 ,南京医科大学基础医学院医学遗传学系,南京 211166
,南京医科大学基础医学院医学遗传学系,南京 211166Seamless genome editing in Drosophila by combining CRISPR/Cas9 and piggyBac technologies
Jue Wang, Juan Huang, Rui Xu,Department of Medical Genetics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing 211166, China通讯作者:
编委: 张雷
收稿日期:2018-12-27修回日期:2019-01-31网络出版日期:2019-05-20
| 基金资助: |
Received:2018-12-27Revised:2019-01-31Online:2019-05-20
| Fund supported: |
作者简介 About authors
王珏,本科,专业方向:果蝇基因打靶技术E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (816KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王珏, 黄娟, 许蕊. 利用CRISPR/Cas9和piggyBac实现果蝇基因组无缝编辑 [J]. 遗传, 2019, 41(5): 422-429 doi:10.16288/j.yczz.18-345
Jue Wang, Juan Huang, Rui Xu.
利用现代生物学技术获得基因突变体,并检测基因突变造成的影响是研究基因功能的重要手段。CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9)技术的出现和应用,为实现基因打靶提供了简单高效的方法[1,2,3]。在sgRNA引导下,Cas9核酸内切酶可作用于特定基因序列,产生DNA双链断裂(double-stranded breaks, DSBs),诱发内源性非同源末端连接(non-homologous end joining, NHEJ)或同源定向修复(homology-directed repair, HDR)。
非同源末端连接可导致修复位点产生小片段的DNA插入或缺失,从而造成目标基因突变。但是,这类突变具有一定的随机性,无法实现对目标基因的特定编辑。如果在DNA双链断裂修复过程中提供外源模板,则在同源定向修复启动时可将模板中的特异性突变引入基因组中,从而实现对目标基因的特定编辑。然而,同源定向修复的效率远远低于非同源末端连接[4]。为了高效筛选到携带特异性突变的转基因果蝇(Drosophila melanogaster)品系,需同时引入筛选标记,以便判断是否发生了同源定向修复。筛选标记的移除通常利用Cre/loxP或FLP/FRT系统进行,但它们都会在基因组中遗留34个碱基的标签序列[5]。如何在对基因组进行精确编辑的同时不引入无关序列,即实现基因组无缝编辑(seamless genome editing)仍有一定难度。
piggyBac转座子系统来源于鳞翅目昆虫,其转座酶识别DNA两端的特定反向末端重复序列(inverted terminal repeat sequences, ITRs),并将其插入染色体中TTAA位点[6,7]。该系统简单高效,早期用在果蝇、蚊子(Aedes aegypti)等昆虫中获得转基因品系[8,9]。近年来研究发现,piggyBac转座子系统在哺乳动物细胞中也具有很高的转座活性[10,11]。除了能够将外源基因转入基因组,piggyBac转座酶还能够将带有特定ITRs的DNA序列从基因组中精确移除。该系统与CRISPR/Cas9技术联用,能够用来移除在人诱导多能干细胞(induced pluripotent stem cells, iPSCs)基因组编辑中引入的药物筛选标记或核酸酶,且不遗留任何外源核酸序列,实现基因组无缝编辑[12,13]。
果蝇CG4894基因位于2号染色体左臂,其编码的Ca-α1D蛋白是L型电压门控钙离子通道(voltage- gating calcium channel, VGCC)的重要组成亚基,参与肌肉收缩、神经重塑、上皮液转运等一系列重要的生命过程[14,15,16,17]。研究Ca-α1D蛋白,尤其是其特定结构域的功能对揭示电压门控钙离子通道的生理功能具有重要意义。
本研究以果蝇为材料,利用CRISPR/Cas9技术和piggyBac转座子系统将CG4894基因第18个外显子上第21位酪氨酸(tyrosine, Y)突变为半胱氨酸(cysteine, C),且没有引入任何外源序列。本研究应用的基因打靶策略为果蝇基因组的精确编辑提供了更多选择。
1 材料与方法
1.1 材料
