 ,浙江大学生命科学学院,生命系统稳态与保护教育部重点实验室,杭州 310058
,浙江大学生命科学学院,生命系统稳态与保护教育部重点实验室,杭州 310058Nonsense mutations and genetic compensation response
Zhipeng Ma, Jun Chen,MOE Laboratory of Biosystems Homeostasis & Protection and Innovation Center for Cell Signaling Network, College of Life Sciences, Zhejiang University, Hangzhou 310058, China通讯作者:
编委: 张博
收稿日期:2019-04-11修回日期:2019-04-18网络出版日期:2019-05-20
| 基金资助: |
Received:2019-04-11Revised:2019-04-18Online:2019-05-20
| Fund supported: |
作者简介 About authors
马志鹏,在站博士后,研究方向:发育生物学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (265KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
马志鹏, 陈军. 无义突变与“遗传补偿效应”[J]. 遗传, 2019, 41(5): 359-364 doi:10.16288/j.yczz.19-101
Zhipeng Ma, Jun Chen.
生命中的遗传物质DNA时时刻刻都会受到损伤的威胁,这些威胁有来自于内在的因素,如细胞分裂、正常细胞代谢产生的活性氧;也有来自于外在因素,如紫外及离子辐射、化学诱变剂等。每个细胞的基因组DNA每天会遇到差不多104次损伤。DNA损伤会产生基因上的变异,这些变异在生命进化中扮演着重要的角色。但其中很多变异会使编码蛋白的基因失去功能,如果变异的基因功能非常重要,机体又不采取措施,它就不能存活,就失去了生命的意义。为了存活,机体就进化出许多机制来应对基因突变这一状况,其中之一就是“遗传补偿效应”(genetic compensation response, GCR)[1,2],即某一基因发生突变彻底失去功能后,机体会采取相应的机制,提高其他基因的表达来代替这一基因的功能,从而能够正常发育存活。但是,其背后的分子机制是什么?在此之前,还不得而知。2019年4月3日,Nature同期在线发表两篇论文:一篇来自于本实验室,另一篇来自于德国马普研究所Stainier实验室,同时对此机制进行了探讨[3,4]。本文将根据这两篇论文所报道的研究结果解读遗传补偿效应的分子机制。
1 无义mRNA降解途径
DNA损伤会造成多种类型的基因突变,包括错义突变、缺失突变、无义突变、染色体重排等。其中最严重的类型之一就是无义突变,即提前终止密码子突变(premature termination codon, PTC)。在细胞中,基因的表达受到严格的监控。如果无义突变的mRNA继续翻译成蛋白质,将会产生一个比正常野生型蛋白短一些的蛋白,通常这种短蛋白不仅没有功能,反而在很多情况下会对细胞产生显性负的作用。因此,真核生物进化了一个mRNA质量监控体系,即无义mRNA介导的降解途径(nonsense mRNA mediated decay pathway, NMD)[5,6],就是当某个基因发生无义突变时,该基因转录出来的无义mRNA就会被NMD降解途径识别,并带到核外进行降解,以消除翻译短蛋白对机体带来的副作用。在NMD降解途径中,首先上游移码蛋白Upf3b与外显子拼接复合体(exon-junction complex, EJC)结合,进一步招募Upf1和Upf2,然后在细胞质中介导无义mRNA降解[7,8,9,10]。有意思的是,进化上与Upf3b蛋白高度同源的上游移码蛋白3a (Upf3a)同样可以与EJC结合,但在无义mRNA降解中作用并不明显[11]。那么无义mRNA降解途径是否参与基因补偿效应,upf3a是否存在新的生理功能?在这两篇论文发表之前还没有明确答案。2 多个斑马鱼遗传突变体具有遗传补偿效应
几年前,本实验室在研究斑马鱼一个钙调蛋白酶Capn3a时观察到采用寡聚吗啉代核苷酸(morpholino)抑制蛋白翻译敲低该基因,斑马鱼胚胎会出现小肝脏表型[12]。为了进一步研究capn3a基因在肝脏发育中的功能,我们运用TALEN方法[13]构建了一个capn3a基因敲除突变体:突变后的capn3a转录本在第60个密码子出现终止密码子,即无义突变。令人疑惑的是,突变体发育正常,没有表现出小肝脏的表型。因此,我们推测capn3a突变可能激活“遗传补偿效应”。通过检测该突变体中19个capn家族基因的表达,发现多个家族基因上调,同时还证明capn3a突变体无表型是由于这些家族基因上调弥补其功能所致[4]。Stainier实验室发现hbegfa、vcla、hif1ab和vegfaa等基因敲除也会激活相应的遗传补偿效应。两个实验室根据各自的遗传突变体分别探讨了遗传补偿效应的分子机制,得出了相似但又有不同的结论,下面将逐一讨论。3 无义突变与核酸序列同源性是激活遗传补偿效应的必要条件
