 ,2
,2Comprehensive re-annotation of protein-coding genes for prokaryotic genomes by Z-curve and similarity-based methods
Shuo Liu1, Zhi Zeng1, Fancai Zeng2, Mengze Du,2通讯作者: 杜萌泽,博士,讲师,研究方向:生物信息学。E-mail:du_mengze@foxmail.com
编委: 包其郁
收稿日期:2020-02-20修回日期:2020-05-11网络出版日期:2020-07-20
| 基金资助: |
Editorial board:
Received:2020-02-20Revised:2020-05-11Online:2020-07-20
| Fund supported: |
作者简介 About authors
刘硕,在读博士研究生,专业方向:微生物基因组学。E-mail:

摘要
随着测序技术的不断发展,产生了海量的基因组测序数据,极大地丰富了公共遗传数据资源。同时为了应对大量基因组数据的产生,基因组比较和注释算法、工具不断更新,使得联合多种注释工具得到更准确的蛋白编码基因的注释信息成为可能。目前公共数据库的原核生物基因组测序和装配有些是10多年前的,存在大量预测的功能未知的编码基因。为了提升美国国家生物信息中心(National Center for Biotechnology Information, NCBI)数据库中基因组的注释质量,本研究联合使用多种原核基因识别算法/软件和基因表达数据重注释1587个细菌和古细菌基因组。首先,利用Z曲线的33个变量从177个基因组原注释中识别获得3092个被过度注释为蛋白编码基因的序列;其次,通过同源比对为939个基因组中的4447个功能未知的蛋白编码基因注释上具体功能;最后,通过联合采用ZCURVE 3.0和Glimmer 3.02以及Prodigal这3种高精度的、广泛使用且基于算法不同而互补的基因识别软件来寻找漏注释基因。最终,从9个基因组中找到了2003个被漏注释的蛋白编码基因,这些基因属于多个蛋白质直系同源簇(clusters of orthologous groups of proteins, COG)。本研究使用新的工具并结合多组学数据重新注释早期测序的细菌和古细菌基因组,不仅为新测序菌株提供注释方法参考,而且这些重注释后得到的细菌基因序列也会对后续基础研究有所帮助。
关键词:
Abstract
The development of sequencing technology has generated huge genomic sequencing information and largely enriched public genetic resources. To analyze such big data, the algorithms and tools for comparison and annotation of genomes are updated continually, enabling genome annotation with higher accuracy via various annotation tools. Many prokaryotic genomes in public database were sequenced and assembled more than a decade ago, and they contained multiple genes with unknown functions. To improve the current annotation for those genomes in NCBI, we re-annotate 1587 bacterial and archaeal genomes using multiple prokaryotic gene recognition algorithms/softwares and gene expression data. The 33 Z-curve variables were applied to recognize sequences that were over-annotated to genes of 1587 bacterial and archaeal genomes deposited in public databases, and a total of 3092 sequences belonging to 177 genomes were recognized as sequences over-annotated as protein-coding genes. Next, 4447 protein-coding genes with unknown functions from 939 genomes were annotated with definite functions by similarity search. Finally, we recognized 2003 missed protein-coding genes that belong to known COG (clusters of orthologous groups of proteins) of nine genomes using three methods (ZCURVE 3.0, Glimmer 3.02 and Prodigal), which are accurate and frequently used for gene finding. Their algorithms are different and complementary. This is a comprehensive study for re-annotation of bacterial and archaeal genomes with new tools combining multi-omics data, which should provide a reference for annotation of newly sequenced strains, and also benefit further fundamental researches with the bacterial gene sequences obtained after re-annotation.
Keywords:
PDF (3018KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘硕, 曾志, 曾凡才, 杜萌泽. 基于序列相似性和Z曲线方法重注释原核生物蛋白编码基因. 遗传[J], 2020, 42(7): 691-702 doi:10.16288/j.yczz.20-022
Shuo Liu.
