 ,1, 潘学峰,1,2,3
,1, 潘学峰,1,2,3SMARCAL1, roles and mechanisms in genome stability maintenance
Yalei Wen1, Kenao Lü2, Xiaokang Xu1, Xin Zhang1, Liang Ding,1, Xuefeng Pan,1,2,3通讯作者: 丁良,博士,教授,研究方向:分析化学。E-mail:345823685@qq.com;潘学峰,博士,教授,研究方向:生物化学、分子遗传学和药理等。E-mail:xuefengpancam@aliyun.com
编委: 卢大儒
收稿日期:2019-07-7修回日期:2019-09-4网络出版日期:2019-12-20
| 基金资助: |
Editorial board:
Received:2019-07-7Revised:2019-09-4Online:2019-12-20
| Fund supported: |
作者简介 About authors
文雅蕾,硕士研究生,专业方向:分子药理学。E-mail:491574395@qq.com。

摘要
SMARCAL1是属于SWI/SNF (SWItch/Sucrose Non-Fermentable)相关、基质相关和激动蛋白依赖的染色质调节因子家族成员ATP驱动的DNA退火解旋酶。SMARCAL1在体外和体内能催化单链结合蛋白RPA结合的DNA单链与其互补链退火成双链DNA。人Smarcal1基因的突变与Schimke免疫骨性发育不良(Schimke immuno-osseous dysplasia, SIOD)所能表现出的临床症状呈高度相关。本文对SMARCAL1在DNA损伤部位DNA复制叉的重塑、在DNA双链断裂(double-stranded DNA, dsDNA)处参与经典的非同源末端连接(non-homologous end joining, NHEJ)修复,以及在人染色体端粒完整性维护等方面的作用与机制进行了梳理,对Smarcal1基因突变类型与SIOD症状之间的对应关系进行了更新,并对SMARCAL1在三核苷酸重复序列扩增关联的神经-肌肉退行性病变过程中的可能作用进行了分析和讨论,旨在更好地理解该退火解旋酶在维持基因组稳定中的作用和机制。
关键词:
Abstract
SMARCAL1 is an ATP-driven DNA annealing helicase that is similar in structure to the chromatin regulators in the subfamily A group of the SWI/SNF-related matrix-associated actin-dependent chromatin regulators. SMARCAL1 catalyzes the formation of dsDNA by annealing the single-stranded binding protein RPA coated ssDNA with its complementary strand both in vitro and in vivo. In humans, different mutations of Smarcal1 gene are found to be closely related to different symptoms shown in individuals with Schimke immuno-osseous dysplasia (SIOD). This paper reviews the recent research progress of SMARCAL1 functions in remodeling DNA replication forks at damaged DNA sites, working in classical non-homologous end joining (NHEJ) repair of DNA double-stranded breaks, and in maintaining chromosomal telomere integrity. The relationships between the mutations of Smarcal1 gene in different SIOD symptoms, and the possible involvements of SMARCAL1 in neuromuscular degenerative diseases associated with trinucleotide repeats expansions are also updated and discussed to better understand the roles and mechanisms of the annealing helicase in genome stability maintenance.
Keywords:
PDF (863KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
文雅蕾, 吕柯孬, 徐小康, 张欣, 丁良, 潘学峰. 退火解旋酶SMARCAL1在维持基因组稳定中的作用与机制. 遗传[J], 2019, 41(12): 1084-1098 doi:10.16288/j.yczz.19-158
Yalei Wen.
基因组不稳定常见于人类遗传疾病、癌症及神经-肌肉退行性疾病的病理过程[1]。常见的基因组不稳定主要包括基因的点突变、插入/缺失突变、DNA链断裂(单链断裂和双链断裂)、DNA链交联、基因组的倍性改变等类型[1,2]。引发基因组不稳定的原因可依据DNA是否受损分为两类:一类源于细胞内源性或细胞外源性因素造成DNA损伤;一类则起因于基因组内特定DNA序列在DNA复制、转录或重组过程中发生了错误折叠[1,2]。据统计,无论是原核细胞还是真核细胞,均需要130种以上的蛋白组份参与基因组稳定维护[1]。其中,退火解旋酶SMARCAL1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A-like protein 1),因广泛参与DNA损伤部位DNA复制叉的重塑、DNA双链断裂部位的非同源末端连接(non-homologous end joining, NHEJ)修复、以及染色体端粒的维护而受到高度重视[3,4,5,6,7]。
SMARCAL1普遍存在于真核生物细胞中,属于一类依赖ATP的染色质“重塑因子”SNF2 (sucrose non-fermenting 2, SNF2)家族的一员[4]。SNF2家族的染色质重塑因子需要利用ATP水解供能,广泛参与细胞周期调控、基因转录、DNA重组、DNA损伤修复和DNA甲基化修饰等过程[5]。除SMARCAL1之外,通过影响DNA复制过程维持基因组稳定性的ZRANB3和HLTF也是SNF2家族的重要成员[6]。这些蛋白均含有一个由7个保守基序(motif)组成的类似“解旋酶”(与常见的DNA和RNA解旋酶类似)的ATPase结构域[7]。尽管SMARCAL1具有DNA“退火解旋酶”(annealing helicase)活性,但目前并未被归类于已有的6个DNA解旋酶超家族(DNA helicase superfamily)中[8]。
人Smarcal1基因定位于2q34-q36区段,含17个外显子,编码一个由954个氨基酸残基组成的蛋白质[7]。在人(Homo sapiens)和小鼠(Mus musculus)的所有组织中均见Smarcal1基因表达,如在人免疫系统中,Smarcal1在单核细胞、B淋巴细胞、CD4+T细胞、CD8+T细胞、NK细胞中的表达量分别为1.259‰、1.584‰、1.259‰、1.995‰和0.100‰;在内分泌系统的胰腺细胞、前列腺细胞中表达量约为0.100‰和0.016‰;在睾丸细胞中表达量则为0.158‰[9,10,11,12,13,14,15,16]。Smarcal1基因突变与Schimke免疫骨性发育不良(Schimke immuno-osseous dysplasia, SIOD)密切相关[17,18]。SIOD是一种累及多系统、进行性加重的罕见常染色体遗传病,主要表现为T细胞免疫缺陷、局灶节段性肾小球硬化、脑发育受损、肾功能衰竭和骨骼发育不良造成的生长迟缓等[8,12,19~21]。除此之外,部分SIOD患者还表现有甲状腺功能减退、骨髓衰竭、头发稀薄、角膜混浊、动脉粥样硬化、中风和偏头痛等[8,12,21]。
本文将对SMARCAL1在DNA损伤部位借退火解旋酶活性重塑DNA复制叉,在DNA双链断裂部位参与非同源末端连接(NHEJ)修复,以及在端粒完整性维护过程等方面的作用与机制进行梳理。同时,对Smarcal1基因突变型与SIOD症状的相关性的最新进展进行了更新,对其在累及多达40余种人神经-肌肉退行性疾病中的三核苷酸重复序列扩增性不稳定和脆性不稳定发生过程中的可能作用进行了分析。
1 SMARCAL1的结构与功能
SMARCAL1含有复制蛋白A(replication protein A , RPA)结合基序、退火解旋酶活性结构域、ATP酶结构域等(图1)[22]。其中,与RPA作用的基序位于N-末端(28个高度保守的氨基酸残基序列);与“退火解旋活性”有关的结构域位于239~307和331~ 4002区间的“HARP”结构域(每个“HARP”各含60个氨基酸残基)[23]。C-末端是解旋酶结构域,具有ATP酶催化活性(由115个氨基酸残基组成的“RecA样”结构域,DEXDc和HELICc)和SWI/SNF“核小体重塑蛋白”结构域(图1)[24]。SMARCAL1的ATP依赖DNA退火解旋酶含有2个HARP结构域组成的ATPase[25,26,27]。当DNA出现损伤时,与单链DNA结合的单链DNA结合蛋白RPA32识别位于SMARCAL1的N端RPA作用结构域(图1),并招募SMARCAL1至dsDNA-ssDNA的单链DNA一侧,这一反应可见于S期细胞周期关卡通路激活、停滞的复制叉重塑、端粒DNA完整性维护以及利用NHEJ机制修复DNA双链断裂损伤等过程[3,24,25,28~31]。
图1
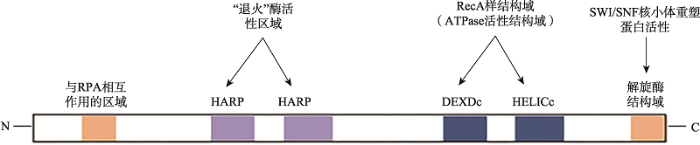 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1退火解旋酶SMARCAL1的功能结构域分布
SMARCAL1在其N端区域包含与RPA相互作用基序和2个退火活性所需的HARP结构域;在其C末端含有解旋酶结构域,为DEXDc和HELICc基序[23,25]。
Fig. 1The functional domains of SMARCAL1 annealing helicase
1.1 SMARCAL1与DNA复制叉的维护
在人体细胞中,Smarcal1缺失常影响细胞对DNA复制逆境(replication stress)应答能力[4]。Smarcal1缺陷的细胞内可见DNA双链断裂,且对多种影响DNA复制的物质敏感,表明SMARCAL1与DNA复制忠实性的监管有关[3,24,28]。研究发现,SMARCAL1可通过与RPA作用参与停滞的DNA复制叉的“重塑”[29],否则停滞的DNA复制叉有可能衍生为DNA双链断裂从而使DNA突变和染色体重排的风险增高[32]。真核生物的RPA是一个由RPA70、RPA32、RPA14亚基组成的异源三聚体,可借4个DNA结合结构域(DNA-binding domain, DBD)与单链DNA结合[33]。RPA异三聚体参与DNA损伤部位修复蛋白的募集和组装,同时规避单链DNA错误折叠出非B型DNA构象[33,34]。SMARCAL1借其N-末端与RPA32的N端结合,从而被快速高效地定位到复制叉的单链DNA部位[3,35~37]。活性氧自由基、紫外辐射及一些化学物质(如放线菌素等)会损伤DNA模板,影响细胞周期S时相的DNA复制[3]。为此,细胞需针对相应的DNA损伤类型做出应答,包括利用ATR对单链空缺(single stranded gap, SSG)损伤应答,利用ATM、DNA-PK等激酶对DNA DSBs应答等[1]。这些激酶对SMARCAL1蛋白的磷酸化修饰有助于有序启动DNA损伤修复[38]。比如,SMARCAL1第652位丝氨酸(S652)残基被ATR磷酸化修饰,可抑制SMARCAL1的ATP酶活性,从而降低其DNA复制叉重塑能力,避免DNA复制叉崩解[39]。
体外研究表明,SMARCAL1的退火解旋酶活性的发挥需要与DNA结合,并由ATP水解供能[22]。在这个过程中,SMARCAL1借RPA结合蛋白与单链DNA结合,并利用ATP水解提供的能量催化彼此互补的单链DNA重新形成双链DNA,并同时解除RPA与单链DNA的结合(图2,A和B)[24]。现已发现,上述反应可以发生在DNA复制叉部位,帮助DNA复制叉内的单链DNA发生“分支迁移”和“分支重塑”[4,22,40],使停滞的DNA复制叉“重塑”出“鸡爪”状四臂交叉中间体结构[41,42](图2C)。图2C中的“鸡爪”结构可以稳定停滞的DNA复制叉,又能使带伤的单链DNA重新“退回”双链DNA状态,可以赢得修复时间,或利用“模板转换”(template switching)使新生DNA链的3 ?生长端“绕过”带伤DNA模板后重启DNA复制[6,27,29,33,43](图2,B和C)。
图2
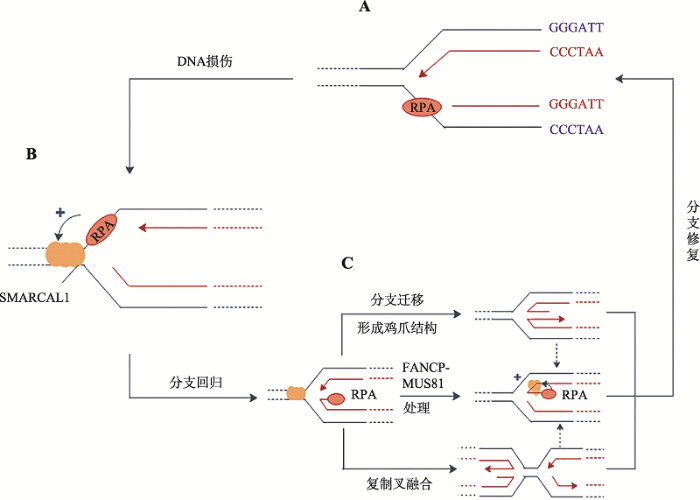 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2SMARCAL1修复受损DNA复制叉的机制
A:DNA链复制遇阻,复制型DNA聚合酶和所“偶联”的DNA解旋酶脱离,导致前导链模板上产生ssDNA空缺;B:在停滞的复制叉上,RPA与单链DNA结合后行成RPA-ssDNA,并招募SMARCAL1,启动复制叉回转;C:SMARCAL1在复制叉回转后以3种可能的方式催化复制叉的修复,包括持续分支迁移产生“鸡爪”状的Holliday结构;与相邻的重新建立的DNA复制叉发生“融合”获得拯救;以及用FANCP-MUS81处理后产生对应于新生前导链的ssDNA链,然后生成单末端DNA双链断裂(DSB)。RPA协助SMARCAL1用互补的模板链与新生的ssDNA前导链“退火”,重构出可正常复制的DNA复制叉[25,39,40]。
Fig. 2Proposed Mechanism of SMARCAL1 in remodeling a damaged DNA replication fork
需要特别指出的是SMARCAL1催化的上述DNA复制叉重塑在体外也可以由大肠杆菌的DNA解旋酶RecG催化[29]。大肠杆菌RecG是一个3?→5?DNA解旋酶,具有催化Holliday中间体(同源重组过程中出现的交叉结构)发生分支迁移能力[29]。在催化DNA复制叉回转时,RecG类似SMARCAL1,需要借DNA单链结合蛋白与DNA复制叉结合。在这个过程中DNA单链结合蛋白还可以进一步稳定RecG与DNA复制叉的结合,有利于RecG催化DNA复制叉回转(图2)[44]。因此,虽然SMARCAL1在结构上与大肠杆菌RNA聚合酶结合蛋白HepA相似(被称为HepA相关蛋白,HARP),但在催化DNA底物结构重塑时则更类似于大肠杆菌的DNA解旋酶RecG[9,10,11]。
1.2 SMARCAL1在维持端粒稳定过程中的作用
人染色体端粒DNA一般由“TTAGGG”六聚体重复序列组成,可形成包括G-四链体、D(t)环等在内的非B型DNA结构。尽管这些DNA结构为端粒稳定所必需,但如果它们出现在DNA复制过程中则会造成DNA复制叉停滞,使端粒变短[45,46,47]。维持端粒稳定是保证细胞增殖能力的关键。肿瘤细胞的端粒长度的维持机制有两种,一是重新激活端粒酶,一是利用同源重组“替补”机制维持(统称为端粒延长替代机制,alterative lengthening of telomere, ALT)[48]。ALT细胞(缺乏端粒酶而需要ALT维持端粒长度的细胞)中可见由端粒DNA的额外染色体形成的C-环(C-circles),可用于ALT活性的标记[43]。DNA损伤修复系统对端粒酶复制稳定端粒发挥着重要的调控作用[49]。Cox等[49]发现SMARCAL1参与端粒的ALT维持机制。在ALT细胞内,SMARCAL1与端粒DNA结合,协助停滞的DNA复制叉重启,应对端粒DNA复制应激[49]。而缺失SMARCAL1的ALT细胞则会出现端粒DNA复制困难,表现为DNA复制叉持续停滞,并最终形成DSBs,表现出高强度染色体融合(图3)[49]。与此同时,SMARCAL1缺失会影响端粒的长度异质性,推测与ALT细胞中SMARCAL1水平过低催生了更多的C-环有关[49]。
此外,Poole等[50]发现SMARCAL1在端粒处发挥作用时无需RPA协助,因为端粒部位重复DNA序列更倾向于形成G-四链体等非B型DNA二级结构,而G-四链体本身就可有效招募SMARCAL1。但是,当SMARCAL1与DNA复制叉内的前导链模板结合时则需要RPA激活[33,50]。
1.3 SMARCAL1与DNA双链断裂修复
真核细胞内与DNA DSBs修复有关的机制有两类,一类是依赖DNA同源性的同源重组修复(homologous recombination, HR),一类则是不严格依赖DNA同源性(无需模板)的非同源末端连接(NHEJ)[51]。HR常用于细胞周期的S、G2等时相,而NHEJ则可用于整个细胞周期,特别当细胞处于G0/G1和S期早期,由于缺乏“同源染色体”,NHEJ对DSBs修复起着关键的作用[52,53,54]。图3
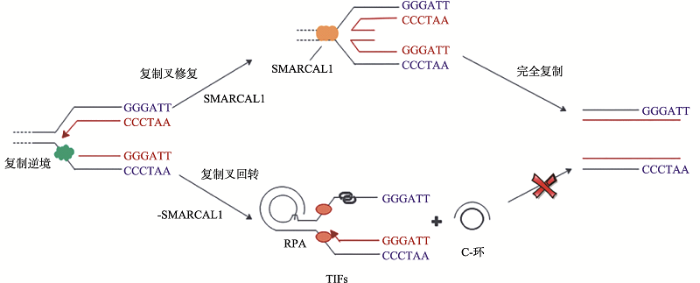 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3SMARCAL1在维持端粒稳定中的作用
ALT细胞内的染色体端粒易出现复制逆境,SMARCAL1帮助重塑端粒DNA上停滞的复制叉,确保端粒序列的完全复制[46,47]。在SMARCAL1缺陷的情况下,ALT端粒上持续停滞的复制叉会催生DNA双链断裂,出现端粒DNA复制障碍及形成C-环,此时,染色体易融合,造成基因组不稳定[49,50]。
Fig. 3The role of SMARCAL1 in telomere maintenance
