 ,1,2,3, 闫世雄4, 孙帅4, 贾俊静4, 胡晓湘,1,2
,1,2,3, 闫世雄4, 孙帅4, 贾俊静4, 胡晓湘,1,2Metabolomics technology and its applications in agricultural animal and plant research
Jing Tian1,2, Yuzhe Wang,1,2,3, Shixiong Yan4, Shuai Sun4, Junjing Jia4, Xiaoxiang Hu,1,2通讯作者: 王宇哲,博士,博士后,研究方向:动物数量遗传与功能基因组。E-mail:yuzhe891@163.com胡晓湘,博士,教授,研究方向:动物遗传育种。E-mail:huxx@cau.edu.cn
责任编辑: 李明洲
收稿日期:2019-11-4修回日期:2020-03-13网络出版日期:2020-05-20
| 基金资助: |
Editor:
Received:2019-11-4Revised:2020-03-13Online:2020-05-20
| Fund supported: |
作者简介 About authors
田菁,在读博士研究生,专业方向:动物遗传育种。E-mail:18604841591@163.com。

摘要
代谢组学是依赖灵敏、稳定的分析流程和数据库,利用色谱-质谱联用、核磁共振技术对生物体内以及生物样品所有的小分子代谢物进行鉴定和定量分析的学科,在医学、食品科学、畜牧学、植物学等领域得到广泛应用。代谢组学方法可将代谢物种类和含量的变化与生物表型变化建立更直接的联系,因此代谢组学逐渐成为继基因组学、转录组学、蛋白组学后对复杂性状系统解析的新的研究手段。本文介绍了代谢组学常用分析策略、检测平台和常用数据库。在此基础上,综述了代谢组学在农业动物重要经济性状代谢分子鉴定、疾病诊断、肉品质及动物制品安全检测等领域取得的进展,并总结了利用代谢组学、转录组学和基因组学等多组学研究在动植物重要性状的发育、形成和解析等领域取得的最新成果。代谢组学与其他多组学方法整合分析,可以更全面地阐述各类复杂性状的遗传机制,有助于完善“突变-基因-表达-代谢-表型”的完整生物学过程,为复杂性状的机理解析提供新方法,为新型农业育种提供新思路。
关键词:
Abstract
Metabolomics is a discipline that uses Chromatography-Mass Spectrometry and Nuclear Magnetic Resonance techniques to identify and quantify all small molecule metabolites in living organisms and biological samples, which relies on sensitive, stable analytical procedures and improving databases. Metabolomics has been widely used in medicine, food science, crop and farm animal research, and other fields. Metabolomics can establish a more direct relationship between changes in the type and content of metabolites and phenotypes. Metabolomics has gradually become a new research method for the analysis of genetic mechanisms of complex traits following genomics, transcriptomics, and proteomics with the advances in omics technology. In this review, we firstly introduce common analytical strategies, metabolomics platforms, and metabolomics databases. Then, we review the application of metabolomics in metabolic molecular identification of important economic traits in agricultural animals, disease diagnosis, meat quality and safety detection of animal products. We also introduce the latest achievements in the development, formation and analysis of important traits of animals and plants by using metabolomics, transcriptome, and genomics. Overall, the integrated analysis of metabolomics and other omics can comprehensively explain the genetic mechanism of all kinds of complex traits and help to improve the complete biological process of “mutation-gene-expression-metabolism-phenotype”. Metabolomics provides a new method for the mechanism analysis of complex characters and a novel idea for new agricultural breeding.
Keywords:
PDF (828KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
田菁, 王宇哲, 闫世雄, 孙帅, 贾俊静, 胡晓湘. 代谢组学技术发展及其在农业动植物研究中的应用. 遗传[J], 2020, 42(5): 452-465 doi:10.16288/j.yczz.19-287
Jing Tian.
代谢组是指生物体或细胞内所有小分子代谢物的集合,其分子量通常小于1000 Da,参与生物体的新陈代谢,维持生物体正常生长发育[1]。代谢组学来源于“代谢组”一词,1997年Oliver等[2]正式提出了代谢组学(metabolomics)概念,主要是研究细胞、生物流体、组织或生物体中发生的代谢过程,是一个静态的概念,对限定条件下的特定生物样品中所有代谢物定性、定量分析,又称为代谢物组学;随后Nicholson等[3]在1999年从另一角度定义代谢组学(metabonomics),即生物体对病理生理应激或遗传修饰引起的代谢反应的定量测定,是生物系统进行整体及其动态变化规律的研究,通常应用于人类营养和对药物或疾病的反应研究中,他们应用代谢组学在疾病诊断和药物筛选等研究[4,5]中取得进展。代谢组学是研究细胞、组织和器官中所有小分子代谢物集合的一门学科,通过对小分子物质进行定性定量分析指示生物体的生化状态,同时也可以寻找代谢物与机体生理病理、遗传环境的变化等之间的相互关联[6]。
