 107]。
机器学习应用于优化CRISPR/Cas9系统尚存在一些问题有待进一步解决。研究者在寻找弥补不足的方法的同时,也在不断拓展该技术的应用领域。深度学习(deep learning)可以避免耗时费力的特征工程提取过程,运用深度学习分析CRISPR/Cas9系统是一个创新可行的思路。DeepCpf1算法[14]基于卷积神经网络(convolutional neural network, CNN)结合染色质易接近性特征,预测CRISPR/Cpf1系统sgRNA的编辑活性。该方法比传统机器学习方法(如L1/L2正则线性回归、梯度上升回归树)预测性能更优。随着研究的不断深入,这些问题将会得到解决,进而推动功能基因组学、基因表观遗传调控、疾病治疗等领域的发展。
6 结语与展望
CRISPR/Cas9系统是由细菌和古细菌等微生物特有的获得性免疫系统发展起来的基因组编辑技术,能够对基因组特定位点进行基因敲除、基因敲入、DNA大片段删除、转录调控等遗传操作。CRISPR/ Cas9技术凭借成本低廉、效率高和易操作性等优点在基因工程领域具有很好的发展潜力。CRISPR/Cas技术处于初步的研究阶段,影响该系统基因组编辑效率和脱靶效应的机制基本明确。但是,仍存在很多问题有待进一步优化,如提高基因组编辑效率与编辑特异性等。如何提高同源重组修复的效率,如何降低非同源重组修复造成的非预期的突变,是当前利用CRISPR/Cas9技术进行基因治疗所面临的技术难题。机器学习为优化CRISPR/Cas9系统所面临的问题提供创新的解决思路。通过对不同实验数据进行整合分析,基于机器学习建立数学模型分析影响CRISPR/Cas9系统编辑效率、编辑特异性的特征,有助于研究者们更深入地理解该系统的作用机制。机器学习在CRISPR/Cas9系统脱靶效应评估、预测sgRNA的活性、设计高效sgRNA、基因敲除、高通量功能基因筛选等研究领域有着日渐广泛的应用。尽管机器学习应用于优化CRISPR/Cas9技术尚处于研究发展阶段,但已显现出广阔的应用前景。通过不断改进,机器学习、深度学习应用于优化CRISPR/ Cas9系统的应用将成为研究热点。今后,机器学习/深度学习优化CRISPR/Cas9系统技术将会为临床医师和科研人员提供更多有价值的信息,进而在基础科学研究、分子生物学研究和基因治疗等诸多领域产生深远的影响。
(责任编委: 谷峰)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 3]
URLPMID:23360965 [本文引用: 1]

Here we use the clustered, regularly interspaced, short palindromic repeats (CRISPR)-associated Cas9 endonuclease complexed with dual-RNAs to introduce precise mutations in the genomes of Streptococcus pneumoniae and Escherichia coli. The approach relies on dual-RNA:Cas9-directed cleavage at the targeted genomic site to kill unmutated cells and circumvents the need for selectable markers or counter-selection systems. We reprogram dual-RNA:Cas9 specificity by changing the sequence of short CRISPR RNA (crRNA) to make single- and multinucleotide changes carried on editing templates. Simultaneous use of two crRNAs enables multiplex mutagenesis. In S. pneumoniae, nearly 100% of cells that were recovered using our approach contained the desired mutation, and in E. coli, 65% that were recovered contained the mutation, when the approach was used in combination with recombineering. We exhaustively analyze dual-RNA:Cas9 target requirements to define the range of targetable sequences and show strategies for editing sites that do not meet these requirements, suggesting the versatility of this technique for bacterial genome engineering.
URLPMID:24906146 [本文引用: 1]
Derived from a microbial defense system, Cas9 can be guided to specific locations within complex genomes by a short RNA. The development, applications, and future directions of the CRISPR-Cas9 system for genome engineering are discussed here.
URLPMID:23792628 [本文引用: 3]
Clustered, regularly interspaced, short palindromic repeat (CRISPR) RNA-guided nucleases (RGNs) have rapidly emerged as a facile and efficient platform for genome editing. Here, we use a human cell-based reporter assay to characterize off-target cleavage of CRISPR-associated (Cas) 9-based RGNs. We find that single and double mismatches are tolerated to varying degrees depending on their position along the guide RNA (gRNA)-DNA interface. We also readily detected off-target alterations induced by four out of six RGNs targeted to endogenous loci in human cells by examination of partially mismatched sites. The off-target sites we identified harbored up to five mismatches and many were mutagenized with frequencies comparable to (or higher than) those observed at the intended on-target site. Our work demonstrates that RGNs can be highly active even with imperfectly matched RNA-DNA interfaces in human cells, a finding that might confound their use in research and therapeutic applications.
URL [本文引用: 5]
URLPMID:4629399 [本文引用: 3]
The CRISPR/Cas9 system has been rapidly adopted for genome editing. However, one major issue with this system is the lack of robust bioinformatics tools for design of single guide RNA (sgRNA), which determines the efficacy and specificity of genome editing. To address this pressing need, we analyze CRISPR RNA-seq data and identify many novel features that are characteristic of highly potent sgRNAs. These features are used to develop a bioinformatics tool for genome-wide design of sgRNAs with improved efficiency. These sgRNAs as well as the design tool are freely accessible via a web server, WU-CRISPR (http://crispr.wustl.edu). The online version of this article (doi:10.1186/s13059-015-0784-0) contains supplementary material, which is available to authorized users.
URLPMID:26579937 [本文引用: 4]
During Cas9 genome editing in eukaryotic cells, the bacterial Cas9 enzyme cleaves DNA targets within chromatin. To understand how chromatin affects Cas9 targeting, we characterized Cas9 activity on nucleosome substrates in vitro. We find that Cas9 endonuclease activity is strongly inhibited when its target site is located within the nucleosome core. In contrast, the nucleosome structure does not affect Cas9 activity at a target site within the adjacent linker DNA. Analysis of target sites that partially overlap with the nucleosome edge indicates that the accessibility of the protospacer-adjacent motif (PAM) is the critical determinant of Cas9 activity on a nucleosome.