Keka等[55]发现SMARCAL1参与G1期DSBs的NHEJ修复过程。SMARCAL1的退火解旋酶活性可以协助DNA末端结合蛋白Ku70/80“获取”双链DNA末端,有利于进一步依序招募参与NHEJ修复的蛋白因子,以完成NHEJ或MMEJ (小同源末端连接,microhomology-mediated end-joining,MMEJ)在G1时相对DNA DSBs修复。在此过程中,SMARCAL1的ATP依赖的退火能力可能避免了RPA在DSB末端与ssDNA结合,因此促进Ku70/Ku80/DNA- PKcs复合体对DNA双链末端识别、结合和保护,为后续XRCC4和DNA连接酶IV在断裂末端的积聚提供便利,从而提供了NHEJ修复的精确度[55~60] (图4)。
图4
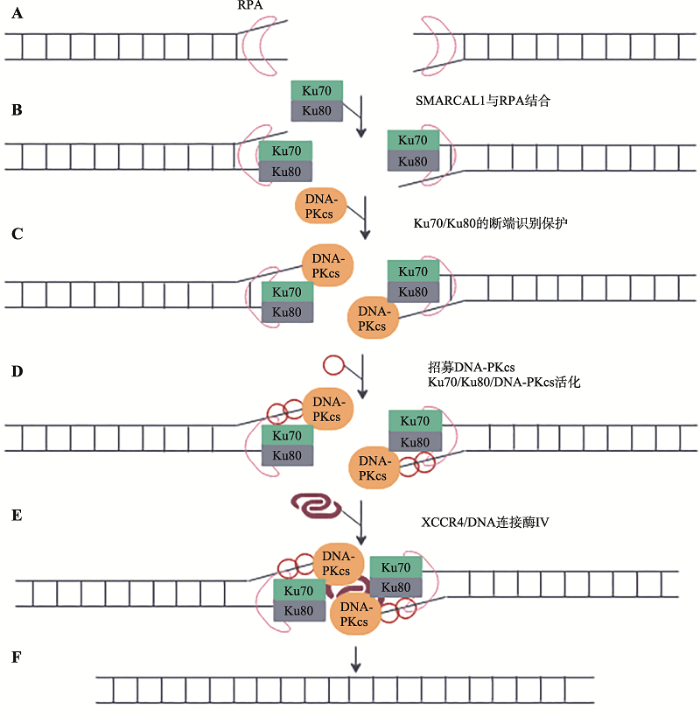 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4SMARCAL1在NHEJ修复DNA双链断裂中的可能作用
A:DSB形成后,RPA识别并结合DNA断端,之后,招募SMARCAL1。SMARCAL1的退火解旋酶活性保证DSB断口DNA呈双链状态;B:Ku70/Ku80组成的异源二聚体与DNA末端结合;C:DNA-Ku复合体招募DNA-PKcs,形成Ku70/Ku80/DNA-PKcs复合体;D:SMARCAL1促进Ku70/Ku80/DNA-PKcs复合体稳定结合在DSB端口,并激活DNA-PKcs。DNA-PKcs对包括自身在内的蛋白进行磷酸化修饰;E:DSB断端的SMARCAL1促进Ku70/Ku80/DNA-PKcs复合体招募DNA连接酶IV和XRCC4复合体,进一步形成Ku70/Ku80/DNA-PKcs/XRCC4/DNA连接酶IV修复复合体;F:含有连接酶的复合体完成两个DNA断端的连接[55,56,57,58,59,60,61,62,63,64]。
Fig. 4Possible role of SMARCAL1 in DNA double-strand break repair using a NHEJ
SMARCAL1在Ku70/Ku80/DNA-PKcs聚积的DNA末端协助Mre11、RAD50、Nbs1组装成MRN复合物(Mre11-RAD50-Nbs1)[65,66]。具体过程如下:DSB信号被ATM“捕获”,ATM将H2AX磷酸化成γ-H2AX。γ-H2AX与Nbs1作用进一步促进RAD50和Mre11在DSB断端处形成MRN复合体(RAD50- Mre11-Nbs1, MRN)[67,68,69]。其中,RAD50二聚体上的ATP酶(Walker A和Walker B)负责与DSB两个末端结合,避免末端错位或漂移。Nbs1则协助SMARCAL1退火可被NHEJ修复的DNA断端,使之保持双链状态,并由RAD50的绞链区将两个断端“固定”[70]。
此外,SMARCAL1也和RAD50一起参与阻止复制叉反转,通过调节Mre11的核酸酶活性,防止Mre11过度降解新生DNA区段[71]。
1.4 SMARCAL1与基因转录
SMARCAL1作为SWI/SNF家族(负责催化核小体重塑)中的成员同时拥有解旋酶(helicase)和ATPase活性,因此有可能参与某些基因转录过程中的核小体重塑。当前研究较多的是SMARCAL1对c-myc基因转录的调节。c-myc编码一种亮氨酸拉链蛋白,参与人类基因组中5%~15%的基因的转录,在细胞增殖、分化、生长和凋亡中发挥重要作用[72,73,74]。SMARCAL1作为辅因子参与c-myc基因的转录[72,73,74]。Heravi等[75]发现SMARCAL1通过ATP依赖性方式改变DNA结构,调节c-myc的转录[75]。Tapan等[76]发现SMARCAL1是c-myc转录的负调控因子。通过与激活蛋白BRG1和RNA聚合酶Ⅱ (RNAPⅡ)“争夺”c-myc基因P1启动子上游的一段富含GC碱基的159 bp DNA区域(Myc-B159),当激活蛋白BRG1和RNAPⅡ占据myc-B159时,c-myc基因转录;当该区域被SMARCAL1占据时,c-myc基因关闭[76]。SMARCAL1与myc-B159结合后使得相应部位的染色质结构更难与BRG1和RNAPⅡ结合,故可抑制c-myc的转录[76]。2 SMARCAL1在三核苷酸重复序列扩增中的潜在作用
人基因组中特定基因部位处的三核苷酸重复序列(CAG)n·(CTG)n的“动态”扩增与多种遗传性脊髓-小脑共济失调、亨廷顿疾病、阿尔兹海默综合征等神经-肌肉系统退行性病变的发生密切相关[77,78,79]。已有的研究结果表明,(CAG)n重复序列的扩增与DNA剪切修复过程中产生的单链DNA错误、DNA复制过程中出现DNA链断裂、DNA复制过程中新生链和模板链间发生“滑动”、链转换(strand switching)、蛋白质与重复DNA序列结合等许多因素有关。上述过程可能起因于(CAG)n重复DNA形成的含错配碱基对的DNA发卡结构,DNA发卡结构可能会直接干扰DNA复制、修复和重组。不仅如此,在基因转录过程中(CAG)n三核苷酸重复序列一旦形成R环结构(DNA杂交体与非模板链形成)也可影响DNA复制叉的移动,导致非模板链DNA断裂,甚至可被用于DNA重新复制的引物[77,78,79]。本实验室已有的研究结果表明,Smarcal1基因的正本基因recG (ΔrecG)功能丧失的大肠杆菌细胞内,疾病相关的(CAG)n·(CTG)n序列呈特异性扩增(待发表)。鉴于SMARCAL1在催化DNA复制叉重塑及避免DNA复制叉崩解的过程中与RecG功能一致,推测出现在人类患者细胞内的(CAG)n·(CTG)n序列扩增有可能与DNA复制叉重塑和DNA复制重起始失败有关(图5A),或出现在(CAG)n重复序列内的R环结构影响了DNA复制,并在RecG/SMARCAL1功能异常的情况下诱生DNA链重排或造成该处DNA局部额外复制[77,78,79](图5B)。最近,类似图5A的工作机制已被Kononenko等[80]的工作证实。他们利用小鼠幼红细胞白血病细胞系验证了可导致人脆性X染色体综合征(马丁-贝尔综合征)的三核苷酸重复序列(CGG)n的扩增不稳定与SMARCAL1所具有的功能直接有关[80]。但是,目前尚无有工作表明SMARCAL1自身是否具有类似于RecG的RNA解旋酶活性或与其他RNA解旋酶一起参与RNA转录,故图5B所描述的机制依然有待进一步检验。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5SMARCAL1及其可能的突变体基因在三核苷酸重复序列稳定维护中的潜在作用
A:SMARCAL1参与三核苷酸重复序列部位处DNA复制叉的重塑。根据参考文献[80]修改绘制;B:SMARCAL1自身或引导其他RNA解旋酶清除在三核苷酸重复序列转录过程中形成的R-环结构[77,78,79]。
Fig. 5Possible roles of SMARCAL1 and its mutants in the stable maintenance of triplet repeats
为了阐释长期困扰国际医学界的亨廷顿疾病、阿尔兹海默综合征和多种遗传性脊髓-小脑共济失调综合征等发病过程中出现的(CAG)n扩增的原因,以及明确其他与三核苷酸重复序列失稳定引发的人类神经-肌肉退行性病变的病理机制,当前亟需深入分析SMARCAL1在包括(CAG)n重复序列在内的三核苷酸重复序列稳定维护过程中可能发挥的作用[77,78,79]。
3 SMARCAL1的基因突变型与SIOD的症状关联
世界范围内SIOD的发病率约为1/300万~1/100万[8,9,81,82]。导致SIOD发生的Smarcal1基因突变多为双等位基因功能缺失、错义突变、插入/缺失(insertion and deletion, Ins/Del)、大片段缺失以及SMARCAL1 mRNA拼接错误(表1)[9,83]。上述基因改变常出现在SMARCAL1的RecA样结构域I中(图1),由于突变影响了SMARCAL1的ATP酶活性,故常见疾病的严重程度与突变体SMARCAL1所表现出的ATP酶活性成反比[82]。SMARCAL1等位基因缺失、无义或移码突变常见于重症患者。重症SIOD患者的症状在孕期外显,表现为胎儿生长迟缓、甲状腺功能减退症、骨髓衰竭、短暂性脑缺血发作、中风和肾衰竭等,一般死于5岁前。SMARCAL1等位基因错义突变患者症状较轻,多数发病较晚,常见8~13岁以后发病,数年后进展至肾衰[84,85](表1)。Table 1
表1
表1Smarcal1基因突变型与SIOD的症状关联
Table 1
| 突变位点 | 突变类型 | 功能 | 症状 | 参考文献 |
|---|---|---|---|---|
| c.1129G>C, p. E377Q | 错义突变 | SNP和非致病突变 | 无 | [86] |
| c.1933C>T, p. R645C | ATP酶活性缺失和解旋酶活性缺失 | T细胞免疫缺陷、肾功能衰竭和生长迟缓 | ||
| c.1334+1G>A | 剪切突变 | 非致病突变 | 无 | |
| c.2142-1G>A | ||||
| NM_014140.3:c.2070t2insT | mRNA剪切突变 | 非致病突变 | 无 | [20] |
| p.[M288_D366del]+[M288_D366del] | 双等位基因突变 | 缺乏解旋酶活性 | 生长迟缓、全血细胞减少症、短暂性脑缺血和肾病综合征 | [12] |
| p.[E377Q]+[I755fsX2] | 双等位基因突变 | 缺乏解旋酶活性 | T细胞免疫缺陷、FSGS和生长迟缓 | |
| p.[Y342X]+[I755S] | 双等位基因突变 | 缺乏解旋酶活性 | 生长迟缓、全血细胞减少症、反复感染和肾病综合征 | |
| c.445C>T(p.Q149X) | 无义突变 | 导致氨基酸的改变 | 生长迟缓、蛋白尿和T细胞免疫缺陷 | [87] |
| p.R817H | 错义突变 | 缺乏解旋酶活性 | 生长迟缓、T细胞免疫缺陷和肾功能不全 | [88] |
| c.1615C> G(p.[Leu539Val]) | 错义突变 | 缺乏解旋酶活性 | 生长迟缓、FSGS、肾衰竭和T细胞免疫缺陷 | [89] |
| p. R247P | 错义突变 | 退火酶活性 | 甲状腺功能不全、骨骼发育不良和蛋白尿 | [90] |
| p. E848* | 无义突变 | 退火酶活性 | ||
| p.L397fsX40 | 移码突变 | 退火酶活性 | FSGS、T细胞免疫缺陷、生长发育不良和鼻窦癌 | [91] |
| p.S859P | 错义突变 | ATP酶活性 | ||
| c.3G> A(p.M1?) | 剪切突变 | 与RPA相互作用 | 生长迟缓、复发感染和中性粒细胞增多症、FSGS,T细胞免疫缺陷和严重腕骨骨质衰老 | [92] |
| c.1682G> A(p. R561H) | 错义突变 | ATP酶活性 | ||
| R561C | 错义突变 | ATP酶活性 | T细胞免疫缺陷、面部畸形、肾病综合征和FSGS | [93] |
| c.2542G> T, p.E848x | 无义突变 | 退火酶活性 | 生长迟缓和慢性肾功能衰竭 | [94] |
| c.1934G> A, p.R645H | 错义突变 | ATP酶活性 | ||
| IVS4-2A>G | 剪接突变 | 非致病突变 | 肾脏畸形、生长迟缓和T细胞免疫缺陷 | [14] |
| 1136A> C, H379P | 无义突变 | 非致病突变 | 生长迟缓、肾衰、FSGS和T细胞免疫缺陷 | [95] |
| 836T> C, F279S | 错义突变 | ATP酶活性 | ||
| 1-BP INS,1849C | 插入 | 生长迟缓和面部畸形 | [96] | |
| 1-BP DEL.2161C | 缺失 | |||
| R586W | 错义突变 | ATP酶活性 | 生长迟缓、蛋白尿和T细胞免疫缺陷 | [97] |
| R644W | 错义突变 | ATP酶活性 | 生长迟缓、FSGS、肾衰竭和T细胞免疫缺陷 | |
| R645C | 错义突变 | ATP酶活性 | 生长迟缓、蛋白尿和T细胞免疫缺陷 | |
| R764Q | 错义突变 | ATP酶活性 | 生长迟缓、FSGS、肾衰竭和T细胞免疫缺陷 | |
| c.797-798delCC | 微缺失突变 | 解旋酶活性 | 骨骼发育不良、蛋白尿和肾病综合征 | [81] |
| IVSA7+1G>7 | 剪切突变 | ATP酶活性 | ||
| A468P | 错义突变 | ATP酶活性 | 生长迟缓、蛋白尿和T细胞免疫缺陷 | [93] |
| I548N | 错义突变 | ATP酶活性 | ||
| S579L | 错义突变 | ATP酶活性 |
新窗口打开|下载CSV
SIOD的共有临床症状是T细胞免疫缺陷。在淋巴细胞发育过程中,编码免疫球蛋白和T细胞受体抗原结合域的功能基因需要经过与NHEJ类似的V(D)J重组才能形成[98,99]。与NHEJ类似,V(D)J重排也需产生DNA双链断裂,并由NHEJ机制完成断链末端的连接[98,99]。而突变体SMARCAL1常影响NHEJ在V(D)J重排重组的连接效率,这可能是SIOD患者常见T细胞免疫缺陷的原因之一[96,100]。此外,突变体SMARCAL1有可能通过影响c-myc基因表达进一步影响SIOD的征候。研究发现成年小鼠的肾脏和脑组织中,c-myc不表达而Smarcal1高表达[13,101]。暗示SIOD患者表现出的肾功能不全和脑发育受损可能与SMARCAL1突变体不能正确调节c-myc表达有关[19]。Tapan等[76]发现SIOD患者的染色质也会出现异常,这种情况暗示SMARCAL1可能依然具有染色质重塑活性。可能由于trxG和PcG的复合体诱导组蛋白的翻译后修饰,影响染色体的结构。而SMARCAL1通过结合染色质直接影响基因表达。
4 结语与展望
目前,DNA退火酶SMARCAL1的功能异常与SIOD之间的关联已经得到了明确,但SIOD患者所能表现的症状征候差异与不同Smarcal1基因突变型的对应关系的细节依然处于不断更新状态。由于临床上SIOD症状可累及人体多个系统,轻重患者之间的临床表现并不尽然一致,也很难呈现一种症状的渐进发展特征,暗示Smarcal1基因突变可能依附携带突变的组织和器官,一种可能的情形是Smarcal1基因突变对不同组织细胞内的染色质结构、基因转录、DNA损伤修复依细胞类型不同而具有差异。因此,目前亟需对有关Smarcal1基因表达的组织特异性机制加深了解。在鉴定出的分子机制方面,SMARCAL1的退火酶活性在复制叉重塑过程中的作用已被体外实验证实,但尚缺乏体内实验证据的支持。不仅如此,Smarcal1不同突变型对其活性的影响呈现多样性(表1),如何关联SIOD症状也需要进一步积累体内实验数据。近年来本实验室对SMARCAL1的原核生物功能类似物DNA解旋酶RecG的研究发现,RecG缺陷突变后的大肠杆菌细胞容许与亨廷顿疾病、阿尔兹海默综合症以及多种遗传型小脑-脊髓共济失调在内的40多种神经-肌肉系统退行性疾病直接有关的三核苷酸重复序列CAG出现特异性扩增,类似的情形是否也有可能出现在携带某些Smarcal1突变类型的患者体内需要给予重视。当前关于SMARCAL1分子机制的研究主要集中于其对DNA复制叉重塑、DNA双链断裂损伤修复及端粒完整性维持等方面。作为SWI/SNF相关、基质相关和激动蛋白依赖的染色质调节因子家族成员的SMARCAL1在染色质重塑、组蛋白修饰编码(histone code)方面是否依然具有作用尚缺乏更多了解。综上所述,深入开展Smarcal1基因及其突变型的分子遗传学和SMARCAL1蛋白的体内分子生物学研究将不仅有助于系统理解SIOD发病机制,而且对破解长期困扰人类健康的40余种进行性神经-肌肉系统退行性疾病的致病原因提供借鉴和参考。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 5]
[本文引用: 5]
DOI:10.1016/j.dnarep.2012.01.007URLPMID:22365498 [本文引用: 2]

Base excision repair (BER) is believed to be the predominant pathway for the repair of oxidative DNA damage. BER is initiated by lesion-specific DNA glycosylases that recognize and remove the damaged base. NEIL1, NEIL2 and NEIL3 are three mammalian members of the Fpg/Nei DNA glycosylase family with similar enzymatic properties. In this study we showed that both the transcription and protein levels of hNEIL3 fluctuated during the cell cycle. Based on predicted promoter elements of cell cycle-regulated genes and microarray data from various reports, we suggest that hNEIL3 repression in quiescent cells might be mediated by the DREAM (DP1, RB p130, E2F4 and MuvB core complex) complex. Release from G0 by mitogenic stimulation showed an induction of hNEIL3 in early S phase under the control of the Ras dependent ERK-MAP kinase pathway. In contrast, the total expression of hNEIL1 was downregulated upon release from quiescence while the expression of hNEIL2 was cell cycle independent. Notably, hNEIL3 showed a similar regulation pattern as the replication protein hFEN1 supporting a function of hNEIL3 in replication associated repair. Thus, it appears that specialized functions of the NEILs are ensured by their expression patterns.
