Fibrillin-2 gene mutations associated with hereditary connective tissue diseases
Furu Xu1,2, Wenjun Jiang2,3, Tao Zhang1,2,3, Qian Jiang1,2,3, Ruixue Zhang1,2,3, Hongsheng Bi,1,2,3,4 1. Shandong University of Traditional Chinese Medicine, Jinan 250014, China 2. Shandong Pvovince Key Laboratory of Integrated Traditional Chinese and Western Medicine for Prevention and Therapy of Ocular Disease, Jinan 250002, China 3. Institute of Shandong University of Traditional Chinese, Jinan 250002, China 4. Affiliated Eye Hospital of Shandong University of Traditional Chinese Medicine, Jinan 250002, China
Supported by the National Natural Science Foundation of China No.(81603421) Shandong Province Key Research and Devlepment Program No.(2016GGH3119) Shandong Province Key Research and Devlepment Program No.(2017CXGC1211)
作者简介 About authors 徐福如,在读硕士研究生,专业方向:屈光不正及白内障E-mail:xufuru1995@163.com。
Abstract Fibrillin-2 (FBN2) is an important component of microfibers which are involved in the formation of elastic fibers in connective tissue throughout the human body. Hereditary connective tissue diseases may result from genetic mutations of FBN2 causing heterogeneity of fibrin. Genetic mutations of FBN2 are associated with a variety of hereditary connective tissue diseases including Congenital Contractural Arachnodactyl (CCA), Macular Degeneration (MD), and myopathy. Studies have shown that the FBN2 gene is recognized as the only pathogenic gene related to CCA and that CCA patients have different clinical presentations depending on the identified genetic mutations at different FBN2 sites. In this review, we summarize the roles of FBN2, its mutations and impact on the physiological and pathological processes of many hereditary connective tissue diseases. We include brief descriptions of clinical manifestations of these diseases providing a basis for further exploration of the specific molecular mechanism of FBN2 gene mutation pathogenesis which provides a theoretical basis for the therapy and medications for refractory diseases caused by FBN2 gene mutation. Keywords:fibrillin-2;gene mutation;congenital contracture arachnoid;macular degeneration;genetic
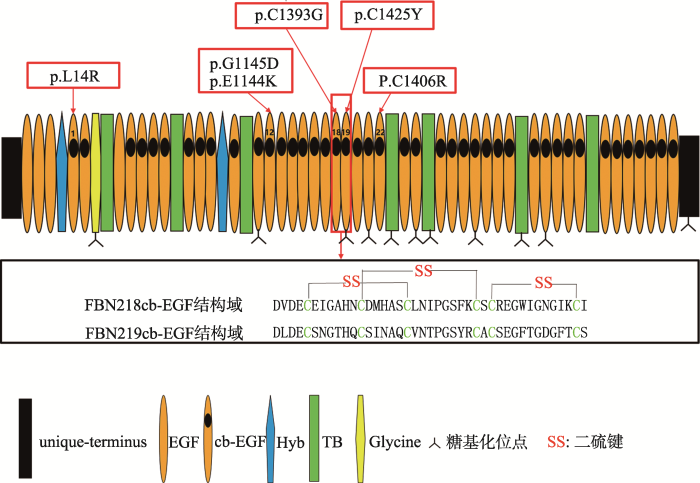
p.G1145D和p.E1144K均为第12cb-EGF结构域的突变位点;p.C1393G为第18cb-EGF结构域的突变位点;p.C1425Y为第19cb-EGF结构域的突变位点;p.C1406R为第22cb-EGF结构域的突变位点。 Fig. 1FBN2 protein domains and CCA-relate mutation sites
DavisMR, SummersKM . Structure and function of the mammalian fibrillin gene family: implications for human connective tissue diseases Mol Genet Metab, 2012,107(4):635-647. [本文引用: 1]
Center for HumanGenetics , DNA test descriptions & CPT codes, http://chginc.org/dna-test-descriptions-cpt-codes/ . URL [本文引用: 1]
LeeB, GodfreyM, VitaleE, HoriH, MatteiMG, SarfaraziM, TsipourasP, RamirezF, HollisterDW . Linkage of Marfan syndrome and a phenotypically related disorder to two different fibrillin genes Nature, 1991,352(6333):330-334. [本文引用: 1]
ZhangH, ApfelrothSD, HuW, DavisEC, SanguinetiC, BonadioJ, MechamRP, RamirezF . Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices J Cell Biol, 1994,124(5):855-863. [本文引用: 1]
TraskTM, RittyTM, BroekelmannT, TisdaleC, MechamRP . N-terminal domains of fibrillin 1 and fibrillin 2 direct the formation of homodimers: a possible first step in microfibril assembly Biochem J, 1999,340(Pt 3):693-701. [本文引用: 1]
