李智1,2,3, 何俊1,3, 蒋隽1,4, Richard G. Tait Jr.3, Stewart Bauck3, 过伟,2, 吴晓林,1,3,41. 湖南农业大学动物科技学院,长沙410128 2. 美国怀俄明大学动物科学系,怀俄明州拉勒米市82071 3. 美国纽勤公司生物信息与生物统计部,内布拉斯加州林肯市68504 4. 美国威斯康星大学动物科学系,美国威斯康星州麦迪逊市53706
Impacts of SNP genotyping call rate and SNP genotyping error rate on imputation accuracy inHolsteincattle
Zhi Li1,2,3, Jun He1,3, Jun Jiang1,4, Richard G. Tait Jr.3, Stewart Bauck3, Wei Guo,2, Xiao-Lin Wu,1,3,41. CollegeofAnimalScienceand Technology, HunanAgricultural University, Changsha 410128, China 2. Department of Animal Science, University of Wyoming, Laramie WY 82071, USA 3. Biostatisticsand Bioinformatics, NeogenGeneSeek, LincolnNE68504, USA 4. Department of Animal Sciences, University of Wisconsin, Madison WI 53706, USA
Supported by Hundred-Talent Project of Hunan Province, Key Researchand Development Program of Hunan Province.2018NK2081 Hunan Innovation Center of Animal Safety Production and Key Researchand Development Program of Changsha City.kq1801014
作者简介 About authors 李智,博士研究生,研究方向:动物遗传育种E-mail:zli13@uwyo.edu。
Abstract Single nucleotide polymorphism (SNP) chips have been widely used in genetic studies and breeding applications in animal and plant species. The quality of SNP genotypes is of paramount importance. More often than not, there are situations in which a number of genotypes may fail, requiring them to be imputed. There are also situations in which ungenotyped loci need to be imputed between different chips, or high-density genotypes need to be imputed based on low-density genotypes. Under these circumstances, the validity and reliability of subsequent data analyses is subject to the accuracy of these imputed genotypes. For justifying a better understanding of factors affecting imputation accuracy, in the present study, the impacts of SNP genotyping call rate and SNP genotyping error rate on the accuracy of genotype imputation were investigated under two scenarios in 20 116 U.S. Holstein cattle, each genotyped with a GGP 50K SNP chip. When the two factors were not correlated in scenario 1, simulated genotyping call rate varied from 50% to 100% and simulated genotyping error rate changed from 0% to 50%, with both factors being independent of each other. In scenario 2, genotyping error rates were correlated with genotyping call rate, and the relationship was set up by fitting a linear regression model between the two variables on a real dataset. That is, the simulated SNP call rate varied from 100% to 50% whereas the SNP genotyping rate changed from 0% to 13.55%. Finally, a 5-fold cross-validation was used to assess the subsequent imputation accuracy. The results showed that when original SNP genotyping call rate were independent of SNP genotyping error rate, the imputation accuracy did not change significantly with the original genotyping call rate (P>0.05), but it decreased significantly as the genotyping error rate increased (P<0.01). However, when original genotyping call rate was negatively correlated with genotyping error rate, the imputation error increased with elevated original genotyping error rate. In both scenarios, genotyping call rate needs to be no less than 0.90 in order to obtain 98% or higher genotype imputation accuracy. The present results can provide guidance for establishing quality assurance criteria for SNP genotyping in practice. Keywords:SNP chip;genotyping;imputation accuracy;call rate;error rate
