Current status and future perspectives in bioinformatical analysis of Hi-C data
Hongqiang Lyu, Lele Hao, Erhu Liu, Zhifang Wu, Jiuqiang Han, Yuan LiuSchool of Electronic and Information Engineering, Xi’an Jiaotong University, Xi’an 710049, China编委: 方向东
收稿日期:2019-07-23修回日期:2019-11-26网络出版日期:2020-01-20
| 基金资助: |
Editorial board:
Received:2019-07-23Revised:2019-11-26Online:2020-01-20
| Fund supported: |
作者简介 About authors
吕红强,博士,副教授,研究方向:生物大数据分析与处理。E-mail:hongqianglv@mail.xjtu.edu.cn。

摘要
染色体的空间交互作用被视为影响基因表达调控的重要因素,高通量染色体构象捕获(high-throughput chromosome conformation capture, Hi-C)技术已成为3D基因组学中探索染色体空间交互作用的主要实验手段之一。随着Hi-C样本数据的持续累积以及分析处理流程复杂度的不断提升,基于生物信息学的Hi-C数据分析对探究基因表达的时空调控机制而言,是机遇也是挑战。本文从生物信息学角度,综合阐述了Hi-C的国内外研究现状及发展动态,包括数据标准化、多级结构分析、数据可视化以及三维建模,重点剖析了多级结构中的A/B区室(A/B compartments)、拓扑相关域(topological associated domains, TADs)和染色质环(chromain looping),在此基础上分析了该方向未来可能的研究热点及发展趋势,以期为将基因表达调控的探索从传统线性空间进一步拓展到三维结构空间提供支持。
关键词:
Abstract
The spatial interaction of chromosomes is regarded as an important issue affecting the regulation of gene expression, and the high-throughput chromosome conformation capture (Hi-C) technology has become the primary tool to explore the temporal and spatial interactions of chromosomes in three-dimensional genomics. With the continuous accumulation of Hi-C samples and the increasing complexity of pipelines, the bioinformatic analysis of Hi-C data has been considered an opportunity and a challenge for understanding the spatial regulation mechanism of gene expression. In this paper, the current status and development outline of bioinformatic methods for Hi-C data are introduced, including data normalization, multi-level structure analysis, data visualization and 3D modeling, especially of multi-level structure at A/B compartments, topological associated domains (TADs) and chromain looping levels. Based on this, we provide the outlook of future hotspots and trends in this area. Hopefully our insight will be beneficial for the exploration of gene expression regulation from the traditional linear model to the 3D mode.
Keywords:
PDF (510KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
吕红强, 郝乐乐, 刘二虎, 吴志芳, 韩九强, 刘源. 基于生物信息学的Hi-C研究现状与发展趋势. 遗传[J], 2020, 42(1): 87-99 doi:10.16288/j.yczz.19-163
Hongqiang Lyu.
基因表达的调控机制是现代分子生物学研究中的重要内容。其所研究的表达调控作用并不局限于传统的以染色体坐标为度量的一维线性结构,染色体的多级空间结构可使在线性坐标空间中的远程调控元件在三维结构空间中近距离调控目标基因的表达水平,因此染色体上各位点在细胞核中的空间交互作用被视为影响基因表达调控的重要因素。随着生物信息学领域相关研究的不断深入,高通量染色体构象捕获(high-throughput chromosome conformation capture, Hi-C)[1]技术逐渐成为探索染色体空间交互作用的主要技术手段[2],以此为核心的3D基因组学被称为基因组学研究的第三次浪潮[3]。当前,以Hi-C为代表的染色体构象捕获(chromosome conformation capture, 3C)[4]技术通过消化和重连空间上接近的染色体片段来确定不同位点之间的空间交互,为分析染色体在细胞核中的空间组织结构提供了有效途径。区别于早期3C技术的单点对单点检测,4C (chromosome conformation capture-on-chip)[5]技术 的单点对多点检测,以及5C (chromosome conformation capture carbon copy)[6]技术的多点对多点检测,Hi-C将高通量测序技术与3C技术相结合,通过全点对全点检测,构建出全基因组范围内无偏的空间交互图谱[7]。正是由于Hi-C技术的这一优越性,才使得研究全基因组范围内的染色体三维结构成为可能[8]。
Hi-C技术通过消化和重连空间上接近的染色体片段,对其进行高通量测序,可确定染色体不同位点之间的空间交互作用。其生物实验的主要步骤包括:(1)交联,用甲醛对细胞进行瞬间固定,使DNA与蛋白,蛋白与蛋白之间相互交联;(2)酶切,利用限制性内切酶(如Hind Ⅲ)对DNA进行切割,使交联两侧产生粘性末端;(3)标记,修复切割末端,并用生物素标记末端;(4)连接,使用DNA连接酶通过平端连接切割末端生成嵌合分子;(5)解交联,对纯化后的DNA嵌合分子进行超声破碎或者利用限制性酶(NheⅠ)进行打断处理,筛选出被生物素标记的DNA片段,获得DNA文库;(6)测序,对DNA文库进行高通量双端测序。近年来,随着Hi-C技术的不断成熟,逐渐发展出系列Hi-C衍生技术,如promoter CHi-C[9]、single-cell Hi-C[10]、BL-Hi-C[11]和DLO Hi-C[12]等。相比于传统的Hi-C技术,promoter CHi-C利用RNA做诱饵筛选出包含启动子的DNA片段,可用于三维结构空间中启动子调控作用的分析;single-cell Hi-C技术用以单细胞染色体构象捕获,使得单细胞水平进行染色体空间交互作用的分析成为可能;BL-Hi-C通过对酶切和连接两个步骤的改进,具备高效和灵敏的结构性和调控性染色体构象捕获能力;DLO Hi-C则通过双交联和免生物素标记的方式,在简化流程的同时,有效降低了实验数据的背景噪音。
Hi-C生物实验产生数以亿计的配对末端序列片段(paired-end sequencing reads),这些两两配对的基因组序列片段,是染色体复杂空间结构在基因组片段水平上两两交互的分解,一般通过二维接触矩阵(contact matrix)的数据组织形式进行可视化和生物信息学分析处理。其中,根据实验数据所生成的二维接触矩阵即Hi-C接触矩阵也称其为交互矩阵,矩阵的行或列代表染色体坐标上固定长度的间隔区间,区间长度被称为分辨率,其值越小,分辨率越高,矩阵元素为落入相应行和列交互区间的配对末端序列片段的数量,称为交互频率(interaction frequency, IF),其值随着行和列之间距离的增加呈指数衰减。不同分辨率下的接触矩阵如图1所示。得益于Hi-C生物实验数据的快速增长,在Gene Expression Omnibus (GEO)[13]和Encyclopedia of DNA Elements (ENCODE)[14]等综合生物数据库以及Juicer[15]等Hi-C专业数据库中,已累积了大量的覆盖多个物种不同细胞系的Hi-C重复性样本数据。
随着Hi-C技术的不断成熟以及染色体各级空间结构的陆续发现,Hi-C数据的分析与处理已成为3D基因组学的研究热点之一。近年来,国内外已有多位专家****对Hi-C方向的研究进展先后进行了综合性阐述,包括从3C到Hi-C的技术与方法的演进[3,16~18]、基于Hi-C技术的染色体多级结构[19]以及用于Hi-C数据分析的方法与工具进展[20]等。本文从生物信息学角度介绍了Hi-C的最新研究现状及发展动态,包括数据标准化、多级结构分析、数据可视化以及三维建模。在此基础上,分析了该方向未来可能的研究热点及发展趋势,以期成为现有Hi-C综述性成果在生物信息学方向的更新与补充,进而为将基因表达调控的探索从传统线性空间进一步拓展到三维结构空间提供支持。
图1
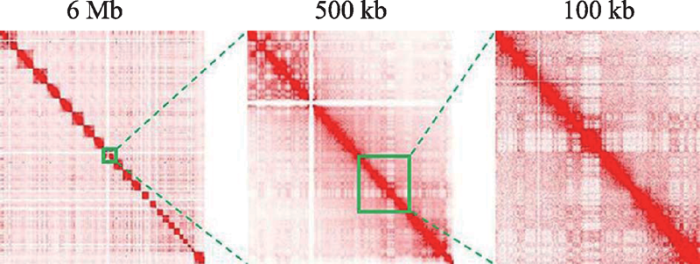 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1Hi-C接触矩阵 数据来源于Juicer数据集。
Fig. 1Hi-C contact matrix
1 Hi-C国内外研究现状
1.1 数据标准化
Hi-C数据标准化用以移除生物实验过程中由各种不可避免的非随机因素所引入的样本间的系统偏差,是后续分析处理的数据质量前提。近年来,诸多Hi-C数据标准化方法陆续被提出。2011年,Yaffe等[21]提出一种基于集成概率模型的标准化方法,其通过序列片段长度、GC含量和序列映射得到先验概率,采用最大似然估计法确定模型参数。2012年,Cournac等[22]提出序列性组件标准化(sequential component normalization, SCN)方法,通过对单染色体接触矩阵的行列归一化产生标准化的双随机矩阵。2012年,Hu等[23]提出基于泊松回归模型的HiCNorm方法,在考虑序列片段长度、GC含量和序列映射3种因素的情况下,将回归后的残差作为标准化后的接触矩阵。2012年,Imakaev等[24]提出了面向全基因组的迭代修正和特征向量分解(iterative correction and eigenvector decomposition, ICE)方法,基于交互频率库规模等量和偏差分解思想进行接触矩阵的快速标准化。2013年,Knight等[25]提出一种矩阵平衡的数学方法(knight-ruiz,KR),后被广泛应用于Hi-C接触矩阵的标准化当中。2016年,Wu等[26]提出一种通过移除拷贝数偏差(copy number bias)对原ICE标准化进行改进的caICB方法。2018年,Stansfield等[27,28]提出基于局部加权线性回归的双样本标准化方法HiCcompare,并在2019年将其升级为有能力处理多组重复性样本的MultiHiCcompare方法。2019年,Spill等[29]提出基于负二项回归模型的Binless方法,其不依赖于接触矩阵分辨率,可在配对末端序列片段水平上进行Hi-C数据标准化。各主要Hi-C数据标准化方法如表1所示。目前,除Binless之外,Hi-C数据的标准化均是在接触矩阵水平上展开。接触矩阵上的标准化方法按照是否考虑系统偏差的具体来源类型可分为显式和隐式标准化,前者如HiCNorm和caICB,后者如SCN、ICE、KR、HiCcompare和MultiHiCcompare,其按照各样本间是否存在数据交互又可分为单样本和跨样本标准化,前者如SCN、HiCNorm、ICE、KR和caICB,后者如HiCcompare和MultiHiCcompare。Table 1
表1
表1Hi-C数据标准化方法
Table 1
| 方法 | 分类 | 特点 | 实现语言 | 典型程序 |
|---|---|---|---|---|
| SCN | 隐式,单样本 | 行列归一化的矩阵平衡 | MATLAB | SCN_sumV2.m |
| HiCNorm | 显式,单样本 | 泊松回归估计系统偏差 | R | HiCNorm.R/HiTC |
| ICE | 隐式,单样本 | 迭代修正的矩阵平衡 | R,C,Python | HiTC/Hi-Corrector |
| KR | 隐式,单样本 | 内外迭代的快速矩阵平衡 | MATLAB | BNEWT.m |
| caICB | 显式,单样本 | 移除拷贝数偏差的改进ICE | R,perl | HiCapp |
| HiCcompare | 隐式,跨样本 | 双样本,局部加权线性回归 | R | HiCcompare |
| MultiHiCcompare | 隐式,跨样本 | 多样本,局部加权线性回归 | R | multiHiCcompare |
| Binless | 隐式,跨样本 | 配对末端序列片段的统计显著性分析 | R | Binless |
新窗口打开|下载CSV
1.2 多级结构分析
染色体的构象具有多个层级结构[30],其结构单元由大到小依次为染色体疆域(chromosome territories)、A/B区室(A/B compartments)、拓扑相关域(topological associated domains, TADs)和染色质环(chromain looping)等(图2)。这些分级结构及其在基因表达调控中的作用,是目前Hi-C生物信息学分析的核心内容。通过对层级结构的鉴别可将模式复杂的交互作用矩阵转化为易于解读的特征信号,既便于样本间的比较,也便于与其他生物特征关联分析[19]。在此关注除染色体疆域(图2A)之外的分级结构。1.2.1 A/B区室