YouG, ZuB, WangB, WangZ, XuY, FuQ . Exome sequencing identified a novel FBN2 mutation in a Chinese family with congenital contractural arachnodactyly Int J Mol Sci, 2017,18(4):626-635. [本文引用: 3]
JensenSA, IqbalS, LoweED, RedfieldC, HandfordPA . Structure and interdomain interactions of a hybrid domain: a disulphide-rich module of the fibrillin/tlbp superfamily of matrix proteins Structure, 2009,17(5):759-768. [本文引用: 1]
MeenaJP, GuptaA, MishraD, JunejaM . Beals-hecht syndrome (congenital contractural arachnodactyly) with additional craniospinal abnormality: a case report J Pediatr Orthop B, 2015,24(3):226-229. [本文引用: 1]
ChenQQ, WuYA, HuangXL, ChenT, HuangY, ChenFL, ChenFW . Analysis of two new mutations in FBN1 gene in Han people with Marfan syndrome (MFS) Genetics, 2010,32(1):49-53. Magsci [本文引用: 1] 为调查马凡综合征(Marfan syndrome, MFS)患者的原纤维蛋白-1(Fibrillin-1, FBN1)基因突变情况, 应用聚合酶链反应(PCR)和变性高效液相色谱法(Denaturing high-performance liquid chromatography, DHPLC)对MFS患者的FBN1基因进行突变筛查, 对DHPLC初筛异常的DNA片段进行测序分析。结果在两个MFS家系中发现FBN1基因两种新的突变: 一种为复合突变包含第55号外显子的缺失突变c.6862_6871delGGCTGTGTAG (p.Gly2288MetfsX109)、同义突变c.6861A>G和内含子的突变c.[6871+1_6871+11delGTAAGAGGATC; 6871+34dupCATCAGAAGTGACAGTGGACA]; 另一种为第20号外显子的错义突变c.2462G>A(p.Cys821Tyr)。研究表明, FBN1基因的缺失突变c.[6862_6871delGGCTGTGTAG; 6871+1_6871+11delGTAAGAGGATC] (p.Gly2288MetfsX109)和错义突变c.2462G>A(p.Cys821Tyr)可能分别是这两个家系患者的致病原因。 陈清泉, 伍严安, 黄肖利, 陈同, 黄毅, 陈发林, 陈发文 . 汉族马凡综合征(MFS)患者FBN1基因两种新发突变分析 遗传, 2010,32(1):49-53. Magsci [本文引用: 1] 为调查马凡综合征(Marfan syndrome, MFS)患者的原纤维蛋白-1(Fibrillin-1, FBN1)基因突变情况, 应用聚合酶链反应(PCR)和变性高效液相色谱法(Denaturing high-performance liquid chromatography, DHPLC)对MFS患者的FBN1基因进行突变筛查, 对DHPLC初筛异常的DNA片段进行测序分析。结果在两个MFS家系中发现FBN1基因两种新的突变: 一种为复合突变包含第55号外显子的缺失突变c.6862_6871delGGCTGTGTAG (p.Gly2288MetfsX109)、同义突变c.6861A>G和内含子的突变c.[6871+1_6871+11delGTAAGAGGATC; 6871+34dupCATCAGAAGTGACAGTGGACA]; 另一种为第20号外显子的错义突变c.2462G>A(p.Cys821Tyr)。研究表明, FBN1基因的缺失突变c.[6862_6871delGGCTGTGTAG; 6871+1_6871+11delGTAAGAGGATC] (p.Gly2288MetfsX109)和错义突变c.2462G>A(p.Cys821Tyr)可能分别是这两个家系患者的致病原因。
GuptaPA, PutnamEA, CarmicalSG, KaitilaI, SteinmannB, ChildA, DanesinoC, MetcalfeK, BerrySA, ChenE, DelormeCV, ThongMK, AdèsLC, MilewiczDM . Ten novel FBN2 mutations in congenital contractural arachnodactyly: delineation of the molecular pathogenesis and clinical phenotype Hum Mutat, 2002,19(1):39-48. [本文引用: 1]
GuoX, SongC, ShiY, LiH, MengW, YuanQ, XueJ, XieJ, LiangY, YuanY, YuB, WangH, ChenY, QiL, LiX . Whole exome sequencing identifies a novel missense FBN2, mutation co-segregating in a four-generation chinese family with congenital contractural arachnodactyly BMC Med Genet, 2016,17(1):91. [本文引用: 2]
DengH, LuQ, XuH, DengX, YuanL, YangZ, GuoY, LinQ, XiaoJ, GuanL, SongZ . Identification of a novel missense FBN2 Mutation in a chinese family with congenital contractural arachnodactyly using exome sequencing PLoS One, 2016,11(5):e0155908. [本文引用: 2]
LiuW, ZhaoN, LiXF, WangH, SuiY, LuYP, FengWH, MaC, HanWT, JiangM . A novel FBN2 mutation in a chinese family with congenital contractural arachnodactyly FEBS Open Bio, 2015,5:163-166. [本文引用: 2]
ZhouS, WangF, DouY, ZhouJ, HaoG, XuC, WangQK, WangH, WangP . A novel mutation cosegregates with congenital contractural arachnodactyly in a five-generation chinese family Clin Case Rep, 2018,6(8):1612-1617. [本文引用: 2]