PDF (451KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 李智, 何俊, 蒋隽, Richard G. Tait Jr., Stewart Bauck, 过伟, 吴晓林. 牛SNP芯片分型检出率和分型错误率对基因型填充准确率的影响[J]. 遗传, 2019, 41(7): 644-652 doi:10.16288/j.yczz.18-319 Zhi Li, Jun He, Jun Jiang, Richard G. Tait Jr., Stewart Bauck, Wei Guo, Xiao-Lin Wu. Impacts of SNP genotyping call rate and SNP genotyping error rate on imputation accuracy inHolsteincattle[J]. Hereditas(Beijing), 2019, 41(7): 644-652 doi:10.16288/j.yczz.18-319
随着高通量DNA测序和基因分型技术水平的不断提高,SNP芯片在各类遗传学研究和动植物育种中均得到了广泛应用[1,2],如全基因组关联分析(genome-wide association study, GWAS)[3,4]、基因组选择(genomic selection)[5,6]、基因组品种鉴定(genomic breed composition)[7]以及基因组选配(genomic mating)[8,9,10]等。SNP芯片在使用过程中,一个重要的数据处理环节是基因型填充(genotype imputation),即利用参考群体提供的各基因座位之间的连锁不平衡和重组率信息,构建彼此连锁的单倍型,然后依据所构建的单倍型信息,对目标个体(测试群体或有缺失基因型的个体)缺失位点上的基因型进行填充(预测)[11,12]。
不同颜色线条代表不同SNP分型错误率水平。 Fig. 2Impact of SNP genotyping call rate on imputation error rate in a Holstein dairy population, genotyped by GGP bovine 50K SNP chips
不同颜色线条代表不同SNP分型检出率水平。 Fig. 3Impact of SNP genotyping error rate on imputation error rate in a Holstein dairy population, genotyped by GGP bovine 50K SNP chips
vanEenennaamAL, WeigelKA, YoungAE, ClevelandMA, DekkersJCM . Applied animal genomics: results from the field Annu Rev AnimBiosci, 2013,2(2):105-139. [本文引用: 1]
WiggansGR, ColeJB, HubbardSM, SonstegardTS . Genomic selection in dairy cattle: the USDA experience Annu Rev AnimBiosci, 2017,5(1):309-327. [本文引用: 1]
AkdemirD, SánchezJI . Efficient breeding by genomic mating Front Genet, 2016,7:210. [本文引用: 1]
MarchiniJ, HowieB . Genotype imputation for genome-wide association studies Nat Rev Genet, 2010,11(7):499-511. [本文引用: 3]
HeS, DingXD, ZhangQ . Comparison of different genotype imputation methods Chin J AnimSci, 2013,49(23):95-100. [本文引用: 1]
AittokallioT . Dealing with missing values in large-scale studies: microarray data imputation and beyond BriefBioinform, 2009,11(2):253-264. [本文引用: 1]
WeigelKA, de los CamposG, González-RecioO, NayaH, WuXL, LongN, RosaGJ, GianolaD . Predictive ability of direct genomic values for lifetime net merit of holstein sires using selected subsets of single nucleotide polymerphism markers J Dairy Sci, 2009,92(10):5248-5257. URL [本文引用: 1]
FelipeVP, OkutH, GianolaD, SilvaMA, RosaGJ . Effect of genotype imputation on genome-enabled prediction of complex traits: an empirical study with mice data BMC Genet, 2014,15(1):149. [本文引用: 1]
ZhangZ, DruetT . Marker imputation with low-density marker panels in dutchholstein cattle J Dairy Sci, 2010,93(11):5487-5494. URL [本文引用: 1]
WuXL, GianolaD, HuZL, ReecyJM . Meta-analysis of quantitative trait association and mapping studies using parametric and non-parametric models J BiomBiostat, 2011,1:1-9. [本文引用: 1]
LopesFB, WuXL, LiH, XuJ, PerkinsT, GenhoJ, FerrettiR, Tait RGJr, BauckS, RosaGJ . Improving accuracy of genomic prediction in Brangus cattle by adding animals with imputed low-density SNP genotypes J Anim Breed Genet, 2018,135(1):14-27. URL [本文引用: 1]