A/B区室代表开放和关闭两种不同状态的染色体区域,A区室富含转录因子结合位点和活性组蛋白标记,属于转录活跃区域,而B区室含有抑制性组蛋白标记,属于转录抑制区域。2009年,Lieberman- Aiden等[1]在首次建立Hi-C技术的同时,利用特定距离上全基因组范围内的平均交互概率因子,对接触矩阵进行标准化,计算出行或列之间的皮尔逊相关系数矩阵,此矩阵的热图呈现出深浅交替的格子状模式(图2B),显示出两种不同结构特性的染色质状态,即A/B区室,通过对矩阵的主成分分析,发现第一主成分中的正负值区间信息分别对应A/B区室,其数值与基因密度、转录因子结合位点以及组蛋白标记等密切相关。2015年,Fortin等[31]提出通过不同类别的表观遗传数据,包括DNA甲基化微阵列、DNase超敏区序列、单细胞ATAC序列和单细胞全基因组亚硫酸氢盐序列等,预测多个细胞系下染色体A/B区室的方法,验证了A/B区室的结构和功能特性。2017年,山东农业大学农学院作物生物学国家重点实验室李平华实验室[32]发现大型植物的染色体可进一步划分为局部的A/B区室,这些区室反映了它们的常染色质、异染色质和多梳结构。在A/B区室识别方法研究及其结构特性分析的基础上,A/B区室与基因表达之间的关系也受到研究者的关注。2018年,Miura等[33]通过对Hi-C接触矩阵的主成分分析生成常染色体和X染色体上的A/B区室图谱数据,在此基础上,进一步分析了A/B区室的空间结构特征,并指出A/B区室的结构特异性及其与不同类型细胞中基因表达模式之间的联系。
图2
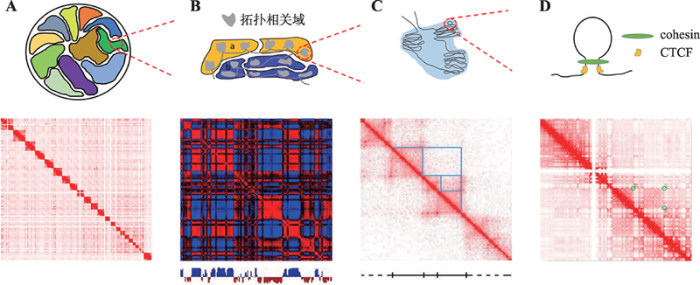 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2染色体多级结构
A:染色质疆域;B:A/B区室;C:拓扑相关域;D:染色质环。数据来源于Juicer数据集。
Fig. 2Multi-level structures of chromosomes
1.2.2 拓扑相关域
拓扑相关域TADs是染色体区域内部交互作用水平远高于相邻区域的染色体结构单元,呈嵌套式(domain-in-domain)层级结构(图2C),已被证实广泛存在于真核生物的染色体当中[34,35]。TADs边界富集染色质调控蛋白CTCF、多种组蛋白修饰和持家基因等,其结构内部的基因持有共同的调控元件,如启动子和增强子等,这些基因在多种细胞系中存在协同表达特征,由此形成一个相对独立的调控单元,被认为是复制时间调控(replication-timing regulation)的稳定结构[34,36,37]。因此,TADs是染色体三维结构中的重要高阶结构单元和基因调控单元,对TADs的识别分析有助于理解染色体的复杂结构及其与生物学功能之间的关系。2012年,Dixon等[34]在最先发现接触矩阵中TADs结构的同时,提出一种互作方向指数(directionality index, DI)识别TAD边界点,并首次分析了TADs边界点附近CTCF和组蛋白修饰的高富集度以及基因的高表达水平特征。2014年,Levy-Leduc等[38]提出采用标准动态规划法,迭代求解TADs边界分割模型以得到TADs边界点的HiCseg方法。2015年,上海交通大学Shi联合美国南加州大学Shin等[39],提出TADs边界点识别方法TopDom,其采用钻石形滑动窗口法,提取接触矩阵对角线附近窗口内交互频率的统计特征,将特征曲线的局部极大值作为TADs边界点。2016年,Weinreb等[40]提出基于TADs内部交互频率的经验分布,进行层级式TADs识别的TADtree方法。2017年,Serra等[41]提出采用基于BIC惩罚的最大似然估计求解接触矩阵交互频率的概率模型,识别TADs边界点的TADbit方法。2017年,华中农业大学彭城等[42]提出层级式TADs识别方法HiTAD,其采用基于适应性交互方向指数的隐马尔科夫模型预测TADs边界点,在此基础上,采用迭代最优化策略搜寻接触矩阵中的层级式TADs。2017年,Haddad等[43]提出采用接触矩阵行或列的层次聚类,识别层级式TADs的IC-Finder方法。2017年,Yu等[44]提出采用高斯混合模型,进行层级式TADs识别的GMAP方法。2018年,Norton等[45]提出基于图理论进行层级式TADs识别的3DNetMod方法。2018年,中国科学院北京基因组研究所张治华团队联合北京航空航天大学计算机科学学院软件开发环境国家重点实验室李昂升团队,提出一种基于图结构熵理论的快速层级式TAD识别方法deDoc[46]。2018年,清华大学生物信息学教育部重点实验室陈阳等、南方科技大学前沿与交叉科学研究院李贵鹏等以及美国德克萨斯大学Zhang等[47],提出结合局部相对隔绝指数和多尺度聚类法进行TADs边界点识别的HiCDB方法。各主要TADs识别方法如表2所示。目前,除HiCDB之外,其他方法均不具备不同条件下TADs边界点差异性分析的能力。各方法按照是否考虑TADs的层级式结构又可分为边界点式和层级式两大类,前者如DI、HiCseg、TopDom和TADbit,后者如TADtree、HiTAD、IC-Finder、GMAP和3DNetMod。
Table 2
表2
表2TADs识别方法
Table 2
| 方法 | 分类 | 特点 | 实现语言 | 典型程序 |
|---|---|---|---|---|
| DI | 边界点,非差异 | 隐马尔科夫模型 | R,Python | HiTC/TADtool |
| HiCseg | 边界点,非差异 | 二维分割矩阵 | R | HiCseg |
| TopDom | 边界点,非差异 | 钻石形滑窗法 | R | TopDom.R |
| TADtree | 层级式,非差异 | 交互频率经验分布 | Python | TADtree |
| TADbit | 边界点,非差异 | 基于BIC惩罚的概率模型 | Python | TADbit |
| HiTAD | 层级式,非差异 | 隐马尔科夫模型 | Python | TADLib |
| IC-Finder | 层级式,非差异 | 层次聚类 | MATLAB | IC-Finder.m |
| GMAP | 层级式,非差异 | 高斯混合模型 | R | GMAP |
| 3DNetMod | 层级式,非差异 | 基于图理论 | Python | 3DNetMod |
| deDoc | 层级式,非差异 | 基于图结构墒理论 | R | deDoc |
| HiCDB | 边界点,差异性 | 局部相对隔绝指数和多尺度聚类 | R,MATLAB | RHiCDB/HiCDB.m |
新窗口打开|下载CSV
1.2.3 染色质环
染色质环(chromatin loops)也可称为交互峰(interaction peaks),由染色体上相距较远的两个片段构成,其在线性空间中虽相距较远,但在三维空间结构中却具有显著的近距交互作用(图2D)。染色质环对理解染色体结构以及基因表达调控具有重要意义。2009年,Sexton等[48]基于3C技术研究了染色体的空间结构及其在基因表达调控中的作用,在分析染色体显著性交互作用的基础上,提出染色质环概念。2013年,复旦大学遗传工程国家重点实验室田卫东团队将Hi-C染色体空间交互数据引入到人类基因组作用元件与目标基因之间关系的预测当中,结果分析表明,基于Hi-C的染色质环信息能有效提升预测结果的生物功能特性及疾病相关性[49]。随后,染色质环的识别方法不断涌现。2014年,Ay等[50]对Hi-C数据中的随机聚合环和技术型系统偏差进行联合建模分析,提出了染色质环的识别方法Fit-Hi-C。2014年,Rao等[51]基于泊松分布模型提出了HiCCUPS方法,在去除TAD结构影响的前提下预测了染色质交互作用。2014年,Hwang等[52]基于负二项分布概率模型提出一种染色质环识别方法HIPPIE。2015年,Lun等[53]提出包括Hi-C配对末端序列片段预处理,数据标准化以及染色质环识别与差异分析的方法包dffHiC。2017年,中国科学院北京基因组研究所张治华[54]团队针对当时因Kbp分辨率Hi-C数据制备成本高昂而造成染色质环精确识别困难的问题,提出一种结合Kbp分辨率MNase- seq数据和低分辨Hi-C数据的染色质环精确识别方法CISD_loop。2018年,Djekidel等[55]基于空间泊松分布模型提出了检测染色质差异交互作用的方法FIND。随着对染色质环结构的深入了解,国内外相关****也针对染色质环与病理之间的关系展开研究。2018年,Manduchi等[56]借助功能基因组学数据,分析了二型糖尿病患者基因组中增强子与启动子之间的空间交互及其与基因表达调控之间的关系,证实了增强子-启动子环在该类疾病发生发展中的作用。主要的染色质环识别方法如表3所示。按照针对显著交互作用还是差异交互作用进行鉴别可以划分为两种类型。其中,针对显著交互的有Fit-HiC、HiCCUPS、HIPPIE和CISD_loop,针对差异交互的有DiffiHiC和FIND。
Table 3
表3
表3染色质环识别方法
Table 3
| 方法 | 分类 | 特点 | 实现语言 | 典型程序 |
|---|---|---|---|---|
| Fit-HiC | 显著交互 | 基于二项分布 | R,Python | Fit-HiC |
| HiCCUPS | 显著交互 | 基于泊松分布 | Java | Juicebox |
| HIPPIE | 显著交互 | 基于负二项分布 | R,Perl | HIPPIE |
| dffHiC | 差异交互 | 基于负二项分布 | R | diffiHic |
| CISD_loop | 显著交互 | 基于支持向量机模型 | R,Python | CISD_loop |
| FIND | 差异交互 | 基于泊松分布 | R | FIND |
新窗口打开|下载CSV
1.3 数据可视化
数据可视化即为Hi-C数据的图形化显示及分析,最初的形式仅为接触矩阵的热图,随着Hi-C数据的不断累积及其分析处理复杂度的不断提升,一些Hi-C可视化平台相继出现。2013年,Zhou等[57]对原有Web Server服务器WashU Epigenome Browser进行升级,在已有不同物种不同组织与细胞系的表观基因组数据和转录组数据基础上,增添了远距基因组交互数据,其可借助三角形热图和两点间弧线对Hi-C和ChIA-PET数据中的空间结构关系进行图形化注解分析。2015年,Akdemir等[58]开发出一款Hi-C专用对比分析工具HiCPlotter,其将不同条件下的Hi-C矩阵热图与多能性因子、长非编码RNA以及结构蛋白等进行图形化并置,极大方便了基于Hi-C技术的染色体结构与功能对比分析。2016年,北京大学生命科学学院李程等[59]开发了一种Web Server服务器3Disease Browser,其实现了Hi-C数据、Chip-seq数据以及疾病相关染色体重排(chromosomal rearrangement, CR)数据的整合与可视化,具备对染色体特定重排区域进行三维立体可视化的能力。同年,Durand等[60]开发了基于云平台的Hi-C可视化软件Juicebox,该软件提供对外数据接口,支持染色体、分辨率和标准化方法选择、热图缩放以及与CTCF和RNA-seq等数据的关联分析等。2017年,Djekidel等[61]开发出的Web Server服务器HiC-3Dviewer,能够在三维空间中对Hi-C接触矩阵映射到染色体的相应区域进行交互式立体可视化,且具备ChIP-Seq和SNP数据标注功能。2017年,中国科学院北京基因组研究所张治华团队[62]开发出的Web Server服务器Delta,实现了Hi-C数据和ChIA-PET数据的可视化及结构分析,包括数据的交互式可视化、TADs和染色质环的结构预测以及基因组的三维建模。2018年,Calandrelli等[63]给出了开源的Hi-C可视化软件工具GITAR,该软件支持Hi-C数据预处理、标准化、TADs分析以及不同样本对比的可视化操作及结果显示。2018年,Wang等[64]开发的三维基因组Web Server服务器3D Genome browser,囊括了人类与小鼠的300多项不同类型数据,包括Hi-C、ChIA-PET、Capture Hi-C、PLAC-Seq、HiChIP、GAM和SPRITE,集成了包括ICE标准化、A/B区室识别和TADs识别工具的分析结果。同年,Wolff等[65]开发的集Hi-C数据预处理、接触矩阵标准化、A/B区室和TADs识别以及基因表达谱数据和Chip-seq数据等辅助分析于一体的可视化Web Server服务器Galaxy HiCExplorer,实现了Hi-C数据分析处理过程中绝大多数流程的数据可视化,人机交互更为友善。各主要Hi-C可视化工具软件如表4所示。Table 4
表4
表4Hi-C数据可视化软件
Table 4
| 方法 | 交互 | 网址 |
|---|---|---|
| WashU Epigenome Browser | 浏览器 | http://epigenomegateway.wustl.edu/ |
| HiCPlotter | Python软件工具 | https://github.com/kcakdemir/HiCPlotter |
| 3Disease Browser | 浏览器 | http://3dgb.cbi.pku.edu.cn/disease/ |
| Juicebox | 浏览器,Java软件工具 | http://aidenlab.org/juicebox |
| HiC-3Dviewer | 浏览器 | http://bioinfo.au.tsinghua.edu.cn/member/nadhir/HiC3DViewer/ |
| Delta | Java软件工具 | http://delta.big.ac.cn |
| GITAR | Python软件工具 | http://genomegitar.org |
| 3D Genome browser | 浏览器 | http://3dgenome.org |
| Galaxy HiCExplorer | 浏览器 | https://hicexplorer.usegalaxy.eu |
新窗口打开|下载CSV
1.4 三维建模
