 ,中山大学生命科学学院,有害生物控制与资源利用国家重点实验室,广州 510006
,中山大学生命科学学院,有害生物控制与资源利用国家重点实验室,广州 510006Generation of cell strains containing point mutations in HPRT1 by CRISPR/Cas9
Kai Zhang, Wei Liu, Xiaofeng Liu, Yaosheng Chen, Xiaohong Liu, Zuyong He,State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou 510006, China通讯作者:
编委: 吴强
收稿日期:2019-05-27修回日期:2019-06-28网络出版日期:2019-10-20
| 基金资助: |
Editorial board:
Received:2019-05-27Revised:2019-06-28Online:2019-10-20
| Fund supported: |
作者简介 About authors
张楷,硕士研究生,专业方向:生物化学与分子生物学E-mail:
刘蔚,硕士研究生,专业方向:生物化学与分子生物学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (832KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
张楷, 刘蔚, 刘小凤, 陈瑶生, 刘小红, 何祖勇. 利用CRISPR/Cas9系统构建人HPRT1基因定点突变细胞株[J]. 遗传, 2019, 41(10): 939-949 doi:10.16288/j.yczz.19-108
Kai Zhang, Wei Liu, Xiaofeng Liu, Yaosheng Chen, Xiaohong Liu, Zuyong He.
CRISPR/Cas9基因编辑系统自问世以来获得了举世瞩目的关注与飞速的发展,被广泛地应用到多个物种,同时因其高效、精准的特点被越来越多地运用到分子治疗领域。利用CRISPR/Cas9系统进行基因编辑时,经由sgRNA对靶位点识别后,Cas9蛋白会结合到靶位点上对其进行切割,产生DNA双链断裂(DNA double-stranded breaks, DSBs)。发生DNA双链断裂的细胞会启动自身的修复机制,主要有两类不同的修复途径:非同源染色体末端连接(nonhomologous end joining, NHEJ)和同源重组(homology-directed repair, HDR)。其中,NHEJ是DNA修复的最主要途径,这一途径常在连接时发生碱基插入或缺失突变,因此往往用于单位点基因敲除或大片段的删除[1,2]。HDR途径则是以另一条染色体或具有同源臂的外源的DNA片段作为模板进行基因重组。这一机制可以用于引入基因单核苷酸突变或片段的插入或删除,从而对基因进行更加可控的、定向的、精确的编辑。通常来说,细胞内HDR发生的频率要小于NHEJ,并且只发生在细胞分裂时,且很大程度上受细胞种类、周期和同源重组模板影响[3]。因此在使用CRISPR/Cas9系统进行基因编辑时,利用NHEJ机制对靶位点进行随机的基因敲除的效率通常较高,但是利用HDR机制对靶位点进行定点突变则相对比较困难。研究表明利用单链寡核苷酸(single-stranded oligo-deoxyribonucleotides, ssODN)做为同源重组的模板,可以增加HDR的频率,诱导细胞向特定的方向突变,从而达到精确的基因编辑的效果[4]。因此,本研究拟以ssODN作为模板,应用CRISPR/Cas9对次黄嘌呤/鸟嘌呤磷酸核糖转移酶1 (hypoxanthine-guanine phosphoribosyl transferase1, HPRT1)基因进行精确的点突变遗传修饰。
