 ,1
,1A method for reliable detection of genomic point mutations based on single-cell target-sequencing
Linan Zhao1, Na Wang1, Guoliang Yang2, Xianbin Su1, Zeguang Han,1通讯作者: 韩泽广,教授,研究方向:肿瘤(肝癌)发病机制研究和单细胞测序。E-mail:hanzg@sjtu.edu.cn
编委: 王晓群
收稿日期:2020-04-26修回日期:2020-06-2网络出版日期:2020-07-20
| 基金资助: |
Editorial board:
Received:2020-04-26Revised:2020-06-2Online:2020-07-20
| Fund supported: |
作者简介 About authors
赵利楠,在读硕士研究生,专业方向:肝癌单细胞测序。E-mail:

摘要
分析基因碱基突变是探究肿瘤细胞克隆演化的研究方法之一。目前,常取组织不同区域的群体细胞测序,基于群体水平的基因碱基突变频率等信息绘制出肿瘤的克隆演化过程。但是,该方法易遗失低频突变,并且组织群体细胞类型不单一,不能代表特定细胞群体的克隆演化过程。本研究以前列腺基底细胞癌(prostate basal cell carcinoma, BCC)为例,建立了一种探究肿瘤单细胞的基因碱基突变方法,实现了基于单细胞基因碱基突变分析肿瘤细胞的克隆演化过程。首先通过HepG2细胞优化了单细胞全基因组扩增方法,其次用Fluidigm公司的微流控芯片捕获BCC单细胞进行全基因组扩增,然后通过外显子组测序获得SCUBE3和MST1L基因碱基突变信息。通过单细胞靶向扩增和Sanger测序,成功在BCC单细胞中检测到SCUBE3和MST1L基因碱基突变信息。本研究建立的方法为肿瘤细胞的克隆演化研究提供了一种可靠的技术方案。
关键词:
Abstract
The analysis of genomic point mutations is one of the research strategies to explore the clonal evolution of tumor cells. At present, clonal evolution of tumor cells is mainly determined by bulk sampling and sequencing of different sections of the tumor. Since this approach analyzes a mixture of different cell types, it may not accurately represent the clonal evolution of specific tumor cell populations and likely miss low frequency mutations, especially when the sequencing depths are not sufficient. To address this issue, we have developed a strategy to analyze genomic point mutations from prostate basal cell carcinoma (BCC) tissues at single-cell resolution. Firstly, we optimized the single-cell whole genome amplification procedure with HepG2 cells. Then the single cells from BCC tissue were captured by a microfluidic chip of Fluidigm and processed for whole-genome amplification. Both SCUBE3 and MST1L genomic mutations were obtained by whole exome sequencing. Finally, we examined the genomic mutations through single-cell targeted amplification and Sanger sequencing. The established method successfully reconfirmed the mutations of SCUBE3 and MST1L in BCC at single cell level. The strategy established in this study could provide a useful tool for determining the clonal evolution of tumor cells based on genomic mutations at single-cell resolution.
Keywords:
PDF (1513KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
赵利楠, 王娜, 杨国良, 苏现斌, 韩泽广. 基于单细胞靶向测序探究基因碱基突变的方法. 遗传[J], 2020, 42(7): 703-712 doi:10.16288/j.yczz.20-046
Linan Zhao.