三维建模是Hi-C的一项重要应用,其通过Hi-C数据的建模分析得到染色体的结构信息,从而在三维立体空间中重现染色体的物理结构,以辅助科学研究。2002年,Dekker等[4]在提出3C技术以及交互频率矩阵概念的基础上,借助聚合体柔度及多种结构参数估算出酵母菌3号染色体上78对位点之间的空间距离,进而首次建立起基于3C数据的染色体空间构象三维模型。2011年,Rousseau等[66]提出一种适用于5C和Hi-C数据的染色体空间结构三维建模方法MCMC5C,该方法给出从染色体交互频率到位点空间距离的概率模型,采用马尔可夫链蒙特卡罗抽样算法进行求解,并将其用在1Mb分辨率的Hi-C数据集上,建立起人类14号染色体的三维模型。2013年,Zhang等[67]提出基于Hi-C数据的染色体三维结构建模方法ChromSDE,其借助黄金分割搜索算法对交互频率与空间距离之间的转换进行参数优化,利用半正定规划技术建立起染色体三维结构模型。2013年,华中农业大学彭城等[68]提出一种基于Hi-C数据的染色体三维结构建模方法AutoChrom3D,其借助测序序列偏置松弛结构参数和分段线性函数实现各位点空间距离的转换,建立起染色体的三维结构模型,不同于以往其它建模方法,该方法考虑了不同实验中测序深度所引发的区域交互作用的偏差。2015年,Trieu等[69]在已有单染色体三维结构建模方法的基础上,提出了基因组三维结构建模软件MOGEN,该软件能够有效处理噪声以及不同染色体间Hi-C数据的差异。2017年,Paulsen等[70]提出了基于Hi-C数据和核纤层蛋白Chip-seq数据的全基因组三维结构建模软件Chrom3D,相比于之前的同类方法,Chrom3D集成了TADs径向位置约束条件,具备在单细胞水平进行全基因组三维空间结构建模的能力。2018年,Segal等[71]针对现有染色体三维结构重建算法准确性难以评估的现状,提出了基于染色体结构图谱的新的精度评估方法。同年,清华大学曾坚阳团队[72]提出一种基于构象能和流行学习的染色体三维结构建模框架GEM,与其它同类建模方法相比,GEM综合考虑了Hi-C交互数据以及染色体的生物物理可行性和结构稳定性,在方法有效性和模型物理生物特性验证中具备优势。可以看出,上述染色体三维空间结构建模方法,大多基于两步走的思路,即首先由交互频率数据推算出染色体各位点之间的空间距离,然后借助空间距离数据构建出染色体的空间结构模型,如MCMC5C、ChromSDE、AutoChrom3D和Chrom3D。与其形成对比的是不需要先行估算出各位点之间的空间距离,而是一种基于交互频率数据的结构模型优化过程,如MOGEN和GEM。2 Hi-C研究发展动态
从2009年Hi-C技术的首次提出,到实验数据的分析处理,基于Hi-C技术的染色体空间结构研究历经了大约10年时间。10年来,Hi-C数据的生物信息学分析进展迅速。在数据标准化方面,2012年到2013年期间,快速涌现出SCN[22]、ICE[24]和KR[25]等多种基于矩阵平衡策略的隐式标准化方法,以及以HiCNorm[23]为代表的基于模型构建策略的显式标准化方法。在随后的几年内,虽然也出现了多种接触矩阵标准化方法,如通过移除拷贝数偏差对ICE进行改进的caICB[26]方法,但这些方法均局限于对单样本Hi-C接触矩阵的处理。直到2018年,HiCcompare[27]方法首次将Hi-C接触矩阵标准化推向了双样本层面。2019年,其又被提出者升级为能够满足重复性样本标准化的MultiHiCcompare[28]方法。同年,出现了在配对末端序列水平上的标准化方法Binless[29]。可以看出,尽管各Hi-C数据标准化方法在显式和隐式,矩阵平衡和模型构建,以及接触矩阵水平和配对末端序列片段水平上有所差异,但基本呈现出由单样本标准化向跨样本标准化推进的发展动态。
在多级结构方面,2009年,Hi-C接触矩阵中的A/B区室被发现,首个A/B区室计算方法被提出[1]。同年,基于3C技术的染色质环结构被发现,其在基因表达调控中的作用被分析。2012年,接触矩阵中的TADs结构被发现,首个TADs边界点识别算法DI被提出[34]。2013年,TADs的层级结构被发现[35]。此后,涌现出多种Hi-C接触矩阵中各级结构的识别分析方法。如2014年的染色质环识别方法Fit-Hi-C[50]、HiCCUPS[51]和HIPPIE[52],2015年的多个细胞系下染色体A/B区室预测方法,同年的首个不同条件下染色质环差异分析方法dffHiC[53],2016年的首个层级式TADs识别方法TADtree[40],2017年的层级式TADs识别方法TADbit[41]、HiTAD[42]、IC-Finder[43]和GMAP[44],以及2018年首个支持TADs边界点差异性分析的方法HiCDB[47]。可以看出,接触矩阵中多级结构的探索大体经历了从结构发现到识别分析的过程。虽然A/B区室结构最早被发现,但对多级结构的分析更多集中在TADs和染色质环上,其中,对TADs的研究已从接触矩阵对角线上TADs边界点的识别,逐步深入到TADs层级式结构及其功能分析,而对染色质环的研究则呈现出由结构识别预测向不同条件下结构差异分析逐步推进的发展态势。
在数据可视化方面,自Hi-C技术提出之时就已使用log比例上的热图来显示接触矩阵数据。此后,在2013年,原Web Server服务器WashU Epigenome Browser[57]通过升级,具备了Hi-C数据热图显示及其功能可视化关联分析的功能。随着Hi-C数据的持续累积及其分析处理复杂度的不断提升,2016年,出现了Hi-C数据云平台及可视化分析软件Juicebox[60],以及集成Hi-C数据、Chip-seq数据和CR数据,且支持重排区域三维可视化的Web Server服务器3Disease Browser[59],为Hi-C相关数据的获取、结果关联分析以及三维可视化提供了软件工具支撑。2017年,出现了交互式三维立体可视化Web Server服务器HiC-3Dviewer[61],以及集成多种Hi-C数据分析工具的Web Server服务器Delta[62]。2018年,进一步涌现出集成多种Hi-C相关数据及其分析工具的可视化软件,如GITAR[63]、3D Genome browser[64]和Galaxy HiCExplorer[65]。可以看出,Hi-C数据可视化软件呈现出数据类型复杂化多样化、视觉交互三维立体化以及分析工具集成化的发展态势。
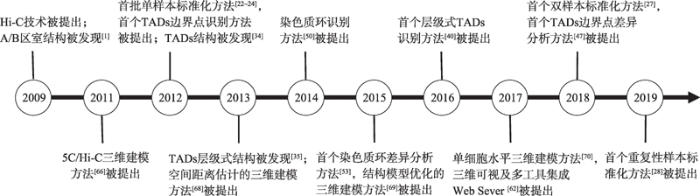
在三维建模方面,2011年,适用于5C和Hi-C数据的染色体空间结构三维建模方法MCMC5C[66]被提出。此后,专注于Hi-C数据的染色体三维建模方法陆续出现,如2013年的ChromSDE[67]和AutoChrom3D[68]。此几种方法虽然在考量因素和具体算法上有所不同,但均遵循了从交互频率到染色体各位点空间距离推算,再到染色体空间结构建模的同一思路。随后,出现了一类无需先行估算各位点空间距离,而是直接基于Hi-C交互频率数据进行染色体空间结构建模的方法,如2015年的全基因组三维结构建模方法MOGEN[69],以及2018年基于构象能和流行学习的建模方法GEM[71]。此外,2017年,出现了支持在单细胞水平上进行全基因组Hi-C三维建模的方法Chrom3D[70]。可以看出,基于Hi-C数据的染色体三维建模方法,经历了由分步计算到直接建模,由单个染色体向全基因组,再向单细胞水平逐步拓展的发展过程(图3)。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Hi-C研究发展动态
Fig. 3Development outline of studies on Hi-C
3 Hi-C研究发展趋势
从上述研究现状和发展动态可以看出,Hi-C技术及实验数据分析已经成为三维基因组学中备受关注的问题。以下仅从4个方面对Hi-C生物信息学方向的研究趋势进行浅析,包括跨样本标准化、多级结构差异及其调控机制分析、单细胞Hi-C数据分析和Hi-C数据可视化平台。3.1 跨样本标准化
Hi-C数据的标准化绝大多数是在接触矩阵水平上展开。接触矩阵上的标准化方法按照是否考虑系统偏差的具体来源类型可分为显式和隐式标准化,按照各样本间是否存在数据交互又可分为单样本和跨样本标准化。标准化是后续分析的数据质量保障,而单样本标准化方法无法保障两组重复性样本之间的统计可比性,仅有的跨样本标准化方法HiCcompare和MultiHiCcompare,在接触矩阵分辨率不断提高、重复性样本数量持续增加和后续分析处理日趋复杂化的情况下,面临质量、效率和方法选择上的多重压力。因此,适用于高分辨率的跨样本高效标准化方法的研究,是Hi-C数据后续分析的结果质量保证和必经之路。3.2 多级结构差异及其调控机制分析
差异分析是研究基因表达调控的重要手段之 一,其通过分析不同条件下两组样本之间的显著性差异,探索基因和表型之间的联系。在基于Hi-C的三维基因组学中,染色体的多级结构与基因表达调控息息相关,使得不同条件下多级结构的差异分析成为此新领域的核心问题之一。如上述Hi-C研究现状和发展动态所述,目前,虽然已涌现出多种用于A/B区室、TADs和染色质环分析的方法软件,但具有差异分析能力的方法却十分缺乏,如可用于Hi-C接触矩阵中染色质环差异分析的方法diffHic。大多差异分析仍停留在单样本实验验证探索或者简单统计分析阶段。例如,Fraser等[73]在2015年给出小鼠胚胎干细胞分化过程中不同时间点上层级式TADs的树形结构,并采用协表相关系数,分析了不同细胞系下TADs树形结构的构造差异。随着接触矩阵分辨率的不断提高,各级结构的可预测数目迅猛增长,再加上为降低随机误差而引入的重复性样本,单靠热图对比和统计检验已远远不能满足后续差异分析的需要,因此,在三维基因组学,研究不同条件下,包括两种正常细胞系之间、正常细胞系与癌变细胞系之间以及同一细胞系不同时间点之间,Hi-C数据中多级结构,包括A/B区室、TADs和染色质环,的差异分析方法,进而探索各级差异性结构在基因表达调控中作用机制,是探索生物体细胞分化、形态产生和疾病发生发展等不可或缺的手段,其必将成为未来Hi-C领域生物信息学的研究热点之一。3.3 单细胞Hi-C数据分析
单细胞Hi-C技术用于稀少细胞或者处于特殊形态细胞的染色体构象捕获。常规Hi-C技术只能借助群体细胞构象数据的平均值来估计染色体交互作用,个别细胞的重要信号往往会被当作异常值受到弱化,而单细胞Hi-C技术可以很好解决细胞群体的异质性问题,其通过对生命活动最小单位的空间构象进行捕获,得到更有针对性的染色体交互信息。自Takashi等[10]于2013年提出单细胞Hi-C技术以来,单细胞Hi-C数据分析也应运而生。例如,Liu等[74]于2018年提出用于消除单细胞Hi-C数据中系统性偏差的软件包scHiCNorm;Liu等[75]于2019年利用单细胞测序揭示了与骨髓基质细胞亚群和培养时间相关的基因表达特征。单细胞Hi-C技术使得在单细胞水平进行染色体空间交互作用的研究成为可能,极大推进了三维基因组学的发展,基于该项技术的单细胞Hi-C数据分析,使得不同条件下各类细胞之间的空间构象得以精细区分,对探究基因表达调控的时空机制意义重大,势必受到专家****的广泛关注与重视。3.4 Hi-C数据可视化平台
随着Hi-C数据中各级结构及其生物学功能分析的不断深入,可视化平台也面临诸多挑战,逐步朝着数据复杂化多样化、视觉交互三维立体化以及分析工具集成化方向发展。Hi-C数据的复杂化多样化,即各物种不同组织和不同细胞系下Hi-C数据和各类组学数据的整合、关联与显示,包括不同分辨率Hi-C数据和ChIP-seq、RNA-seq、SNP以及疾病相关CR等数据;视觉交互三维立体化,即交互作用数据的可视化已不再局限于传统热图形式,呈现出与三维建模相结合的交互式三维立体显示趋势;分析工具集成化,即各类用于Hi-C数据分析的基础性方法工具逐渐被集成到系统平台当中,如标准化方法以及A/B区室、TADs和染色质环预测方法等。此外,Hi-C数据的集约型分析显示方法也日趋重要。得益于Hi-C技术的进步,接触矩阵的分辨率得到了显著提高,从原来的Mb级别发展到现今的1 Kb甚至200 bp[76],这使得高分辨率条件下Hi-C数据的处理显示面临计算资源不足的压力,因此,高效快速的Hi-C数据组织、分析及可视化方法工具在平台集成中将更具优势。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1126/science.1181369URLPMID:19815776 [本文引用: 3]

We describe Hi-C, a method that probes the three-dimensional architecture of whole genomes by coupling proximity-based ligation with massively parallel sequencing. We constructed spatial proximity maps of the human genome with Hi-C at a resolution of 1 megabase. These maps confirm the presence of chromosome territories and the spatial proximity of small, gene-rich chromosomes. We identified an additional level of genome organization that is characterized by the spatial segregation of open and closed chromatin to form two genome-wide compartments. At the megabase scale, the chromatin conformation is consistent with a fractal globule, a knot-free, polymer conformation that enables maximally dense packing while preserving the ability to easily fold and unfold any genomic locus. The fractal globule is distinct from the more commonly used globular equilibrium model. Our results demonstrate the power of Hi-C to map the dynamic conformations of whole genomes.
