Analysis of rice root bacterial microbiota of Nipponbare and IR24
Yali Hu1,2,3, Rui Dai2,3,4,5, Yongxin Liu2,3,4, Jingying Zhang2,3,4, Bin Hu2, Chengcai Chu2, Huaibo Yuan,1, Yang Bai,2,3,4,5 1. School of Food and Biological Engineering, Hefei University of Technology, Hefei 230009, China 2. State Key Laboratory of Plant Genomics, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China 3. CAS Center for Excellence in Biotic Interactions, University of Chinese Academy of Sciences, Beijing 100049, China 4. CAS-JIC Centre of Excellence for Plant and Microbial Science, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China 5. College of Advanced Agricultural Sciences, University of Chinese Academy of Sciences, Beijing 100101, China
Supported by the Key Research Program of Frontier Sciences of the Chinese Academy of Science No.QYZDB-SSW-SMC021 the National Natural Science Foundation of China No.31772400
作者简介 About authors 胡雅丽,在读硕士研究生,专业方向:根系微生物组。E-mail:2017111262@mail.hfut.edu.cn。
Abstract The root-associated bacterial microbiota is closely related to life activities of land plants, and its composition is affected by geographic locations and plant genotypes. However, the influence of plant genotypes on root microbiota in rice grown in northern China remains to be explained. In this study, we performed 16S rRNA gene amplicon sequencing to generate bacterial community profiles of two representative rice cultivars, Nipponbare and IR24. They are planted in Changping and Shangzhuang farms in Beijing and have reached the reproductive stage. We compared their root microbiota in details by Random Forest machine learning algorithm and network analysis. We found that the diversity of rice root microbiota was significantly affected by geographic locations and rice genotypes. Nipponbare and IR24 showed distinct taxonomic composition of the root microbiota and the interactions between different bacteria. Moreover, the root bacteria could be used as biomarkers to distinguish Nipponbare from IR24 across regions. Our study provides a theoretical basis for the in-depth understanding of rice root microbiota in Northern China and the improvement of rice breeding from the perspective of the interaction between root microorganisms and plants. Keywords:rice root microbiota;diversity analysis;taxonomic composition;machine learning;network analysis
PDF (1366KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 胡雅丽, 戴睿, 刘永鑫, 张婧赢, 胡斌, 储成才, 袁怀波, 白洋. 水稻典型品种日本晴和IR24根系微生物组的解析. 遗传[J], 2020, 42(5): 506-518 doi:10.16288/j.yczz.20-070 Yali Hu. Analysis of rice root bacterial microbiota of Nipponbare and IR24. Hereditas(Beijing)[J], 2020, 42(5): 506-518 doi:10.16288/j.yczz.20-070