肿瘤组织内部除了含有血管、基质细胞和浸润的免疫细胞外,还包含具有不同遗传变异的肿瘤细胞,故而肿瘤是一种高度异质性的疾病[1,2]。此外,肿瘤在形成过程中,其内部的肿瘤细胞因含有不同的突变组合而形成不同的亚克隆群体,且克隆群体会不断进化以适应环境[3]。遗传异质性结合表观遗传异质性等因素会引起肿瘤细胞的表型异质性,进而形成了不同肿瘤细胞对治疗的反应或本身的转移能力不同,最终导致抗药性或复发[4]。为此,揭示肿瘤内部的克隆结构演变规律,将有助于理解其发生发展机制,为今后癌症患者的个性化治疗提供理论基础。
目前,研究肿瘤组织内部细胞克隆结构演化的方法之一是对不同区域取样进行群体水平的外显子测序,通过检测基因突变的发生频率(variant allele frequency, VAF)从而探索肿瘤细胞的发生发展过程[5,6]。该方法存在的不足之处在于,群体细胞中不仅含有肿瘤细胞,而且同时混有其他类型细胞,这很容易导致肿瘤细胞中特有的所占比重较少的突变信息被掩盖[7]。单细胞全外显子测序可以有效解决这一问题,但考虑到测序成本问题一般难以大规模应用,部分研究者以单细胞靶向测序作为替代方案[8,9,10]。
利用单细胞靶向测序技术,本研究建立了一种优化的有效靶向分析大规模单细胞中特定碱基突变的方法。通过结合组织酶解方法、微流控单细胞分离、多重置换扩增(multiple displacement amplification, MDA)、外显子组测序和单细胞靶向扩增技术组建了探究肿瘤单细胞水平基因碱基突变的方法。首先以HepG2细胞为例优化MDA技术,并将建立的方法应用到一例人类前列腺基底细胞癌(prostate basal cell carcinoma, BCC)患者的肿瘤样本,通过对混合单细胞样品全外显子测序结果分析找到BCC肿瘤细胞中存在的克隆碱基突变点,并在单细胞水平上通过靶向扩增及Sanger测序验证该突变的存在,从而为绘制肿瘤细胞克隆演化过程奠定基础(图1)。
图1
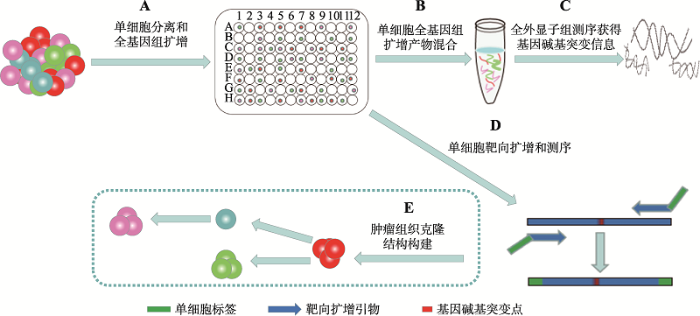 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1单细胞水平靶向检测肿瘤基因碱基突变的技术流程
A:肿瘤组织单细胞分离和单细胞全基因组扩增;B:取等体积肿瘤单细胞基因组放大产物获得混合样品;C:混合基因组产物进行全外显子测序经生物信息分析获得基因碱基突变列表;D:选取目的基因并根据碱基突变位点设计引物,以肿瘤单细胞基因组产物为模板进行靶向扩增和测序;E:基于单细胞碱基突变信息绘制肿瘤细胞的克隆演化过程。
Fig. 1Workflow of targeted detection of genomic mutations in tumor cells at single-cell resolution
1 材料与方法
1.1 实验材料
HepG2肝细胞癌细胞系来自ATCC。人类前列腺基底细胞癌(prostate basal cell carcinoma, BCC)样本来自上海交通大学医学院附属仁济医院。1.2 单细胞悬液的制备
