 ,2,3,4
,2,3,4The roles and regulation mechanism of transcription factor GATA6 in cardiovascular diseases
Zhaoqing Sun1, Bo Yan,2,3,4通讯作者:
编委: 刘峰
收稿日期:2019-02-21修回日期:2019-03-18网络出版日期:2019-05-20
| 基金资助: |
Received:2019-02-21Revised:2019-03-18Online:2019-05-20
| Fund supported: |
作者简介 About authors
孙兆庆,在读硕士研究生,专业方向:心血管病专业E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (379KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
孙兆庆, 闫波. 转录因子GATA6在心血管疾病中的作用及其调控机制[J]. 遗传, 2019, 41(5): 375-383 doi:10.16288/j.yczz.19-044
Zhaoqing Sun, Bo Yan.
目前,心血管疾病(cardiovascular disease, CVD)已经成为一类严重威胁全球人类生命健康的疾病。在中国,随着社会人口老龄化以及多种危险因素水平上升致使CVD患病率持续增长,且发病年龄提前[1]。如今,我国CVD患者已超过 2.9 亿例,因其死亡人数占居民疾病死亡构成40%以上,高于肿瘤及其他疾病,居死因谱第一位[2],给社会发展和家庭带来了沉重的负担。目前,针对CVD的发病机制的研究主要包括遗传和后天环境两大方面,而基因组测序技术的提高极大地推动了CVD分子遗传学方面的相关研究,加深了人们对基因表达失调在CVD发病过程中作用的认识。转录因子GATA6从属于GATA家族,在哺乳动物的心脏分化发育过程中起着重要的作用[3],且基因表达有时空特异性。近年来,有关GATA6基因序列和功能的变异与人类疾病相关的报道越来越多。本文将对转录因子GATA6在CVD中的研究进展进行综述,以期为未来的个体化基因治疗提供遗传学基础,并促进基础研究向临床医学转化的发展。
1 GATA转录因子家族与GATA6
GATA转录因子家族由6个成员组成(GATA1~ GATA6),每个成员都含有一个高度保守的DNA结合域,可优先结合靶基因启动子的核苷酸序列片段为5°-(A/T) GATA(A/GA)-3°,并因此而得名[4]。此家族不仅能调节多种细胞的生长和分化,还对细胞的存活以及机体功能的维持有着至关重要的作用[5]。GATA家族中某些基因碱基位点的突变的与人类发育障碍疾病有关,如贫血、甲状旁腺功能减退、耳聋和不孕以及肾和心脏缺陷等[6]。人类GATA6的基因克隆最初是于1996年从胎心cDNA文库中分离得到,其基因定位于染色体 18q11.1-q11.2,由7个外显子(5°端第一个为非编码外显子)和6个内含子相互间隔组成,序列长34 812 bp,共计编码595个氨基酸[7]。GATA6基因启动区或编码区碱基的突变可通过影响转录和翻译水平,来导致人类某些心血管疾病的发生。2 GATA6在心血管疾病中的作用
近年来,随着转录因子GATA6在人类心血管疾病方面的深入研究,越来越多的报道表明,GATA6基因的变异(以启动子区和外显子区突变为主)与多种心血管疾病的发生及发展密切相关(表1)。Table 1
表1
表1 GATA6基因变异在不同心血管疾病中的发病机制
Table 1
| 类型 | 疾病 | 变异位点或区域 | 分子机制 | 文献 |
|---|---|---|---|---|
| CHD | ASD | 外显子区c.551G>A | 编码氨基酸改变(p.Ser184Asn)体外的转录活性明显降低,影响下游ANF启动子的激活 | [11] |
| VSD | 启动子区g.22169190A>T和 g.22169311C>G | 启动子区突变致使基因转录活性降低,影响基因正常功能,影响心脏发育 | [12] | |
| BAV | 外显子区c.1156G>T | 产生仅含385个氨基酸的截短蛋白质(p.Glu386X),破坏GATA6和GATA4之间的协同转录激活,致使GATA6转录活性大大下降 | [14] | |
| TOF | 外显子区c.1099G>T和 c.1180G>T | 编码氨基酸改变(p.Glyg367X和p.Gly394Cys)致使转录活性降低,影响基因正常功能 | [17] | |
| PTA | Exon4:1396A>C和 Exon5:1456-1457delGA | GATA6突变蛋白未能激活血管引导分子信号素3c(sema3c)及其受体丛蛋白A2(plxna2)的基因 | [18] | |
| 心律失常 | CCS阻滞 | 心肌特异性缺失GATA6 羧基锌蛋白 | 导致超极化环核苷酸门控通道丢失,致使转录抑制因子ID2和心脏钠钙交换物NCx1的下调 | [26] |
| AF | 外显子区 | p.Pro91Ser, Ala177Thr,和Ala543Gly改变转录活性,影响基因正常功能,影响心脏发育 | [27] | |
| 心肌病 | DCM | ZF2:c.1340G>A和 NLS:c.1424A>G | 分别导致p.Cys447Tyr和p.His475Arg,可能通过干扰GATA 6与靶DNA的结合或其核分布而对GATA 6的转录活性产生影响 | [33] |
| HCM | Exon7:c.1663C>G | 编码的氨基酸改变(p.Pro555Ala),可能通过影响GATA6蛋白的稳定性而产生损害作用 | [35] |
新窗口打开|下载CSV
2.1 GATA6与先天性心脏病
先天性心脏病(congenital heart disease, CHD)是常见的出生缺陷之一,是多基因参与和相关环境因素共同作用的复杂性先天性畸形疾病,每1000名活产婴儿中有5~15名患病[8]。目前,已有大量的研究证实GATA6在胚胎期心脏的发育和分化中表达,CHD中与GATA6基因突变相关的表征主要包括:房间隔缺损(atrial septal defect, ASD)、室间隔缺损(ventricular septal defect, VSD)、二叶式主动脉瓣(bicuspid aortic valve, BAV)、动脉导管未闭(patent ductus arteriosus, PDA)、法洛四联症(tetralogy of Fallot, TOF)和永久性动脉干(permanent trunk of artery, PTA)等[9,10,11,12,13,14,15,16,17,18,19]。这些研究,大多是发现GATA6基因中某一关键位点发生突变,从而导致其转录活性下降,进而影响下游基因转录而致病。另外,由于体内存在多基因联合作用方式,GATA6也可与其他基因,如GATA4、TBX5和MADS家族和NF-AT家族等一起发挥调控心脏发育的作用[20]。例如,转录因子GATA6和GATA4因具有高度同源性,在联合作用后可经Ca2+依赖通道与MEF2C的增强子区域直接结合进而调控心脏发育[21,22]。已有研究发现,两者之间可能存在某种互促作用,即单纯的GATA6和GATA4基因突变并不引起表型变化,但当两者同时发生突变后能导致转录活性下降,而引起先天性心脏病[23]。除了互促增效的作用外,转录因子GATA6与其他转录因子结合作用还具有多样性,如GATA6基因的N段结构域发生突变后可以通过其锌指结构正常与TBX5结合,但会抑制TBX5下游靶基因的表达,从而间接调控心脏发育[24]。这些围绕转录因子GATA6的研究不仅对人类先天性心脏病多种类型,也对体内其他转录因子之间的相互作用进行了分子遗传学方面的探讨,为揭示遗传表型和疾病表型的关系提供了新的见解。2.2 GATA6与心脏传导系统
心脏传导系统(cardiac conduction system, CCS)是由位于心肌内能够产生和传导冲动的特殊心肌细胞构成,其系统中任何一部位或环节出现了问题均有可能引起心律失常症状。临床中,常见的水电解质失衡、结构性心脏畸形和药物不良副作用等情况均可能促进心律失常的发生。此外,也可能由于基因遗传突变而导致心律失常,如长QT综合征[25]。先前的研究已经证实GATA6在心脏早期发育中表达,如房室管心肌细胞、心脏神经嵴(neural crest,NC)细胞和心脏成纤维细胞等。在Liu等[26]的一项研究中,不仅发现GATA6除了在心脏形态发生中的关键作用外,还需要用于房室结(atrioventricular node,AVN)的正常发育和功能维持。为确定GATA6是否有助于CCS,他们建立了一个在肌球蛋白轻链2V(mlc2v)启动子控制下产生心肌特异性缺失GATA6羧基锌指域的小鼠模型,导致GATA6在心室心肌、右心室流出道和房室环中被截断。对幼年动物进行的心电图分析结果显示,这些突变小鼠的P-R间期延长,AVN缺损。这项研究工作首次提供了将GATA6与AVN形成和功能联系起来的实验证据。在另外一项临床研究中[27],研究者评估了1个有16个家庭成员的谱系,其中1个患有ASD,1个患有VSD,3个患有房颤(atrial fibrillation, AF),对3个受影响的家庭成员的基因组进行了全外显子测序,并采用检测荧光素酶活性的方法对得到的GATA6变异体进行了功能鉴定,最终发现在所有的研究对象中,以及在散发性、早发型房颤中,GATA6都有功能突变。此外,据报道GATA4/5/6蛋白可调控许多传导相关基因,如缝隙连接和离子通道的基因。它们还与NKx2.5和TBX2/3/5在物理和功能上相互作用,调节其对靶基因的活性,因此可作为引起心律失常的基因的遗传修饰剂[28,29,30]。目前,对人类GATA6基因与CCS的相关研究,不仅让人们在疾病方面对CCS的认识和理解更上一层楼,并为进一步阐明心律失常的发病机制提供了帮助,也为未来的个体化的基因治疗打下基础。2.3 GATA6与心肌病
心肌病(cardiomyopathies)是一组异质性心肌疾病,由不同病因引起心脏机械和电活动的异常,严重心肌病会引起心血管性死亡或进展性心力衰竭。其中,扩张型心肌病(dilated cardiomyopathy, DCM)是最常见的原发性心肌疾病,是心力衰竭的第三大常见原因,也是心脏移植最常见的原因[31]。DCM的病因是多种多样的,其中就包括环境和遗传两方面的致病因素。有数据显示,大约25%~50%的DCM患者有家族性疾病,表明遗传缺陷在DCM发病机制中的重要作用[32]。此前,有研究者对140例不相关的DCM患者的GATA6基因编码外显子和侧翼内含子进行了序列测定,并在2例DCM患者中分别鉴定出两个新的杂合子突变p.Cys447Tyr和p.His475Arg。他们在对家系的分析中发现,在每个家族中,与DCM共分离的突变以常染色体显性模式传播,具有完全外显率[33]。在对GATA6变异体进行功能分析后表明,突变的GATA6蛋白与野生型的GATA6蛋白相比,与转录激活显著降低相关。这是关于GATA6功能缺失突变与家族性DCM易感性增强之间关系的首次研究,此报道不仅丰富了DCM发病的分子遗传学机制,也为DCM的产前预防和等位基因特异性治疗提供了新的思路。另外,肥厚性心肌病(hypertrophic cardiomyopathy, HCM)是遗传性心肌病最常见的形式,且相关基因的突变可以解释约60%病例[34]。先前有报道称GATA6基因敲除可以抑制心脏肥大的发生,而另有研究者在512份临床样本中(HCM组212例,健康组300例)把GATA6作为筛选基因之一,进行分析研究以检测其遗传变异是否与HCM相关,并试图发现转录因子中可能与该疾病的病理生理学或表型异质性相关的遗传变异[35]。最终研究结果表明,GATA6中的罕见变异p.Pro555Ala (rs146243018)与HCM的较大的最大后壁厚度相关,该变异体可改变其相关靶基因的表达而改变肥厚反应。此项研究既增加了人们对HCM发病的相关分子机制的新认识,又丰富了GATA6与心肌病相关的群体遗传学资料。2.4 GATA6与心血管疾病相关危险因素