URL [本文引用: 8]
[本文引用: 1]
URLPMID:4880442 [本文引用: 2]
10.7554/eLife.13450.001The CRISPR-Cas9 bacterial surveillance system has become a versatile tool for genome editing and gene regulation in eukaryotic cells, yet how CRISPR-Cas9 contends with the barriers presented by eukaryotic chromatin is poorly understood. Here we investigate how the smallest unit of chromatin, a nucleosome, constrains the activity of the CRISPR-Cas9 system. We find that nucleosomes assembled on native DNA sequences are permissive to Cas9 action. However, the accessibility of nucleosomal DNA to Cas9 is variable over several orders of magnitude depending on dynamic properties of the DNA sequence and the distance of the PAM site from the nucleosome dyad. We further find that chromatin remodeling enzymes stimulate Cas9 activity on nucleosomal templates. Our findings imply that the spontaneous breathing of nucleosomal DNA together with the action of chromatin remodelers allow Cas9 to effectively act on chromatin in vivo.DOI: http://dx.doi.org/10.7554/eLife.13450.001
[本文引用: 1]
URL [本文引用: 2]
Category: GA Bibtype: Book Author: David E. Goldberg Title: Genetic Algorithm in Search, Optimization and Machine Learning Year: 1989 Address: Reading, MA Publisher: Addison-Wesley
URL [本文引用: 12]
Tools to predict single guide RNA (sgRNA) activity in the CRISPR/Cas9 system derived from S. pyogenes have been possible due to the large amount of data that have been generated in sgRNA library screens. However, with the discovery of additional CRISPR systems from different bacteria, which show potent activity in eukaryotic cells, the approach of generating large datasets for each of these... [Show full abstract]
URL [本文引用: 6]
The bacteria-derived clustered regularly interspaced short palindromic repeat (CRISPR)–Cas systems are powerful tools for genome engineering. Recently, in addition to Cas protein engineering, the improvement of guide RNAs are also performed, contributing to broadening the research area of CRISPR–Cas9 systems. Here we develop a fusion guide RNA (fgRNA) that functions with both Cas9 and Cpf1... [Show full abstract]
URL [本文引用: 1]
URLPMID:29251726 [本文引用: 2]
Abstract Combinatorial genetic screening using CRISPR-Cas9 is a useful approach to uncover redundant genes and to explore complex gene networks. However, current methods suffer from interference between the single-guide RNAs (sgRNAs) and from limited gene targeting activity. To increase the efficiency of combinatorial screening, we employ orthogonal Cas9 enzymes from Staphylococcus aureus and Streptococcus pyogenes. We used machine learning to establish S. aureus Cas9 sgRNA design rules and paired S. aureus Cas9 with S. pyogenes Cas9 to achieve dual targeting in a high fraction of cells. We also developed a lentiviral vector and cloning strategy to generate high-complexity pooled dual-knockout libraries to identify synthetic lethal and buffering gene pairs across multiple cell types, including MAPK pathway genes and apoptotic genes. Our orthologous approach also enabled a screen combining gene knockouts with transcriptional activation, which revealed genetic interactions with TP53. The "Big Papi" (paired aureus and pyogenes for interactions) approach described here will be widely applicable for the study of combinatorial phenotypes.
URLPMID:24837660 [本文引用: 4]
RNA-guided genome editing with the CRISPR-Cas9 system has great potential for basic and clinical research, but the determinants of targeting specificity and the extent of off-target cleavage remain insufficiently understood. Using chromatin immunoprecipitation and high-throughput sequencing (ChIP-seq), we mapped genome-wide binding sites of catalytically inactive Cas9 (dCas9) in HEK293T cells, in combination with 12 different single guide RNAs (sgRNAs). The number of off-target sites bound by dCas9 varied from 10 to >1,000 depending on the sgRNA. Analysis of off-target binding sites showed the importance of the PAM-proximal region of the sgRNA guiding sequence and that dCas9 binding sites are enriched in open chromatin regions. When targeted with catalytically active Cas9, some off-target binding sites had indels above background levels in a region around the ChIP-seq peak, but generally at lower rates than the on-target sites. Our results elucidate major determinants of Cas9 targeting, and we show that ChIP-seq allows unbiased detection of Cas9 binding sites genome-wide.
URLPMID:28655737 [本文引用: 11]
The adaptation of CRISPR/SpCas9 technology to mammalian cell lines is transforming the study of human functional genomics. Pooled libraries of CRISPR guide RNAs (gRNAs) targeting human protein-coding genes and encoded in viral vectors have been used to systematically create gene knockouts in a variety of human cancer and immortalized cell lines, in an effort to identify whether these knockouts cause cellular fitness defects. Previous work has shown that CRISPR screens are more sensitive and specific than pooled-library shRNA screens in similar assays, but currently there exists significant variability across CRISPR library designs and experimental protocols. In this study, we reanalyze 17 genome-scale knockout screens in human cell lines from three research groups, using three different genome-scale gRNA libraries. Using the Bayesian Analysis of Gene Essentiality algorithm to identify essential genes, we refine and expand our previously defined set of human core essential genes from 360 to 684 genes. We use this expanded set of reference core essential genes, CEG2, plus empirical data from six CRISPR knockout screens to guide the design of a sequence-optimized gRNA library, the Toronto KnockOut version 3.0 (TKOv3) library. We then demonstrate the high effectiveness of the library relative to reference sets of essential and nonessential genes, as well as other screens using similar approaches. The optimized TKOv3 library, combined with the CEG2 reference set, provide an efficient, highly optimized platform for performing and assessing gene knockout screens in human cell lines.
URL [本文引用: 3]
URLPMID:4833918 [本文引用: 9]
The adaptation of the CRISPR-Cas9 system to pooled library gene knockout screens in mammalian cells represents a major technological leap over RNA interference, the prior state of the art. New methods for analyzing the data and evaluating results are needed. We offer BAGEL (Bayesian Analysis of Gene EssentiaLity), a supervised learning method for analyzing gene knockout screens. Coupled with gold-standard reference sets of essential and nonessential genes, BAGEL offers significantly greater sensitivity than current methods, while computational optimizations reduce runtime by an order of magnitude. Using BAGEL, we identify ~2000 fitness genes in pooled library knockout screens in human cell lines at 5 % FDR, a major advance over competing platforms. BAGEL shows high sensitivity and specificity even across screens performed by different labs using different libraries and reagents.
URLPMID:27992409
A lentiviral library expressing Cpf1 guide RNAs and containing target sequences allows high-throughput profiling of highly active guide RNAs and is the basis for cindel, a webtool to predict the activity at any given target sequence.
URLPMID:4744125 [本文引用: 11]
CRISPR-Cas9-based genetic screens are a powerful new tool in biology. By simply altering the sequence of the single-guide RNA (sgRNA), Cas9 can be reprogrammed to target different sites in the genome with relative ease, but the on-target activity and off-target effects of individual sgRNAs can vary widely. Here, we use recently-devised sgRNA design rules to create human and mouse genome-wide libraries, perform positive and negative selection screens and observe that the use of these rules produced improved results. Additionally, we profile the off-target activity of thousands of sgRNAs and develop a metric to predict off-target sites. We incorporate these findings from large-scale, empirical data to improve our computational design rules and create optimized sgRNA libraries that maximize on-target activity and minimize off-target effects to enable more effective and efficient genetic screens and genome engineering.
URLPMID:5461693 [本文引用: 3]
CRISPR is a versatile gene editing tool which has revolutionized genetic research in the past few years. Optimizing sgRNA design to improve the efficiency of target/DNA cleavage is critical to ensure the success of CRISPR screens. By borrowing knowledge from oligonucleotide design and nucleosome occupancy models, we systematically evaluated candidate features computed from a number of nucleic acid, thermodynamic and secondary structure models on real CRISPR datasets. Our results showed that taking into account position-dependent dinucleotide features improved the design of effective sgRNAs with area under the receiver operating characteristic curve (AUC) >0.8, and the inclusion of additional features offered marginal improvement (2% increase in AUC). Using a machine-learning approach, we proposed an accurate prediction model for sgRNA design efficiency. An R package predictSGRNA implementing the predictive model is available athttp://www.ams.sunysb.edu/~pfkuan/softwares.html#predictsgrna. The online version of this article (doi:10.1186/s12859-017-1697-6) contains supplementary material, which is available to authorized users.