组织样本用剪刀剪碎,加入胶原酶IV于37℃孵育30~120 min,经70 μm滤膜除去消化不完全组织和裂解红细胞后以100 g离心5 min,再进行洗涤重悬获得单细胞悬液。制备的单细胞悬液经计数及细胞直径测量后用于后续的单细胞分离、捕获、基因组扩增、生物信息分析和靶基因扩增及验证等。1.3 单细胞捕获及全基因组扩增
单细胞由美国Fluidigm公司的C1芯片捕获,单次最多可捕获96个单细胞。单个细胞的DNA不易于目的基因的扩增,采用MDA技术对芯片捕获到的单细胞进行全基因组放大可以有效用于靶向扩增。1.4 外显子组测序与基因碱基突变分析
C1芯片捕获单个BCC细胞后,首先完成单细胞的全基因组放大,再分别吸取2 μL单细胞的基因组放大产物进行混合,可获得单细胞水平的群体基因产物样本。将捕获到的BCC单细胞全基因组扩增产物混合样品进行外显子组测序(whole exome sequencing, WES) (测序深度~200×)[11],且测序数据采用本课题组构建的分析流程处理获得基因突变列表[12]。测序数据已上传至NCBI SRA数据库,登录号:PRJNA607168,https://www.ncbi.nlm.nih.gov/sra/ PRJNA607168。1.5 引物序列与合成
利用Primer3网站设计引物,用于扩增检测SCUBC3基因第35213742位碱基和MST1L基因第17084321位碱基的突变情况。引物设计原则是:在碱基突变位点的上下游约100 bp设计引物。采用Primer Premier 5.0软件设计ACTB基因的实时荧光定量PCR (quantitative real time PCR, q-PCR)引物。本文所使用引物的相关信息如表1所示。Table 1
表1
表1引物序列信息
Table 1
| 引物名称 | 引物序列(5ʹ→3ʹ) | 扩增产物长度(bp) | 用途 |
|---|---|---|---|
| SCUBE3 | F:AGGGTGTGTGTATGCGAAGG | 253 | 碱基突变验证 |
| R:GGCCTCGCTTGTCTTGAAGT | |||
| MST1L | F:TTACCCGTACCTGCAGTGAG | 287 | 碱基突变验证 |
| R:GACAACCTGGGAGGATGTGA | |||
| ACTB | F:TCTCGCAGCTCACCATGGAT | 363 | 内参基因 |
| R:TGCTCGATGGGGTACTTCAG |
新窗口打开|下载CSV
1.6 目的基因靶向扩增与测序分析
通过对混合样品的全外显子测序数据分析获得突变基因列表信息,选取目的基因SCUBC3和MST1L进行单细胞靶向扩增与测序分析验证。以BCC单细胞基因组扩增产物为模板,用设计的基因引物分别扩增目的片段即包含SCUBC3基因第35213742位碱基和MST1L基因第17084321位碱基。根据琼脂糖凝胶电泳的结果判断扩增效果,然后将每3个单细胞的目的基因扩增产物混合一起,进行Sanger测序。通过将测序片段序列与目的基因的匹配参考片段序列比对,若发现目的基因的特定位点碱基发生突变,则分别将该样品的3个单细胞靶向扩增产物送去Sanger测序,最终确定基因碱基突变具体发生在哪些单细胞中。