随着组学时代的到来,代谢组学已经成为除基因组学、转录组学、蛋白质组学外的重要组学分支,共同组成系统生物学[7]。与其他组学相比,代谢组学具有以下特点:(1)小分子代谢物是基因组下游产物[8],可以直接、准确地反映生物体的生理病理状态;(2)代谢组是与表型组最接近的组学,可以利用代谢物的变化表征生物体不同的表型特征,得到更精确的表型数据;(3)代谢物在各个生物体中是类似的,所以代谢组学研究中采用的技术更容易在各个领域中通用。本文将从代谢组学的常用分析策略、检测平台的发展、代谢组学在农业动物不同研究领域中的应用及代谢组与其他多组学联合应用等4个方面进行综述,以期更好地理解代谢组学的发展现状和应用前景。
1 代谢组学常用分析策略
对代谢物进行检测的目的不同,选择的策略也存在较大差异,目前常用的代谢组学研究策略包括非靶向代谢组学(non-targeted metabolomics)、靶向代谢组学(targeted metabolomics)和拟靶向/广泛靶向代谢组学(pseudotargeted/widely targeted metabolomics)等。1.1 非靶向代谢组学
非靶向代谢组学又被称为发现代谢组学,对生物样本的内源性物质进行全面系统的测定分析。主要特点为对样本中代谢物分析的无偏向性,可以对样本小分子物质整体轮廓进行表征和筛选,通过生物信息方法进行差异分析或通路分析。目前非靶向代谢组学被广泛应用于生物标志物发现[9]、疾病诊断[10]、食品风味和安全[11,12]等多个领域。常见的代谢组学分析样本包括血浆、血清、尿液、组织和细胞等[7],往往需要使用多个生物学重复来减小样本个体差异带来的误差。样品前处理即代谢物的提取对代谢物的检测分析尤为重要,其中对小分子代谢物的累计富集和去除干扰测定的杂质物质是关键步骤[13]。目前主要的样本前处理方法包括液-液萃取(liquid-liquid extraction, LLE)、固相萃取(solid-phase extraction, SPE)、超临界流体萃取(supercritical-fluid extraction, SFE)、加速溶剂萃取(pressurized-liquid extraction, PLE)、微波辅助萃取(microwave-assisted extraction, MAE)和蛋白质沉淀等[14]。
代谢组数据一般采用多元数据分析策略,通常包括主成分分析(principal-component analysis, PCA)、偏最小二乘判别分析(partial least squares discrimination, PLS-DA)和正交偏最小二乘判别分析(orthogonal partial least squares discrimination, OPLS-DA)等[15],结合t检验和变量权重重要性排序(variable importance projection, VIP)值筛选差异代谢物或潜在生物标志物[16],同时利用筛选得到的代谢物可以进行代谢通路富集、聚类等分析开展后续研究。其主要分析流程如图1所示。
由于复杂样本中代谢物质种类多、每种代谢物含量差异大且性质复杂,很难有一种提取方法可以将其中所有的代谢物提取出来[17],同时非靶向代谢组学通常使用低分辨检测平台进行检测。因此,受到以上条件的限制,某些代谢物信息容易丢失(尤其低丰度代谢物),代谢物的鉴定和定量的准确性有待验证,需要开发适用性更强的流程来满足目前对于代谢组学研究中的要求。
1.2 靶向代谢组学
靶向代谢组学分析是对关注的几个、几十个或者几类已知的目标化合物进行测定的技术。通常是利用非靶向代谢组学发现差异代谢物,再使用靶向代谢组学进一步系统验证[18]。其主要特点是对关注代谢物进行准确鉴定和绝对定量,在分析上更具有针对性,与非靶向代谢组学优势互补,被主要应用于药物开发[7]、代谢疾病诊断及机制研究[19,20]等多图1
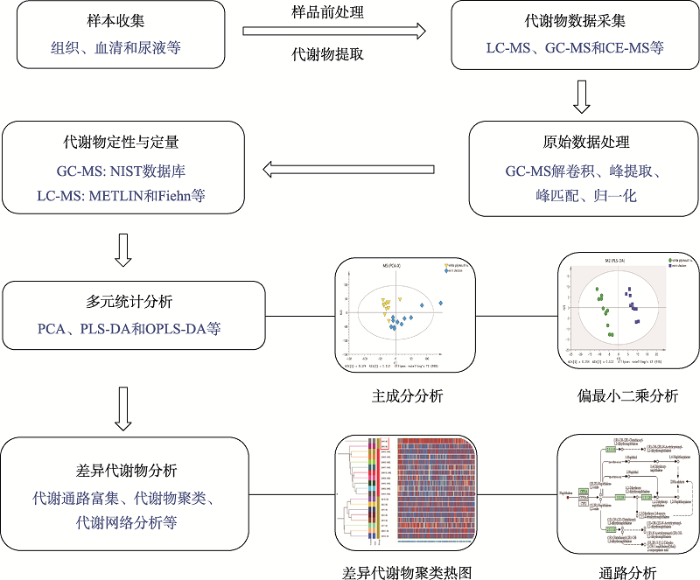 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1非靶向代谢组学主要分析流程
Fig. 1The platform of non-targeted metabolomics
个研究领域。
靶向代谢组学分析样本与非靶向代谢组学类似,但在前处理时可以根据要检测代谢物的特性选择合适的代谢物提取方法,使得检测更具可靠性和准确性。由于靶向代谢组学中只对关注的已知代谢物进行分析及生物学功能阐述,因此数据处理流程相对于非靶向代谢组学来说更加简单方便。在检测到关注的代谢物后,进行通路富集及该代谢物在生物体中合成机制等研究。
1.3 拟靶向/广泛靶向代谢组学
拟靶向/广泛靶向代谢组学[21]分析是一种结合非靶向代谢组学和靶向代谢组学优点的新方法,可以同时定性和精确定量数百种已知代谢物并对数千种已知及未知代谢物进行定量。通常使用超高相液相质谱-高分辨质谱联(ultra performance liquid chromatography high resolution mass spectrometer, UPLC- HRMS)和超相液相质谱-三重四级杆质谱联用(UPLC-MS/MS)平台实现代谢物信息采集。其主要由两部分搭建而成:第一部分是通过传统质谱或高分辨质谱进行代谢物采集并建立二级谱图(MS/MS)数据库,目前主要包括全扫描和ddMS2模式结合方法[22]、SWATH-MS方法[23]和基于多离子监控模式-增强碎片离子(multiple ion monitoring enhanced product ions, MIM-EPI)方法[24,25]等;第二部分是将待测样本进行多反应监测(multiple reaction monitoring, MRM)模 式[26]扫描后与第一部分中建立的数据库进行比对,对待测样本中代谢物进行定性及准确定量,实现拟靶向/广泛靶向代谢组的目的。该方法兼具了非靶向代谢组学和靶向代谢组学各自的优点,已经被广泛用于代谢组学研究中。其主要分析流程如图2所示。图2
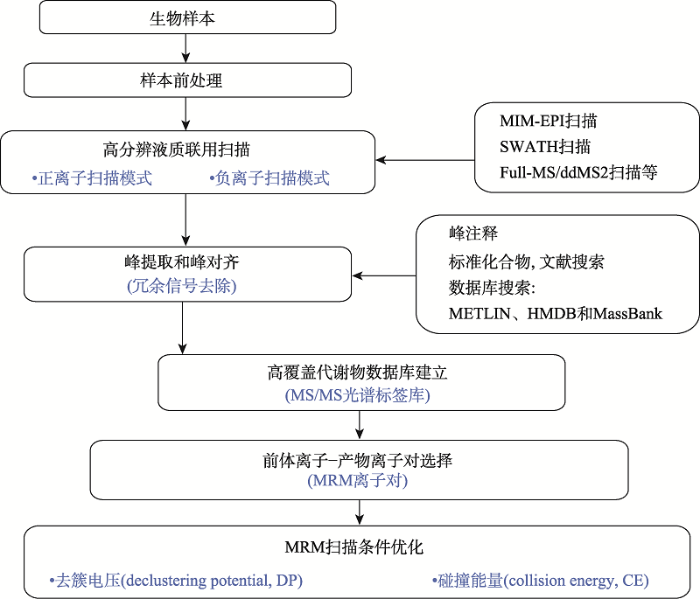 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2广泛靶向代谢组一般分析流程
Fig. 2The platform of widely targeted metabolomics
2 代谢组学检测平台发展及常用数据库
由于代谢物具有种类繁多、性质差异大、浓度范围分布广等特点,代谢组学发展出不同的检测平台[27],例如核磁共振(nuclear magnetic resonance, NMR)平台、液相色谱-质谱联用(liquid chromatography-mass spectrometry, LC-MS)平台、气相色谱-质谱联用(gas chromatography-mass spetrometry, GC- MS)平台、毛细管电泳-质谱联用(capillary electrophoresis-mass spectrometry, CE-MS)平台等。NMR是最早应用于代谢组学研究的平台,主要应用于代谢物结构的解析,但其检测灵敏度低,目前已经不能满足现在代谢组学研究。质谱具有高灵敏性的特点,已经成为代谢组学研究的主要分析技术,LC-MS[28]、GC-MS[29]及CE-MS[30]已经普遍应用于代谢组学研究。近年来随着高分辨质谱仪的推出及应用,对代谢物检测荷质比(m/z)可以精确到小数点后第4位,同时进行二级谱MS/MS信息的采集,利用同位素峰比例信息,进一步提升了代谢物定性的准确性,为代谢组学发展提供更好的技术平台。目前市场上主流的高分辨质谱仪主要分为四级杆飞行时间质谱(time of flight mass spectrometer, QTOF-MS)和静电场轨道阱质谱(electrostatic obitrap mass spectrometer, Obitrap-MS),其性能特点见表1。Table 1
表1
表1不同高分辨质谱仪性能特点
Table 1
| 质谱名称 | 检测质量范围 (质荷比: m/z,单位: Da) | 分辨率 | 优点 | 缺点 |