A:在两个种植地点中日本晴根系富集的OTUs。与IR24相比,日本晴在昌平(CP)和上庄(SZ)分别有166个和207个OTU富集,且其中有66个OTU在这两个种植地区都富集(Wilcoxon秩和检验用于比较差异OTUs,当P < 0.05、FDR < 0.2且差异倍数 > 1.2时,认为该OTU在两组样品间存在差异)。B:日本晴在两个种植地区均富集的根系OTUs的分类组成。共同富集的66个OTU大部分属于拟杆菌门,厚壁菌门和gamma-变形菌纲。C:在两个种植地点中IR24根系富集的OTUs。与日本晴相比,IR24在昌平和上庄分别有172个和135个富集OTUs,且其中有45个OTUs在这两个种植地区都富集。D:IR24在两个种植地区均富集的根系OTU的分类组成。共同富集的45个OTU大部分属于beta-变形菌纲,厚壁菌门和delta-变形菌纲。从图中可知,水稻基因型影响了微生物在根系的富集。Actinobacteria:放线菌门;Bacteroidetes:拟杆菌门;Firmicutes:厚壁菌门;Spirochaetes:螺旋体门;其余为alpha-、beta-、delta-、gamma-变形菌纲。 Fig. 3The overlap and taxonomic composition of OTUs enriched in Nipponbare/IR24 of two fields
HirumaK, GerlachN, SacristánS, NakanoRT, HacquardS, KracherB, NeumannU, RamírezD, BucherM, O’Connell RJ, Schulze-LefertP,. Root endophyte colletotrichum tofieldiae confers plant fitness benefits that are phosphate status dependent Cell, 2016,165(2):464-474. DOI:10.1016/j.cell.2016.02.028URL [本文引用: 1]
WangXL, WangET . NRT1.1B connects root microbiota and nitrogen use in rice Bull Bot, 2019,54(3):285-287. DOI:10.11983/CBB19060URL [本文引用: 1] Root-associated microbial communities in the soil play fundamental roles in plant nutrition uptake and fitness. However, how plants shape root microbial communities and how the microbes affect the fitness of their hosts remain elusive. Recently, Chinese scientists have made a breakthrough discovery that the nitrogen-use efficiency between indica and japonica rice varieties is associated with different root microbiota in rice. Nitrogen metabolism is greatly enriched in indica-enriched bacteria as compared with japonica-enriched bacteria. Rice NRT1.1B, a nitrogen sensor contributing to nitrogen use divergence between rice subspecies, is associated with the recruitment of these bacterial taxa. Inoculation of the japonica variety with indica-enriched bacteria can improve rice growth in organic nitrogen conditions in the SynCom experimental system. This work highlights the links between root microbiota and nitrogen use in rice and could be exploited to modulate the root microbiota that increase crop productivity and sustainability. 王孝林, 王二涛 . 根际微生物促进水稻氮利用的机制 植物学报, 2019,54(3):285-287. DOI:10.11983/CBB19060URL [本文引用: 1] Root-associated microbial communities in the soil play fundamental roles in plant nutrition uptake and fitness. However, how plants shape root microbial communities and how the microbes affect the fitness of their hosts remain elusive. Recently, Chinese scientists have made a breakthrough discovery that the nitrogen-use efficiency between indica and japonica rice varieties is associated with different root microbiota in rice. Nitrogen metabolism is greatly enriched in indica-enriched bacteria as compared with japonica-enriched bacteria. Rice NRT1.1B, a nitrogen sensor contributing to nitrogen use divergence between rice subspecies, is associated with the recruitment of these bacterial taxa. Inoculation of the japonica variety with indica-enriched bacteria can improve rice growth in organic nitrogen conditions in the SynCom experimental system. This work highlights the links between root microbiota and nitrogen use in rice and could be exploited to modulate the root microbiota that increase crop productivity and sustainability.
