 ,1,2
,1,2Effects of CSN4 knockdown on proliferation and apoptosis of breast cancer MDA-MB-231 cells
Tonglu YU1,2, Dongliang Cai1,2, Genfeng Zhu1,2, Xiaojuan Ye1,2, Taishan Min1,2, Hongyan Chen1,2, Daru Lu1,2, Haoming Chen,1,2通讯作者:
编委: 周钢桥
收稿日期:2018-12-11修回日期:2019-02-18网络出版日期:2019-04-20
| 基金资助: |
Received:2018-12-11Revised:2019-02-18Online:2019-04-20
| Fund supported: |
作者简介 About authors
余同露,硕士研究生;专业方向:生物工程E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (722KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
余同露, 蔡栋梁, 朱根凤, 叶晓娟, 闵太善, 陈红岩, 卢大儒, 陈浩明. CSN4基因干扰对乳腺癌MDA-MB-231细胞增殖和凋亡的影响[J]. 遗传, 2019, 41(4): 318-326 doi:10.16288/j.yczz.18-278
Tonglu YU, Dongliang Cai, Genfeng Zhu, Xiaojuan Ye, Taishan Min, Hongyan Chen, Daru Lu, Haoming Chen.
乳腺癌是危害女性健康最常见的恶性肿瘤之一,其发病率逐年上升。据GLOBOCAN (global cancer observatory)的数据显示,乳腺癌高居中国女性癌症发病率首位,发病率为21.6/10万[1]。2015年,中国新发乳腺癌高达27万例,严重威胁女性健康[2]。乳腺癌细胞的恶性增殖和转移是患者死亡的主要原因,尤其是乳腺癌中恶性程度最高的三阴性乳腺癌[3],具有高转移性、强侵袭性和致死性强等特点,因此,揭示乳腺癌发生发展的分子机制对于科学研究和临床治疗均具有十分重要的意义。
CSN4基因编码COP9信号复合体(COP9 signalosome complex, CSN)第4亚基。COP9信号复合体在真核生物中高度保守,对于细胞增殖、生长和生存都具有重要作用[4,5,6,7,8]。COP9复合体参与蛋白质泛素化过程,对泛素降解的蛋白质如p27、p53、Mdm2、Smad7和Skp2等具有一定的调控作用[9,10,11,12,13,14,15,16],同时研究发现COP9复合体中的各个亚基的表达与很多恶性肿瘤的发生发展密切相关[17,18,19,20,21,22,23,24]。
CSN4基因曾被发现在前列腺癌细胞中,干扰CSN4的表达能够抑制前列腺癌细胞的增殖[25]。CSN4亚基对整个复合体的稳定性以及复合体的其它亚基都具有重要的调节作用。本实验室前期通过TCGA数据库分析CSN4基因在肿瘤和正常组织样本的mRNA存在差异。在本实验室的前期研究工作中初步探索了CSN5在三阴性乳腺癌TNBC (triple- negative breast cancer)中的发生发展机制,且大量文献也揭示CSN5确实影响乳腺癌的发生发展。文献中报道,在CSN复合体的结构中,CSN复合物在泛素化途径中需要连接cullin-RING E3连接酶发挥作用,而CSN5和CSN6在泛素化途径中共同发挥去NEDD化作用是需要CSN4协同作用。因此,为进一步探讨CSN4是否在三阴性乳腺癌中发挥重要作用,本研究在高转移性的三阴性乳腺癌细胞系MDA- MB-231中建立稳定干扰CSN4的表达体系,通过慢病毒介导的RNA干扰技术(RNA interference,RNAi),探讨了在三阴性乳腺癌细胞中抑制CSN4表达后,对肿瘤细胞的细胞增殖、细胞周期和细胞凋亡的影响,初步揭示CSN4参与调控CDK6和Caspase3的分子机制,为进一步制定CSN4基因的临床治疗方案提供参考。
1 材料与方法
1.1 材料
人乳腺癌细胞MDA-MB-231和293T细胞为本实验室保存;慢病毒包装相关质粒VSVG、PLP1和PLP2为本实验室保存;Trizol和Lipofectamin2000试剂购自Invitrogen公司(美国);RNA反转录试剂盒和定量PCR试剂购自南京诺维赞生物科技有限公司;HRP标记的Western blot二抗均购自上海碧云天公司;细胞凋亡检测试剂盒Annexin V-PE/7- AAD购自美国BD公司;CSN4抗体购自美国Novus公司,Caspase3、β-actin和CDK6抗体均购自美国Cell signaling公司;细胞培养试剂均购自Gibco品牌赛默飞世尔科技(中国)有限公司。1.2 细胞培养
MDA-MB-231和293T细胞培养使用DMEM高糖培养基,培养基中含有10%胎牛血清(FBS)和1%的青霉素-链霉素双抗,37℃置于CO2培养箱。1.3 慢病毒包装
干扰CSN4表达的慢病毒shRNA (对照),CSN4sh1和CSN4sh3 (干扰组)慢病毒包装时,在10 cm培养皿的生长状态较好的293T细胞中,加入40 μL的Lipofectin2000转染试剂、7.5 μg表达shRNA的慢病毒载体质粒以及3种辅助质粒VSVG、PLP1和PLP2各2.5βμg,12 h后换新鲜培养基,再培养48 h后收集病毒上清,2 000 r/min离心10 min,上清用0.45 μm滤器过滤分装保存在-80℃冰箱;感染时,取适量慢病毒加入到悬浮的乳腺癌细胞中,并按照(1∶1000)体积加入10 mg/mL polybrene。干扰CSN4基因表达的慢病毒shRNA序列:CSN4sh1:5°-GCCCAAGTGTTGGTGGGAATTT-3°;CSN4sh3:5°-GCTCTCTTACAAGACAATAGT-3°。对照组慢病毒shRNA序列:5°-CGTACGCGGAATACTTCGA-3°。
1.4 实时定量PCR
收集慢病毒感染的细胞,弃去上层培养基后用PBS洗涤3次,加入TRIzol裂解5 min,加入三氯甲烷震荡15 s后静置2 min,4℃离心后取上清,再加入等量异丙醇充分混匀后放置-20℃静置10 min,弃上清后留下沉淀并用75%乙醇洗涤2次,4℃离心弃上清后晾干10~20 min,加入适量DEPC水溶解并测完浓度后保存于-80℃冰箱。根据反转录试剂盒合成cDNA,实时荧光定量采用SYBR Green I荧光染料检测,在ABI公司7900HT的定量PCR仪检测。GAPDH基因作为内参,实验进行3组重复;CSN4上游引物:5°-GTAAGCCTCTGCCTGGACTG-3°,CSN4下游引物:5°-AGGAGCAGGTTGCTTCCATA- 3°。根据样本的Ct值计算2-ΔΔCt值,计算每个肿瘤样品中的目的基因对于正常样品的倍数差异。首先计算出每个样品每个基因的Ct平均值,然后计算ΔCt = Ct (目标基因) - Ct(管家基因)。本实验采取GAPDH作为管家基因,首先计算每个样品中目的基因的相对Ct值,然后用公式ΔΔCt=ΔCt(实验样本) - ΔCt(对照样本)。重复3次试验计算P值。1.5 Western blot