|---|---|---|---|---|
| 傅里叶变换离子回旋 共振质谱 (Fourier transform Ion cyclotron resonance, FT-ICR MS) | 100~10,000 | 100,000~1,000,000以上 | 分辨率和灵敏度最高 | 仪器体积大; 售价高; 维护费用昂贵; 扫描速度慢 |
| 静电场轨道阱质谱 (Orbitrap-MS) | LC-MS 50~6000 GC-MS 30~3000 | 140,000(在m/z =272时) 100,000(在m/z =272时) | 分辨率和灵敏度仅次于FT-ICR; 价格比FT-ICR低 | 扫描速度慢; 不能单独做串极质谱 |
| 四级杆飞行时间质谱 (QTOF-MS) | LC-MS 20~4000 GC-MS 20~3000 | 最高可达40,000以上 | 提供高分辨谱图; 速度快; 适合复杂样品分析; 对更小分子量代谢物检测较好 | 分辨力和灵敏度不及 Orbitrap |
新窗口打开|下载CSV
代谢物定性的准确性不仅取决于质谱仪的检测分辨能力,对应的代谢物数据库也至关重要。随着代谢组学研究的发展,代谢物数据库的构建逐渐完善。GC-MS检测到的代谢物通常参考NIST和FiehnLib数据库进行代谢物定性;LC-MS通常使用METLIN、HMBD数据库及部分自建库;脂质物质的种类繁多、结构特殊,通常使用脂质组专用的数据库LipidSearch、LMSD等进行比对确认。通过多元统计分析得到关注的代谢物或差异代谢物后往往还需要进行通路富集分析,揭示影响复杂生物过程的多种因素,通常使用KEGG和Reactome等数据库进行分析。随着代谢组学逐步发展,将多种数据库资源整合已经成为趋势,进一步提高数据资源利用率,其中跨库原始数据检索平台-MetabolomeXchange (http://www.metabolomexchange.org/site)于2015年由多家机构共同建立,为数据库资源的整合和扩展提供快捷途径。代谢组学中常用数据库见表2。
Table 2
表2
表2代谢组学中常见数据库特点
Table 2
| 数据库 | 优点 | 缺点 | 文献 |
|---|---|---|---|
| HMDB | (1)公开数据库,免费公开下载; (2)检测定量代谢物18,557个; (3)包含MS/MS谱 GC-MS谱 (4)预测代谢物MS/MS谱图、GC-MS谱图 | MS/MS检测中对前体离子大小有限制 (< 20 Da) | [31] |
| METLIN | (1)公开数据库; (2)包含超过13,000个标准品的高分辨质谱图; (3)不同碰撞能量下的高分辨的MS/MS谱图 >68,000个 | (1)所有数据均采自于QTOF质谱; (2)数据不可以下载 | [32,33] |
| NIST v17 | (1)公开数据库; (2)包含267,376个化合物的EI 谱图; (3)包含13,808个化合物的574,826个MS/MS谱图 | 对外部数据来源标识不清 | [34] |
| FiehnLib | (1)包含超过2200个EI谱图,覆盖超过1000种 代谢物 | (1)数据来源于Angilent公司气相色谱-单四级杆质谱和LECO公司GC-TOF; (2)数据主要来源于植物材料 | [35] |
| LipidSearch | (1)包括超过150万个脂质离子及其预测离子碎片; (2)包括脂质加合离子和MSn信息 | (1)商业数据库; (2)数据使用Thermo公司orbitrap类仪器采集; (3)MS/MS数据由计算机算法得到 | [32] |
| LMSD | (1)包含43,402个脂质结构; (2)谱图数据可以下载 | (1)在正负电离模式下预测MS/MS谱; (2)MS/MS光谱仅适用于每种脂质的一种加合物 | [36] |
| MassBank | (1)公开数据库; (2)支持输入文本格式的质谱搜索; (3)所有质谱图均是由标准品得到的; (4)有19,000个MS1和28,000个 MS2及MSn谱图 | (1)数据库中信息未经充分筛选; (2)某些谱图信息包含噪音信号 | [37,38] |
| KEGG | (1)综合性数据库; (2)包含代谢通路和互作网络信息; (3)包含17,268种代谢物和460条通路 | 图片分辨率较低,不美观 | [39] |
| Reactome | (1)公开数据库; (2)包含1419个化合物及人类2287条通路 | 数据来源较为单一,主要收集人体主要代谢 通路信息 | [40] |
| MetabolomeXchange | (1)公开数据库; (2)包含的数据来源于4个不同数据库; (3)包含代谢物结构及参考光谱、生物学作用、组 织和浓度等信息 | 数据库建设完善难度大 |
新窗口打开|下载CSV
3 代谢组学在农业动物研究领域中的应用
农业动物具有重要的经济价值,为人类提供肉、蛋、奶等农副产品,是人类日常营养物质获取的重要来源。随着代谢组学技术的发展与应用,目前已经在农业动物差异性状表征与重要经济性状代谢分子鉴定、农业动物疾病诊断、畜禽肉质风味及动物制品安全检测等多个领域中得到广泛应用。3.1 代谢组在农业动物差异性状表征、重要经济性状代谢分子鉴定中的应用
通过对不同品种(系)、性别畜禽的血清、尿液和组织等生物样本进行代谢物检测,可以筛选出一些小分子作为区分该品种(系)、性别的生物标志物,为不同品种(系)、性别畜禽差异性状表征提供理论依据。Bovo等[41]利用靶向代谢组学方法检测来自意大利的两个商品猪品种—大白猪(Large White)和杜洛克猪(Duroc)血浆和血清中180种代谢物,其中鞘磷脂、生物胺(犬尿氨酸和乙酰鸟氨酸)等代谢物在两个猪种中有很大差异,可以作为区别两品种猪的生物标记物;He等[42]检测了肥胖型猪—宁乡猪(Ningxiang strain)和瘦肉型杂交猪(Duroc×Landrace×Large Yorkshire strain)的血清代谢物,在肥胖型猪中胰岛素、胰高血糖素、脂质、肌醇和丙氨酸等代谢物含量均高于瘦肉型猪,但其血清中尿素和葡糖糖的含量较低;Ji等[43]利用LC-MS检测了来航鸡(Leghorn)、Fayoumi瘦肉型鸡和商业肉鸡的脂肪组织中92种代谢物,与商业肉鸡相比,其他两个品种鸡脂肪组织中有47种代谢物发生变化,其中肉碱、乙酰肉碱、鸟苷、胞嘧啶、腺苷、磷酸戊糖等多种代谢物含量有2倍以上的改变;Beauclercq等[44]对肌糖原含量不同(pHu-和pHu+)的两个品系鸡的血清和肌肉进行代谢物检测,在pHu-中鉴定到碳水化合物丰度高,pHu+的代谢物与肌糖原分解和氧化应激相关,最终鉴定到15个代谢标志物用于鸡肉质鉴定;Bovo等[45]对阉割公猪和母猪的血浆进行靶向代谢组学检测,共检测到132个代谢物,其中85种代谢物可以体现出两组猪之间的差异,在阉割公猪中酰基肉碱、生物胺、氨基酸分解等与脂质沉积相关的代谢物丰度高。代谢小分子物质可以反映生物体的生理状态和表型,利用代谢物表型可以更加准确地表征复杂表型。Rohart等[46]利用NMR对大白猪、长白猪(Landrace)和皮特兰猪(Pietrain)的血浆进行代谢物检测,结果显示肌酸酐、缬氨酸、柠檬酸、β-丙氨酸、乳酸、丙氨酸和异亮氨酸等代谢物与猪的瘦肉率性状相关,可以用于猪瘦肉率预测;Picone等[47]对大白猪、长白猪、杜洛克母猪的初乳代谢物进行检测,乳糖可以区分3种母猪品种,醋酸盐、牛磺酸、二甲胺和顺式-酸酯与仔猪体重增加和存活率有关;Karisa等[48]利用NMR测定了16头纯种安格斯牛(Angus)和10头杂交肉牛血浆代谢组,共检测到45种代谢物,其中肌酸、肉毒碱和马尿酸与肉牛剩余采食量(residual feed intake, FRI)性状相关,解释了32%的FRI表型变异,可以作为肉牛选育的分子标志物;Sun等[49]对饲喂苜蓿和玉米秸秆的泌乳牛的瘤胃液、牛奶、血清和尿液进行代谢物检测,在每种生物流体中分别发现55、8、28和31种差异代谢物,其差异代谢物主要参与甘氨酸、丝氨酸、苏氨酸、酪氨酸等氨基酸代谢通路,可以作为提高奶牛产奶量和乳蛋白质质量的生物标记物;Beauclercq等[50]使用NMR检测鸡血清、回肠和盲肠代谢物,利用氮校正表观代谢能(apparent metabolisable energy corrected to zero nitrogen retention, AMEn)来评估鸡对饲料的消化效率(digestible energy, DE),发现与AMEn关联最强的代谢物在血清中是脯氨酸,回肠中是富马酸盐,盲肠中是葡糖糖,为鸡DE的预测提供新的见解。对农业动物重经济性状进行标志代谢物筛选,将标志代谢物应用于标记辅助选择可提高选择准确性。3.2 代谢组在农业动物疾病诊断中的应用