HPRT1基因位于X染色体末端q26-27区域,编码序列长44 kb,其结构包括8个内含子和9个外显子,共编码217个氨基酸,蛋白质大小为25 kDa。次黄嘌呤/鸟嘌呤磷酸核糖转移酶1可以促进次黄嘌呤或鸟嘌呤与磷酸核糖焦磷酸进行转磷酸核糖基作用,生成相应的核苷-5-磷酸。HPRT1蛋白参与核苷酸合成的补救途径,对于维持细胞内嘌呤核苷酸的浓度具有重要意义。HPRT1蛋白的功能障碍会导致细胞内尿酸过量生成并排出,引起血液内尿酸浓度过高,从而导致高尿酸血症的发生。大约10%的高尿酸血症患者会出现尿酸盐的结晶在关节附近的沉积从而引起痛风[5,6,7]。严重的HPRT1基因突变会引起大脑基底神经节的功能障碍,从而表现出一些神经行为疾病症状,如自残、脑发育不足、智力低下等,被称为Lesch-Nyhan综合征(又称自毁容貌症)[5]。研究发现,HPRT1基因存在多个遗传变异位点,且这些突变位点不完全是随机分布的,存在某些位点出现突变的频率很高,因此这些高突变频率位点很可能与疾病的发生密切相关。Jinnah等[5]对271个HPRT1基因突变病例的统计分析表明,不同突变可以导致氨基酸单位点替换、翻译提前终止、mRNA剪接错误或大片段删除。不同突变导致的症状也不尽相同,部分症状轻微的突变可能仅仅表现为高尿酸血症或轻度的神经疾病,但有一部分(196个病例)表现为严重的Lesch-Nyhan综合征。在该196个病例当中,有25个病例为翻译提前终止突变的病例,其中有11个病例发生了c.151 C>T突变,使该位点编码精氨酸的密码子变为终止密码子;有10个病例发生了c.508 C>T突变,同样使精氨酸密码子突变为终止密码子。在这些突变病例当中,除1例症状未知,1例表现为轻微神经障碍以外,其余均呈现严重的Lesch-Nyhan综合征[6],由此推测这两个位点的突变可能和Lesch-Nyhan综合征的发病机制有密切的关联。因此,本研究在HEK293T和HeLa细胞中,以ssODN作为同源重组模板,利用CRISPR/Cas9系统技术构建了这两种突变的单克隆细胞株。进一步通过Western blot以及6-TG毒性实验分别对点突变的单克隆细胞的HPRT1蛋白表达水平和次黄嘌呤/鸟嘌呤磷酸核糖转移酶活性进行检测,结果表明纯合突变的单细胞克隆不能正常表达HPRT1蛋白,其次黄嘌呤/鸟嘌呤磷酸核糖转移酶活性丧失;杂合突变的单细胞克隆的HPRT1蛋白表达量降低,仍具有部分次黄嘌呤/鸟嘌呤磷酸核糖转移酶活性。
1 材料与方法
1.1 材料
HEK293T细胞和HeLa细胞由本实验室保存,pX458质粒(Cas9/sgRNA/EGFP共同表达质粒)购于美国Addgene公司,引物和Oligo、ssODN由生工生物工程(上海)股份有限公司合成,BbsⅠ、NlaⅢ、T7E1等核酸内切酶购于美国NEB公司,HPRT抗体和6-TG购于德国Sigma-Aldrich公司。1.2 sgRNA设计和CRISPR/Cas9表达质粒构建
