Methods and applications for microbiome data analysis
Yongxin Liu1,2, Yuan Qin1,2,3, Xiaoxuan Guo1,2, Yang Bai,1,2,3 1. State Key Laboratory of Plant Genomics, Institute of Genetics and Developmental Biology, the Innovative Academy of Seed Design, Chinese Academy of Sciences, Beijing 100101, China 2. CAS-JIC Centre of Excellence for Plant and Microbial Science, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China 3. College of Advanced Agricultural Sciences, University of Chinese Academy of Sciences, Beijing 100101, China
Supported by the Key Research Program of Frontier Sciences of the Chinese Academy of Science No(QYZDB-SSW-SMC021) the National Natural Science Foundation of China No(31772400) the Key Research Program of the Chinese Academy of Sciences No(KFZD-SW-219)
作者简介 About authors 刘永鑫,博士,工程师,研究方向:生物信息学、宏基因组学E-mail:yxliu@genetics.ac.cn。
Abstract Development of high-throughput sequencing stimulates a series of microbiome technologies, such as amplicon sequencing, metagenome, metatranscriptome, which have rapidly promoted microbiome research. Microbiome data analysis involves a lot of basic knowledge, software and databases, and it is difficult for peers to learn and select proper methods. This review systematically outlines the basic ideas of microbiome data analysis and the basic knowledge required to conduct analysis. In addition, it summarizes the advantages and disadvantages of commonly used software and databases used in the comparison, visualization, network, evolution, machine learning and association analysis. This review aims to provide a convenient and flexible guide for selecting analytical tools and suitable databases for mining the biological significance of microbiome data. Keywords:microbiome;data analysis;amplicon;metagenome;pipeline
PDF (557KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 刘永鑫, 秦媛, 郭晓璇, 白洋. 微生物组数据分析方法与应用[J]. 遗传, 2019, 41(9): 845-862 doi:10.16288/j.yczz.19-222 Yongxin Liu, Yuan Qin, Xiaoxuan Guo, Yang Bai. Methods and applications for microbiome data analysis[J]. Hereditas(Beijing), 2019, 41(9): 845-862 doi:10.16288/j.yczz.19-222
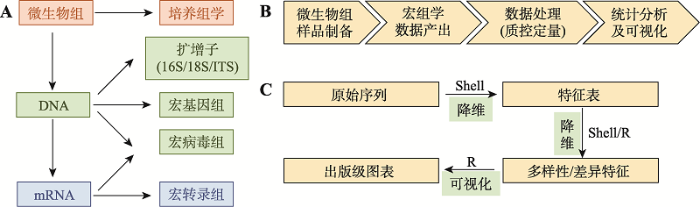
A:微生物组常用的研究层面和对应方法。微生物组按研究层面主要分为微生物培养、DNA和mRNA等3个层面;按研究技术主要包括培养组学(culturome)、扩增子(amplicon)、宏基因组(metagenome)、宏病毒组(metavirome) 和宏转录组(metatranscriptome)等测序技术[1,12]。B:微生物组研究的基本步骤。基于测序技术为基础的微生物组研究,主要分为样本制备、测序、数据处理和统计分析4个阶段。C:微生物组数据分析的基本步骤、常用环境和思想。组学数据分析主要分3步,图中箭头上描述了实现分析的常用语言环境Shell和/或R;图中箭头下展示各步分析的目的,即通过降维和可视化的基本思想,实现将大数据转化为可读图表。 Fig. 1Methods in microbiome research
(1) mothur:由美国密歇根大学的Patrick D. Schloss教授团队在2009年发布的首个扩增子分析流程[16]。它整合了之前发表的OTU定义软件DOTUR[17]、OTU差异比较工具SONS[18]以及其他可用工具,实现了第一套较完整的分析流程,让广大研究者开展扩增子分析成为可能(图2)。