收集慢病毒感染后的细胞,加入RIPA裂解液冰上裂解1 h,4℃、12 000 r/min离心15 min。吸上清转移入新的Eppendorf管,测定蛋白浓度,然后加入适量的蛋白上样缓冲液混匀后煮沸5~10 min,上样30~100 μg的总蛋白进行SDS-PAGE电泳,恒流转膜2 h,转膜条件:250 V,0.3 A。封闭1 h后一抗二抗孵育,加入Thermo显色剂,在G-BOX显影仪曝光显影。1.6 CCK-8细胞增殖实验
将慢病毒感染后的各组细胞,计数细胞后调整细胞密度,按每孔1000个细胞接种于96孔板中,每组6个重复,细胞培养一定时间后CCK-8处理,利用美国Bio-Rad公司酶标仪在450 nm波长下检测OD值。1.7 流式细胞仪检测细胞周期
收集慢病毒感染后的状态较好的细胞,1 000 r/min离心3 min收集细胞,PBS洗涤2次。每组细胞加入500 μL PBS,包含10 μg/mL碘化丙啶(PI) 5 μL,0.2% Tritonx-100 75 μL和1 μg/mL RNase A的染色液,室温避光染色15 min后检测细胞Sub G1期,通过DNA含量分析法分析结果。1.8 克隆形成实验
将慢病毒感染后的细胞计数后,按同等数目1000个细胞每孔传代到六孔板,7~15 d左右随时观察。待细胞长成肉眼可见的细胞集落,开始处理细胞,PBS洗涤细胞2次,再用甲醇容易固定细胞20 min,PBS洗净,加上Gimasa染色液染色30 min。然后统计集落数。1.9 统计分析
本研究中数值用“均值±标准差”表示,所有实验重复3次以上,组间通过SPSS17.0软件进行t检验,用统计学分析方法检测差异是否具有统计学意义。2 结果与分析
2.1 慢病毒介导的CSN4基因shRNA稳定干扰体系的构建
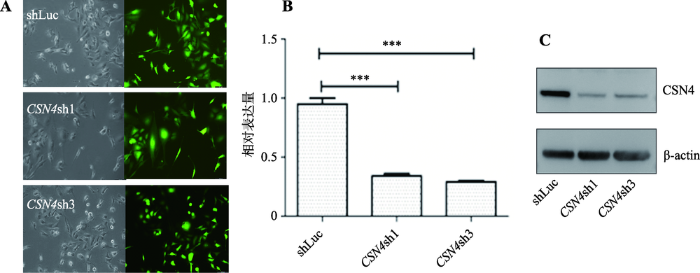
首先,将CSN4基因的shRNA干扰慢病毒(CSN4sh1和CSN4sh3)和对照慢病毒(shLuc)分别感染MDA-MB-231细胞,96βh后用荧光显微镜观察慢病毒感染效率。然后,分别提取感染后的细胞总RNA和总蛋白质,通过实时定量PCR和Western blot方法检测细胞内CSN4的mRNA和蛋白的表达水平。结果表明,根据可见光视野和荧光视野对比,对照组和CSN4干扰组慢病毒感染乳腺癌细胞MDA-MB-231,感染效率达到90%以上,表明感染效率高(图1A);实时定量PCR分析结果显示,相比较对照组shLuc,CSN4干扰后的实验组的mRNA表达量显著低于用shLuc对照慢病毒感染的乳腺癌细胞,t检验统计分析差异极显著,即P<0.001 (图1B)。Western blot实验结果显示,CSN4干扰组 的蛋白表达翻译水平显著低于对照组 慢病毒感染的乳腺癌细胞(图1C)。上述研究结果表明,慢病毒介导的shRNA干扰能有效抑制CSN4基因的mRNA 和蛋白水平的表达,本研究成功构建了乳腺癌细胞 MDA-MB-231中CSN4的表达干扰系统。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1MDA-MB-231细胞中慢病毒介导的CSN4干扰表达的检测
A:慢病毒感染MDA-MB-231细胞荧光拍摄图。左侧为可见光视野图片,右侧为荧光视野图片。B:乳腺癌细胞系MDA-MB-231中CSN4干扰后的mRNA的相对表达水平变化。t检验方法分析差异具有统计学意义,***表示P<0.001,存在极显著差异。C:MDA-MB-231细胞中慢病毒介导的CSN4干扰表达后的CSN4蛋白的表达水平。β-actin蛋白为内参。
Fig. 1Knock down of CSN4 expression by Lentivirus-mediated shRNA in MDA-MB-231 cells
2.2 CSN4表达干扰对MDA-MB-231细胞增殖的影响
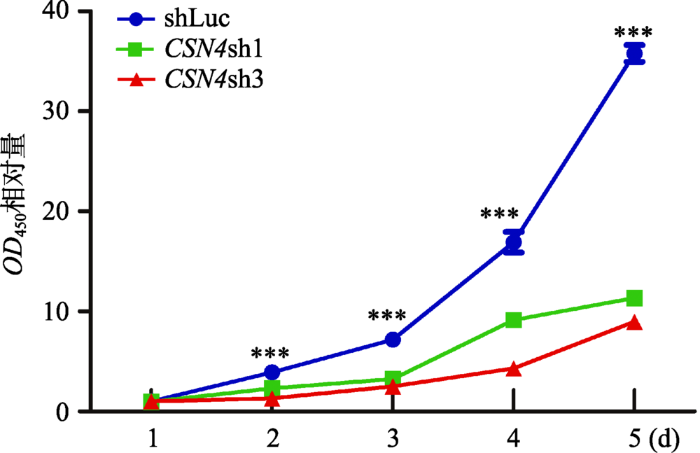
使用对照组 慢病毒和CSN4干扰组慢病毒 分别感染MDA-MB-231细胞,确认感染效率后,使用CCK-8细胞生长速率试剂盒检测对照组 和CSN4干扰组 的细胞增殖速率。结果显示,在MDA-MB- 231乳腺癌细胞中,相比较对照组,CSN4干扰组的生长速率均显著低于对照shLuc。统计分析表明,CSN4干扰组和对照组相比差异具有统计学意义(P< 0.001) (图2)。这表明MDA-MB-231细胞中CSN4基因的表达干扰使得细胞的增殖能力发生了显著的下降,即CSN4基因对于MDA-MB-231的细胞增殖能力具有重要作用。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2CCK8试剂盒检测CSN4表达干扰对MDA-MB- 231细胞增殖的影响
各组细胞在CCK8细胞增殖实验中450 nm处的平均吸光值±SD (n=6),代表细胞相对的增殖能力。两组干扰组(CSN4sh1和CSN4sh3)与对照组相比差异均具有统计学意义,***表示P<0.001,存在极显著差异。
Fig. 2CSN4 knockdown regulates proliferation of MDA-MB-231 cells as detected by CCK8 Kits
2.3 CSN4表达干扰对MDA-MB-231细胞克隆形成能力的影响
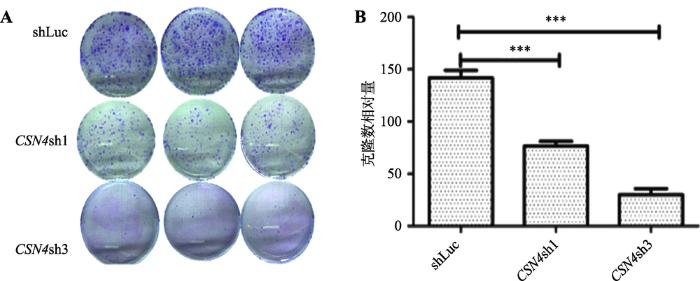
为进一步检测CSN4对乳腺癌细胞增殖能力的影响,本研究检测了干扰CSN4后对乳腺癌细胞克隆形成能力的影响。结果发现,CSN4基因干扰实验组 的克隆细胞数相比对照组 细胞数目少(图3A);重复3次实验后的统计结果表明,差异具有极其显著的统计学意义(图3B)。这说明CSN4基因表达被干扰后能够抑制了乳腺癌细胞MDA-MB-231的细胞克隆形成能力,进一步证明CSN4对于乳腺癌细胞的增殖起到很重要的作用。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3CSN4表达干扰对于MDA-MB-231细胞克隆形成能力的影响
A:CSN4干扰组和对照组的慢病毒感染MDA-MB-231形成的细胞克隆对比。B:重复3次实验的统计分析结果。***表示P<0.001,存在极显著差异。
Fig. 3Effects of CSN4 knockdown on colony formation ability of MDA-MB-231 cells
2.4 CSN4表达干扰对于MDA-MB-231细胞凋亡的影响