基因组分析是生命科学研究中非常关键的基础工作,只有获得准确的基因组注释才能为后续的分析和研究提供有力支撑并得到有意义的结果。目前,美国国家生物信息中心(National Center for Biotechnology Information, NCBI)积累了大量10多年前完成测序的基因组。受限于早期有限的基因注释工具[1],这些基因组注释的精确度有待于进一步提升。例如,在原核生物基因组中寻找小的基因时容易出现此类基因被漏注释而丢失的问题[2]。随着测序效率的提升,几何级数增长的测序量意味着这些注释错误将会通过同源搜索等方式被放大[3]。Breitwieser等[4]检查了所有的细菌和古细菌基因组后发现2250个原核生物基因组居然包含了人类基因组的信息。为了确保基因组信息的准确性[3,5],相关数据库需要时常更新原始的基因组注释信息。当前随着各种组学数据的日益完善,通过联合多工具多组学的方式对基因组精确重注释也具备可行性。
自1995年第1个原核生物流感嗜血杆菌(Haemophilus influenzae)基因组被注释以来,大量的原核生物基因组陆续完成测序和注释[5]。根据GenBank[6]的记录,自1999年4月至2019年10月,被测序序列的数量从3,525,418增长至216,763,706,增长倍数约为60倍。近年来基因识别软件,尤其是识别原核生物中基因的新方法和软件在被陆续开发和不断升级[7,8,9,10,11,12,13,14],其中较广泛使用的有IPred[9]、GeneMarkS[11]、Glimmer[12]、EasyGene[13]和ZCURVE[14]等。这些新的基因探测工具不仅可以用于注释基因组从而获得更精确的结果,而且还可赋予假定蛋白详尽的功能[15,16,17,18,19]。这些工具中基于Z-curve理论的软件ZCURVE 3.0[8]具有93.7%的准确率,与具有93.0%准确率的Glimmer 3.02[20]相当,而且Z-curve通过把单核苷酸、双核苷酸和三核苷酸的碱基组成转换成33个空间变量来表示DNA序列的特征,并通过对正样本和负样本的学习来对未知序列进行编码蛋白能力的预测[15,16]。该方法在原核生物的编码蛋白的预测上准确度高、应用广[21]。这两个软件联合使用可以获得互相补充的结果。更准确的细菌和古细菌基因组信息有助于更精确地推导生命的起源和演化,如Weiss等[22]对原核生物基因组的610万个蛋白编码基因的全部簇和进化树进行研究以推断最后的共同祖先(the last universal common ancestor, LUCA),而不准确的注释会影响该研究结论的可靠性。
本研究对1587个测序和注释较早的细菌和古细菌菌株的基因组进行了重新注释,这些基因组包含功能已知的开放阅读框(open reading frame, ORF)和假定ORFs (功能未知的ORFs),而假定ORFs中有些实际上是非蛋白编码基因的序列,此外基因组还包含被漏注释掉但实际上是可编码蛋白基因的那些序列。参考基因表达数据并联合3个注释准确率较高的原核生物识别软件(ZCURVE 3.0[8]、Glimmer 3.02[20]以及Prodigal[23]),移除掉一些之前被过度注释为基因的序列,同时给功能未知的ORFs注释了新的功能,并获得了之前漏注释的基因。
1 材料与方法
1.1 数据的选取
选用来自30个门(phylum)1587个20年内被陆续测序的细菌和古细菌的基因组来进行重注释(附表1),基因组来自992个物种(附表2)。基因组的装配序列和注释信息均于2019年1月获取自NCBI数据库,它们的测序时间为1999年~2019年。在使用33个Z-curve变量去除1587个基因组的过度注释基因时,选取它们的一些具有已知功能的ORFs作为正样本,并选取每个基因组的此类ORFs随机打乱1000次后产生的完全随机的序列作为负样本。
为判断计算识别的序列为真正的非蛋白编码序列,从GEO[24]和paxdb[25]下载了蛋白丰度或者RNA丰度数据。原基因组中第二类功能不确定的ORFs即假定ORFs如果被Z-curve 33变量方法识别为可能的非蛋白编码序列,同时其没有对应的蛋白或者mRNA被检测到,即认定其为过度注释为基因的序列,继而从基因组基因列表中移除。
本研究对1587个基因组中的9个模式物种基因组进行新基因的注释。它们分别是嗜酸氧化亚铁硫杆菌(Acidithiobacillus ferrooxidans ATCC 23270)、炭疽芽孢杆菌(Bacillus anthracis str. Ames)、枯草芽孢杆菌(B. subtilis subsp. subtilis str. 168)、大肠杆菌(E. coli str. K-12 substr. MG1655)、流感嗜血杆菌(Haemophilus influenzae Rd KW20)、脑膜炎奈瑟球菌(Neisseria meningitidis MC58)、肠道沙门氏菌(Salmonella enterica subsp. enterica serovar Typhi str. CT18)、金黄色葡萄球菌(Staphylococcus aureus subsp. aureus NCTC 8325)和酿脓链球菌(Streptococcus pyogenes SF370)。
1.2 重注释流程
整个重注释的流程分为3个部分:(1)识别过度注释为基因的序列;(2)未知功能ORFs的功能注释;(3)识别欠注释的基因(图1),其具体的方法学细节部分见1.3、1.4、1.5和1.6。图1