URL [本文引用: 10]
The CRISPR/Cas9 system is a creative and innovative gene editing biotechnology tool in genetic engineering. Although several achievements have been attained using the CRISPR/Cas9 system, it is still a challenge to avoid off-target effects and improve the editing efficacy. Previous efforts on evaluating the efficacy and designing the guide RNA mainly focused on DNA properties. However, some DNA features have not been characterized but can be reflected by protein properties such as the disorder features and the sequence conservation. In this study, we provided a computational framework to identify important features related to the efficacy of CRISPR/Cas9 focusing on the properties of the proteins encoded by the target DNA fragments. The feature selection method, maximal-relevance-minimal-redundancy (mRMR), was adopted to analyze these features. And incremental feature selection (IFS) together with support vector machine, were employed to extract optimal features, on which an optimal classifier can be constructed. As a result, 152 important features were extracted, with which an optimal classifier based on support vector machine was built. This classifier obtained the highest MCC value of 0.355. Finally, a series of detailed biological analyses were performed on the optimal features. From the results, we found that some key factors may differentially affect the binding activity of sgRNAs to their targets. Among them, the disorder status of the target protein sequences was found to be a major factor that is related to the efficacy of sgRNAs, suggesting the DNA features associated with the protein disorder status could also affect the CRISPR/Cas9 efficacy.
URL [本文引用: 2]
Examines a Swiss herdsman with an unusual hand condition. Swiss alpine valley general practitioner who makes house call to mountain farm; Hand and ankle oedema; Impeded blood flow from tight shirts which may cause swelling in hands; No information found on condition.
URL [本文引用: 1]
URL [本文引用: 2]
URLPMID:21048762 [本文引用: 1]
Bacteria and Archaea have developed several defence strategies against foreign nucleic acids such as viral genomes and plasmids. Among them, clustered regularly interspaced short palindromic repeats (CRISPR) loci together with cas (CRISPR-associated) genes form the CRISPR/Cas immune system, which involves partially palindromic repeats separated by short stretches of DNA called spacers, acquired from extrachromosomal elements. It was recently demonstrated that these variable loci can incorporate spacers from infecting bacteriophages and then provide immunity against subsequent bacteriophage infections in a sequence-specific manner. Here we show that the Streptococcus thermophilus CRISPR1/Cas system can also naturally acquire spacers from a self-replicating plasmid containing an antibiotic-resistance gene, leading to plasmid loss. Acquired spacers that match antibiotic-resistance genes provide a novel means to naturally select bacteria that cannot uptake and disseminate such genes. We also provide in vivo evidence that the CRISPR1/Cas system specifically cleaves plasmid and bacteriophage double-stranded DNA within the proto-spacer, at specific sites. Our data show that the CRISPR/Cas immune system is remarkably adapted to cleave invading DNA rapidly and has the potential for exploitation to generate safer microbial strains.
URL [本文引用: 1]
URLPMID:19246744 [本文引用: 1]
Abstract Clustered regularly interspaced short palindromic repeats (CRISPR) and their associated CRISPR-associated sequence (CAS) proteins constitute a novel antiviral defence system that is widespread in prokaryotes. Repeats are separated by spacers, some of them homologous to sequences in mobile genetic elements. Although the whole process involved remains uncharacterized, it is known that new spacers are incorporated into CRISPR loci of the host during a phage challenge, conferring specific resistance against the virus. Moreover, it has been demonstrated that such interference is based on small RNAs carrying a spacer. These RNAs would guide the defence apparatus to foreign molecules carrying sequences that match the spacers. Despite this essential role, the spacer uptake mechanism has not been addressed. A first step forward came from the detection of motifs associated with spacer precursors (proto-spacers) of Streptococcus thermophilus, revealing a specific recognition of donor sequences in this species. Here we show that the conservation of proto-spacer adjacent motifs (PAMs) is a common theme for the most diverse CRISPR systems. The PAM sequence depends on the CRISPR-CAS variant, implying that there is a CRISPR-type-specific (motif-directed) choice of the spacers, which subsequently determines the interference target. PAMs also direct the orientation of spacers in the repeat arrays. Remarkably, observations based on such polarity argue against a recognition of the spacer precursors on transcript RNA molecules as a general rule.
URLPMID:3070239 [本文引用: 1]
Abstract CRISPR/Cas systems constitute a widespread class of immunity systems that protect bacteria and archaea against phages and plasmids, and commonly use repeat/spacer-derived short crRNAs to silence foreign nucleic acids in a sequence-specific manner. Although the maturation of crRNAs represents a key event in CRISPR activation, the responsible endoribonucleases (CasE, Cas6, Csy4) are missing in many CRISPR/Cas subtypes. Here, differential RNA sequencing of the human pathogen Streptococcus pyogenes uncovered tracrRNA, a trans-encoded small RNA with 24-nucleotide complementarity to the repeat regions of crRNA precursor transcripts. We show that tracrRNA directs the maturation of crRNAs by the activities of the widely conserved endogenous RNase III and the CRISPR-associated Csn1 protein; all these components are essential to protect S. pyogenes against prophage-derived DNA. Our study reveals a novel pathway of small guide RNA maturation and the first example of a host factor (RNase III) required for bacterial RNA-mediated immunity against invaders.
URLPMID:24529477 [本文引用: 1]
Crystal structure of Cas9 in complex with single guide RNA and target DNA elucidates the molecular mechanism of RNA-guided DNA targeting by Cas9.
URLPMID:25713109 [本文引用: 2]
First introduced into mammalian organisms in 2013, the RNA-guided genome editing tool CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9) offers several advantages over conventional ones, such as simple-to-design, easy-to-use and multiplexing (capable of editing multiple genes simultaneously). Consequently, it has become a cost-effective and convenient tool for various genome editing purposes including gene therapy studies. In cell lines or animal models, CRISPR-Cas9 can be applied for therapeutic purposes in several ways. It can correct the causal mutations in monogenic disorders and thus rescue the disease phenotypes, which currently represents the most translatable field in CRISPR-Cas9-mediated gene therapy. CRISPR-Cas9 can also engineer pathogen genome such as HIV for therapeutic purposes, or induce protective or therapeutic mutations in host tissues. Moreover, CRISPR-Cas9 has shown potentials in cancer gene therapy such as deactivating oncogenic virus and inducing oncosuppressor expressions. Herein, we review the research on CRISPR-mediated gene therapy, discuss its advantages, limitations and possible solutions, and propose directions for future research, with an emphasis on the opportunities and challenges of CRISPR-Cas9 in cancer gene therapy.