冠状动脉粥样硬化心脏病简称冠心病(coronary artery disease, CAD)是临床上极为常见的,由遗传因素和后天环境共同作用的一类复杂心血管疾病[36]。后天环境中重要的因素有高血压、血脂异常、糖代谢异常、吸烟、肥胖、缺少运动和心理压力等[37]。此前,有研究者在探讨组蛋白脱乙酰基酶(histone deacetylase, HDAC)选择性抑制剂是否能调节高血压及潜在作用机制中发现,血管紧张素II (angiotensin II, Ang II)增强了磷酸化HDAC4和GATA6蛋白的表达,这些蛋白特异性地定位在肾脏动脉和主动脉的细胞胞质中[38]。HDAC4的强制表达或敲除分别会增加或减少血管平滑肌细胞(vascular smooth muscle cell, VSMC)的增殖,而GATA6是HDAC4的一个新的结合伴侣,能显著促进VSMCs增殖。最终研究结果表明,HDAC抑制剂可通过Ca2+/钙调蛋白依赖激酶IIa/蛋白激酶d1/HDAC4/GATA6途径负性调节室间隔细胞肥大和增生,从而减轻高血压。这为治疗高血压,预防冠心病提供了新的临床治疗思路。另外,动脉粥样硬化是动脉壁的一种慢性炎症反应,是心血管疾病的主要病理基础[39]。血管细胞粘附分子-1 (vascular cell adhesion molecule-1, VCAM-1)可通过与α4β1整合素结合,介导活化单核细胞与动脉壁的牢固粘附,从而在动脉粥样硬化的发生中起到中心作用[40,41]。一项关于雷帕霉素(mammalian target of rapamycin, mTOR)在TNFα诱导的血管内皮细胞VCAM-1表达中的作用机制研究表明,mTOR通过转录因子GATA6的作用后,mTOR的抑制显著抑制了TNF诱导的VCAM-1转录[42]。也有研究报道mTOR可抑制VSMCs增生,促进VSMCs分化,并需要转录因子GATA6[43]。雷帕霉素复合物1 ( mechanistic target of rapamycin complex 1, mTORC1)的抑制稳定了GATA6,促进了GATA6的在细胞核中的积累、与DNA的结合、编码收缩蛋白的启动子的激活和抑制增殖。此项研究还表明,GATA6和AKT2 (一种在VSMCs中被激活的激酶)参与了mTORC1介导的VSMCs增殖和分化的调节。若能确定mTORC1的下游转录靶点并能提供细胞类型特异性药物靶点,来对抗与过度增殖VSMCs相关的心血管疾病,这将大大促进基础医学向临床转化的发展。然而,转录因子GATA6在CAD发生过程中的作用是不唯一的。众所周知,糖尿病(diabetes mellitus, DM)是CAD致病的高危因素。而胰腺发育不全是一种罕见的人类疾病,由胚胎发育过程中胰腺形成缺陷引起,其患者出生时没有胰腺,可发展为永久性新生儿糖尿病和胰腺酶功能不全[44]。大量研究已经证实,GATA锌指转录家族成员对人和小鼠胰腺器官发生至关重要[45,46],而外显子测序研究表明杂合子GATA6突变是胰腺发育不全的最常见原因[47,48]。据报道,GATA6转录因子的表达并不局限于胰腺形成的胚胎阶段,GATA6突变也与亚临床或无外分泌不足的成年发病糖尿病有关,在成年期胰腺功能中也起着重要作用[49]。因此,GATA6基因突变不仅在心脏发育过程中致病,也能影响新生儿或成人胰腺的发育和功能,以DM的形式增加着患者患CAD的风险。2.5 GATA6在其他类心血管疾病中的相关研究
在人类心脏分化发育的初期阶段,不同多能干细胞(pluripotent stem cells, PSC)之间的差异会造成心脏分化效率的显著不同。据报道,GATA6是唯一一个在PSC向心肌细胞分化早期阶段被大量诱导的基因,其水平与心肌细胞分化效率呈正相关,PSC中GATA6的基因敲除降低了心肌细胞的产生效率[50]。在心脏与系统循环的联系中,心脏间隔形成以及胚胎循环的复杂重塑是两个关键的发育事件。其中转录因子GATA6作为关键调节剂参与了主动脉弓周围的NC细胞分化成平滑肌细胞的过程,同时还促进应退化的主动脉弓的保存[51]。这些发现为GATA6突变如何导致人类先天性心脏病提供了一个新的框架。另外,在关于中国草药(参附和参麦注射液)联合用药于eNOS基因敲除小鼠的心血管保护作用效果检测的研究中发现,参附注射液可通过GATA4、GATA6和COL3A1途径,在治疗早期表现出较好的疗效,改善了心肌功能,同时参麦注射液在慢性期具有较好的保护作用[52]。3 结语与展望
纵观目前转录因子GATA6与心血管病的报道,多集中于CHD的相关研究。GATA6基因的启动子区或外显子区内碱基突变除可产生多种类型的CHD外,还能在CCS中致病引起多种心律失常疾病,同时与心肌病和CAD的发病也有着密切的联系,具体的信号通路图见图1。虽然人们对GATA6转录因子的结构特征及其在心脏发育过程中的表达调控有了较深认识[53],但CHD是一个由多基因参与的复杂先天性畸形,GATA6与其他转录因子间相互作用的仍不完全清楚,能否进一步阐明心脏基因调控网络及GATA6基因缺陷与表型间的关系还需要更多的基础和临床研究来证明。另外,GATA6基因在CCS、心肌病和CAD的研究虽有报道,但更多的分子遗传学发病机制仍待探讨。例如,未来的研究需要确定GATA6是否直接调节GATA6基因敲除小鼠的高血压;转录因子GATA6虽已被证明能诱导病理性心脏肥大,但在VSMCs中,GATA6的作用是存在争议的;GATA6基因可在成年人心肌细胞中和VSMCs表达,但至今与成人心脏疾病,如CAD和急性心肌梗死(acute myocardial infarction, AMI)等的研究报道相对较少,且转录因子GATA6能通过多种方式参与到CAD危险因素中,其作用也存在争议,所以更多的GATA6基因变异在成人心肌细胞及VSMCs中表达和发病机制的研究亟待探讨,这不仅可以丰富人类对基因功能的认识,也可以促进分子遗传学的发展以及精准医学-基因治疗时代的到来!图1
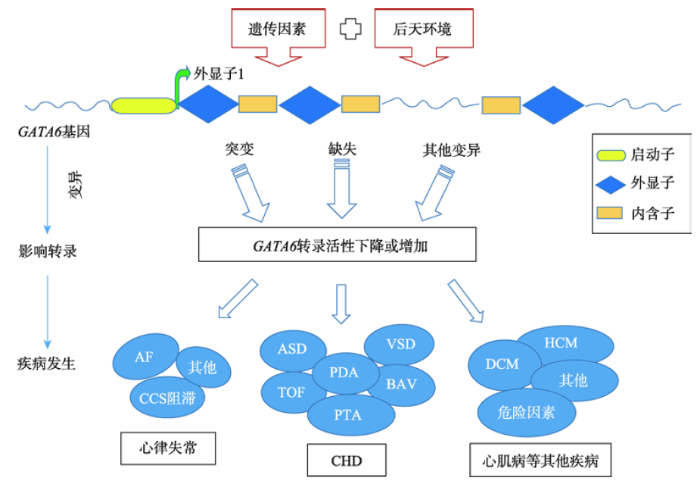 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1GATA6基因变异在心血管相关疾病中的信号通路图
CHD:先天性心脏病;ASD:房间隔缺损;VSD:室间隔缺损;BAV:二叶式主动脉瓣;PDA:动脉导管未闭;TOF:法洛四联症;PTA:永久性动脉干;AF:房颤;CCS:心脏传导系统;DCM:扩张性心肌病;HCM:肥厚性心肌病。
Fig. 1Signaling pathway of GATA6 Gene variations in cardiovascular related diseases
(责任编委: 刘峰)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]

我国心血管病(CVD)防治工作已取得初步成效,但仍面临严峻挑战。总体上看,中国CVD患病率及死亡率仍处于上升阶段。推算CVD现患人数2.9亿,其中脑卒中1 300万,冠心病1 100万,肺原性心脏病500万,心力衰竭450万,风湿性心脏病250万,先天性心脏病200万,高血压2.7亿。CVD死亡占居民疾病死亡构成40%以上,居首位,高于肿瘤及其他疾病。近几年来农村CVD死亡率持续高于城市水平。2004年至今,心脑血管病住院费用年均增速远高于国内生产总值增速。中国CVD负担日渐加重,已成为重大的公共卫生问题,防治CVD刻不容缓。
URL [本文引用: 1]
我国心血管病(CVD)防治工作已取得初步成效,但仍面临严峻挑战。总体上看,中国CVD患病率及死亡率仍处于上升阶段。推算CVD现患人数2.9亿,其中脑卒中1 300万,冠心病1 100万,肺原性心脏病500万,心力衰竭450万,风湿性心脏病250万,先天性心脏病200万,高血压2.7亿。CVD死亡占居民疾病死亡构成40%以上,居首位,高于肿瘤及其他疾病。近几年来农村CVD死亡率持续高于城市水平。2004年至今,心脑血管病住院费用年均增速远高于国内生产总值增速。中国CVD负担日渐加重,已成为重大的公共卫生问题,防治CVD刻不容缓。
.
URL [本文引用: 1]
The transcription factors GATA4, GATA5 and GATA6 play important roles in heart muscle differentiation. The data presented in this article are related to the research article entitled “Genome-wide transcriptomics analysis identifies sox7 and sox18 as specifically regulated by gata4 in cardiomyogenesis” (Afouda et al., 2017)[1]. The present study identifies genes regulated by these individual cardiogenic GATA factors using genome-wide transcriptomics analysis. We have presented genes that are specifically regulated by each of them, as well those regulated by either of them. The gene ontology terms (GO) associated with the genes differentially affected are also presented. The data set will allow further investigations on the gene regulatory network downstream of individual cardiogenic GATA factors during cardiac muscle formation.
.
URLPMID:10684344 [本文引用: 1]