野生型果蝇w1118、平衡染色体品系Bc/CyO和piggyBac转座酶表达品系BL#8284均来自于Bloomington果蝇库,BL#8284果蝇品系2号染色体携带hsp70-PBac并与CyO标记相偶联,可内源性表达piggyBac转座酶。Nos-Cas9[w-](3rd chr)果蝇品系为本实验室果蝇库所有,该品系3号染色体attP2位置插入了Nos-Cas9基因,胚胎细胞能够内源性表达Cas9核酸内切酶。U6b-sgRNA-short载体由清华大学医学院倪建泉实验室构建[18],pGX-attP载体为本实验室构建[19]。1.2 果蝇基因组测序
在Flybase数据库中查询CG4894基因序列信息,设计引物对Nos-Cas9[w-](3rd chr)果蝇品系CG4894基因第18外显子(长度为108 bp)上下游各约300 bp进行PCR扩增及测序(引物序列见表1),扩增片段大小为700 bp,经测序,结果表明序列与Flybase数据库中相同。这一步骤的目的是确保sgRNA能顺利与基因组中相应序列匹配,并正确引导Cas9切割靶序列。由于基因内含子区域常存在比较大的变异性,当sgRNA序列位于内含子区域时,这一步尤为重要。Table 1
表1
表1 引物序列信息
Table 1
| 名称 | 序列(5′→3′) | 用途 |
|---|---|---|
| FW1 | CTTGAACGTGTTTCGGTTGT | PCR扩增及测序 |
| RV1 | CTGAACTGAGCTGGGTGG | PCR扩增及测序 |
| FW2 | TTCGCACCAACATGATATTACCAA | sgRNA质粒构建 |
| RV2 | AAACTTGGTAATATCATGTTGGTG | sgRNA质粒构建 |
| FW3 | GGGCGCGTACTCCACGAATTCAGAACGTTATAGTCTTGTAGCAGGG | 5′同源臂构建 |
| RV3 | ATAATGTTTCCAATGGTTTTGATTGCGAC | 5′同源臂构建 |
| FW4 | AAAACCATTGGAAACATTATGTTGGTGACATGTTTGTTACAATTCATGTTCG | 5′同源臂构建 |
| RV4 | ATATGATTATCTTTCTAGGGTTAAAGCTGTCTTCAGATAC | 5′同源臂构建 |
| FW5 | CGCAGACTATCTTTCTAGGGTTAAGTGTGCGCTTTTATTT | 3′同源臂构建 |
| RV5 | GAAGTTATGGTACCGGGTACCACATCATCGAAGTGGAAGCG | 3′同源臂构建 |
| FW6 | ACTCAAGCGTAGGGAAACAC | 跨5′同源臂PCR扩增 |
| RV6 | TGAATTGTCGCTCCGTAGAC | 跨5′同源臂PCR扩增 |
| FW7 | AAATGCACAGCGACGGATTC | 跨3′同源臂PCR扩增 |
| RV7 | CAGCCCTCAAATGTGGACAC | 跨3′同源臂PCR扩增 |
新窗口打开|下载CSV
1.3 sgRNA质粒构建
首先在目标突变位点附近寻找带有PAM (protospacer adjacent motif)的序列作为sgRNA序列,将对应序列(5′-CACCAACATGATATTACCAA-3′)在果蝇CRISPR网站(http://flycrispr.molbio.wisc.edu)上检测,没有发现可能的脱靶序列。根据sgRNA序列设计2条5′端分别带有TTCG和AAAC序列的互补引物FW2和RV2 (序列见表1)。将2种引物分别配制成100 μmol/L溶液,各取10 μL,加入2.5 μL 10×的T4 DNA连接酶缓冲液,并补水2.5 μL至终体积25 μL,在PCR仪上复性(复性条件为100℃ 10 min,之后每分钟温度降1℃至最终温度25℃)形成5′末端带有粘性末端的双链DNA片段,通过T4 DNA连接酶(NEB公司,美国)连接在经BbsⅠ酶切后回收的载体U6b-sgRNA-short上,获得sgRNA表达质粒,方法见参考文献[20]。1.4 供体DNA质粒构建
将pGX-attP载体(载体详情见参考文献[19])上两个多克隆位点之间的attP-hsp70-white (从Acc65Ⅰ位点至SpeⅠ位点)替换为两端带有piggyBac转座酶识别位点ITRs的DsRed标记基因,获得载体pGX- DsRed。在目标突变位点附近寻找TTAA位点作为标记基因插入位置,在此序列前后各约1.2 kb和1.1 kb分别设计5′同源臂和3′同源臂,同源臂扩增使斜体下划线分别为酶切位点EcoRⅠ和Acc65Ⅰ序列,仅下划线为同义突变碱基,粗体下划线为点突变碱基,粗体为TTAA序列。用的PCR酶为Phanta高保真酶(南京诺唯赞公司)。扩增5′同源臂的引物为FW3、RV3、FW4和RV4(序列见表1),为了引入突变碱基,本研究将5′同源臂分成2部分进行扩增。由于供体DNA质粒中包含sgRNA序列,为防止供体质粒注入果蝇胚胎后被Cas9切割,本研究对这部分序列中的3个氨基酸进行了同义突变(GGT→GGA,AAT→AAC,ATC→ ATT),同时扩增过程中还在FW3引物中引入了点突变(TAT→TGT),编码氨基酸由酪氨酸变成半胱氨酸。3′同源臂扩增引物为FW4和RV4(序列见表1)。为保证DsRed标记在后续实验过程中顺利移除,5′同源臂的3′端和3′同源臂的5′端均带有TTAA序列,这样经piggyBac转座酶处理后才能得到无缝效果。扩增得到的片段使用多片段同源重组酶(南京诺唯赞公司)进行连接。