DOI:10.1080/10409238.2017.1380597URLPMID:28954549 [本文引用: 5]
A large number of SNF2 family, DNA and ATP-dependent motor proteins are needed during transcription, DNA replication, and DNA repair to manipulate protein-DNA interactions and change DNA structure. SMARCAL1, ZRANB3, and HLTF are three related members of this family with specialized functions that maintain genome stability during DNA replication. These proteins are recruited to replication forks through protein-protein interactions and bind DNA using both their motor and substrate recognition domains (SRDs). The SRD provides specificity to DNA structures like forks and junctions and confers DNA remodeling activity to the motor domains. Remodeling reactions include fork reversal and branch migration to promote fork stabilization, template switching, and repair. Regulation ensures these powerful activities remain controlled and restricted to damaged replication forks. Inherited mutations in SMARCAL1 cause a severe developmental disorder and mutations in ZRANB3 and HLTF are linked to cancer illustrating the importance of these enzymes in ensuring complete and accurate DNA replication. In this review, we examine how these proteins function, concentrating on their common and unique attributes and regulatory mechanisms.
DOI:10.1042/BSR20180568URLPMID:29748240 [本文引用: 4]
ATP-dependent chromatin remodeling proteins use the energy released from ATP hydrolysis to reposition nucleosomes in DNA-dependent processes. These proteins are classified as SF2 helicases. SMARCAL1, a member of this protein family, is known to modulate both DNA repair and transcription by specifically recognizing DNA molecules possessing double-strand to single-strand transition regions. Mutations in this gene cause a rare autosomal recessive disorder known as Schimke Immuno-Osseous Dysplasia (SIOD).Structural studies have shown that the ATP-dependent chromatin remodeling proteins possess two RecA-like domains termed as RecA-like domain 1 and RecA-like domain 2. Using Active DNA-dependent ATPase A domain (ADAAD), the bovine homolog of SMARCAL1, as a model system we had previously shown that the RecA-like domain 1 containing helicase motifs Q, I, Ia, II, and III are sufficient for ligand binding; however, the Rec A-like domain 2 containing motifs IV, V, and VI are needed for ATP hydrolysis. In the present study, we have focused on the motifs present in the RecA-like domain 2. Our studies demonstrate that the presence of an aromatic residue in motif IV is needed for interaction with DNA in the presence of ATP. We also show that the motif V is required for the catalytic efficiency of the protein and motif VI is needed for interaction with DNA in the presence of ATP. Finally, we show that the SIOD-associated mutation, R820H, present in motif VI results in loss of ATPase activity, and therefore, reduced response to DNA damage.
DOI:10.1101/gad.1839909URLPMID:19793861 [本文引用: 2]
Mutations in SMARCAL1 (HARP) cause Schimke immunoosseous dysplasia (SIOD). The mechanistic basis for this disease is unknown. Using functional genomic screens, we identified SMARCAL1 as a genome maintenance protein. Silencing and overexpression of SMARCAL1 leads to activation of the DNA damage response during S phase in the absence of any genotoxic agent. SMARCAL1 contains a Replication protein A (RPA)-binding motif similar to that found in the replication stress response protein TIPIN (Timeless-Interacting Protein), which is both necessary and sufficient to target SMARCAL1 to stalled replication forks. RPA binding is critical for the cellular function of SMARCAL1; however, it is not necessary for the annealing helicase activity of SMARCAL1 in vitro. An SIOD-associated SMARCAL1 mutant fails to prevent replication-associated DNA damage from accumulating in cells in which endogenous SMARCAL1 is silenced. Ataxia-telangiectasia mutated (ATM), ATM and Rad3-related (ATR), and DNA-dependent protein kinase (DNA-PK) phosphorylate SMARCAL1 in response to replication stress. Loss of SMARCAL1 activity causes increased RPA loading onto chromatin and persistent RPA phosphorylation after a transient exposure to replication stress. Furthermore, SMARCAL1-deficient cells are hypersensitive to replication stress agents. Thus, SMARCAL1 is a replication stress response protein, and the pleiotropic phenotypes of SIOD are at least partly due to defects in genome maintenance during DNA replication.
DOI:10.1101/gad.178459.111URLPMID:22279047 [本文引用: 3]
SMARCAL1 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A-like1) maintains genome integrity during DNA replication. Here we investigated its mechanism of action. We found that SMARCAL1 travels with elongating replication forks, and its absence leads to MUS81-dependent double-strand break formation. Binding to specific nucleic acid substrates activates SMARCAL1 activity in a reaction that requires its HARP2 (Hep-A-related protein 2) domain. Homology modeling indicates that the HARP domain is similar in structure to the DNA-binding domain of the PUR proteins. Limited proteolysis, small-angle X-ray scattering, and functional assays indicate that the core enzymatic unit consists of the HARP2 and ATPase domains that fold into a stable structure. Surprisingly, SMARCAL1 is capable of binding three-way and four-way Holliday junctions and model replication forks that lack a designed ssDNA region. Furthermore, SMARCAL1 remodels these DNA substrates by promoting branch migration and fork regression. SMARCAL1 mutations that cause Schimke immunoosseous dysplasia or that inactivate the HARP2 domain abrogate these activities. These results suggest that SMARCAL1 continuously surveys replication forks for damage. If damage is present, it remodels the fork to promote repair and restart. Failures in the process lead to activation of an alternative repair mechanism that depends on MUS81-catalyzed cleavage of the damaged fork.
DOI:10.1016/j.dnarep.2017.06.015URLPMID:28623093 [本文引用: 3]
SMARCAL1 (SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily A-Like 1), also known as HARP, is an ATP-dependent annealing helicase that stabilizes replication forks during DNA damage. Mutations in this gene are the cause of Schimke immune-osseous dysplasia (SIOD), an autosomal recessive disorder characterized by T-cell immunodeficiency and growth dysfunctions. In this review, we summarize the main roles of SMARCAL1 in DNA repair, telomere maintenance and replication fork stability in response to DNA replication stress.
DOI:10.1007/s00234-018-2052-yURLPMID:29978300 [本文引用: 4]
DOI:10.1101/gad.1860609URLPMID:19833762 [本文引用: 4]
In this issue of Genes & Development, four papers report that the annealing helicase HepA-related protein (HARP, also known as SMARCAL1 [SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a-like 1]) binds directly to the ssDNA-binding protein Replication protein A (RPA) and is recruited to sites of replicative stress. Knockdown of HARP results in hypersensitivity to multiple DNA-damaging agents and defects in fork stability or restart. These exciting insights reveal a key new player in the S-phase DNA damage response.
DOI:10.1074/jbc.M114.618801URLPMID:26272746 [本文引用: 2]
Members of the Swi2/Snf2 (switch/sucrose non-fermentable) family depend on their ATPase activity to mobilize nucleic acid-protein complexes for gene expression. In bacteria, RapA is an RNA polymerase (RNAP)-associated Swi2/Snf2 protein that mediates RNAP recycling during transcription. It is known that the ATPase activity of RapA is stimulated by its interaction with RNAP. It is not known, however, how the RapA-RNAP interaction activates the enzyme. Previously, we determined the crystal structure of RapA. The structure revealed the dynamic nature of its N-terminal domain (Ntd), which prompted us to elucidate the solution structure and activity of both the full-length protein and its Ntd-truncated mutant (RapAΔN). Here, we report the ATPase activity of RapA and RapAΔN in the absence or presence of RNAP and the solution structures of RapA and RapAΔN either ligand-free or in complex with RNAP. Determined by small-angle x-ray scattering, the solution structures reveal a new conformation of RapA, define the binding mode and binding site of RapA on RNAP, and show that the binding sites of RapA and σ(70) on the surface of RNAP largely overlap. We conclude that the ATPase activity of RapA is inhibited by its Ntd but stimulated by RNAP in an allosteric fashion and that the conformational changes of RapA and its interaction with RNAP are essential for RNAP recycling. These and previous findings outline the functional cycle of RapA, which increases our understanding of the mechanism and regulation of Swi2/Snf2 proteins in general and of RapA in particular. The new structural information also leads to a hypothetical model of RapA in complex with RNAP immobilized during transcription.
DOI:10.1038/cr.2011.32URL [本文引用: 2]
Macromolecular assemblies that regulate chromatin structure using the energy of ATP hydrolysis have critical roles in development, cancer, and stem cell biology. The ATPases of this family are encoded by 27 human genes and are usually associated with several other proteins that are stable, non-exchangeable subunits. One fundamental mechanism used by these complexes is thought to be the movement or exchange of nucleosomes to regulate transcription. However, recent genetic studies indicate that chromatin remodelers may also be involved in regulating other aspects of chromatin structure during many cellular processes. The SWI/SNF family in particular appears to have undergone a substantial change in subunit composition and mechanism coincident with the evolutionary advent of multicellularity and the appearance of linking histones. The differential usage of this greater diversity of mammalian BAF subunits is essential for the development of specific cell fates, including the progression from pluripotency to multipotency to committed neurons. Recent human genetic screens have revealed that BRG1, ARID1A, BAF155, and hSNF5 are frequently mutated in tumors, indicating that BAF complexes also play a critical role in the initiation or progression of cancer. The mechanistic bases underlying the genetic requirements for BAF and other chromatin remodelers in development and cancer are relatively unexplored and will be a focus of this review.
DOI:10.1016/j.ejmg.2016.06.002URLPMID:27282802 [本文引用: 4]
Schimke immunoosseous dysplasia (SIOD) is an autosomal recessive disease characterized by skeletal dysplasia, focal segmental glomerulosclerosis, renal failure and immunodeficiency. In this work, we report the molecular studies undertaken in a severely affected SIOD patient that died at six years old due to nephropathy. The patient was screened for mutations using a targeted skeletal dysplasias panel. A homozygous novel missense mutation was identified, c.1615C?>?G (p.[Leu539Val]) that was predicted as mildly pathogenic by in silico pathogenicity prediction tools. However, splicing prediction software suggested that this variant may create a new splicing donor site in exon 9, which was subsequently confirmed using a minigene assay in HEK293?cells. Thus, the splicing alteration, c.1615C?>?G; r.1615c?>?g, 1615_1644del; (p.[Leu539_Ile548del]), results in the loss of 10 amino acids of the HARP-ATPase catalytic domain and the RPA-binding domain. Several studies have demonstrated a weak genotype-phenotype correlation among such patients. Thus, the molecular characterization has helped us to understand why a predicted weakly pathogenic missense mutation results in severe SIOD and should be considered in similar scenarios.
DOI:10.1002/ajmg.a.35532URL [本文引用: 2]
Schimke immuno-osseous dysplasia (SIOD) is a multisystemic disorder with prominent skeletal, renal, immunological, and ectodermal abnormalities. It is caused by mutations of SMARCAL1 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a-like 1), which encodes a DNA stress response protein. To determine the relationship of this function to the SIOD phenotype, we profiled the cancer prevalence in SIOD and assessed if defects of nucleotide excision repair (NER) and nonhomologous end joining (NHEJ), respectively, explained the ectodermal and immunological features of SIOD. Finally, we determined if Smarcal1(del/del) mice had hypersensitivity to irinotecan (CPT-11), etoposide, and hydroxyurea (HU) and whether exposure to these agents induced features of SIOD. Among 71 SIOD patients, three had non-Hodgkin lymphoma (NHL) and one had osteosarcoma. We did not find evidence of defective NER or NHEJ; however, Smarcal1-deficient mice were hypersensitive to several genotoxic agents. Also, CPT-11, etoposide, and HU caused decreased growth and loss of growth plate chondrocytes. These data, which identify an increased prevalence of NHL in SIOD and confirm hypersensitivity to DNA damaging agents in vivo, provide guidance for the management of SIOD patients. (C) 2012 Wiley Periodicals, Inc.
DOI:10.1203/PDR.0b013e31816721ccURLPMID:18356746 [本文引用: 2]
Schimke immuno-osseous dysplasia (SIOD) is an autosomal recessive disorder caused by loss-of-function mutations in SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a-like 1 (SMARCAL1), with clinical features of growth retardation, spondylo-epiphyseal dysplasia, nephrotic syndrome, and immunodeficiency. We report a patient with SIOD and SMARCAL1 splice mutation (IVS4-2 A>G) in a nonconsanguineous Ashkenazi family, who came to our attention at 1 mo of age due to renal malformation and only later developed signs compatible with Schimke. Interestingly, residual SMARCAL1 mRNA levels in the patient's peripheral blood were lower compared with those observed in both asymptomatic brothers' carrying the same bi-allelic mutation, whereas the latter had levels similar to those found in heterozygous carriers (parents and sister). Examination of the carrier frequency of the splice mutation in the Ashkenazi population demonstrated 1 carrier in 760 DNA samples. In situ localization of SMARCAL1 in human kidneys as well as analysis of its temporal expression during murine nephrogenesis and in the metanephric organ culture suggested a role in the early renal progenitor population and after renal maturation. Thus, disease severity within the same family might be modified by the splicing machinery. The renal expression pattern of SMARCAL1 explains a broader spectrum of renal disease in SIOD than previously described.
[本文引用: 1]
DOI:10.1136/jmg.2008.060095URLPMID:18805831 [本文引用: 1]
Schimke immuno-osseous dysplasia (SIOD) is an autosomal recessive pleiotropic disorder caused by mutations in SMARCAL1. SMARCAL1 encodes an enzyme with homology to the SNF2 chromatin remodelling proteins.