The GATA-binding transcription factors comprise a protein family whose members contain either one or two highly conserved zinc finger DNA-binding domains. Members of this group have been identified in organisms ranging from cellular slime mold to vertebrates, including plants, fungi, nematodes, insects, and echinoderms. While much work has been done describing the expression patterns, functional aspects, and target genes for many of these proteins, an evolutionary analysis of the entire family has been lacking. Herein we show that,only the C-terminal zinc finger (Cf) and basic domain, which together constitute the GATA-binding domain, are conserved throughout this protein family. Phylogenetic analyses of amino acid sequences demonstrate distinct evolutionary pathways. Analysis of GATA factors isolated from vertebrates suggests that the six distinct vertebrate GATAs are descended from a common ancestral sequence, while those isolated from nonvertebrates (with the exception of the fungal AREA orthologues and Arabidopsis paralogues) appear to be related only within the DNA-binding domain and otherwise provide little insight into their evolutionary history. These results suggest multiple modes of evolution, including gene duplication and modular evolution of GATA factors based upon inclusion of a class IV zinc finger motif. As such, GATA transcription factors represent a group of proteins related solely by their homologous DNA-binding domains. Further analysis of this domain examines the degree of conservation at each amino acid site using the Boltzmann entropy measure, thereby identifying residues critical to preservation of structure and function, Finally, we construct a predictive motif that can accurately identify potential GATA proteins. [References: 67]
URLPMID:28566565 [本文引用: 1]
Abstract The development of mature blood cell from hematopoietic stem cells is regulated by transcription factors that coordinate the expression of lineage-specific genes. GATA transcription factors are zinc finger DNA-binding proteins that play crucial roles in various biological processes, including hematopoiesis. Among GATA family proteins, GATA-1, GATA-2, and GATA-3 are essential for hematopoiesis. GATA-1 functions to promote development of erythrocytes, megakaryocytes, eosinophils, and mast cells. Mutations in GATA-1 are associated with acute megakaryoblastic leukemia (AMKL), congenital erythroid hypoplasia (Diamond-Blackfan anemia; DBA), and X-linked anemia and/or thrombocytopenia. Conversely, GATA-2 functions early in hematopoiesis and is required for maintenance and expansion of hematopoietic stem cells (HSCs) and/or multipotent progenitors. GATA-2 mutations are associated with immunodeficiency, lymphedema, myelodysplastic syndrome (MDS), and leukemia. Furthermore, decreased GATA-2 expression may contribute to the pathophysiology of aplastic anemia. GATA-3 has an important role in T cell development, and has been suggested to be involved in the pathophysiology of acute lymphoblastic leukemias. This review summarizes current knowledge on hematological disorders associated with GATA-1 and GATA-2 mutations.
[本文引用: 1]
.
URL [本文引用: 1]
.
URLPMID:5135520 [本文引用: 1]
Abstract Background: Information about known risk factors for congenital heart disease is scarce. In this population-based study, we aimed to investigate the relation between maternal chronic disease and congenital heart disease in offspring. Methods: The study cohort consisted of 1 387 650 live births from 2004 to 2010. We identified chronic disease in mothers and mild and severe forms of congenital heart disease in their offspring from Taiwan's National Health Insurance medical claims. We used multivariable logistic regression analysis to assess the associations of all cases and specific types of congenital heart disease with various maternal chronic diseases. Results: For mothers with the following chronic diseases, the overall prevalence of congenital heart disease in their children was significantly higher than for mothers without these diseases: diabetes mellitus type 1 (adjusted odds ratio [OR] 2.32, 95% confidence interval [CI] 1.66-3.25), diabetes mellitus type 2 (adjusted OR 2.85, 95% CI 2.60-3.12), hypertension (adjusted OR 1.87, 95% CI 1.69-2.07), congenital heart defects (adjusted OR 3.05, 95% CI 2.45-3.80), anemia (adjusted OR 1.31, 95% CI 1.25-1.38), connective tissue disorders (adjusted OR 1.39, 95% CI 1.19-1.62), epilepsy (adjusted OR 1.37, 95% CI 1.08-1.74) and mood disorders (adjusted OR 1.25, 95% CI 1.11-1.41). The same pattern held for mild forms of congenital heart disease. A higher prevalence of severe congenital heart disease was seen only among offspring of mothers with congenital heart defects or type 2 diabetes. Interpretation: The children of women with several kinds of chronic disease appear to be at risk for congenital heart disease. Preconception counselling and optimum treatment of pregnant women with chronic disease would seem prudent.