1.7 统计分析
数据分析均由 GraphPad Prism 6.0 软件完成统计并作图。2 结果与分析
2.1 单细胞全基因组扩增条件优化及验证
2.1.1 单细胞全基因组扩增条件优化MDA技术是一种进行单细胞全基因组扩增放大的方法,它能对全基因组进行高保真的均匀扩增,扩增出10~100 kb大小的片段,常用于单碱基变异(single nucleotide variation, SNV)分析研究肿瘤克隆结构[13,14,15]。但是MDA技术也有一些缺点,特别是显著的非特异扩增,空白对照也会产生核酸产物。
此前有报道通过紫外光照射可以有效消除试剂及外界环境中的外源核酸污染[16,17],为确定最适且有效降低外源核酸污染的紫外光照射时间,本研究以试剂盒中gDNA (人基因组DNA)为扩增模板即实验组,等体积纯净水为空白对照组进行MDA技术优化实验。通过紫外光照射MDA试剂0 min、15 min、30 min、60 min探索减少非特异性核酸扩增的最佳条件。结果显示,实验组的核酸扩增产物浓度远远大于空白对照组(图2A),并且当用紫外光照射MDA相关试剂30 min时,可以有效显著地减少非特异性核酸扩增(图2B)。
图2
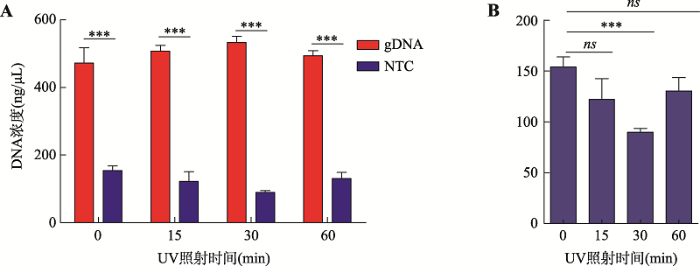 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2通过紫外照射降低外源核酸污染对MDA扩增条件进行优化
A:MDA扩增结果,gDNA表示人基因组DNA为模板,NTC表示以等体积水为模板;B:紫外光(UV)照射MDA试剂不同时间段非特异性扩增结果。*** P<0.001;ns:表明无显著差异。
Fig. 2Optimization of MDA by UV exposure to reduce contaminating nucleic acids
2.1.2 HepG2单细胞全基因组扩增
通过对HepG2单细胞全基因组扩增验证优化条件的准确性,首先将HepG2细胞制备成单细胞悬液,并以166~250 cells/μL的浓度加入C1芯片进行单细胞捕获。单次C1芯片具有96个单细胞捕获小室,可制备96个单细胞。为避免C1芯片细胞小室捕获到多个细胞,利用较低的起始细胞密度,并通过显微镜观察确认每个小室捕获到单个细胞,本次共捕获到47个HepG2单细胞。
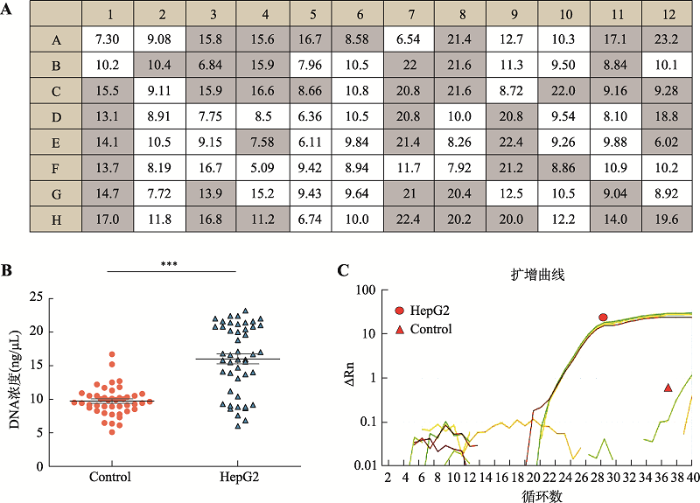
随后将经紫外光照射30 min后的MDA试剂输入C1芯片的单细胞捕获小室进行核酸扩增,利用Qubit测得每个单细胞捕获小室的核酸扩增产物浓度(图3A)。扩增结果显示捕获到单细胞的核酸扩增产物浓度(有细胞组)显著性高于无单细胞存在的核酸扩增产物浓度(无细胞组) (图3B)。本研究选取人类内参基因ACTB设计引物,利用 q-PCR对有细胞组和无细胞组ACTB基因的核酸模板量进行分析。扩增曲线结果显示,无细胞组的q-PCR反应结束后无扩增曲线出现,可能是由于无细胞组的核酸浓度偏低或者模板为降解的核酸片段导致(图3C)。以上实验结果证实,本研究优化的MDA方法可以显著的降低外源核酸的干扰,并对单细胞基因组进行有效的扩增。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3HepG2单细胞全基因组放大结果