在畜禽养殖业发展过程中,疾病的发生往往对畜禽生长、经济效益产生很大的影响,但是很多疾病的防治及治疗难度很大,而机体在发生疾病前后往往会引起体内代谢物水平的改变,应用代谢组学可以进行早期诊断的特点,通过检测血液、尿液等生物样本鉴定与疾病相关的生物标志物。Gong等[51]对感染猪瘟后3天和7天龄的断奶仔猪血清进行代谢物检测,代谢物主要富集在色氨酸分解代谢、苯丙氨酸代谢、脂肪代谢、核苷酸代谢等关键代谢通路上,同时观察到与肠道微生物相关的代谢物,对仔猪猪瘟疾病认识提供新的见解;Welle等[52]将猪结肠与猪流行性乙型痢疾杆菌共培养以模拟猪感染痢疾杆菌的肠道环境,并对暴露8 h的结肠外植体进行代谢物检测,结果显示感染流行性乙型痢疾的外植体上皮细胞明显坏死,l-瓜氨酸和IL-1α水平升高,为猪痢疾病理生理机制解析提供科学参考;Hailemariam等[53]对12头奶牛过渡期的4个时间点的120种血浆代谢物进行测定,肉毒碱、丙酰肉碱、溶血磷脂酰胆碱酰基(C14: 0)3种代谢物发生显著改变,利用这3种血浆代谢物就可以对在围产期可能患病的奶牛进行预测;Zhang等[54]对生产前8周和4周奶牛、产后患有酮症的奶牛以及生产后4周和8周的奶牛靶向检测检测128种血清代谢物,在整个检测周期中氨基酸、甘油磷脂、鞘脂、酰基肉碱和生物胺等代谢物水平发生显著变化,鉴定到可能作为酮症风险预测性生物标志物,为奶牛酮症预测提供科学依据;Shen等[55]利用LC-MS对患有腹水综合征的鸡和健康鸡的肝脏代谢物进行检测,结合血液生化和病理学结果表明,牛磺酸脱氧胆酸、胆酸葡萄糖醛酸、甘胆酸、LysoPC(15: 0)和牛磺胆酸被鉴定为腹水综合征的潜在生物标志物,且代谢物与脂质代谢紊乱有关,有助于了解鸡腹水综合征疾病的机制;Lu等[56]对处于热应激状态的鸡和健康鸡的257种血清代谢物进行了检测,其中78种代谢物存在显著差异且热应激肉鸡处于负能量平衡状态,不能有效的调动脂肪,影响肉鸡生长。以上研究证明代谢组可以对农业动物生产疾病引起的机体代谢物变化进行综合分析,筛选到的标志代谢物可用于疾病诊断和预测等。3.3 代谢组在畜禽肉质风味及动物制品安全检测中的应用
通过动物育种后产生的优良品种主要为人类提供丰富的食物,而影响食物风味的挥发性物质主要是分子量小于1000 Da的小分子物质。以往宏观的表型测定方法无法表征这类特殊表型,随着代谢组检测平台以及代谢物数据库的日益完善,可以对该类物质进行准确测定并深入研究,为动物产品风味改良提供代谢物表型。Romarathnam等[12]研究表明醛类(烯醛、二烯醛)是鸡脂肪受热时的特征香味物;Wasseriwan等[57]研究认为杂环化合物风味阈值低,是烤肉中最重要的风味呈味物,主要包括呋喃、噻吩、噻唑、吡啶、吡咯和吡嗪类化合物;Liu等[58]利用GC-O-MS对4种北京烤鸭的鸭皮和鸭胸肉检测到42种挥发性香味物质,主要分为醛、酮、醇、酸、酚、含硫化合物和含氮化合物7类,通过香气重组和遗漏实验及感官评价最终确定了9种对烤鸭风味有显著贡献的香味化合物:2-糠基硫醇、二甲基三硫醚、己醛、庚醛、辛醛、壬醛、甲硫氨酸、1-辛烯-3-醇和(E, E)-2,4-癸二烯醛。食品品质与人类健康密切相关,食品安全越来越受到人们关注,代谢组便可以对食品中含量甚微或难分离的化学污染物以及肉品掺假进行快速、准确的检测。Liu等[59]利用HPLC-MS/MS建立了快速检测猪肉、鸡蛋、牛奶等多种农产品中丙硫菌唑-硫脲的方法;Yin等[60]利用HPLC-MS/MS开发建立了快速简便检测猪肉中210种药物的残留方法,对食品安全检测和兽药使用具有重要意义;Trivedi等[61]对市场购买的猪肉碎、牛肉碎馅进行不同百分比混合样制备并进行代谢组测定,鉴定到15种差异代谢物用于鉴定区分猪肉和牛肉不同掺假比例的混合样品。4 代谢组与其他组学联合分析研究应用
代谢物是一切生命活动的物质基础,代谢组被认为是与表型组最为接近的组学,可以利用代谢物的变化对复杂的性状进行量化表征,形成代谢物分子表型。目前主要的农业经济性状大多为复杂的数量性状,而传统的全基因组关联分析(genome wide association study, GWAS)对这类主效基因的鉴定往往停留在QTL的水平,其中一个关键因素就是生长表型为典型复合性状,其基础调控途径和代谢通路相对应的一级表型在几年前难以评估和测定,导致基因型-表型无法直接对应。使用代谢物含量作为分子表型与基因组进行关联分析可以提高检测效力和检测精度,有望对影响复杂性状主效基因的致因突变的发掘提供新的解决手段。此外,利用转录组技术对不同时空状态下的基因表达进行检测,可以得到大量差异基因和众多调控网络,同时差异积累代谢物信息可以辅助时序表达的基因进行共表达分析,对基因功能解析、代谢通路和分子生化机制的完善提供研究基础。截至目前,利用代谢组作为基因组、转录组等其他组学与表型之间的桥梁,形成“突变-基因-调控-表达-代谢-表型”的关系网络,已经在番茄(Solanum lycopersicum)[62]、黄瓜(Cucumis sativus L.)[63]、水稻(Oryza sativa L.)[64]等农艺植物性状研究中被广泛应用。在农艺作物番茄中,Tieman等[62]利用GC-MS对398份番茄检测影响其风味的代谢物35个并与全基因组200多万个SNP进行mGWAS分析,找到影响番茄果糖、葡萄糖、类胡萝卜衍生挥发物的多个位点,同时提出利用具有甜味感知的挥发性物质进行番茄风味改良的建议;Zhu等[65]利用番茄的代谢组(442份材料、980种代谢物)、基因组(610份材料,2000多万SNP)、转录组(399份材料)数据进行多组学联合分析,利用鉴定到的3526个mGWAS信号和9万多个eQTL位点构建“代谢物-SNP-基因”网络,对番茄果实重量、甾体糖生物碱生物合成驯化、番茄颜色差异等进行综合解析,揭示了番茄以外观/味觉为导向的育种对番茄代谢组成的影响。总而言之,研究者找到了可以表征番茄风味的代谢物,同时也鉴定了影响上述代谢物的SNP位点,为番茄风味丢失历史以及风味改良提供了科学依据,而番茄风味的多组学分析也为农业动物的遗传育种提供了很好的借鉴。在农业动物上利用代谢组学方法进行复杂性状遗传机制的研究正在兴起,目前主要集中在猪、牛等大型家养动物重要经济性状研究中[66,67],通过使用代谢物小分子作为表型后与基因组、转录组等多组学进行关联分析,可以更加精确地鉴定影响肉质、乳质或生长性能相关的遗传标记或者遗传规律,为农业动物分子育种提供理论支持。