CarriónVJ, Perez-JaramilloJ, CordovezV, Tracanna V, de Hollander M, Ruiz-BuckD, MendesLW, van Ijcken WFJ, Gomez-ExpositoR, ElsayedSS, MohanrajuP, ArifahA, vander Oost J, PaulsonJN, MendesR, van Wezel GP, MedemaMH, RaaijmakersJM. Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome Science, 2019,366(6465):606-612. [本文引用: 1]
ZhangJY, LiuYX, ZhangN, HuB, JinT, XuHR, QinY, YanPX, ZhangXN, GuoXX, HuiJ, CaoSY, WangX, WangC, WangH, QuBY, FanGY, YuanLX, Garrido-OterR, ChuCC, BaiY . NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice Nat Biotechnol, 2019,37(6):676-684. [本文引用: 4]
CastrilloG, TeixeiraPJPL, ParedesSH, Law TF, de Lorenzo L, FeltcherME, FinkelOM, BreakfieldNW, MieczkowskiP, JonesCD, Paz-AresJ, DanglJL. Root microbiota drive direct integration of phosphate stress and immunity Nature, 2017,543(7646):513-518. [本文引用: 1]
Van DeynzeA, ZamoraP, DelauxPM, HeitmannC, JayaramanD, RajasekarS, GrahamD, MaedaJ, GibsonD, SchwartzKD, BerryAM, BhatnagarS, JospinG, DarlingA, JeannotteR, LopezJ, WeimerBC, EisenJA, ShapiroHY, AnéJM, BennettAB . Nitrogen fixation in a landrace of maize is supported by a mucilage-associated diazotrophic microbiota PLoS Biol, 2018,16(8):e2006352. [本文引用: 1]
WangC, BaiY . Maize aerial roots fix atmospheric N2 by interacting with nitrogen fixing bacteria (in Chinese) Sci Sin Vitae, 2019,49(1):89-90. DOI:10.1360/N052018-00215URL [本文引用: 1]
WangW, YangJ, ZhangJ, LiuYX, TianCP, QuBY, GaoCL, XinPY, ChengSJ, ZhangWJ, MiaoP, LiL, ZhangXJ, ChuJF, ZuoJR, LiJY, BaiY, LeiXG, ZhouJM . An Arabidopsis secondary metabolite directly targets expression of the bacterial type III secretion system to inhibit bacterial virulence Cell Host Microbe, 2020,27(4):601-613. [本文引用: 1]
ChenT, NomuraK, WangXL, SohrabiR, XuJ, YaoYL, PaaschBC, MaL, KremerJ, ChengYT, ZhangL, WangN, WangET, XinXF, HeSY . A plant genetic network for preventing dysbiosis in the phyllosphere Nature, 2020,580(7804):653-657. [本文引用: 1]
EdwardsJ, JohnsonC, Santos-MedellínC, LurieE, PodishettyNK, BhatnagarS, EisenJA, SundaresanV . Structure, variation, and assembly of the root-associated microbiomes of rice Proc Natl Acad Sci USA, 2015,112(8):E911-E920. [本文引用: 4]
ZhangJY, ZhangN, LiuYX, ZhangXN, HuB, QinY, XuHR, WangH, GuoXX, QianJM, WangW, ZhangPF, JinT, ChuCC, BaiY . Root microbiota shift in rice correlates with resident time in the field and developmental stage Science China Life Sciences, 2018,61(6):613-621. [本文引用: 5]