1.5 基因敲入果蝇突变体的获得
取sgRNA表达质粒5 μg、供体DNA质粒10 μg,混合后用酚/氯仿抽提纯化,并用20 μL注射缓冲液(5 mmol/L氯化钾,0.1 mmol/L磷酸钠,pH 6.8)溶解质粒,使sgRNA质粒和供体DNA质粒终浓度分别为250 ng/μL和500 ng/μL。收集Nos-Cas9[w-](3rd chr)果蝇胚胎进行显微注射,孵化出的雌蝇和雄蝇分别与w1118的雄蝇和处女蝇杂交,筛选后代携带红色荧光标记的果蝇,并与平衡染色体品系Bc/CyO杂交。对筛选到的转基因果蝇进行鉴定时,分别在基因组和插入的标记基因上设计引物用于跨同源臂PCR鉴定。跨5′同源臂PCR产物大小约为1.8 kb,上游引物FW6位于基因组5′同源臂上游,下游引物RV6位于DsRed基因近5′端;跨3′同源臂PCR产物大小约为1.2 kb,上游引物FW7位于基因组3′同源臂上游,下游引物RV7位于DsRed基因近3′端。引物序列见表1。经1%琼脂糖凝胶电泳检测确认扩增条带大小是否正确。1.6 筛选标记基因的切除
将鉴定正确的转基因果蝇品系与表达piggyBac转座酶的果蝇品系BL#8284杂交,在杂交后代中挑选基因型为cg4894[DsRed]*/CyO,hsp70-PBac的杂合子,该基因型的杂合子部分细胞中的DsRed标记被转座酶切除而显示出镶嵌表型,可通过观察复眼的镶嵌型红色荧光表型挑选该基因型的果蝇,并根据复眼中红色荧光存留比例估算大致移除效率。将该杂合子与平衡染色体品系Bc/CyO杂交,在后代中挑选不带DsRed标记且带有CyO标记的果蝇,并与平衡染色体品系Bc/CyO杂交,并使其纯合。提取纯合果蝇基因组DNA,对CG4894基因第18个外显子附近进行PCR扩增及测序,确定纯合品系是否携带点突变。2 结果与分析
2.1 CG4894敲入突变体的获得及鉴定
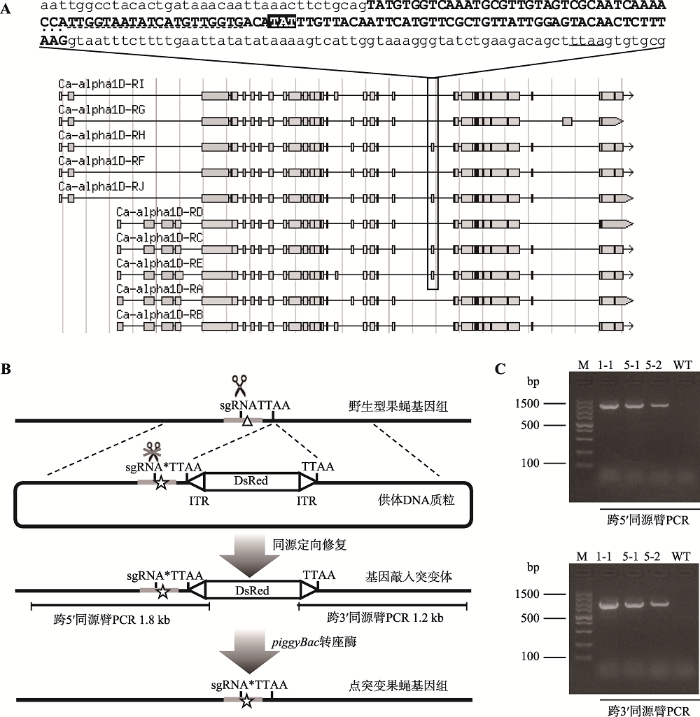
本研究通过查询Flybase数据库获得CG4894基因序列,发现目标氨基酸—酪氨酸位于第18外显子第21位,此外显子为转录本C、E、H和J共有,其序列见图1A (大写粗体部分)。本研究通过PCR扩增及测序,验证了第18外显子上下游约300 bp的序列与数据库中查询序列相同,在目标氨基酸附近选取合适的sgRNA序列和TTAA序列。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1基因敲入突变体的设计及鉴定
A:CG4894基因转录本第18外显子示意图。下方为CG4894基因转录本示意图(引自Flybase数据库),方框标记表示部分转录本C、E、H和J共有的第18外显子,上方为这一外显子及其上下游序列,阴影部分为目标突变位点,实线下划线为TTAA序列,虚线下划线为sgRNA序列,加点CCA为PAM序列;B:CG4894基因点突变体构建示意图。三角形标记为基因组中目标点突变位置,五角星标记为供体DNA质粒携带的点突变,sgRNA*表示供体DNA质粒上的sgRNA携带同义突变;C:CG4894基因敲入果蝇跨同源臂PCR鉴定结果。M:100 bp DNA maker;1-1、5-1、5-2为携带DsRed筛选标记的转基因果蝇品系;WT:野生型果蝇。
Fig. 1The design and molecular verification of CG4894 knock-in flies
在构建供体DNA质粒时,本研究以目标突变位点附近的TTAA序列为界,分别选取TTAA上游1.2 kb序列作为5′同源臂,TTAA下游1.1 kb序列作为3′同源臂。由于5′同源臂包含了sgRNA序列,为防止Cas9剪切,本研究将sgRNA序列中3个氨基酸进行了同义突变。5′同源臂分为2个部分进行扩增,并通过引物引入sgRNA中的同义突变和21位氨基酸的点突变(图1B),成功进行同源重组后两端带有ITRs的DsRed基因被插入基因组TTAA位置。后续实验中,piggyBac转座酶可将2个TTAA之间的DsRed标记移除,恢复为原基因组中的1个TTAA序列,实现基因组的无缝编辑。