Son等[68]利用靶向方法测定了480只商业杜洛克猪的皮下脂肪中不同长链脂肪酸,与全基因组3.9万个SNP进行关联分析后,在14号染色体117.6~124.6 Mb检测到一个显著关联区间,其中与不同脂肪酸关联SNP位于CPN1、PKD2L1和PAX2等多个基因上,可用于脂肪酸去饱和水平的遗传选择;Welzenbach等[69]对97头杜洛克和皮特兰杂交猪背最长肌的126个代谢物和35种蛋白质进行检测同时利用60 K Illumina芯片进行基因分型,富集分析发现共有10条通路,其中鞘脂代谢和糖酵解/糖异生通路对肉滴水损失有影响,GWAS分析发现在18号染色体中与滴水损失、蛋白质“磷酸甘油酸突变酶2”和代谢产物甘氨酸相关的SNP,同时鉴定到与滴水损失相关的候选基因区间;Melzer等[70]利用GC-MS对1305头荷斯坦奶牛(Holstein)泌乳开始21天和120天的乳汁进行代谢组测定,共检测到187种已知代谢物和3种未知代谢物,然后利用全基因组4万多个SNP进行关联分析,分别鉴定到与牛奶脂肪含量、pH值和蛋白质分别相关的11种、10种和16种代谢物,同时找到与上述性状相关的具有遗传效应的SNP位点,为牛奶品质改良提供理论基础;Widmann等[71]对237头F2代牛的血浆进行221种代谢物的靶向检测,同时测定了牛出生后6~9月的剩余采食量(RFI)、饲料转化率(feed conversion ratio, FCR)、日常能量摄入(daily energy intake, dEI)等评估饲料效率的指标,与全基因组4万多个SNP进行GWAS分析,发现rs109570900位点是影响RFI最显著的位点且与精氨酸含量显著相关,通过构建“表型-代谢型-基因型”网络寻找到对RFI有显著调控作用的候选基因(TP53和TGFB1),为农业动物饲料效率研究提供新的研究思路;Shi等[72]为研究影响鸡腹水综合征(broiler ascites syndrome, AS)的代谢标志物和差异表达基因,选择抗AS鸡和AS鸡利用LC-MS测定其血清代谢组及肝脏转录组,在实验组和对照组中鉴定到15个生物标志物,结合转录组数据表明甘油磷脂的代谢在抗AS肉鸡发育中起重要作用。更多相关研究见表3。Table 3
表3
表3代谢组与多组学联合分析在农业动物研究结果汇总
Table 3
| 物种 | 研究群体 | 样本类型 | 研究性状 | 代谢物分子 | 研究结论 | 文献 |
|---|---|---|---|---|---|---|
| 猪 | 白杜洛克猪×二花脸猪 (n=591)、苏太猪(n=282) | 背最长肌 腹部脂肪 | 脂肪酸组成 | 长链脂肪酸 | 腹部脂肪中C20:0与16号染色体中SNP显著关联;肌肉中C18:0与14号染色体中SNP显著关联;脂肪酸组成的候选基因:SCD和ELOVL7 | [73] |
| 杜洛克猪×长白猪× 约克夏猪DLY(n=610) | 背最长肌 | 酸肉 | 肌糖原、葡萄糖 (RG)和乳酸 | PRKAG3基因中低频错义突变R200Q和PHKG1g.8283C> A引起猪RG含量显著升高,导致DLY猪酸肉性状 | [74] | |
| 未阉割公猪(n=1282) | 颈下脂肪 | 公猪肉膻味 | 雄烯酮 | SULT2A1、SULT2B1、HSD17B14和CYP2A19基因与公猪膻味性状有关 | [75] | |
| 牛 | 挪威红牛(n=878) | 牛乳 | 牛乳脂肪酸 组成 | 短链、中长链和 长链脂肪酸 | 在BTA1、BTA13和BTA15中检测到重要的与牛奶脂肪酸相关QTL,鉴定到脂肪酸从头合成重要候选基因NCOA6 | [76] |
| 意大利西门塔尔牛(n=416) 意大利荷斯坦牛(n=436) | 牛乳 | 牛乳脂肪酸 组成 | 脂肪酸 | 在西门塔尔牛中鉴定到ECI2、PCYT2、DCXR、G6PC3、PYCR1和ALG12与牛乳脂肪酸组成相关基因;在荷斯坦牛中CYP17A1、ACO2、PI4K2A、GOT1、GPT、NT5C2、PDE6G、POLR3H和COX15与牛乳脂肪酸组成相关基因 | [77] | |
| 荷斯坦-弗里斯兰奶牛 (n=248) | 牛乳 | 奶牛酮症预后 研究 | 磷酸胆碱和 甘油磷酸胆碱 | 位于25号染色体的APOBR基因中QTL与代谢物显著相关 | [78] | |
| 荷斯坦牛(n=1217) | 血清 | 血清电解质 QTL鉴定 | 钙、氯、钠、 钾和镁离子 | GATA2、TMEM123和SCN5A基因与牛电解质平衡相关 | [79] | |
| 中国西门塔尔牛(n=723) | 最长肌 | 脂肪酸组成 | 中链脂肪酸 | FASN和ELOVL5基因组脂肪酸有关 | [80] | |
| 内洛尔肉牛(表型n=963; 基因型n=1616) | 最长肌 | 脂肪酸含量 | 中链和长链脂肪酸 | 鉴定到与脂肪酸相关基因ELOVL5、ESSRG、PCYT1A、ABC5、ABC6和ABC10 | [81] | |
| 日本黑牛(n=574) | 最长肌 | 牛肉鲜味 | 牛磺酸、肌苷和 次黄嘌呤 | 鉴定到与牛磺酸相关基因SLC6A6、RAB7A、RPN1和CCDC12;与肌苷和次黄嘌呤相关基因NT5E | [82] | |
| 日本黑牛(n=1836) | 最长肌 | 氨基酸组成 | β-丙氨酸和 牛磺酸 | 鉴定到与氨基酸有关基因STT3B、SUV420H1、CPT1A、MRPL21和IGHMBP2 | [83] | |
| 鸡 | 伊朗Urmia鸡×AA肉鸡 F2群体(n=270) | 血浆 | 代谢性状相关 基因座鉴定 | 甘油三酯、 胆固醇和葡糖糖 | 鉴定到与甘油三酯相关基因DOCK10和AP1S3 | [84] |
新窗口打开|下载CSV
总之,通过使用代谢物小分子作为表型与基因组、转录组等多组学进行联合关联分析,可以更加精确地鉴定影响畜禽重要经济性状,例如肉质、乳质或生长性能相关的功能基因或遗传标记,进一步阐明复杂数量性状的遗传机制,为新型农业动物分子育种提供全面的理论支持。
5 结语与展望
在代谢组学近20年的发展过程中,已经被广泛应用于环境污染物识别、疾病诊断、医药研制开发、食品营养科学等领域[85]。代谢组在动植物育种研究中也发挥了巨大功效,随着近年来代谢物检测仪器的发展、代谢物数据库的完善以及不同代谢组分析策略的开发应用,代谢组学必将会在农业动植物重要复杂经济性状解析中占有不可或缺的地位。目前,代谢组研究仍有许多问题亟待解决。就研究对象而言,人们已经将目光从较为简单地研究个体转向大样本复杂性状研究,由于代谢组会受到遗传、不同生理状态、环境刺激的影响,选择能准确表征当下状态的样本进行研究尤为重要;就代谢物检测而言,虽然近年来代谢组学技术平台得到长足进步,但是其在代谢物检测灵敏度及覆盖度上仍存在局限性,因此需要发展多平台联合检测技术实现全代谢谱的获取;就数据处理而言,不同于其他组学,代谢组数据为高维数据,需要使用多元统计分析进行信息挖掘,但是其数据处理能力还是有限,随着人工智能的兴起,有助于降低整体数据的维度从而对代谢组数据信息深入解读提供新思路。
组学时代的到来,代谢组学已经发展成了除基因组、转录组和蛋白组以外不可或缺的部分,多组学整合分析逐渐成为研究热点,有助于建立的“突变-基因-表达-代谢-表型”完整生物学网络,成为人类复杂疾病解析、动植物重要经济性状研究以及生命活动过程中复杂调控网络研究等多领域研究中新的工具和方法。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
DOI:10.1099/00221287-143-5-1483URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nrd1157URL [本文引用: 1]
[本文引用: 3]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
DOI:10.1073/pnas.1607571113URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
DOI:10.1080/10408347.2015.1079475URL [本文引用: 1]