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 4]
URLPMID:24476820 [本文引用: 2]
Abstract The clustered regularly interspaced short palindromic repeats (CRISPR)-associated enzyme Cas9 is an RNA-guided endonuclease that uses RNA-DNA base-pairing to target foreign DNA in bacteria. Cas9-guide RNA complexes are also effective genome engineering agents in animals and plants. Here we use single-molecule and bulk biochemical experiments to determine how Cas9-RNA interrogates DNA to find specific cleavage sites. We show that both binding and cleavage of DNA by Cas9-RNA require recognition of a short trinucleotide protospacer adjacent motif (PAM). Non-target DNA binding affinity scales with PAM density, and sequences fully complementary to the guide RNA but lacking a nearby PAM are ignored by Cas9-RNA. Competition assays provide evidence that DNA strand separation and RNA-DNA heteroduplex formation initiate at the PAM and proceed directionally towards the distal end of the target sequence. Furthermore, PAM interactions trigger Cas9 catalytic activity. These results reveal how Cas9 uses PAM recognition to quickly identify potential target sites while scanning large DNA molecules, and to regulate scission of double-stranded DNA.
URLPMID:4066725 [本文引用: 3]
Abstract CRISPR/Cas9-mediated DNA cleavage (CCMDC) is becoming increasingly used for efficient genome engineering. Proto-spacer adjacent motif (PAM) adjacent to target sequence is one of the key components in the design of CCMDC strategies. It has been reported that NAG sequences are the predominant non-canonical PAM for CCMDC at the human EMX locus, but it is not clear whether it is universal at other loci. In the present study, we attempted to use a GFP-reporter system to comprehensively and quantitatively test the efficiency of CCMDC with non-canonical PAMs in human cells. The initial results indicated that the effectiveness of NGA PAM for CCMDC is much higher than that of other 14 PAMs including NAG. Then we further designed another three pairs of NGG, NGA and NAG PAMs at different locations in the GFP gene and investigated the corresponding DNA cleavage efficiency. We observed that one group of NGA PAMs have a relatively higher DNA cleavage efficiency, while the other groups have lower efficiency, compared with the corresponding NAG PAMs. Our study clearly demonstrates that NAG may not be the universally predominant non-canonical PAM for CCMDC in human cells. These findings raise more concerns over off-target effects in CRISPR/Cas9-mediated genome engineering.
URLPMID:3969858 [本文引用: 11]
The Streptococcus pyogenes Cas9 (SpCas9) nuclease can be efficiently targeted to genomic loci by means of single-guide RNAs (sgRNAs) to enable genome editing(1-10). Here, we characterize SpCas9 targeting specificity in human cells to inform the selection of target sites and avoid off-target effects. Our study evaluates >700 guide RNA variants and SpCas9-induced indel mutation levels at >100 predicted genomic off-target loci in 293T and 293FT cells. We find that SpCas9 tolerates mismatches between guide RNA and target DNA at different positions in a sequence-dependent manner, sensitive to the number, position and distribution of mismatches. We also show that SpCas9-mediated cleavage is unaffected by DNA methylation and that the dosage of SpCas9 and sgRNA can be titrated to minimize off-target modification. To facilitate mammalian genome engineering applications, we provide a web-based software tool to guide the selection and validation of target sequences as well as off-target analyses.
URLPMID:4540238 [本文引用: 1]
Engineered CRISPR-Cas9 nucleases with altered and improved PAM specificities and their use in genomic engineering, epigenomic engineering, and genome targeting.
URL [本文引用: 1]
URL [本文引用: 6]
URLPMID:4145672 [本文引用: 5]
Bacterial type II CRISPR-Cas9 systems have been widely adapted for RNA-guided genome editing and transcription regulation in eukaryotic cells, yet their in vivo target specificity is poorly understood. Here we mapped genome-wide binding sites of a catalytically inactive Cas9 (dCas9) from Streptococcus pyogenes loaded with single guide RNAs (sgRNAs) in mouse embryonic stem cells (mESCs). Each of the four sgRNAs we tested targets dCas9 to between tens and thousands of genomic sites, frequently characterized by a 5-nucleotide seed region in the sgRNA and an NGG protospacer adjacent motif (PAM). Chromatin inaccessibility decreases dCas9 binding to other sites with matching seed sequences; thus 70% of off-target sites are associated with genes. Targeted sequencing of 295 dCas9 binding sites in mESCs transfected with catalytically active Cas9 identified only one site mutated above background levels. We propose a two-state model for Cas9 binding and cleavage, in which a seed match triggers binding but extensive pairing with target DNA is required for cleavage.
URLPMID:24972169 [本文引用: 1]
A letter to the editor is presented which describes a computer program using microhomology prediction to assist in the choice of Cas9 nuclease, zinc-finger nuclease and transcription activator-like effector nuclease (TALEN) target sites.
URLPMID:4262738 [本文引用: 3]
Components of the prokaryotic clustered, regularly interspaced, short palindromic repeats (CRISPR) loci have recently been repurposed for use in mammalian cells. The CRISPR-associated (Cas)9 can be programmed with a single guide RNA (sgRNA) to generate site-specific DNA breaks, but there are few known rules governing on-target efficacy of this system. We created a pool of sgRNAs, tiling across all possible target sites of a panel of six endogenous mouse and three endogenous human genes and quantitatively assessed their ability to produce null alleles of their target gene by antibody staining and flow cytometry. We discovered sequence features that improved activity, including a further optimization of the protospacer-adjacent motif (PAM) of Streptococcus pyogenes Cas9. The results from 1,841 sgRNAs were used to construct a predictive model of sgRNA activity to improve sgRNA design for gene editing and genetic screens. We provide an online tool for the design of highly active sgRNAs for any gene of interest.
URLPMID:26322839 [本文引用: 1]
CRISPR/Cas9 technology provides a powerful system for genome engineering. However, variable activity across different single guide RNAs (sgRNAs) remains a significant limitation. We have analyzed the molecular features that influence sgRNA stability, activity and loading into Cas9in vivo. We observe that guanine enrichment and adenine depletion increase sgRNA stability and activity, while loading, nucleosome positioning and Cas9 off-target binding are not major determinants. We additionally identified truncated and 5′ mismatch-containing sgRNAs as efficient alternatives to canonical sgRNAs. Based on these results, we created a predictive sgRNA-scoring algorithm (CRISPRscan.org) that effectively captures the sequence features affecting Cas9/sgRNA activityin vivo. Finally, we show that targeting Cas9 to the germ line using a Cas9-nanos-3′-UTR fusion can generate maternal-zygotic mutants, increase viability and reduce somatic mutations. Together, these results provide novel insights into the determinants that influence Cas9 activity and a framework to identify highly efficient sgRNAs for genome targetingin vivo.
URLPMID:24336569 [本文引用: 3]
Abstract The bacterial clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system for genome editing has greatly expanded the toolbox for mammalian genetics, enabling the rapid generation of isogenic cell lines and mice with modified alleles. Here, we describe a pooled, loss-of-function genetic screening approach suitable for both positive and negative selection that uses a genome-scale lentiviral single-guide RNA (sgRNA) library. sgRNA expression cassettes were stably integrated into the genome, which enabled a complex mutant pool to be tracked by massively parallel sequencing. We used a library containing 73,000 sgRNAs to generate knockout collections and performed screens in two human cell lines. A screen for resistance to the nucleotide analog 6-thioguanine identified all expected members of the DNA mismatch repair pathway, whereas another for the DNA topoisomerase II (TOP2A) poison etoposide identified TOP2A, as expected, and also cyclin-dependent kinase 6, CDK6. A negative selection screen for essential genes identified numerous gene sets corresponding to fundamental processes. Last, we show that sgRNA efficiency is associated with specific sequence motifs, enabling the prediction of more effective sgRNAs. Collectively, these results establish Cas9/sgRNA screens as a powerful tool for systematic genetic analysis in mammalian cells.