在靶位点附近寻找PAM序列(5°NGG3°),在PAM序列上游选取20 bp序列即为sgRNA结合序列。对于起始碱基不是G的gRNA需要在5°端加G。随后在正反向gRNA的5°端分别添加BbsⅠ酶在pX458质粒上的粘性末端CACC (加在正向序列前)和AAAC (加在反向序列前)即可。本研究针对c.508C> T位点设计了4条gRNA,针对c.151C>T位点设计1条gRNA (表1),5条gRNA在HPRT1基因上的位置如图1A所示。将合成的gRNA正负链分别加入超纯水溶解稀释到100 μL进行磷酸化和退火,使之成为带有BbsⅠ酶切位点的双链DNA片段,克隆到pX458质粒载体中。将构建完成的质粒进行转化,涂平板,过夜培养后挑取单克隆,测序验证表达质粒是否构建成功。Table 1
表1
表1 CRISPR/Cas9基因编辑质粒Oligo序列
Table 1
| 名称 | gRNA序列(5°→3°) |
|---|---|
| HPRT1 508-1 top oligo | caccgAAAAGGACCCCACGAAGTGT |
| HPRT1 508-1 bottom oligo | aaacACACTTCGTGGGGTCCTTTTc |
| HPRT1 508-2 top oligo | caccGGCTTATATCCAACACTTCG |
| HPRT1 508-2 bottom oligo | aaacCGAAGTGTTGGATATAAGCC |
| HPRT1 508-3 top oligo | caccGCTTATATCCAACACTTCGT |
| HPRT1 508-3 bottom oligo | aaacACGAAGTGTTGGATATAAGC |
| HPRT1 508-4 top oligo | caccgCTTATATCCAACACTTCGTG |
| HPRT1 508-4 bottom oligo | aaacCACGAAGTGTTGGATATAAGc |
| HPRT1 151-1 top oligo | caccgTCTTGCTCGAGATGTGATGA |
| HPRT1 151-1 bottom oligo | aaacTCATCACATCTCGAGCAAGAc |
新窗口打开|下载CSV
1.3 ssODN设计
选择突变位点左右各40 bp序列作为同源臂,合成含有突变位点的反义链作为同源重组的ssODN (表2)。设计ssODN时,在不引起氨基酸移码突变或错义突变的前提下,为防止基因组DNA成功突变后CRISPR/Cas9系统在突变位点继续发挥切割活性,同时对PAM位点进行突变以终止其切割活性。同时为方便后续的鉴定,在突变位点附近引入一个新的限制酶识别位点(?图1B):(1)在c.508C>T突变位点前4个碱基处进行c.504C>A的同义突变,使该处的PAM位点GGG突变为GGT,重组后即可令此处失去Cas9蛋白的切割活性;c.508C>T突变本身向该位点引入了限制性内切酶NlaⅢ的识别位点(CATG),因而无需另外引入其他酶切位点;(2)在c.151C>T突变位点下游第15~16位的GA碱基突变为AC碱基(模板上的反向互补序列为TC>GT),使该处的PAM位点失活的同时引入了BbsⅠ酶切位点(GAAGAC)。图1