DOI:10.1016/j.bbagrm.2017.07.003URLPMID:28716689 [本文引用: 1]
Recent investigations have emphasized the role of miRNA biogenesis proteins in the synthesis of non-coding RNA when double-strand DNA breaks are induced by ionizing radiations. However, the role of these non-coding RNA and their regulation in response to doxorubicin-induced DNA damage is not known. In this paper, BRG1 and SMARCAL1, members of the ATP-dependent chromatin remodelling family, are shown to co-regulate the transcription of DROSHA, DGCR8, and DICER in response to double-strand DNA breaks induced by doxorubicin. Both BRG1 and SMARCAL1 are needed for the upregulation of the three miRNA biogenesis genes as absence of BRG1 results in downregulation of DGCR8 and DICER while absence of SMARCAL1 results in downregulation of DROSHA. These two proteins act in coordination to upregulate expression of DROSHA, DGCR8, and DICER when cells are treated with doxorubicin. This transcriptional regulation of the miRNA biogenesis proteins is needed for the formation of 53BP1 foci as downregulation of either BRG1 or SMARCAL1 reduced the number of 53BP1 foci in DNA damaged cells. The foci formation was restored when the downregulated cells were treated with ncRNA purified from doxorubicin treated HeLa cells. From the results obtained, we conclude that the regulation of miRNA biogenesis proteins by SMARCAL1 and BRG1 is needed for the formation of non-coding RNA and thus, 53BP1 foci in response to doxorubicin-induced DNA damage.
DOI:10.1002/ajmg.a.36111URLPMID:23950031 [本文引用: 1]
Schimke immuno-osseous dysplasia (SIOD, OMIM 242900) is a rare autosomal recessive multisystem childhood disorder characterized by short stature, renal failure, T-cell immunodeficiency, and hypersensitivity to genotoxic agents. SIOD is associated with biallelic mutations in SMARCAL1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin, subfamily a-like 1), which encodes a DNA stress response enzyme with annealing helicase activity. Two features of SIOD causing much morbidity and mortality are bone marrow failure and T-cell deficiency with the consequent opportunistic infections. To address the safety and efficacy of bone marrow transplantation (BMT) in SIOD, we reviewed the outcomes of the only five SIOD patients known to us in whom bone marrow or hematopoietic stem cell transplantation has been attempted. We find that only one patient survived the transplantation procedure and that the existing indicators of a good prognosis for bone marrow transplantation were not predictive in this small cohort. Given these observations, we also discuss some considerations for the poor outcomes.
DOI:10.1016/j.bbrc.2012.09.060URLPMID:22995303 [本文引用: 2]
SMARCAL1 is a SNF2 chromatin-remodeling protein with ATP-dependent annealing helicase activity. Recent studies have shown that SMARCAL1 is involved in DNA damage repair and cell cycle progression. Deficiency of SMARCAL1 enhances the anticancer activity of chemotherapy agents and reverses cancer cell resistance to these agents. Therefore, targeting SMARCAL1 is an attractive therapeutic approach for cancers with defects in DNA damage repair or cell cycle checkpoints. Here, we review advances in our understanding of the biochemical and cellular functions of SMARCAL1 made over the recent years and discuss the rationale for development of SMARCAL1 inhibitors as novel anticancer therapies.
DOI:10.1002/ajmg.a.37146URLPMID:25943327 [本文引用: 1]
Schimke Immunoosseous Dysplasia (SIOD) is a rare, autosomal recessive disorder of childhood characterized by spondyloepiphyseal dysplasia, focal segmental glomerulosclerosis and renal failure, T-cell immunodeficiency, and cancer in certain instances. Approximately half of patients with SIOD are reported to have biallelic mutations in SMARCAL1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin, subfamily a-like 1), which encodes a DNA translocase that localizes to sites of DNA replication and repairs damaged replication forks. We present a novel mutation (NM_014140.3:c.2070+2insT) that results in defective SMARCAL1 mRNA splicing in a child with SIOD. This mutation, within the donor site of intron 12, results in the skipping of exon 12, which encodes part of a critical hinge region connecting the two lobes of the ATPase domain. This mutation was not recognized as deleterious by diagnostic SMARCAL1 sequencing, but discovered through next generation sequencing and found to result in absent SMARCAL1 expression in patient-derived lymphoblasts. The splicing defect caused by this mutation supports the concept of exon definition. Furthermore, it illustrates the need to broaden the search for SMARCAL1 mutations in patients with SIOD lacking coding sequence variants.
URLPMID:28256679 [本文引用: 2]
DOI:10.1126/science.1161233URLPMID:18974355 [本文引用: 3]
DNA-dependent adenosine triphosphatases (ATPases) participate in a broad range of biological processes including transcription, DNA repair, and chromatin dynamics. Mutations in the HepA-related protein (HARP) ATPase are responsible for Schimke immuno-osseous dysplasia (SIOD), but the function of the protein is unknown. We found that HARP is an ATP-dependent annealing helicase that rewinds single-stranded DNA bubbles that are stably bound by replication protein A. Other related ATPases, including the DNA translocase Rad54, did not exhibit annealing helicase activity. Analysis of mutant HARP proteins suggests that SIOD is caused by a deficiency in annealing helicase activity. Moreover, the pleiotropy of HARP mutations is consistent with the function of HARP as an annealing helicase that acts throughout the genome to oppose the action of DNA-unwinding activities in the nucleus.
DOI:10.1038/embor.2011.74URL [本文引用: 2]
Mutations in HepA-related protein (HARP, or SMARCAL1) cause Schimke immunoosseous dysplasia (SIOD). HARP has ATP-dependent annealing helicase activity, which helps to stabilize stalled replication forks and facilitate DNA repair during replication. Here, we show that the conserved tandem HARP (2HP) domain dictates this annealing helicase activity. Furthermore, chimeric proteins generated by fusing the 2HP domain of HARP with the SNF2 domain of BRG1 or HELLS show annealing helicase activity in vitro and, when targeted to replication forks, mimic the functions of HARP in vivo. We propose that the HARP domain endows HARP with this ATP-driven annealing helicase activity.
DOI:10.1101/gad.1832309URLPMID:19793862 [本文引用: 4]
The integrity of genomic DNA is continuously challenged by the presence of DNA base lesions or DNA strand breaks. Here we report the identification of a new DNA damage response protein, SMARCAL1 (SWI/SNF-related, matrix associated, actin-dependent regulator of chromatin, subfamily a-like 1), which is a member of the SNF2 family and is mutated in Schimke immunoosseous dysplasia (SIOD). We demonstrate that SMARCAL1 directly interacts with Replication protein A (RPA) and is recruited to sites of DNA damage in an RPA-dependent manner. SMARCAL1-depleted cells display sensitivity to DNA-damaging agents that induce replication fork collapse, and exhibit slower fork recovery and delayed entry into mitosis following S-phase arrest. Furthermore, SIOD patient fibroblasts reconstituted with SMARCAL1 exhibit faster cell cycle progression after S-phase arrest. Thus, the symptoms of SIOD may be caused, at least in part, by defects in the cellular response to DNA replication stress.
DOI:10.1016/j.sbi.2011.09.003URLPMID:21996440 [本文引用: 4]
This review focuses on recent structural insights into regulation and nucleic acid binding of Superfamily 2 (SF2)-type helicases as they relate to chromatin remodelers. We review structural features of the Chd1 chromatin remodeler regarding regulation of the ATPase motor, and discuss related strategies observed for other SF2 ATPases. Since no SWI2/SNF2 ATPases have yet been captured bound to DNA in a state competent for ATP hydrolysis, we turn to structural examples from the DEAD-box RNA helicase family, and suggest that SWI2/SNF2-specific inserts may be poised to alter canonical duplex DNA structure.
DOI:10.4161/15384101.2014.956551URL [本文引用: 1]
Regulation of chromatin structure is an essential component of the DNA damage response (DDR), which effectively preserves the integrity of DNA by a network of multiple DNA repair and associated signaling pathways. Within the DDR, chromatin is modified and remodeled to facilitate efficient DNA access, to control the activity of repair proteins and to mediate signaling. The mammalian ISWI family has recently emerged as one of the major ATP-dependent chromatin remodeling complex families that function in the DDR, as it is implicated in at least 3 major DNA repair pathways: homologous recombination, non-homologous end-joining and nucleotide excision repair. In this review, we discuss the various manners through which different ISWI complexes regulate DNA repair and how they are targeted to chromatin containing damaged DNA.
DOI:10.1097/PAS.0000000000001011URLPMID:29309303 [本文引用: 2]
The SWI/SNF chromatin-remodeling complex, which is composed of evolutionarily conserved core subunits such as SMARCB1/INI1 (INI1), SMARCA4/BRG1 (BRG1), SMARCC1/BAF155 (BAF155), and SMARCC2/BAF170 (BAF170), can be viewed as the prototype of an epigenetic regulator of gene expression that is involved in tumor suppression. Epithelioid sarcoma, which classified as a tumor of uncertain differentiation, shows an almost complete loss of INI1. However, some cases of epithelioid sarcoma have preserved INI1, and the clinicopathologic features of these cases are uncertain. To date, there has been no investigation focused on the SWI/SNF chromatin-remodeling complex in INI1-preserved epithelioid sarcoma cases. First, an investigation of INI1 immunoexpression statuses in 60 formalin-fixed paraffin-embedded epithelioid sarcoma specimens (proximal type, 29 cases; conventional type, 31 cases) was performed. In the available INI1-preserved epithelioid sarcoma cases, we analyzed the BRG1, BAF155, and BAF170 protein expressions. INI1 preservation was observed in 6 of 29 (21%) proximal-type and 2 of 31 (6%) conventional-type epithelioid sarcoma cases. Six cases of INI1-preserved epithelioid sarcomas of proximal type were available for further immunohistochemical study. One proximal type showed loss of BAF170, and 2 proximal-type cases revealed loss of BRG1 with preservation of the other remaining core subunit proteins. One proximal-type case showed a mosaic pattern of BRG1 and loss of BAF155. However, in the remaining 2 proximal-type cases, all core subunit proteins were preserved. Overall, these results suggest that loss of expression of SWI/SNF chromatin-remodeling complex proteins has an important role in tumorigenesis. The remaining 2 INI1-preserved epithelioid sarcoma cases may have had other abnormalities causing dysfunction of SWI/SNF chromatin remodeling.
DOI:10.1111/nph.15311URLPMID:29974976 [本文引用: 2]
DNA replication is a fundamental process for the faithful transmission of genetic information in all living organisms. Many endogenous and environmental signals impede fork progression during DNA synthesis, which induces replication errors and DNA replication stress. Chromatin remodeling factors regulate nucleosome occupancy and the histone composition of the nucleosome in chromatin; however, whether chromatin remodeling factors are involved in the DNA replication stress response in plants is unknown. We reveal that chromatin remodeling factor CHR18 plays important roles in DNA replication stress in Arabidopsis thaliana by interacting with the DNA replication protein RPA1A. According to the genetic analysis, the loss of function of either CHR18 or RPA1A confers a high sensitivity to DNA replication stress in Arabidopsis. CHR18 interacts with RPA1A in both yeast cells and tobacco epidermal cells. The coexpression of RPA1A and CHR18 enhances the accumulation of CHR18 in nuclear foci in plants. CHR18 is a typical nuclear-localized chromatin remodeling factor with ATPase activity. Our results demonstrate that during DNA synthesis in plants, RPA1A interacts with CHR18 and recruits CHR18 to nuclear foci to resolve DNA replication stress, which is important for cell propagation and root growth in Arabidopsis plants.
DOI:10.1016/j.celrep.2013.05.002URLPMID:23746452 [本文引用: 4]
Stalled replication forks are sources of genetic instability. Multiple fork-remodeling enzymes are recruited to stalled forks, but how they work to promote fork restart is poorly understood. By combining ensemble biochemical assays and single-molecule studies with magnetic tweezers, we show that SMARCAL1 branch migration and DNA-annealing activities are directed by the single-stranded DNA-binding protein RPA to selectively regress stalled replication forks caused by blockage to the leading-strand polymerase and to restore normal replication forks with a lagging-strand gap. We unveil the molecular mechanisms by which RPA enforces SMARCAL1 substrate preference. E.?coli RecG acts similarly to SMARCAL1 in the presence of E.?coli SSB, whereas the highly related human protein ZRANB3 has different substrate preferences. Our findings identify the important substrates of SMARCAL1 in fork repair, suggest that RecG and SMARCAL1 are functional orthologs, and provide a comprehensive model of fork repair by these DNA translocases.
DOI:10.1038/cr.2014.147URLPMID:25403473 [本文引用: 1]
The Replication Protein A (RPA) complex is an essential regulator of eukaryotic DNA metabolism. RPA avidly binds to single-stranded DNA (ssDNA) through multiple oligonucleotide/oligosaccharide-binding folds and coordinates the recruitment and exchange of genome maintenance factors to regulate DNA replication, recombination and repair. The RPA-ssDNA platform also constitutes a key physiological signal which activates the master ATR kinase to protect and repair stalled or collapsed replication forks during replication stress. In recent years, the RPA complex has emerged as a key target and an important regulator of post-translational modifications in response to DNA damage, which is critical for its genome guardian functions. Phosphorylation and SUMOylation of the RPA complex, and more recently RPA-regulated ubiquitination, have all been shown to control specific aspects of DNA damage signaling and repair by modulating the interactions between RPA and its partners. Here, we review our current understanding of the critical functions of the RPA-ssDNA platform in the maintenance of genome stability and its regulation through an elaborate network of covalent modifications.
DOI:10.1534/genetics.117.200238URLPMID:28258182 [本文引用: 1]
DNA double-strand breaks (DSBs) pose a serious threat to genomic integrity. If unrepaired, they can lead to chromosome fragmentation and cell death. If repaired incorrectly, they can cause mutations and chromosome rearrangements. DSBs are repaired using end-joining or homology-directed repair strategies, with the predominant form of homology-directed repair being synthesis-dependent strand annealing (SDSA). SDSA is the first defense against genomic rearrangements and information loss during DSB repair, making it a vital component of cell health and an attractive target for chemotherapeutic development. SDSA has also been proposed to be the primary mechanism for integration of large insertions during genome editing with CRISPR/Cas9. Despite the central role for SDSA in genome stability, little is known about the defining step: annealing. We hypothesized that annealing during SDSA is performed by the annealing helicase SMARCAL1, which can anneal RPA-coated single DNA strands during replication-associated DNA damage repair. We used unique genetic tools in Drosophila melanogaster to test whether the fly ortholog of SMARCAL1, Marcal1, mediates annealing during SDSA. Repair that requires annealing is significantly reduced in Marcal1 null mutants in both synthesis-dependent and synthesis-independent (single-strand annealing) assays. Elimination of the ATP-binding activity of Marcal1 also reduced annealing-dependent repair, suggesting that the annealing activity requires translocation along DNA. Unlike the null mutant, however, the ATP-binding defect mutant showed reduced end joining, shedding light on the interaction between SDSA and end-joining pathways.
DOI:10.1074/jbc.M114.627083URLPMID:25552480 [本文引用: 4]
SMARCAL1 catalyzes replication fork remodeling to maintain genome stability. It is recruited to replication forks via an interaction with replication protein A (RPA), the major ssDNA-binding protein in eukaryotic cells. In addition to directing its localization, RPA also activates SMARCAL1 on some fork substrates but inhibits it on others, thereby conferring substrate specificity to SMARCAL1 fork-remodeling reactions. We investigated the mechanism by which RPA regulates SMARCAL1. Our results indicate that although an interaction between SMARCAL1 and RPA is essential for SMARCAL1 activation, the location of the interacting surface on RPA is not. Counterintuitively, high-affinity DNA binding of RPA DNA-binding domain (DBD) A and DBD-B near the fork junction makes it easier for SMARCAL1 to remodel the fork, which requires removing RPA. We also found that RPA DBD-C and DBD-D are not required for SMARCAL1 regulation. Thus, the orientation of the high-affinity RPA DBDs at forks dictates SMARCAL1 substrate specificity.
DOI:10.2741/3652URLPMID:20515732 [本文引用: 1]
Since the initial discovery of replication protein A (RPA) as a DNA replication factor, much progress has been made on elucidating critical roles for RPA in other DNA metabolic pathways. RPA has been shown to be required for DNA replication, DNA repair, DNA recombination, and the DNA damage response pathway with roles in checkpoint activation. This review summarizes the current understanding of RPA structure, phosphorylation and protein-protein interactions in mediating these DNA metabolic processes.