URL [本文引用: 4]
URLPMID:24463574 [本文引用: 2]
Abstract Clustered, regularly interspaced, short palindromic repeat (CRISPR) RNA-guided nucleases (RGNs) are highly efficient genome editing tools. CRISPR-associated 9 (Cas9) RGNs are directed to genomic loci by guide RNAs (gRNAs) containing 20 nucleotides that are complementary to a target DNA sequence. However, RGNs can induce mutations at sites that differ by as many as five nucleotides from the intended target. Here we report that truncated gRNAs, with shorter regions of target complementarity <20 nucleotides in length, can decrease undesired mutagenesis at some off-target sites by 5,000-fold or more without sacrificing on-target genome editing efficiencies. In addition, use of truncated gRNAs can further reduce off-target effects induced by pairs of Cas9 variants that nick DNA (paired nickases). Our results delineate a simple, effective strategy to improve the specificities of Cas9 nucleases or paired nickases.
URL [本文引用: 1]
URLPMID:23992846 [本文引用: 1]
Targeted genome editing technologies have enabled a broad range of research and medical applications. The Cas9 nuclease from the microbial CRISPR-Cas system is targeted to specific genomic loci by a 20 nt guide sequence, which can tolerate certain mismatches to the DNA target and thereby promote undesired off-target mutagenesis. Here, we describe an approach that combines a Cas9 nickase mutant with paired guide RNAs to introduce targeted double-strand breaks. Because individual nicks in the genome are repaired with high fidelity, simultaneous nicking via appropriately offset guide RNAs is required for double-stranded breaks and extends the number of specifically recognized bases for target cleavage. We demonstrate that using paired nicking can reduce off-target activity by 50- to 1,500-fold in cell lines and to facilitate gene knockout in mouse zygotes without sacrificing on-target cleavage efficiency. This versatile strategy enables a wide variety of genome editing applications that require high specificity.
URL [本文引用: 13]
URL [本文引用: 1]
URLPMID:26121414 [本文引用: 1]
The article discusses the disadvantages of using the online tool, CRISPR-Cas9 endonuclease system, for single guide RNA (sgRNA) which include limited flexibility and throughput, its availability for only few organisms, and the limited mismatches on the number of target-sgRNA. The flexibility and speed provided by offline solutions such as sgRNAcas9 is stated. A chart is presented depicting the offline software, Protospacer Workbench, for rapid, flexible design of Cas9 sgRNA.
URLPMID:24199189 [本文引用: 1]
Cas9/CRISPR has been reported to efficiently induce targeted gene disruption and homologous recombination in both prokaryotic and eukaryotic cells. Thus, we developed a Guide RNA Sequence Design Platform for the Cas9/CRISPR silencing system for model organisms. The platform is easy to use for gRNA design with input query sequences. It finds potential targets by PAM and ranks them according to factors including uniqueness, SNP, RNA secondary structure, and AT content. The platform allows users to upload and share their experimental results. In addition, most guide RNA sequences from published papers have been put into our database.
URL [本文引用: 2]
URL [本文引用: 1]
URLPMID:26735016 [本文引用: 1]
CRISPR as9 nucleases are widely used for genome editing but can induce unwanted off-target mutations. Existing strategies for reducing genome-wide off-target effects of the widely used Streptococcus pyogenes Cas9 (SpCas9) are imperfect, possessing only partial or unproven efficacies and other limitations that constrain their use. Here we describe SpCas9-HF1, a high-fidelity variant harbouring alterations designed to reduce non-specific DNA contacts. SpCas9-HF1 retains on-target activities comparable to wild-type SpCas9 with >85% of single-guide RNAs (sgRNAs) tested in human cells. Notably, with sgRNAs targeted to standard non-repetitive sequences, SpCas9-HF1 rendered all or nearly all off-target events undetectable by genome-wide break capture and targeted sequencing methods. Even for atypical, repetitive target sites, the vast majority of off-target mutations induced by wild-type SpCas9 were not detected with SpCas9-HF1. With its exceptional precision, SpCas9-HF1 provides an alternative to wild-type SpCas9 for research and therapeutic applications. More broadly, our results suggest a general strategy for optimizing genome-wide specificities of other CRISPR-RNA-guided nucleases.
URLPMID:25503383 [本文引用: 1]
Although great progress has been made in the characterization of the off-target effects of engineered nucleases, sensitive and unbiased genome-wide methods for the detection of off-target cleavage events and potential collateral damage are still lacking. Here we describe a linear amplification-mediated modification of a previously published high-throughput, genome-wide, translocation sequencing (HTGTS) method that robustly detects DNA double-stranded breaks (DSBs) generated by engineered nucleases across the human genome based on their translocation to other endogenous or ectopic DSBs. HTGTS with different Cas9:sgRNA or TALEN nucleases revealed off-target hotspot numbers for given nucleases that ranged from a few or none to dozens or more, and extended the number of known off-targets for certain previously characterized nucleases more than tenfold. We also identified translocations between bona fide nuclease targets on homologous chromosomes, an undesired collateral effect that has not been described previously. Finally, HTGTS confirmed that the Cas9D10A paired nickase approach suppresses off-target cleavage genome-wide.
URL [本文引用: 8]
URLPMID:28732207 [本文引用: 5]
We have combined a machine-learning approach with other strategies to optimize knockout-efficiency with the CRISPR/Cas9 system. In addition, we have developed a multiplexed sgRNA expression strategy that promotes the functional ablation of single genes, and allows for combinatorial targeting. These strategies have been combined to design and construct a genome-wide, sequence verified, arrayed CRISPR library. This resource allows single-target or combinatorial genetic screens to be carried out at scale, in a multiplexed or arrayed format. By conducting parallel loss-of-function screens, we compare our approach to existing sgRNA design and expression strategies.
URLPMID:28146356 [本文引用: 2]
It has been possible to create tools to predict single guide RNA (sgRNA) activity in the CRISPR/Cas9 system derived from Streptococcus pyogenes due to the large amount of data that has been generated in sgRNA library screens. However, with the discovery of additional CRISPR systems from different bacteria, which show potent activity in eukaryotic cells, the approach of generating large data sets for each of these systems to predict their activity is not tractable. Here, we present a new guide RNA tool that can predict sgRNA activity across multiple CRISPR systems. In addition to predicting activity for Cas9 from S. pyogenes and Streptococcus thermophilus CRISPR1, we experimentally demonstrate that our algorithm can predict activity for Cas9 from Staphylococcus aureus and S. thermophilus CRISPR3. We also have made available a new version of our software, sgRNA Scorer 2.0, which will allow users to identify sgRNA sites for any PAM sequence of interest.