URLPMID:2940936 [本文引用: 1]
Abstract Although the etiology for the majority of congenital heart disease (CHD) remains poorly understood, the known genetic causes are often the result of mutations in cardiac developmental genes. GATA6 encodes for a cardiac transcription factor, which is broadly expressed in the developing heart and is critical for normal cardiac morphogenesis, making it a candidate gene for congenital heart defects in humans. The objective of this study was to determine the frequency of GATA6 sequence variants in a population of individuals with a spectrum of cardiac malformations. The coding regions of GATA6 were sequenced in 310 individuals with CHD. We identified two novel sequence variations in GATA6 that altered highly conserved amino acid residues (A178V and L198V) and were not found in a control population. These variants were identified in two individuals (one with tetralogy of Fallot and the other with an atrioventricular septal defect in the setting of complex CHD). Biochemical studies demonstrate that the GATA6 A178V mutant protein results in increased transactivation ability when compared with wild-type GATA6. These data suggest that nonsynonymous GATA6 sequence variants are infrequently found in individuals with CHD.
.
URLPMID:3955383 [本文引用: 1]
Background Congenital diaphragmatic hernia (CDH) is a common birth defect affecting 1 in 3000 births. It is characterised by herniation of abdominal viscera through an incompletely formed diaphragm. Although chromosomal anomalies and mutations in several genes have been implicated, the cause for most patients is unknown. Methods We used whole exome sequencing in two families with CDH and congenital heart disease, and identified mutations in GATA6 in both. Results In the first family, we identified a de novo missense mutation (c.1366C>T, p.R456C) in a sporadic CDH patient with tetralogy of Fallot. In the second, a nonsense mutation (c.712G>T, p.G238") was identified in two siblings with CDH and a large ventricular septal defect. The G238" mutation was inherited from their mother, who was clinically affected with congenital absence of the pericardium, patent ductus arteriosus and intestinal malrotation. Deep sequencing of blood and saliva-derived DNA from the mother suggested somatic mosaicism as an explanation for her milder phenotype, with only approximately 15% mutant alleles. To determine the frequency of GATA6 mutations in CDH, we sequenced the gene in 378 patients with CDH. We identified one additional de novo mutation (c.1071delG, p.V358Cfs34"). Conclusions Mutations in GATA6 have been previously associated with pancreatic agenesis and congenital heart disease. We conclude that, in addition to the heart and the pancreas, GATA6 is involved in development of two additional organs, the diaphragm and the pericardium. In addition, we have shown that de novo mutations can contribute to the development of CDH, a common birth defect.
.
URLPMID:20631719 [本文引用: 1]
Journal of Human Genetics, official journal of the Japan Society of Human Genetics, publishes original articles and reviews on all aspects of human genetics, including medical genetics and genomics
.
URLPMID:4139867 [本文引用: 1]
Congenital heart disease (CHD) is the most common birth defect in humans. Genetic causes and underlying molecular mechanisms for isolated CHD remain largely unknown. Studies have demonstrated that GATA transcription factor 6 (GATA6) plays an essential role in the heart development. Mutations in GATA6 gene have been associated with diverse types of CHD. As GATA6 functions in a dosage-dependent manner, we speculated that changed GATA6 levels, resulting from DNA sequence variants (DSVs) within the gene regulatory regions, may mediate the CHD development. In the present study, GATA6 gene promoter was genetically and functionally analyzed in large groups of patients with ventricular septal defect (VSD) (n = 359) and ethnic-matched healthy controls (n = 365). In total, 11 DSVs, including four SNPs, were identified in VSD patients and controls. Two novel and heterozygous DSVs, g.22169190Agt;T and g.22169311Cgt;G, were identified in two VSD patients, but in none of controls. In cultured cardiomyocytes, the activities of the GATA6 gene promoter were significantly reduced by the DSVs g.22169190Agt;T and g.22169311Cgt;G. Therefore, our findings suggested that the DSVs within the GATA6 gene promoter identified in VSD patients may change GATA6 levels, contributing to the VSD development as a risk factor.
.
PMID:22407241 [本文引用: 1]
Ventricular septal defect (VSD) is the most common form of congenital cardiovascular malformation and an important contributor to the substantially increased morbidity and mortality in infants. Emerging evidence indicates the genetic basis for the pathogenesis of congenital VSD in a significant proportion of patients. However, congenital VSD is a genetically heterogeneous disease and the genetic defects responsible for VSD in the overwhelming majority of cases remain unclear. In this study, the entire coding region of the GATA6 gene, which encodes a zinc-finger transcription factor crucial to normal cardiogenesis, was sequenced in 130 unrelated patients with congenital VSD. The available relatives of the index patient carrying the identified mutation and 200 unrelated ethnically matched healthy individuals used as controls were subsequently genotyped. The functional characteristics of the mutant GATA6 were assessed in contrast to its wild-type counterpart using a luciferase reporter assay system. As a result, a novel heterozygous missense GATA6 mutation, p.G220S, was identified in a proband with VSD. The variation was absent in 400 control chromosomes and the altered amino acid was highly conserved evolutionarily across species. Genetic analysis of the family members of the mutation carrier showed that the substitution co-segregated with VSD was inherited as an autosomal dominant trait. Functional analysis demonstrated that the p.G220S mutation of GATA6 was associated with significantly decreased transcriptional activity. The findings provide novel insight into the molecular mechanism involved in VSD, implying the potential clinical implications in the gene-specific prophylaxis and therapy of this common developmental abnormality in neonates.
.
URLPMID:29653232 [本文引用: 1]
.
URLPMID:29567669 [本文引用: 1]
Abstract Background -Bicuspid aortic valve (BAV), the most common congenital heart defect affecting 1-2% of the population, is a major risk factor for premature aortic valve disease and accounts for the majority of valve replacement. The genetic basis and the mechanisms of BAV etiology and pathogenesis remain largely undefined. Methods -Cardiac structure and function was assessed in mice lacking a Gata6 allele. Human GATA6 gene variants were analyzed in 452 BAV cases from the BAV consortium and 1849 controls from the Framingham GWAS study. GATA6 expression was determined in mice and human tissues using qRT-PCR and immunohistochemistry. Mechanistic studies were carried out in cultured cells. Results - Gata6 heterozygous mice have highly penetrant RL type BAV, the most frequent type in human. GATA6 transcript levels are lower in human BAV as compared to normal tricuspid valves. Mechanistically, Gata6 haploinsufficiency disrupts valve remodeling and extracellular matrix composition through dysregulation of important signaling molecules including matrix metalloproteinase 9. Cell-specific inactivation of Gata6 reveals an essential role for GATA6 in secondary heart field myocytes as loss of one Gata6 allele from Isl-1 positive cells- but not from endothelial or neural crest cells-recapitulates the phenotype of Gata6 heterozygous mice. Conclusions -The data identify a new cellular and molecular mechanism underlying BAV. The availability of an animal model for the most frequent human BAV opens the way for the elucidation of BAV pathogenesis and the development of much needed therapies.
.
URLPMID:23020118 [本文引用: 1]
Abstract Congenital heart disease (CHD) is the most common form of developmental malformation and is the leading noninfectious cause of infant mortality. Emerging evidence indicates that genetic defects are involved in the pathogenesis of CHD. Nevertheless, CHD is genetically heterogeneous, and the molecular basis for CHD in a majority of patients remains unknown. In this study, the whole coding region of GATA6, a gene encoding a zinc-finger transcription factor crucial for normal cardiogenesis, was sequenced in 380 unrelated patients with CHD. The relatives of the index patients harboring the identified mutations and 200 unrelated control individuals were subsequently genotyped. The functional effect of the mutations was characterized using a luciferase reporter assay system. As a result, two novel heterozygous GATA6 mutations, p.D404Y and p.E460X, were identified in two families with ventricular septal defect and tetralogy of Fallot, respectively. The mutations co-segregated with CHD in the families with complete penetrance, and were absent in 400 control chromosomes. Functional analysis demonstrated that the mutated GATA6 proteins were associated with significantly decreased transactivational activity in comparison with their wild-type counterpart. These findings provide novel insight into the molecular mechanism implicated in CHD, suggesting potential implications for the early prophylaxis and personalized treatment of CHD.
.
URLPMID:23175051 [本文引用: 1]
Tetralogy of Fallot (TOE) is the most common cyanotic congenital heart disease associated with significant morbidity and mortality in humans. However, the molecular etiology underlying TOF in most patients remains largely unknown. In the present study, sequence analysis of the GATA6 gene was performed from fresh-frozen cardiac tissues and matched blood samples of 52 unrelated patients who underwent surgical repair of TOF. The cardiac tissues and matched blood specimens from 46 patients who underwent cardiac valve replacement due to rheumatic heart disease and blood samples from 200 healthy individuals as controls were genotyped. The functional characteristics of the mutations were assessed using a luciferase reporter assay system. Based on the results, two novel heterozygous GATA6 mutations, p.G367X and p.G394C, were identified in the cardiac tissues of 2 TOE patients, respectively. No mutations were found in the cardiac tissues from 46 patients with rheumatic heart disease and in the blood samples from the 298 participants. Functional analysis demonstrated that the GATA6 mutants were consistently associated with significantly reduced transcriptional activation compared with their wildtype counterpart. This is the first report on the link of somatic GATA6 mutation to TOF, providing novel insight into the molecular mechanism involved in TOF.