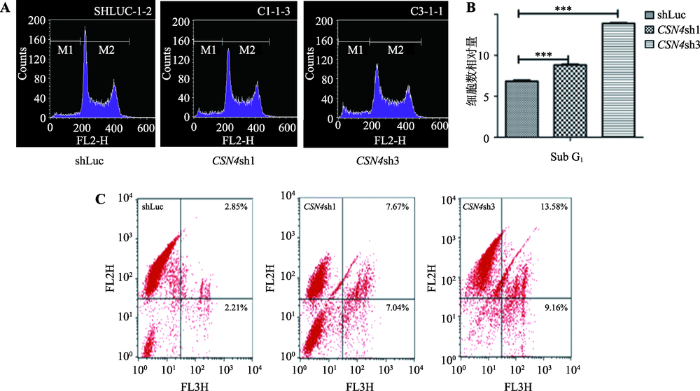
将CSN4干扰表达实验组和shLuc对照组的乳腺癌MDA-MB-231细胞进行PI染色后用流式细胞仪检测Sub G1期比例,结果显示,CSN4干扰表达实验组的Sub G1期细胞比例相较于shLuc对照组显著增加,CSN4sh3组细胞相比对照组shLuc细胞上调近50%,统计分析表明两者差异极显著(P<0.001),具有统计学意义(图4,A和B)。这表明在乳腺癌细胞中干扰CSN4的表达会明显增加细胞凋亡的比例。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4流式细胞仪测定CSN4表达干扰对MDA-MB-231细胞凋亡的影响
A:CSN4干扰表达后 MDA-MB-231细胞 Sub G1期比例检测。B:重复3次实验的统计分析结果。t检验结果表明差异极显著(***表示P<0.001),具有统计学意义。C:Annexin V-PE/7-AAD 细胞凋亡试剂盒检测结果。在每组4个象限中,左上:死亡细胞;右上:晚期凋亡细胞;左下:正常细胞;右下:早期凋亡细胞。
Fig. 4Effects of CSN4 knockdown on apoptosis as detected by flow cytometry
同时,将CSN4干扰实验组和对照组shLuc的乳腺癌细胞进行Annexin V-PE/7-AAD细胞凋亡试剂盒检测。结果显示,在MDA-MB-231细胞中,当CSN4的表达被干扰后,与对照组相比,早期凋亡的细胞(右下象限)和晚期凋亡细胞(右上象限)都有显著性增加(图4C),统计分析表明差异极其显著(P<0.001)。这进一步证明CSN4基因在乳腺癌细胞凋亡过程中起到很关键的作用。
2.5 CSN4表达干扰对于MDA-MB-231细胞中CDK6和Caspase3蛋白表达的影响
上述研究结果表明,CSN4的干扰表达能够影响乳腺癌细胞的增殖和凋亡。进一步利用显微镜观察到CSN4干扰表达的实验组乳腺癌细胞会发生大量死亡的现象,细胞状态明显变差,细胞皱缩,有漂浮现象;而对照组细胞状态较好,细胞舒展,形态正常(图5A)。图5
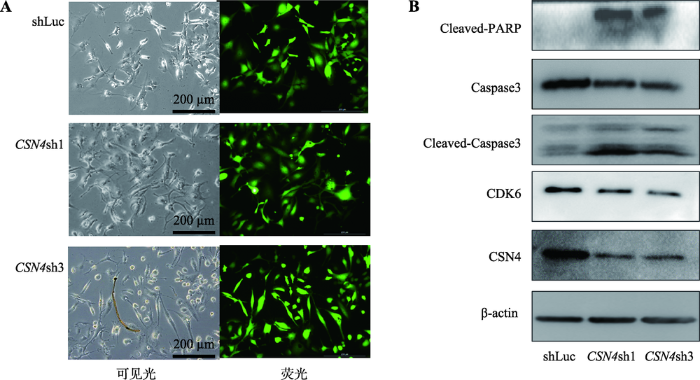 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5CSN4表达干扰对MDA-MB-231细胞中CDK6和Caspase3蛋白表达的影响
A:慢病毒感染MDA-MB-231细胞荧光拍摄图。左侧为可见光视野,右侧为荧光视野;B:Western blot检测不同实验组细胞中CSN4、CDK6和Caspase3蛋白的表达水平。β-actin为内参。
Fig. 5Effects of CSN4 knockdown on CDK6 and Caspase3 protein levels in MDA-MB-231 cells
对照组慢病毒和CSN4干扰组慢病毒感染后,分别抽提慢病毒感染后乳腺癌MDA-MB-231细胞的总蛋白,Western blot 检测CSN4被干扰后的Caspase3、CDK6和PARP的蛋白表达水平。结果表明,通过慢病毒感染干扰细胞内CSN4蛋白的表达,即CSN4的蛋白表达水平下降后,同时检测的CDK6蛋白表达的水平发生了显著的下调(图5B)。这说明干扰CSN4的表达能够引起乳腺癌细胞 关键性周期蛋白CDK6表达量发生变化。同时,干扰细胞内CSN4蛋白表达水平后,Caspase3总蛋白的表达水平发生了下调,而被剪切的cleaved-caspase3蛋白量发生上调,被剪切的PARP蛋白出现上调,表明Caspase3被激活,作用于其底物PARP,将PARP剪切,剪切的PARP蛋白出现显著上调。上述结果充分说明干扰CSN4蛋白的表达后,Caspase3的信号通路被激活。
3 讨论
三阴性乳腺癌的恶性程度较高,表现在侵袭性强、易远处转移以及预后较差。采用内分泌治疗及靶向治疗的方式均对其没有明显的效果。目前,化疗是TNBC乳腺癌患者的主要治疗手段,但是TNBC的治疗后存活率依然很低。由于缺乏特定的分子靶点,TNBC的诊断和治疗都受到极大的挑战。因此亟需探索TNBC的肿瘤分子生物学机制,寻找有效生物分子靶点,为临床诊断以及治疗发挥作用。目前的研究表明,CSN4作为COP9复合体中的一个亚基,对整个复合体的稳定性以及复合体的其他亚基都具有重要的调节作用[26]。而且有报道称CSN4基因能够调控CSN5蛋白水平从而影响sGC1和p53的表达水平,并且能够影响肿瘤细胞的增殖和迁移[25]。根据COP9复合体的重要性,以及CSN4在COP9复合体的结构,提示CSN4是COP9复合体的重要成员,但是CSN4基因在肿瘤的发生发展中的功能与分子机制研究较少,尚没有文献报道CSN4基因在乳腺癌细胞中的功能研究以及对细胞凋亡方面的调控的具体机制。
为探索CSN4的表达在乳腺癌细胞的主要生物学功能,本研究利用慢病毒感染的方法在乳腺癌细胞系MDA-MB-231构建了稳定的干扰CSN4表达的体系。通过CCK-8和克隆形成实验,结果显示干扰CSN4基因可以抑制乳腺癌细胞的生长增殖和克隆形成能力。通过流式细胞仪检测和凋亡试剂盒检测表明干扰CSN4的表达能够显著增加细胞凋亡的比例,可诱导细胞进入凋亡。研究结果为深入揭示CSN4在乳腺癌细胞中的分子机制打下了基础,同时也具有一定的临床应用潜力。
本研究中,干扰CSN4基因可以抑制乳腺癌细胞的生长增殖和克隆形成能力,同时检测出细胞周期相关蛋白CDK6,干扰CSN4基因的表达能够引起CDK6的蛋白表达水平下调,CDK6是细胞周期的重要调控因子,主要促进细胞周期由G1期进展到S期。细胞周期的失控是人类肿瘤发生的主要原因之一[27,28,29],揭示干扰CSN4基因可能导致乳腺癌的细胞周期发生异常,从而导致细胞生长阻滞,甚至发生凋亡现象。
已有研究报道,COP9复合体主要是以Caspases依赖性形式影响细胞凋亡[30]。为进一步探索CSN4基因是如何促进细胞凋亡,通过Western blot检测干扰CSN4基因后导致细胞的细胞凋亡基因Caspase3的前体蛋白总量表达下调,而被活化的cleaved- Caspase3的蛋白量明显上调,继而又同时检测出被剪切的PARP的蛋白表达水平也上调,因此推测CSN4基因可能是直接或间接激活Caspase3的活性进而引起乳腺癌细胞的凋亡。CSN4作为COP9复合体的一员,其对复合体的稳定性调节起到很关键的作用。在COP9复合体中,尤其是CSN5,已经有很多研究表明CSN5在细胞凋亡中发挥重要的作用。在胚胎细胞中CSN5敲除可诱导细胞凋亡,抑制细胞增殖,上调p27、p53和细胞周期蛋白E[31]。除了CSN5外,COP9复合体的其他亚基也被报道与凋亡有关。有研究表明[32],CSN6能够被caspase8剪切,从而影响CSN复合物的完整性,继而在凋亡级联反应过程中影响CSN复合物的活性。在肺癌和肝癌细胞中,敲低CSN3能够提高细胞凋亡水平[33,34]。CSN8在肝癌细胞中敲除后,同样也会引起细胞发生大量凋亡现象,导致促凋亡因子Bax,抗凋亡因子Bcl-2和Bcl-xl发生异常表达[33]。而且还有研究显示[34],CSN1能够在紫外线照射处理下诱导细胞发生凋亡。在K562细胞系中敲低CSN2或者CSN5,会导致不同的细胞模式死亡,分别是自噬或者细胞凋亡[35]。综上所述,COP9复合物在细胞凋亡过程中发挥重要作用。目前CSN4有可能是作为单体独立发挥作用于肿瘤细胞从而促进细胞凋亡,同时也有可能是与复合体其他亚基相互作用影响复合体的稳定性,从而影响其他亚基的表达。但是至于CSN4是作为单体还是复合亚基发挥作用,目前尚不清楚,且相关文献报道较少。因此,未来需要更进一步的探索CSN4的分子机制。