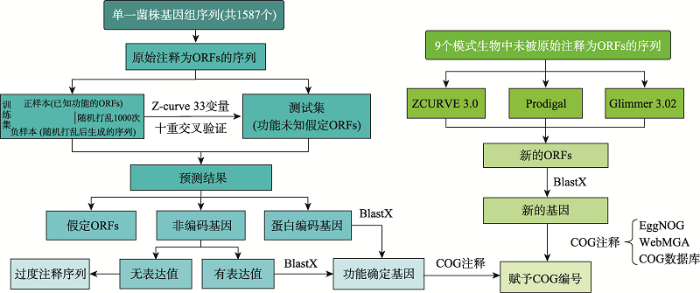 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1重注释流程图
Fig. 1The pipeline for re-annotation
1.3 寻找过度注释为基因的序列
Z-curve把DNA序列中出现在第一(1、4、7...)、第二(2、5、8...)和第三(3、6、9...)密码子位的A、G、C、T的碱基的频率分别用a1、g1、c1、t1;a2、 g2、c2、t2;a3、g3、c3、t3来表示,根据Z曲线理论,一条DNA序列可以用三维平面中的点Pi (i=1、2、3)来表示,而P1、P2、P3所对应的xi、yi、zi可以通过下边的公式来计算得出,据此得出Z-curve的单核苷酸的9个变量(9=3×3)。$\begin{cases}x_{i}=(a_{i}+g_{i})-(c_{i}+t_{i})\\ y_{i}=(a_{i}+c_{i})-(g_{i}+t_{i})\\z_{i}=(a_{i}+t_{i})-(g_{i}+c_{i}) \end{cases}$
当用p12(XY)或p23(XY)来表示DNA序列的第一第二密码子位或第二第三密码子位时,上边的公式可以衍生出以下公式,其中X代表A、G、C或者T中的一种碱基。k表示密码子相位组合,k=12表示第一第二相位,同理k=23表示第二第三相位,可以得到24个变量(24=3×4×2)。
$\begin{cases} x_{k}^{X}=(Pk(XA)+Pk(XG))-(Pk(XC)+Pk(XT)) \\ y_{k}^{X}=(Pk(XA)+Pk(XC))-(Pk(XG)+Pk(XT)) \\ z_{k}^{x}=(Pk(XA)+Pk(XT))-(Pk(XG)+(XC)) \end{cases}$
得到以上Z-curve 33变量后,文章使用每个基因组对应的正样本和负样本进行训练。从正样本和负样本中随机抽取1/10的样本作为测试集,并进行十次交叉验证。最终平均预测准确率均大于99%,证明该预测方法稳定可靠。
将没有提供明确功能的ORFs作为测试集可寻找过度注释为基因的序列。预测为非蛋白编码基因的序列且同时没有在公共数据库中找到表达数据的假定ORFs将被归为过度注释为基因的序列。基因的表达数据来自不同的RNA测序集和GEO数据集(附表3),这些数据可以帮助排除掉具有表达数据的基因。
1.4 给功能未知ORFs注释功能
假如一些假定/功能未知的ORFs没有被识别为非蛋白编码序列而是被预测为蛋白编码基因,研究就会通过同源搜索赋予其功能,使用的工具为BLAST[26],赋予基因功能的同源搜索的参数设置为E值(E-value) < 1e-20,覆盖率(Coverage) > 80%以及一致性(Identity) > 70%。而假如被预测为非蛋白编码基因的序列其RNA-seq的数据显示为Count/TPM/ RPKM/CPM > 0,即存在,可以认为该序列具有表达数据,则需通过BLAST来注释其功能,根据经验参数,把赋予基因功能的同源搜索的参数同样设置为E值 < 1e-20,覆盖率> 80%以及一致性 > 70%。1.5 补充漏注释基因
基因组注释经常会丢掉一些真正的基因[2,27],使用多种基因搜索工具可以尽可能减少该错误的发生。此处联合ZCURVE 3.0[8]、Glimmer 3.02[20]和Prodigal[23]来注释9种重要的模式生物的代表株。这3个软件识别出的不在原基因组基因列表中的ORFs将被全部加入候选基因序列集。对此集合中的序列用BLAST[26] (https://blast.ncbi.nlm.nih.gov/Blast. cgi)进行同源比对后只保留那些与公共数据库中功能已知的基因有显著相似性的候选基因序列。由于此处目的为注释被遗漏的基因,研究根据经验参数把同源比对的参数设置为比1.4部分的阈值稍宽松的值,即E < 1e-20,覆盖率 > 60%,一致性 > 60%,并用2个阈值对模式生物大肠杆菌的代表株E. coli str. K-12 substr. MG1655被注释后得到的246个新基因进行了2次同源比对,通过对比,发现宽松的阈值更能确保注释得到的新基因的完整性,详细结果参见2.4。1.6 同源簇注释
结构和功能相似的蛋白编码序列属于同一个蛋白簇。在此,为注释得到的新基因进行同源簇COG注释可以帮助更好去理解基因组如何正常行使功能。研究同时联合使用了EggNOG[28]、WebMGA[29]和NCBI中的COG数据库[30]来注释这些基因序列的同源簇,其对应的COG编号可在附表7中找到。EggNOG包含2031个物种和19万个同源簇和对应功能注释数据。序列分析器WebMGA[29]和NCBI上的COG数据库[30]用于进一步完善同源簇功能注释。2 结果与分析
2.1 1587个物种的注释现状
根据这1587个物种的具体测序时间,绘制了同一年份被测序的基因组数目占全部基因组比例的柱形图(图2,附表1),其中2012年之前被测序的基因组占整体基因组的97%以上。根据CVTree[31]列出的门(phylum)的数量,与细菌和古细菌相关的门(phylum)为41个,选取的细菌和古细菌的基因组在门(phylum)水平的覆盖度为73%,研究选取的基因组尽可能包含了模式微生物的代表菌株如大肠杆菌和枯草芽孢杆菌等属于同一物种的注释时间不同的多种血清型,整体来说,选取的基因组在Refseq数据库中装配和注释的时间较早。图2
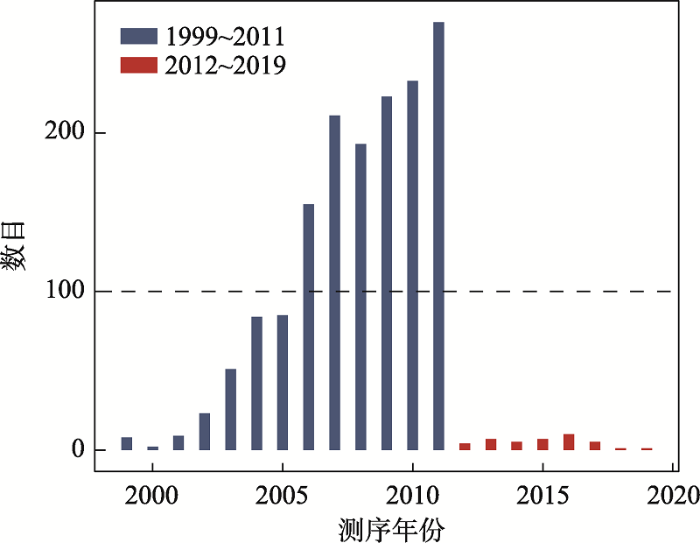 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图21587个基因组的测序时间分布