为了证实是否只有无义突变才能激活补偿效应,本实验室采用CRISPR/Cas9方法构建了多个capn3a遗传突变体携带不同类型的突变,如:同框缺失突变(缺失序列长度是3倍数);不同位置无义突变;提前终止密码子在最后一个外显子上的突变。在另一项研究中,Stainier实验室采用同样的技术路线也获得了不同类型的遗传突变体,如:同框缺失突变;提前终止密码子在最后一个外显子上的突变体;启动子缺失突变体。两个实验室针对不同基因遗传突变体的分析结果同时显示只有无义突变才能激活遗传补偿效应。此外,我们观察到在补偿基因和无义突变基因之间存在着核酸序列的同源性。基于这些结果,我们提出了一个假设即无义突变mRNA通过提高同源基因的表达来实现补偿效应。如果这个假设是正确的,那么具有这些特点的转基因也能激活内源基因的表达。于是我们设计了不同的转基因载体,利用这些载体构建了一系列转基因斑马鱼。结果显示,只有同时携带提前终止密码子(无义突变)和核酸同源序列的转基因载体才能诱导提高内源同源基因的表达。Stainier实验室通过转录本测序,发现在分别敲除Fermt2、Actg1和Actb等基因的小鼠细胞系中也会出现相应同源基因上调的现象;在斑马鱼中,他们采用体外注射的方法,证明只有注射与内源基因同源无帽结构的RNA才可以激活相应内源基因的上调。斑马鱼和小鼠的实验结果揭示无义突变与核酸序列同源性是激活遗传补偿效应的两个必要条件。
4 无义mRNA降解途径参与遗传补偿效应
既然只有无义突变才能诱导遗传补偿效应,NMD降解途径是目前已知的细胞内监控无义突变的唯一机制,那么NMD是否参与遗传补偿效应呢?我们在capn3a突变体中通过基因特异反义寡聚吗啉代核苷酸来分别敲低NMD降解途径中几个关键因子upf1、upf2、upf3a和upf3b,分析敲低这些基因对补偿基因表达的影响。为了进一步验证这些基因在遗传补偿效应中作用,我们利用CRISPR/Cas9方法构建upf1、upf3a和upf3b突变体;在此基础上,与capn3a突变体杂交,获得各个基因与capn3a双敲除突变体。非常有意思的是,我们发现遗传补偿效应不需要参与无义突变mRNA降解的因子,如upf1、upf2和upf3b;相反,无论是敲低还是敲除功能尚未阐明的因子upf3a,都会阻止capn3a突变体中的遗传补偿效应,使其出现小肝脏的表型;同时,我们还利用另一个具有遗传补偿效应的nid1a基因突变体来验证这些NMD途径中的关键因子在遗传补偿效应中的作用,获得类似实验结果。这些实验数据充分证明,upf3a是参与遗传补偿效应的关键因子。但是与我们研究结果不同的是,Stainier实验室在hbegfa、vcla和vegfaa等基因突变体的基础上敲除upf1,发现遗传补偿效应降低,于是他们提出无义mRNA的降解过程参与了遗传补偿效应。综合两个实验室的结果可以得出这样结论:NMD途径参与了遗传补偿效应的激活。5 遗传补偿效应与组蛋白修饰
综合上面的实验结果,接下来我们通过一系列的实验证明无义突变mRNA与以前想象的并不一样,一无用处,无义突变mRNA的转录翻译,以及它的完整性对于“遗传补偿效应”都是必需的。这些实验结果让我们联想到无义突变mRNA可能与一些长链非编码RNA (long non-coding RNA, lncRNA)有类似的功能,即与COMPASS (complex of proteins associated with Set1)复合体结合促进一些基因的表达。COMPASS复合体主要负责组蛋白3上第4位赖氨酸的三甲基化修饰(histone H3 lysine 4 trimethylation, H3K4me3),改变染色质结构,促进基因的表达[14,15,16]。为了证实这一联想,我们采用染色体免疫共沉淀技术(chromatin immunoprecipitation assay, ChIP)检测在补偿效应基因的启动子区域H3K4me3修饰的程度,发现在capn3a突变体中,H3K4me3修饰显著增加;并且,敲低upf3a会降低capn3a突变体中补偿效应基因启动子区域H3K4me3的修饰。最后,我们通过蛋白互作实验证明Upf3a可以直接与COMPASS复合体中类似于支架蛋白Wdr5结合;并且敲低或敲除wdr5,capn3a突变体的遗传补偿效应被阻断。这些实验结果充分显示补偿效应基因的启动子区域H3K4me3修饰在遗传补偿效应中起着重要的作用。Stainier实验室发现在敲除基因的小鼠细胞系中,补偿效应基因的启动子区域H3K4me3修饰显著增加,敲低Wdr5会削弱遗传补偿效应;同时,敲低Upf1也会降低相应补偿基因启动子区域的H3K4me3修饰。两个实验室同时证实遗传补偿效应与染色质的表观遗传学修饰密不可分。
6 结语与展望
根据以上两个实验室的研究结果,我们提出了“遗传补偿效应”的分子机制模型:转录后无义突变mRNA可以与Upf3b结合,在细胞质中通过NMD途径降解;同时,未降解的无义mRNA或降解后的片段都可以与Upf3a结合,在Upf3a蛋白的帮助下,这些RNA重新进入细胞核;Upf3a招募COMPASS复合体,在无义突变mRNA的帮助下,靶向与无义mRNA具有核酸序列同源性的基因附近,COMPASS复合体通过改变基因启动子区域表观遗传学修饰改变染色质构象,促进同源基因表达,弥补缺失基因的功能,完成补偿效应(图1)。图1
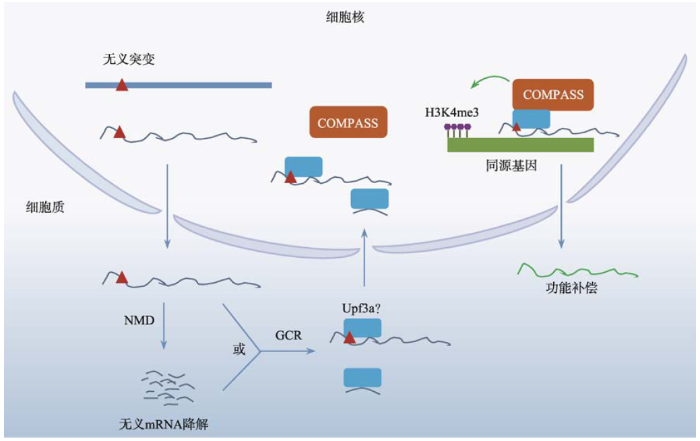 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1无义mRNA介导的遗传补偿效应机制
NMD:无义mRNA介导的降解途径;GCR:遗传补偿效应;Upf3a:上游移码蛋白3a;COMPASS:与Set1蛋白结合的蛋白复合体;H3K4me3:组蛋白3第4位赖氨酸的三甲基化修饰。