DOI:10.1038/nrm.2016.104URLPMID:27580841 [本文引用: 1]
Chromosomes of eukaryotes adopt highly dynamic and complex hierarchical structures in the nucleus. The three-dimensional (3D) organization of chromosomes profoundly affects DNA replication, transcription and the repair of DNA damage. Thus, a thorough understanding of nuclear architecture is fundamental to the study of nuclear processes in eukaryotic cells. Recent years have seen rapid proliferation of technologies to investigate genome organization and function. Here, we review experimental and computational methodologies for 3D genome analysis, with special focus on recent advances in high-throughput chromatin conformation capture (3C) techniques and data analysis.
DOI:10.1360/N972014-00163URL [本文引用: 2]
DOI:10.1360/N972014-00163URL [本文引用: 2]
DOI:10.1126/science.1067799URLPMID:11847345 [本文引用: 2]
We describe an approach to detect the frequency of interaction between any two genomic loci. Generation of a matrix of interaction frequencies between sites on the same or different chromosomes reveals their relative spatial disposition and provides information about the physical properties of the chromatin fiber. This methodology can be applied to the spatial organization of entire genomes in organisms from bacteria to human. Using the yeast Saccharomyces cerevisiae, we could confirm known qualitative features of chromosome organization within the nucleus and dynamic changes in that organization during meiosis. We also analyzed yeast chromosome III at the G1 stage of the cell cycle. We found that chromatin is highly flexible throughout. Furthermore, functionally distinct AT- and GC-rich domains were found to exhibit different conformations, and a population-average 3D model of chromosome III could be determined. Chromosome III emerges as a contorted ring.
DOI:10.1038/ng1891URLPMID:17033624 [本文引用: 1]
Accumulating evidence converges on the possibility that chromosomes interact with each other to regulate transcription in trans. To systematically explore the epigenetic dimension of such interactions, we devised a strategy termed circular chromosome conformation capture (4C). This approach involves a circularization step that enables high-throughput screening of physical interactions between chromosomes without a preconceived idea of the interacting partners. Here we identify 114 unique sequences from all autosomes, several of which interact primarily with the maternally inherited H19 imprinting control region. Imprinted domains were strongly overrepresented in the library of 4C sequences, further highlighting the epigenetic nature of these interactions. Moreover, we found that the direct interaction between differentially methylated regions was linked to epigenetic regulation of transcription in trans. Finally, the patterns of interactions specific to the maternal H19 imprinting control region underwent reprogramming during in vitro maturation of embryonic stem cells. These observations shed new light on development, cancer epigenetics and the evolution of imprinting.
DOI:10.1101/gr.5571506URLPMID:16954542 [本文引用: 1]
Physical interactions between genetic elements located throughout the genome play important roles in gene regulation and can be identified with the Chromosome Conformation Capture (3C) methodology. 3C converts physical chromatin interactions into specific ligation products, which are quantified individually by PCR. Here we present a high-throughput 3C approach, 3C-Carbon Copy (5C), that employs microarrays or quantitative DNA sequencing using 454-technology as detection methods. We applied 5C to analyze a 400-kb region containing the human beta-globin locus and a 100-kb conserved gene desert region. We validated 5C by detection of several previously identified looping interactions in the beta-globin locus. We also identified a new looping interaction in K562 cells between the beta-globin Locus Control Region and the gamma-beta-globin intergenic region. Interestingly, this region has been implicated in the control of developmental globin gene switching. 5C should be widely applicable for large-scale mapping of cis- and trans- interaction networks of genomic elements and for the study of higher-order chromosome structure.
DOI:10.16288/j.yczz.17-152URLPMID:28936982 [本文引用: 1]
Highest-throughput chromosome conformation capture (Hi-C) is one of the key assays for genome- wide chromatin interaction studies. It is a time-consuming process that involves many steps and many different kinds of reagents, consumables, and equipments. At present, the reproducibility is unsatisfactory. By optimizing the key steps of the Hi-C experiment, such as crosslinking, pretreatment of digestion, inactivation of restriction enzyme, and in situ ligation etc., we established a robust Hi-C procedure and prepared two biological replicates of Hi-C libraries from the GM12878 cells. After preliminary quality control by Sanger sequencing, the two replicates were high-throughput sequenced. The bioinformatics analysis of the raw sequencing data revealed the mapping-ability and pair-mate rate of the raw data were around 90% and 72%, respectively. Additionally, after removal of self-circular ligations and dangling-end products, more than 96% of the valid pairs were reached. Genome-wide interactome profiling shows clear topological associated domains (TADs), which is consistent with previous reports. Further correlation analysis showed that the two biological replicates strongly correlate with each other in terms of both bin coverage and all bin pairs. All these results indicated that the optimized Hi-C procedure is robust and stable, which will be very helpful for the wide applications of the Hi-C assay.
DOI:10.16288/j.yczz.17-152URLPMID:28936982 [本文引用: 1]
Highest-throughput chromosome conformation capture (Hi-C) is one of the key assays for genome- wide chromatin interaction studies. It is a time-consuming process that involves many steps and many different kinds of reagents, consumables, and equipments. At present, the reproducibility is unsatisfactory. By optimizing the key steps of the Hi-C experiment, such as crosslinking, pretreatment of digestion, inactivation of restriction enzyme, and in situ ligation etc., we established a robust Hi-C procedure and prepared two biological replicates of Hi-C libraries from the GM12878 cells. After preliminary quality control by Sanger sequencing, the two replicates were high-throughput sequenced. The bioinformatics analysis of the raw sequencing data revealed the mapping-ability and pair-mate rate of the raw data were around 90% and 72%, respectively. Additionally, after removal of self-circular ligations and dangling-end products, more than 96% of the valid pairs were reached. Genome-wide interactome profiling shows clear topological associated domains (TADs), which is consistent with previous reports. Further correlation analysis showed that the two biological replicates strongly correlate with each other in terms of both bin coverage and all bin pairs. All these results indicated that the optimized Hi-C procedure is robust and stable, which will be very helpful for the wide applications of the Hi-C assay.
DOI:10.1101/gad.179804.111URLPMID:22215806 [本文引用: 1]
Over the past 10 years, the development of chromosome conformation capture (3C) technology and the subsequent genomic variants thereof have enabled the analysis of nuclear organization at an unprecedented resolution and throughput. The technology relies on the original and, in hindsight, remarkably simple idea that digestion and religation of fixed chromatin in cells, followed by the quantification of ligation junctions, allows for the determination of DNA contact frequencies and insight into chromosome topology. Here we evaluate and compare the current 3C-based methods (including 4C [chromosome conformation capture-on-chip], 5C [chromosome conformation capture carbon copy], HiC, and ChIA-PET), summarize their contribution to our current understanding of genome structure, and discuss how shape influences genome function.
DOI:10.1101/gr.185272.114URLPMID:25752748 [本文引用: 1]
The mammalian genome harbors up to one million regulatory elements often located at great distances from their target genes. Long-range elements control genes through physical contact with promoters and can be recognized by the presence of specific histone modifications and transcription factor binding. Linking regulatory elements to specific promoters genome-wide is currently impeded by the limited resolution of high-throughput chromatin interaction assays. Here we apply a sequence capture approach to enrich Hi-C libraries for >22,000 annotated mouse promoters to identify statistically significant, long-range interactions at restriction fragment resolution, assigning long-range interacting elements to their target genes genome-wide in embryonic stem cells and fetal liver cells. The distal sites contacting active genes are enriched in active histone modifications and transcription factor occupancy, whereas inactive genes contact distal sites with repressive histone marks, demonstrating the regulatory potential of the distal elements identified. Furthermore, we find that coregulated genes cluster nonrandomly in spatial interaction networks correlated with their biological function and expression level. Interestingly, we find the strongest gene clustering in ES cells between transcription factor genes that control key developmental processes in embryogenesis. The results provide the first genome-wide catalog linking gene promoters to their long-range interacting elements and highlight the complex spatial regulatory circuitry controlling mammalian gene expression.
DOI:10.1038/nature12593URL [本文引用: 2]
Large-scale chromosome structure and spatial nuclear arrangement have been linked to control of gene expression and DNA replication and repair. Genomic techniques based on chromosome conformation capture (3C) assess contacts for millions of loci simultaneously, but do so by averaging chromosome conformations from millions of nuclei. Here we introduce single-cell Hi-C, combined with genome-wide statistical analysis and structural modelling of single-copy X chromosomes, to show that individual chromosomes maintain domain organization at the megabase scale, but show variable cell-to-cell chromosome structures at larger scales. Despite this structural stochasticity, localization of active gene domains to boundaries of chromosome territories is a hallmark of chromosomal conformation. Single-cell Hi-C data bridge current gaps between genomics and microscopy studies of chromosomes, demonstrating how modular organization underlies dynamic chromosome structure, and how this structure is probabilistically linked with genome activity patterns.
DOI:10.1038/s41467-017-01754-3URLPMID:29158486 [本文引用: 1]
In human cells, DNA is hierarchically organized and assembled with histones and DNA-binding proteins in three dimensions. Chromatin interactions play important roles in genome architecture and gene regulation, including robustness in the developmental stages and flexibility during the cell cycle. Here we propose in situ Hi-C method named Bridge Linker-Hi-C (BL-Hi-C) for capturing structural and regulatory chromatin interactions by restriction enzyme targeting and two-step proximity ligation. This method improves the sensitivity and specificity of active chromatin loop detection and can reveal the regulatory enhancer-promoter architecture better than conventional methods at a lower sequencing depth and with a simpler protocol. We demonstrate its utility with two well-studied developmental loci: the beta-globin and HOXC cluster regions.