EdwardsJA, Santos-MedellínCM, LiechtyZS, NguyenB, LurieE, EasonS, PhillipsG, SundaresanV . Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice PLoS Biol, 2018,16(2):e2003862. [本文引用: 3]
ChenSM, WaghmodeTR, SunRB, KuramaeEE, HuCS, LiuBB . Root-associated microbiomes of wheat under the combined effect of plant development and nitrogen fertilization Microbiome, 2019,7(1):136. [本文引用: 1]
ShiY, LiYT, XiangXJ, SunRB, YangT, HeD, ZhangKP, NiYY, ZhuYG, AdamsJM, ChuHY . Spatial scale affects the relative role of stochasticity versus determinism in soil bacterial communities in wheat fields across the North China Plain Microbiome, 2018,6(1):27. DOI:10.1186/s40168-018-0409-4URL [本文引用: 1]
SunRB, LiWY, DongWX, TianYP, HuCS, LiuBB . Tillage changes vertical distribution of soil bacterial and fungal communities Front Microbiol, 2018,9:699. [本文引用: 1]
WaltersWA, JinZ, YoungblutN, WallaceJG, SutterJ, ZhangW, González-Pe?aA, PeifferJ, KorenO, ShiQJ, KnightR, Glavina del Rio T, TringeSG, BucklerES, DanglJL, LeyRE,. Large-scale replicated field study of maize rhizosphere identifies heritable microbes Proc Natl Acad Sci USA, 2018,115(28):7368-7373. [本文引用: 2]
WeiZ, GuY, FrimanVP, KowalchukGA, XuYC, ShenQR , Jousset A. Initial soil microbiome composition and functioning predetermine future plant health Sci Adv, 2019,5(9): eaaw0759. [本文引用: 1]
XiongW, SongYQ, YangKM, GuY, WeiZ, KowalchukGA, XuYC, JoussetA, ShenQR, GeisenS . Rhizosphere protists are key determinants of plant health Microbiome, 2020,8(1):27. [本文引用: 1]
LiuYX, QinY, BaiY . Reductionist synthetic community approaches in root microbiome research Curr Opin Microbiol, 2019,49:97-102. [本文引用: 2]
FanKK, WeisenhornP, GilbertJA, ShiY, BaiY, ChuHY . Soil pH correlates with the co-occurrence and assemblage process of diazotrophic communities in rhizosphere and bulk soils of wheat fields Soil Biol Biochem, 2018,121:185-192. [本文引用: 3]
MoriA, KirkGJD, LeeJS, MoreteMJ, NandaAK, Johnson-BeeboutSE, WissuwaM . Rice genotype differences in tolerance of zinc-deficient soils: evidence for the importance of root-induced changes in the rhizosphere Front Plant Sci, 2016,6:1160. [本文引用: 2]
FanKK, Delgado-BaquerizoM, GuoXS, WangDZ, WuYY, ZhuM, YuW, YaoHY, ZhuYG, ChuHY . Suppressed N fixation and diazotrophs after four decades of fertilization Microbiome, 2019,7(1):143. [本文引用: 1]
ZhengMS, ZhouN, LiuSF, DangCY, LiuYX, HeSS, ZhaoYJ, LiuW, WangXK . N2O and NO emission from a biological aerated filter treating coking wastewater: main source and microbial community J Clean Prod, 2019,213:365-374. [本文引用: 1]
OksanenJ, KindtR, LegendreP, O’Hara B, StevensMHH, OksanenMJ, SuggestsM,. The vegan package Community Ecology Package, 2007,10:631-637. [本文引用: 1]