将sgRNA质粒和供体DNA质粒混合物注入约1000个Nos-Cas9[w-](3rd chr)果蝇胚胎中,最终得到257只雄蝇和240只雌蝇,注射成活率为49.7%。这些雄蝇和雌蝇分别和w1118处女蝇、雄蝇进行杂交,后代经过红色荧光筛选,获得3个携带DsRed筛选标记的果蝇品系。杂交实验表明,3个品系中DsRed标记均位于目标基因所在的2号染色体上,其编号分别为1-1、5-1和5-2。此外,如果发生了同源重组,利用FW6、RV6引物扩增可得到1.8 kb大小、跨5′同源臂的PCR产物,利用FW7、RV7引物扩增可得到1.2 kb大小、跨3′同源臂的PCR产物,电泳结果显示3个品系的基因组DNA均能扩增得到预期大小的条带,证实本研究已成功构建了敲入突变体(图1C)。
2.2 DsRed荧光标记的去除和鉴定
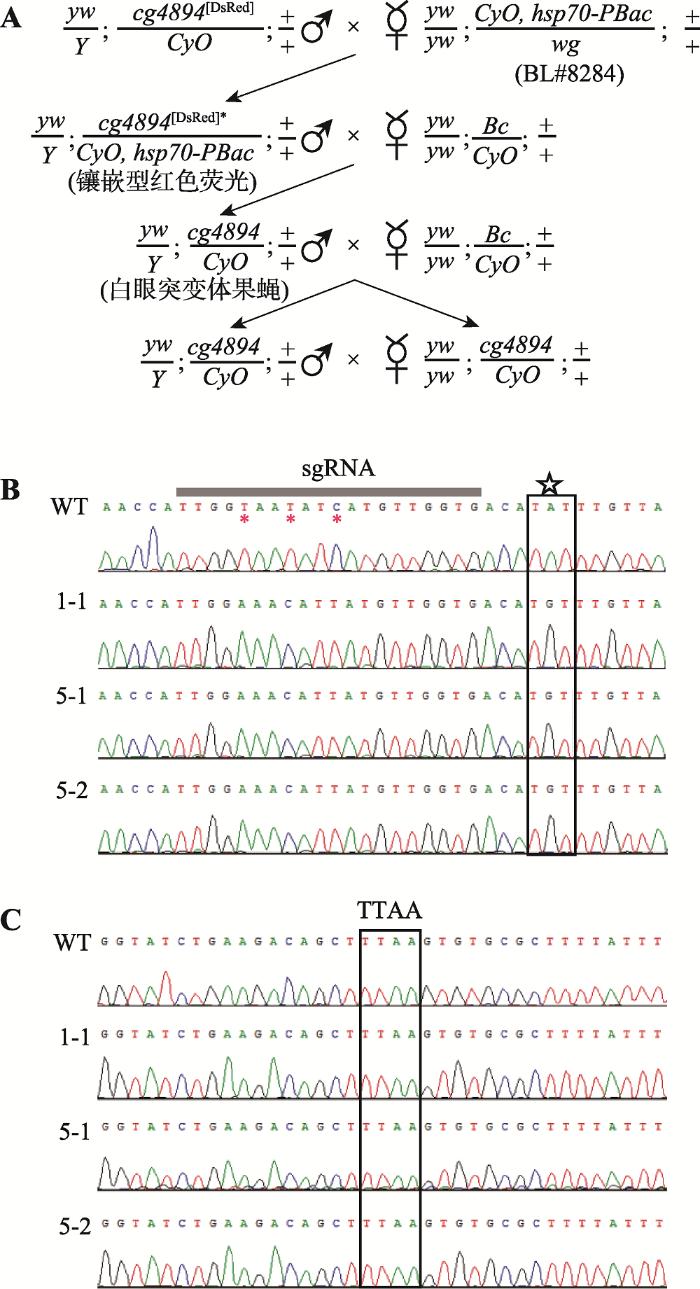
为了移除上述基因敲入品系中的筛选标记,本研究通过杂交方式引入piggyBac转座酶。BL#8284果蝇品系2号染色体上带有由hsp70启动子驱动表达的piggyBac转座酶,将这一品系与带有DsRed标记的转基因品系杂交,后代基因型为cg4894[DsRed]*/ CyO, hsp70-PBac的杂合体表现出镶嵌表型,即部分细胞中DsRed标记被piggyBac转座酶切除,且红色荧光存留比例取决于piggyBac的表达和剪切效率。本研究杂合体果蝇复眼中约有50%的小眼无红色荧光,据此估算转座酶的切除效率大概为50%左右。将镶嵌表型果蝇与Bc/CyO平衡系果蝇杂交,后代出现的无红色荧光果蝇(图2A)与平衡染色体品系Bc/CyO杂交,并使其纯合。3个品系中携带DsRed筛选标记的转基因果蝇1-1、5-1和5-2经过杂交,均获得了不携带荧光标记的白眼点突变纯合果蝇,这些品系未出现明显表型。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2标记基因的去除和点突变体的测序鉴定
A:去除标记基因DsRed的杂交流程。星号表示部分DsRed标记基因移除;B:野生型果蝇和CG4894基因点突变体在目标突变位点处的序列比较。五角星标记表示目标突变位点,星号表示同义突变位点;C:野生型果蝇和CG4894基因点突变体在TTAA位点处的序列比较。WT:野生型果蝇;1-1、5-1、5-2为白眼点突变纯合果蝇品系。
Fig. 2Removal of marker genes and identification of point mutants by sequencing
理论上,同源重组可以发生在同源臂的任意位置。在本研究中,如果同源重组发生在突变位点和TTAA之间,则突变不被引入基因组。因此,本研究对获得的白眼点突变纯合果蝇品系进行了测序,验证突变是否引入。测序结果表明,3个品系中均存在Y→C突变及sgRNA部分的同义突变(图2B),且TTAA位点附近没有引入任何突变和外源序列(图2C)。
综上所述,本研究成功在果蝇CG4894基因中引入了点突变,并且没有任何外源序列遗留,证实了CRISPR/Cas9和piggyBac转座子系统联用策略用于果蝇基因组精确编辑的可行性。
3 讨论
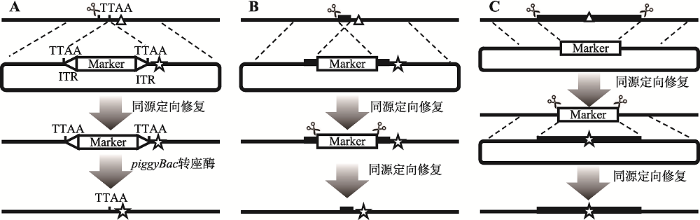
基因组编辑技术能够精确地实现生物体基因组定点突变、插入或敲除,人为改变生物遗传信息。20世纪末人们就开始对基因编辑技术进行探索,从第一代DNA核酸酶编辑系统ZFN (zinc finger nuclease)、第二代TALEN (transcription activator-like effector nuclease)到第三代CRISPR/Cas9系统,基因编辑效率不断提高、成本逐渐降低,应用范围不断扩大[21]。近几年CRISPR/Cas9技术的迅速发展,及其在各种模式生物中的广泛应用,使得突变体的获得变得更加简单高效。与此同时,人们对突变体的特异性也提出了越来越高的要求。如前所述,NHEJ容易引起随机插入和缺失,它导致的突变带有很大的随机性,想要获得“设计”的突变必须通过同源定向修复。但在这一策略中,提高突变体筛选的效率和不引入无关序列就成了一对矛盾,这一矛盾在果蝇中尤为突出。由于果蝇体型较小,无法使用部分身体组织提取DNA,只能在子代产出之后再将亲本处死,提取DNA进行基因型鉴定,因此在果蝇基因编辑过程中多引入各种筛选标记,通过对后代进行表型筛选提高突变体的筛选效率。为了精确去除筛选标记,在人诱导多能干细胞的基因组编辑中,piggyBac转座酶被尝试用于去除基因编辑过程中引入的药物筛选标记,以实现基因无缝编辑,并获得了成功(图3A)。除此以外,还有2种策略已被报道。单链退火(single-strand annealing, SSA)介导的DNA双链断裂修复(SSA-mediated DNA double-strand break repair)[22]在HEK293T细胞CCR5基因编辑过程中得到应用,该策略是在同源重组阶段通过带有重叠的同源臂引入染色体内重复,然后介导染色体内同源重组将标记基因去除(图3B)。两步同源重组实现的无缝基因置换[23]被用于果蝇tan基因的编辑,该策略利用第一次同源重组将目标基因替换为标记基因,再利用第二次同源重组将标记基因替换为突变后基因(图3C)。但这两种方法与piggyBac转座酶法相比,筛选标记的去除都需通过同源重组,因此稍显复杂。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图33种基因组编辑策略的比较