Fig. 1Model of genetic compensation response
“遗传补偿效应”并不是斑马鱼所独有的现象,在其他模式生物中同样存在,如小鼠、拟南芥[17,18]。来自于不同实验室的人类基因组测序结果显示,在正常人群的基因组中存在着大量携带有纯合无义突变的基因,其中有些基因的错义突变会引起严重的人类遗传疾病[19,20]。我们推测“遗传补偿效应”是产生这一现象的重要原因。该研究发现不仅仅在理论上取得了大的突破,而且还有实践价值。例如,“遗传补偿效应”对机体存活具有重要意义,但对于基因功能研究是一个巨大的障碍。斑马鱼超过80%的基因被敲除后没有表型[21],所以很难研究这些基因的功能,这其中大部分是由于“遗传补偿效应”导致。针对这一问题,目前就可以根据这两项研究发现的机理,用不同方法阻断“遗传补偿效应”即可。比如,开展基因敲低实验或直接在相应的突变体中敲低/除Upf3a;同时在构建突变体时,尽量避免构建导致无义突变的突变体,如敲除基因启动子,以减少基因补偿效应的发生。
(责任编委: 张博)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:29169924 [本文引用: 1]

正To decode biological processes,interfering with gene functions is a widely used strategy.There are a number of ways to interfere with gene functions.For instance,small interfering RNAs(siRNAs)are used to induce mRNA degradation;morpholinos are used to block protein translation(Nasevicius and Ekker,2000);CRISPR interference/inactive CRISPR-associated 9 nuclease(CRISPR-i/dCas9)is used to block mRNA transcription(Qi et al.,2013);genetic
URLPMID:26168398 [本文引用: 1]
Abstract Cells sense their environment and adapt to it by fine-tuning their transcriptome. Wired into this network of gene expression control are mechanisms to compensate for gene dosage. The increasing use of reverse genetics in zebrafish, and other model systems, has revealed profound differences between the phenotypes caused by genetic mutations and those caused by gene knockdowns at many loci, an observation previously reported in mouse and Arabidopsis. To identify the reasons underlying the phenotypic differences between mutants and knockdowns, we generated mutations in zebrafish egfl7, an endothelial extracellular matrix gene of therapeutic interest, as well as in vegfaa. Here we show that egfl7 mutants do not show any obvious phenotypes while animals injected with egfl7 morpholino (morphants) exhibit severe vascular defects. We further observe that egfl7 mutants are less sensitive than their wild-type siblings to Egfl7 knockdown, arguing against residual protein function in the mutants or significant off-target effects of the morpholinos when used at a moderate dose. Comparing egfl7 mutant and morphant proteomes and transcriptomes, we identify a set of proteins and genes that are upregulated in mutants but not in morphants. Among them are extracellular matrix genes that can rescue egfl7 