DOI:10.1111/febs.12867URL [本文引用: 1]
Replication protein A subunit RPA32 contains a C-terminal domain that interacts with a variety of DNA damage response proteins including SMARCAL1, Tipin, UNG2 and XPA. We have solved the high-resolution crystal structure of RPA32 C-terminal domain (RPA32C) in complex with a 26-amino-acid peptide derived from the N-terminus of SMARCAL1 (SMARCAL1N). The RPA32C-SMARCAL1N structure reveals a 1 : 1 binding stoichiometry and displays a well-ordered binding interface. SMARCAL1N adopts a long a-helical conformation with the highly conserved 11 residues aligned on one face of the a-helix showing extensive interactions with the RPA32C domain. Extensive mutagenesis experiments were performed to corroborate the interactions observed in crystal structure. Moreover, the alpha 1/alpha 2 loop of the RPA32C domain undergoes a conformational rearrangement upon SMARCAL1N binding. NMR study has further confirmed that the RPA32C-SMARCAL1N interaction induces conformational changes in RPA32C. Isothermal titration calorimetry studies have also demonstrated that the conserved alpha-helical motif defined in the current study is required for sufficient binding of RPA32C. Taken together, our study has provided convincing structural information that redefines the common recognition pattern shared by RPA32C interacting proteins.
DOI:10.1016/j.molcel.2016.10.038URLPMID:27984746
Human cancers are characterized by the presence of?oncogene-induced DNA replication stress (DRS), making them dependent on repair pathways such as break-induced replication (BIR) for damaged DNA replication forks. To better understand BIR, we performed a targeted siRNA screen for genes whose depletion inhibited G1 to S phase progression when oncogenic cyclin E was overexpressed. RAD52, a gene dispensable for normal development in mice, was among the top hits. In cells in which fork collapse was induced by oncogenes or chemicals, the Rad52 protein localized to DRS foci. Depletion of Rad52 by siRNA or knockout of the gene by CRISPR/Cas9 compromised restart of collapsed forks and led to DNA damage in cells experiencing DRS. Furthermore, in cancer-prone, heterozygous APC mutant mice, homozygous deletion of the Rad52 gene suppressed tumor growth and prolonged lifespan. We therefore propose that mammalian RAD52 facilitates repair of collapsed DNA replication forks in cancer cells.
DOI:10.1021/bi500252wURL [本文引用: 1]
SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A-like l (SMARCAL1) is a recently identified DNA damage response protein involved in remodeling stalled replication forks. The eukaryotic single-strand DNA binding protein replication protein A (RPA) recruits SMARCAL1 to stalled forks in vivo and facilitates regression of forks containing leading strand gaps. Both activities are mediated by a direct interaction between an RPA binding motif (RBM) at the N-terminus of SMARCAL1 and the C-terminal winged-helix domain of the RPA 32 kDa subunit (RPA32C). Here we report a biophysical and structural characterization of the SMARCAL1-RPA interaction. Isothermal titration calorimetry and circular dichroism spectroscopy revealed that RPA32C binds SMARCAL1-RBM with a K,, of 2.5 mu M and induces a disorder-to-helix transition. The crystal structure of RPA32C was refined to 1.4 angstrom resolution, and the SMARCAL1-RBM binding site was mapped on the structure on the basis of nuclear magnetic resonance chemical shift perturbations. Conservation of the interaction surface to other RBM-containing proteins allowed construction of a model for the RPA32C/SMARCAL1-RBM complex. The implications of our results are discussed with respect to the recruitment of SMARCAL1 and other DNA damage response and repair proteins to stalled replication forks.
DOI:10.1016/j.dnarep.2009.04.006URL [本文引用: 1]
Abstract
Kinases in the phosphoinositide three-kinase-related kinase (PIKK) family include ATM (ataxia-telangiectasia mutated), ATR (ATM- and Rad3-related), DNA-PKcs (DNA-dependent protein kinase catalytic subunit), mTOR (mammalian target of rapamycin), and SMG1 (suppressor with morphological effect on genitalia family member). These atypical protein kinases regulate DNA damage responses, nutrient-dependent signaling, and nonsense-mediated mRNA decay. This review focuses on the mechanisms regulating the PIKK family with a strong emphasis on the DNA damage regulated kinases. We outline common regulatory themes and suggest how discoveries about the regulation of one PIKK can be informative for the other family members.DOI:10.1101/gad.214080.113URLPMID:23873943 [本文引用: 2]
The DNA damage response kinase ataxia telangiectasia and Rad3-related (ATR) coordinates much of the cellular response to replication stress. The exact mechanisms by which ATR regulates DNA synthesis in conditions of replication stress are largely unknown, but this activity is critical for the viability and proliferation of cancer cells, making ATR a potential therapeutic target. Here we use selective ATR inhibitors to demonstrate that acute inhibition of ATR kinase activity yields rapid cell lethality, disrupts the timing of replication initiation, slows replication elongation, and induces fork collapse. We define the mechanism of this fork collapse, which includes SLX4-dependent cleavage yielding double-strand breaks and CtIP-dependent resection generating excess single-stranded template and nascent DNA strands. Our data suggest that the DNA substrates of these nucleases are generated at least in part by the SMARCAL1 DNA translocase. Properly regulated SMARCAL1 promotes stalled fork repair and restart; however, unregulated SMARCAL1 contributes to fork collapse when ATR is inactivated in both mammalian and Xenopus systems. ATR phosphorylates SMARCAL1 on S652, thereby limiting its fork regression activities and preventing aberrant fork processing. Thus, phosphorylation of SMARCAL1 is one mechanism by which ATR prevents fork collapse, promotes the completion of DNA replication, and maintains genome integrity.
DOI:10.1371/journal.pone.0063149URLPMID:23671665 [本文引用: 2]
SMARCAL1 is an ATPase in the SNF2 family that functions at damaged replication forks to promote their stability and restart. It acts by translocating on DNA to catalyze DNA strand annealing, branch migration, and fork regression. Many SNF2 enzymes work as motor subunits of large protein complexes. To determine if SMARCAL1 is also a member of a protein complex and to further understand how it functions in the replication stress response, we used a proteomics approach to identify interacting proteins. In addition to the previously characterized interaction with replication protein A (RPA), we found that SMARCAL1 forms complexes with several additional proteins including DNA-PKcs and the WRN helicase. SMARCAL1 and WRN co-localize at stalled replication forks independently of one another. The SMARCAL1 interaction with WRN is indirect and is mediated by RPA acting as a scaffold. SMARCAL1 and WRN act independently to prevent MUS81 cleavage of the stalled fork. Biochemical experiments indicate that both catalyze fork regression with SMARCAL1 acting more efficiently and independently of WRN. These data suggest that RPA brings a complex of SMARCAL1 and WRN to stalled forks, but that they may act in different pathways to promote fork repair and restart.
DOI:10.1093/nar/gkp244URLPMID:19406929 [本文引用: 1]
The progress of replication forks is often threatened in vivo, both by DNA damage and by proteins bound to the template. Blocked forks must somehow be restarted, and the original blockage cleared, in order to complete genome duplication, implying that blocked fork processing may be critical for genome stability. One possible pathway that might allow processing and restart of blocked forks, replication fork reversal, involves the unwinding of blocked forks to form four-stranded structures resembling Holliday junctions. This concept has gained increasing popularity recently based on the ability of such processing to explain many genetic observations, the detection of unwound fork structures in vivo and the identification of enzymes that have the capacity to catalyse fork regression in vitro. Here, we discuss the contexts in which fork regression might occur, the factors that may promote such a reaction and the possible roles of replication fork unwinding in normal DNA metabolism.
DOI:10.1038/nrm3935URLPMID:25714681 [本文引用: 1]
The remodelling of replication forks into four-way junctions following replication perturbation, known as fork reversal, was hypothesized to promote DNA damage tolerance and repair during replication. Albeit conceptually attractive, for a long time fork reversal in vivo was found only in prokaryotes and specific yeast mutants, calling its evolutionary conservation and physiological relevance into question. Based on the recent visualization of replication forks in metazoans, fork reversal has emerged as a global, reversible and regulated process, with intriguing implications for replication completion, chromosome integrity and the DNA damage response. The study of the putative in vivo roles of recently identified eukaryotic factors in fork remodelling promises to shed new light on mechanisms of genome maintenance and to provide novel attractive targets for cancer therapy.
DOI:10.1016/j.cell.2017.11.047URLPMID:29290468 [本文引用: 2]
Telomere maintenance critically depends on the distinct activities of telomerase, which adds telomeric repeats to solve the end replication problem, and RTEL1, which dismantles DNA secondary structures at telomeres to facilitate replisome progression. Here, we establish that reversed replication forks are?a pathological substrate for telomerase and the source of telomere catastrophe in Rtel1-/- cells. Inhibiting telomerase recruitment to telomeres, but not its activity, or blocking replication fork reversal through PARP1 inhibition or depleting UBC13 or ZRANB3 prevents the rapid accumulation of dysfunctional telomeres in RTEL1-deficient cells. In this context, we establish that telomerase binding to reversed replication forks inhibits telomere replication, which can be mimicked by preventing replication fork restart through depletion of RECQ1 or PARG. Our results lead us to propose that telomerase inappropriately binds to and inhibits restart of reversed replication forks within telomeres, which compromises replication and leads to critically short telomeres.
DOI:10.1002/pro.3114URLPMID:28078722 [本文引用: 1]
In E. coli, the regression of stalled DNA replication forks is catalyzed by the DNA helicase RecG. One means of gaining access to the fork is by binding to the single strand binding protein or SSB. This interaction occurs via the wedge domain of RecG and the intrinsically disordered linker (IDL) of SSB, in a manner similar to that of SH3 domains binding to PXXP motif-containing ligands in eukaryotic cells. During loading, SSB remodels the wedge domain so that the helicase domains bind to the parental, duplex DNA, permitting the helicase to translocate using thermal energy. This translocation may be used to clear the fork of obstacles, prior to the initiation of fork regression.
DOI:10.3724/SP.J.1005.2009.01185URL [本文引用: 1]
The maintenance of the length and normal structure of telomeres is highly related to the development of senescence and tumorigenesis. The mechanisms of maintaining telomere are essential for cell growth and the reactivation of these mechanisms is an important step in tumor progression. The mechanism of telomere maintenance might be the reactivation of telomerase. In the case of telomerase deficiency, the mechanisms for maintaining the lengths of telomeres are referred to as alternative lengthening of telomere (ALT). The characteristics of the ALT cells include great heterogeneity of telomere size in individual cells, ALT-associated PML (promyelocytic leukemia) bodies, and evident homologous recombination. The ALT-related proteins and elevated homologous recombination found in ALT cells provide a possible mechanism for the alternative lengthening of telomere. The study of ALT provides a new view of crosstalk between senescence and tumori-genesis.
DOI:10.3724/SP.J.1005.2009.01185URL [本文引用: 1]
The maintenance of the length and normal structure of telomeres is highly related to the development of senescence and tumorigenesis. The mechanisms of maintaining telomere are essential for cell growth and the reactivation of these mechanisms is an important step in tumor progression. The mechanism of telomere maintenance might be the reactivation of telomerase. In the case of telomerase deficiency, the mechanisms for maintaining the lengths of telomeres are referred to as alternative lengthening of telomere (ALT). The characteristics of the ALT cells include great heterogeneity of telomere size in individual cells, ALT-associated PML (promyelocytic leukemia) bodies, and evident homologous recombination. The ALT-related proteins and elevated homologous recombination found in ALT cells provide a possible mechanism for the alternative lengthening of telomere. The study of ALT provides a new view of crosstalk between senescence and tumori-genesis.
DOI:10.16288/j.yczz.15-356URLPMID:27103453 [本文引用: 2]
Telomerase is composed of the catalytic subunit TERT (Telomerase reverse transcriptase),RNA subunit TERC (Telomerase RNA component) and other telomerase associated proteins. These two compartments with other telomerase subunits can assemble a holoenzyme, which maintain the length of telomeres. Telomerase plays important roles in cell senescence and tumor formation. The molecular mechanisms of the regulation of telomerase are very complicated. These processes comprise the regulation of transcription, post-transcription, post-translation and subcellular localization. Trafficking and assemble of TERT and TERC, as well as recruitment to telomeres, are also involved in this process. Here we review the regulation mechanism of telomerase from these aspects, and aim to laid a foundation for telomerase associated research and the drug targeting the telomerase.
DOI:10.16288/j.yczz.15-356URLPMID:27103453 [本文引用: 2]
Telomerase is composed of the catalytic subunit TERT (Telomerase reverse transcriptase),RNA subunit TERC (Telomerase RNA component) and other telomerase associated proteins. These two compartments with other telomerase subunits can assemble a holoenzyme, which maintain the length of telomeres. Telomerase plays important roles in cell senescence and tumor formation. The molecular mechanisms of the regulation of telomerase are very complicated. These processes comprise the regulation of transcription, post-transcription, post-translation and subcellular localization. Trafficking and assemble of TERT and TERC, as well as recruitment to telomeres, are also involved in this process. Here we review the regulation mechanism of telomerase from these aspects, and aim to laid a foundation for telomerase associated research and the drug targeting the telomerase.
DOI:10.1016/j.cell.2009.06.021URLPMID:19596237 [本文引用: 2]
Telomeres protect chromosome ends through the interaction of telomeric repeats with shelterin, a protein complex that represses DNA damage signaling and DNA repair reactions. The telomeric repeats are maintained by telomerase, which solves the end replication problem. We report that the TTAGGG repeat arrays of mammalian telomeres pose a challenge to the DNA replication machinery, giving rise to replication-dependent defects that resemble those of aphidicolin-induced common fragile sites. Gene deletion experiments showed that efficient duplication of telomeres requires the shelterin component TRF1. Without TRF1, telomeres activate the ATR kinase in S phase and show a fragile-site phenotype in metaphase. Single-molecule analysis of replicating telomeres showed that TRF1 promotes efficient replication of TTAGGG repeats and prevents fork stalling. Two helicases implicated in the removal of G4 DNA structures, BLM and RTEL1, were required to repress the fragile-telomere phenotype. These results identify a second telomere replication problem that is solved by the shelterin component TRF1.
DOI:10.1016/j.tig.2017.09.003URLPMID:28969871 [本文引用: 1]
Telomeres shorten during each cellular division, with cumulative attrition resulting in telomeric damage and replicative senescence. Bypass of replicative senescence precipitates catastrophic telomere shortening or crisis, and is characterized by widespread genomic instability. Activation of a telomere maintenance mechanism (TMM) is necessary to stabilise the genome and establish cellular immortality through the reconstitution of telomere capping function. The alternative lengthening of telomeres (ALT) pathway is a TMM frequently activated in tumors of mesenchymal or neuroepithelial origin. ALT is a homology-directed recombination-dependent replication pathway that utilizes telomeric templates for synthesis; however, its precise protein requirements have remained elusive. Recently, several developments have shed light on the DNA repair pathways that become engaged at ALT telomeres, implicating ALT telomeres as DNA repair hot spots. Here, we review recent discoveries regarding the ALT mechanism, and discuss how DNA repair pathways converge to maintain the length and functional integrity of telomeres in ALT cancers.
DOI:10.1016/j.celrep.2016.01.011URLPMID:26832416 [本文引用: 6]
Cancer cells overcome replicative senescence by exploiting mechanisms of telomere elongation, a process often accomplished by reactivation of the enzyme telomerase. However, a subset of cancer cells lack telomerase activity and rely on the alternative lengthening of telomeres (ALT) pathway, a recombination-based mechanism of telomere elongation. Although the mechanisms regulating ALT are not fully defined, chronic replication stress at telomeres might prime these fragile regions for recombination. Here, we demonstrate that the replication stress response protein SMARCAL1 is a critical regulator of ALT activity. SMARCAL1 associates with ALT telomeres to resolve replication stress and ensure telomere stability. In the absence of SMARCAL1, persistently stalled replication forks at ALT telomeres deteriorate into DNA double-strand breaks promoting the formation of chromosome fusions. Our studies not only define a role for SMARCAL1 in ALT telomere maintenance, but also demonstrate that resolution of replication stress is a crucial step in the ALT mechanism.
DOI:10.1080/19491034.2016.1179413URLPMID:27355316 [本文引用: 3]
DNA replication is constantly challenged by both endogenous and exogenous sources of replication stress. SMARCAL1, an SNF2 family DNA translocase, functions in the DNA damage response to address these obstacles and promote the completion of replication. Most studies examining the function of SMARCAL1 and related enzymes have relied on the addition of exogenous genotoxic agents, but SMARCAL1 is needed even in the absence of these drugs to maintain genome stability during DNA replication. We recently determined that SMARCAL1 functions to limit DNA damage during replication of difficult-to-replicate telomere sequences. SMARCAL1-deficient cells display several markers of telomere instability including extrachromosomal telomere circles and co-localization with DNA damage markers. Furthermore, cells lacking the highly related proteins ZRANB3 and HLTF do not exhibit similar problems suggesting a unique function for SMARCAL1. These studies identified the first source of endogenous replication stress that SMARCAL1 resolves and provide insight into the mechanism of SMARCAL1 function in maintaining genome stability.