A:HepG2单细胞全基因组扩增产物核酸浓度(ng/μL),灰色背景:有细胞(47个孔),白色背景:无细胞(49个孔);B:有细胞组(HepG2)与无细胞组(Control)核酸浓度比较;C:ACTB基因q-PCR扩增曲线图结果展示。*** P<0.001。
Fig. 3Single-cell whole genome amplification of HepG2
2.2 BCC单细胞的全基因组扩增和碱基突变分析
2.2.1 BCC单细胞捕获及全基因组扩增将计划进行单细胞分析的BCC组织剪碎,用胶原酶 IV 处理并通过膜过滤、离心及细胞重悬获得单细胞悬液(图4A)。利用C1芯片同样以166~ 250 cells/μL的浓度对细胞悬液进行单细胞捕获(图4B)。本次捕获到53个BCC单细胞,且在显微镜下观看Fluidigm C1芯片捕获到的单细胞大部分形态正常,加入紫外光照射处理的MDA试剂进行单细胞全基因组扩增,保证有效减少非特异性核酸扩增的干扰。最后通过Qubit测得每个单细胞的基因组扩增产物浓度,结果显示捕获到BCC单细胞的基因组被成功扩增放大(图4C),而扩增产物浓度的差异可能由于裂解或扩增效率不同造成。
图4
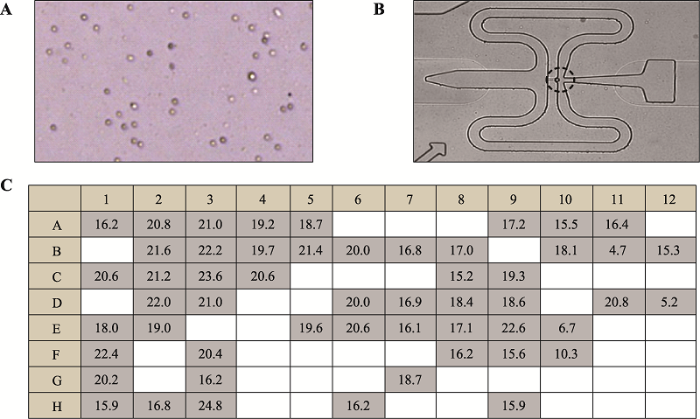 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4BCC单细胞全基因组扩增
A:BCC单细胞悬液;B:C1芯片捕获BCC单细胞;C:BCC单细胞全基因组扩增产物浓度(ng/μL)。
Fig. 4Single-cell whole genome amplification of BCC
2.2.2 BCC外显子组测序及生物信息分析
分别吸取扩增的BCC单细胞全基因组扩增产物各2 μL混合,并开展外显子组测序(测序深度~ 200×)[11]。测序数据用本课题组构建的成熟分析流程处理,以Burrows-WheelerAligner (BWA) 进行序列比对,以Genome Analysis Toolkit (GATK)检测遗传变异,以 SnpEff 对遗传变异进行注释,最后获得突变基因列表[18,19,20]。通过生物信息分析发现SCUBE3和MST1L基因均发生碱基突变(表2),且SCUBE3测序深度为281×,MST1L测序深度为493×。
Table 2
表2
表2基因突变信息
Table 2
| 基因名称 | 染色体 | 突变位点 | 参考碱基 | 突变碱基 | 突变发生频率(VAF) |
|---|---|---|---|---|---|
| SCUBE3 | 6 | 35213742 | C | T | 0.302491103 |