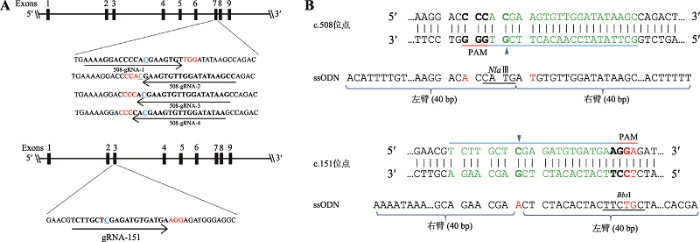 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1gRNA和ssODN位点示意图
A:HPRT1基因gRNA位置(蓝色表示突变位点,红色表示PAM序列);B:ssODN设计示意图(箭头表示靶位点,红色字母表示为破坏PAM序列或引入酶切位点而引入的无义突变)。
Fig. 1Target sites of gRNA and ssODN
Table 2
表2
表2 HPRT1基因的同源重组寡核苷酸序列
Table 2
| 名称 | 序列(5°→3°) |
|---|---|
| ssODN-508 | acattttgtaattaacagcttgctggtgaaaaggacAccaTgaagtgttggatataagccagactgtaagtgaattacttttt |
| ssODN-151 | agcacacagagggctacaatgtgatggcctcccatcGTcttcatcacatctcAagcaagacgttcagtcctacagaaataaaatcagga |
新窗口打开|下载CSV
1.4 细胞转染
转染前24 h将细胞接种至六孔板中,用含有10%胎牛血清的DMEM培养基进行传代培养。待细胞密度达到70%~80%进行转染操作。用Opti-MEM减血清培养基分别将3 μL待转染质粒和5 μL PEI-MAX溶液(2 μg/μL)稀释至150 μL,轻轻吹打混匀,室温静置5 min后混合,再次吹打混匀后室温静置15 min。将混合液缓缓加入6孔板中,摇匀,37℃培养过夜,24 h后更换新鲜培养基并在荧光显微镜下观察转染情况并拍照。1.5 流式细胞分选
细胞转染72 h后,弃培养基并用PBS洗细胞两次后用胰酶消化,200×g离心5 min,弃上清,PBS洗一次,弃上清,用600 μL PBS重悬,经孔径为40 μm的细胞网筛筛入流式管中,分选GFP阳性较强的部分细胞。使用单细胞模式直接分选到96孔板,每孔1个细胞,96孔板中预加200 μL培养基(新鲜培养基和旧培养基1∶1混合后经0.22 μm滤膜过滤后使用)。另外使用富集模式分选一管用于基因组DNA提取和T7E1实验。1.6 T7E1实验
T7核酸内切酶1 (T7E1)可以识别DNA上的茎环结构并切开DNA双链,即由基因编辑产生的含有杂交异源双链DNA,酶切产物中包括扩增主带以及酶切形成的两条较小的DNA条带,通过聚丙烯酰胺凝胶电泳分离3条大小不一的条带,即可根据亮度估算CRISPR/Cas9的编辑效率。提取分选后的细胞的基因组DNA,设计包含突变位点且不位于扩增片段中央的扩增长度约500 bp的PCR引物(表3)并进行扩增,用胶回收试剂盒进行PCR产物纯化。纯化后的PCR产物经变性和退火后即可形成异源双链DNA,在经变性退火的产物中加入0.5 μL T7E1酶,37℃水浴30 min。在新鲜配置的10% PAGE胶中120 V恒压电泳90 min。电泳结束后小心取出PAGE胶,EB溶液染色15 min后用蒸馏水洗3次,置于凝胶成像仪中拍照,反色后用ImageJ软件计算每个条带的灰度,根据公式:突变率=1-[c/(a+b+c)]0.5(其中a,b,c分别为峰面积,c为最大峰面积)估算切割效率。Table 3
表3
表3 T7E1酶切实验所用引物
Table 3
| 名称 | 序列(5°→3°) |
|---|---|
| HPRT1 508-F | gcacggatgaaatgaaacag |