A:piggyBac介导的基因组无缝编辑。标记基因两端带有转座酶特异识别序列(ITRs),CRISPR/Cas9介导的同源重组使其随着突变插入染色体TTAA位点,后经piggyBac转座酶切除,只在染色体中保留突变;B:单链退火(single-strand annealing, SSA)介导的DNA双链断裂修复。两条同源臂在基因组中有重叠,因此第一次同源重组导致染色体上携带重复片段,且重复片段位于标记基因两端,再次利用CRISPR/Cas9制造染色体断裂可以诱导染色体内同源重组的发生,将标记基因去除;C:通过两步同源重组实现的无缝基因置换。第一次同源重组将目标基因替换为标记基因,第二次同源重组将标记基因替换为带有突变的目标基因。其中,目标基因两端可以携带不同于基因组剪切所用的sgRNA序列,以保证Cas9不会作用于同源臂载体。图根据文献[13,22,23]修改绘制,三角形标记表示基因组中目标点突变位置,五角星标记表示供体DNA质粒携带的点突变。
Fig. 3The comparison of three genome editing strategies
piggyBac转座子系统简单高效,不需二次基因打靶就能将标记基因精确切除。而且,由于这一系统早就用于果蝇的基因编辑,果蝇中已发展了一系列相关工具,如本研究中用到的内源表达piggyBac转座酶的果蝇品系。这一品系使得标记基因的去除可以通过简单的杂交进行,而不像其他系统,需要通过注射或其他复杂操作将转座酶引入突变体。当然,该系统对基因序列有一定要求,标记基因需要插入内源的TTAA序列,如果其距离目标突变位点较远则会降低突变引入的效率。理论上,TTAA在基因组中出现的概率为1/256,由于TTAA只需在目标区段附近即可,因而不难找到。
综上所述,CRISPR/Cas9和piggyBac转座子系统联用策略在果蝇中的可行性为果蝇基因组的精确编辑提供了更多选择。
(责任编委: 张雷)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]
URLPMID:22745249 [本文引用: 1]

Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems provide bacteria and archaea with adaptive immunity against viruses and plasmids by using CRISPR RNAs (crRNAs) to guide the silencing of invading nucleic acids. We show here that in a subset of these systems, the mature crRNA that is base-paired to trans-activating crRNA (tracrRNA) forms a two-RNA structure that directs the CRISPR-associated protein Cas9 to introduce double-stranded (ds) breaks in target DNA. At sites complementary to the crRNA-guide sequence, the Cas9 HNH nuclease domain cleaves the complementary strand, whereas the Cas9 RuvC-like domain cleaves the noncomplementary strand. The dual-tracrRNA:crRNA, when engineered as a single RNA chimera, also directs sequence-specific Cas9 dsDNA cleavage. Our study reveals a family of endonucleases that use dual-RNAs for site-specific DNA cleavage and highlights the potential to exploit the system for RNA-programmable genome editing.