.
URLPMID:19666519 [本文引用: 1]
Congenital heart diseases (CHD) occur in nearly 1 % of all live births and are the major cause of infant mortality and morbidity. Although an improved understanding of the genetic causes of CHD would provide insight into the underlying pathobiology, the genetic etiology of most CHD remains unknown. Here we show that mutations in the gene encoding the transcription factor GATA6 cause CHD characteristic of a severe form of cardiac outflow tract (OFT) defect, namely persistent truncus arteriosus (PTA). Two different GATA6 mutations were identified by systematic genetic analysis using DNA from patients with PTA. Genes encoding the neurovascular guiding molecule semaphorin 3C (SEMA3C) and its receptor plexin A2 (PLXNA2) appear to be regulated directly by GATA6, and both GATA6 mutant proteins failed to transactivate these genes. Transgenic analysis further suggests that, in the developing heart, the expression of SEMA3C in the OFT/subpulmonary myocardium and PLXNA2 in the cardiac neural crest contributing to the OFT is dependent on GATA transcription factors. Together, our data implicate mutations in GATA6 as genetic causes of CHD involving OFT development as a result of the disruption of the direct regulation of semaphorin-plexin signaling.
.
URLPMID:23639568 [本文引用: 1]
Permanent neonatal diabetes mellitus is a rare condition mostly due to heterozygous mutations in the KCNJ11, ABCC8 and INS genes. Neonatal diabetes due to pancreatic agenesis is extremely rare. Mutations in PDX1, PTF1A, HNF1B, EIF2AK3, RFX6 and GATA6 genes have been shown to result in pancreatic agenesis or hypoplasia. This report describes a 40-day-old male infant diagnosed with permanent neonatal diabetes associated with atrial septal defect, pulmonary stenosis, patent ductus arteriosus and a novel de novo heterozygous missense mutation (p.N466S) in the GATA6 gene with no evidence of exocrine pancreas insufficiency. In addition to permanent neonatal diabetes, the patient had transient idiopathic neonatal cholestasis and hypoglycaemic episodes unrelated to insulin treatment, features that are rarely described in children with permanent neonatal diabetes. (C) 2013 Elsevier Masson SAS. All rights reserved.
URLMagsci [本文引用: 1]
转录因子GATA6属于GATA转录因子家族,具有2个保守的锌指结构域。它不仅存在于胚胎组织,并且对心脏、肺和胰腺中细胞功能的维持起重要作用。该文对转录因子GATA6在心脏发育以及先天性房间隔缺损中的作用作一综述,探讨其对人类房间隔缺损疾病诊断和治疗的价值。
URLMagsci [本文引用: 1]
转录因子GATA6属于GATA转录因子家族,具有2个保守的锌指结构域。它不仅存在于胚胎组织,并且对心脏、肺和胰腺中细胞功能的维持起重要作用。该文对转录因子GATA6在心脏发育以及先天性房间隔缺损中的作用作一综述,探讨其对人类房间隔缺损疾病诊断和治疗的价值。
.
URLPMID:15253934 [本文引用: 1]
The vertebrate heart forms initially as a linear tube derived from a primary heart field in the lateral mesoderm. Recent studies in and chick have demonstrated that the outflow tract and right ventricle originate from a separate source of mesoderm that is anterior to the primary heart field. The discovery of this anterior, or secondary, heart field has led to a greater understanding of the morphogenetic events involved in heart formation; however, many of the underlying molecular events controlling these processes remain to be determined. The domain factor is required for proper formation of the cardiac outflow tract and right ventricle, suggesting a key role in anterior heart field . Therefore, as a first step toward identifying the transcriptional pathways upstream of , we introduced a reporter gene into a bacterial artificial (BAC) encompassing the locus and used this recombinant to generate . This BAC transgene was sufficient to recapitulate endogenous expression, and comparative sequence analyses revealed multiple regions of significant conservation in the noncoding regions of the BAC. We show that one of these conserved noncoding regions represents a transcriptional enhancer that is sufficient to direct expression of exclusively to the anterior heart field throughout . This conserved enhancer contains two consensus sites that are efficiently bound by the zinc finger and are completely required for enhancer function in vivo. This enhancer also contains two perfect consensus sites for the -homeodomain protein . We show that these elements are specifically bound by and are essential for enhancer function in transgenic embryos. Thus, these findings establish as the first direct transcriptional target of in the anterior heart field and support a model in which factors and serve as the earliest controlling outflow tract and right ventricle .
.
URLPMID:512972 [本文引用: 1]
Despite significant advances in identifying signaling molecules that induce cardiogenesis in mammals, the transcription factors that control the onset of cardiac myocyte gene expression have remained elusive. Candidates include the zinc finger transcription factors GATA binding proteins 4 and 6 (GATA4, GATA6). The individual loss of either protein in mice results in lethality prior to the onset of heart development due to defects in the extra-embryonic endoderm; however, when this extra-embryonic deficiency is circumvented using tetraploid embryo complementation, cardiac myocyte differentiation initiates normally. Here we show that these factors have redundant roles in controlling the onset of cardiac myocyte differentiation. As a consequence, Gata461/61Gata661/61 embryos completely lack hearts, although second heart field progenitor cells are still generated. Our data support a model whereby GATA4 or GATA6 are essential for expression of the network of transcription factors that regulate the onset of cardiac myocyte gene expression during mammalian development.
.
URLPMID:22498567 [本文引用: 1]
Abstract BACKGROUND: The genetic basis of most congenital heart defects (CHDs), especially non-syndromic and non-familial conditions, remains largely unknown. METHODS AND RESULTS: DNA samples were collected from immortalized cell lines and original genomes of 256 non-syndromic, non-familial patients with cardiac outflow tract (OFT) defects. Genes encoding NKX2.5, GATA4, GATA6, MEF2C, and ISL1, essential for heart development, were analyzed using PCR-based bidirectional sequencing. The transcriptional activity of proteins with identified sequence variations was analyzed using a luciferase assay. A novel sequence variant (A103V in MEF2C) was identified, in addition to 4 unreported non-synonymous sequence variants in 3 known causative genes (A6V in NKX2.5, T330R and S339R in GATA4, and E142K in GATA6) in 5 individuals. None of these was found in 500 controls without CHDs. In vitro functional assay showed that all proteins with identified sequence variations exhibited significant changes in transcriptional activity and/or synergistic activity with other transcription factors. Furthermore, overexpression of the A103V MEF2C variant in a fish system disturbed early cardiac development. CONCLUSIONS: New mutations in the transcription factors NKX2.5, GATA4, GATA6, and MEF2C that affect their protein function were identified in 2.3% (6/256) of patients with OFT defects. Our results provide the first demonstration of MEF2C mutation and suggest that disturbances in the regulatory circuits involving these cardiac transcription factors may cause a subset of non-syndromic and non-familial CHDs.
.
URLPMID:2651674 [本文引用: 1]
Congenital heart disease is the most common type of birth defect with an incidence of 1%. Previously, we described a point mutation in GATA4 that segregated with cardiac defects in a family with autosomal dominant disease. The mutation (G296S) exhibited biochemical deficits and disrupted a novel interaction between Gata4 and Tbx5. To determine if Gata4 and Tbx5 genetically interact in vivo, we generated mice heterozygous for both alleles. We found that nearly 100% of mice heterozygous for Gata4 and Tbx5 were embryonic or neonatal lethal and had complete atrioventricular (AV) septal defects with a single AV valve and myocardial thinning. Consistent with this phenotype, Gata4 and Tbx5 are co-expressed in the developing endocardial cushions and myocardium. In mutant embryos, cardiomyocyte proliferation deficits were identified compatible with the myocardial hypoplasia. Similar to Gata4, Gata6 and Tbx5 are co-expressed in the embryonic heart, and the transcription factors synergistically activate the atrial natiuretic factor promoter. We demonstrate a genetic interaction between Gata6 and Tbx5 with an incompletely penetrant phenotype of neonatal lethality and thin myocardium. Gene expression analyses were performed on both sets of compound heterozygotes and demonstrated downregulation of -myosin heavy chain only in Gata4/Tbx5 heterozygotes. These findings highlight the unique genetic interactions of Gata4 and Gata6 with Tbx5 for normal cardiac morphogenesis in vivo.
.