DOI:10.1038/s41588-018-0111-2URLPMID:29700467 [本文引用: 1]
Chromosome conformation capture (3C) technologies can be used to investigate 3D genomic structures. However, high background noise, high costs, and a lack of straightforward noise evaluation in current methods impede the advancement of 3D genomic research. Here we developed a simple digestion-ligation-only Hi-C (DLO Hi-C) technology to explore the 3D landscape of the genome. This method requires only two rounds of digestion and ligation, without the need for biotin labeling and pulldown. Non-ligated DNA was efficiently removed in a cost-effective step by purifying specific linker-ligated DNA fragments. Notably, random ligation could be quickly evaluated in an early quality-control step before sequencing. Moreover, an in situ version of DLO Hi-C using a four-cutter restriction enzyme has been developed. We applied DLO Hi-C to delineate the genomic architecture of THP-1 and K562 cells and uncovered chromosomal translocations. This technology may facilitate investigation of genomic organization, gene regulation, and (meta)genome assembly.
DOI:10.1016/S0076-6879(06)11019-8URLPMID:16939800 [本文引用: 1]
The Gene Expression Omnibus (GEO) repository at the National Center for Biotechnology Information archives and freely distributes high-throughput molecular abundance data, predominantly gene expression data generated by DNA microarray technology. The database has a flexible design that can handle diverse styles of both unprocessed and processed data in a Minimum Information About a Microarray Experiment-supportive infrastructure that promotes fully annotated submissions. GEO currently stores about a billion individual gene expression measurements, derived from over 100 organisms, submitted by over 1500 laboratories, addressing a wide range of biological phenomena. To maximize the utility of these data, several user-friendly web-based interfaces and applications have been implemented that enable effective exploration, query, and visualization of these data at the level of individual genes or entire studies. This chapter describes how data are stored, submission procedures, and mechanisms for data retrieval and query. GEO is publicly accessible at http://www.ncbi.nlm.nih.gov/projects/geo/.
DOI:10.1016/j.gpb.2013.05.001URLPMID:23722115 [本文引用: 1]
The ENCyclopedia Of DNA Elements (ENCODE) project is an international research consortium that aims to identify all functional elements in the human genome sequence. The second phase of the project comprised 1640 datasets from 147 different cell types, yielding a set of 30 publications across several journals. These data revealed that 80.4% of the human genome displays some functionality in at least one cell type. Many of these regulatory elements are physically associated with one another and further form a network or three-dimensional conformation to affect gene expression. These elements are also related to sequence variants associated with diseases or traits. All these findings provide us new insights into the organization and regulation of genes and genome, and serve as an expansive resource for understanding human health and disease.
[本文引用: 1]
DOI:10.1101/gad.179804.111URLPMID:22215806 [本文引用: 1]
Over the past 10 years, the development of chromosome conformation capture (3C) technology and the subsequent genomic variants thereof have enabled the analysis of nuclear organization at an unprecedented resolution and throughput. The technology relies on the original and, in hindsight, remarkably simple idea that digestion and religation of fixed chromatin in cells, followed by the quantification of ligation junctions, allows for the determination of DNA contact frequencies and insight into chromosome topology. Here we evaluate and compare the current 3C-based methods (including 4C [chromosome conformation capture-on-chip], 5C [chromosome conformation capture carbon copy], HiC, and ChIA-PET), summarize their contribution to our current understanding of genome structure, and discuss how shape influences genome function.
DOI:10.1093/bib/bbv085URLPMID:26433013
Chromosome conformation capture techniques are producing a huge amount of data about the architecture of our genome. These data can provide us with a better understanding of the events that induce critical regulations of the cellular function from small changes in the three-dimensional genome architecture. Generating a unified view of spatial, temporal, genetic and epigenetic properties poses various challenges of data analysis, visualization, integration and mining, as well as of high performance computing and big data management. Here, we describe the critical issues of this new branch of bioinformatics, oriented at the comprehension of the three-dimensional genome architecture, which we call 'Nucleome Bioinformatics', looking beyond the currently available tools and methods, and highlight yet unaddressed challenges and the potential approaches that could be applied for tackling them. Our review provides a map for researchers interested in using computer science for studying 'Nucleome Bioinformatics', to achieve a better understanding of the biological processes that occur inside the nucleus.
DOI:10.1038/nrm.2016.104URLPMID:27580841 [本文引用: 1]
Chromosomes of eukaryotes adopt highly dynamic and complex hierarchical structures in the nucleus. The three-dimensional (3D) organization of chromosomes profoundly affects DNA replication, transcription and the repair of DNA damage. Thus, a thorough understanding of nuclear architecture is fundamental to the study of nuclear processes in eukaryotic cells. Recent years have seen rapid proliferation of technologies to investigate genome organization and function. Here, we review experimental and computational methodologies for 3D genome analysis, with special focus on recent advances in high-throughput chromatin conformation capture (3C) techniques and data analysis.
DOI:10.1016/j.tibs.2018.03.006URLPMID:29685368 [本文引用: 2]
Chromosomes are folded and compacted in interphase nuclei, but the molecular basis of this folding is poorly understood. Chromosome conformation capture methods, such as Hi-C, combine chemical crosslinking of chromatin with fragmentation, DNA ligation, and high-throughput DNA sequencing to detect neighboring loci genome-wide. Hi-C has revealed the segregation of chromatin into active and inactive compartments and the folding of DNA into self-associating domains and loops. Depletion of CTCF, cohesin, or cohesin-associated proteins was recently shown to affect the majority of domains and loops in a manner that is consistent with a model of DNA folding through extrusion of chromatin loops. Compartmentation was not dependent on CTCF or cohesin. Hi-C contact maps represent the superimposition of CTCF/cohesin-dependent and -independent folding states.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/ng.947URLPMID:22001755 [本文引用: 1]
Hi-C experiments measure the probability of physical proximity between pairs of chromosomal loci on a genomic scale. We report on several systematic biases that substantially affect the Hi-C experimental procedure, including the distance between restriction sites, the GC content of trimmed ligation junctions and sequence uniqueness. To address these biases, we introduce an integrated probabilistic background model and develop algorithms to estimate its parameters and renormalize Hi-C data. Analysis of corrected human lymphoblast contact maps provides genome-wide evidence for interchromosomal aggregation of active chromatin marks, including DNase-hypersensitive sites and transcriptionally active foci. We observe extensive long-range (up to 400 kb) cis interactions at active promoters and derive asymmetric contact profiles next to transcription start sites and CTCF binding sites. Clusters of interacting chromosomal domains suggest physical separation of centromere-proximal and centromere-distal regions. These results provide a computational basis for the inference of chromosomal architectures from Hi-C experiments.
DOI:10.1093/nar/gkx644URLPMID:28973466 [本文引用: 2]
Hi-C experiments generate data in form of large genome contact maps (Hi-C maps). These show that chromosomes are arranged in a hierarchy of three-dimensional compartments. But to understand how these compartments form and by how much they affect genetic processes such as gene regulation, biologists and bioinformaticians need efficient tools to visualize and analyze Hi-C data. However, this is technically challenging because these maps are big. In this paper, we remedied this problem, partly by implementing an efficient file format and developed the genome contact map explorer platform. Apart from tools to process Hi-C data, such as normalization methods and a programmable interface, we made a graphical interface that let users browse, scroll and zoom Hi-C maps to visually search for patterns in the Hi-C data. In the software, it is also possible to browse several maps simultaneously and plot related genomic data. The software is openly accessible to the scientific community.
DOI:10.1093/bioinformatics/bts570URL [本文引用: 2]
We propose a parametric model, HiCNorm, to remove systematic biases in the raw Hi-C contact maps, resulting in a simple, fast, yet accurate normalization procedure. Compared with the existing Hi-C normalization method developed by Yaffe and Tanay, HiCNorm has fewer parameters, runs>1000 times faster and achieves higher reproducibility.
DOI:10.1038/NMETH.2148URL [本文引用: 2]
Extracting biologically meaningful information from chromosomal interactions obtained with genome-wide chromosome conformation capture (3C) analyses requires the elimination of systematic biases. We present a computational pipeline that integrates a strategy to map sequencing reads with a data-driven method for iterative correction of biases, yielding genome-wide maps of relative contact probabilities. We validate this ICE (iterative correction and eigenvector decomposition) technique on published data obtained by the high-throughput 3C method Hi-C, and we demonstrate that eigenvector decomposition of the obtained maps provides insights into local chromatin states, global patterns of chromosomal interactions, and the conserved organization of human and mouse chromosomes.
DOI:10.1093/imanum/drs019URL [本文引用: 2]
As long as a square non-negative matrix A has total support then it can be balanced, that is, we can find a diagonal scaling of A that has row and column sums equal to one. A number of algorithms have been proposed to achieve the balancing, the most well known of these being Sinkhorn-Knopp. In this paper, we derive new algorithms based on inner-outer iteration schemes. We show that Sinkhorn-Knopp belongs to this family, but other members can converge much more quickly. In particular, we show that while stationary iterative methods offer little or no improvement in many cases, a scheme using a preconditioned conjugate gradient method as the inner iteration converges at much lower cost (in terms of matrix-vector products) for a broad range of matrices; and succeeds in cases where the Sinkhorn-Knopp algorithm fails.
DOI:10.1093/bioinformatics/btw540URLPMID:27531101 [本文引用: 2]
The Hi-C technology was designed to decode the three-dimensional conformation of the genome. Despite progress towards more and more accurate contact maps, several systematic biases have been demonstrated to affect the resulting data matrix. Here we report a new source of bias that can arise in tumor Hi-C data, which is related to the copy number of genomic DNA. To address this bias, we designed a chromosome-adjusted iterative correction method called caICB. Our caICB correction method leads to significant improvements when compared with the original iterative correction in terms of eliminating copy number bias.
DOI:10.1186/s12859-018-2288-xURLPMID:30064362 [本文引用: 2]
Changes in spatial chromatin interactions are now emerging as a unifying mechanism orchestrating the regulation of gene expression. Hi-C sequencing technology allows insight into chromatin interactions on a genome-wide scale. However, Hi-C data contains many DNA sequence- and technology-driven biases. These biases prevent effective comparison of chromatin interactions aimed at identifying genomic regions differentially interacting between, e.g., disease-normal states or different cell types. Several methods have been developed for normalizing individual Hi-C datasets. However, they fail to account for biases between two or more Hi-C datasets, hindering comparative analysis of chromatin interactions.
DOI:10.1093/bioinformatics/btz048URLPMID:30668639 [本文引用: 2]
With the development of chromatin conformation capture technology and its high-throughput derivative Hi-C sequencing, studies of the three-dimensional interactome of the genome that involve multiple Hi-C datasets are becoming available. To account for the technology-driven biases unique to each dataset, there is a distinct need for methods to jointly normalize multiple Hi-C datasets. Previous attempts at removing biases from Hi-C data have made use of techniques which normalize individual Hi-C datasets, or, at best, jointly normalize two datasets.
DOI:10.1038/s41467-019-09907-2URLPMID:31028255 [本文引用: 2]
Chromosome conformation capture techniques, such as Hi-C, are fundamental in characterizing genome organization. These methods have revealed several genomic features, such as chromatin loops, whose disruption can have dramatic effects in gene regulation. Unfortunately, their detection is difficult; current methods require that the users choose the resolution of interaction maps based on dataset quality and sequencing depth. Here, we introduce Binless, a resolution-agnostic method that adapts to the quality and quantity of available data, to detect both interactions and differences. Binless relies on an alternate representation of Hi-C data, which leads to a more detailed classification of paired-end reads. Using a large-scale benchmark, we demonstrate that Binless is able to call interactions with higher reproducibility than other existing methods. Binless, which is freely available, can thus reliably be used to identify chromatin loops as well as for differential analysis of chromatin interaction maps.
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s13059-015-0741-yURLPMID:26316348 [本文引用: 1]
Analysis of Hi-C data has shown that the genome can be divided into two compartments called A/B compartments. These compartments are cell-type specific and are associated with open and closed chromatin. We show that A/B compartments can reliably be estimated using epigenetic data from several different platforms: the Illumina 450 k DNA methylation microarray, DNase hypersensitivity sequencing, single-cell ATAC sequencing and single-cell whole-genome bisulfite sequencing. We do this by exploiting that the structure of long-range correlations differs between open and closed compartments. This work makes A/B compartment assignment readily available in a wide variety of cell types, including many human cancers.