morphants, indicating that they could be compensating for the loss of Egfl7 function in the phenotypically wild-type egfl7 mutants. Moreover, egfl7 CRISPR interference, which obstructs transcript elongation and causes severe vascular defects, does not cause the upregulation of these genes. Similarly, vegfaa mutants but not morphants show an upregulation of vegfab. Taken together, these data reveal the activation of a compensatory network to buffer against deleterious mutations, which was not observed after translational or transcriptional knockdown.
[本文引用: 1]
[本文引用: 2]
URLPMID:17352659 [本文引用: 1]
Nonsense-mediated mRNA decay (NMD) is a quality-control mechanism that selectively degrades mRNAs harboring premature termination (nonsense) codons. If translated, these mRNAs can produce truncated proteins with dominant-negative or deleterious gain-of-function activities. In this review, we describe the molecular mechanism of NMD. We first cover conserved factors known to be involved in NMD in all eukaryotes. We then describe a unique protein complex that is deposited on mammalian mRNTAs during splicing, which defines a stop codon as premature. Interaction between this exon-junction complex (EJC) and NMD factors assembled at the upstream stop codon triggers a series of steps that ultimately lead to mRNA decay. We discuss whether these proofreading events preferentially occur during a "pioneer" round of translation in higher and lower eukaryotes, their cellular location, and whether they can use alternative EJC factors or act independent of the EJC.
URLPMID:15901503 [本文引用: 1]
Nonsense-mediated mRNA decay (NMD) is an mRNA surveillance pathway that ensures the rapid degradation of mRNAs containing premature translation termination codons (PTCs), thereby preventing the synthesis of truncated and potentially harmful proteins. In addition, this pathway regulates the expression of 鈭10% of the transcriptome and is essential in mice. Although NMD is conserved in eukaryotes, recent studies in several organisms have revealed that different mechanisms have evolved to discriminate natural from premature stop codons and to degrade the targeted mRNAs. With the elucidation of the first crystal structures of components of the NMD machinery, the way is paved towards a molecular understanding of the protein interaction network underlying this process.