Fig. 2The distribution of sequencing time for 1587 genomes
2.2 原注释中3092个假定ORFs为非蛋白编码序列
在这1587个基因组的原始注释中,平均有1.26%的假定基因重注释后信息发生改变(图3A)。使用功能已知的ORFs作为正样本,选用对正样本序列随机打乱1000次后产生的序列作为负样本,进行训练后通过十重交叉验证后得到预测序列编码能力的平均准确率为0.9985 (图3B)。接着,研究依次对1587个细菌基因组中的功能未知ORFs进行预测,其中56,462个ORFs被预测为可能的非蛋白编码序列(附表4)。图3
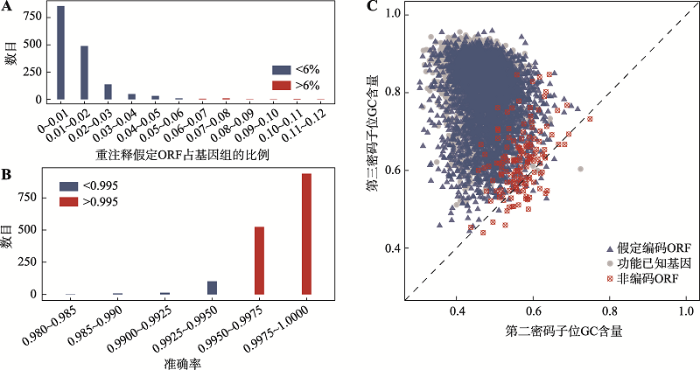 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3假定ORFs的比率及不同准确率对应的基因组数量及大豆根瘤菌(B. japonicum USDA 110)3类序列两个密码子位GC含量
A:不同比率的重注释假定ORFs对应基因组的数目;B:不同识别非编码ORFs的准确率对应基因组的数目;C:大豆根瘤菌3类序列对应的第2和第3位密码子GC含量。
Fig. 3The numbers of genomes according to ratio of hypothetical ORFs and accuracy for recognizing non-coding ORFs, and the GC contents of two codon positions of three types of sequences of B. japonicum USDA 110
由于计算方法本身的偏差,本研究使用实验数据进一步确定这些序列是否为非蛋白编码序列。当且仅当这些序列在公共数据库中查询不到表达数据,才认为它们确实不具有编码功能。研究查询了GEO等数据库并最终确定177个基因组中共3092个ORFs为过度注释为基因的序列(附表5)。需要重点提及的是其中43个基因组有20个以上的假定ORFs被识别为这类序列(表1)。
之前的研究已经报道过编码的ORFs在第二和第三密码子位的位置上有着不同的GC含量分布[16],即大多数功能已知的ORFs的第三密码子位的GC含量比其第二密码子位的GC含量都高,而对于非编码ORFs,第二密码子位的GC含量则是接近第三密码子位的GC含量。在43个基因组中,大豆根瘤菌(Bradyrhizobium japonicum USDA 110)基因组的假定ORFs被识别到了最多的非蛋白编码序列(147条),其第二密码子位和第三密码子位的GC含量的分布和以上的论断是类似的(图3C),这验证了本研究鉴定出的非编码ORFs具有可信性。
Table 1
表1
表1识别出过度注释的ORFs多于20的菌株基因组的信息
Table 1
| 菌株 | NC序列号 | 数量 | 菌株 | NC序列号 | 数量 |
|---|---|---|---|---|---|
| 慢性型大豆根瘤菌(B. japonicum USDA 110) | NC_004463 | 147 | 金黄色葡萄球菌 (S. aureus subsp. Aureus MW2) | NC_003923 | 36 |
| 哈氏弧菌(Vibrio harveyi ATCC BAA-1116) | NC_009784 | 133 | 黑海甲烷袋状菌 (Methanoculleus marisnigri JR1) | NC_009051 | 34 |
| 大肠杆菌(E. coli CFT073) | NC_004431 | 119 | 双叶钩端螺旋体血清型Patoc菌株 (Leptospira biflexa serovar Patoc strain 'Patoc 1) | NC_010602 | 34 |
| 织片草螺菌 (Herbaspirillum seropedicae SmR1) | NC_014323 | 109 | 拟杆菌属 (Bacteroides salanitronis DSM 18170) | NC_015164 | 34 |
| 结核分枝杆菌 (Mycobacterium tuberculosis CDC1551) | NC_002755 | 98 | 巴尔通体杆菌(Bartonella clarridgeiae 73) | NC_014932 | 33 |
| 多形类杆菌 (Bacteroides thetaiotaomicron VPI-5482) | NC_004663 | 84 | 梅毒螺旋体梅毒亚种 (Treponema pallidum subsp. pallidum SS14) | NC_010741 | 32 |
| 鞘脂菌(Sphingobium japonicum UT26S) | NC_014006 | 80 | 梅毒螺旋体 (Treponema paraluiscuniculi Cuniculi A) | NC_015714 | 32 |
| 生丝微菌属(Hyphomicrobium sp. MC1) | NC_015717 | 70 | 鼠疫杆菌(Yersinia pestis CO92) | NC_003143 | 31 |
| 长双歧杆菌(B. longum NCC2705) | NC_004307 | 69 | 台湾贪铜菌 (Cupriavidus taiwanensis LMG 19424) | NC_010528 | 31 |