(2) QIIME:2010年,美国科罗拉多大学的Rob Knight教授(现单位美国加州大学圣地亚哥分校)团队发布QIIME (发音同chime)分析流程[19]。该流程可在Linux或Mac系统中运行,相比mothur具有更多的优点,主要包括:整合了200多款相关软件和包,实现每个步骤更多软件和方法的选择;提供150多个脚本,实现各种个性化分析,并可以应对不同类型数据和实验设计;流程开放程度高,容易整合新软件和方法;增强统计和可视化,实现多样性、物种组成、差异比较和网络等众多方法和出版级图表绘制。由于QIIME允许同领域研究者较自主地开展扩增子数据的个性化分析和可视化,逐渐成为本领域最受欢迎的软件(图2)。为了满足日益增长的测序数据量和可重复计算的要求,Gregory J. Caporaso教授于2016年起发起了基于Python 3语言从头编写的QIIME 2项目[20]。该项目实现了分析流程的可追溯以满足科研可重复计算的要求;同时推出了一系列新算法,如基于进化距离的快速算法条型(Striped) UniFrac[21]、物种分类新方法2-feature-classifier[22]等;更重要的是软件的可扩展性和得到了同际同行的广泛支持,如接头和引物序列去除工具cutadapt[23]、序列质量控制R包DADA2[24]、聚类和去冗余的软件VSEARCH[25]、纵向和成对样本分析工具longitudinal[26]等,甚至包括宏基因组、宏代谢组分析和中文帮助文档,极大了提高了流程的适用范围和易用性。
图中橙色为Patrick D. Schloss教授开发的分析流程mothur,绿色为Rob Knight教授主持开发的QIIME系列分析流程,蓝色显示Robert Edgar独立研究员编写的相关软件和算法。 Fig. 2Important software and algorithms of microbiome in the past decade
Table 1 表1 表1 扩增子分析常用软件和数据库 Table 1 Software and databases for amplicon analysis
许多其他领域的分析方法在微生物组中也得到了推广和应用。全基因组关联分析(genome-wide association study, GWAS)[116]在鉴定人类疾病相关基因中发挥了巨大作用,目前也应用于微生物组领域来大规模探索人类与微生物组间的调控规律[117,118]、植物微生物组与产量[119]等。环境因子关联分析也有较多的分析方法在微生物生态学中得到广泛应用,如揭示温度[120]、pH[121]和盐分[122]等在不同环境中是微生物群落结构的决定因素。更多关于微生物组下游分析工具的介绍,详见表3。
BaiY, MüllerDB, SrinivasG, Garrido-OterR, PotthoffE, RottM, DombrowskiN, MünchPC, SpaepenS, Remus-EmsermannM, HüttelB,McHardyAC,VorholtJA, Schulze-LefertP. Functional overlap of the Arabidopsis leaf and root microbiota Nature, 2015,528(7582):364-369. [本文引用: 1]
ZhangJ, LiuYX, ZhangN, HuB, JinT, XuH, QinY, YanP, ZhangX, GuoX, HuiJ, CaoS, WangX, WangC, WangH, QuB, FanG, YuanL, Garrido-OterR, ChuC, BaiY . NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice Nat Biotechnol, 2019,37(6):676-684. [本文引用: 2]
ShiW, LiM, WeiG, TianR, LiC, WangB, LinR, ShiC, ChiX, ZhouB, GaoZ . The occurrence of potato common scab correlates with the community composition and function of the geocaulosphere soil microbiome Microbiome, 2019,7(1):14. [本文引用: 1]
MaY, YouX, MaiG, TokuyasuT, LiuC . A human gut phage catalog correlates the gut phageome with type 2 diabetes Microbiome, 2018,6(1):24. [本文引用: 1]
YuK, YiS, LiB, GuoF, PengX, WangZ, WuY, Alvarez-CohenL, ZhangT . An integrated meta-omics approach reveals substrates involved in synergistic interactions in a bisphenol A (BPA)-degrading microbial community Microbiome, 2019,7(1):16. [本文引用: 2]
LiJ, JiaH, CaiX, ZhongH, FengQ, SunagawaS, ArumugamM, KultimaJR, PriftiE, NielsenT, JunckerAS, ManichanhC, ChenB, ZhangW, LevenezF, WangJ, XuX, XiaoL, LiangS, ZhangD, ZhangZ, ChenW, ZhaoH, Al-AamaJY, EdrisS, YangH, WangJ, HansenT, NielsenHB, BrunakS, KristiansenK, GuarnerF, PedersenO, DoréJ, EhrlichSD, MetaHITConsortium,BorkP,WangJ,PonsN,Le Chatelier E,BattoJM,KennedyS,HaimetF,WinogradskiY,PelletierE,LePaslierD,ArtiguenaveF,BrulsT,WeissenbachJ,TurnerK,ParkhillJ,AntolinM,CasellasF,BorruelN,VarelaE,TorrejonA,DenariazG,DerrienM,van Hylckama Vlieg JET,ViegaP,OozeerR,KnollJ,RescignoM,BrechotC,M'RiniC,MérieuxA,YamadaT,TimsS,ZoetendalEG,KleerebezemM,de Vos WM,CultroneA,LeclercM,JusteC,GuedonE,DelormeC,LayecS,KhaciG,van de GuchteM,VandemeulebrouckG,JametA,DervynR,SanchezN,BlottièreH,MaguinE,RenaultP,TapJ,MendeDR. An integrated catalog of reference genes in the human gut microbiome Nat Biotechnol, 2014,32(8):834-841. [本文引用: 1]