URLPMID:11163187 [本文引用: 1]
Nonsense-mediated decay (NMD) rids eukaryotic cells of aberrant mRNAs containing premature termination codons. These are discriminated from true termination codons by downstream cis-elements, such as exon–exon junctions. We describe three novel human proteins involved in NMD, hUpf2, hUpf3a, and hUpf3b. While in HeLa cell extracts these proteins are complexed with hUpf1, in intact cells hUpf3a and hUpf3b are nucleocytoplasmic shuttling proteins, hUpf2 is perinuclear, and hUpf1 cytoplasmic. hUpf3a and hUpf3b associate selectively with spliced β-globin mRNA in vivo, and tethering of any hUpf protein to the 3′UTR of β-globin mRNA elicits NMD. These data suggest that assembly of a dynamic hUpf complex initiates in the nucleus at mRNA exon–exon junctions and triggers NMD in the cytoplasm when recognized downstream of a translation termination site.
URLPMID:16601204 [本文引用: 1]
The exon鈥搄unction complex (EJC) components hUpf3a and hUpf3b serve a dual function: They promote nonsense-mediated mRNA decay (NMD), and they also regulate translation efficiency. Whether these two functions are interdependent or independent of each other is unknown. We characterized the function of the hUpf3 proteins in a 位N/boxB-based tethering system. Despite the high degree of sequence similarity between hUpf3b and hUpf3a, hUpf3a is much less active than hUpf3b to induce NMD and to stimulate translation. We show that induction of NMD by hUpf3 proteins requires interaction with Y14, Magoh, BTZ, and eIF4AIII. The protein region that mediates this interaction and discriminates between hUpf3a and hUpf3b in NMD function is located in the C-terminal domain and fully contained within a small sequence that is highly conserved in Upf3b but not Upf3a proteins. Stimulation of translation is independent of this interaction and is determined by other regions of the hUpf3 protein, indicating the presence of different downstream pathways of hUpf3 proteins either in NMD or in translation.
URLPMID:11546873 [本文引用: 1]
Nonsense-mediated messenger RNA (mRNA) decay, or NMD, is a critical process of selective degradation of mRNAs that contain premature stop codons. NMD depends on both pre-mRNA splicing and translation, and it requires recognition of the position of stop codons relative to exon-exon junctions. A key factor in NMD is hUpf3, a mostly nuclear protein that shuttles between the nucleus and cytoplasm and interacts specifically with spliced mRNAs. We found that hUpf3 interacts with Y14, a component of post-splicing mRNA-protein (mRNP) complexes, and that hUpf3 is enriched in Y14-containing mRNP complexes. The mRNA export factors Aly/REF and TAP are also associated with nuclear hUpf3, indicating that hUpf3 is in mRNP complexes that are poised for nuclear export. Like Y14 and Aly/REF, hUpf3 binds to spliced mRNAs specifically ( approximately 20 nucleotides) upstream of exon-exon junctions. The splicing-dependent binding of hUpf3 to mRNAs before export, as part of the complex that assembles near exon-exon junctions, allows it to serve as a link between splicing and NMD in the cytoplasm.
URLPMID:19503078 [本文引用: 1]
Nature Structural & Molecular Biology is an integrated forum for structural and molecular studies. The journal places a strong emphasis on functional and mechanistic understanding of how molecular components in a biological process work together. Structural data may provide such insights, but they are not a pre-requisite for publication in the journal.
URLPMID:27040500 [本文引用: 1]
Nonsense-mediated decay (NMD) of mRNA needs to be downregulated for key developmental processes. One means by which this may be accomplished is through a NMD inhibitor encoded by the sister paralog of a NMD activator.
URLPMID:3641591 [本文引用: 1]
p53 protein turnover through the ubiquitination pathway is a vital mechanism in the regulation of its transcriptional activity; however, little is known about p53 turnover through proteasome-independent pathway(s). The digestive organ expansion factor (Def) protein is essential for the development of digestive organs. In zebrafish, loss of function of def selectively upregulates the expression of p53 response genes, which raises a question as to what is the relationship between Def and p53. We report here that Def is a nucleolar protein and that loss of function of def leads to the upregulation of p53 protein, which surprisingly accumulates in the nucleoli. Our extensive studies have demonstrated that Def can mediate the degradation of p53 protein and that this process is independent of the proteasome pathway, but dependent on the activity of Calpain3, a cysteine protease. Our findings define a novel nucleolar pathway that regulates the turnover function of p53, which will advance our understanding of p53's role in organogenesis and tumorigenesis.