综上所述,本研究在乳腺癌MDA-MB-231细胞中建立了CSN4稳定干扰表达系统,探索了CSN4基因对乳腺癌细胞的增殖能力和凋亡造成的影响,以及进一步研究了干扰CSN4的表达在乳腺癌细胞发生凋亡的分子机制。但是,CSN4是如何通过Caspase3调控细胞具体凋亡的分子机制尚不清楚,仍有待进一步的研究去揭示。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:25116958 [本文引用: 1]

We report herein the synthesis and photophysical studies on a new multicomponent chemosensor dyad comprising two fluorescing units, dansylamide (DANS) and nitrobenzoxadiazole (NBD). The system has been developed to investigate receptor-analyte binding interactions in the presence of both cations and anions in a single molecular system. A dimethyl amino (in the DANS unit) group is used as a receptor for cations, and acidic hydrogens of sulfonamide and the NBD group are used as receptors for anions. The system is characterized by conventional analytical techniques. The photophysical properties of this supramolecular system in the absence and presence of various metal ions and nonmetal ions as additives are investigated in an acetonitrile medium. Utility of this system in an aqueous medium has also been demonstrated. The absorption and fluorescence spectrum of the molecular system consists of a broad band typical of an intramolecular charge-transfer (ICT) transition. A low quantum yield and lifetime of the NBD moiety in the present dyad indicates photoinduced electron transfer (PET) between DANS and the NBD moiety. The fluorescence intensity of the system is found to decrease in the presence of fluoride and acetate anions; however, the quenching is found to be much higher for fluoride. This quenching behavior is attributed to the enhanced PET from the anion receptor to the fluorophore moiety. The mechanistic aspect of the fluoride ion signaling behavior has also been studied by infrared (IR) and (1)H NMR experiments. The hydrogen bonding interaction between the acidic NH protons of the DPN moiety and F(-) is found to be primarily responsible for the fluoride selective signaling behavior. While investigating the cation signaling behavior, contrary to anions, significant fluorescence enhancement has been observed only in the presence of transition-metal ions. This behavior is rationalized by considering the disruption of PET communication between DANS and the NBD moiety due to transition-metal ion binding. Theoretical (density functional theory) studies are also performed for the better understanding of the receptor-analyte interaction. Interestingly, negative cooperativity in binding is observed when the interaction of this system is studied in the presence of both Zn(2+) and F(-). Fluorescence microscopy studies also revealed that the newly developed fluorescent sensor system can be employed as an imaging probe in live cells.