[本文引用: 1]
URLPMID:4814844 [本文引用: 1]
The clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) system has been broadly adopted for highly efficient genome editing in a variety of model organisms and human cell types. Unlike previous genome editing technologies such as zinc finger nucleases (ZFNs) and transcription activator61like effector nucleases (TALENs), CRISPR/Cas technology does not... [Show full abstract]
URLPMID:12464689 [本文引用: 1]
In the postgenomic era the mouse will be central to the challenge of ascribing a function to the 40,000 or so genes that constitute our genome. In this review, we summarize some of the classic and modern approaches that have fueled the recent dramatic explosion in mouse genetics. Together with the sequencing of the mouse genome, these tools will have a profound effect on our ability to generate new and more accurate mouse models and thus provide a powerful insight into the function of human genes during the processes of both normal development and disease.
URLPMID:2549707 [本文引用: 1]
The transposable IFP2 element of Trichoplusia ni was originally isolated as a host DNA insertion in spontaneous FP mutants of Galleria mellonella or Autographa californica nuclear polyhedrosis viruses (NPVs). The termini of IFP2 insertions from five independently isolated FP mutants were sequenced. In all cases IFP2 is flanked by 13-bp terminal inverted repeats and has additional inverted repeats of 19 by in length located asymmetrically with respect to the ends of the element. Insertion of IFP2 into the viral genome always generated a duplication of the tetranucleotide target site, TTAA. There was an apparent preference for insertion within a 12-bp A+T-rich imperfect palindromic sequence surrounding the target site. Sequence analysis of three independent IFP2 elements revealed an internal domain of 2.475 kb containing an RNA polymerase II promoter region and two large open reading frames. Primer extension analysis of IFP2-specific mRNA positioned the 5鈥 terminus of the transcript. The element is present in DNA isolated from T. ni cell lines TN-368 and TN-5131, but is not apparent in DNAs isolated from the TN-R 2 cell line or our laboratory colony of T. ni larvae, suggesting IFP2 was recently introduced into the T. ni genome.
URLPMID:2553540 [本文引用: 1]
We report the complete sequences of two representatives of the TFP3 transposable element family of the lepidopteran, Trichoplusia ni. These elements were isolated as insertions mobilized from the Lepidopteran host genome into two closely related nuclear polyhedrosis viruses (NPV) during infection. Both elements inserted within the 500-bp FP locus of the respective viral genomes (map units 36.0 to 37.0), causing a distinctive plaque morphology phenotype and the loss of a 25-kDa viral-specific protein. Both insertions occurred at the identical TTAA target site in the respective genomes, in the same relative orientation, and are flanked by 15-bp imperfect inverted repeats. The inserted elements interrupt the 25 K open reading frame (ORF). One of these FP mutants undergoes spontaneous reversion. Sequence analysis at the excision site of a spontaneous revertant demonstrates that the TFP3 elements are capable of precise excision, restoring the expression of the 25-kDa protein. We also compare the sequences of the 25 K genes of the Autographa californica and Galleria mellonella viruses (AcMNPV and GmMNPV, respectively). The 25 K gene sequences diverge in five areas, resulting in an additional EcoRV and TaqI site within the GmMNPV 25 K gene, and extension of the ORF for an additional 2 amino acids at the C-terminus of the predicted GmMNPV 25 kDa protein. The phenomenon of transposon mutagenesis of Baculovirus genomes provides a unique opportunity for analysis of transposon mobility.
[本文引用: 1]
[本文引用: 1]
URLPMID:16096065 [本文引用: 1]
Transposable elements have been routinely used for genetic manipulation in lower organisms, including generating transgenic animals and insertional mutagenesis. In contrast, the usage of transposons in mice and other vertebrate systems is still limited due to the lack of an efficient transposition system. We have tested the ability of piggyBac ( PB), a DNA transposon from the cabbage looper moth Trichoplusia ni, to transpose in mammalian systems. We show that PB elements carrying multiple genes can efficiently transpose in human and mouse cell lines and also in mice. PB permits the expression of the marker genes it carried. During germline transposition, PB could excise precisely from original insertion sites and transpose into the mouse genome at diverse locations, preferably transcription units. These data provide a first and critical step toward a highly efficient transposon system for a variety of genetic manipulations including transgenesis and insertional mutagenesis in mice and other vertebrates.
URLPMID:17005721 [本文引用: 1]
A nonviral vector for highly efficient site-specific integration would be desirable for many applications in transgenesis, including gene therapy. In this study we directly compared the genomic integration efficiencies of piggyBac, hyperactive Sleeping Beauty (SB11), Tol2, and Mos1 in four mammalian cell lines. piggyBac demonstrated significantly higher transposition activity in all cell lines whereas Mos1 had no activity. Furthermore, piggyBac transposase coupled to the GAL4 DNA-binding domain retains transposition activity whereas similarly manipulated gene products of Tol2 and SB11 were inactive. The high transposition activity of piggyBac and the flexibility for molecular modification of its transposase suggest the possibility of using it routinely for mammalian transgenesis.
URLPMID:27929521 [本文引用: 1]
Abstract Genome editing of human induced pluripotent stem cells (hiPSCs) offers unprecedented opportunities for in vitro disease modeling and personalized cell replacement therapy. The introduction of Cas9-directed genome editing has expanded adoption of this approach. However, marker-free genome editing using standard protocols remains inefficient, yielding desired targeted alleles at a rate of 1-5%. We developed a protocol based on a doxycycline-inducible Cas9 transgene carried on a piggyBac transposon to enable robust and highly efficient Cas9-directed genome editing, so that a parental line can be expeditiously engineered to harbor many separate mutations. Treatment with doxycycline and transfection with guide RNA (gRNA), donor DNA and piggyBac transposase resulted in efficient, targeted genome editing and concurrent scarless transgene excision. Using this approach, in 7 weeks it is possible to efficiently obtain genome-edited clones with minimal off-target mutagenesis and with indel mutation frequencies of 40-50% and homology-directed repair (HDR) frequencies of 10-20%.