| MST1L | 1 | 17084321 | A | G | 0.170385396 |
新窗口打开|下载CSV
2.2.3 靶向扩增及Sanger测序验证
为了在单细胞水平进一步验证混合样品外显子组测序发现的基因碱基突变点,分别以每个BCC单细胞全基因组为模板,利用PCR对SCUBE3和MST1L基因进行引物扩增,电泳结果显示分别成功扩增出43个和44个BCC单细胞的SCUBE3和MST1L基因目的条带(图5A,图6A)。
按顺序将扩增产物每3个样品取等体积混合一起作为一个样品进行Sanger测序。测序结果显示SCUBE3基因扩增产物的第5个混合样品中SCUBE3基因第35213742位碱基有较高的C→T突变,但第6个混合样品的相同位点没有突变发生(图5B)。为了进一步在单细胞水平验证,分别将混合样品的3个单细胞目的片段扩增产物进行Sanger测序,测序图谱显示第5个混合样品的第2个BCC单细胞(4A)发生C→T突变,而第6个混合样品的第1个BCC单细胞(4C)SCUBE3基因第35213742位碱基未发生碱基突变(图5C)。
同时,本研究也检测到MST1L基因扩增产物的第6个混合样品中MST1L基因第17084321位碱基发生A→G突变,但第4个混合样品的相同位点没有突变发生(图6B)。单细胞测序结果显示,第6个混合样品的第3个BCC单细胞(5A)发生A→G突变,而第4个混合样品的第1个BCC单细胞(3A)未发生碱基突变(图6C)。
为了排除本文检测到的基因碱基突变由基因靶向扩增和Sanger测序过程引入,进一步观察其他两个单细胞的目的基因碱基突变情况,发现SCUBE3基因在Mix-5-1-SCUBE3细胞,MST1L基因在Mix-6-MST1L其他两个单细胞中的特定位点碱基也分别发生相同突变,但它们相互对照无突变组的其他两个单细胞没有发生突变(附图1,附图2)。同时,考虑到基因靶向扩增和Sanger测序过程引入碱基突变是随机的,因此同时在多个单细胞中同时引入特定位点相同碱基突变的可能性不大。以上结果说明基于单细胞水平靶向扩增与测序,本研究建立的方法可以有效检测肿瘤单细胞水平的基因碱基突变。
图5
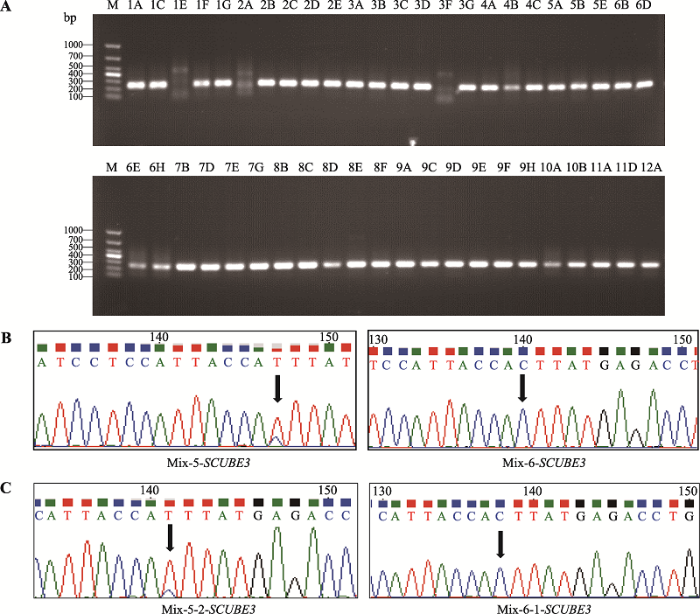 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5SCUBE3基因靶向扩增结果和Sanger测序图谱
A:SCUBE3基因靶向扩增产物结果。泳道1A~12A:BCC单细胞96孔板位置编号;M:DL1000 marker。B、C:SCUBE3基因靶向扩增产物Sanger测序图谱。Mix-X-Y-基因:Mix代
Fig. 5Target amplification and Sanger sequencing of SCUBE3 gene
3 讨论