ChenL, LiC, SargolzaeiM, SchenkelF . Impact of genotype imputation on the performance of GBLUP and Bayesian methods for genomic prediction PLoS One, 2014,9(7):e101544. URL [本文引用: 1]
BrowningBL, BrowningSR . A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals Am J Hum Genet, 2009,84(2):210-223. URL [本文引用: 1]
VenturaRV, LuD, SchenkelFS, WangZ, LiC, MillerSP . Impact of reference population on accuracy of imputation from 6K to 50K single nucleotide polymorphism chips in purebred and crossbreed beef cattle J AnimSci, 2014,92(4):1433-1444. [本文引用: 1]
RoshyaraNR, ScholzM . Impact of genetic similarity on imputation accuracy BMC Genet, 2015,16(1):90. [本文引用: 1]
PurfieldDC, McClureM, BerryDP . Justification for setting the individual animal genotype call rate threshold at eighty-five percent J AnimSci, 2016,94(11):4558-4569. [本文引用: 1]
BoisonSA, SantosDJA, UtsunomiyaAHT, CarvalheiroR, NevesHHR, O’BrienAMP, GarciaJF, SölknerJ, da SilvaMVGB . Strategies for single nucleotide polymorphism (SNP) genotyping to enhance genotype imputation in Gyr (Bosindicus) dairy cattle: comparison of commercially available SNP chips J Dairy Sci, 2015,98(7):4969-4989. URL [本文引用: 1]
VenturaRV, MillerSP, DoddsKG, AuvrayB, LeeM, BixleyM, ClarkeSM, McEwanJC . Assessing accuracy of imputation using different SNP panel densities in a multi-breed sheep population Genet SelEvol, 2016,48(1):71. [本文引用: 1]
HessMA, RhydderchJG, LeClairLL, BuckleyRM, KawaseM, HauserL . Estimation of genotyping error rate from repeat genotyping, unintentional recaptures and known parent-offspring comparisons in 16 microsatellite loci for brown rockfish (Sebastesauriculatus) MolEcolResour, 2012,12(6):1114-1123. [本文引用: 1]
WangJ . Estimating genotyping errors from genotype and reconstructed pedigree data Methods EcolEvol, 2018,9(1):109-120. [本文引用: 1]
SargolzaeiM, ChesnaisJP, SchenkelFS . A new approach for efficient genotype imputation using information from relatives BMC Genomics, 2014,15(1):478. URL [本文引用: 1]
CalusMP, BouwmanAC, HickeyJM, VeerkampRF, MulderHA . Evaluation of measures of correctness of genotype imputation in the context of genomic prediction: a review of livestock applications Animal, 2014,8(11):1743-1753. URL [本文引用: 1]
WuXL, XuJ, FengG, WiggansGR, TaylorJF, HeJ, QianC, QiuJ, SimpsonB, WalkerJ, BauckS . Optimal design of low-density SNP arrays for genomic prediction: algorithm and applications PLoS One, 2016,11(9):e0161719. URL [本文引用: 1]
ZhangB, ZhiD, ZhangK, GaoG, LimdiNN, LiuN . Practical consideration of genotype imputation: sample size, window size, reference choice, and untyped rate Stat Interface, 2011,4(3):339-352. URL [本文引用: 3]
SpitsC, Le CaignecC, de RyckeM, van HauteL, van SteirteghemA, LiebaersI, SermonK . Whole-genome multiple displacement amplification from single cells Nat Protoc, 2006,1(4):1965-1970. [本文引用: 1]
HaoK, LiC, RosenowC, WongWH . Estimation of genotype error rate using samples with pedigree information—an application on the GeneChip Mapping 10K array Genomics, 2004,84(4):623-630. URL [本文引用: 1]
 ,2, 吴晓林
,2, 吴晓林
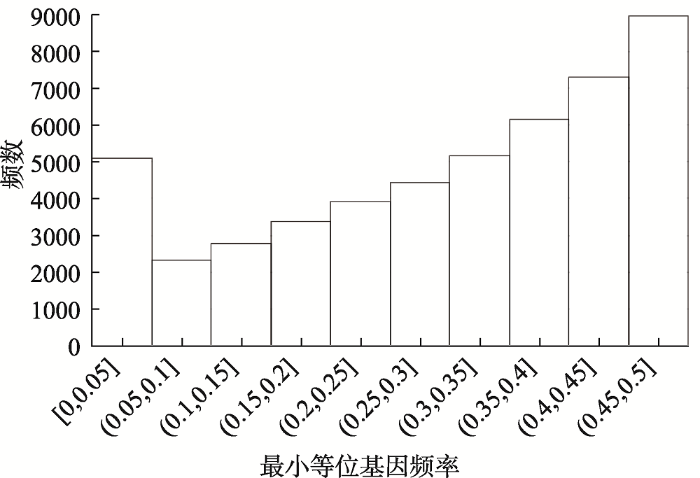 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT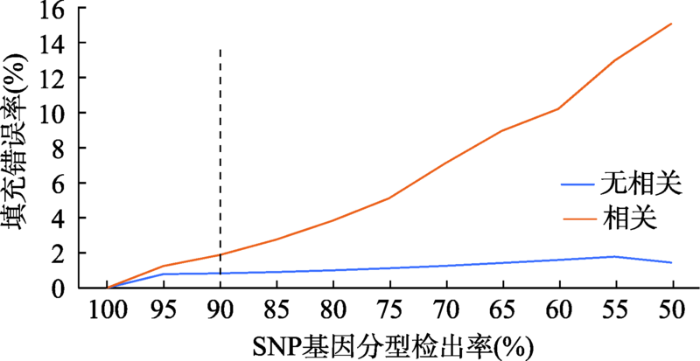 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}