| 大肠杆菌(E. coli O157:H7 str. Sakai) | NC_002695 | 55 | 噬纤维素菌属 (Cellulophaga algicola DSM 14237) | NC_014934 | 31 |
| 哈维弧菌(V. harveyi ATCC BAA-1116) | NC_009783 | 53 | 结核分枝杆菌(M. tuberculosis H37Rv) | NC_000962 | 30 |
| 溶血葡萄球菌(S. haemolyticus JCSC1435) | NC_007168 | 51 | 沙漠自然球菌(Deinococcus deserti VCD115) | NC_012526 | 30 |
| 缓纤维梭菌 (Clostridium lentocellum DSM 5427) | NC_015275 | 51 | 溃疡拟杆菌(Bacteroides helcogenes P 36-108) | NC_014933 | 27 |
| 表皮葡萄球菌(S. epidermidis RP62A) | NC_002976 | 47 | 金黄色葡萄球菌 (S. aureus subsp. Aureus Mu50) | NC_002758 | 26 |
| 鼠疫杆菌(Y. pestis KIM10+) | NC_004088 | 46 | 金黄色葡萄球菌 (S. aureus subsp. Aureus N315) | NC_002745 | 25 |
| 海单孢菌属 (Marinomonas mediterranea MMB-1) | NC_015276 | 43 | 内脏臭气杆菌 (Odoribacter splanchnicus DSM 20712) | NC_015160 | 25 |
| 红球菌(Rhodococcus jostii RHA1) | NC_008268 | 40 | 嗜热盐碱细菌 (Natranaerobius thermophilus JW/NM-WN-LF) | NC_010718 | 23 |
| 大肠杆菌(E. coli O157:H7 str. EDL933) | NC_002655 | 39 | 圆柱杆菌(Teredinibacter turnerae T7901) | NC_012997 | 23 |
| 固氮密螺旋体(T. azotonutricium ZAS-9) | NC_015577 | 39 | 盐孢菌属(Salinispora tropica CNB-440) | NC_009380 | 20 |
| 白蚁塞巴鲁德氏菌 (Sebaldella termitidis ATCC 33386) | NC_013517 | 38 | 巴西浮霉状菌 (Planctomyces brasiliensis DSM 5305) | NC_015174 | 20 |
| 金黄色葡萄球菌(S. aureus subsp.COL) | NC_002951 | 37 | 苜蓿根瘤菌(Sinorhizobium meliloti AK83) | NC_015590 | 20 |
| 肺炎衣原体 (Chlamydophila pneumoniae AR39) | NC_002179 | 36 |
新窗口打开|下载CSV
2.3 4447个功能未知ORFs被注释上确定的 功能
本研究用同源比对的方法为那些功能未知的ORFs注释上功能。最终,939个基因组中共计4447个ORFs被注释了确切功能(附表6)。其中在33个基因组中,有超过20个假定ORFs被注释上具体功能(表2)。Table 2
表2
表2含有20个以上的假定ORFs被注释上准确功能的基因组的信息
Table 2
| 菌株 | NC序列号 | 数量 | 菌株 | NC序列号 | 数量 |
|---|---|---|---|---|---|
| 大肠杆菌(E. coli O111:H-str. 11128) | NC_013364 | 80 | 炭疽芽孢杆菌(B. anthracis str. CDC 684) | NC_012581 | 23 |
| 肠道沙门氏菌(S. enterica subsp. enterica serovar Paratyphi B str. SPB7) | NC_010102 | 64 | 肠道沙门氏菌(S. enterica subsp. enterica serovar Paratyphi C strain RKS4594) | NC_012125 | 23 |
| 鲍氏志贺菌(Shigella boydii CDC 3083-94) | NC_010658 | 52 | 金黄色葡萄球菌(S. aureus subsp. aureus MW2) | NC_003923 | 23 |
| 肠道沙门氏菌(S. enterica subsp. arizonae serovar 62:z4, z23:- str. RSK2980) | NC_010067 | 45 | 蜡状芽孢杆菌(Bacillus cereus ATCC 10987) | NC_003909 | 22 |
| 枯草芽孢杆菌(B. subtilis subsp. subtilis str. 168) | NC_000964 | 42 | 大肠杆菌(E. coli O26:H11 str. 11368) | NC_013361 | 22 |
| 鼠李糖乳杆菌(Lactobacillus rhamnosus Lc 705) | NC_013199 | 38 | 肺炎链球菌(Streptococcus pneumoniae P1031) | NC_012467 | 22 |