GrüningB, DaleR, Sj?dinA, ChapmanBA, RoweJ, Tomkins-TinchCH, ValierisR, K?sterJ, BiocondaTeam . Bioconda: sustainable and comprehensive software distribution for the life sciences Nat Methods, 2018,15(7):475-476. [本文引用: 1]
WickhamH . ggplot2: elegant graphics for data analysis Springer, 2016. [本文引用: 1]
SchlossPD, WestcottSL, RyabinT, HallJR, HartmannM, HollisterEB, LesniewskiRA, OakleyBB, ParksDH, RobinsonCJ, SahlJW, StresB, ThallingerGG, van HornDJ, WeberCF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities Appl Environ Microb, 2009,75(23):7537-7541. [本文引用: 1]
SchlossPD, HandelsmanJ . Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness Appl Environ Microb, 2005,71(3):1501-1506. [本文引用: 1]
SchlossPD, HandelsmanJ . Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures Appl Environ Microb, 2006,72(10):6773-6779. [本文引用: 1]
CaporasoJG, KuczynskiJ, StombaughJ, BittingerK, BushmanFD, CostelloEK, FiererN, Pe?aAG, GoodrichJK, GordonJI, HuttleyGA, KelleyST, KnightsD, KoenigJE, LeyRE, LozuponeCA,McDonaldD,MueggeBD,PirrungM,ReederJ,SevinskyJR,TurnbaughPJ,WaltersWA,WidmannJ,YatsunenkoT,ZaneveldJ,KnightR. QIIME allows analysis of high-throughput community sequencing data Nat Methods, 2010,7(5):335-336. [本文引用: 1]
CallahanBJ , McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data Nat Methods, 2016,13(7):581-583. [本文引用: 1]
RognesT, FlouriT, NicholsB, QuinceC, MahéF . VSEARCH: a versatile open source tool for metagenomics PeerJ, 2016,4:e2584. [本文引用: 4]
BokulichNA, DillonMR, ZhangY, RideoutJR, BolyenE, LiH, AlbertPS, CaporasoJG . Q2-longitudinal: longitudinal and paired-sample analyses of microbiome data mSystems, 2018,3(6):e00219-00218. [本文引用: 1]
EdgarRC . Search and clustering orders of magnitude faster than BLAST Bioinformatics, 2010,26(19):2460-2461. [本文引用: 1]
EdgarRC, HaasBJ, ClementeJC, QuinceC, KnightR . UCHIME improves sensitivity and speed of chimera detection Bioinformatics, 2011,27(16):2194-2200. [本文引用: 1]
EdgarRC . UPARSE: highly accurate OTU sequences from microbial amplicon reads Nature Methods, 2013,10(10):996-998. [本文引用: 1]
EdgarRC, FlyvbjergH . Error filtering, pair assembly and error correction for next-generation sequencing reads Bioinformatics, 2015,31(21):3476-3482. [本文引用: 1]
OksanenJ, KindtR, LegendreP , O’Hara B, Stevens MHH, Oksanen MJ, Suggests M. The vegan package Community Ecology Package, 2007,10:631-637. [本文引用: 2]
McMurdiePJ, HolmesS . Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data PLoS One, 2013,8(4):e61217. [本文引用: 2]
MitchellAL, ScheremetjewM, DeniseH, PotterS, TarkowskaA, QureshiM, SalazarGA, PesseatS, BolandMA, HunterFMI, Ten HoopenP, AlakoB, AmidC, WilkinsonDJ, CurtisTP, CochraneG, FinnRD . EBI Metagenomics in 2017: enriching the analysis of microbial communities, from sequence reads to assemblies Nucleic Acids Res, 2018,46(D1):D726-D735.