URL [本文引用: 1]
URLPMID:25478862 [本文引用: 1]
The possible role of viruses in breast cancer etiology remains an unresolved question. We hypothesized that if some viruses are involved, it may be in a subgroup of breast cancers only. Epidemiological arguments drove our interest in breast cancer subgroups that are more frequent in Africa, namely inflammatory breast cancer (IBC) and triple-negative breast cancer. We tested whether viral prevalence was significantly higher in these subgroups.One hundred fifty-five paraffin-embedded malignant breast tumors were randomly selected at the pathology laboratory of the University Hospital of Annaba (Algeria) to include one third of IBC and two thirds of non-IBC. They were tested for the presence of DNA from 61 viral agents (46 human papillomaviruses, 10 polyomaviruses, and 5 herpesviruses) using type-specific multiplex genotyping assays, which combine multiplex PCR and bead-based Luminex technology.Viral DNA was found in 22 (17.9%) of 123 tumors. The most prevalent viruses were EBV1 and HPV16. IBC tumors carried significantly more viruses (any type) than non-IBC tumors (30% vs. 13%, p<0.04). Similarly, triple-negative tumors displayed higher virus-positivity than non-triple-negative tumors (44% vs. 14%, p<0.009).Our results suggest an association between the presence of viral DNA and aggressive breast cancer phenotypes (IBC, triple-negative). While preliminary, they underline the importance of focusing on subgroups when studying viral etiology in breast cancer. Further studies on viruses in breast cancer should be conducted in much larger samples to confirm these initial findings.
URLPMID:12672462 [本文引用: 1]
The COP9 signalosome (CSN), the lid subcomplex of the proteasome and translational intitation factor 3 (eIF3) share structural similarities and are often referred to as the PCI family of complexes. In multicellular eukaryotes, the CSN is highly conserved as an 8-subunit complex but in Saccharomyces cerevisiae the complex is rather divergent. We further characterize the composition and properties of the CSN in budding yeast and its interactions with these related complexes. Using the generalized profile method we identified CSN candidates, four with PCI domains: Csn9, Csn10, Pci8/Csn11, and Csn12, and one with an MPN domain, Csn5/Rri1. These proteins and an additional interactor, Csi1, were tested for pairwise interactions by yeast two-hybrid and were found to form a cluster surrounding Csn12. Csn5 and Csn12 cofractionate in a complexed form with an apparent molecular weight of roughly 250 kDa. However, Csn5 migrates as a monomer in csn12 supporting the pivotal role of Csn12 in stabilizing the complex. Confocal fluorescence microscopy detects GFP-tagged Csn5 preferentially in the nucleus, whereas in absence of Csn12, Csn10, Pci8/Csn11, or Csi1, Csn5 is delocalized throughout the cell, indicating that multiple subunits are required for nuclear localization of Csn5. Two CSN subunits, Csn9 and Csi1, interact with the proteasome lid subunit Rpn5. Pci8/Csn11 has previously been shown to interact with eIF3. Together, these results point to a network of interactions between these three structurally similar, yet functionally diverse, complexes.
URLPMID:19787423 [本文引用: 1]
The COP9 signalosome (CSN) is a multi-subunit protein complex conserved in plants and animals. CSN subunits have been identified as light-mediated master regulators of eukaryotic circadian clocks from fungi to animals. The Indian false vampire bat Megaderma lyra is completely adapted to an anthropic biotope and behavioral studies have reported that M. lyra exhibits light-sampling behavior to assess environmental light. LC-MS-MS results for a 36 kDa protein were analyzed using the Sequest search engine, and COP9 signalosome subunit 5 (CSN5) was pinpointed as having the highest score with 6 matching peptides. To confirm the presence of CSN5, up-regulated cDNA was amplified, sequenced, and identified as CSN5. Furthermore, semi-quantitative RT-PCR analysis demonstrated that the level of induction of CSN5 was regulated by environmental light. We estimated the level of expression across a light-dark cycle and observed a higher level of expression at the end of the light phase. Similarly, when the animal was shifted from continuous dark to light, CSN5 expression was induced. Correspondingly, we detected the similar pattern of translated protein with JAB1 antibody. Knowledge about the circadian rhythm and its molecular mechanism in Chiroptera is very limited and this study suggests that CSN5 might be involved in the M. lyra light-signaling process.
URLPMID:26096786 [本文引用: 1]
The COP9 signalosome (CSN) is a regulator of the ubiquitin (Ub) proteasome system (UPS). It interacts with hundreds of cullin-RING ubiquitin E3 ligases (CRLs) and regulates their activity by removing the Ub-like protein Nedd8 from cullins. In mammalian cells 7 different cullins exist which form CRLs with adaptor proteins and with a large number of substrate recognition subunits such as F-box and BTB proteins. This large variety of CRL-complexes is deneddylated by the CSN. The capacity of the CSN to interact with numerous types of CRL complexes can be explained by its structural diversity, which allows different CSN variants to interact with different binding partners and substrates and enables different subunit expression profiles. Diversity of CSN complexes presumably occurs by: (1) flexibility of CSN holo complex structure; (2) formation of CSN mini complexes and free CSN subunits and (3) generation of CSN variants via integration of CSN subunit isoforms. In this review we will discuss the structural diversity of the CSN complex and possible functional consequences.
URLPMID:25043011 [本文引用: 1]
Abstract Ubiquitination is a crucial cellular signalling process, and is controlled on multiple levels. Cullin-RING E3 ubiquitin ligases (CRLs) are regulated by the eight-subunit COP9 signalosome (CSN). CSN inactivates CRLs by removing their covalently attached activator, NEDD8. NEDD8 cleavage by CSN is catalysed by CSN5, a Zn(2+)-dependent isopeptidase that is inactive in isolation. Here we present the crystal structure of the entire 65350-kDa human CSN holoenzyme at 3.80203 resolution, detailing the molecular architecture of the complex. CSN has two organizational centres: a horseshoe-shaped ring created by its six proteasome lid-CSN-initiation factor 3 (PCI) domain proteins, and a large bundle formed by the carboxy-terminal α-helices of every subunit. CSN5 and its dimerization partner, CSN6, are intricately embedded at the core of the helical bundle. In the substrate-free holoenzyme, CSN5 is autoinhibited, which precludes access to the active site. We find that neddylated CRL binding to CSN is sensed by CSN4, and communicated to CSN5 with the assistance of CSN6, resulting in activation of the deneddylase.
URLPMID:22767593 [本文引用: 1]
Background:A detailed description of the kinetics of deneddylation of cullin by CSN has been lacking. Results:Selected factors and SCF subunits are able to inhibit deneddylation to varying degrees. CSN interferes with SCF-mediated ubiquitination through a noncatalytic mechanism. Conclusion:Deneddylation of Cul1 by CSN is regulated by F-box protein, substrate, and other factors. Significance:Our work reported here could facilitate the development of directed therapies. COP9 signalosome (CSN) mediates deconjugation of the ubiquitin-like protein Nedd8 from the cullin subunits of SCF and other cullin-RING ubiquitin ligases (CRLs). This process is essential to maintain the proper activity of CRLs in cells. Here, we report a detailed kinetic characterization of CSN-mediated deconjugation of Nedd8 from SCF. CSN is an efficient enzyme, with akcatof 651 s611andKmfor neddylated Cul1-Rbx1 of 65200 nm, yielding akcat/Kmnear the anticipated diffusion-controlled limit. Assembly with an F-box-Skp1 complex markedly inhibited deneddylation, although the magnitude varied considerably, with Fbw7-Skp1 inhibiting by 655-fold but Skp2-Cks1-Skp1 by only 6515%. Deneddylation of both SCFFbw7and SCFSkp2-Cks1was further inhibited 652.5-fold by the addition of substrate. Combined, the inhibition by Fbw7-Skp1 plus its substrate cyclin E was greater than 10-fold. Unexpectedly, our results also uncover significant product inhibition by deconjugated Cul1, which results from the ability of Cul1 to bind tightly to CSN. Reciprocally, CSN inhibits the ubiquitin ligase activity of deneddylated Cul1. We propose a model in which assembled CRL complexes engaged with substrate are normally refractory to deneddylation. Upon consumption of substrate and subsequent deneddylation, CSN can remain stably bound to the CRL and hold it in low state of reduced activity.