URLPMID:27402906 [本文引用: 2]
Abstract MOTIVATION: Despite the growing popularity in using CRISPR/Cas9 technology for genome editing and gene knockout, its performance still relies on well-designed single guide RNAs (sgRNA). In this study, we propose a web application for the Design and Optimization (CRISPR-DO) of guide sequences that target both coding and non-coding regions in spCas9 CRISPR system across human, mouse, zebrafish, fly and worm genomes. CRISPR-DO uses a computational sequence model to predict sgRNA efficiency, and employs a specificity scoring function to evaluate the potential of off-target effect. It also provides information on functional conservation of target sequences, as well as the overlaps with exons, putative regulatory sequences and single-nucleotide polymorphisms (SNPs). The web application has a user-friendly genome-browser interface to facilitate the selection of the best target DNA sequences for experimental design. AVAILABILITY AND IMPLEMENTATION: CRISPR-DO is available at http://cistrome.org/crispr/ CONTACT: qiliu@tongji.edu.cn or hanxu@jimmy.harvard.edu or xsliu@jimmy.harvard.eduSupplementary information: Supplementary data are available at Bioinformatics online. The Author 2016. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
URL [本文引用: 4]
URLPMID:26063738
Abstract The CRISPR/Cas9 system has revolutionized mammalian somatic cell genetics. Genome-wide functional screens using CRISPR/Cas9-mediated knockout or dCas9 fusion-mediated inhibition/activation (CRISPRi/a) are powerful techniques for discovering phenotype-associated gene function. We systematically assessed the DNA sequence features that contribute to single guide RNA (sgRNA) efficiency in CRISPR-based screens. Leveraging the information from multiple designs, we derived a new sequence model for predicting sgRNA efficiency in CRISPR/Cas9 knockout experiments. Our model confirmed known features and suggested new features including a preference for cytosine at the cleavage site. The model was experimentally validated for sgRNA-mediated mutation rate and protein knockout efficiency. Tested on independent data sets, the model achieved significant results in both positive and negative selection conditions and outperformed existing models. We also found that the sequence preference for CRISPRi/a is substantially different from that for CRISPR/Cas9 knockout and propose a new model for predicting sgRNA efficiency in CRISPRi/a experiments. These results facilitate the genome-wide design of improved sgRNA for both knockout and CRISPRi/a studies. 2015 Xu et al.; Published by Cold Spring Harbor Laboratory Press.
URLPMID:26167643 [本文引用: 2]
We develop anin vivolibrary-on-library methodology to simultaneously assess single guide RNA (sgRNA) activity across ~1,400 genomic loci. Assaying across multiple human cell types, end-processing enzymes, and two Cas9 orthologs, we unravel underlying nucleotide sequence and epigenetic parameters. Our results enable improved design of reagents, shed light on mechanisms of genome targeting, and provide a generalizable framework to study nucleic acid-nucleic acid interactions and biochemistry in high throughput.
URLPMID:29868757 [本文引用: 3]
Abstract Motivation: Genome-wide CRISPR-Cas9 screen has been widely used to interrogate gene functions. However, the rules to design better libraries beg further refinement. Results: We found sgRNA outliers are characterized by higher G-nucleotide counts, especially in regions distal from the PAM motif, and are associated with stronger off-target activities. Furthermore, using non-targeting sgRNAs as negative controls lead to strong bias, which can be mitigated by using sgRNAs targeting multiple "safe harbor" regions. Custom-designed screens confirmed our findings and further revealed that 19nt sgRNAs consistently gave the best signal-to-noise ratio. Collectively, our analysis motivated the design of a new genome-wide CRISPR/Cas9 screen library and uncovered some intriguing properties of the CRISPR-Cas9 system. Availability: The MAGeCK workflow is available open source at https://bitbucket.org/liulab/mageck_nest under the MIT license. Contact: xsliu@jimmy.harvard.edu. Supplementary information: Supplementary data are available at Bioinformatics online.
[本文引用: 1]
[本文引用: 1]
URLPMID:4714946 [本文引用: 1]
The RNA-guided endonuclease Cas9 is a versatile genome-editing tool with a broad range of applications from therapeutics to functional annotation of genes. Cas9 creates double-strand breaks (DSBs) at targeted genomic loci complementary to a short RNA guide. However, Cas9 can cleave off-target sites that are not fully complementary to the guide, which poses a major challenge for genome editing. Here, we use structure-guided protein engineering to improve the specificity of Streptococcus pyogenes Cas9 (SpCas9). Using targeted deep sequencing and unbiased whole-genome off-target analysis to assess Cas9-mediated DNA cleavage in human cells, we demonstrate that “enhanced specificity” SpCas9 (eSpCas9) variants reduce off-target effects and maintain robust on-target cleavage. Thus, eSpCas9 could be broadly useful for genome-editing applications requiring a high level of specificity. Authors: Ian M. Slaymaker, Linyi Gao, Bernd Zetsche, David A. Scott, Winston X. Yan, Feng Zhang
URLPMID:4320685 [本文引用: 1]
Abstract CRISPR RNA-guided nucleases (RGNs) are widely used genome-editing reagents, but methods to delineate their genome-wide, off-target cleavage activities have been lacking. Here we describe an approach for global detection of DNA double-stranded breaks (DSBs) introduced by RGNs and potentially other nucleases. This method, called genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq), relies on capture of double-stranded oligodeoxynucleotides into DSBs. Application of GUIDE-seq to 13 RGNs in two human cell lines revealed wide variability in RGN off-target activities and unappreciated characteristics of off-target sequences. The majority of identified sites were not detected by existing computational methods or chromatin immunoprecipitation sequencing (ChIP-seq). GUIDE-seq also identified RGN-independent genomic breakpoint 'hotspots'. Finally, GUIDE-seq revealed that truncated guide RNAs exhibit substantially reduced RGN-induced, off-target DSBs. Our experiments define the most rigorous framework for genome-wide identification of RGN off-target effects to date and provide a method for evaluating the safety of these nucleases before clinical use.
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
Different classification tasks require different learning schemes to be satisfactorily solved. Most real-world datasets can be modeled only by complex structures resulting from deep data exploration with a number of different classification and data transformation methods. The search through the space of complex structures must be augmented with reliable validation strategies. All these techniques were necessary to build accurate models for the five high-dimensional datasets of the NIPS 2003 Feature Selection Challenge. Several feature selection algorithms (e.g. based on variance, correlation coefficient, decision trees) and several classification schemes (e.g. nearest neighbors, Normalized RBF, Support Vector Machines) were used to build complex models which transform the data and then classify. Committees of feature selection models and ensemble classifiers were also very helpful to construct models of high generalization abilities.
URL [本文引用: 1]
Variable and feature selection have become the focus of much research in areas of application for which datasets with tens or hundreds of thousands of variables are available. These areas include text processing of internet documents, gene expression array analysis, and combinatorial chemistry. The objective of variable selection is three-fold: improving the prediction performance of the predictors, providing faster and more cost-effective predictors, and providing a better understanding of the underlying process that generated the data. The contributions of this special issue cover a wide range of aspects of such problems: providing a better definition of the objective function, feature construction, feature ranking, multivariate feature selection, efficient search methods, and feature validity assessment methods.