ShiW, QiH, SunQ, FanG, LiuS, WangJ, ZhuB, LiuH, ZhaoF, WangX, HuX, LiW, LiuJ, TianY, WuL, MaJ . GcMeta: a global catalogue of metagenomics platform to support the archiving, standardization and analysis of microbiome data Nucleic Acids Res, 2018,47(D1):D637-D648.
McDonaldD, PriceMN, GoodrichJ, NawrockiEP, DeSantisTZ, ProbstA, AndersenGL, KnightR, HugenholtzP . An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea ISME J, 2012,6(3):610-618.
QuastC, PruesseE, YilmazP, GerkenJ, SchweerT, YarzaP, PepliesJ, Gl?cknerFO . The SILVA ribosomal RNA gene database project: improved data processing and web-based tools Nucleic Acids Res, 2013,41(Database issue):D590-596.
ColeJR, WangQ, FishJA, ChaiB, McGarrellDM,SunY,BrownCT,Porras-AlfaroA,KuskeCR,TiedjeJM. Ribosomal Database Project: data and tools for high throughput rRNA analysis Nucleic Acids Res, 2014,42(Database issue):D633-D642.
NilssonRH, LarssonK-H, TaylorAFS, Bengtsson- PalmeJ, JeppesenTS, SchigelD, KennedyP, PicardK, Gl?cknerFO, TedersooL, SaarI, K?ljalgU, AbarenkovK . The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications Nucleic Acids Res, 2019,47(D1):D259-D264.
LangilleMGI, ZaneveldJ, CaporasoJG, McDonaldD,KnightsD,ReyesJA,ClementeJC,BurkepileDE,Vega ThurberRL,KnightR,BeikoRG,HuttenhowerC. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences Nat Biotechnol, 2013,31(9):814-821.
A?hauerKP, WemheuerB, DanielR, MeinickeP . Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data Bioinformatics, 2015,31(17):2882-2884.
LoucaS, ParfreyLW, DoebeliM . Decoupling function and taxonomy in the global ocean microbiome Science, 2016,353(6305):1272-1277.
NguyenNH, SongZ, BatesST, BrancoS, TedersooL, MenkeJ, SchillingJS, KennedyPG . FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild Fungal Ecol, 2016,20:241-248.
LomsadzeA, GemayelK, TangS, BorodovskyM . Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes Genome Res, 2018,28(7):1079-1089. [本文引用: 1]
FuL, NiuB, ZhuZ, WuS, LiW . CD-HIT: accelerated for clustering the next-generation sequencing data Bioinformatics, 2012,28(23):3150-3152. [本文引用: 1]
LiuB, ZhengD, JinQ, ChenL, YangJ . VFDB 2019: a comparative pathogenomic platform with an interactive web interface Nucleic Acids Res, 2019,47(D1):D687-D692. [本文引用: 1]
KangD, LiF, KirtonE, ThomasA, EganR, AnH, WangZ . MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies PeerJ, 2019,7:e7359. [本文引用: 1]
WuYW, SimmonsBA, SingerSW . MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets Bioinformatics, 2015,32(4):605-607. [本文引用: 1]
AlnebergJ, BjarnasonBS,deBruijn I,SchirmerM,QuickJ,IjazUZ,LahtiL,LomanNJ,AnderssonAF,QuinceC,. Binning metagenomic contigs by coverage and composition Nat Methods, 2014,11(11):1144-1146. [本文引用: 1]
UritskiyGV, DiRuggieroJ, TaylorJ. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis Microbiome, 2018,6(1):158. [本文引用: 1]
SieberCMK, ProbstAJ, SharrarA, ThomasBC, HessM, TringeSG, BanfieldJF . Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy Nat Microbiol, 2018,3(7):836-843. [本文引用: 1]
JiP, ZhangY, WangJ, ZhaoF . MetaSort untangles metagenome assembly by reducing microbial community complexity Nat Commun, 2017,8:14306. [本文引用: 1]
BisharaA, MossEL, KolmogorovM, ParadaAE, WengZ, SidowA, DekasAE, BatzoglouS, BhattAS . High- quality genome sequences of uncultured microbes by assembly of read clouds Nat Biotechnol, 2018,36(11):1067-1075. [本文引用: 1]