URLPMID:28663329 [本文引用: 1]
Abstract Long QT syndrome (LQTS) is an inheritable primary electric disease of the heart characterised by abnormally long QT intervals and a propensity to develop atrial and ventricular tachyarrhythmias. It is caused by an inherited channelopathy responsible for sudden cardiac death in individuals with structurally normal hearts. Long QT syndrome can present early in life, and some studies suggest that it may be associated with up to 20% of sudden unexplained infant death (SUID), particularly when associated with external stressors such as asphyxia, which is commonly seen in many infant death scenes. With an understanding of the genetic defects, it has now been possible to retrospectively analyse samples from infants who have presented to forensic pathology services with a history of unexplained sudden death, which may, in turn, enable the implementation of preventative treatment for siblings previously not known to have pathogenic genetic variations. In this viewpoint article, we will discuss SUID, LQTS and postmortem genetic analysis. Article author(s) (or their employer(s) unless otherwise stated in the text of the article) 2017. All rights reserved. No commercial use is permitted unless otherwise expressly granted.
.
URLPMID:25613430 [本文引用: 1]
http://circgenetics.ahajournals.org/cgi/doi/10.1161/CIRCGENETICS.113.000587
.
URLPMID:27756709 [本文引用: 1]
The genetic basis of atrial fibrillation (AF) and congenital heart disease remains incompletely understood. We sought to determine the causative mutation in a family with AF, atrial septal and ventricular septal defects. We evaluated a pedigree with 16 family members, one with an atrial septal defect, one with a ventricular septal defect and three with AF; we performed whole exome sequencing in three affected family members. Given that early-onset AF was prominent in the family, we then screened individuals with early-onset AF, defined as an age of onset < 66 years, for mutations inGATA6. Variants were functionally characterized using reporter assays in a mammalian cell line. Exome sequencing in three affected individuals identified a conserved mutation, R585L, in the transcription factor gene,GATA6. In the MGH AF Study the mean age of AF onset was 47.1 卤 10.9 years, 79% of the participants were male, and there was no evidence of structural heart disease. We identified three GATA6 variants (P91S, A177T, and A543G). Using wild-type and mutantGATA6constructs driving NPPA promoter reporter, we found that three of the four variants had a marked upregulation of luciferase activity (R585L; 4.1 fold, p<0.0001; P91S; 2.5 fold, p=0.0002; A177T; 1.7 fold, p=0.03). Additionally, when co-overexpressed with GATA4 and MEF2C, GATA6 variants exhibited upregulation of the 伪MHC and NPPA activity. Overall, we found gain-of-function mutations in GATA6 in both a family with early-onset AF and atrioventricular septal defects as well as in sporadic, early-onset AF.
.
URLPMID:4863647 [本文引用: 1]
Techniques for the separate, consecutive and simultaneous demonstration of acetylcholinesterase and norepinephrine in cryostat sections are described and discussed. The results, obtained in a variety of tissues from different animal species, are as satisfactory—in terms of sensitivity and sharpness of localization—as those obtained by previously described methods. The simultaneous visualization of both acetylcholinesterase and norepinephrine in the same section provides hitherto unavailable data relative to the exact interrelationship of cholinergic and adrenergic components of the peripheral autonomic nervous system.
.
URL [本文引用: 1]
[本文引用: 1]
.
URLPMID:5498329 [本文引用: 1]
Heart failure (HF) is a complex clinical syndrome defined by the inability of the heart to pump enough blood to meet the body metabolic demands. Major causes of HF are cardiomyopathies (diseases of the myocardium associated with mechanical and/or electrical dysfunction), among which the most common form is dilated cardiomyopathy (DCM). DCM is defined by ventricular chamber enlargement and systolic dysfunction with normal left ventricular wall thickness, which leads to progressive HF. Over 60 genes are linked to the etiology of DCM. Titin (TTN) is the largest known protein in biology, spanning half the cardiac sarcomere and, as such, is a basic structural and functional unit of striated muscles. It is essential for heart development as well as mechanical and regulatory functions of the sarcomere. Next-generation sequencing (NGS) in clinical DCM cohorts implicated truncating variants in titin (TTNtv) as major disease alleles, accounting for more than 25% of familial DCM cases, but these variants have also been identified in 2鈥3% of the general population, where these TTNtv blur diagnostic and clinical utility. Taking into account the published TTNtv and their association to DCM, it becomes clear that TTNtv harm the heart with position-dependent occurrence, being more harmful when present in the A-band TTN, presumably with dominant negative/gain-of-function mechanisms. However, these insights are challenged by the depiction of position-independent toxicity of TTNtv acting via haploinsufficient alleles, which are sufficient to induce cardiac pathology upon stress. In the current review, we provide an overview of TTN and discuss studies investigating various TTN mutations. We also present an overview of different mechanisms postulated or experimentally validated in the pathogenicity of TTNtv. DCM-causing genes are also discussed with respect to non-truncating mutations in the etiology of DCM. One way of understanding pathogenic variants is probably to understand the context in which they may or may not affect protein鈥損rotein interactions, changes in cell signaling, and substrate specificity. In this regard, we also provide a brief overview of TTN interactions in situ. Quantitative models in the risk assessment of TTNtv are also discussed. In summary, we highlight the importance of gene鈥揺nvironment interactions in the etiology of DCM and further mechanistic studies used to delineate the pathways which could be targeted in the management of DCM.
.
URLPMID:23281406 [本文引用: 1]
Genetic mutations account for a significant percentage of cardiomyopathies, which are a leading cause of congestive heart failure. In hypertrophic cardiomyopathy (HCM), cardiac output is limited by the thickened myocardium through impaired filling and outflow. Mutations in the genes encoding the thick filament components myosin heavy chain and myosin binding protein C (MYH7 and MYBPC3) together explain 75% of inherited HCMs, leading to the observation that HCM is a disease of the sarcomere. Many mutations are "private" or rare variants, often unique to families. In contrast, dilated cardiomyopathy (DCM) is far more genetically heterogeneous, with mutations in genes encoding cytoskeletal, nudeoskeletal, mitochondrial, and calcium-handling proteins. DCM is characterized by enlarged ventricular dimensions and impaired systolic and diastolic function. Private mutations account for most DCMs, with few hotspots or recurring mutations. More than 50 single genes are linked to inherited DCM, including many genes that also link to HCM. Relatively few clinical clues guide the diagnosis of inherited D CM, but emerging evidence supports the use of genetic testing to identify those patients at risk for faster disease progression, congestive heart failure, and arrhythmia.
.
URLPMID:25119427 [本文引用: 1]
Dilated cardiomyopathy (DCM), the most prevalent form of primary heart muscle disease, is the third most common cause of heart failure and the most frequent reason for cardiac transplantation. Mounting evidence has demonstrated that genetic risk factors are crucial in the pathogenesis of DCM. However, DCM is genetically heterogeneous, and the genetic basis of DCM in a large majority of cases remains unclear. In the current study, the coding exons and flanking introns of the GATA6 gene, which encodes a zinc62finger transcription factor essential for cardiogenesis, was sequenced in 14002unrelated patients with DCM, and two02novel heterozygous mutations, p.C447Y and p.H475R, were identified in two02index patients with DCM, respectively. Analysis of the pedigrees showed that in each family the mutation co-segregated with DCM transmitted in an autosomal-dominant pattern, with complete penetrance. The missense mutations were absent in 40002control chromosomes and predicted to be disease-causing by MutationTaster or probably damaging by PolyPhen-2. The alignment of multiple GATA6 proteins across species revealed that the altered amino acids were completely conserved evolutionarily. The functional assays showed that the mutated GATA6 proteins were associated with significantly reduced transcriptional activation in comparison with their wild-type counterpart. To the best of our knowledge, this is the first study on the association of GATA6 loss-of-function mutations with enhanced susceptibility to familial DCM, which provides novel insight into the molecular mechanism of DCM and suggests potential implications for the antenatal prophylaxis and allele-specific treatment of DCM.
.
URL [本文引用: 1]
.
URLPMID:28381408 [本文引用: 1]
Hypertrophic cardiomyopathy (HCM) is a very heterogeneous disease. Although primarily caused by mutations in genes encoding sarcomeric proteins, other genes might explain that heterogeneity. Potential candidate genes are GATA transcription factors that regulate the expression of proteins associated with HCM. Exons of GATA2, GATA4, and GATA6 genes were sequenced in 212 patients with unrelated HCM previously analyzed for genes encoding the most frequently mutated sarcomeric proteins. Functional effects of variants were predicted by in silico analyses. 3 potentially pathogenic variants were identified: c.-77G>A in GATA2, p.Ala343Thr (rs370588269) in GATA4, and p.Pro555Ala (rs146243018) in GATA6. Multivariate analyses showed that angina was more frequent in patients carrying sarcomeric and GATA rare variants (55% vs 23.2% in non-carriers of GATA rare variants, OR (95% CI) 7.12 (1.23 to 41.27), p=0.029). Among patients without a known causal mutation, GATA rare variants were associated with a greater maximum posterior wall thickness (16.4卤4.4 vs 14.0卤3.1鈥卪m in non-carriers, p=0.021). Thus, variants having a putative effect on GATA genes would alter the expression of their target genes and could modify the hypertrophic response. Therefore, although relatively infrequent in patients with HCM, they may represent a novel insight into the molecular mechanisms related to the pathogenesis of HCM.