URLPMID:22237956 [本文引用: 1]
Abstract PURPOSE: Csn3 (or CSN3) encodes the third subunit of an eight-subunit complex, the COP9 signalosome (CSN), which acts as a protein kinase and a deneddylase in mammalian cells. Previous studies have shown that Csn3 is essential for maintenance of cell proliferation in the mouse embryonic epiblast and associated with the tumorigenesis process in osteosarcoma. However, its correlation with hepatocellular carcinoma (HCC) has not been explored yet. METHODS: The expression of Csn3 in HCC (n = 30), cirrhosis (n = 30), and normal tissues (n = 30) was detected using immunohistochemical analysis. The impacts of lentivirus-mediated inhibition of Csn3 on HCC cells were detected using MTT, BrdU incorporation assay, and flow cytometric analysis. In addition, the colony formation and tumor growth ability in nude mice were detected to define the role of Csn3 in tumorigenesis. RESULTS: Knockdown of Csn3 expression in HCC cell lines (SMMC-7721 and Hep3B) significantly inhibits the tumor growth both in vitro and in vivo. Further investigation indicates that this growth inhibition effect may be mediated through cell cycle arrest in G0/G1 phase and inductions of pro-apoptotic proteins BIK and Caspase-8. In addition, knockdown of Csn3 expression evidently suppresses tumor growth in a xenograft nude mice model. CONCLUSION: Collectively, this study demonstrates Csn3 as an oncogene that regulates the tumorigenesis process in HCC cells.
URLPMID:29148631 [本文引用: 1]
Abstract Retraction: Retracted: Silencing of the COPS3 Gene by siRNA Reduces Proliferation of Lung Cancer Cells Most Likely via Induction of Cell Cycle Arrest and Apoptosis Asian Pacific Journal of Cancer Prevention has retracted the article titled 090008Silencing of the COPS3 Gene by siRNA Reduces Proliferation of Lung Cancer Cells Most Likely via Induction of Cell Cycle Arrest and Apoptosis090009(1) for reason of similarity with a series of articles identified by Byrne and Labb0108 (2). Xue-Mei Wang, Jiu-Wei Cui1&, Wei Li , Lu Cai, Wei Song , Guan-Jun Wang 1. Xue-Mei Wang, Jiu-Wei Cui1&, Wei Li , Lu Cai, Wei Song , Guan-Jun Wang. Silencing of the COPS3 Gene by siRNA Reduces Proliferation of Lung Cancer Cells Most Likely via Induction of Cell Cycle Arrest and Apoptosis. Asian Pac J Cancer Prev. 2012;13(5):1823-7. 2. J. A. Byrne and C. Labb0108, 090008Striking similarities between publications from China describing single gene knockdown experiments in human cancer cell lines,090009 Scientometrics, vol. 110, no. 3, pp. 14710900091493, 2017. Authors did not respond to request for comment.
URLPMID:3049400 [本文引用: 1]
The mammalian constitutive photomorphogenesis 9 (COP9) signalosome (CSN), a protein complex involved in embryonic development, is implicated in cell cycle regulation and the DNA damage response. Its role in tumor development, however, remains unclear. Here, we have shown that the COP9 subunit 6 (CSN6) gene is amplified in human breast cancer specimens, and the CSN6 protein is upregulated in human breast and thyroid tumors. CSN6 expression positively correlated with expression of murine double minute 2 (MDM2), a potent negative regulator of the p53 tumor suppressor. Expression of CSN6 appeared to prevent MDM2 autoubiquitination at lysine 364, resulting in stabilization of MDM2 and degradation of p53. Mice in which Csn6 was deleted died early in embryogenesis (E7.5). Embryos lacking both Csn6 and p53 survived to later in embryonic development (E10.5), which suggests that loss of p53 could partially rescue the effect of loss of Csn6. Mice heterozygous for Csn6 were sensitized to irradiation-induced, p53-dependent apoptosis in both the thymus and the developing CNS. These mice were also less susceptible than wild-type mice to -irradiation-induced tumorigenesis. These results suggest that loss of CSN6 enhances p53-mediated tumor suppression in vivo and that CSN6 plays an important role in regulating DNA damage-associated apoptosis and tumorigenesis through control of the MDM2-p53 signaling pathway.
URLPMID:17879958 [本文引用: 1]
The 5th subunit of COP9 signalosome (CSN5, also known as Jab1 or COPS5) is implicated in regulating p53 activity and is overexpressed in various tumors. However, the precise roles of CSN5 in p53 network and tumorigenesis are not well characterized. Here we show that CSN5 is a critical regulator of both p53 and MDM2. We show that curcumin, an important inhibitor of CSN-associated kinases, can downregulate not only CSN5 but also MDM2, which results in p53 stabilization. Importantly, CSN5 interacts with p53. CSN5 expression leads to p53 degradation, facilitating MDM2-mediated p53 ubiquitination, and promoting p53 nuclear export. Additionally, CSN5 expression results in stabilization of MDM2 through reducing MDM2 self-ubiquitination and decelerating turnover rate of MDM2. Significantly, we further show that CSN5 antagonizes the transcriptional activity of p53. These results demonstrate that CSN5 is a pivotal regulator for both p53 and MDM2. Our studies may pave the way for targeting CSN5 for anti-cancer drug development.
URLPMID:10086358 [本文引用: 1]
The proliferation of mammalian cells is under strict control, and the cyclin-dependent-kinase inhibitory protein p27Kip1 is an essential participant in this regulation both in vitro and in vivo. Although mutations in p27Kip1 are rarely found in human tumours, reduced expression of the protein correlates well with poor survival among patients with breast or colorectal carcinomas, suggesting that disruption of the p27Kip1 regulatory mechanisms contributes to neoplasia. The abundance of p27Kip1 in the cell is determined either at or after translation, for example as a result of phosphorylation by cyclinE/Cdk2 complexes, degradation by the ubiquitin/proteasome pathway, sequestration by unknown Myc-inducible proteins, binding to cyclinD/Cdk4 complexes, or inactivation by the viral E1A oncoprotein. We have found that a mouse 38K protein (p38) encoded by the Jab1 gene interacts specifically with p27Kip1 and show here that overexpression of p38 in mammalian cells causes the translocation of p27Kip1 from the nucleus to the cytoplasm, decreasing the amount of p27Kip1 in the cell by accelerating its degradation. Ectopic expression of p38 in mouse fibroblasts partially overcomes p27Kip1-mediated arrest in the G1 phase of the cell cycle and markedly reduces their dependence on serum. Our findings indicate that p38 functions as a negative regulator of p27Kip1 by promoting its degradation.