URLPMID:24071911 [本文引用: 1]
I report here a detailed protocol for seamless genome editing using the piggyBac transposon in human pluripotent stem cells (hPSCs). Recent advances in custom endonucleases have enabled us to routinely perform genome editing in hPSCs. Conventional approaches use the Cre/loxP system that leaves behind residual sequences in the targeted genome. I used the piggyBac transposon to seamlessly remove a drug selection cassette and demonstrated safe genetic correction of a mutation causing alpha-1 antitrypsin deficiency in patient-derived hPSCs. An alternative approach to using the piggyBac transposon to correct mutations involves using single-stranded oligonucleotides, which is a faster process to complete. However, this experimental procedure is rather complicated and it may be hard to achieve homozygous modifications. In contrast, using the piggyBac transposon with drug selection-based enrichment of genetic modifications, as described here, is simple and can yield multiple correctly targeted clones, including homozygotes. Although two rounds of genetic manipulation are required to achieve homozygote modifications, the entire process takes similar to 3 months to complete.
URLPMID:7869089 [本文引用: 1]
Abstract We report the complete sequence of a calcium channel alpha 1 subunit cDNA cloned from a Drosophila head cDNA library. This cDNA encodes a deduced protein containing 2516 amino acids with a predicted molecular weight of 276,493. The deduced protein shares many features with vertebrate homologs, including four repeat structures, each containing six transmembrane domains, a conserved ion selectivity filter region between transmembrane domains 5 and 6, and an EF hand in the carboxy tail. The Drosophila subunit has unusually long initial amino and terminal carboxy tails. The region corresponding to the last transmembrane domain (IVS6) and the adjacent cytoplasmic domain has been postulated to form a phenylalkylamine-binding site in vertebrate calcium channels. This region is conserved in the Drosophila sequence, while domains thought to be involved in dihydropyridine binding show numerous changes. The Drosophila subunit exhibits 78.3% sequence similarity to the rat brain type D calcium channel alpha 1 subunit, and so has been designated as a Drosophila melanogaster calcium channel alpha 1 type D subunit (Dmca1D). In situ hybridization shows that Dmca1D is highly expressed in the embryonic nervous system. Northern analysis shows that Dmca1D cDNA hybridizes to three size classes of mRNA (9.5, 10.2, and 12.5 kb) in heads, but only two classes (9.5 and 12.5 kb) in bodies and legs. PCR analysis suggests that the Dmca1D message undergoes alternative splicing with more heterogeneity appearing in head and embryonic extracts than in bodies and legs.
[本文引用: 1]
URLPMID:2600454412 [本文引用: 1]
We studied, in a genetic model organism, Drosophila melanogaster, the channel mechanisms underlying membrane excitation in the embryonic body wall muscle whose biophysical properties have been poorly characterized. The inward current underlying the action potential was solely mediated by a high-threshold class of voltage-gated Ca2+ channels, which exhibited slow inactivation, Ca2+ permeability with saturation at high [Ca2+]OUT, and sensitivity to a Ca2+ channel blocker, Cd2+. The Ca2+ current in the embryonic muscle was completely eliminated in Dmca1D mutants, indicating that the Dmca1D-encoded Ca2+ channel is the major mediator of inward currents in the body wall muscles throughout the embryonic and larval stages.
URL [本文引用: 1]
Dysregulation of L-type Ca 2+ channels (LTCCs) underlies numerous cardiac pathologies. Understanding their modulation with high fidelity relies on investigating LTCCs in their native environment with intact interacting proteins. Such studies benefit from genetic manipulation of endogenous channels in cardiomyocytes, which often proves cumbersome in mammalian models. Drosophila melanogaster , however, offers a potentially efficient alternative as it possesses a relatively simple heart, is genetically pliable, and expresses well-conserved genes. Fluorescence in situ hybridization confirmed an abundance of Ca-1D and Ca-1T mRNA in fly myocardium, which encode subunits that specify hetero-oligomeric channels homologous to mammalian LTCCs and T-type Ca 2+ channels, respectively. Cardiac-specific knockdown of Ca-伪1D via interfering RNA abolished cardiac contraction, suggesting Ca-1D represents the primary functioning Ca 2+ channel in Drosophila hearts. Moreover, we successfully isolated viable single cardiomyocytes and recorded Ca 2+ currents via patch clamping, a feat never before accomplished with the fly model. The profile of Ca 2+ currents recorded in individual cells when Ca 2+ channels were hypomorphic or absent, or under selective LTCC blockage by nifedipine, additionally confirmed the predominance of Ca-伪1D current across all activation voltages. T-type currents, activated at more negative voltages, were also detected. Lastly, Ca-伪1D channels displayed Ca 2+ -dependent inactivation, a critical negative feedback mechanism of LTCCs, and the current through them was augmented by forskolin, an activator of the protein kinase A pathway. In sum, the Drosophila heart possesses a conserved compendium of Ca 2+ channels, suggesting that the fly may serve as a robust and effective platform for studying cardiac channelopathies.