URLMagsci [本文引用: 1]
人工构建的序列特异性核酸内切酶能够识别并切割特定的DNA靶序列, 造成双链断裂, 从而引起基因组结构的定点改变。人工核酸内切酶技术使得研究人员有可能对任意物种的基因组进行定点修饰, 开启了反向遗传学研究的新天地。类转录激活因子效应物核酸酶(Transcription activator-like effector nuclease, TALEN)自2010年底开始成功应用于基因打靶, 很快成为一种比锌指核酸酶(Zinc-finger nuclease, ZFN)更容易设计、特异性更高和毒性更低的人工核酸内切酶。文章综述了TALEN技术的研究进展及应用前景, 重点介绍TALEN的结构、作用机制与构建方法和利用TALEN进行基因组定点修饰的策略, 以及目前利用这一技术已成功实现突变的物种及内源基因, 特别是在斑马鱼中的应用, 为开展这一领域的研究工作提供参考。
URLMagsci [本文引用: 1]
人工构建的序列特异性核酸内切酶能够识别并切割特定的DNA靶序列, 造成双链断裂, 从而引起基因组结构的定点改变。人工核酸内切酶技术使得研究人员有可能对任意物种的基因组进行定点修饰, 开启了反向遗传学研究的新天地。类转录激活因子效应物核酸酶(Transcription activator-like effector nuclease, TALEN)自2010年底开始成功应用于基因打靶, 很快成为一种比锌指核酸酶(Zinc-finger nuclease, ZFN)更容易设计、特异性更高和毒性更低的人工核酸内切酶。文章综述了TALEN技术的研究进展及应用前景, 重点介绍TALEN的结构、作用机制与构建方法和利用TALEN进行基因组定点修饰的策略, 以及目前利用这一技术已成功实现突变的物种及内源基因, 特别是在斑马鱼中的应用, 为开展这一领域的研究工作提供参考。
URLPMID:4010150 [本文引用: 1]
The Saccharomyces cerevisiae Set1/COMPASS was the first histone H3 lysine 4 (H3K4) methylase identified over 10 years ago. Since then, it has been demonstrated that Set1/COMPASS and its enzymatic product, H3K4 methylation, is highly conserved across the evolutionary tree. Although there is only one COMPASS in yeast, Drosophila possesses three and humans bear six COMPASS family members, each capable of methylating H3K4 with nonredundant functions. In yeast, the histone H2B monoubiquitinase Rad6/Bre1 is required for proper H3K4 and H3K79 trimethylations. The machineries involved in this process are also highly conserved from yeast to human. In this review, the process of histone H2B monoubiquitination-dependent and -independent histone H3K4 methylation as a mark of active transcription, enhancer signatures, and developmentally poised genes is discussed. The misregulation of histone H2B monoubiquitination and H3K4 methylation result in the pathogenesis of human diseases, including cancer. Recent findings in ...
URLPMID:3805109 [本文引用: 1]
Promoters of many developmentally regulated genes, in the embryonic stem cell state, have a bivalent mark of H3K27me3 and H3K4me3, proposed to confer precise temporal activation upon differentiation. Although Polycomb repressive complex 2 is known to implement H3K27 trimethylation, the COMPASS family member responsible for H3K4me3 at bivalently marked promoters was previously unknown. Here, we identify Mll2 (KMT2b) as the enzyme catalyzing H3K4 trimethylation at bivalently marked promoters in embryonic stem cells. Although H3K4me3 at bivalent genes is proposed to prime future activation, we detected no substantial defect in rapid transcriptional induction after retinoic acid treatment in Mll2-depleted cells. Our identification of the Mll2 complex as the COMPASS family member responsible for H3K4me3 marking bivalent promoters provides an opportunity to reevaluate and experimentally test models for the function of bivalency in the embryonic stem cell state and in differentiation.