URLPMID:11967155 [本文引用: 1]
The COP9 signalosome (CSN) is a conserved protein complex with homologies to the lid subcomplex of the 26S proteasome [1]. It promotes cleavage of the Nedd8 conjugate (deneddylation) from the cullin component of SCF ubiquitin ligases [2]. We provide evidence that cullin neddylation and deneddylation is highly dynamic, that its equilibrium can be effectively modulated by CSN, and that neddylation allows Cul1 to form larger protein complexes. CSN2 integrates into the CSN complex via its C-terminal region and its N-terminal half region is necessary for direct interaction with Cul1. The polyclonal antibodies against CSN2 but not other CSN subunits cause accumulation of neddylated Cul1/Cul2 in HeLa cell extract, indicating that CSN2 is essential in cullin deneddylation. Further, CSN inhibits ubiquitination and degradation of the cyclin-dependent kinase inhibitor p27kip1 in vitro. Microinjection of the CSN complex impeded the G1 cells from entering the S phase. Moreover, anti-CSN2 antibodies negate the CSN-dependent p27 stabilization and the G1/S blockage, suggesting that these functions require the deneddylation activity. We conclude that CSN inhibits SCF ubiquitin ligase activity in targeting p27 proteolysis and negatively regulates cell cycle at the G1 phase by promoting deneddylation of Cul1.
URLPMID:11285227 [本文引用: 1]
In higher eukaryotic cells, the p53 protein is degraded by the ubiquitin09000926S proteasome system mediated by Mdm2 or the human papilloma virus E6 protein. Here we show that COP9 signalosome (CSN)-specific phosphorylation targets human p53 to ubiquitin09000926S proteasome-dependent degradation. As visualized by electron microscopy, p53 binds with high affinity to the native CSN complex. p53 interacts via its N-terminus with CSN subunit 5/Jab1 as shown by far-western and pull-down assays. The CSN-specific phosphorylation sites were mapped to the core domain of p53 including Thr155. A phosphorylated peptide, 0200p53(145090009164), specifically inhibits CSN-mediated phosphorylation and p53 degradation. Curcumin, a CSN kinase inhibitor, blocks E6-dependent p53 degradation in reticulocyte lysates. Mutation of Thr155 to valine is sufficient to stabilize p53 against E6-dependent degradation in reticulocyte lysates and to reduce binding to Mdm2. The p53T155V mutant accumulates in both HeLa and HL 60 cells and exhibits a mutant (PAb 240+) conformation. It induces the cyclin-dependent inhibitor p21. In HeLa and MCF-7 cells, inhibition of CSN kinase by curcumin or 0200p53(145090009164) results in accumulation of endogenous p53.
[本文引用: 1]
URLPMID:17356713 [本文引用: 1]
DNA microarrays have been widely applied to cancer transcriptome analysis; however, the majority of such data are not easily accessible or comparable. Furthermore, several important analytic approaches have been applied to microarray analysis; however, their application is often limited. To overcome these limitations, we have developed Oncomine, a bioinformatics initiative aimed at collecting, standardizing, analyzing, and delivering cancer transcriptome data to the biomedical research community. Our analysis has identified the genes, pathways, and networks deregulated across 18,000 cancer gene expression microarrays, spanning the majority of cancer types and subtypes. Here, we provide an update on the initiative, describe the database and analysis modules, and highlight several notable observations. Results from this comprehensive analysis are available at http://www.oncomine.org.
URLPMID:15920519 [本文引用: 1]
DNA microarrays have been widely applied to transcriptome analysis. The Oncomine database contains a large collection of such data, as well as hundreds of derived gene-expression signatures. We studied the regulatory mechanisms responsible for gene deregulation in these signatures by searching for the coordinate regulation of genes with common sites. We found that genes with sites for the archetypal factor, E2F, were disproportionately overexpressed in a wide variety of , whereas genes with sites for other factors, such as -Max, c-and ATF, were disproportionately overexpressed in specific types. These results suggest that alterations in pathways activating these factors may be responsible for the observed gene deregulation and .
URL [本文引用: 1]
URLPMID:11731421 [本文引用: 1]
Angiogenesis is a prerequisite for solid tumor growth and metastasis. Elucidation of the signaling pathways that control tumor angiogenesis constitutes the basis for a rational antiangiogenic tumor therapy. Here we show that the production of vascular endothelial growth factor (VEGF) in HeLa and HL-60 cells is directed by the constitutive photomorphogenesis 9 signalosome (CSN). The CSN is a kinase complex that cooperates with the ubiquitin/26S proteasome system in regulating the stability of proteins involved in signal transduction. VEGF expression is controlled by the transcription factors activator protein (AP)-1, AP-2, SP-1, and hypoxia-inducible factor 1. Inhibition of CSN kinase activity by 50 curcumin for 2 h decreases the cellular c-Jun concentration, resulting in a reduction of the VEGF production by approximately 75%. The removal of the inhibitor from the cells led to a time-dependent recovery of endogenous c-Jun that is paralleled by increasing VEGF production. Elevated cellular CSN activity induced by CSN subunit 2 overexpression causes increased VEGF production in HeLa cells. A competitor of CSN-dependent c-Jun phosphorylation, the NH-terminal c-Jun fragment c-Jun(1 226), inhibits VEGF production in HeLa cells. The transcription factors AP-2 and SP-1 act independently of the CSN. They contribute less than a quarter to basal VEGF production. Under our experimental conditions, hypoxia-inducible factor 1 protein was not detected. Overexpression of the tumor suppressor p53 reduces VEGF production in HeLa cells. p53 competes with c-Jun for CSN-specific phosphorylation with the consequence of c-Jun destabilization. We conclude that CSN-directed c-Jun signaling mediates high VEGF production in HeLa and HL-60 cells. The data provide an explanation for the known antiangiogenic and antitumorigenic activities of curcumin. Because the CSN regulates the major part of VEGF production in the tested tumor cells, it constitutes a potentially important target for tumor therapy.
URLPMID:19535905 [本文引用: 1]
The COP9 complex (signalosome) is a known regulator of the proteasome/ubiquitin pathway. Furthermore it regulates the activity of the cullin-RING ligase (CRL) families of ubiquitin E3-complexes. Besides the CRL family, the anaphase-promoting complex (APC/C) is a major regulator of the cell cycle. To investigate a possible connection between both complexes we assessed interacting partners of COP9 using an in vivo protein-protein interaction assay. Hereby, we were able to show for the first time that CSN2, a subunit of the COP9 signalosome, interacts physically with APC/C. Furthermore, we detected a functional influence of the COP9 complex regarding the stability of several targets of the APC/C. Consistent with these data we showed a genetic instability of cells over-expressing CSN2.