DOI:10.1016/j.molp.2017.11.005URLPMID:29175436 [本文引用: 1]
The spatial organization of the genome plays an important role in the regulation of gene expression. However, the core structural features of animal genomes, such as topologically associated domains (TADs) and chromatin loops, are not prominent in the extremely compact Arabidopsis genome. In this study, we examine?the chromatin architecture, as well as their DNA methylation, histone modifications, accessible chromatin, and gene expression, of maize, tomato, sorghum, foxtail millet, and rice with genome sizes ranging from 0.4?to 2.4 Gb. We found that these plant genomes can be divided into mammalian-like A/B compartments. At higher resolution, the chromosomes of these plants can be further partitioned to local A/B compartments that reflect their euchromatin, heterochromatin, and polycomb status. Chromatins in all these plants are organized into domains that are not conserved across species. They show similarity to the Drosophila compartment domains, and are clustered into active, polycomb, repressive, and intermediate types based on their transcriptional activities and epigenetic signatures, with domain border overlaps with the local A/B compartment junctions. In the large maize and tomato genomes, we observed extensive chromatin loops. However, unlike the mammalian chromatin loops that are enriched at the TAD border, plant chromatin loops are often formed between gene islands outside the repressive domains?and are closely associated with active compartments. Our study indicates that plants have complex and unique 3D chromatin architectures, which require?further study to elucidate their biological functions.
DOI:10.1007/978-1-0716-0247-8_19URLPMID:31939184 [本文引用: 1]
Expression of programmed death ligand-1 (PD-L1, CD274) on cancer cells is regulated by interferon-γ (IFNγ) signaling as well as by epigenetic mechanisms. By binding to PD-1 on cytotoxic T cells, PD-L1 inhibits T cell-mediated antitumor responses, resulting in immune escape. This chapter describes analysis of the surface PD-L1 expression in ovarian cancer (OC) cells using flow cytometry (FC). Our data demonstrate that the surface PD-L1 expression in OC cells is induced by IFNγ as well as by the class I histone deacetylase (HDAC) inhibition by romidepsin, suggesting that class I HDAC inhibition might provide a useful strategy to modulate the PD-L1 levels on OC cells.
DOI:10.1038/nature11082URL [本文引用: 4]
The spatial organization of the genome is intimately linked to its biological function, yet our understanding of higher order genomic structure is coarse, fragmented and incomplete. In the nucleus of eukaryotic cells, interphase chromosomes occupy distinct chromosome territories, and numerous models have been proposed for how chromosomes fold within chromosome territories(1). These models, however, provide only few mechanistic details about the relationship between higher order chromatin structure and genome function. Recent advances in genomic technologies have led to rapid advances in the study of three-dimensional genome organization. In particular, Hi-C has been introduced as a method for identifying higher order chromatin interactions genome wide(2). Here we investigate the three-dimensional organization of the human and mouse genomes in embryonic stem cells and terminally differentiated cell types at unprecedented resolution. We identify large, megabase-sized local chromatin interaction domains, which we term 'topological domains', as a pervasive structural feature of the genome organization. These domains correlate with regions of the genome that constrain the spread of heterochromatin. The domains are stable across different cell types and highly conserved across species, indicating that topological domains are an inherent property of mammalian genomes. Finally, we find that the boundaries of topological domains are enriched for the insulator binding protein CTCF, housekeeping genes, transfer RNAs and short interspersed element (SINE) retrotransposons, indicating that these factors may have a role in establishing the topological domain structure of the genome.
DOI:10.1016/j.cell.2013.04.053URL [本文引用: 2]
Understanding the topological configurations of chromatin may reveal valuable insights into how the genome and epigenome act in concert to control cell fate during development. Here, we generate high-resolution architecture maps across seven genomic loci in embryonic stem cells and neural progenitor cells. We observe a hierarchy of 3D interactions that undergo marked reorganization at the submegabase scale during differentiation. Distinct combinations of CCCTC-binding factor (CTCF), Mediator, and cohesin show widespread enrichment in chromatin interactions at different length scales. CTCF/cohesin anchor long-range constitutive interactions that might form the topological basis for invariant subdomains. Conversely, Mediator/cohesin bridge short-range enhancer-promoter interactions within and between larger subdomains. Knockdown of Smc1 or Med12 in embryonic stem cells results in disruption of spatial architecture and downregulation of genes found in cohesin-mediated interactions. We conclude that cell-type-specific chromatin organization occurs at the submegabase scale and that architectural proteins shape the genome in hierarchical length scales.
DOI:10.1038/nature13986URL [本文引用: 1]
Eukaryotic chromosomes replicate in a temporal order known as the replication-timing program(1). In mammals, replication timing is cell-type-specific with at least half the genome switching replication timing during development, primarily in units of 400-800 kilobases (replication domains'), whose positions are preserved in different cell types, conserved between species, and appear to confine long-range effects of chromosome rearrangements(2-7). Early and late replication correlate, respectively, with open and closed three-dimensional chromatin compartments identified by high-resolution chromosome conformation capture (Hi-C), and, to a lesser extent, late replication correlates with lamina-associated domains (LADs)(4,5,8,9). Recent Hi-C mapping has unveiled substructure within chromatin compartments called topologically associating domains (TADs) that are largely conserved in their positions between cell types and are similar in size to replication domains(8,10). However, TADs can be further sub-stratified into smaller domains, challenging the significance of structures at any particular scale(11,12). Moreover, attempts to reconcile TADs and LADs to replication-timing data have not revealed a common, underlying domain structure(8,9,13). Here we localize boundaries of replication domains to the early-replicating border of replication-timing transitions and map their positions in 18 human and 13 mouse cell types. We demonstrate that, collectively, replication domain boundaries share a near one-to-one correlation with TAD boundaries, whereas within a cell type, adjacent TADs that replicate at similar times obscure replication domain boundaries, largely accounting for the previously reported lack of alignment. Moreover, cell-type-specific replication timing of TADs partitions the genome into two large-scale sub-nuclear compartments revealing that replication-timing transitions are indistinguishable from late-replicating regions in chromatin composition and lamina association and accounting for the reduced correlation of replication timing to LADs and heterochromatin. Our results reconcile cell-type-specific sub-nuclear compartmentalization and replication timing with developmentally stable structural domains and offer a unified model for large-scale chromosome structure and function.
DOI:10.1101/gad.288324.116URLPMID:28087711 [本文引用: 1]
The genome is organized into repeating topologically associated domains (TADs), each of which is spatially isolated from its neighbor by poorly understood boundary elements thought to be conserved across cell types. Here, we show that deletion of CTCF (CCCTC-binding factor)-binding sites at TAD and sub-TAD topological boundaries that form within the HoxA and HoxC clusters during differentiation not only disturbs local chromatin domain organization and regulatory interactions but also results in homeotic transformations typical of Hox gene misregulation. Moreover, our data suggest that CTCF-dependent boundary function can be modulated by competing forces, such as the self-assembly of polycomb domains within the nucleus. Therefore, CTCF boundaries are not merely static structural components of the genome but instead are locally dynamic regulatory structures that control gene expression during development.
DOI:10.1093/bioinformatics/btu443URL [本文引用: 1]
Motivation: The spatial conformation of the chromosome has a deep influence on gene regulation and expression. Hi-C technology allows the evaluation of the spatial proximity between any pair of loci along the genome. It results in a data matrix where blocks corresponding to (self-) interacting regions appear. The delimitation of such blocks is critical to better understand the spatial organization of the chromatin. From a computational point of view, it results in a 2D segmentation problem.
Results: We focus on the detection of cis-interacting regions, which appear to be prominent in observed data. We define a block-wise segmentation model for the detection of such regions. We prove that the maximization of the likelihood with respect to the block boundaries can be rephrased in terms of a 1D segmentation problem, for which the standard dynamic programming applies. The performance of the proposed methods is assessed by a simulation study on both synthetic and resampled data. A comparative study on public data shows good concordance with biologically confirmed regions.
DOI:10.1093/nar/gkv1505URLPMID:26704975 [本文引用: 1]
Genome-wide proximity ligation assays allow the identification of chromatin contacts at unprecedented resolution. Several studies reveal that mammalian chromosomes are composed of topological domains (TDs) in sub-mega base resolution, which appear to be conserved across cell types and to some extent even between organisms. Identifying topological domains is now an important step toward understanding the structure and functions of spatial genome organization. However, current methods for TD identification demand extensive computational resources, require careful tuning and/or encounter inconsistencies in results. In this work, we propose an efficient and deterministic method, TopDom, to identify TDs, along with a set of statistical methods for evaluating their quality. TopDom is much more efficient than existing methods and depends on just one intuitive parameter, a window size, for which we provide easy-to-implement optimization guidelines. TopDom also identifies more and higher quality TDs than the popular directional index algorithm. The TDs identified by TopDom provide strong support for the cross-tissue TD conservation. Finally, our analysis reveals that the locations of housekeeping genes are closely associated with cross-tissue conserved TDs. The software package and source codes of TopDom are available athttp://zhoulab.usc.edu/TopDom/.
DOI:10.1093/bioinformatics/btv485URLPMID:26315910 [本文引用: 2]
The three-dimensional structure of the genome is an important regulator of many cellular processes including differentiation and gene regulation. Recently, technologies such as Hi-C that combine proximity ligation with high-throughput sequencing have revealed domains of self-interacting chromatin, called topologically associating domains (TADs), in many organisms. Current methods for identifying TADs using Hi-C data assume that TADs are non-overlapping, despite evidence for a nested structure in which TADs and sub-TADs form a complex hierarchy.
DOI:10.1371/journal.pcbi.1005665URLPMID:28723903 [本文引用: 2]
The sequence of a genome is insufficient to understand all genomic processes carried out in the cell nucleus. To achieve this, the knowledge of its three-dimensional architecture is necessary. Advances in genomic technologies and the development of new analytical methods, such as Chromosome Conformation Capture (3C) and its derivatives, provide unprecedented insights in the spatial organization of genomes. Here we present TADbit, a computational framework to analyze and model the chromatin fiber in three dimensions. Our package takes as input the sequencing reads of 3C-based experiments and performs the following main tasks: (i) pre-process the reads, (ii) map the reads to a reference genome, (iii) filter and normalize the interaction data, (iv) analyze the resulting interaction matrices, (v) build 3D models of selected genomic domains, and (vi) analyze the resulting models to characterize their structural properties. To illustrate the use of TADbit, we automatically modeled 50 genomic domains from the fly genome revealing differential structural features of the previously defined chromatin colors, establishing a link between the conformation of the genome and the local chromatin composition. TADbit provides three-dimensional models built from 3C-based experiments, which are ready for visualization and for characterizing their relation to gene expression and epigenetic states. TADbit is an open-source Python library available for download from https://github.com/3DGenomes/tadbit.
DOI:10.1093/nar/gkx735URLPMID:28977529 [本文引用: 2]
A current question in the high-order organization of chromatin is whether topologically associating domains (TADs) are distinct from other hierarchical chromatin domains. However, due to the unclear TAD definition in tradition, the structural and functional uniqueness of TAD is not well studied. In this work, we refined TAD definition by further constraining TADs to the optimal separation on global intra-chromosomal interactions. Inspired by this constraint, we developed a novel method, called HiTAD, to detect hierarchical TADs from Hi-C chromatin interactions. HiTAD performs well in domain sensitivity, replicate reproducibility and inter cell-type conservation. With a novel domain-based alignment proposed by us, we defined several types of hierarchical TAD changes which were not systematically studied previously, and subsequently used them to reveal that TADs and sub-TADs differed statistically in correlating chromosomal compartment, replication timing and gene transcription. Finally, our work also has the implication that the refinement of TAD definition could be achieved by only utilizing chromatin interactions, at least in part. HiTAD is freely available online.