DOI:10.1074/jbc.TM117.000374URLPMID:29247009 [本文引用: 1]
Nonhomologous DNA end-joining (NHEJ) is the predominant double-strand break (DSB) repair pathway throughout the cell cycle and accounts for nearly all DSB repair outside of the S and G2 phases. NHEJ relies on Ku to thread onto DNA termini and thereby improve the affinity of the NHEJ enzymatic components consisting of polymerases (Pol μ and Pol λ), a nuclease (the Artemis·DNA-PKcs complex), and a ligase (XLF·XRCC4·Lig4 complex). Each of the enzymatic components is distinctive for its versatility in acting on diverse incompatible DNA end configurations coupled with a flexibility in loading order, resulting in many possible junctional outcomes from one DSB. DNA ends can either be directly ligated or, if the ends are incompatible, processed until a ligatable configuration is achieved that is often stabilized by up to 4 bp of terminal microhomology. Processing of DNA ends results in nucleotide loss or addition, explaining why DSBs repaired by NHEJ are rarely restored to their original DNA sequence. Thus, NHEJ is a single pathway with multiple enzymes at its disposal to repair DSBs, resulting in a diversity of repair outcomes.
DOI:10.1126/science.1248024URLPMID:24652939 [本文引用: 1]
Mitotic cells inactivate DNA double-strand break (DSB) repair, but the rationale behind this suppression remains unknown. Here, we unravel how mitosis blocks DSB repair and determine the consequences of repair reactivation. Mitotic kinases phosphorylate the E3 ubiquitin ligase RNF8 and the nonhomologous end joining factor 53BP1 to inhibit their recruitment to DSB-flanking chromatin. Restoration of RNF8 and 53BP1 accumulation at mitotic DSB sites activates DNA repair but is, paradoxically, deleterious. Aberrantly controlled mitotic DSB repair leads to Aurora B kinase-dependent sister telomere fusions that produce dicentric chromosomes and aneuploidy, especially in the presence of exogenous genotoxic stress. We conclude that the capacity of mitotic DSB repair to destabilize the genome explains the necessity for its suppression during mitosis, principally due to the fusogenic potential of mitotic telomeres.
URLPMID:31826966 [本文引用: 1]
Rapid pollen tube growth requires uptake of sucrose or its hydrolytic products, hexoses, from the apoplast of surrounding tissues in the style. Due to species-specific sugar requirements, reliance of pollen germination and tube growth on cell wall invertase and sucrose or hexose transporters varies between species, but it is not known if there exists a sugar transporter that mediates the uptake of both hexose and sucrose for pollen tube growth. Here, we show that a sugar transporter protein in apple (Malus domestica), MdSTP13a, takes up both hexose and sucrose when expressed in yeast, and is essential for pollen tube growth on glucose and sucrose but not on maltose. MdSTP13a-mediated direct uptake of sucrose is primarily responsible for apple pollen tube growth on sucrose medium. Sorbitol, a major photosynthate and transport carbohydrate in apple, modulates pollen tube growth via a MYB transcription factor, MdMYB39L, which binds to the promoter of MdSTP13a to activate its expression. Antisense repression of MdSTP13a blocks the sorbitol-modulated pollen tube growth. These findings demonstrate that MdSTP13a takes up both hexose and sucrose for sorbitol-modulated pollen tube growth in apple, revealing a situation where acquisition of sugars for pollen tube growth is regulated by a sugar alcohol.
DOI:10.1038/s41467-017-02146-3URLPMID:29229926 [本文引用: 1]
Pathway choice within DNA double-strand break (DSB) repair is a tightly regulated process to maintain genome integrity. RECQL4, deficient in Rothmund-Thomson Syndrome, promotes the two major DSB repair pathways, non-homologous end joining (NHEJ) and homologous recombination (HR). Here we report that RECQL4 promotes and coordinates NHEJ and HR in different cell cycle phases. RECQL4 interacts with Ku70 to promote NHEJ in G1 when overall cyclin-dependent kinase (CDK) activity is low. During S/G2 phases, CDK1 and CDK2 (CDK1/2) phosphorylate RECQL4 on serines 89 and 251, enhancing MRE11/RECQL4 interaction and RECQL4 recruitment to DSBs. After phosphorylation, RECQL4 is ubiquitinated by the DDB1-CUL4A E3 ubiquitin ligase, which facilitates its accumulation at DSBs. Phosphorylation of RECQL4 stimulates its helicase activity, promotes DNA end resection, increases HR and cell survival after ionizing radiation, and prevents cellular senescence. Collectively, we propose that RECQL4 modulates the pathway choice of NHEJ and HR in a cell cycle-dependent manner.
DOI:10.1093/nar/gkv621URLPMID:26089390 [本文引用: 3]
Smarcal1 is a SWI/SNF-family protein with an ATPase domain involved in DNA-annealing activities and a binding site for the RPA single-strand-DNA-binding protein. Although the role played by Smarcal1 in the maintenance of replication forks has been established, it remains unknown whether Smarcal1 contributes to genomic DNA maintenance outside of the S phase. We disrupted the SMARCAL1 gene in both the chicken DT40 and the human TK6 B cell lines. The resulting SMARCAL1(-/-) clones exhibited sensitivity to chemotherapeutic topoisomerase 2 inhibitors, just as nonhomologous end-joining (NHEJ) null-deficient cells do. SMARCAL1(-/-) cells also exhibited an increase in radiosensitivity in the G1 phase. Moreover, the loss of Smarcal1 in NHEJ null-deficient cells does not further increase their radiosensitivity. These results demonstrate that Smarcal1 is required for efficient NHEJ-mediated DSB repair. Both inactivation of the ATPase domain and deletion of the RPA-binding site cause the same phenotype as does null-mutation of Smarcal1, suggesting that Smarcal1 enhances NHEJ, presumably by interacting with RPA at unwound single-strand sequences and then facilitating annealing at DSB ends. SMARCAL1(-/-)cells showed a poor accumulation of Ku70/DNA-PKcs and XRCC4 at DNA-damage sites. We propose that Smarcal1 maintains the duplex status of DSBs to ensure proper recruitment of NHEJ factors to DSB sites.
DOI:10.1038/emboj.2010.193URLPMID:20729809 [本文引用: 1]
In this study, we investigate the interplay between Ku, a central non-homologous end-joining component, and the Mre11-Rad50-Xrs2 (MRX) complex and Sae2, end-processing factors crucial for initiating 5'-3' resection of double-strand break (DSB) ends. We show that in the absence of end protection by Ku, the requirement for the MRX complex is bypassed and resection is executed by Exo1. In contrast, both the Exo1 and Sgs1 resection pathways contribute to DSB processing in the absence of Ku and Sae2 or when the MRX complex is intact, but functionally compromised by elimination of the Mre11 nuclease activity. The ionizing radiation sensitivity of a mutant defective for extensive resection (exo1Δ sgs1Δ) cannot be suppressed by the yku70Δ mutation, indicating that Ku suppression is specific to the initiation of resection. We provide evidence that replication-associated DSBs need to be processed by Sae2 for repair by homologous recombination unless Ku is absent. Finally, we show that the presence of Ku exacerbates DNA end-processing defects established in the sae2Δ sgs1Δ mutant, leading to its lethality.
DOI:10.1073/pnas.1720962115URLPMID:29472448 [本文引用: 1]
Activation-induced cytidine deaminase (AID) inflicts DNA damage at Ig genes to initiate class switch recombination (CSR) and chromosomal translocations. However, the DNA lesions formed during these processes retain an element of randomness, and thus knowledge of the relationship between specific DNA lesions and AID-mediated processes remains incomplete. To identify necessary and sufficient DNA lesions in CSR, the Cas9 endonuclease and nickase variants were used to program DNA lesions at a greater degree of predictability than is achievable with conventional induction of CSR. Here we show that Cas9-mediated nicks separated by up to 250 nucleotides on opposite strands can mediate CSR. Staggered double-stranded breaks (DSBs) result in more end resection and junctional microhomology than blunt DSBs. Moreover, Myc-Igh chromosomal translocations, which are carried out primarily by alternative end joining (A-EJ), were preferentially induced by 5' DSBs. These data indicate that DSBs with 5' overhangs skew intrachromosomal and interchromosomal end-joining toward A-EJ. In addition to lending potential insight to AID-mediated phenomena, this work has broader carryover implications in DNA repair and lymphomagenesis.
DOI:10.1016/j.pbiomolbio.2014.12.003URLPMID:25550082 [本文引用: 1]
The DNA-dependent protein kinase (DNA-PK) is a serine/threonine protein kinase composed of a large catalytic subunit (DNA-PKcs) and the Ku70/80 heterodimer. Over the past two decades, significant progress has been made in elucidating the role of DNA-PK in non-homologous end joining (NHEJ), the major pathway for repair of ionizing radiation-induced DNA double strand breaks in human cells and recently, additional roles for DNA-PK have been reported. In this review, we will describe the biochemistry, structure and function of DNA-PK, its roles in DNA double strand break repair and its newly described roles in mitosis and other cellular processes.
DOI:10.1038/nrm.2017.48URLPMID:28512351 [本文引用: 1]
DNA double-strand breaks (DSBs) are the most dangerous type of DNA damage because they can result in the loss of large chromosomal regions. In all mammalian cells, DSBs that occur throughout the cell cycle are repaired predominantly by the non-homologous DNA end joining (NHEJ) pathway. Defects in NHEJ result in sensitivity to ionizing radiation and the ablation of lymphocytes. The NHEJ pathway utilizes proteins that recognize, resect, polymerize and ligate the DNA ends in a flexible manner. This flexibility permits NHEJ to function on a wide range of DNA-end configurations, with the resulting repaired DNA junctions often containing mutations. In this Review, we discuss the most recent findings regarding the relative involvement of the different NHEJ proteins in the repair of various DNA-end configurations. We also discuss the shunting of DNA-end repair to the auxiliary pathways of alternative end joining (a-EJ) or single-strand annealing (SSA) and the relevance of these different pathways to human disease.
DOI:10.1074/jbc.M116.751867URLPMID:27875301 [本文引用: 2]
DNA double-strand break (DSB) repair by non-homologous end joining (NHEJ) in human cells is initiated by Ku heterodimer binding to a DSB, followed by recruitment of core NHEJ factors including DNA-dependent protein kinase catalytic subunit (DNA-PKcs), XRCC4-like factor (XLF), and XRCC4 (X4)-DNA ligase IV (L4). Ku also interacts with accessory factors such as aprataxin and polynucleotide kinase/phosphatase-like factor (APLF). Yet, how these factors interact to tether, process, and ligate DSB ends while allowing regulation and chromatin interactions remains enigmatic. Here, small angle X-ray scattering (SAXS) and mutational analyses show APLF is largely an intrinsically disordered protein that binds Ku, Ku/DNA-PKcs (DNA-PK), and X4L4 within an extended flexible NHEJ core complex. X4L4 assembles with Ku heterodimers linked to DNA-PKcs via flexible Ku80 C-terminal regions (Ku80CTR) in a complex stabilized through APLF interactions with Ku, DNA-PK, and X4L4. Collective results unveil the solution architecture of the six-protein complex and suggest cooperative assembly of an extended flexible NHEJ core complex that supports APLF accessibility while possibly providing flexible attachment of the core complex to chromatin. The resulting dynamic tethering furthermore, provides geometric access of L4 catalytic domains to the DNA ends during ligation and of DNA-PKcs for targeted phosphorylation of other NHEJ proteins as well as trans-phosphorylation of DNA-PKcs on the opposing DSB without disrupting the core ligation complex. Overall the results shed light on evolutionary conservation of Ku, X4, and L4 activities, while explaining the observation that Ku80CTR and DNA-PKcs only occur in a subset of higher eukaryotes.
DOI:10.1016/j.bpj.2014.05.027URLPMID:24988341 [本文引用: 1]
Chromatin dynamics modulate DNA repair factor accessibility throughout the DNA damage response. The spatiotemporal scale upon which these dynamics occur render them invisible to live cell imaging. Here we present a believed novel assay to monitor the in?vivo structural rearrangements of chromatin during DNA repair. By pair correlation analysis of EGFP molecular flow into chromatin before and after damage, this assay measures millisecond variations in chromatin compaction with submicron resolution. Combined with laser microirradiation we employ this assay to monitor the real-time accessibility of DNA at the damage site. We find from comparison of EGFP molecular flow with a molecule that has an affinity toward double-strand breaks (Ku-EGFP) that DNA damage induces a transient decrease in chromatin compaction at the damage site and an increase in compaction to adjacent regions, which together facilitate DNA repair factor recruitment to the lesion with high spatiotemporal control.
DOI:10.1126/science.aak9654URLPMID:28154079 [本文引用: 1]
DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is a central component of nonhomologous end joining (NHEJ), repairing DNA double-strand breaks that would otherwise lead to apoptosis or cancer. We have solved its structure in complex with the C-terminal peptide of Ku80 at 4.3 angstrom resolution using x-ray crystallography. We show that the 4128-amino acid structure comprises three large structural units: the N-terminal unit, the Circular Cradle, and the Head. Conformational differences between the two molecules in the asymmetric unit are correlated with changes in accessibility of the kinase active site, which are consistent with an allosteric mechanism to bring about kinase activation. The location of KU80ct194 in the vicinity of the breast cancer 1 (BRCA1) binding site suggests competition with BRCA1, leading to pathway selection between NHEJ and homologous recombination.
DOI:10.1074/jbc.M507113200URLPMID:16093244 [本文引用: 1]
Artemis protein has irreplaceable functions in V(D)J recombination and nonhomologous end joining (NHEJ) as a hairpin and 5' and 3' overhang endonuclease. The kinase activity of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is necessary in activating Artemis as an endonuclease. Here we report that three basal phosphorylation sites and 11 DNA-PKcs phosphorylation sites within the mammalian Artemis are all located in the C-terminal domain. All but one of these phosphorylation sites deviate from the SQ or TQ motif of DNA-PKcs that was predicted previously from in vitro phosphorylation studies. Phosphatase-treated mammalian Artemis and Artemis that is mutated at the three basal phosphorylation sites still retain DNA-PKcs-dependent endonucleolytic activities, indicating that basal phosphorylation is not required for the activation. In vivo studies of Artemis lacking the C-terminal domain have been reported to be sufficient to complement V(D)J recombination in Artemis null cells. Therefore, the C-terminal domain may have a negative regulatory effect on the Artemis endonucleolytic activities, and phosphorylation by DNA-PKcs in the C-terminal domain may relieve this inhibition.
DOI:10.1093/nar/gkt1309URLPMID:24362840 [本文引用: 1]
5' strand resection at DNA double strand breaks (DSBs) is critical for homologous recombination (HR) and genomic stability. Here we develop a novel method to quantitatively measure single-stranded DNA intermediates in human cells and find that the 5' strand at endonuclease-generated break sites is resected up to 3.5 kb in a cell cycle-dependent manner. Depletion of CtIP, Mre11, Exo1 or SOSS1 blocks resection, while depletion of 53BP1, Ku or DNA-dependent protein kinase catalytic subunit leads to increased resection as measured by this method. While 53BP1 negatively regulates DNA end processing, depletion of Brca1 does not, suggesting that the role of Brca1 in HR is primarily to promote Rad51 filament formation, not to regulate end resection.
DOI:10.1038/cr.2017.110URLPMID:28840859 [本文引用: 1]
DNA-dependent protein kinase (DNA-PK) is a serine/threonine protein kinase complex composed of a catalytic subunit (DNA-PKcs) and KU70/80 heterodimer bound to DNA. DNA-PK holoenzyme plays a critical role in non-homologous end joining (NHEJ), the major DNA repair pathway. Here, we determined cryo-electron microscopy structure of human DNA-PK holoenzyme at 6.6 ? resolution. In the complex structure, DNA-PKcs, KU70, KU80 and DNA duplex form a 650-kDa heterotetramer with 1:1:1:1 stoichiometry. The N-terminal α-solenoid (~2 800 residues) of DNA-PKcs adopts a double-ring fold and connects the catalytic core domain of DNA-PKcs and KU70/80-DNA. DNA-PKcs and KU70/80 together form a DNA-binding tunnel, which cradles ~30-bp DNA and prevents sliding inward of DNA-PKcs along with DNA duplex, suggesting a mechanism by which the broken DNA end is protected from unnecessary processing. Structural and biochemical analyses indicate that KU70/80 and DNA coordinately induce conformational changes of DNA-PKcs and allosterically stimulate its kinase activity. We propose a model for activation of DNA-PKcs in which allosteric signals are generated upon DNA-PK holoenzyme formation and transmitted to the kinase domain through N-terminal HEAT repeats and FAT domain of DNA-PKcs. Our studies suggest a mechanism for recognition and protection of broken DNA ends and provide a structural basis for understanding the activation of DNA-PKcs and DNA-PK-mediated NHEJ pathway.