| 鼠李糖乳杆菌(L. rhamnosus GG) | NC_013198 | 36 | 金黄色葡萄球菌(S. aureus subsp. aureus Mu3) | NC_009782 | 21 |
| 肠道沙门氏菌(S. enterica subsp. enterica serovar Paratyphi A str. AKU_12601) | NC_011147 | 35 | 金黄色葡萄球菌(S. aureus subsp. aureus JH9) | NC_009487 | 21 |
| 大肠杆菌(E. coli O157:H7 str. Sakai) | NC_002695 | 34 | 结核分枝杆菌(M. tuberculosis CDC1551) | NC_002755 | 20 |
| 大肠杆菌(E. coli O157:H7 str. EDL933) | NC_002655 | 30 | 炭疽芽孢杆菌(B. anthracis str. A0248) | NC_012659 | 20 |
| 肠道沙门氏菌(S. enterica subsp. enterica serovar Paratyphi A str. ATCC 9150) | NC_006511 | 30 | 大肠杆菌(E. coli O157:H7 str. EC4115) | NC_011353 | 20 |
| 大肠杆菌(E. coli SE11) | NC_011415 | 28 | 炭疽芽孢杆菌(B. anthracis str. Ames) | NC_003997 | 20 |
| 大肠杆菌(E. coli ED1a) | NC_011745 | 27 | 哈维氏弧菌(V. harveyi ATCC BAA-1116) | NC_009783 | 20 |
| 大肠杆菌(E. coli UTI89) | NC_007946 | 26 | 结核分枝杆菌(M. tuberculosis KZN 1435) | NC_012943 | 20 |
| 枯草芽孢杆菌(B. subtilis BSn5) | NC_014976 | 25 | 大肠杆菌(E. coli O55:H7 str. CB9615) | NC_013941 | 20 |
| 肠道沙门氏菌 (S. enterica subsp. enterica serovar Typhi str. Ty2) | NC_004631 | 25 | 牛型分枝杆菌 (Mycobacterium bovis AF2122/97) | NC_002945 | 20 |
| 大肠杆菌(E. coli CFT073) | NC_004431 | 23 |
新窗口打开|下载CSV
这些新注释的功能对生命活动非常重要。例如本研究发现1587个基因组中的601个功能未知的ORFs与一些质膜蛋白基因序列相似。质膜蛋白控制细胞内外物质的交换和信息的传递,参与信号通路的调控[32]。其中沙眼衣原体(C. trachomatis D/UW-3/ CX)中共有5个功能未知的蛋白被注释为质膜蛋白,沙眼衣原体可以引发炎症病变[33],如它与子宫颈癌直接或间接相关。这些被注释上新功能的假定ORFs的详细信息请见附表6。
2.4 对新基因选取宽松阈值和严格阈值进行同源比对的结果
鉴于1.4和1.5部分进行同源比对的阈值不同,研究对模式生物大肠杆菌的代表株E. coli str. K-12 substr. MG1655注释后得到的246个新基因进行了第二次同源比对,选择的阈值为E值 < 1e-20,覆盖率 > 80%以及一致性 > 70%,对两次比对做比较后发现有9个基因满足宽松的阈值(E值 < 1e-20,覆盖率 > 60%,一致性 > 60%),而当设置严格的阈值(E值 < 1e-20,覆盖率 > 80%以及一致性 > 70%)时会被丢失掉。这9个基因的功能和对应值在表3中被列出,这些功能没有出现在E. coli str. K-12substr. MG1655的现有注释中,因此认为它们可能对菌株发挥功能起着不可或缺的作用,而这恰恰是对该菌株注释新基因的意义所在。Table 3
表3
表3大肠杆菌(E.coli str. K-12 substr. MG1655)满足宽松阈值的新基因
Table 3
| 正/负链(在基因组上位置) | 同源序列来源 | 功能 | E值 | 覆盖度 (%) | 一致性 (%) |
|---|---|---|---|---|---|
| 负链(190551~191603) | 肠道沙门菌 (S. enterica subsp. enterica) | 亮氨酸操纵子先导肽 (leu operon leader peptide) | 8e-139 | 79 | 74.01 |
| 负链(2228549~2228758) | 志贺氏菌属(Shigella) | 多药耐药外膜蛋白MdtQ (multidrug resistance outer membrane protein MdtQ) | 1e-27 | 72 | 100 |
| 负链(4322661~4323281) | 志贺氏菌属(Shigella) | Pn转运体膜通道蛋白组分(membrane channel protein component of Pn transporter) | 4e-108 | 77 | 98.74 |
| 负链(4500432~4500791) | 猪布鲁氏杆菌(Brucella suis) | 磷酸乙醇胺转移酶(MULTISPECIES: phosphoethanolamine transferase) | 3e-45 | 69 | 91.57 |