[本文引用: 1]
[本文引用: 2]
URL [本文引用: 1]
生殖系统功能的正常维持是物种繁衍的基础,需要多基因协同作用,但其中许多基因的具体功能和作用机制并不清楚.本研究选取了果蝇(Drosophila melanogaster)中 8 个在睾丸中表达、功能未知且与人(Homo sapiens)和小鼠(Mus musculus)高度同源的基因(CG4161、CG11475、CG2921、CG10541、CG7276、CG3800、CG8117和CG16779),分析了它们在不同组织中的表达水平,并分别检测了它们在雄性生殖系统中的功能.在这8个基因中,前5个为睾丸优势表达基因,其余3个为全身性表达.首先,利用CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9)技术结合同源定向修复(homology-directed re-pair, HDR)在果蝇中对8个候选基因逐一进行敲除,建立了纯合的基因敲除突变品系;然后对这些品系的雄蝇进行了生育力测试及睾丸细胞学观察.结果显示,CG7276 和 CG3800 基因敲除果蝇出现部分雄蝇不育且可育雄蝇后代数量较野生型显著下降.睾丸解剖观察显示CG7276和CG3800的功能缺失可导致雄蝇分别出现不同程度的精囊缩小及精原干细胞减少和细胞分布混乱;染色结果也提示CG7276和CG3800在精子的成熟过程中发挥一定作用.其他6个基因突变并未导致雄蝇育性变化或睾丸形态异常.这些突变体的获得及表型的初步分析为进一步研究基因功能及机制提供了良好的动物模型及基础.
URL [本文引用: 1]
生殖系统功能的正常维持是物种繁衍的基础,需要多基因协同作用,但其中许多基因的具体功能和作用机制并不清楚.本研究选取了果蝇(Drosophila melanogaster)中 8 个在睾丸中表达、功能未知且与人(Homo sapiens)和小鼠(Mus musculus)高度同源的基因(CG4161、CG11475、CG2921、CG10541、CG7276、CG3800、CG8117和CG16779),分析了它们在不同组织中的表达水平,并分别检测了它们在雄性生殖系统中的功能.在这8个基因中,前5个为睾丸优势表达基因,其余3个为全身性表达.首先,利用CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9)技术结合同源定向修复(homology-directed re-pair, HDR)在果蝇中对8个候选基因逐一进行敲除,建立了纯合的基因敲除突变品系;然后对这些品系的雄蝇进行了生育力测试及睾丸细胞学观察.结果显示,CG7276 和 CG3800 基因敲除果蝇出现部分雄蝇不育且可育雄蝇后代数量较野生型显著下降.睾丸解剖观察显示CG7276和CG3800的功能缺失可导致雄蝇分别出现不同程度的精囊缩小及精原干细胞减少和细胞分布混乱;染色结果也提示CG7276和CG3800在精子的成熟过程中发挥一定作用.其他6个基因突变并未导致雄蝇育性变化或睾丸形态异常.这些突变体的获得及表型的初步分析为进一步研究基因功能及机制提供了良好的动物模型及基础.
URL [本文引用: 1]
基因编辑技术是以特异性改变遗传物质靶向序列为目标的技术。近年来,锌指核酸酶(zinc finger nuclease,ZFN)、类转录激活因子效应核酸酶(transcription activator-like effector nuclease, TALEN)、规律成簇的间隔短回文重复(regular clustering of short palindrome repeats, CRISPR)和单碱基编辑(base editing, BE)技术的相继出现,不仅为基因功能研究提供了有力的工具,还为生命医学提供了新的治疗方案。基因编辑技术已经大范围应用于动物细胞模型的构建、药物靶点的筛查和基因功能研究等,在基因治疗领域也展现出广阔的应用前景。本文就基因编辑技术的研究进展及其在基因治疗中的应用进行了概述,并对基因编辑技术的的原理、发展史、优缺点以及在基因治疗中的应用前景和机遇挑战进行了讨论,以期为基因编辑技术的临床转化提供参考。
URL [本文引用: 1]
基因编辑技术是以特异性改变遗传物质靶向序列为目标的技术。近年来,锌指核酸酶(zinc finger nuclease,ZFN)、类转录激活因子效应核酸酶(transcription activator-like effector nuclease, TALEN)、规律成簇的间隔短回文重复(regular clustering of short palindrome repeats, CRISPR)和单碱基编辑(base editing, BE)技术的相继出现,不仅为基因功能研究提供了有力的工具,还为生命医学提供了新的治疗方案。基因编辑技术已经大范围应用于动物细胞模型的构建、药物靶点的筛查和基因功能研究等,在基因治疗领域也展现出广阔的应用前景。本文就基因编辑技术的研究进展及其在基因治疗中的应用进行了概述,并对基因编辑技术的的原理、发展史、优缺点以及在基因治疗中的应用前景和机遇挑战进行了讨论,以期为基因编辑技术的临床转化提供参考。
URL [本文引用: 1]
URLPMID:27494619 [本文引用: 1]
Genome editing via the CRISPR/Cas9 RNA-guided nuclease system has opened up exciting possibilities for genetic analysis. However, technical challenges associated with homology-directed repair have proven to be roadblocks for producing changes in the absence of unwanted, secondary mutations commonly known as 090008scars.090009 To address these issues, we developed a 2-stage, marker-assisted strategy to facilitate precise, 090008scarless090009 edits in Drosophila with a minimal requirement for molecular screening. Using this method, we modified 2 base pairs in a gene of interest without altering the final sequence of the CRISPR cut sites. We executed this 2-stage allele swap using a novel transformation marker that drives expression in the pupal wings, which can be screened for in the presence of common eye-expressing reporters. The tools we developed can be used to make a single change or a series of allelic substitutions in a region of interest in any D. melanogaster genetic background as well as in other Drosophila species.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}