DOI:10.1002/(ISSN)1098-2787URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1158/0008-5472.CAN-14-0109URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/pcp/pcn183URL [本文引用: 1]
DOI:10.1021/acs.analchem.8b01331URL [本文引用: 1]
DOI:10.1021/acs.analchem.7b05318URL [本文引用: 1]
DOI:10.1002/jms.v43:10URL [本文引用: 1]
DOI:10.1093/mp/sst080URL [本文引用: 1]

Liquid chromatography-mass spectrometry (LC-MS)-based metabolomics has been facilitated by the construction of MS2 spectral tag (MS2T) library from the total scan ESI MS/MS data, and the development of widely targeted metabolomics method using MS/MS data gathered from authentic standards. In this report, a novel strategy called stepwise multiple ion monitoring-enhanced product ions (stepwise MIM-EPI) was developed to construct the MS2T library, in which stepwise MIM was used as survey scans to trigger the acquisition of EPI. A total number of 698 (almost) non-redundant metabolites with MS2 spectra were obtained, of which 135 metabolites were identified/annotated. Integrating the data gathered from our MS2T library and other available multiple reaction monitoring (MRM) information, a widely targeted metabolomics method was developed to quantify 277 metabolites, including some phytohormones. Evaluation of the dehydration responses and natural variations of these metabolites in rice leaf not only suggested the coordinated regulation of abscisic acid (ABA) with metabolites such as serotonin derivative(s), polyamine conjugates under drought stress, but also revealed some C-glycosylated flavones as the potential markers for the discrimination of indica and japonica rice subspecies. The new MS2T library construction and widely targeted metabolomics strategy could be used as a tool for rice functional genomics.
DOI:10.1111/j.1365-313X.2012.04903.xURL [本文引用: 1]
A comprehensive and large-scale metabolome quantitative trait loci (mQTL) analysis was performed to investigate the genetic backgrounds associated with metabolic phenotypes in rice grains. The metabolome dataset consisted of 759 metabolite signals obtained from the grains of 85 lines of rice (Oryza sativa, Sasanishiki x Habataki back-crossed inbred lines). Metabolome analysis was performed using four mass spectrometry pipelines to enhance detection of different classes of metabolites. This mQTL analysis of a wide range of metabolites highlighted an uneven distribution of 802 mQTLs on the rice genome, as well as different modes of metabolic trait (m-trait) control among various types of metabolites. The levels of most metabolites within rice grains were highly sensitive to environmental factors, but only weakly associated with mQTLs. Coordinated control was observed for several groups of metabolites, such as amino acids linked to the mQTL hotspot on chromosome 3. For flavonoids, m-trait variation among the experimental lines was tightly governed by genetic factors that alter the glycosylation of flavones. Many loci affecting levels of metabolites were detected by QTL analysis, and plausible gene candidates were evaluated by in silico analysis. Several mQTLs profoundly influenced metabolite levels, providing insight into the control of rice metabolism. The genomic region and genes potentially responsible for the biosynthesis of apigenin-6,8-di-C-a-l-arabinoside are presented as an example of a critical mQTL identified by the analysis.