肿瘤内部的高度异质性是导致高致死率和耐药性的一个关键因素[21]。因此,在单细胞水平探究肿瘤内部的克隆演化可以有效地帮助人们了解肿瘤的发生发展,为肿瘤的早期诊断和干预提供理论基础。随着单细胞测序技术的出现,在单细胞水平分析肿瘤的演变过程可以提高人们对肿瘤内部细胞克隆亚群的了解,但测序成本限制了单细胞测序技术的大规模应用。针对上述问题,本研究基于单细胞靶向扩增和测序技术建立探究肿瘤单细胞水平基因碱基突变的方法,其优点在于:(1)可以减少研究样本中低频突变的损失;(2)可以靶向分析大规模单细胞中特定点突变;(3)有效降低外源核酸非特异性扩增产物的干扰。目前,单细胞全基因组扩增有多种方法,MDA由于使用的Phi29聚合酶具有3ʹ→5ʹ的外切酶活性及校正功能,是所有扩增方法中公认的保真性最好和最适合进行点突变检测的方法[22,23,24,25]。本研究所使用的人类BCC单细胞MDA扩增产物的混合全外显子组测序分析显示突变的VAF数值在0.5左右波动[11],说明MDA过程中不太可能通过非特异扩增丢失关键的突变,但有可能影响细胞突变频率低的突变。同时,本研究展示的BCC单细胞SCUBE3基因和MST1L基因的Sanger测序图谱中除突变点外的其他位点可作为技术对照,证明MDA扩增过程没有引入非特异的假阳性突变。此外,已有研究表明紫外光照射60 min可以有效降低MDA扩增试剂中残留DNA的污染[26],本研究对MDA的条件进行优化,实验结果提示紫外光照射MDA试剂30 min为最佳条件。分析实验结果产生差异的原因可能是:(1)不同批次单细胞扩增试剂中残留的DNA污染量不同。Woyke 等[26]以E. coli DNA进行条件优化,而本研究以illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare, 25660031)中自带gDNA (人基因组DNA)进行条件优化;(2)实验操作流程或者环境中的DNA污染浓度不同。本研究以肝癌细胞系HepG2为例进一步验证了MDA优化条件的可靠性,结果依旧支持本文所得结论,即紫外光照射MDA试剂30 min可以有效减少外源核酸非特异性扩增的影响(图3B)。以上结果提示,紫外光照射MDA试剂主要是为了去除扩增试剂中或环境中的DNA污染,与单细胞类型关系不大。
图6
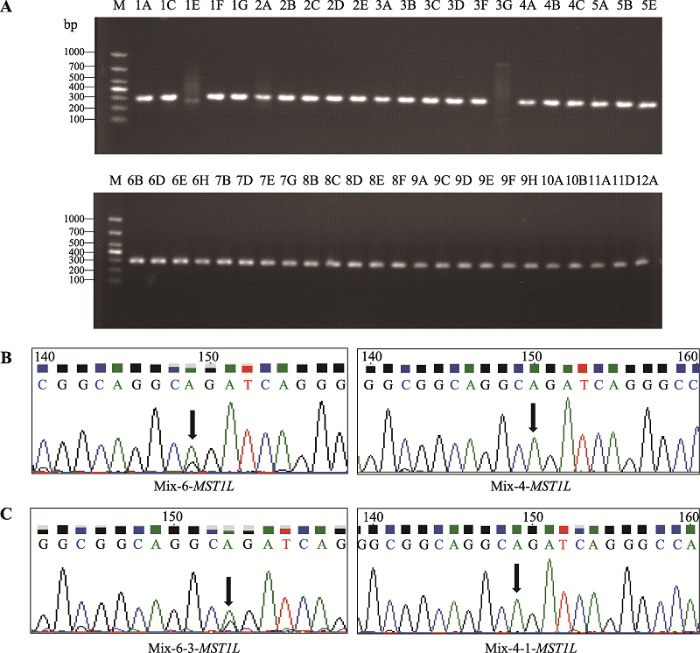 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6MST1L基因靶向扩增结果和Sanger测序图谱
A:MST1L基因靶向扩增结果,泳道1A~12A:BCC单细胞96孔板位置编号,M:DL1000 marker;B~C:MST1L基因靶向扩增产物Sanger测序图谱,Mix-X-Y-基因:Mix代
Fig. 6Target amplification and Sanger sequencing map of MST1L gene
本研究建立的基于微流控芯片MDA技术、全外显子测序和Sanger测序技术相结合的方法可以有效针对肿瘤单细胞进行基因点突变检测,有助于肿瘤异质性问题的研究。另外,本研究所建立的方法可以结合流式分选等技术进行细胞类型挑选(肿瘤细胞、肿瘤干/祖细胞等),可以有效地避免因其他类型细胞的干扰降低对低频突变的检测,有利于对特定细胞群体的克隆演化过程进行探索。此外,该方法可以通过扩大样本量,例如设计带有细胞标签的靶向扩增引物并结合第二代测序技术,实现高通量的靶向分析大规模单细胞中的特定点突变以达到对肿瘤克隆结构进行重构的目的,为肿瘤克隆演化的研究提供了切实可行的方案。