URLPMID:12369084 [本文引用: 1]
Abstract The area under the receiver operating characteristic curve is frequently used as a measure for the effectiveness of diagnostic markers. In this paper we discuss and compare estimation procedures for this area. These are based on (i) the Mann鈥揥hitney statistic; (ii) kernel smoothing; (iii) normal assumptions; (iv) empirical transformations to normality. These are compared in terms of bias and root mean square error in a large variety of situations by means of an extensive simulation study. Overall we find that transforming to normality usually is to be preferred except for bimodal cases where kernel methods can be effective. Copyright 2002 John Wiley & Sons, Ltd.
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:25748654 [本文引用: 1]
Using an in02vivo genome-wide CRISPR/Cas9 screen, loss-of-function mutations that drive tumor growth and metastasis to the lung have been identified, demonstrating Cas9-based screening as a robust method to systematically assay gene phenotypes in cancer evolution.
URLPMID:24584096 [本文引用: 1]
Abstract Targeted genome editing using engineered nucleases has rapidly gone from being a niche technology to a mainstream method used by many biological researchers. This widespread adoption has been largely fueled by the emergence of the clustered, regularly interspaced, short palindromic repeat (CRISPR) technology, an important new approach for generating RNA-guided nucleases, such as Cas9, with customizable specificities. Genome editing mediated by these nucleases has been used to rapidly, easily and efficiently modify endogenous genes in a wide variety of biomedically important cell types and in organisms that have traditionally been challenging to manipulate genetically. Furthermore, a modified version of the CRISPR-Cas9 system has been developed to recruit heterologous domains that can regulate endogenous gene expression or label specific genomic loci in living cells. Although the genome-wide specificities of CRISPR-Cas9 systems remain to be fully defined, the power of these systems to perform targeted, highly efficient alterations of genome sequence and gene expression will undoubtedly transform biological research and spur the development of novel molecular therapeutics for human disease.
URLPMID:26612492 [本文引用: 1]
CRISPR-based approaches have quickly become a favored method to perturb genes to uncover their functions. Here, we review the key considerations in the design of genome editing experiments, and...
URLPMID:23849981 [本文引用: 1]
Catalytically inactive CRISPR can be targeted to specific loci in human and yeast cells to specifically repress and activate transcription. The study demonstrates the potential for adapting CRISPRi for multiple modes of transcriptional control, chromatin modification, and regulatory element mapping in a broad range of eukaryotes.
URLPMID:23892895 [本文引用: 1]
Technologies for engineering synthetic transcription factors have enabled many advances in medical and scientific research. In contrast to existing methods based on engineering of DNA-binding proteins, we created a Cas9-based transactivator that is targeted to DNA sequences by guide RNA molecules. Coexpression of this transactivator and combinations of guide RNAs in human cells induced specific expression of endogenous target genes, demonstrating a simple and versatile approach for RNA-guided gene activation.
URLPMID:27418421 [本文引用: 3]
CRISPR-based genome editing is widely implemented in various cell types and organisms. This rapidly advancing technology mainly comprises the following types of genetic perturbations: gene knockout (KO), gene knockin (KI) for genome editing, and inhibition or activation of gene expression (CRISPRi/a). A major challenge in effectively applying the CRISPR/Cas9 endonuclease system to all of these genetic perturbations is designing highly-efficient single-guide (sg)RNA with minimal off-target cleavage. Because the CRISPR technique has quickly advanced, increasing volumes of genome-editing data have accumulated and challenging computational problems have emerged.In silicosgRNA design has become a key issue for successful gene-editing experiments and will allow CRISPR studies to take advantage of various bioinformatics and computational techniques.
URLPMID:16990807 [本文引用: 1]
Abstract Large-scale RNA interference (RNAi)-based analyses, very much as other 'omic' approaches, have inherent rates of false positives and negatives. The variability in the standards of care applied to validate results from these studies, if left unchecked, could eventually begin to undermine the credibility of RNAi as a powerful functional approach. This Commentary is an invitation to an open discussion started among various users of RNAi to set forth accepted standards that would insure the quality and accuracy of information in the large datasets coming out of genome-scale screens.
URL [本文引用: 1]
URLPMID:5424143 [本文引用: 1]
CRISPR-Cas9 screens are powerful tools for high-throughput interrogation of genome function, but can be confounded by nuclease-induced toxicity at both on- and off-target sites, likely due to DNA damage. Here, to test potential solutions to this issue, we design and analyse a CRISPR-Cas9 library with 10 variable-length guides per gene and thousands of negative controls targeting non-functional, non-genic regions (termed safe-targeting guides), in addition to non-targeting controls. We find this library has excellent performance in identifying genes affecting growth and sensitivity to the ricin toxin. The safe-targeting guides allow for proper control of toxicity from on-target DNA damage. Using this toxicity as a proxy to measure off-target cutting, we demonstrate with tens of thousands of guides both the nucleotide position-dependent sensitivity to single mismatches and the reduction of off-target cutting using truncated guides. Our results demonstrate a simple strategy for high-throughput evaluation of target specificity and nuclease toxicity in Cas9 screens.
URLPMID:28203699 [本文引用: 1]
Abstract CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-based gene editing has been widely implemented in various cell types and organisms. A major challenge in the effective application of the CRISPR system is the need to design highly efficient single-guide RNA (sgRNA) with minimal off-target cleavage. Several tools are available for sgRNA design, while limited tools were compared. In our opinion, benchmarking the performance of the available tools and indicating their applicable scenarios are important issues. Moreover, whether the reported sgRNA design rules are reproducible across different sgRNA libraries, cell types and organisms remains unclear. In our study, a systematic and unbiased benchmark of the sgRNA predicting efficacy was performed on nine representative on-target design tools, based on six benchmark data sets covering five different cell types. The benchmark study presented here provides novel quantitative insights into the available CRISPR tools. The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
URL [本文引用: 1]
URLPMID:26564855 [本文引用: 1]
The RNA-guided CRISPR-associated protein Cas9 is used for genome editing, transcriptional modulation, and live-cell imaging. Cas9-guide RNA complexes recognize and cleave double-stranded DNA sequences on the basis of 20-nucleotide RNA-DNA complementarity, but the mechanism of target searching in mammalian cells is unknown. Here, we use single-particle tracking to visualize diffusion and chromatin binding of Cas9 in living cells. We show that three-dimensional diffusion dominates Cas9 searching in vivo, and off-target binding events are, on average, short-lived (<1 second). Searching is dependent on the local chromatin environment, with less sampling and slower movement within heterochromatin. These results reveal how the bacterial Cas9 protein interrogates mammalian genomes and navigates eukaryotic chromatin structure.
URLPMID:4529991 [本文引用: 1]
CRISPR-Cas9 genome editing technology holds great promise for discovering therapeutic targets in cancer and other diseases. Current screening strategies target CRISPR-induced mutations to the 5’ exons of candidate genes1–5, but this approach often produces in-frame variants that retain functionality, which can obscure even strong genetic dependencies. Here we overcome this limitation by targeting CRISPR mutagenesis to exons encoding functional protein domains. This generates a higher proportion of null mutations and substantially increases the potency of negative selection. We show that the magnitude of negative selection reports the functional importance of individual protein domains of interest. A screen of 192 chromatin regulatory domains in murine acute myeloid leukemia cells identifies six known drug targets and 19 additional dependencies. A broader application of this approach may allow comprehensive identification of protein domains that sustain cancer cells and are suitable for drug targeting.