[本文引用: 1]
[本文引用: 1]
DOI:10.1021/pr4007624URL [本文引用: 1]
Acute respiratory distress syndrome (ARDS) remains a significant hazard to human health and is clinically challenging because there are no prognostic biomarkers and no effective pharmacotherapy. The lung compartment metabolome may detail the status of the local environment that could be useful in ARDS biomarker discovery and the identification of drug target opportunities. However, neither the utility of bronchoalveolar lavage fluid (BALF) as a biofluid for metabolomics nor the optimal analytical platform for metabolite identification is established. To address this, we undertook a study to compare metabolites in BALF samples from patients with ARDS and healthy controls using a newly developed liquid chromatography (LC)-mass spectroscopy (MS) platform for untargeted metabolomics. Following initial testing of three different high-performance liquid chromatography (HPLC) columns, we determined that reversed phase (RP)-LC and hydrophilic interaction chromatography (HILIC) were the most informative chromatographic methods because they yielded the most and highest quality data. Following confirmation of metabolite identification, statistical analysis resulted in 37 differentiating metabolites in the BALF of ARDS compared with health across both analytical platforms. Pathway analysis revealed networks associated with amino acid metabolism, glycolysis and gluconeogenesis, fatty acid biosynthesis, phospholipids, and purine metabolism in the ARDS BALF. The complementary analytical platforms of RPLC and HILIC-LC generated informative, insightful metabolomics data of the ARDS lung environment.
DOI:10.1021/pr3008946URL [本文引用: 1]
Ozonated autohemotherapy (O-3-AHT) is a medical approach during which blood obtained from the patient is ozonated and injected back into the body. Despite an increasing number of evidence that O-3-AHT is safe, this type of therapy remains controversial. To extend knowledge about the changes in blood evoked by O-3-AHT, LC-MS- and GC-MS-based metabolic fingerprinting was used to compare plasma samples obtained from blood before and after the treatment with potentially therapeutic concentrations of ozone. The procedure was performed in PVC bags utilized for blood storage to study also possible interactions between ozone and plastic. By use of GC-MS, an increase in lactic acid and pyruvic acid was observed, which indicated an increased rate of glycolysis. With LC-MS, changes in plasma antioxidants were observed. Moreover, concentrations of lipid oxidation products (LOP) and lysophospholipids were increased after ozone treatment. This is the first report of increased LOPs metabolites after ozonation of blood. Seven metabolites detected by LC-QTOF-MS only in ozonated samples could be considered as novel biomarkers of oxidative stress. Several plasticizers have been detected by both techniques in blood stored in PVC bags. PVC is known to be an ozone resistant material, but ozonation of blood in PVC bags stimulates leaching of plasticizers into the blood.
DOI:10.1002/elps.v30:1URL [本文引用: 1]
DOI:10.1093/nar/gkx1089URL
DOI:10.1016/j.trac.2015.09.005URL
DOI:10.1097/01.ftd.0000179845.53213.39URL
DOI:10.1021/ac9019522URL
DOI:10.1093/nar/gkl838URL
DOI:10.1002/jms.v45:7URL
DOI:10.1017/S1751731116000483URL [本文引用: 1]
DOI:10.1016/j.jnutbio.2010.11.007URL [本文引用: 1]
Childhood obesity has become a prevalent risk to health of children and teenagers. To develop biomarkers in serum for altered lipid metabolism, genetically obese (Ningxiang strain) and lean (DurocxLandracexLarge Yorkshire strain) growing pigs were used as models to identify potential differences in the serum metabonome between the two strains of pigs after consuming the same diet for 46 days. At the end of the study, pigs were euthanized for analysis of the serum metabonome and determination of body composition. Obese pigs had higher fat mass (42.3 +/- 8.8% vs. 21.9 +/- 4.5%) and lower muscle mass (35.4 +/- 4.5% vs. 58.9 +/- 2.5%) than lean pigs (P< 0.01). Serum concentrations of insulin and glucagon were higher (P<0.02) in obese than in lean pigs. With the use of an NMR-based metabonomic technology, orthogonal projection to latent structure with discriminant analysis showed that serum HDL, VLDL, lipids, unsaturated lipids, glycoprotein, myo-inositol, pyruvate, threonine, tyrosine and creatine were higher in obese than in lean pigs (P<0.05), while serum glucose and urea were lower in obese pigs (P< 0.05). In addition, changes in gut microbiota-related metabolites, including trimethylamine-N-oxide and choline, were observed in sera of obese pigs relatively to lean pigs (P<0.05). These novel findings indicate that obese pigs have distinct metabolism, including lipogenesis, lipid oxidation, energy utilization and partition, protein and amino acid metabolism, and fermentation of gastrointestinal microbes, compared with lean pigs. The obese Ningxiang pig may be a useful model for childhood obesity research. (C) 2012 Elsevier Inc.
DOI:10.1152/physiolgenomics.00163.2013.URL [本文引用: 1]
Domestic broiler chickens rapidly accumulate fat and are naturally hyperglycemic and insulin resistant, making them an attractive model for studies of human obesity. We previously demonstrated that short-term (5 h) fasting rapidly upregulates pathways of fatty acid oxidation in broiler chickens and proposed that activation of these pathways may promote leanness. The objective of the current study was to characterize adipose tissue from relatively lean and fatty lines of chickens and determine if heritable leanness in chickens is associated with activation of some of the same pathways induced by fasting. We compared adipose gene expression and metabolite profiles in white adipose tissue of lean Leghorn and Fayoumi breeds to those of fattier commercial broiler chickens. Both lipolysis and expression of genes involved in fatty acid oxidation were upregulated in lean chickens compared with broilers. Although there were strong similarities between the lean lines compared with broilers, distinct expression signatures were also found between Fayoumi and Leghorn, including differences in adipogenic genes. Similarities between genetically lean and fasted chickens suggest that fatty acid oxidation in white adipose tissue is adaptively coupled to lipolysis and plays a role in heritable differences in fatness. Unique signatures of leanness in Fayoumi and Leghorn lines highlight distinct pathways that may provide insight into the basis for leanness in humans. Collectively, our results provide a number of future directions through which to fully exploit chickens as unique models for the study of human obesity and adipose metabolism.
DOI:10.1021/acs.jproteome.5b01050URL [本文引用: 1]
DOI:10.2527/jas.2015-9528URL [本文引用: 1]
DOI:10.2527/jas.2012-5338URL [本文引用: 1]
Predicting phenotypes is a statistical and biotechnical challenge, both in medicine (predicting an illness) and animal breeding (predicting the carcass economical value on a young living animal). High-throughput fine phenotyping is possible using metabolomics, which describes the global metabolic status of an individual, and is the closest to the terminal phenotype. The purpose of this work was to quantify the prediction power of metabolomic profiles for commonly used production phenotypes from a single blood sample from growing pigs. Several statistical approaches were investigated and compared on the basis of cross validation: raw data vs. signal preprocessing (wavelet transformation), with a single-feature selection method. The best results in terms of prediction accuracy were obtained when data were preprocessed using wavelet transformations on the Daubechies basis. The phenotypes related to meat quality were not well predicted because the blood sample was taken some time before slaughter, and slaughter is known to have a strong influence on these traits. By contrast, phenotypes of potential economic interest (e.g., lean meat percentage and ADFI) were well predicted (R-2 = 0.7; P < 0.0001) using metabolomic data.