DOI:10.1016/j.dnarep.2010.09.018URL [本文引用: 1]
Abstract
The repair of DNA double strand breaks (dsb) is important for maintaining the physical and genetic integrity of the genome. Moreover, in humans it is associated with the prevention of diseases such as immune deficiencies and cancer. This review briefly explores the fundamental strategies for repairing dsb, examines how cells maximize the fidelity of dsb repair in the cell cycle and discusses the requirements for dsb repair in the context of chromatin.DOI:10.1016/j.molcel.2013.11.003URLPMID:24316220 [本文引用: 1]
MRE11 within the MRE11-RAD50-NBS1 (MRN) complex acts in DNA double-strand break repair (DSBR), detection, and signaling; yet, how its endo- and exonuclease activities regulate DSBR by nonhomologous end-joining (NHEJ) versus homologous recombination (HR) remains enigmatic. Here, we employed structure-based design with a focused chemical library to discover specific MRE11 endo- or exonuclease inhibitors. With these inhibitors, we examined repair pathway choice at DSBs generated in G2 following radiation exposure. While nuclease inhibition impairs radiation-induced replication protein A (RPA) chromatin binding, suggesting diminished resection, the inhibitors surprisingly direct different repair outcomes. Endonuclease inhibition promotes NHEJ in lieu of HR, while exonuclease inhibition confers a repair defect. Collectively, the results describe nuclease-specific MRE11 inhibitors, define distinct nuclease roles in DSB repair, and support a mechanism whereby MRE11 endonuclease initiates resection, thereby licensing HR followed by MRE11 exonuclease and EXO1/BLM bidirectional resection toward and away from the DNA end, which commits to HR.
DOI:10.3389/fmolb.2019.00043URLPMID:31231660 [本文引用: 1]
DNA double-strand breaks (DSBs) are highly cytotoxic lesions that must be repaired to ensure genomic stability and avoid cell death. The cellular response to DSBs is initiated by the evolutionarily conserved Mre11-Rad50-Xrs2/NBS1 (MRX/MRN) complex that has structural and catalytic functions. Furthermore, it is responsible for DSB signaling through the activation of the checkpoint kinase Tel1/ATM. Here, we review functions and regulation of the MRX/MRN complex in DSB processing in a chromatin context, as well as its interplay with Tel1/ATM.
DOI:10.1093/nar/gkz518URLPMID:31184714 [本文引用: 1]
The nucleolus is a nuclear sub-domain containing the most highly transcribed genes in the genome. Hundreds of human ribosomal RNA (rRNA) genes, located in the nucleolus, rely on constant maintenance. DNA double-strand breaks (DSBs) in rRNA genes activate the ATM kinase, repress rRNA transcription and induce nucleolar cap formation. Yet how ribosomal-DNA (rDNA) lesions are detected and processed remains elusive. Here, we use CRISPR/Cas9-mediated induction of DSBs and report a chromatin response unique to rDNA depending on ATM-phosphorylation of the nucleolar protein TCOF1 and recruitment of the MRE11-RAD50-NBS1 (MRN) complex via the NBS1-subunit. NBS1- and MRE11-depleted cells fail to suppress rRNA transcription and to translocate rDNA into nucleolar caps. Furthermore, the DNA damage response (DDR) kinase ATR operates downstream of the ATM-TCOF1-MRN interplay and is required to fully suppress rRNA transcription and complete DSB-induced nucleolar restructuring. Unexpectedly, we find that DSBs in rDNA neither activate checkpoint kinases CHK1/CHK2 nor halt cell-cycle progression, yet the nucleolar-DDR protects against genomic aberrations and cell death. Our data highlight the concept of a specialized nucleolar DNA damage response (n-DDR) with a distinct protein composition, spatial organization and checkpoint communication. The n-DDR maintains integrity of ribosomal RNA genes, with implications for cell physiology and disease.
DOI:10.1126/science.1164382URLPMID:18772396 [本文引用: 1]
Glioblastoma multiforme (GBM) is the most common and lethal type of brain cancer. To identify the genetic alterations in GBMs, we sequenced 20,661 protein coding genes, determined the presence of amplifications and deletions using high-density oligonucleotide arrays, and performed gene expression analyses using next-generation sequencing technologies in 22 human tumor samples. This comprehensive analysis led to the discovery of a variety of genes that were not known to be altered in GBMs. Most notably, we found recurrent mutations in the active site of isocitrate dehydrogenase 1 (IDH1) in 12% of GBM patients. Mutations in IDH1 occurred in a large fraction of young patients and in most patients with secondary GBMs and were associated with an increase in overall survival. These studies demonstrate the value of unbiased genomic analyses in the characterization of human brain cancer and identify a potentially useful genetic alteration for the classification and targeted therapy of GBMs.
DOI:10.1016/j.molcel.2017.07.001URLPMID:28757209 [本文引用: 1]
Brca2 deficiency causes Mre11-dependent degradation of nascent DNA at stalled forks, leading to cell lethality. To understand the molecular mechanisms underlying this process, we isolated Xenopus laevis Brca2. We demonstrated that Brca2 protein prevents single-stranded DNA gap accumulation at replication fork junctions and behind them by promoting Rad51 binding to replicating DNA. Without Brca2, forks with persistent gaps are converted by Smarcal1 into reversed forks, triggering extensive Mre11-dependent nascent DNA degradation. Stable Rad51 nucleofilaments, but not RPA or Rad51T131P mutant proteins, directly prevent Mre11-dependent DNA degradation. Mre11 inhibition instead promotes reversed fork accumulation in the absence of Brca2. Rad51 directly interacts with the Pol α N-terminal domain, promoting Pol α and δ binding to stalled replication forks. This?interaction?likely promotes replication fork restart?and gap avoidance. These results indicate that Brca2?and Rad51 prevent formation of abnormal DNA replication intermediates, whose processing by?Smarcal1 and Mre11 predisposes to genome instability.
DOI:10.1101/gad.1095203URLPMID:12730130 [本文引用: 2]
DOI:10.1038/nature07449URLPMID:19011615 [本文引用: 2]
The Myc oncogene regulates the expression of several components of the protein synthetic machinery, including ribosomal proteins, initiation factors of translation, RNA polymerase III and ribosomal DNA. Whether and how increasing the cellular protein synthesis capacity affects the multistep process leading to cancer remains to be addressed. Here we use ribosomal protein heterozygote mice as a genetic tool to restore increased protein synthesis in Emu-Myc/+ transgenic mice to normal levels, and show that the oncogenic potential of Myc in this context is suppressed. Our findings demonstrate that the ability of Myc to increase protein synthesis directly augments cell size and is sufficient to accelerate cell cycle progression independently of known cell cycle targets transcriptionally regulated by Myc. In addition, when protein synthesis is restored to normal levels, Myc-overexpressing precancerous cells are more efficiently eliminated by programmed cell death. Our findings reveal a new mechanism that links increases in general protein synthesis rates downstream of an oncogenic signal to a specific molecular impairment in the modality of translation initiation used to regulate the expression of selective messenger RNAs. We show that an aberrant increase in cap-dependent translation downstream of Myc hyperactivation specifically impairs the translational switch to internal ribosomal entry site (IRES)-dependent translation that is required for accurate mitotic progression. Failure of this translational switch results in reduced mitotic-specific expression of the endogenous IRES-dependent form of Cdk11 (also known as Cdc2l and PITSLRE), which leads to cytokinesis defects and is associated with increased centrosome numbers and genome instability in Emu-Myc/+ mice. When accurate translational control is re-established in Emu-Myc/+ mice, genome instability is suppressed. Our findings demonstrate how perturbations in translational control provide a highly specific outcome for gene expression, genome stability and cancer initiation that have important implications for understanding the molecular mechanism of cancer formation at the post-genomic level.
DOI:10.1021/jacs.6b09196URLPMID:27669098 [本文引用: 2]
MYC is overexpressed in many different cancer types and is an intensively studied oncogene because of its contributions to tumorigenesis. The regulation of MYC is complex, and the NHE III1 and FUSE elements rely upon noncanonical DNA structures and transcriptionally induced negative superhelicity. In the NHE III1 only the G-quadruplex has been extensively studied, whereas the role of the i-motif, formed on the opposite C-rich strand, is much less understood. We demonstrate here that the i-motif is formed within the 4CT element and is recognized by hnRNP K, which leads to a low level of transcription activation. For maximal hnRNP K transcription activation, two additional cytosine runs, located seven bases downstream of the i-motif-forming region, are also required. To access these additional runs of cytosine, increased negative superhelicity is necessary, which leads to a thermodynamically stable complex between hnRNP K and the unfolded i-motif. We also demonstrate mutual exclusivity between the MYC G-quadruplex and i-motif, providing a rationale for a molecular switch mechanism driven by SP1-induced negative superhelicity, where relative hnRNP K and nucleolin expression shifts the equilibrium to the on or off state.
DOI:10.1093/hmg/dds083URL [本文引用: 2]
Biallelic mutations of the DNA annealing helicase SMARCAL1 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a-like 1) cause Schimke immuno-osseous dysplasia (SIOD, MIM 242900), an incompletely penetrant autosomal recessive disorder. Using human, Drosophila and mouse models, we show that the proteins encoded by SMARCAL1 orthologs localize to transcriptionally active chromatin and modulate gene expression. We also show that, as found in SIOD patients, deficiency of the SMARCAL1 orthologs alone is insufficient to cause disease in fruit flies and mice, although such deficiency causes modest diffuse alterations in gene expression. Rather, disease manifests when SMARCAL1 deficiency interacts with genetic and environmental factors that further alter gene expression. We conclude that the SMARCAL1 annealing helicase buffers fluctuations in gene expression and that alterations in gene expression contribute to the penetrance of SIOD.
DOI:10.1038/srep17910URLPMID:26648259 [本文引用: 4]
SMARCAL1, a member of the SWI2/SNF2 protein family, stabilizes replication forks during DNA damage. In this manuscript, we provide the first evidence that SMARCAL1 is also a transcriptional co-regulator modulating the expression of c-Myc, a transcription factor that regulates 10-15% genes in the human genome. BRG1, SMARCAL1 and RNAPII were found localized onto the c-myc promoter. When HeLa cells were serum starved, the occupancy of SMARCAL1 on the c-myc promoter increased while that of BRG1 and RNAPII decreased correlating with repression of c-myc transcription. Using Active DNA-dependent ATPase A Domain (ADAAD), the bovine homolog of SMARCAL1, we show that the protein can hydrolyze ATP using a specific region upstream of the CT element of the c-myc promoter as a DNA effector. The energy, thereby, released is harnessed to alter the conformation of the promoter DNA. We propose that SMARCAL1 negatively regulates c-myc transcription by altering the conformation of its promoter region during differentiation.
DOI:10.1016/S0379-4172(06)60001-2URLPMID:16450581 [本文引用: 5]
The mechanism underlying CAG.CTG CGG.CCG and GAA.TTC trinucleotide repeats expansion and contraction instabilities has not been clearly understood. Investigations in vitro have demonstrated that the disease causing repeats are capable of adopting non-B secondary structures that mediate repeats expansion. However, in vivo, similar observations have not been easily made so far. Investigations on the non-B secondary structure formation using E.coli, yeast etc cannot simulate the suggested repeats expansion instability. These could leave a space to infer a disassociation of the suggested repeats non-B secondary structure formation and the repeats expansion in vivo. Although longer trinucleotide repeats may be theoretically easier to form non-B DNA secondary structures in replication or in post-replication, however such non-B secondary structures are likely to cause repeat fragility rather than repeat expansion. In fact, repeat expansion as seen in patients may not necessarily require trinucleotide repeats to form non-B secondary structures, instead the repeat expansions can be produced through a RNA transcription-stimulated local repeat DNA replication and a subsequent DNA rearrangement.
DOI:10.3724/SP.J.1005.2014.1185URLPMID:25487262 [本文引用: 5]
R-loop is a type of three-stranded nucleic acid structure that is made up of an RNA:DNA hybrid, formed due to failing separation of a nascent RNA molecule with transcripting template in transcription or by the re-annealing of RNA molecule with one of the two strands in a double stranded DNA molecule, along with the single stranded DNA, which is either the non-template strand in the transcription bubble or the RNA substituted DNA strand. Formation of R-loops can occur when transcription goes through a genomic DNA region having a tract of G bases in the non-template strand in the transcription bubble or through a type of triplet microsatellite DNA sequences that are known to be associated with certain human diseases. The negative supercoiling forces accumulated in the transcription bubble, and the misprocessing of RNA precursors, as well as the delayed utilizations and transportations of RNA molecules to cytoplasm promote R loop formation. Many studies show that cells can manage R loop formation with efficiency, and can also process the R-loops already formed in the cell, and by which, the bad effects of R-loops on DNA replication, gene mutation and homologous recombination can be regulated. In this review, we summarize the formation and the impacts of R-loops on DNA replication, mutation rates and the frequencies of homologous recombination, and also discusse the possible role of the R-loop induced DNA replication in mediating trinucleotide repeat expansions as seen with those frequently associated with human neuromuscular degenerative diseases.
DOI:10.3724/SP.J.1005.2014.1185URLPMID:25487262 [本文引用: 5]
R-loop is a type of three-stranded nucleic acid structure that is made up of an RNA:DNA hybrid, formed due to failing separation of a nascent RNA molecule with transcripting template in transcription or by the re-annealing of RNA molecule with one of the two strands in a double stranded DNA molecule, along with the single stranded DNA, which is either the non-template strand in the transcription bubble or the RNA substituted DNA strand. Formation of R-loops can occur when transcription goes through a genomic DNA region having a tract of G bases in the non-template strand in the transcription bubble or through a type of triplet microsatellite DNA sequences that are known to be associated with certain human diseases. The negative supercoiling forces accumulated in the transcription bubble, and the misprocessing of RNA precursors, as well as the delayed utilizations and transportations of RNA molecules to cytoplasm promote R loop formation. Many studies show that cells can manage R loop formation with efficiency, and can also process the R-loops already formed in the cell, and by which, the bad effects of R-loops on DNA replication, gene mutation and homologous recombination can be regulated. In this review, we summarize the formation and the impacts of R-loops on DNA replication, mutation rates and the frequencies of homologous recombination, and also discusse the possible role of the R-loop induced DNA replication in mediating trinucleotide repeat expansions as seen with those frequently associated with human neuromuscular degenerative diseases.
[本文引用: 5]
[本文引用: 5]
DOI:10.1038/s41594-018-0094-9URLPMID:30061600 [本文引用: 3]
We developed an experimental system for studying genome instability caused by fragile X (CGG)n repeats in mammalian cells. Our method uses a selectable cassette carrying the HyTK gene under the control of the FMR1 promoter with (CGG)n repeats in its 5' UTR, which is integrated into the unique RL5 site in murine erythroid leukemia cells. Carrier-size (CGG)n repeats markedly elevated the frequency of reporter inactivation, making cells ganciclovir resistant. These resistant clones had a unique mutational signature: a change in repeat length concurrent with mutagenesis in the reporter gene. Inactivation of genes implicated in break-induced replication, including Pold3, Pold4, Rad52, Rad51, and Smarcal1, reduced the frequency of ganciclovir-resistant clones to the baseline level that was observed in the absence of (CGG)n repeats. We propose that replication fork collapse at carrier-size (CGG)n repeats can trigger break-induced replication, which results in simultaneous repeat length changes and mutagenesis at a distance.
[本文引用: 2]
DOI:10.1542/peds.2005-2614URLPMID:16816006 [本文引用: 2]
Schimke-immuno-osseous dysplasia is a rare autosomal-recessive multisystem disorder with the main clinical features of disproportionate growth deficiency, defective cellular immunity, and progressive renal disease. It is caused by mutations of SMARCAL1, a gene encoding a putative chromatin remodeling protein of unknown function. Because a detailed description of the clinical features is an essential first step in elucidating the function of SMARCAL1, we present the first detailed anthropometric data for Schimke-immuno-osseous dysplasia patients. By comprehensive anthropometric examination (28 parameters) of 8 patients (3 females) with the typical findings of Schimke-immuno-osseous dysplasia (mean age: 14.8 years; range: 4.9-30.5 years) and 304 patients (117 females) with congenital and hereditary chronic kidney disease (mean age: 10.7 +/- 4.8 years; range: 3-21.8 years), we show that Schimke-immuno-osseous dysplasia patients differ significantly from those with other forms of chronic kidney disease. z scores were calculated with reference limits derived from 5155 healthy children (2591 females) aged 3 to 18 years. The key finding was that, in the latter group, median leg length was significantly more reduced than sitting height, whereas in Schimke-immuno-osseous dysplasia patients, the reduction of sitting height was significantly more pronounced than for leg length. Therefore, the ratio of sitting height/leg length might be a simple tool for the clinician to distinguish Schimke-immuno-osseous dysplasia from other chronic kidney disease patients. Schimke-immuno-osseous dysplasia is very likely if this ratio is < 0.83. However, other forms of chronic kidney disease have to be discussed in case of a ratio > 1.01.