URL [本文引用: 1]
URLPMID:20160482 [本文引用: 1]
Skp2 is the substrate binding subunit of the SCFSkp2 ubiquitin ligase, which plays a key role in the regulation of cell cycle progression. The activity of Skp2 is regulated by the APCCdh1, which targets Skp2 for degradation in early G1 and prevent premature S phase entry. Overexpression of Skp2 leads to dysregulation of the cell cycle and is commonly observed in human cancers. We have previously shown that Skp2 is phosphorylated on Ser64 and Ser72 in vivo, and that these modifications regulate its stability. Recently, two studies have proposed a role for Ser72 phosphorylation in the cytosolic relocalization of Skp2 and in the assembly and activity of SCFSkp2 ubiquitin ligase complex. We have revisited this question and analyzed the impact of Ser72 phosphorylation site mutations on the biological activity and subcellular localization of Skp2. We show here that phosphorylation of Ser72 does not control Skp2 binding to Skp1 and Cul1, has no influence on SCFSkp2 ubiquitin ligase activity, and does not affect the subcellular localization of Skp2 in a panel of cell lines.
URL [本文引用: 1]
URL [本文引用: 2]
URLPMID:25043011 [本文引用: 1]
Abstract Ubiquitination is a crucial cellular signalling process, and is controlled on multiple levels. Cullin-RING E3 ubiquitin ligases (CRLs) are regulated by the eight-subunit COP9 signalosome (CSN). CSN inactivates CRLs by removing their covalently attached activator, NEDD8. NEDD8 cleavage by CSN is catalysed by CSN5, a Zn(2+)-dependent isopeptidase that is inactive in isolation. Here we present the crystal structure of the entire 65350-kDa human CSN holoenzyme at 3.80203 resolution, detailing the molecular architecture of the complex. CSN has two organizational centres: a horseshoe-shaped ring created by its six proteasome lid-CSN-initiation factor 3 (PCI) domain proteins, and a large bundle formed by the carboxy-terminal α-helices of every subunit. CSN5 and its dimerization partner, CSN6, are intricately embedded at the core of the helical bundle. In the substrate-free holoenzyme, CSN5 is autoinhibited, which precludes access to the active site. We find that neddylated CRL binding to CSN is sensed by CSN4, and communicated to CSN5 with the assistance of CSN6, resulting in activation of the deneddylase.
URLPMID:23948297 [本文引用: 1]
In contrast to its close homolog CDK4, the cell cycle kinase CDK6 is expressed at high levels in lymphoid malignancies. In a model for p185(BCR-ABL+) B-acute lymphoid leukemia, we show that CDK6 is part of a transcription complex that induces the expression of the tumor suppressor p16(INK4a) and the pro-angiogenic factor VEGF-A. This function is independent of CDK6's kinase activity. High CDK6 expression thus suppresses proliferation by upregulating p16(INK4a), providing an internal safeguard. However, in the absence of p16(INK4a), CDK6 can exert its full tumor-promoting function by enhancing proliferation and stimulating angiogenesis. The finding that CDK6 connects cell-cycle progression to angiogenesis confirms CDK6's central role in hematopoietic malignancies and could underlie the selection pressure to upregulate CDK6 and silence p16(INK4a).
URLPMID:10384057 [本文引用: 1]
Mamm Genome. 1999 Jul;10(7):764-7. Comparative Study
URLPMID:11828325 [本文引用: 1]
http://www.nature.com/doifinder/10.1038/nsb756
URLPMID:18060501 [本文引用: 1]
In concert with the ubiquitin (Ub) proteasome system (UPS) the COP9 signalosome (CSN) controls the stability of cellular regulators. The CSN interacts with cullin-RING Ub ligases (CRLs) consisting of a specific cullin, a RING protein as Rbx1 and substrate recognition proteins. The Ub-like protein Nedd8 is covalently linked to cullins and removed by the CSN-mediated deneddylation. Cycles of neddylation and deneddylation regulate CRLs. Apoptotic stimuli cause caspase-dependent modifications of the UPS. However, little is known about the CSN during apoptosis. We demonstrate in02vitro and in02vivo that CSN6 is cleaved most effectively by caspase 3 at D 23 after 2–302h of apoptosis induced by anti-Fas-Ab or etoposide. CSN6 processing occurs in CSN–CRL complexes and is followed by the cleavage of Rbx1, the direct interaction partner of CSN6. Caspase-dependent cutting of Rbx1 is accompanied by decrease of neddylated proteins in Jurkat T cells. Another functional consequence of CSN6 cleavage is the enhancement of CSN-mediated deneddylating activity causing deneddylation of cullin 1 in cells. The CSN-associated deubiquitinating as well as kinase activity remained unchanged in presence of active caspase 3. The cleavage of Rbx1 and increased deneddylation of cullins inactivate CRLs and presumably stabilize pro-apoptotic factors for final apoptotic steps.
[本文引用: 1]
[本文引用: 1]
URLPMID:3698095 [本文引用: 2]
The COP9 signalosome (CSN), an evolutionally highly conserved protein complex composed of 8 unique subunits (CSN1 through CSN8) in higher eukaryotes, is purported to modulate protein degradation mediated by the ubiquitin-proteasome system (UPS) but this has not been demonstrated in a critical mitotic parenchymal organ of vertebrates. Hepatocyte-specific knockout of the Cops8 gene (HS-Csn8KO) was shown to cause massive hepatocyte apoptosis and liver malfunction but the underlying mechanism remains unclear. Here, we report that Csn8/CSN exerts profound impacts on hepatic UPS function and is critical to the stability of the pro-apoptotic protein Bim. Significant decreases in CIS (cytokine-inducible Src homology 2 domain-containing protein), a Bim receptor of a cullin2-based ubiquitin ligase, were found to co-exist with a marked increase of Bim proteins. Csn8 deficiency also significantly decreased 19S proteasome subunit Rpt5 and markedly increased high molecular weight neddylated and ubiquitinated proteins. The use of a surrogate UPS substrate further reveals severe impairment of UPS-mediated proteolysis in HS-Csn8KO livers. Inclusion body-like materials were accumulated in Csn8 deficient hepatocytes. In addition to Bim, massive hepatocyte apoptosis in HS-Csn8KO livers is also associated with elevated expression of other members of the Bcl2 family, including pro-apoptotic Bax as well as anti-apoptotic Bcl2 and Bcl-XL. Increased interaction between Bcl2 and Bim, but not between Bcl2 and Bax, was detected. Hence, it is concluded that hepatic CSN8 deficiency impairs the UPS in the liver and the resultant Bim upregulation likely plays an important role in triggering hepatocyte apoptosis via sequestering Bcl2 away from Bax.
URLPMID:26986008 [本文引用: 2]
Abstract The COP9/signalosome (CSN) multi-protein complex regulates the activity of cullin-RING ubiquitin ligases (CRLs), including the DDB2 and CSA CRL4 ligases (CRL4DDB2 and CRL4CSA), which are involved in the repair of UV-induced DNA damages. In the present study, we demonstrated that the protein kinase ATM, a key component of the DNA damage response (DDR), phosphorylates CSN1 and CSN7a, two subunits of the CSN complex, in a UV-dependent manner. The phosphorylation of CSN1 on serine02474 was detected as early as 302h after UV-exposure, peaked at 802h and persisted until 4802h post-UV irradiation. Such a time course suggests a role in late DDR rather than in DNA repair. Consistently, overexpression of a phosphorylation-resistant S474A CSN1 mutant reduced UV-induced apoptosis. Thus, CSN1 appears to play a role not only in DNA repair but also in UV-induced apoptosis.
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}