| HPRT1 508-R | tatgaggtgctggaaggaga |
| HPRT1 151-F | caaaggatgtgttacgtggaag |
| HPRT1 151-R | gtcataggaatggatctatcac |
新窗口打开|下载CSV
1.7 单克隆培养和鉴定
分选至96孔板的细胞培养7 d后在显微镜下观察形态和状态。选取只有单个细胞团的孔的细胞进行传代并扩大培养。当细胞形成一个单克隆细胞团时弃去培养基,用PBS洗两遍后加入20 μL胰酶消化,终止消化后吹散并转移至48孔板再扩大培养,以此类推。在从48孔板向24孔板转移时取少量细胞提取基因组DNA,扩增含有突变位点的片段并克隆到pMD-18T载体中,转化涂板,培养过夜后挑单克隆进行测序鉴定基因型。1.8 Western blot检测
将细胞接种于六孔板中,待生长至汇合度达到约90%时,弃培养基并用PBS洗2遍,加入100 μL含有1 mmol/L PMSF的RIPA细胞裂解液中,冰上裂解2 min后收集悬液至1.5 mL离心管中。加入5×SDS Loading Buffer,100℃水浴10 min,14000×g离心5 min,在12%浓度分离胶的SDS-PAGE胶中电泳。电泳结束后小心剥下凝胶,采用半干转法将蛋白转移到PVDF膜上,室温封闭1 h,4℃孵育一抗过夜。孵育完毕用TBST洗膜3次,随后室温孵育二抗1 h,孵育完毕用TBST洗膜3次,采用ECL法在化学发光自显影仪检测蛋白条带并拍照。1.9 6-TG毒性实验
将细胞接种于6孔板中传代培养,待细胞进入对数期时,加入6-TG至终浓度为5 μL/mL,48 h后在显微镜白光下观察细胞状态。2 结果与分析
2.1 gRNA的活性鉴定
将构建成功的5个pX458-gRNA载体分别转染到HEK293T细胞,48 h后收集细胞并提取基因组DNA,PCR扩增切割位点两侧片段,通过T7E1实验检测gRNA对HPRT1基因相应位点的切割活性。另将PCR扩增产物纯化后连接pMD18-T载体,转化涂板并过夜培养,16 h后在各板上挑选50个单克隆测序,以鉴定gRNA的实际切割效率。T7E1实验结果显示5条gRNA均表现出切割活性,活性分别为21.2%、27.6%、31.2%、29.8%和35.2% (图2A)。靶位点测序结果表明,151位点的gRNA有约31.7%的切割活性,对于508位点,活性最高的一条gRNA是3号gRNA,其切割活性达到约30.2% (图2B)。后续的基因定点突变实验选用以上两条gRNA进行。图2
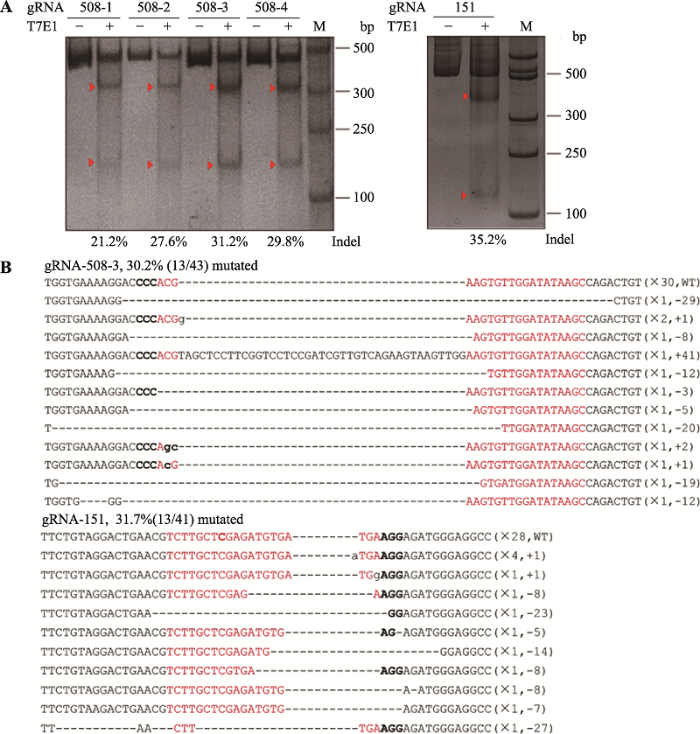 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2gRNA活性检测和切割效率验证
A:T7E1实验(红色箭头示T7E1酶切条带);B:靶位点测序结果比对(红色字体示gRNA识别序列,黑色加粗字体示PAM位点)。
Fig. 2Detection of gRNA activity and knock out efficiency
2.2 流式分选富集HPRT1基因点突变细胞