DOI:10.1111/febs.13382URLPMID:26195148 [本文引用: 1]
Mutations and deletions in SMARCAL1, an SWI2/SNF2 protein, cause Schimke immuno-osseous dysplasia (SIOD). SMARCAL1 preferentially binds to DNA molecules possessing double-stranded to single-stranded transition regions and mediates annealing helicase activity. The protein is critical for alleviating replication stress and maintaining genome integrity. In this study, we have analysed the ATPase activity of three mutations – A468P, I548N and S579L – present in SIOD patients. These mutations are present in RecA-like domain I of the protein. Analysis using active DNA-dependent ATPase A domain (ADAAD), an N-terminal deleted construct of bovine SMARCAL1, showed that all three mutants were unable to hydrolyse ATP. Conformational studies indicated that the α-helix and β-sheet content of the mutant proteins was altered compared to the wild-type protein. Molecular simulation studies confirmed that major structural changes had occurred in the mutant proteins. These changes included alteration of a loop region connecting motif Ia and II. As motif Ia has been implicated in DNA binding, ligand binding studies were done using fluorescence spectroscopy. These studies revealed that the Kd for protein-DNA interaction in the presence of ATP was indeed altered in the case of mutant proteins compared to the wild-type. Finally, in vivo studies were done to complement the in vitro and in silico studies. The results from these experiments demonstrate that mutations in human SMARCAL1 that result in loss in ATPase activity lead to increased replication stress and therefore possibly manifestation of SIOD.
DOI:10.1136/jmg.36.10.786URLPMID:10528861 [本文引用: 1]
Immuno-osseous dysplasia is characterised by spondyloepiphyseal dysplasia, lymphopenia with defective cellular immunity, and progressive renal disease. We describe a patient with a severe form of the disease, review the features of another 24 patients, and discuss the previous classification. The differences between the two groups are not striking, and although similarities are greater between affected sibs, the same diagnosis of Schimke immuno-osseous dysplasia should apply to them all. The aetiology and physiopathology of this rare osteochondrodysplasia of presumed autosomal recessive inheritance remain unknown.
DOI:10.1007/s004310050001URL [本文引用: 1]
Schimke immuno-osseous dysplasia (SIOD) is a rare autosomal recessive spondylo-epiphyseal dysplasia. The characteristic features of SIOD include 1) short stature with hyperpigmented macules and an unusual facies, 2) proteinuria with progressive renal failure, 3) lymphopenia with recurrent infections, and 4) cerebral ischaemia. Although 25 patients have been reported with this disorder, the clinical course and phenotype of SIOD are not well characterized. This report summarizes the clinical findings, course and treatment of reported patients and includes 14 additional patients with SIOD. We emphasize the high incidence of cerebral ischaemia and ocular abnormalities, define the high incidence of thyroid dysfunction and blood cytopenia, and confirm the absence of effective and durable medical therapies. Conclusion Schimke immuno-osseous dysplasia is a multi-system autosomal recessive disorder with variable expression that affects the skeletal, renal, immune, vascular, and haematopoietic systems. Medical therapy is limited especially for more severely affected individuals.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12887-017-0968-8URLPMID:29282041 [本文引用: 1]
Schimke immune-osseous dysplasia (SIOD, OMIM 242900) is characterized by spondyloepiphyseal dysplasia, T-cell deficiency, renal dysfunction and special facial features. SMARCAL1 gene mutations are determined in approximately 50% of patients diagnosed with SIOD.
DOI:10.1016/j.ejmg.2016.06.002URLPMID:27282802 [本文引用: 1]
Schimke immunoosseous dysplasia (SIOD) is an autosomal recessive disease characterized by skeletal dysplasia, focal segmental glomerulosclerosis, renal failure and immunodeficiency. In this work, we report the molecular studies undertaken in a severely affected SIOD patient that died at six years old due to nephropathy. The patient was screened for mutations using a targeted skeletal dysplasias panel. A homozygous novel missense mutation was identified, c.1615C?>?G (p.[Leu539Val]) that was predicted as mildly pathogenic by in silico pathogenicity prediction tools. However, splicing prediction software suggested that this variant may create a new splicing donor site in exon 9, which was subsequently confirmed using a minigene assay in HEK293?cells. Thus, the splicing alteration, c.1615C?>?G; r.1615c?>?g, 1615_1644del; (p.[Leu539_Ile548del]), results in the loss of 10 amino acids of the HARP-ATPase catalytic domain and the RPA-binding domain. Several studies have demonstrated a weak genotype-phenotype correlation among such patients. Thus, the molecular characterization has helped us to understand why a predicted weakly pathogenic missense mutation results in severe SIOD and should be considered in similar scenarios.
DOI:10.1186/1471-2369-15-41URLPMID:24589093 [本文引用: 1]
Schimke immuno-osseous dysplasia (SIOD, OMIM #242900) is an autosomal-recessive pleiotropic disorder characterized by spondyloepiphyseal dysplasia, renal dysfunction and T-cell immunodeficiency. SIOD is caused by mutations in the gene SMARCAL1.
DOI:10.1002/pbc.24542URL [本文引用: 1]
Schimke Immunoosseous Dysplasia (SIOD) is a rare, autosomal recessive disorder of childhood with classical features of spondyloepiphyseal dysplasia, renal failure, and T cell immunodeficiency. SIOD has been associated with several malignancies, including non-Hodgkin lymphoma and osteosarcoma. About half of SIOD patients have biallelic mutations in SMARCAL1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin, subfamily a-like 1). This gene encodes an annealing helicase and replication stress response protein that localizes to damage-stalled DNA replication forks. We report a child with SIOD and a novel S859P missense mutation in SMARCAL1 who developed undifferentiated carcinoma of the sinus. Pediatr Blood Cancer 2013;60:E88-E90. (c) 2013 Wiley Periodicals, Inc.
DOI:10.1093/ndt/gfq071URLPMID:20179009 [本文引用: 1]
Schimke immuno-osseous dysplasia (SIOD) is a rare autosomal recessive pleiotropic disease caused by mutations in the SMARCAL1 gene. To date there have been no data from the Chinese population. Here, we report the first SIOD case in the Chinese population. No case with gross carpal bone age retardation has been reported previously.
DOI:10.1007/s00467-005-2047-xURL [本文引用: 2]
Autosomal-recessive Schimke immuno-osseous dysplasia (SIOD) characterized by spondyloepiphyseal dysplasia, focal-segmental glomerulosclerosis (FSGS), T-cell immunodeficiency and facial dysmorphism is caused by defects in the SMARCAL1 gene. The gene product is involved in the transcriptional regulation of other genes. A 12-year-old boy of consanginous Turkish descent developed disproportionate short stature from spondyloepiphyseal dysplasia at the age of 6 and nephrotic syndrome at the age of 10years. Renal biopsy revealed FSGS, the kidney function was normal, T-lymphocytes were diminished without infectious complications, and he has had no cerebral ischemia. Analysis of the patient
 s SMARCAL1 gene revealed a novel homozygous C1798T transition leading to a R561C substitution. The parents and two healthy sisters were found to be heterozygous. A younger brother, who is also homozygous for the mutation, is clinically asymptomatic and has no proteinuria at the age of 18months. Still, his CD4 cells are diminished. For SMARCAL1 mutations a clear genotype-phenotype correlation has been reported: severe SIOD with in utero or early-childhood onset leading to end-stage renal disease within a few years is caused by nonsense, frame shift or splice mutations. Many patients die from infections and cerebrovascular insults during childhood. Mild SIOD manifests later and progresses more slowly without infectious or cerebral vascular complications—the underlying defect being missense mutations in all three patients reported so far. The novel R561C missense mutation in our patient with mild SIOD is additional evidence for the genotype-phenotype correlation reported for SMARCAL1 mutations.
s SMARCAL1 gene revealed a novel homozygous C1798T transition leading to a R561C substitution. The parents and two healthy sisters were found to be heterozygous. A younger brother, who is also homozygous for the mutation, is clinically asymptomatic and has no proteinuria at the age of 18months. Still, his CD4 cells are diminished. For SMARCAL1 mutations a clear genotype-phenotype correlation has been reported: severe SIOD with in utero or early-childhood onset leading to end-stage renal disease within a few years is caused by nonsense, frame shift or splice mutations. Many patients die from infections and cerebrovascular insults during childhood. Mild SIOD manifests later and progresses more slowly without infectious or cerebral vascular complications—the underlying defect being missense mutations in all three patients reported so far. The novel R561C missense mutation in our patient with mild SIOD is additional evidence for the genotype-phenotype correlation reported for SMARCAL1 mutations.DOI:10.1203/PDR.0b013e3181998a74URLPMID:19127206 [本文引用: 1]
Schimke immuno-osseous dysplasia (SIOD) is a rare autosomal-recessive multisystem disorder with disproportionate growth failure, impaired T cell function, and steroid-resistant nephrotic syndrome. Recently, we presented the typical anthropometric features of SIOD. We now present data on two siblings who were initially classified as suffering from familial steroid-resistant nephrotic syndrome of unknown genetic origin. Apart from growth failure, no syndrome-specific symptoms were found until the age of 10 y. However, serial anthropometric examinations showed the development of a SIOD-like pattern with a decreased ratio of trunk to leg length in early adolescence. The growth pattern was significantly different from that seen in children with chronic renal failure of other origins. In prepuberty the siblings had proportionate short stature but developed disproportion only during adolescence. Molecular genetic analysis revealed compound heterozygosity for a known and a new mutation in the SMARCAL1 gene.
DOI:10.1002/ajmg.a.30692URLPMID:15884045 [本文引用: 1]
Schimke immuno-osseous dysplasia (SIOD) is characterized by spondyloepiphyseal dysplasia, nephropathy, and T-cell deficiency. SIOD is caused by mutations in the putative chromatin remodeling protein SMARCAL1. We report an 8-year-old boy with SIOD and recurrent, severe, refractory migraine-like headaches. Through a retrospective questionnaire-based study, we found that refractory and severely disabling migraine-like headaches occur in nearly half of SIOD patients. We have also found that the vasodilator minoxidil provided symptomatic relief for one patient. We hypothesize that these headaches may arise from an intrinsic vascular, neuroimmune, or neurovascular defect resulting from loss of SMARCAL1 function.
DOI:10.1007/s10875-013-9957-3URL [本文引用: 2]
Chromosomal instability syndromes include a group of rare diseases characterized by defective DNA-damage-response and increased risk of chromosomal breakage. Patients display defects in the recognition and/or repair of DNA damage, with a subsequent high rate of malignancies and abnormal gene rearrangements. Other clinical manifestations, such as immunodeficiency, neurodevelopmental delay and skeletal abnormalities, are present in some of these syndromes. We studied a patient with profound T-lymphocyte defect, neurodevelopmental delay, facial dysmorphism, nephrotic syndrome and spondyloepiphyseal bone dysplasia typical of SIOD.
Karyotype and chromosome fragility assays on patients' peripheral blood mononuclear cells showed an abnormal rate of spontaneous breaks. Cell cycle analysis of patient's fibroblasts following replication stress induced by hydroxyhurea revealed a delay in their release from S-phase to G2. When using higher concentrations of hydroxyhurea no patient fibroblast colonies could survive, compared with control fibroblasts. Whole-exome sequencing revealed novel compound heterozygote mutations in SMARCAL1 gene, resulting in putative frame shifts of encoded SMARCAL1 from each allele and no detected protein in patient's cells. The patient's youngest brother was found to have similar manifestations of SIOD but of less severity, including short stature, facial dysmorphism and typical osseous dysplasia, but no clinical findings suggestive of immunodeficiency and no chromosomal fragility. Similar to his sister, the brother carries both bi-allelic mutations in SMARCAL1 gene.
We present here the first evidence of intrinsic chromosomal instability in a severe SMARCAL1-deficient patient with a clinical picture of SIOD. Our results are consistent with the recently outlined role of SMARCAL1 protein in DNA damage response.
DOI:10.1038/ng821URLPMID:11799392 [本文引用: 1]
Schimke immuno-osseous dysplasia (SIOD, MIM 242900) is an autosomal-recessive pleiotropic disorder with the diagnostic features of spondyloepiphyseal dysplasia, renal dysfunction and T-cell immunodeficiency. Using genome-wide linkage mapping and a positional candidate approach, we determined that mutations in SMARCAL1 (SWI/SNF2-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a-like 1), are responsible for SIOD. Through analysis of data from persons with SIOD in 26 unrelated families, we observed that affected individuals from 13 of 23 families with severe disease had two alleles with nonsense, frameshift or splicing mutations, whereas affected individuals from 3 of 3 families with milder disease had a missense mutation on each allele. These observations indicate that some missense mutations allow retention of partial SMARCAL1 function and thus cause milder disease.
DOI:10.1111/j.0105-2896.2004.00162.xURLPMID:15242401 [本文引用: 2]
Efficient repair of DNA double-strand breaks is essential for the maintenance of chromosomal integrity. In higher eukaryotes, non-homologous end-joining (NHEJ) DNA is the primary pathway that repairs these breaks. NHEJ also functions in developing lymphocytes to repair strand breaks that occur during V(D)J recombination, the site-specific recombination process that provides for the assembly of functional antigen-receptor genes. If V(D)J recombination is impaired, B- and T-lymphocyte development is blocked resulting in severe combined immunodeficiency disease. In the last decade, an intensive research effort has focused on NHEJ resulting in a reasonable understanding of how double-strand breaks are resolved. Six distinct gene products have been identified that function in this pathway (Ku70, Ku86, XRCC4, DNA ligase IV, Artemis, and DNA-PKcs). Three of these comprise one complex, the DNA-dependent protein kinase (DNA-PK). This protein complex is central during NHEJ, because DNA-PK initially recognizes and binds to the damaged DNA and then targets the other repair activities to the site of DNA damage. In this review, we discuss recent developments that have provided insight into how DNA-PK functions, once bound to DNA ends.
DOI:10.1002/eji.200535401URLPMID:16358361 [本文引用: 2]
DNA double-strand breaks (dsb) during V(D)J recombination of T and B lymphocyte receptor genes are resolved by the non-homologous DNA end joining pathway (NHEJ) including at least six factors: Ku70, Ku80, DNA-PK(cs), Artemis, Xrcc4, and DNA ligase IV (Lig4). Artemis and Lig4 are the only known V(D)J/NHEJ factors found deficient in human genetic disorders. Null mutations of the Artemis gene result in a complete absence of T and B lymphocytes and increased cellular sensitivity to ionizing radiations, causing radiosensitive-SCID. Mutations of Lig4 are exclusively hypomorphic and have only been described in six patients, four exhibiting mild immunodeficiency associated with microcephaly and developmental delay, while two patient had leukemia. Here we report a SCID associated with microcephaly caused by compound heterozygous hypomorphic mutations in Lig4. Residual activity of Lig4 in these patients is underscored by a normal pattern of TCR-alpha and -beta junctions in the T cells of the patients and a moderate impairment of V(D)J recombination as tested in vitro. These observations contrast with the severity of the clinical immunodeficiency, suggesting that Lig4 may have additional critical roles in lymphocyte survival beyond V(D)J recombination.
DOI:10.1016/j.clim.2009.08.017URLPMID:19796992 [本文引用: 1]
Schimke immuno-osseous dysplasia (SIOD) is caused by SMARCAL1 deficiency and characterized by defective T-cell immunity. The immunodeficiency and the role of thymic function in SIOD patients are not clearly understood. We performed thymic evaluations by assessing T-cell receptor (TCR) diversity, rearrangement, and excision circles in family members with different disease severity carrying the same bi-allelic mutation and in a heterozygous carrier. The expression of SMARCAL1 mRNA in a normal thymic sample was measured using real-time quantitative polymerase chain reaction. Thymus functions were significantly reduced in SIOD patients, and these findings were highly correlated with the clinical phenotype. Quantification of SMARCAL1 mRNA transcript was 3.86-fold higher than normal values for adult kidneys. Genotype alone apparently does not define phenotype, and analysis of TCR diversity, rearrangement, and thymus output can quantify the extent of T-cell immunodeficiency. High thymic expression of SMARCAL1 mRNA raises the possibility of its importance in thymus maintenance and function.
DOI:10.1371/journal.pone.0049822URLPMID:23209606 [本文引用: 1]
Previously, we showed that aminoglycoside phosphotransferases catalyze the formation of a specific inhibitor of the SWI2/SNF2 proteins. Aminoglycoside phosphotransferases, for example neomycin-resistant genes, are used extensively as selection markers in mammalian transfections as well as in transgenic studies. However, introduction of the neomycin-resistant gene is fraught with variability in gene expression. We hypothesized that the introduction of neomycin-resistant genes into mammalian cells results in inactivation of SWI2/SNF2 proteins thereby leading to global epigenetic changes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}