BertrandD, ShawJ, KalathiyappanM, NgAHQ, KumarMS, LiC, DvornicicM, SoldoJP, KohJY, TongC, NgOT, BarkhamT, YoungB, MarimuthuK, ChngKR, SikicM, NagarajanN . Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes Nat Biotechnol, 2019,37(8):937-944. [本文引用: 2]
StewartRD, AuffretMD, WarrA, WalkerAW, RoeheR, WatsonM . Compendium of 4,941 rumen metagenome- assembled genomes for rumen microbiome biology and enzyme discovery Nat Biotechnol, 2019,37(8):953-961. [本文引用: 1]
EwelsP, MagnussonM, LundinS, K?llerM . MultiQC: summarize analysis results for multiple tools and samples in a single report Bioinformatics, 2016,32(19):3047-3048.
BolgerAM, LohseM, UsadelB . Trimmomatic: a flexible trimmer for Illumina sequence data Bioinformatics, 2014,30(15):2114-2120.
LangmeadB, SalzbergSL . Fast gapped-read alignment with Bowtie 2 Nat Methods, 2012,9(4):357-359.
SuzekBE, WangY, HuangH, McGarveyPB, WuCH, UniProtConsortium. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches Bioinformatics, 2015,31(6):926-932.
LiD, LiuCM, LuoR, SadakaneK, LamTW . MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph Bioinformatics, 2015,31(10):1674-1676. [本文引用: 1]
ComeauAM, DouglasGM, LangilleMGI . Microbiome helper: a custom and streamlined workflow for microbiome research mSystems, 2017,2(1):e00127-00116.
RobinsonMD, McCarthyDJ, SmythGK. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data Bioinformatics, 2010,26(1):139-140. [本文引用: 1]
LoveMI, HuberW, AndersS . Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 Genome Biol, 2014,15(12):550. [本文引用: 1]
AsnicarF, WeingartG, TickleTL, HuttenhowerC, SegataN . Compact graphical representation of phylogenetic data and metadata with GraPhlAn PeerJ, 2015,3:e1029. [本文引用: 2]
DhariwalA, ChongJ, HabibS, KingIL, AgellonLB, XiaJ . MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data Nucleic Acids Res, 2017,45(W1):W180-W188. [本文引用: 1]
R?ttjersL, FaustK . From hairballs to hypotheses- biological insights from microbial networks FEMS Microbiol Rev, 2018,42(6):761-780. [本文引用: 1]
BanerjeeS, SchlaeppiK, van der HeijdenMGA. Keystone taxa as drivers of microbiome structure and functioning Nat Rev Microbiol, 2018,16(9):567-576. [本文引用: 1]
CsardiG, NepuszT . The igraph software package for complex network research. InterJournal, Complex Systems, 2006,1695(5):1-9. [本文引用: 1]
BastianM, HeymannS, JacomyM . Gephi: an open source software for exploring and manipulating networks In Third international AAAI conference on weblogs and social media:2009. [本文引用: 1]
FanK, WeisenhornP, GilbertJA, ShiY, BaiY, ChuH . Soil pH correlates with the co-occurrence and assemblage process of diazotrophic communities in rhizosphere and bulk soils of wheat fields Soil Biol Biochem, 2018,121:185-192. [本文引用: 1]
WangJ, ZhengJ, ShiW, DuN, XuX, ZhangY, JiP, ZhangF, JiaZ, WangY, ZhengZ, ZhangH, ZhaoF . Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus Gut, 2018,67(9):1614-1625. [本文引用: 1]
WangJ, JiaZ, ZhangB, PengL, ZhaoF. Tracing the accumulation of in vivo human oral microbiota elucidates microbial community dynamics at the gateway to the GI tract Gut, 2019: gutjnl-2019-318977. [本文引用: 1]
KatohK, StandleyDM . MAFFT Multiple sequence alignment software version 7: improvements in performance and usability Mol Biol Evol, 2013,30(4):772-780. [本文引用: 1]