DOI:10.1186/s40104-018-0237-1URL [本文引用: 1]
DOI:10.1016/j.livsci.2014.03.002URL [本文引用: 1]
DOI:10.1021/pr501305gURL [本文引用: 1]
DOI:10.1038/s41598-018-24978-9URL [本文引用: 1]
DOI:10.3389/fmicb.2017.00731URL [本文引用: 1]
DOI:10.1007/s11306-017-1219-6URL [本文引用: 1]
DOI:10.3168/jds.2013-6803URL [本文引用: 1]
In dairy cows, periparturient disease states, such as metritis, mastitis, and laminitis, are leading to increasingly significant economic losses for the dairy industry. Treatments for these pathologies are often expensive, ineffective, or not cost-efficient, leading to production losses, high veterinary bills, or early culling of the cows. Early diagnosis or detection of these conditions before they manifest themselves could lower their incidence, level of morbidity, and the associated economic losses. In an effort to identify predictive biomarkers for postpartum or periparturient disease states in dairy cows, we undertook a cross-sectional and longitudinal metabolomics study to look at plasma metabolite levels of dairy cows during the transition period, before and after becoming ill with postpartum diseases. Specifically we employed a targeted quantitative metabolomics approach that uses direct flow injection mass spectrometry to track the metabolite changes in 120 different plasma metabolites. Blood plasma samples were collected from 12 dairy cows at 4 time points during the transition period (-4 and 1 wk before and 1 and 4 wk after parturition). Out of the 12 cows studied, 6 developed multiple periparturient disorders in the post-calving period, whereas the other 6 remained healthy during the entire experimental period. Multivariate data analysis (principal component analysis and partial least squares discriminant analysis) revealed a clear separation between healthy controls and diseased cows at all 4 time points. This analysis allowed us to identify several metabolites most responsible for separating the 2 groups, especially before parturition and the start of any postpartum disease. Three metabolites, carnitine, propionyl carnitine, and lysophosphatidylcholine acyl C14:0, were significantly elevated in diseased cows as compared with healthy controls as early as 4 wk before parturition, whereas 2 metabolites, phosphatidylcholine acyl-alkyl C42:4 and phosphatidylcholine diacyl C42:6, could be used to discriminate healthy controls from diseased cows 1 wk before parturition. A 3-metabolite plasma biomarker profile was developed that could predict which cows would develop periparturient diseases, up to 4 wk before clinical symptoms appearing, with a sensitivity of 87% and a specificity of 85%. This is the first report showing that periparturient diseases can be predicted in dairy cattle before their development using a multimetabolite biomarker model. Further research is warranted to validate these potential predictive biomarkers.
DOI:10.1007/s11306-017-1180-4URL [本文引用: 1]
DOI:10.1039/c4mb00137kURL [本文引用: 1]
Ascites is a major problem for both human health and animal production, due to its association with high rates of morbidity and mortality, low efficiency of nutrient utilization, and permanent adverse effects on performance. Although it is one of the three major metabolic diseases in poultry production, the underlying mechanisms are largely unknown. In this study, six ascites syndrome (AS) chickens and six normal chickens were obtained from each group (108 chickens) at 21 and 35 days. A liver metabolomics method based on ultra-performance liquid chromatography/quadruple time-of-flight mass spectrometry (UPLC/Q-TOF/MS) was used to explore the metabolic pattern of low molecular mass metabolites in chickens with low-temperature-induced AS. Coupled with blood biochemistry and histopathology results, the significant difference in metabolic profiling between the AS group and the control group, as determined through pattern recognition analysis, indicated changes in global tissue metabolites. The results showed that a primary bile acid synthesis disorder and inflammation had occurred by 21 days and that lysophospholipid metabolism was disrupted by 35 days with the continuation of low temperatures. Several metabolites, including taurodeoxycholic acid, cholic acid glucuronide, glycocholic acid, LysoPC(15:0) and taurocholic acid, were identified as the potential and proper biomarkers of AS. These biochemical changes in tissue metabolites are related to perturbations of lipid metabolism, which may be helpful to further understand the AS mechanisms. This work shows that the metabolomics is a valuable tool for studying metabolic diseases.
DOI:10.1017/S0007114518000247URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1021/acs.jafc.7b00062URL [本文引用: 1]
DOI:10.1016/j.chroma.2016.08.001URL [本文引用: 1]
DOI:10.1039/C6AN00108DURL [本文引用: 1]
DOI:10.1126/science.aal1556URL [本文引用: 2]
DOI:10.1126/science.1259215URL [本文引用: 1]
DOI:10.1038/ng.3007URL [本文引用: 1]
DOI:10.1016/j.cell.2017.12.019URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12864-017-3752-0URL [本文引用: 1]
DOI:10.3390/ijms17091426URL [本文引用: 1]
DOI:10.1371/journal.pone.0070256URL [本文引用: 1]
DOI:10.1371/journal.pone.0124574URL [本文引用: 1]
DOI:10.1038/s41598-017-02492-8URL [本文引用: 1]
DOI:10.1371/journal.pone.0065554URL
DOI:10.1186/s12711-019-0488-0URL
DOI:10.2527/jas2014-7863URL
Androstenone is one of the compounds causing boar taint of pork and is highly heritable (approximately 0.6). Recently, indirect genetic effects (IGE; also known as associative effects or social genetic effects) were found for androstenone, meaning that pen mates (boars) affect each other's androstenone level genetically. Similar to estimating variance components with a direct-indirect animal model, direct and indirect genetic SNP effects can be estimated for androstenone. This study aims to detect SNP with significant direct genetic effects and IGE on androstenone. The dataset consisted of 1,282 noncastrated boars (993 boars genotyped) from 184 groups of pen members. After quality control, 46,421 SNP were included in the analysis. One model for single-SNP regression was fitted, where both the direct SNP effect of the individual itself and the indirect SNP effects of its pen mates were included. None of the SNP (direct or indirect) were found genomewide significant. One QTL on SSC6 was chromosome-wide significant for the direct effect. A single SNP on SSC9 and 2 regions and a single SNP on SSC14 were found for the indirect effect. A backwards elimination method and haplotype analysis were used to quantify the variance explained by the SNP. The backwards elimination method identified 4 independent regions affecting androstenone. The QTL on SSC6 explained 2.1 and 2.6% of the phenotypic variance using the backwards elimination method or the haplotype analysis. The QTL on SSC14 explained 3.4 and 2.7% of the phenotypic variance using the backwards elimination method or the haplotype analysis. The single association on SSC9 explained 2.2% of the phenotypic variance. All significant QTL together explained 7 to 8% of phenotypic variance and 40 to 44% of the total genetic variance available for response to selection. Besides the newly discovered QTL and the confirmation of known QTL, this study also presents a methodology to model SNP for IGE.
DOI:10.1186/s12711-017-0294-5URL
DOI:10.3168/jds.2018-14413URL
DOI:10.1152/physiolgenomics.00126.2014URL
DOI:10.1111/age.v50.6URL
DOI:10.1186/s12864-017-3847-7URL
DOI:10.1186/s12864-016-2511-yURL
DOI:10.1186/s12864-017-4275-4URL
DOI:10.1111/asj.2018.89.issue-8URL
DOI:10.1080/00071668.2018.1472743URL
DOI:10.1371/journal.pone.0177675URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}