URLPMID:26278888 [本文引用: 1]
Summary Atherosclerotic cardiovascular disease (CVD) is a leading cause of mortality and morbidity worldwide, with coronary artery disease (CAD) being the single leading cause of death. Better control of risk factors, enhanced diagnostic techniques and improved medical therapies have all substantially decreased the mortality of CAD in developed countries. However, CAD and other forms of atherosclerotic CVD are projected to remain the leading cause of death by 2030 and we face a number of challenges if the outcomes of CAD are to be further improved. The fact that a substantial fraction of high-risk subjects do not reach treatment goals for important risk factors is one of these challenges. At the same time, there is also a non-negotiable fraction of oncealed high-risk subjects who are not detected by current risk algorithms and diagnostic modalities. In recent years, we have started to rapidly increase our knowledge of the framework of common genetics underlying CAD and atherosclerotic CVD in the population. In conjunction with modern diagnostic and therapeutic options, this new genetic knowledge may provide a valuable tool for further improvements in prevention. This review summarizes the recent findings from the search for common genetic risk factors for CAD. Furthermore, the author discusses how such recent findings could potentially be used in a number of clinical applications within CAD prevention, including in clinical risk stratification, in prediction of drug treatment response and in the search for targets for novel preventive therapies.
.
URLPMID:29702990 [本文引用: 1]
Abstract BACKGROUND: Well-developed coronary collateral circulation usually results in fewer infarct size, improved cardiac function, and fewer mortality. Traditional coronary risk factors (diabetes, hypertension, and smoking) have some effects on coronary collateral circulation. However, the association between these risk factors and coronary collateral circulation are controversial. Given the confusing evidences regarding traditional cardiovascular risk factors on coronary collateral circulation, we performed this meta-analysis protocol to investigate the relationship between traditional risk factors of coronary artery disease and coronary collateral circulation. METHODS: MEDINE, EMBASE, and Science Citation Index will be searched to identify relevant studies. The primary outcomes of this meta-analysis are well-developed coronary collateral circulation. Meta-analysis was performed to calculate the odds ratio (OR) and 95% confidence interval (CI) of traditional coronary risk factors (diabetes, smoking, hypertriton). Pooled ORs were computed as the Mantel-Haenszel-weighted average of the ORs for all included studies. Sensitivity analysis, quality assessment, publication bias analysis, and the Grading of Recommendations Assessment, Development and Evaluation approach (GRADE) will be performed to ensure the reliability of our results. RESULTS: This study will provide a high-quality synthesis of current evidence of traditional risk factors on collateral circulation. CONCLUSION: This conclusion of our systematic review and meta-analysis will provide evidence to judge whether traditional risk factors affects coronary collateral circulation.Ethics and dissemination: Ethical approval is not required because our systematic review and meta-analysis will be based on published data without interventions on patients. The findings of this study will be published in a peer-reviewed journal.
.
URLPMID:27512969 [本文引用: 1]
Histone deacetylase (HDAC) inhibitors have been reported to improve essential and secondary hypertension. However, the specific HDAC that might serve as a therapeutic target and the associated upstream and downstream molecules involved in regulating hypertension remain unknown. Our study was aimed at investigating whether a selective inhibitor of class II HDAC (MC1568) modulates hypertension, elucidating the underlying mechanism. Hypertension was established by administering angiotensin II (Ang II) to mice before treatment with MC1568. SBP was measured. Treatment with MC1568 reduced elevated SBP; attenuated arterial remodeling in the kidney's small arteries and thoracic aorta; and inhibited cell cycle regulatory gene expression, vascular smooth muscle cell (VSMC) proliferation, DNA synthesis, and VSMC hypertrophy in vivo and in vitro. Ang II enhanced the expression of phosphorylated HDAC4 and GATA-binding factor 6 (GATA6) proteins, which were specifically localized in the cytoplasm of cells in the arteries of kidneys and in aortas. Forced expression and knockdown of HDAC4 increased and decreased, respectively, the proliferation and expression of cell cycle genes in VSMCs. GATA6, a newly described binding partner of HDAC4, markedly enhanced the size and number of VSMCs. Calcium/calmodulin-dependent kinase II (CaMKII), but not HDAC4, translocated from the nucleus to the cytoplasm in response to Ang II. CaMKII and protein kinase D1 were associated with VSMC hypertrophy and hyperplasia via direct interaction with HDAC4. MC1568 treatment weakened the association between HDAC4 and CaMKII. These results suggest that class II HDAC inhibition attenuates hypertension by negatively regulating VSMC hypertrophy and hyperplasia via the CaMKII/protein kinase D1/HDAC4/GATA6 pathway.
.
[本文引用: 1]
URL [本文引用: 1]
.
URLPMID:17717539 [本文引用: 1]
Abstract Neutrophil recruitment, lymphocyte recirculation and monocyte trafficking all require adhesion and transmigration through blood-vessel walls. The traditional three steps of rolling, activation and firm adhesion have recently been augmented and refined. Slow rolling, adhesion strengthening, intraluminal crawling and paracellular and transcellular migration are now recognized as separate, additional steps. In neutrophils, a second activation pathway has been discovered that does not require signalling through G-protein-coupled receptors and the signalling steps leading to integrin activation are beginning to emerge. This Review focuses on new aspects of one of the central paradigms of inflammation and immunity--the leukocyte adhesion cascade.
[本文引用: 1]
URLPMID:25969542 [本文引用: 1]
Abstract Vascular smooth muscle cells (VSMCs) undergo transcriptionally regulated reversible differentiation in growing and injured blood vessels. This dedifferentiation also contributes to VSMC hyperplasia after vascular injury, including that caused by angioplasty and stenting. Stents provide mechanical support and can contain and release rapamycin, an inhibitor of the mechanistic target of rapamycin complex 1 (mTORC1). Rapamycin suppresses VSMC hyperplasia and promotes VSMC differentiation. We report that rapamycin-induced differentiation of VSMCs required the transcription factor GATA-6. Inhibition of mTORC1 stabilized GATA-6 and promoted the nuclear accumulation of GATA-6, its binding to DNA, its transactivation of promoters encoding contractile proteins, and its inhibition of proliferation. These effects were mediated by phosphorylation of GATA-6 at Ser(290), potentially by Akt2, a kinase that is activated in VSMCs when mTORC1 is inhibited. Rapamycin induced phosphorylation of GATA-6 in wild-type mice, but not in Akt2(-/-) mice. Intimal hyperplasia after arterial injury was greater in Akt2(-/-) mice than in wild-type mice, and the exacerbated response in Akt2(-/-) mice was rescued to a greater extent by local overexpression of the wild-type or phosphomimetic (S290D) mutant GATA-6 than by that of the phosphorylation-deficient (S290A) mutant. Our data indicated that GATA-6 and Akt2 are involved in the mTORC1-mediated regulation of VSMC proliferation and differentiation. Identifying the downstream transcriptional targets of mTORC1 may provide cell type-specific drug targets to combat cardiovascular diseases associated with excessive proliferation of VSMCs. Copyright 2015, American Association for the Advancement of Science.
.
URL [本文引用: 1]
Abstract Haploinsufficient GATA6 mutations are associated with human pancreatic agenesis. Shi et al. (2017) in this issue of Cell Stem Cell and Tiyaboonchai et al. (2017) in a recent issue of Cell Reports utilize hPSCs to characterize GATA6 function during human pancreas development. One functional copy of GATA6 is sufficient for definitive endoderm development and pancreas formation, but it is inadequate for functional 尾 cell differentiation.
.
URLPMID:23006330 [本文引用: 1]
Recently, heterozygous mutations in GATA6 have been found in neonatal diabetic patients with failed pancreatic organogenesis. To investigate the roles of GATA4 and GATA6 in mouse pancreas organogenesis, we conditionally inactivated these genes within the pancreas. Single inactivation of either gene did not have a major impact on pancreas formation, indicating functional redundancy. However, double Gata4/Gata6 mutant mice failed to develop pancreata, died shortly after birth, and displayed hyperglycemia. Morphological defects in Gata4/Gata6 mutant pancreata were apparent during embryonic development, and the epithelium failed to expand as a result of defects in cell proliferation and differentiation. The number of multipotent pancreatic progenitors, including PDX1+ cells, was reduced in the Gata4/Gata6 mutant pancreatic epithelium. Remarkably, deletion of only 1 Gata6 allele on a Gata4 conditional knockout background severely reduced pancreatic mass. In contrast, a single WT allele of Gata4 in Gata6 conditional knockout mice was sufficient for normal pancreatic development, indicating differential contributions of GATA factors to pancreas formation. Our results place GATA factors at the top of the transcriptional network hierarchy controlling pancreas organogenesis.