WickhamH . Ggplot2: Elegant Graphics for Data Analysis Springer, 2016. [本文引用: 1]
LiawA, WienerM . Classification and regression by randomForest R News, 2002,2(3):18-22. [本文引用: 1]
QianXB, LiuYX, YeXH, ZhengWJ, LvSX, MoMJ, LinJJ, WangWQ, WangWH, ZhangXN, LuMP . Gut microbiota in children with juvenile idiopathic arthritis: characteristics, biomarker identification, and usefulness in clinical prediction BMC Genomics, 2020,21(1):286. [本文引用: 1]
CsardiG, NepuszT . The igraph software package for complex network research InterJ Complex Systems, 2006,1695(5):1-9. [本文引用: 1]
WangJF, ZhengJY, ShiWY, DuN, XuXM, ZhangYM, JiPF, ZhangFY, JiaZ, WangYP, ZhengZ, ZhangHP, ZhaoFQ . Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus Gut, 2018,67(9):1614-1625. [本文引用: 1]
National Genomics Data Center Members and Partners. Database resources of the national genomics data center in 2020 Nucleic Acids Res, 2020,48(D1):D24-D33. [本文引用: 1]
LiuBB, ZhangXJ, BakkenLR, SnipenL, Frosteg?rd? . Rapid succession of actively transcribing denitrifier populations in agricultural soil during an anoxic spell Front Microbiol, 2019,9:3208. [本文引用: 1]
ZhangKP, ShiY, CuiXQ, YueP, LiKH, LiuXJ, TripathiBM, ChuHY . Salinity is a key determinant for soil microbial communities in a desert ecosystem mSystems, 2019,4(1):e00225-00218. [本文引用: 1]
LebeisSL, ParedesSH, LundbergDS, BreakfieldN, GehringJ, McDonald M, MalfattiS, Glavina del Rio T, JonesCD, TringeSG, DanglJL. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa Science, 2015,349(6250):860-864. DOI:10.1126/science.aaa8764URL [本文引用: 1]
VogesMJEEE, BaiY, Schulze-LefertP, SattelyES . Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc Natl Acad Sci USA, 2019,116(25):12558-12565.
ZamioudisC, KortelandJ, Van Pelt JA, van Hamersveld M, DombrowskiN, BaiY, HansonJ, Van Verk MC, LingHQ, Schulze-LefertP, PieterseCMJ,. Rhizobacterial volatiles and photosynthesis-related signals coordinate MYB72 expression in Arabidopsis roots during onset of induced systemic resistance and iron-deficiency responses Plant J, 2015,84(2):309-322. [本文引用: 1]
NiuB, PaulsonJN, ZhengX, KolterR . Simplified and representative bacterial community of maize roots Proc Natl Acad Sci USA, 2017,114(12):E2450-E2459. [本文引用: 1]
 ,1, 白洋
,1, 白洋
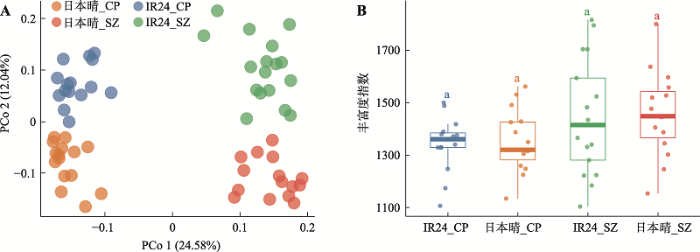 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT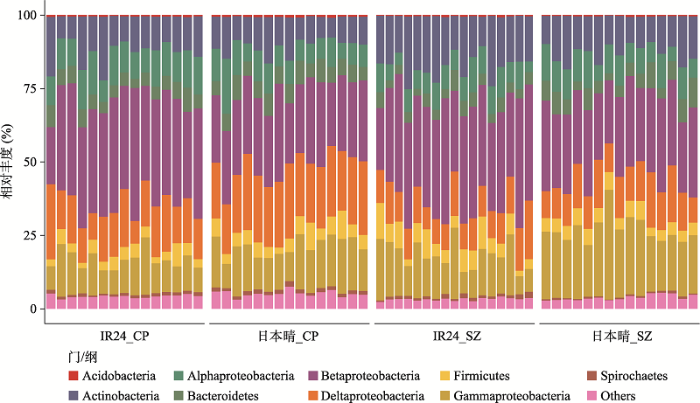 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT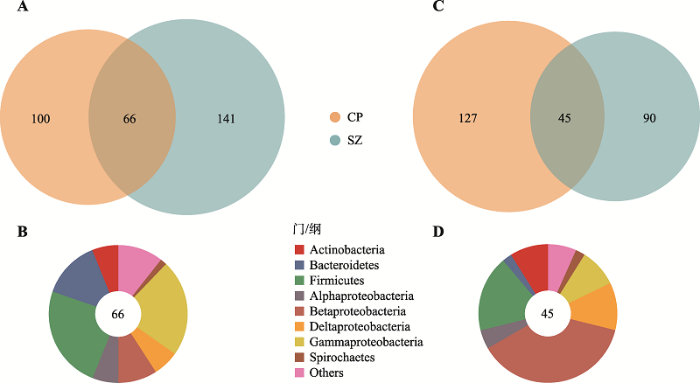 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT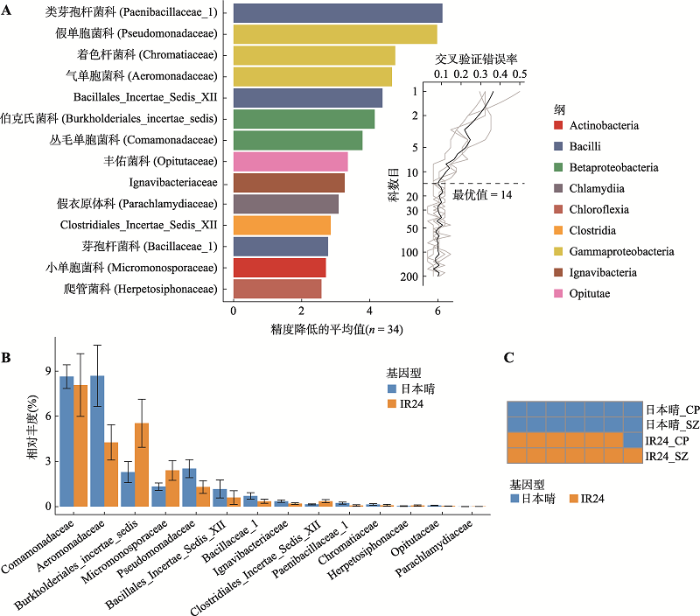 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT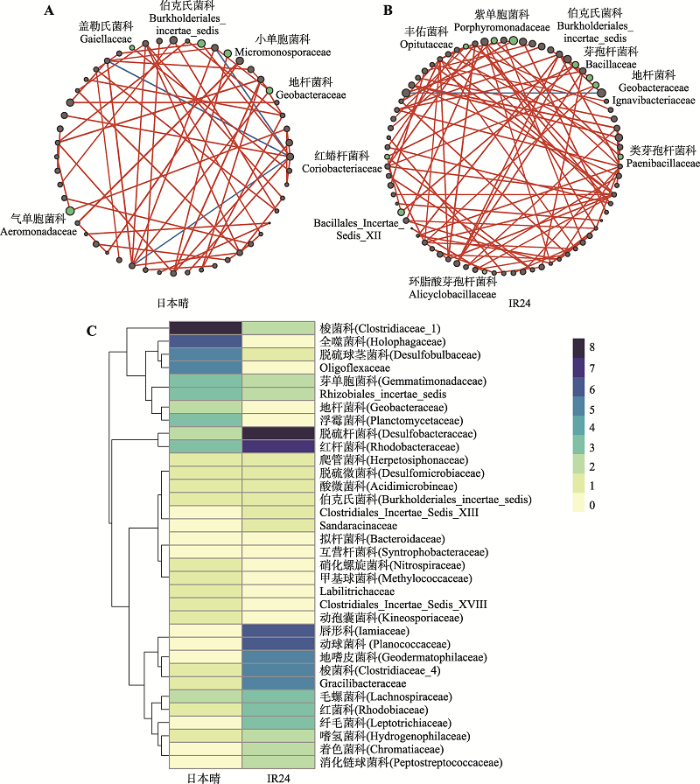 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}