URLPMID:26627737 [本文引用: 1]
CRISPR knockout screens for essential genes reveal oncogenic drivers specific to different cancer cell lines as well as unexpected metabolic and signaling dependencies that may guide future therapeutic targeting.
URLPMID:5547152 [本文引用: 1]
Genome-wide CRISPR-based knockout (CRISPR-KO) screening is an emerging technique which enables systematic genetic analysis of a cellular or molecular phenotype in question. Continuous improvements, such as modifications to the guide RNA (gRNA) scaffold and the development of gRNA on-target prediction algorithms, have since been made to increase their screening performance. We compared the performance of three available second-generation human genome-wide CRISPR-KO libraries that included at least one of the improvements, and examined the effect of gRNA scaffold, number of gRNAs per gene and number of replicates on screen performance. We identified duplicated screens using a library with 6 gRNAs per gene as providing the best trade-off. Despite the improvements, we found that each improved library still has library-specific false negatives and, for the first time, estimated the false negative rates of CRISPR-KO screens, which are between 10% and 20%. Our newly-defined optimal screening parameters would be helpful in designing screens and constructing bespoke gRNA libraries.
URLPMID:27111720 [本文引用: 1]
Abstract High-throughput genetic screens have become essential tools for studying a wide variety of biological processes. Here we experimentally compare systems based on clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) or its transcriptionally repressive variant, CRISPR-interference (CRISPRi), with a traditional short hairpin RNA (shRNA)-based system for performing lethality screens. We find that the CRISPR technology performed best, with low noise, minimal off-target effects and consistent activity across reagents.
URLPMID:24360272 [本文引用: 1]
The spatiotemporal organization and dynamics of chromatin play critical roles in regulating genome function. However, visualizing specific, endogenous genomic loci remains challenging in living cells. Here, we demonstrate such an imaging technique by repurposing the bacterial CRISPR/Cas system. Using an EGFP-tagged endonuclease-deficient Cas9 protein and a structurally optimized small guide (sg) RNA, we show robust imaging of repetitive elements in telomeres and coding genes in living cells. Furthermore, an array of sgRNAs tiling along the target locus enables the visualization of nonrepetitive genomic sequences. Using this method, we have studied telomere dynamics during elongation or disruption, the subnuclear localization of the MUC4 loci, the cohesion of replicated MUC4 loci on sister chromatids, and their dynamic behaviors during mitosis. This CRISPR imaging tool has potential to significantly improve the capacity to study the conformation and dynamics of native chromosomes in living human cells.
URL [本文引用: 1]
URLPMID:4861601 [本文引用: 2]
10.7554/eLife.12677.001The prokaryotic CRISPR (clustered regularly interspaced palindromic repeats)-associated protein, Cas9, has been widely adopted as a tool for editing, imaging, and regulating eukaryotic genomes. However, our understanding of how to select single-guide RNAs (sgRNAs) that mediate efficient Cas9 activity is incomplete, as we lack insight into how chromatin impacts Cas9 targeting. To address this gap, we analyzed large-scale genetic screens performed in human cell lines using either nuclease-active or nuclease-dead Cas9 (dCas9). We observed that highly active sgRNAs for Cas9 and dCas9 were found almost exclusively in regions of low nucleosome occupancy. In vitro experiments demonstrated that nucleosomes in fact directly impede Cas9 binding and cleavage, while chromatin remodeling can restore Cas9 access. Our results reveal a critical role of eukaryotic chromatin in dictating the targeting specificity of this transplanted bacterial enzyme, and provide rules for selecting Cas9 target sites distinct from and complementary to those based on sequence properties.DOI: http://dx.doi.org/10.7554/eLife.12677.001
[本文引用: 1]
URLPMID:27918548 [本文引用: 1]
Targeting of multiple genomic loci with Cas9 is limited by the need for multiple or large expression constructs. Here we show that the ability of Cpf1 to process its own CRISPR RNA (crRNA) can be used to simplify multiplexed genome editing. Using a single customized CRISPR array, we edit up to four genes in mammalian cells and three in the mouse brain, simultaneously.
URL [本文引用: 1]
URLPMID:26786045 [本文引用: 1]
We present multiplex Digenome-seq to profile genome-wide specificities of up to 11 CRISPR-Cas9 nucleases simultaneously, saving time and reducing cost. Cell-free human genomic DNA was digested using multiple sgRNAs combined with the Cas9 protein and then subjected to whole-genome sequencing. In vitro cleavage patterns, characteristic of on- and off-target sites, were computationally identified across the genome using a new DNA cleavage scoring system. We found that many false-positive, bulge-type off-target sites were cleaved by sgRNAs transcribed from an oligonucleotide duplex but not by those transcribed from a plasmid template. Multiplex Digenome-seq captured many bona fide off-target sites, missed by other genome-wide methods, at which indels were induced at frequencies <0.1%. After analyzing 964 sites cleaved in vitro by these sgRNAs and measuring indel frequencies at hundreds of off-target sites in cells, we propose a guideline for the choice of target sites for minimizing CRISPR-Cas9 off-target effects in the human genome.
URL [本文引用: 1]
URL [本文引用: 2]
Nucleotide base editors in plants have been limited to conversion of cytosine to thymine. Here, we describe a new plant adenine base editor based on an evolved tRNA adenosine deaminase fused to the nickase CRISPR/Cas9, enabling A61T to G61C conversion at frequencies up to 7.5% in protoplasts and 59.1% in regenerated rice and wheat plants. An endogenous gene is also successfully modified through introducing a gain-of-function point mutation to directly produce an herbicide-tolerant rice plant. With this new adenine base editing system, it is now possible to precisely edit all base pairs, thus expanding the toolset for precise editing in plants. The online version of this article (10.1186/s13059-018-1443-z) contains supplementary material, which is available to authorized users.
[本文引用: 1]
[本文引用: 1]
URLPMID:29512652 [本文引用: 3]
Programmable DNA nucleases have provided scientists with the unprecedented ability to probe, regulate, and manipulate the human genome. Zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeat-Cas9 system (CRISPR-Cas9) represent a powerful array of tools that can bind to and cleave a specified DNA... [Show full abstract]
URLPMID:29070703 [本文引用: 1]
Abstract Nucleic acid editing holds promise for treating genetic disease, particularly at the RNA level, where disease-relevant sequences can be rescued to yield functional protein products. Type VI CRISPR-Cas systems contain the programmable single-effector RNA-guided RNases Cas13. Here, we profile Type VI systems to engineer a Cas13 ortholog capable of robust knockdown and demonstrate RNA editing by using catalytically-inactive Cas13 (dCas13) to direct adenosine to inosine deaminase activity by ADAR2 to transcripts in mammalian cells. This system, referred to as RNA Editing for Programmable A to I Replacement (REPAIR), which has no strict sequence constraints, can be used to edit full-length transcripts containing pathogenic mutations. We further engineer this system to create a high specificity variant and minimize the system to facilitate viral delivery. REPAIR presents a promising RNA editing platform with broad applicability for research, therapeutics, and biotechnology. Copyright 2017, American Association for the Advancement of Science.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}