| 正链(1465410~1467950) | 宋内志贺菌(Shigella sonnei) | 包含蛋白质的自转运体结构域 (autotransporter domain-containing protein) | 0.0 | 74 | 87.03 |
| 正链(1470858~1474013) | 福氏志贺氏菌 (Shigella flexneri) | 自转运体外膜β管(MULTISPECIES: autotransporter outer membrane beta-barrel) | 0.0 | 76 | 100 |
| 正链(2070501~2071211) | 双歧杆菌 (Bifidobacterium longum) | GTPase家族蛋白(GTPase family protein) | 4e-129 | 77 | 99.45 |
| 正链(3993850~3994335) | 痢疾志贺氏菌 (Shigella dysenteriae 1617) | CyaX蛋白(CyaX protein) | 9e-67 | 63 | 96.12 |
| 负链(4506626~4506883) | 红树杆菌属 (Mangrovibacter plantisponsor) | 表皮粘着蛋白E (surface-adhesin protein E) | 1e-28 | 98 | 60 |
新窗口打开|下载CSV
2.5 9个基因组中新识别出2003个新基因
ZCURVE 3.0具有93.7%的准确率[8],Glimmer 3.02具有93.0%的准确率[20],这两个软件和假阳性率较低的Prodigal[23]被一起用来识别漏注释的基因,预测出的新的编码ORFs形成的并集被用来同源比对。最终,在9个基因组中识别到了2003个新基因(表4,附表7)。在最初的注释中基因数量相对较少的酿脓链球菌和流感嗜血杆菌分别得到了104和123个新基因,新基因的数量大约是原始基因数目的6%。脑膜炎奈瑟球菌和肠道沙门氏菌获得了很多新注释到的基因,它们占据了其原始基因数目的14%。这些被新识别的基因的名字和其起始位置的信息存放在附表7中。Table 4
表4
表49个菌株的名称、NC序列号、基因组大小、基因总数和新注释基因的数目
Table 4
| 菌株 | NC序列号 | 基因组大小(bp) | 基因总数 | 新基因的数目 |
|---|---|---|---|---|
| 枯草芽孢杆菌(B. subtilis subsp. subtilis str. 168) | NC_000964 | 4215606 | 4175 | 52 |
| 金黄色葡萄球菌(S. aureus subsp. aureus NCTC 8325) | NC_007795 | 2821361 | 2767 | 61 |
| 酿脓链球菌(S. pyogenes SF370) | NC_002737 | 1852441 | 1696 | 104 |
| 流感嗜血杆菌(H. influenzae Rd KW20) | NC_000907 | 1830138 | 1610 | 123 |
| 嗜酸氧化亚铁硫杆菌(A. ferrooxidans ATCC 23270) | NC_011761 | 2982397 | 3147 | 143 |
| 大肠杆菌(E. coli str. K-12 substr. MG1655) | NC_000913 | 4641652 | 4140 | 246 |
| 脑膜炎奈瑟球菌(N. meningitidis MC58) | NC_003112 | 2272360 | 1953 | 279 |
| 炭疽芽孢杆菌(B. anthracis str. Ames) | NC_003997 | 5227293 | 5039 | 418 |
| 肠道沙门氏菌(S. enterica subsp. enterica serovar Typhi str. CT18) | NC_003198 | 4809037 | 4111 | 577 |
新窗口打开|下载CSV
注释同源蛋白簇对研究具有相似功能的基因帮助颇多。通过综合EggNOG[28]、WebMGA[29]和COG数据库[30],共有1073个新识别的基因被注释到对应的COGs(图4),其丰度就是某蛋白质直系同源簇所包含的新识别的基因占全部新识别的基因的比例。这1073条序列的详细COG注释可以在附表7中查询到,而所有1587个基因组重注释的具体信息则可以在附表8中被查到,该表列出的具体信息包含其基本信息即NC_ID和基因组大小(bp),还包含其原始注释信息即蛋白质数目、功能已知的基因和假定基因,并列出了通过重注释得到的注释信息即假定ORFs中被注释上新功能的ORFs数目和非编码ORFs的数目,还提供了9个基因组新基因的数量,并提供了通过Z-curve 33变量方法识别非编码ORFs的准确率。
图4
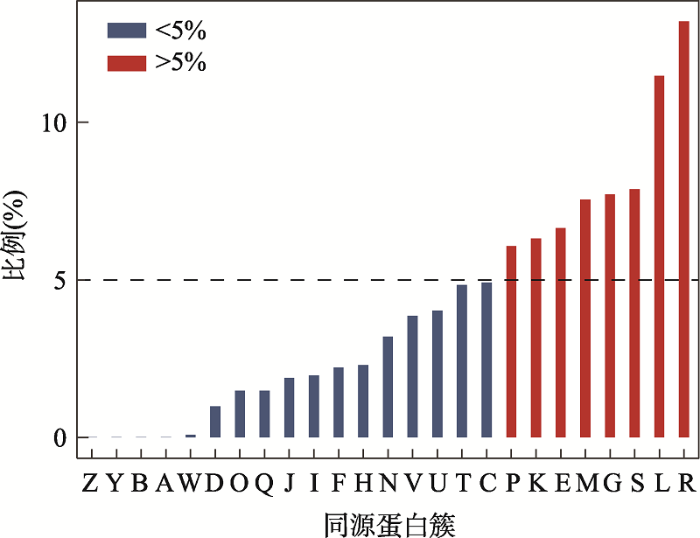 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4特定同源簇对应的新基因占全部新基因的比例