ChenY, LeiYP, ZhengHX, WangW, ChengHB, ZhangJ, WangHY, JinL, LiH . A novel mutation (C1425Y) in the FBN2 gene in a father and son with congenital contractural arachnodactyly Genet Test Mol Bioma, 2009,13(3):295-300. [本文引用: 2]
GürlerAI, YükselZ, KaraerK . A novel FBN2 mutation in a turkish case with congenital contractural arachnodactyly Clin Dysmorphol, 2018,27(3):109-111. [本文引用: 2]
RennerS, SchülerH, AlawiM, KolbeV, RybczynskiM, WoitschachR, SheikhzadehS, StarkVC, OlfeJ, RoserE, SeggewiesFS, MahlmannA, HempelM, HartmannMJ, HillebrandM, WieczorekD, VolkAE, KlothK, Koch-HogrebeM, Abou JamraR, MitterD, AltmüllerJ, Wey-FabriziusA, PetersenC, RauI, BorckG, KubischC, MirTS, von Kodolitsch Y, KutscheK, RosenbergerG . Next-generation sequencing of 32 genes associated with hereditary aortopathies and related disorders of connective tissue in a cohort of 199 patients Genet Med, 2019. doi: 10.1038/s41436-019-0435-z [本文引用: 2]
TakedaN, MoritaH, FujitaD, InuzukaR, TaniguchiY, ImaiY, HirataY, KomuroI . Congenital contractural arachnodactyly complicated with aortic dilatation and dissection: case report and review of literature Am J Med Genet A, 2015,167(10):2382-2387. [本文引用: 2]
MeharV, YadavD, KumarR, YadavS, SinghK, CallewaertB, PathanS, De PaepeA, CouckePJ . Congenital contractural arachnodactyly due to a novel splice site mutation in the FBN2 gene. J Pediatr Genet, 2014,03(03):163-166. [本文引用: 3]
LavillaureixA, HeideS, Chantot-BastaraudS, MareyI, KerenB, GrigorescuR, JouannicJM, GelotA, WhalenS, HéronD, SiffroiJP . Mosaic intragenic deletion of FBN2 and severe congenital contractural arachnodactyly Clin Genet, 2017,92(5):556-558. [本文引用: 2]
AggarwalS, Das BhowmikA, TandonA, DalalA . Exome sequencing reveals blended phenotype of double heterozygous FBN1 and FBN2 variants in a fetus Eur J Med Genet, 2018,61(7):399-402. [本文引用: 2]
DuvvariMR, van de VenJP, GeerlingsMJ, SaksensNT, BakkerB, HenkesA, NevelingK, del RosarioM, WestraD, van den HeuvelLP, SchickT, FauserS, BoonCJ, HoyngCB, de JongEK, den HollanderAI . Whole exome sequencing in patients with the cuticular drusen subtype of age-related macular degeneration PLoS One, 2016,11(3):e0152047. [本文引用: 1]
ShiY, TuY, MechamRP, BassnettS . Ocular phenotype of fbn2-null mice Invest Ophth Vis Sci, 2013,54(12):7163-7173. [本文引用: 1]
KunnasT, SolakiviT, NikkariST . Gene polymorphisms of fibronectin rs2289202 and fibrillin 2 rs331069 associate with vascular disease, the TAMRISK study Biomed Rep, 2018,8(1):65-68. [本文引用: 1]
KhouryLE, PosthumusM, CollinsM, Collins M, van der Merwe W, HandleyC, CookJ, RaleighSM . ELN and FBN2 gene variants as risk factors for two sports-related musculoskeletal injuries Int J Sports Med, 2015,36(4):333-337. [本文引用: 1]
BuchanJG, AlvaradoDM, HallerGE, CruchagaC, HarmsMB, ZhangT, WillingMC, GrangeDK, BravermanAC, MillerNH, MorcuendeJA, TangNL, LamTP, NgBK, ChengJC, DobbsMB, GurnettCA . Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis Hum Mol Genet, 2014,23(19):5271-5282. [本文引用: 1]
RatschillerT, MüllerH, SchachnerT, FellnerF, SulzbacherG, ZiererA . Femoral artery aneurysm repair in a patient with a fibrillin-2 mutation Vasc Endovascular Surg, 2018,52(7):583-586. [本文引用: 1]
ZhangJH, GuoQ, ZhouJZ, ZhangY . Study on the correlation between FBN1/FBN2 gene methylation and esophageal carcinoma J Mod Oncol, 2018,26(4):523-526. [本文引用: 1]
 ,1,2,3,4
,1,2,3,4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT

{kind=link}
{kind=link}