将pX458-gRNA载体和对应位点的ssODN共转染到HEK293T细胞,培养24 h后在倒置荧光显微镜下观察带有绿色荧光的细胞并拍照(图3A)。继续培养至细胞汇合度达到90%以上进行流式分析,结果表明pX458-508gRNA3和pX458-151gRNA的阳性率分别为41.0%和38.5% (图3B)。对两个样品分别将荧光强度最强的24.5%和21.4%的细胞分选出来用于提取基因组DNA,同时提取未经分选的细胞基因组DNA,通过T7E1实验和靶位点测序结果分析对比分析两者基因组DNA发生编辑的效率。经分选的细胞用T7E1和各自通过ssODN引入的酶切位点的酶,其酶切条带亮度都更高于未分选的细胞,灰度扫描结果显示,c.508位点的T7E1酶切和NlaⅢ酶切DNA量分别提高了1.5倍和4倍,c.151位点的T7E1酶切和BbsⅠ酶切DNA量分别提高了2.4倍和1.5倍(图3C)。说明经流式分选后的细胞群表现了更强的DNA双链切割效率和更高的基因组DNA重组效率。与酶切实验结论相同,靶位点测序结果也证明经过流式分选富集后的细胞,其总体突变效率和基因组DNA发生重组的概率都高于未分选的细胞,c.508位点分选前的点突变和总体突变率分别为2.0%和32%,经分选后分别提高到16.3%和56.3%,c.151位点分选前的点突变和总体突变率分别为2.22%和28.9%,分选后分别提高到10%和56.7% (图3D)。图3
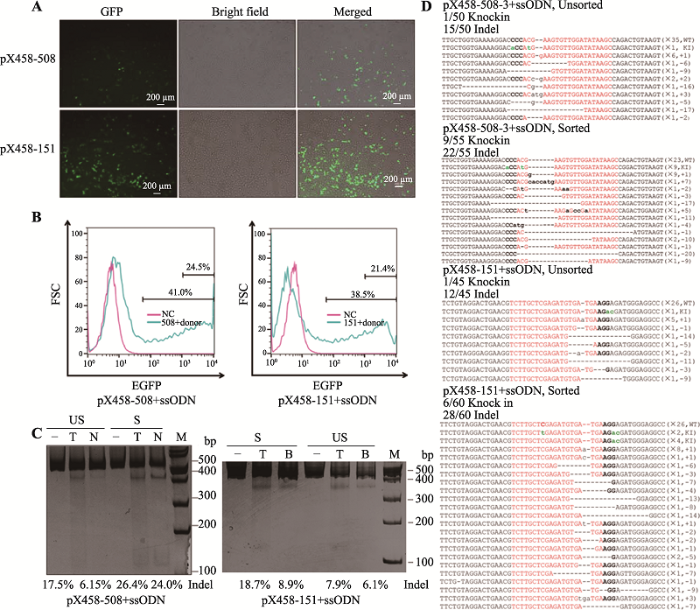 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3流式分选富集GFP阳性细胞提高突变频率和重组效率
A:转染24 h后荧光拍照;B:流式分选较强GFP阳性细胞;C:酶切实验(US: unsorted;S: sorted;T: T7E1 cleaved;N: NlaⅢ cleaved;B: BbsⅠ cleaved;M: 100 bp DNA marker);D:流式分选前后靶位点测序结果对比。
Fig. 3Enhanced mutation rate and HDR efficiency with FACS
2.3 HPRT1基因点突变单克隆细胞株构建
重新将pX458-508gRNA3和pX458-151gRNA与相应的ssODN分别共转到HEK293T细胞和HeLa细胞,48 h后以单细胞模式分选到96孔板,待细胞形成单克隆(图4A)时进行传代扩大培养。细胞扩大到24孔板时收集部分细胞提取基因组DNA,PCR扩增含有靶位点的片段,对PCR产物直接测序。选择目标位点出现双峰且双峰包括基因突变后的T碱基的细胞株(图4B),将其PCR产物纯化并连接T载体,转化涂板,过夜培养后挑取10个菌落单克隆测序,以鉴定该单克隆细胞株的基因型(图4C)。c.508位点基因编辑的HEK293T细胞最终得到43个正常增殖的单克隆细胞株,其中3个克隆(#1、#2和#3)的PCR产物测序结果在靶位点显示双峰。细胞单克隆靶位点测序结果显示:#1单克隆细胞株的靶位点测序的10个菌落单克隆中有3个发生了定点突变,7个为野生型,因HEK293T细胞含有3条X染色体[7],故可认为HPRT1的一个等位基因发生了c.508C>T突变,另外两个等基因未发生编辑,仍为野生型;#2单克隆细胞株的靶位点测序的10个菌落单克隆中有7个发生了定点突变,3个在切割位点下游发生一段长为27 bp的DNA片段插入,该插入片段在原读码框下游8个氨基酸处引入了一个终止密码子(TGA),该条染色体的转录本也会使蛋白翻译提前终止,推测该株细胞的3个等位基因中2个发生了定点突变,另一个等位基因发生翻译提前终止的突变,该株细胞近似看作翻译提前终止的纯合细胞株;#3单克隆细胞株的靶位点测序的10个菌落单克隆中有3个发生了定点突变,3个为野生型,4个发生了17 bp的删除突变,该删除突变造成移码,突变位点的第4个密码子为TAA,同样造成翻译提前终止,推测该株细胞的3个等位基因的2个发生了预期的定点突变和删除突变,1个为野生型(图4C,表4)。