DOI:10.1093/nar/gkx036URLPMID:28130423 [本文引用: 2]
The spatial organization of the genome plays a crucial role in the regulation of gene expression. Recent experimental techniques like Hi-C have emphasized the segmentation of genomes into interaction compartments that constitute conserved functional domains participating in the maintenance of a proper cell identity. Here, we propose a novel method, IC-Finder, to identify interaction compartments (IC) from experimental Hi-C maps. IC-Finder is based on a hierarchical clustering approach that we adapted to account for the polymeric nature of chromatin. Based on a benchmark of realistic in silico Hi-C maps, we show that IC-Finder is one of the best methods in terms of reliability and is the most efficient numerically. IC-Finder proposes two original options: a probabilistic description of the inferred compartments and the possibility to explore the various hierarchies of chromatin organization. Applying the method to experimental data in fly and human, we show how the predicted segmentation may depend on the normalization scheme and how 3D compartmentalization is tightly associated with epigenomic information. IC-Finder provides a robust and generic 'all-in-one' tool to uncover the general principles of 3D chromatin folding and their influence on gene regulation. The software is available at http://membres-timc.imag.fr/Daniel.Jost/DJ-TIMC/Software.html.
DOI:10.1038/s41467-017-00478-8URLPMID:28912419 [本文引用: 2]
The spatial organization of the genome plays a critical role in regulating gene expression. Recent chromatin interaction mapping studies have revealed that topologically associating domains and subdomains are fundamental building blocks of the three-dimensional genome. Identifying such hierarchical structures is a critical step toward understanding the three-dimensional structure-function relationship of the genome. Existing computational algorithms lack statistical assessment of domain predictions and are computationally inefficient for high-resolution Hi-C data. We introduce the Gaussian Mixture model And Proportion test (GMAP) algorithm to address the above-mentioned challenges. Using simulated and experimental Hi-C data, we show that domains identified by GMAP are more consistent with multiple lines of supporting evidence than three state-of-the-art methods. Application of GMAP to normal and cancer cells reveals several unique features of subdomain boundary as compared to domain boundary, including its higher dynamics across cell types and enrichment for somatic mutations in cancer.Spatial organization of the genome plays a crucial role in regulating gene expression. Here the authors introduce GMAP, the Gaussian Mixture model And Proportion test, to identify topologically associating domains and subdomains in Hi-C data.
DOI:10.1038/nmeth.4560URLPMID:29334377 [本文引用: 1]
Mammalian genomes are folded in a hierarchy of compartments, topologically associating domains (TADs), subTADs and looping interactions. Here, we describe 3DNetMod, a graph theory-based method for sensitive and accurate detection of chromatin domains across length scales in Hi-C data. We identify nested, partially overlapping TADs and subTADs genome wide by optimizing network modularity and varying a single resolution parameter. 3DNetMod can be applied broadly to understand genome reconfiguration in development and disease.
DOI:10.1038/s41467-018-05691-7URLPMID:30111883 [本文引用: 1]
Submegabase-size topologically associating domains (TAD) have been observed in high-throughput chromatin interaction data (Hi-C). However, accurate detection of TADs depends on ultra-deep sequencing and sophisticated normalization procedures. Here we propose a fast and normalization-free method to decode the domains of chromosomes (deDoc) that utilizes structural information theory. By treating Hi-C contact matrix as a representation of a graph, deDoc partitions the graph into segments with minimal structural entropy. We show that structural entropy can also be used to determine the proper bin size of the Hi-C data. By applying deDoc to pooled Hi-C data from 10 single cells, we detect megabase-size TAD-like domains. This result implies that the modular structure of the genome spatial organization may be fundamental to even a small cohort of single cells. Our algorithms may facilitate systematic investigations of chromosomal domains on a larger scale than hitherto have been possible.
DOI:10.1093/nar/gky789URLPMID:30184171 [本文引用: 2]
Contact domains are closely linked to gene regulation and lineage commitment, while current understanding of contact domains and their boundaries is still limited. Here, we present a novel method HiCDB, which is constructively based on local relative insulation metric and multi-scale aggregation approach to detect contact domain boundaries (CDBs) on Hi-C maps. Compared with other 'state-of-art' methods, HiCDB shows improved sensitivity and specificity in determining CDBs at various Hi-C resolutions. The superiority of HiCDB enabled us to study the epigenetic features of detected CDBs and showed enrichment of architectural proteins and cell-type-specific transcription factor binding sites at CDBs. The further comparison of GM12878 and IMR90 Hi-C datasets suggested that cell-type-specific CDBs are marked by active regulatory signals and correlate with activation of nearby cell identity genes.
DOI:10.1016/j.semcdb.2009.06.004URL [本文引用: 1]
Abstract
The chromosome conformation capture (3C) technique and its genome-wide applications (‘4C’) have identified a plethora of distal DNA sequences that are frequently in close spatial proximity. In many cases, these have been correlated with transcriptional regulation of the interacting genes, but the functional significance of many of the extreme long-range and interchromosomal interactions remains unclear. This review summarises our current understanding of how chromatin conformation can impinge on gene expression, the major questions that need to be addressed to understand this more fully, and how these questions may be answered in the near future.DOI:10.1093/nar/gkt785URLPMID:24003029 [本文引用: 1]
Defining the target genes of distal regulatory elements (DREs), such as enhancer, repressors and insulators, is a challenging task. The recently developed Hi-C technology is designed to capture chromosome conformation structure by high-throughput sequencing, and can be potentially used to determine the target genes of DREs. However, Hi-C data are noisy, making it difficult to directly use Hi-C data to identify DRE-target gene relationships. In this study, we show that DREs-gene pairs that are confirmed by Hi-C data are strongly phylogenetic correlated, and have thus developed a method that combines Hi-C read counts with phylogenetic correlation to predict long-range DRE-target gene relationships. Analysis of predicted DRE-target gene pairs shows that genes regulated by large number of DREs tend to have essential functions, and genes regulated by the same DREs tend to be functionally related and co-expressed. In addition, we show with a couple of examples that the predicted target genes of DREs can help explain the causal roles of disease-associated single-nucleotide polymorphisms located in the DREs. As such, these predictions will be of importance not only for our understanding of the function of DREs but also for elucidating the causal roles of disease-associated noncoding single-nucleotide polymorphisms.
DOI:10.1101/gr.160374.113URL [本文引用: 2]
Our current understanding of how DNA is packed in the nucleus is most accurate at the fine scale of individual nucleosomes and at the large scale of chromosome territories. However, accurate modeling of DNA architecture at the intermediate scale of similar to 50 kb-10 Mb is crucial for identifying functional interactions among regulatory elements and their target promoters. We describe a method, Fit-Hi-C, that assigns statistical confidence estimates to mid-range intra-chromosomal contacts by jointly modeling the random polymer looping effect and previously observed technical biases in Hi-C data sets. We demonstrate that our proposed approach computes accurate empirical null models of contact probability without any distribution assumption, corrects for binning artifacts, and provides improved statistical power relative to a previously described method. High-confidence contacts identified by Fit-Hi-C preferentially link expressed gene promoters to active enhancers identified by chromatin signatures in human embryonic stem cells (ESCs), capture 77% of RNA polymerase II-mediated enhancer-promoter interactions identified using ChIA-PET in mouse ESCs, and confirm previously validated, cell line-specific interactions in mouse cortex cells. We observe that insulators and heterochromatin regions are hubs for high-confidence contacts, while promoters and strong enhancers are involved in fewer contacts. We also observe that binding peaks of master pluripotency factors such as NANOG and POU5F1 are highly enriched in high-confidence contacts for human ESCs. Furthermore, we show that pairs of loci linked by high-confidence contacts exhibit similar replication timing in human and mouse ESCs and preferentially lie within the boundaries of topological domains for human and mouse cell lines.
DOI:10.1016/j.cell.2014.11.021URL [本文引用: 2]
We use in situ Hi-C to probe the 3D architecture of genomes, constructing haploid and diploid maps of nine cell types. The densest, in human lymphoblastoid cells, contains 4.9 billion contacts, achieving 1 kb resolution. We find that genomes are partitioned into contact domains (median length, 185 kb), which are associated with distinct patterns of histone marks and segregate into six subcompartments. We identify similar to 10,000 loops. These loops frequently link promoters and enhancers, correlate with gene activation, and show conservation across cell types and species. Loop anchors typically occur at domain boundaries and bind CTCF. CTCF sites at loop anchors occur predominantly (>90%) in a convergent orientation, with the asymmetric motifs "facing'' one another. The inactive X chromosome splits into two massive domains and contains large loops anchored at CTCF-binding repeats.
DOI:10.1093/bioinformatics/btu801URLPMID:25480377 [本文引用: 2]
We implemented a high-throughput identification pipeline for promoter interacting enhancer element to streamline the workflow from mapping raw Hi-C reads, identifying DNA-DNA interacting fragments with high confidence and quality control, detecting histone modifications and DNase hypersensitive enrichments in putative enhancer elements, to ultimately extracting possible intra- and inter-chromosomal enhancer-target gene relationships.
DOI:10.1186/s12870-016-0945-7URLPMID:27905870 [本文引用: 2]
During the photosynthesis, two isoforms of the fructose-1,6-bisphosphatase (FBPase), the chloroplastidial (cFBP1) and the cytosolic (cyFBP), catalyse the first irreversible step during the conversion of triose phosphates (TP) to starch or sucrose, respectively. Deficiency in cyFBP and cFBP1 isoforms provokes an imbalance of the starch/sucrose ratio, causing a dramatic effect on plant development when the plastidial enzyme is lacking.
DOI:10.1093/nar/gkx885URLPMID:29036650 [本文引用: 1]
High-throughput chromosome conformation capture (3C) technologies, such as Hi-C, have made it possible to survey 3D genome structure. However, obtaining 3D profiles at kilobase resolution at low cost remains a major challenge. Therefore, we herein present an algorithm for precise identification of chromatin interaction sites at kilobase resolution from MNase-seq data, termed chromatin interaction site detector (CISD), and a CISD-based chromatin loop predictor (CISD_loop) that predicts chromatin-chromatin interactions (CCIs) from low-resolution Hi-C data. We show that the predictions of CISD and CISD_loop overlap closely with chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) anchors and loops, respectively. The validity of CISD/CISD_loop was further supported by a 3C assay at about 5 kb resolution. Finally, we demonstrate that only modest amounts of MNase-seq and Hi-C data are sufficient to achieve ultrahigh resolution CCI maps. Our results suggest that CCIs may result in characteristic nucleosomes arrangement patterns flanking the interaction sites, and our algorithms may facilitate precise and systematic investigations of CCIs on a larger scale than hitherto have been possible.
DOI:10.1101/gr.212241.116URLPMID:29440282 [本文引用: 1]
Polymer-based simulations and experimental studies indicate the existence of a spatial dependency between the adjacent DNA fibers involved in the formation of chromatin loops. However, the existing strategies for detecting differential chromatin interactions assume that the interacting segments are spatially independent from the other segments nearby. To resolve this issue, we developed a new computational method, FIND, which considers the local spatial dependency between interacting loci. FIND uses a spatial Poisson process to detect differential chromatin interactions that show a significant difference in their interaction frequency and the interaction frequency of their neighbors. Simulation and biological data analysis show that FIND outperforms the widely used count-based methods and has a better signal-to-noise ratio.
URLPMID:29218913 [本文引用: 1]
We utilized evidence for enhancer-promoter interactions from functional genomics data in order to build biological filters to narrow down the search space for two-way Single Nucleotide Polymorphism (SNP) interactions in Type 2 Diabetes (T2D) Genome Wide Association Studies (GWAS). This has led us to the identification of a reproducible statistically significant SNP pair associated with T2D. As more functional genomics data are being generated that can help identify potentially interacting enhancer-promoter pairs in larger collection of tissues/cells, this approach has implications for investigation of epistasis from GWAS in general.
DOI:10.1038/nmeth.2440URLPMID:23629413 [本文引用: 2]
DOI:10.1186/s13059-015-0767-1URLPMID:26392354 [本文引用: 1]
Metazoan genomic material is folded into stable non-randomly arranged chromosomal structures that are tightly associated with transcriptional regulation and DNA replication. Various factors including regulators of pluripotency, long non-coding RNAs, or the presence of architectural proteins have been implicated in regulation and assembly of the chromatin architecture. Therefore, comprehensive visualization of this multi-faceted structure is important to unravel the connections between nuclear architecture and transcriptional regulation. Here, we present an easy-to-use open-source visualization tool, HiCPlotter, to facilitate juxtaposition of Hi-C matrices with diverse genomic assay outputs, as well as to compare interaction matrices between various conditions. https://github.com/kcakdemir/HiCPlotter.