URLPMID:5404503 [本文引用: 1]
Abstract The spatiotemporal regulation of gene expression is central for cell-lineage specification during embryonic development and is achieved through the combinatorial action of transcription factors/co-factors and epigenetic states at cis-regulatory elements. Here, we show that in addition to implementing H3K4me3 at promoters of bivalent genes, Mll2 (KMT2B)/COMPASS can also implement H3K4me3 at a subset of non-TSS regulatory elements, a subset of which shares epigenetic signatures of active enhancers. Our mechanistic studies reveal that association of Mll2's CXXC domain with CpG-rich regions plays an instrumental role for chromatin targeting and subsequent implementation of H3K4me3. Although Mll2/COMPASS is required for H3K4me3 implementation on thousands of loci, generation of catalytically mutant MLL2/COMPASS demonstrated that H3K4me3 implemented by this enzyme was essential for expression of a subset of genes, including those functioning in the control of transcriptional programs during embryonic development. Our findings suggest that not all H3K4 trimethylations implemented by MLL2/COMPASS are functionally equivalent. Copyright 2017 Elsevier Inc. All rights reserved.
URLPMID:16945951 [本文引用: 1]
RNA interference (RNAi) has great potential as a tool for studying gene function in mammals. However, the specificity and magnitude of the in vivo response to RNAi remains to be fully characterized. A molecular and phenotypic comparison of a genetic knockout mouse and the corresponding knockdown version would help clarify the utility of the RNAi approach. Here, we used hydrodynamic delivery of small interfering RNA (siRNA) to knockdown peroxisome proliferator activated receptor alpha (Ppara), a gene that is central to the regulation of fatty acid metabolism. We found that Ppara knockdown in the liver results in a transcript profile and metabolic phenotype that is comparable to those of Ppara(-/-) mice. Combining the profiles from mice treated with the PPAR alpha agonist fenofibrate, we confirmed the specificity of the RNAi response and identified candidate genes proximal to PPAR alpha regulation. Ppara knockdown animals developed hypoglycemia and hypertriglyceridemia, phenotypes observed in Ppara(-/-) mice. In contrast to Ppara(-/-) mice, fasting was not required to uncover these phenotypes. Together, these data validate the utility of the RNAi approach and suggest that siRNA can be used as a complement to classical knockout technology in gene function studies.
URL [本文引用: 1]
URLPMID:27065010 [本文引用: 1]
Abstract Genetic studies of human disease have traditionally focused on the detection of disease-causing mutations in afflicted individuals. Here we describe a complementary approach that seeks to identify healthy individuals resilient to highly penetrant forms of genetic childhood disorders. A comprehensive screen of 874 genes in 589,306 genomes led to the identification of 13 adults harboring mutations for 8 severe Mendelian conditions, with no reported clinical manifestation of the indicated disease. Our findings demonstrate the promise of broadening genetic studies to systematically search for well individuals who are buffering the effects of rare, highly penetrant, deleterious mutations. They also indicate that incomplete penetrance for Mendelian diseases is likely more common than previously believed. The identification of resilient individuals may provide a first step toward uncovering protective genetic variants that could help elucidate the mechanisms of Mendelian diseases and new therapeutic strategies.
URLPMID:26940866 [本文引用: 1]
Abstract Examining complete gene knockouts within a viable organism can inform on gene function. We sequenced the exomes of 3222 British adults of Pakistani heritage with high parental relatedness, discovering 1111 rare-variant homozygous genotypes with predicted loss of function (knockouts) in 781 genes. We observed 13.7% fewer homozygous knockout genotypes than we expected, implying an average load of 1.6 recessive-lethal-equivalent loss-of-function (LOF) variants per adult. When genetic data were linked to the individuals' lifelong health records, we observed no significant relationship between gene knockouts and clinical consultation or prescription rate. In this data set, we identified a healthy PRDM9-knockout mother and performed phased genome sequencing on her, her child, and control individuals. Our results show that meiotic recombination sites are localized away from PRDM9-dependent hotspots. Thus, natural LOF variants inform on essential genetic loci and demonstrate PRDM9 redundancy in humans. Copyright 2016, American Association for the Advancement of Science.
URLPMID:25533206 [本文引用: 1]
Kok et al. report that their frameshift mutations seldom replicate published morphant phenotypes. Neither maternal effects nor failures to follow standard knockdown guidelines readily account for the discrepancy. While mutants may be slightly more prone to amelioration by long-term compensatory mechanisms, many morphant phenotypes are likely attributable to off-target effects.

{kind=link}
{kind=link}