EdgarRC . MUSCLE: multiple sequence alignment with high accuracy and high throughput Nucleic Acids Res, 2004,32(5):1792-1797. [本文引用: 1]
PriceMN, DehalPS, ArkinAP . FastTree 2-approximately maximum-likelihood trees for large alignments PLoS One, 2010,5(3):e9490. [本文引用: 1]
NguyenLT, SchmidtHA, von HaeselerA, MinhBQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies Mol Biol Evol, 2015,32(1):268-274. [本文引用: 1]
TrifinopoulosJ, NguyenLT, von HaeselerA, MinhBQ. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis Nucleic Acids Res, 2016,44(W1):W232-W235. [本文引用: 1]
SubramanianB, GaoS, LercherMJ, HuS, ChenWH . Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees Nucleic Acids Res, 2019,47(W1):W270-W275. [本文引用: 1]
LetunicI, BorkP . Interactive Tree Of Life (iTOL) v4: recent updates and new developments Nucleic Acids Res, 2019,47(W1):W256-W259. [本文引用: 1]
YuG, SmithDK, ZhuH, GuanY, LamTTY . Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data Methods Ecol Evol, 2017,8(1):28-36. [本文引用: 1]
LeCunY, BengioY, HintonG . Deep learning Nature, 2015,521:436-444. [本文引用: 1]
RenZ, LiA, JiangJ, ZhouL, YuZ, LuH, XieH, ChenX, ShaoL, ZhangR, XuS, ZhangH, CuiG, ChenX, SunR, WenH, LerutJP, KanQ, LiL, ZhengS . Gut microbiome analysis as a tool towards targeted non- invasive biomarkers for early hepatocellular carcinoma Gut, 2019,68(6):1014-1023. [本文引用: 1]
MetcalfJL, XuZZ, WeissS, LaxS,van TreurenW,HydeER,SongSJ,AmirA,LarsenP,SangwanN,HaarmannD,HumphreyGC,AckermannG,ThompsonLR,LauberC,BibatA,NicholasC,GebertMJ,PetrosinoJF,ReedSC,GilbertJA,LynneAM,BucheliSR,CarterDO,KnightR. Microbial community assembly and metabolic function during mammalian corpse decomposition Science, 2016,351(6269):158-162. [本文引用: 1]
ZhangJ, ZhangN, LiuYX, ZhangX, HuB, QinY, XuH, WangH, GuoX, QianJ, WangW, ZhangP, JinT, ChuC, BaiY . Root microbiota shift in rice correlates with resident time in the field and developmental stage Sci China Life Sci, 2018,61(6):613-621. [本文引用: 1]
LiawA, WienerM . Classification and regression by randomForest R News, 2002,2(3):18-22. [本文引用: 1]
GalkinF, AliperA, PutinE, KuznetsovI, GladyshevVN, ZhavoronkovA . Human microbiome aging clocks based on deep learning and tandem of permutation feature importance and accumulated local effects bioRxiv, 2018,507780. [本文引用: 1]
YangC, YangRF, CuiYJ . Bacterial genome-wide association study: methodologies and applications Hereditas(Beijing), 2018,40(1):57-65. [本文引用: 1]
WangZ, LuG, YuanM, YuH, WangS, LiX, DengY . Elevated temperature overrides the effects of N amendment in Tibetan grassland on soil microbiome Soil Biology and Biochemistry, 2019,136:107532. [本文引用: 1]
ShiY, LiY, XiangX, SunR, YangT, HeD, ZhangK, NiY, ZhuYG, AdamsJM, ChuH . Spatial scale affects the relative role of stochasticity versus determinism in soil bacterial communities in wheat fields across the North China Plain Microbiome, 2018,6(1):27. [本文引用: 1]
ZhangK, ShiY, CuiX, YueP, LiK, LiuX, TripathiBM, ChuH . Salinity is a key determinant for soil microbial communities in a desert ecosystem mSystems, 2019,4(1):e00225-00218. [本文引用: 1]
DohertyMK, DingT, KoumpourasC, TelescoSE, MonastC, DasA, BrodmerkelC, SchlossPD . Fecal microbiota signatures are associated with response to ustekinumab therapy among crohn’s disease patients mBio, 2018,9(2):e02120-02117.