DOI:10.1038/srep34651URLPMID:27734896 [本文引用: 2]
Chromosomal rearrangement (CR) events have been implicated in many tumor and non-tumor human diseases. CR events lead to their associated diseases by disrupting gene and protein structures. Also, they can lead to diseases through changes in chromosomal 3D structure and gene expression. In this study, we search for CR-associated diseases potentially caused by chromosomal 3D structure alteration by integrating Hi-C and ChIP-seq data. Our algorithm rediscovers experimentally verified disease-associated CRs (polydactyly diseases) that alter gene expression by disrupting chromosome 3D structure. Interestingly, we find that intellectual disability may be a candidate disease caused by 3D chromosome structure alteration. We also develop a Web server (3Disease Browser, http://3dgb.cbi.pku.edu.cn/disease/) for integrating and visualizing disease-associated CR events and chromosomal 3D structure.
DOI:10.1016/j.cels.2015.07.012URLPMID:27467250 [本文引用: 2]
Hi-C experiments study how genomes fold in 3D, generating contact maps containing features as small as 20?bp and as large as 200 Mb. Here we introduce Juicebox, a tool for exploring Hi-C and other contact map data. Juicebox allows users to zoom in and out of Hi-C maps interactively, just as a user of Google Earth might zoom in and out of a geographic map. Maps can be compared to one another, or to 1D tracks or 2D feature sets.
DOI:10.1007/s40484-017-0091-8URL [本文引用: 2]
DOI:10.1093/bioinformatics/btx805URLPMID:29253110 [本文引用: 2]
Delta is an integrative visualization and analysis platform to facilitate visually annotating and exploring the 3D physical architecture of genomes. Delta takes Hi-C or ChIA-PET contact matrix as input and predicts the topologically associating domains and chromatin loops in the genome. It then generates a physical 3D model which represents the plausible consensus 3D structure of the genome. Delta features a highly interactive visualization tool which enhances the integration of genome topology/physical structure with extensive genome annotation by juxtaposing the 3D model with diverse genomic assay outputs. Finally, by visually comparing the 3D model of the β-globin gene locus and its annotation, we speculated a plausible transitory interaction pattern in the locus. Experimental evidence was found to support this speculation by literature survey. This served as an example of intuitive hypothesis testing with the help of Delta.
DOI:10.1016/j.cbd.2020.100654URLPMID:31954363 [本文引用: 2]
Histia rhodope Cramer (Lepidoptera: Zygaenidae) is one of the most destructive defoliators of landscape tree Bischofia polycarpa (Levl.) Airy Shaw in China stretching to other Southeast Asia regions. Olfactory genes, encoding proteins such as odorant carrier proteins believed to initiate olfactory signal transduction in insects, have been acknowledged to be novel targets for pest control. In this study, we established antennal transcriptome of H. rhodope and ultimately identified 19 odorant binding proteins (OBPs), 23 chemosensory proteins (CSPs) and 4 Niemann-Pick type C2 proteins (NPC2s). The 19 OBPs, 6 CSPs and 4 NPC2s were assessed to validate the differential expressions between sexes, and between olfactory and non-olfactory tissues. 8 OBPs and 2 CSPs exhibited male-biased antennae expression, while 6 OBPs, 2 CSPs and HrhoNPC2a exhibited female-biased antennae expression. Moreover, 17 OBPs, 4 CSPs and 2 NPC2s were predominantly expressed in the antennae compared with non-olfactory tissues. HrhoOBP1 and HrhoOBP8 were predominantly expressed in the antennae and heads, HrhoCSP8 and HrhoCSP14 were highly expressed in abdomens and legs, HrhoNPC2c was highly expressed in abdomens, while HrhoNPC2d was expressed in all tissues. Phylogenetic analysis revealed that most H. rhodope proteins were closely related to proteins from other moths. Moreover, compared with other nocturnal moths, acting as a diurnal moth, we found that H. rhodope may have lost a PBP gene. Our results provide important molecular information for further studies on olfactory mechanisms of H. rhodope.
DOI:10.1186/s13059-018-1519-9URLPMID:30286773 [本文引用: 2]
Here, we introduce the 3D Genome Browser, http://3dgenome.org , which allows users to conveniently explore both their own and over 300 publicly available chromatin interaction data of different types. We design a new binary data format for Hi-C data that reduces the file size by at least a magnitude and allows users to visualize chromatin interactions over millions of base pairs within seconds. Our browser provides multiple methods linking distal cis-regulatory elements with their potential target genes. Users can seamlessly integrate thousands of other omics data to gain a comprehensive view of both regulatory landscape and 3D genome structure.
DOI:10.1093/nar/gky504URLPMID:29901812 [本文引用: 2]
Galaxy HiCExplorer is a web server that facilitates the study of the 3D conformation of chromatin by allowing Hi-C data processing, analysis and visualization. With the Galaxy HiCExplorer web server, users with little bioinformatic background can perform every step of the analysis in one workflow: mapping of the raw sequence data, creation of Hi-C contact matrices, quality assessment, correction of contact matrices and identification of topological associated domains (TADs) and A/B compartments. Users can create publication ready plots of the contact matrix, A/B compartments, and TADs on a selected genomic locus, along with additional information like gene tracks or ChIP-seq signals. Galaxy HiCExplorer is freely usable at: https://hicexplorer.usegalaxy.eu and is available as a Docker container: https://github.com/deeptools/docker-galaxy-hicexplorer.
DOI:10.1186/1471-2105-12-414URLPMID:22026390 [本文引用: 2]
Long-range interactions between regulatory DNA elements such as enhancers, insulators and promoters play an important role in regulating transcription. As chromatin contacts have been found throughout the human genome and in different cell types, spatial transcriptional control is now viewed as a general mechanism of gene expression regulation. Chromosome Conformation Capture Carbon Copy (5C) and its variant Hi-C are techniques used to measure the interaction frequency (IF) between specific regions of the genome. Our goal is to use the IF data generated by these experiments to computationally model and analyze three-dimensional chromatin organization.
[本文引用: 2]
DOI:10.1093/nar/gkt745URLPMID:23965308 [本文引用: 2]
The 3D chromatin structure modeling by chromatin interactions derived from Hi-C experiments is significantly challenged by the intrinsic sequencing biases in these experiments. Conventional modeling methods only focus on the bias among different chromatin regions within the same experiment but neglect the bias arising from different experimental sequencing depth. We now show that the regional interaction bias is tightly coupled with the sequencing depth, and we further identify a chromatin structure parameter as the inherent characteristics of Hi-C derived data for chromatin regions. Then we present an approach for chromatin structure prediction capable of relaxing both kinds of sequencing biases by using this identified parameter. This method is validated by intra and inter cell-line comparisons among various chromatin regions for four human cell-lines (K562, GM12878, IMR90 and H1hESC), which shows that the openness of chromatin region is well correlated with chromatin function. This method has been executed by an automatic pipeline (AutoChrom3D) and thus can be conveniently used.
DOI:10.1093/bioinformatics/btv754URLPMID:26722115 [本文引用: 2]
The three-dimensional (3D) conformation of chromosomes and genomes play an important role in cellular processes such as gene regulation, DNA replication and genome methylation. Several methods have been developed to reconstruct 3D structures of individual chromosomes from chromosomal conformation capturing data such as Hi-C data. However, few methods can effectively reconstruct the 3D structures of an entire genome due to the difficulty of handling noisy and inconsistent inter-chromosomal contact data.
DOI:10.1186/s13059-016-1146-2URLPMID:28137286 [本文引用: 2]
Current three-dimensional (3D) genome modeling platforms are limited by their inability to account for radial placement of loci in the nucleus. We present Chrom3D, a user-friendly whole-genome 3D computational modeling framework that simulates positions of topologically-associated domains (TADs) relative to each other and to the nuclear periphery. Chrom3D integrates chromosome conformation capture (Hi-C) and lamin-associated domain (LAD) datasets to generate structure ensembles that recapitulate radial distributions of TADs detected in single cells. Chrom3D reveals unexpected spatial features of LAD regulation in cells from patients with a laminopathy-causing lamin mutation. Chrom3D is freely available on github.
DOI:10.1186/s12859-018-2214-2URLPMID:29848293 [本文引用: 2]
Three dimensional (3D) genome spatial organization is critical for numerous cellular functions, including transcription, while certain conformation-driven structural alterations are frequently oncogenic. Genome conformation had been difficult to elucidate but the advent chromatin conformation capture assays, notably Hi-C, has transformed understanding of chromatin architecture and yielded numerous biological insights. Although most of these findings have flowed from analysis of proximity data produced by these assays, added value in generating 3D reconstructions has been demonstrated, deriving, in part, from superposing genomic features on the reconstruction. However, advantages of 3D structure-based analyses are clearly conditional on the accuracy of the attendant reconstructions, which is difficult to assess. Proponents of competing reconstruction algorithms have evaluated their accuracy by recourse to simulation of toy structures and/or limited fluorescence in situ hybridization (FISH) imaging that features a handful of low resolution probes. Accordingly, new methods of reconstruction accuracy assessment are needed.
DOI:10.1093/nar/gky065URLPMID:29408992 [本文引用: 1]
Decoding the spatial organizations of chromosomes has crucial implications for studying eukaryotic gene regulation. Recently, chromosomal conformation capture based technologies, such as Hi-C, have been widely used to uncover the interaction frequencies of genomic loci in a high-throughput and genome-wide manner and provide new insights into the folding of three-dimensional (3D) genome structure. In this paper, we develop a novel manifold learning based framework, called GEM (Genomic organization reconstructor based on conformational Energy and Manifold learning), to reconstruct the three-dimensional organizations of chromosomes by integrating Hi-C data with biophysical feasibility. Unlike previous methods, which explicitly assume specific relationships between Hi-C interaction frequencies and spatial distances, our model directly embeds the neighboring affinities from Hi-C space into 3D Euclidean space. Extensive validations demonstrated that GEM not only greatly outperformed other state-of-art modeling methods but also provided a physically and physiologically valid 3D representations of the organizations of chromosomes. Furthermore, we for the first time apply the modeled chromatin structures to recover long-range genomic interactions missing from original Hi-C data.
DOI:10.15252/msb.20156492URLPMID:26700852 [本文引用: 1]
Mammalian chromosomes fold into arrays of megabase-sized topologically associating domains (TADs), which are arranged into compartments spanning multiple megabases of genomic DNA. TADs have internal substructures that are often cell type specific, but their higher-order organization remains elusive. Here, we investigate TAD higher-order interactions with Hi-C through neuronal differentiation and show that they form a hierarchy of domains-within-domains (metaTADs) extending across genomic scales up to the range of entire chromosomes. We find that TAD interactions are well captured by tree-like, hierarchical structures irrespective of cell type. metaTAD tree structures correlate with genetic, epigenomic and expression features, and structural tree rearrangements during differentiation are linked to transcriptional state changes. Using polymer modelling, we demonstrate that hierarchical folding promotes efficient chromatin packaging without the loss of contact specificity, highlighting a role far beyond the simple need for packing efficiency.
DOI:10.1093/bioinformatics/btx747URLPMID:29186290 [本文引用: 1]
We build a software package scHiCNorm that uses zero-inflated and hurdle models to remove biases from single-cell Hi-C data. Our evaluations prove that our models can effectively eliminate systematic biases for single-cell Hi-C data, which better reveal cell-to-cell variances in terms of chromosomal structures.
.
DOI:10.1186/s12967-018-1766-2URLPMID:30635013 [本文引用: 1]
Bone marrow stromal cells (BMSCs) are a heterogeneous population that participates in wound healing, immune modulation and tissue regeneration. Next generation sequencing was used to analyze transcripts from single BMSCs in order to better characterize BMSC subpopulations.
DOI:10.1038/s41467-017-02526-9URLPMID:29335463 [本文引用: 1]
Topologically associating domains (TADs) are fundamental elements of the eukaryotic genomic structure. However, recent studies suggest that the insulating complexes, CTCF/cohesin, present at TAD borders in mammals are absent from those in Drosophila melanogaster, raising the possibility that border elements are not conserved among metazoans. Using in situ Hi-C with sub-kb resolution, here we show that the D. melanogaster genome is almost completely partitioned into >4000 TADs, nearly sevenfold more than previously identified. The overwhelming majority of these TADs are demarcated by the insulator complexes, BEAF-32/CP190, or BEAF-32/Chromator, indicating that these proteins may play an analogous role in flies as that of CTCF/cohesin in mammals. Moreover, extended regions previously thought to be unstructured are shown to consist of small contiguous TADs, a property also observed in mammals upon re-examination. Altogether, our work demonstrates that fundamental features associated with the higher-order folding of the genome are conserved from insects to mammals.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}