附录:
附图详见文章电子版1、SCUBE3: Chr.6:35,213,612~35,213,872 Chr.6:35213742 C→T
AGGGTGTGTGTATGCGAAGGgggagcctttgtcagaatgattctcttgccctatacccccaccaccagctgtacagcccacgttctaggagggcttgggtagcctgccctgctgctctactgacctgctgcttgccttcccagcatccccatcctccattaccaCttatgagacctgccagacctacgagcgtcccattgccttcactgcccgttccaggaagctctggatcaACTTCAAGACAAGCGAGGCC
2、MST1L: Chr.1:17,084,143~17,084,429 Chr.1:17084321 A→G
TTACCCGTACCTGCAGTGAGgggaatggggagaaggagacggtcctggaggaagatccagggctgggcctcctggccaccagcagtcctgtgcactatgctcttacctttggtctcaccccagcctgcaatctcacacttggtccctgga
ggcaccacataccattcaggcggcaggcAgatcagggccacacgctggttcagggtcacagatctttaacaagaatgggggcactcagggtctgaggccacaaggctcagccccaccTCACATCCTCCCAGGTTGTC
附图1
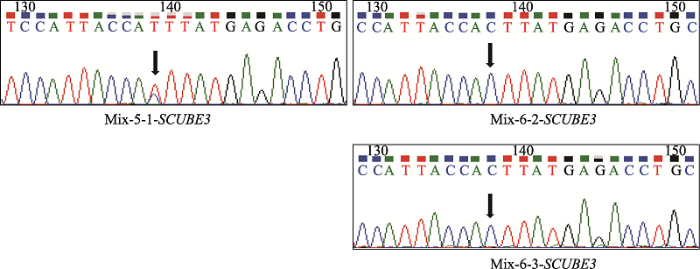 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图1SCUBE3基因靶向扩增结果和Sanger测序图谱
SCUBE3基因靶向扩增产物Sanger测序图谱,Mix-X-Y-基因:Mix代
Supplemental Fig. 1Target Amplification and Sanger sequencing of SCUBE3 gene
附图2
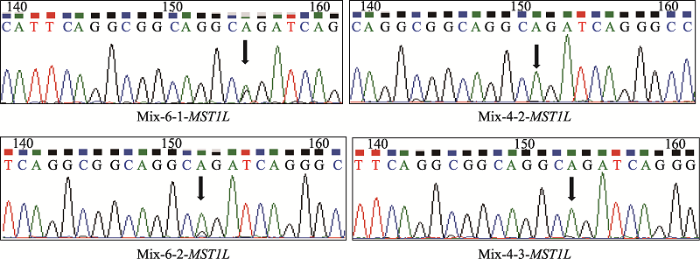 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图2MST1L基因靶向扩增结果和Sanger测序图谱
MST1L基因靶向扩增产物Sanger测序图谱,Mix-X-Y-基因:Mix代
Supplemental Fig. 2Target Amplification and Sanger sequencing map of MST1L gene
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}