DiGiulioDB, CallahanBJ, McMurdiePJ, CostelloEK, LyellDJ, RobaczewskaA, SunCL, GoltsmanDSA, WongRJ, ShawG, StevensonDK, HolmesSP, RelmanDA . Temporal and spatial variation of the human microbiota during pregnancy Proc Natl Acad Sci USA, 2015,112(35):11060-11065.
Garrido-OterR, NakanoRT, DombrowskiN, MaKW, McHardyAC, Schulze-LefertP. Modular traits of the Rhizobiales root microbiota and their evolutionary relationship with symbiotic Rhizobia Cell Host Microbe, 2018, 24(1): 155-167. e5.
CastrilloG, TeixeiraPL, ParedesSH, LawTF,de Lorenzo L,FeltcherME,FinkelOM,BreakfieldNW,MieczkowskiP,JonesCD,Paz-AresJ, Dangl JL. Root microbiota drive direct integration of phosphate stress and immunity Nature, 2017,543(7646):513-518.
Herrera ParedesS, GaoT, LawTF, FinkelOM, MucynT, TeixeiraPJPL, Salas GonzálezI, FeltcherME, PowersMJ, ShankEA, JonesCD, JojicV, DanglJL, CastrilloG . Design of synthetic bacterial communities for predictable plant phenotypes PLoS Biol, 2018,16(2):e2003962.
AlmeidaA, MitchellAL, BolandM, ForsterSC, GloorGB, TarkowskaA, LawleyTD, FinnRD . A new genomic blueprint of the human gut microbiota Nature, 2019,568(7753):499-504.
VandeputteD, KathagenG, D’hoeK, Vieira-SilvaS, Valles-ColomerM, SabinoJ, WangJ, TitoRY, De CommerL, DarziY, VermeireS, FalonyG, RaesJ . Quantitative microbiome profiling links gut community variation to microbial load Nature, 2017,551(7681):507-511.
StewartCJ, AjamiNJ, O’BrienJL,HutchinsonDS,SmithDP,WongMC,RossMC,LloydRE,DoddapaneniH,MetcalfGA,MuznyD,GibbsRA,VatanenT,HuttenhowerC,XavierRJ,RewersM,HagopianW,ToppariJ,ZieglerAG,SheJX,AkolkarB,LernmarkA,HyotyH,VehikK,KrischerJP,PetrosinoJF. Temporal development of the gut microbiome in early childhood from the TEDDY study Nature, 2018,562(7728):583-588.
MeadowJF, AltrichterAE, KembelSW, MoriyamaM, O’ConnorTK,WomackAM,BrownGZ,GreenJL,BohannanBJM. Bacterial communities on classroom surfaces vary with human contact Microbiome, 2014,2(1):7.
BradleyP, den BakkerHC, RochaEPC, McVeanG, IqbalZ . Ultrafast search of all deposited bacterial and viral genomic data Nat Biotechnol, 2019,37(2):152-159. [本文引用: 1]
 ,1,2,3
,1,2,3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT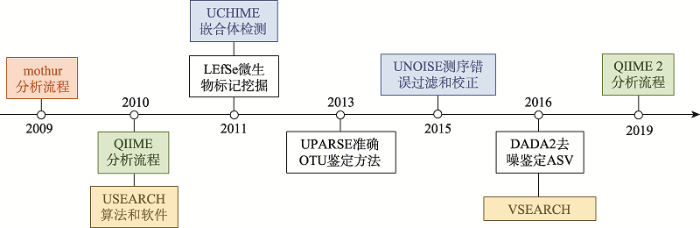 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}
{kind=link}
{kind=link}