c.151位点突变的HeLa细胞最终得到60个正常增殖的细胞株,其中2个克隆(#4和#5)的PCR产物测序结果在靶位点显示双峰。细胞单克隆测序结果显示:#4单克隆细胞株的靶位点测序的10个菌落单克隆中5个为野生型,5个同时发生了C>T和GA>AC突变,该株细胞为定点突变的杂合子;#5单克隆细胞株的靶位点测序的10个菌落单克隆中10个单克隆中4个同时发生了C>T和GA>AC突变,6个仅发生GA>AC突变,使该处由谷氨酸突变为苏氨酸,但不影响该条染色体转录合成完整的HPRT1肽链,该株细胞同样视为定点突变的杂合子(图4C,表4)。图4
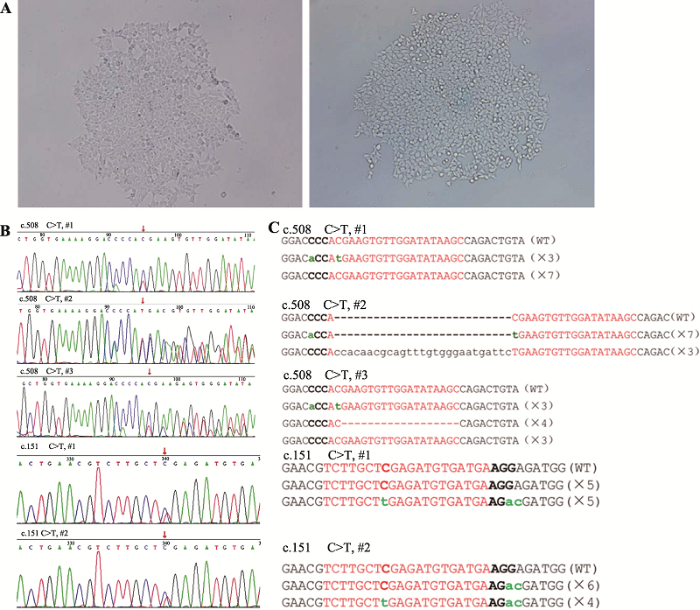 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4细胞单克隆基因型鉴定
A:细胞单克隆左:HEK29T细胞,508位点;右:HeLa细胞,151位点);B:PCR产物测序结果;C:细胞单克隆T-A克隆测序结果(红色字体示gRNA识别序列,黑色加粗字体示PAM位点,绿色字体示ssODN引入的无义突变)。
Fig. 4Genotyping of monoclonal cells
Table 4
表4
表4 单克隆细胞基因型鉴定
Table 4
| 细胞编号 | 编辑位点 | T-A克隆测序结果 | 基因型 |
|---|---|---|---|
| #1 | c.508C>T | 点突变∶野生型=3∶7 | 突变杂合子 |
| #2 | c.508C>T | 点突变∶提前终止=7∶3 | 突变纯合子 |
| #3 | c.508C>T | 点突变∶野生型∶提前终止=3∶3∶4 | 突变杂合子 |
| #4 | c.151C>T | 点突变∶野生型=5∶5 | 突变杂合子 |
| #5 | c.151C>T | 点突变∶野生型=4∶6 | 突变杂合子 |
新窗口打开|下载CSV
2.4 Western blot验证突变株的HPRT1蛋白表达情况
培养上述5株细胞,提取蛋白进行Western Blot实验,使用抗原结合位点为HPRT1蛋白N端序列的抗体,以野生型细胞为对照,检测基因突变的细胞株的HPRT1蛋白表达情况。在508位点突变的HEK293T细胞株中,#1细胞克隆可以正常表达大小为25 kDa的完整的HPRT1蛋白,3条染色体均发生转录提前终止的#2细胞克隆则完全不表达正常的HPRT1蛋白,2条X染色体发生翻译提前终止的#3细胞克隆的HPRT1蛋白表达量明显下降。另外,在预期的17 kDa位置未检测到翻译提前终止产生的截短的蛋白。在151位点突变的2个HeLa细胞株中,HPRT1蛋白表达量均下调,同样地,在预期的5 kDa的位置也未检测到截短的蛋白条带(图5A)。这表明两种突变可能意味着嘌呤磷酸核糖转移酶功能的完全丧失。2.5 6-TG实验检测突变株的次黄嘌呤/鸟嘌呤磷酸核糖转移酶活性