A:RNA加工和修饰;B:染色质结构和动力学;Y:核结构;Z:细胞骨架;W:细胞外结构;D:细胞周期控制,细胞分裂,染色体分裂;O:翻译后修饰,蛋白反转,伴侣;Q:次生代谢产物的生物合成、运输和分解代谢;I:脂质转运与代谢;J:翻译,核糖体结构和生物发生;H:辅酶运输与代谢;F:核苷酸转运与代谢;V:防卫机制;N:细胞迁移;C:能量产生和转化;U:细胞内传输,分泌和囊泡转运;P:无机离子转运与代谢;T:信号转导机制;K:转录;E:氨基酸转运和代谢;M:细胞壁和细胞膜的生物发生;G:碳水化合物运输和代谢;S:功能未知;R:只能预测大致功能;L:复制重组和修复。
Fig. 4The ratio of the newly recognized genes of certain COG to all new genes
3 讨论
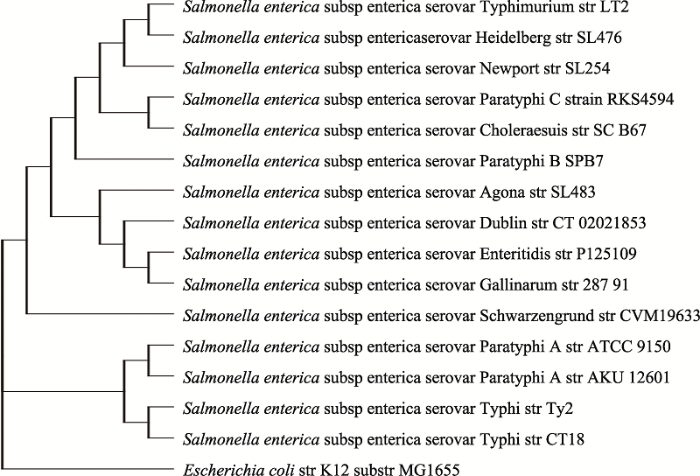
本研究结合3种原核基因组注释软件和表达数据重注释了1587个细菌和古细菌的基因组。首先,识别了过度注释为基因的序列,隶属于177个基因组的3092个假定ORFs被识别为非蛋白编码序列;接下来,还赋予了假定ORFs以功能,939个基因组的4447个基因被通过相似性搜索而注释获得准确功能,并且在9个基因组中识别了2003个属于多个蛋白质直系同源簇的新基因。重注释可以保证进一步分析的数据的准确性。在真核生物中有研究表明多拷贝的基因会出现功能的分化[34],原核生物也存在多拷贝的基因,它们是否也存在这种分化,可以通过开展重注释后获得准确的信息来进一步研究。而正如前文所提及的那样,伴随着测序数据的增加和技术的不断进步,对已测序菌株的基因组进行重新注释的必要性也在增大,同时根据Breitwieser的研究[4],微生物基因组可能会被人类基因组所污染而导致基因的丢失,这就更加要求研究者们用多种方法来更新注释。本次进行重新注释的1587个菌株可归为992个物种,每个物种涵盖多种血清型,例如沙门氏菌根据抗原的不同可以分为不同的血清型[35],其基因组层面可能会存在差异。Liu等[33]通过全基因组序列对6个沙门氏菌(Salmonella)菌株建立进化树显示S. enterica亚种的相同血清型的S. paratyphi C和S. paratyphi A进化距离较远,却与不同血清型的S. choleraesuis进化距离较近。研究通过CVTree[31]对15个S. enterica亚种构建进化树发现血清型之间会呈现不同的聚集,与Liu等[33]绘制的进化树一致(附图1)。因此,通过重注释不同血清型的菌株来使得基因组信息更完善,从而帮助深入探索血清型之间更准确的演化关系。而研究参考的数据集为1587个基因组中已经被注释为具有明确功能的ORFs,为了验证这些基因具有表达数据,针对大肠杆菌的功能已知的基因进行验证,研究下载了对应该物种的表达数据并寻找这些功能已知的基因中有多少具有表达数据,通过与GEO数据库中GSE56133[36]和GSE118058[37]的表达数据比较,发现2835个功能已知的基因中有2806个在这两个数据集中有表达数据,这进一步证实了现有明确功能注释的基因可以作为参考集来进行预测。
得益于众多基因识别软件的升级如ZCURVE 3.0[8]和不断更新的公共数据库如NCBI的GEO[24],使得寻找新的编码蛋白ORFs和用表达数据验证成为可能。在识别新的基因方面,本研究联合ZCURVE 3.0[8]、Glimmer 3.02[20]和Prodigal[23]对原核基因组重新注释,其中Glimmer 3.02是基于隐马尔可夫模型,识别准确率高达93.0%,但是它对序列的局部特征有着强烈的依赖性,而Prodigal是通过对现有的细菌和古细菌基因组注释信息进行训练,因此对已知基因/保守基因的识别效果优于其余的软件,但预测未报道的基因时会有些许偏差。而ZCURVE 3.0是基于DNA序列的全局统计特征,它将Fisher线性判别替换为支持向量机(SVM)[38]以提高灵敏度,并且鉴于在预测过程中由于负样本的随机性容易产生假阳性的基因,ZCURVE 3.0依据了ORFs核酸分布的花瓣模型[39]在训练集中产生5类负样本并逐次分类,然后保留在多次分类中均被预测为基因的那些ORFs,并且算法还把33个包含零阶和一阶的Z曲线变量增加为额外包含二阶和三阶Z曲线变量的765个变量,可以最大化地优化程序的预测效果。选取三个程序混合预测更能保证预测的准确率。
总之,本研究基于序列的全局特征和局部特征,结合同源比对对多个物种的多种菌株的基因组进行了全面的重新注释。未来,伴随着基因组多组学数据的增加以及相应的注释工具和数据库的完善[40,41,42,43],公共数据库里基因组的注释将会更加准确。
致谢
感谢电子科技大学生命科学与技术学院的郭锋彪教授在研究开展中给予的帮助。
附录:
附图和附表详见文章电子版附图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图1S. enterica亚种不同血清型的系统发育树
Supplementary Fig. 1 The phylogenetic tree of different serological types of S. enterica sub-species
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 7]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 5]
[本文引用: 1]
[本文引用: 1]
[本文引用: 4]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 3]
[本文引用: 3]
[本文引用: 2]
[本文引用: 1]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}