.
URLPMID:3461916 [本文引用: 1]
Abstract Pancreatic agenesis is a human disorder caused by defects in pancreas development. To date, only a few genes have been linked to pancreatic agenesis in humans, with mutations in pancreatic and duodenal homeobox 1 (PDX1) and pancreas-specific transcription factor 1a (PTF1A) reported in only 5 families with described cases. Recently, mutations in GATA6 have been identified in a large percentage of human cases, and a GATA4 mutant allele has been implicated in a single case. In the mouse, Gata4 and Gata6 are expressed in several endoderm-derived tissues, including the pancreas. To analyze the functions of GATA4 and/or GATA6 during mouse pancreatic development, we generated pancreas-specific deletions of Gata4 and Gata6. Surprisingly, loss of either Gata4 or Gata6 in the pancreas resulted in only mild pancreatic defects, which resolved postnatally. However, simultaneous deletion of both Gata4 and Gata6 in the pancreas caused severe pancreatic agenesis due to disruption of pancreatic progenitor cell proliferation, defects in branching morphogenesis, and a subsequent failure to induce the differentiation of progenitor cells expressing carboxypeptidase A1 (CPA1) and neurogenin 3 (NEUROG3). These studies address the conserved and nonconserved mechanisms underlying GATA4 and GATA6 function during pancreas development and provide a new mouse model to characterize the underlying developmental defects associated with pancreatic agenesis.
.
URLPMID:4062962 [本文引用: 1]
Abstract Understanding the regulation of pancreatic development is key for efforts to develop new regenerative therapeutic approaches for diabetes. Rare mutations in PDX1 and PTF1A can cause pancreatic agenesis, however, most instances of this disorder are of unknown origin. We report de novo heterozygous inactivating mutations in GATA6 in 15/27 (56%) individuals with pancreatic agenesis. These findings define the most common cause of human pancreatic agenesis and establish a key role for the transcription factor GATA6 in human pancreatic development.
URLPMID:3581234 [本文引用: 1]
We recently reported de novoGATA6mutations as the most common cause of pancreatic agenesis, accounting for 15 of 27 (56%) patients with insulin-treated neonatal diabetes and exocrine pancreatic insufficiency requiring enzyme replacement therapy. We investigated the role ofGATA6mutations in 171 subjects with neonatal diabetes of unknown genetic etiology from a cohort of 795 patients with neonatal diabetes. Mutations in known genes had been confirmed in 624 patients (including 15GATA6mutations). Sequencing of the remaining 171 patients identified nine new case subjects (24 of 795, 3%). Pancreatic agenesis was present in 21 case subjects (six new); two patients had permanent neonatal diabetes with no enzyme supplementation and one had transient neonatal diabetes. Four parents with heterozygousGATA6mutations were diagnosed with diabetes outside the neonatal period (12鈥46 years). Subclinical exocrine insufficiency was demonstrated by low fecal elastase in three of four diabetic patients who did not receive enzyme supplementation. One parent with a mosaic mutation was not diabetic but had a heart malformation. Extrapancreatic features were observed in all 24 probands and three parents, with congenital heart defects most frequent (83%). HeterozygousGATA6mutations cause a wide spectrum of diabetes manifestations, ranging from pancreatic agenesis to adult-onset diabetes with subclinical or no exocrine insufficiency.
URL [本文引用: 1]
URLPMID:29335067 [本文引用: 1]
Pluripotent stem cell (PSC) variations can cause significant differences in the efficiency of cardiac differentiation. This process is unpredictable, as there is not an adequate indicator at the undifferentiated stage of the PSCs. We compared global gene expression profiles of two PSCs showing significant differences in cardiac differentiation potential. We identified 12 up-regulated genes related to heart development, and we found that 4 genes interacted with multiple genes. Among these genes, Gata6 is the only gene that was significantly induced at the early stage of differentiation of PSCs to cardiomyocytes. Gata6 knock-down in PSCs decreased the efficiency of cardiomyocyte production. In addition, we analyzed 6 mESC lines and 3 iPSC lines and confirmed that a positive correlation exists between Gata6 levels and efficiency of differentiation into cardiomyocytes. In conclusion, Gata6 could be utilized as a biomarker to select the best PSC lines to produce PSC-derived cardiomyocytes for therapeutic purposes. [BMB Reports 2018; 51(2): 85-91]
URLPMID:28952437 [本文引用: 1]
10.7554/eLife.31362.001Connection of the heart to the systemic circulation is a critical developmental event that requires selective preservation of embryonic vessels (aortic arches). However, why some aortic arches regress while others are incorporated into the mature aortic tree remains unclear. By microdissection and deep sequencing in mouse, we find that neural crest (NC) only differentiates into vascular smooth muscle cells (SMCs) around those aortic arches destined for survival and reorganization, and identify the transcription factor Gata6 as a crucial regulator of this process. Gata6 is expressed in SMCs and its target genes activation control SMC differentiation. Furthermore, Gata6 is sufficient to promote SMCs differentiation in vivo, and drive preservation of aortic arches that ought to regress. These findings identify Gata6-directed differentiation of NC to SMCs as an essential mechanism that specifies the aortic tree, and provide a new framework for how mutations in GATA6 lead to congenital heart disorders in humans.
URLPMID:29133875 [本文引用: 1]
Abstract Ginseng, a popular herbal remedy, is often used in combination with other drugs to achieve the maximum therapeutic response. Shenfu (SFI) and Shenmai injection (SMI) have been widely used to treat cardiovascular disease in China. Our study explored the cardiovascular protection of SFI and SMI in eNOS knockout mice to investigate the differences and similarities of the two ginseng-combinations. Transthoracic echocardiography was performed to evaluate the left ventricular structure and function at baseline and 3, 7, and 14 days after drug administration. Agilent Gene Expression microarrays were used to demonstrate the gene expression profiling of the thoracic aorta. Ingenuity Pathway Analysis was performed to evaluate the mechanism improved by SFI and SMI in eNOS knockout mice. Both SFI and SMI could modulate Gadd45 Signaling from TOP15 canonical pathways. Moreover, SFI showed a better effect in the early treatment stage and improved myocardial function via GATA4, GATA6 and COL3A1. Meanwhile, SMI exerted better protective effects at the chronic stage, which may be related to endothelium protection by VEGFA and ACE. The advantage of multi-target by drug combination in progression of complex diseases should be noticed. The appropriate adjustment of drug combination could lead to a better accurate medical care in clinic.
URLPMID:29229250 [本文引用: 1]
The transcription factors GATA4, GATA5 and GATA6 are important regulators of heart muscle differentiation (cardiomyogenesis), which function in a partially redundant manner. We identified genes specifically regulated by individual cardiogenic GATA factors in a genome-wide transcriptomics analysis. The genes regulated bygata4are particularly interesting because GATA4 is able to induce differentiation of beating cardiomyocytes in Xenopus and in mammalian systems. Among the specificallygata4-regulated transcripts we identified two SoxF family members,sox7andsox18. Experimental reinstatement of gata4 restoressox7andsox18expression, and loss of cardiomyocyte differentiation due to gata4 knockdown is partially restored by reinstatingsox7orsox18expression, while (as previously reported) knockdown ofsox7orsox18interferes with heart muscle formation. In order to test for conservation in mammalian cardiomyogenesis, we confirmed in mouse embryonic stem cells (ESCs) undergoing cardiomyogenesis that knockdown ofGata4leads to reducedSox7(andSox18) expression and that Gata4 is also uniquely capable of promptly inducingSox7expression. Taken together, we identify an important and conserved gene regulatory axis fromgata4to the SoxF paralogssox7andsox18and further to heart muscle cell differentiation. fx1 61Gata 4, 5 and 6 have redundant and non-redundant functions in heart development.61RNA-seq analysis of Gata4, 5 and 6 knockdown experiments was carried out.61Genes specifically regulated by Gata4, 5 and 6 were identified.61The SoxF genes sox7 and sox18 were identified as specifically regulated by Gata4.61Epistasis demonstrates a regulatory axis from Gata4 to Sox7/18 to cardiomyogenesis. Gata 4, 5 and 6 have redundant and non-redundant functions in heart development. RNA-seq analysis of Gata4, 5 and 6 knockdown experiments was carried out. Genes specifically regulated by Gata4, 5 and 6 were identified. The SoxF genes sox7 and sox18 were identified as specifically regulated by Gata4. Epistasis demonstrates a regulatory axis from Gata4 to Sox7/18 to cardiomyogenesis.

{kind=link}
{kind=link}