6-TG是一种嘌呤类似物,经HPRT1酶催化可以在DNA复制过程中掺入DNA双链中,诱导DNA的双链断裂,破坏DNA结构的稳定性并产生细胞毒性。HPRT1基因的功能缺陷会使体外培养的细胞能够抵抗6-TG的毒性从而在含有6-TG的培养基中正常存活。因此6-TG实验是一种普遍使用的筛选HPRT1突变细胞的方式。在细胞达到对数生长期时加入6-TG继续培养48 h后,光镜下可见,5株细胞中仅有#2细胞克隆可以存活,其他4株细胞均大量死亡,这表明仅有#2细胞克隆由于完全不表达正常的HPRT1蛋白,所以几乎完全丧失了HPRT1蛋白的次黄嘌呤/鸟嘌呤磷酸核糖转移酶活性,其他4株细胞由于仍能表达少量的HPRT1蛋白,其次黄嘌呤/鸟嘌呤磷酸核糖转移酶活性未完全丧失,因而导致细胞死亡(图5B)。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5Western blot和6-TG实验结果
A:Western blot结果。HPRT1蛋白仅在1条等位基因翻译提前终止的#1细胞株中正常表达,其他4株细胞中都不能正常表达,且在预测的可能出现截短肽链的位置均未能检测到截短的肽链;B:6-TG实验结果。仅全部HPRT1等位基因翻译提前终止的#2细胞株可以在添加6-TG的培养体系中存活,其他4株细胞都死亡。
Fig. 5Results of Western blot and 6-TG
3 讨论
本研究针对HPRT1基因,利用CRISPR/Cas9系统结合流式细胞分选的富集手段,成功进行了c.508C>T和c.151C>T的点突变,经流式分选后细胞总体的突变频率从2%提高到了10%~20%,说明流式分选富集GFP阳性细胞是一种有效的筛选和提高突变频率的手段。此外,在对c.151位点进行突变时,本研究同时突变了其gRNA对应的PAM序列,T-A克隆测序结果表明:#5细胞克隆的10个T-A克隆产物均发生了GA>AC的突变,仅有4个单克隆出现了C>T的突变。这可能是由于DNA双链断裂修复中HDR的效率受位点与DSB位点的距离有关,在更靠近DSB位点的位置上HDR的效率更高[8]。本研究对这些突变的细胞株进行了Western blot实验检测其HPRT1蛋白表达情况,发现除了3个位于不同X染色体上的等位基因全部发生翻译提前终止突变的508位点#2号细胞克隆株完全不表达HPRT1蛋白以外,其余4个杂合的突变细胞株均表达部分正常的HPRT1蛋白。但在翻译提前终止的#3细胞克隆、#5细胞克隆中未检测到被截短的肽链,说明这两个位点的翻译提前终止突变导致了最终细胞内该蛋白水平的降低甚至完全消失,而不是存在具有部分次黄嘌呤/鸟嘌呤磷酸核糖转移酶功能的蛋白质残基,推测这可能是由于真核生物广泛存在的无义介导的mRNA降解(nonsense-mediated mRNA decay, NMD)机制的存在,由于突变引入了一个新的提前终止密码子,引起Upf蛋白复合物识别并降解了突变的mRNA序列[9,10]。与此同时,只有508位点的#2细胞克隆能在含有6-TG的培养基中存活,由此推测HPRT1蛋白的翻译提前终止导致HPRT1不能正常而稳定地表达,细胞内次黄嘌呤/鸟嘌呤磷酸核糖转移酶活性几乎完全消失,是该位点突变导致严重的Lesch-Nyhan疾病的主要原因。HPRT1基因定点突变细胞模型的建立可为今后建立其他HPRT1基因突变的细胞系或动物模型提供借鉴,为深入研究Lesch-Nyhan致病机制提供基础。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}