Brain developmental diseases and pathogenic mechanisms
Yisheng Jiang, Zhiheng Xu,Department of Developmental Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China
Supported by the National Natural Science Foundation of China Nos(31430037) Supported by the National Natural Science Foundation of China Nos(31730108) Strategic Priority Research Program and Innovation Program of the Chinese Academy of Sciences Nos(XDB32020100) Strategic Priority Research Program and Innovation Program of the Chinese Academy of Sciences Nos(QYZDJ-SSW-SMC007) Strategic Priority Research Program and Innovation Program of the Chinese Academy of Sciences Nos(GJHZ1827)
作者简介 About authors 姜义圣,博士研究生,专业方向:细胞生物学E-mail:ysjiang@genetics.ac.cn。
摘要 大脑发育是一个极其复杂又被精确调控的过程,主要包括神经前体细胞增殖和分化、神经元迁移和形态发生(包括轴、树突发育)、突触形成与修剪、轴突髓鞘化、神经网络的形成与重塑等过程,最终形成功能完善的神经系统。其中的任何过程出现问题都有可能导致大脑发育异常,造成大脑功能障碍,即脑发育疾病。儿童脑发育疾病在医疗总负担中占比最高,因此被广泛关注。脑发育疾病通常被划分为两类:一类以大脑形态结构异常为指标,即大脑皮层发育畸形(malformation of cortical development, MCD);另一类以大脑功能障碍为指标,即神经精神疾病(neuropsychopathy)。大脑皮层发育畸形中的小颅畸形(microcephaly)和神经精神疾病中的孤独症谱系障碍(autism spectrum disorder, ASD)这两种疾病具有许多共同之处,例如小颅畸形致病基因的突变高频地出现在ASD病人中。本文针对这两类具有代表性的脑发育疾病,从症状、病因、机制和相关基因等方面展开介绍,以期为疾病的基础研究和治疗提供理论指导。 关键词:脑发育疾病;小颅畸形;孤独症;致病基因
Abstract Development of the human brain is a strictly complex and precisely regulated process. Brain development includes the proliferation and differentiation of neural progenitor cells, migration and maturation of neurons, myelination of neuronal axons, synaptogenesis and organization of the neural circuits. Abnormalities of these developmental processes can lead to severe malformation and dysfunction of the brain, which may result in brain developmental diseases which have a high medical burden and have attracted global attention. Brain developmental diseases are typically divided into two categories according to abnormal brain morphology and dysfunction: malformation of cortical development (MCD) and neuropsychopathy. Microcephaly and autism spectrum disorder (ASD) are representative disorders of MCD and neuropsychopathy respectively. In this review, we summarize the progresses of these two typical and relevant brain developmental diseases including the mechanism and etiology of their development, gene expression, symptoms, and related to provide theoretical guidance for basic research and management and treatment. Keywords:brain developmental diseases;microcephaly;autism;disease associated genes
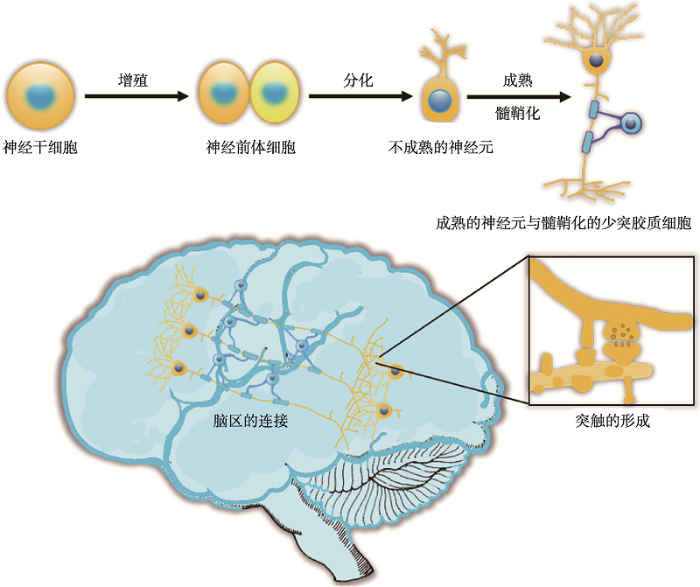
神经干细胞通过增殖和分化生成不成熟神经元。之后不成熟神经元发生一系列的形态变化,包括神经元轴突和树突发育、轴突髓鞘化和树突棘发生等,逐渐成为成熟的神经元。神经元之间通过轴突末梢和树突棘两者共同形成的突触相互联系,构成复杂的神经网络。 Fig. 1The diagram of neurogenesis and construction of neural network
人类大脑发育是一个极其复杂的过程,大约有1/3的基因在大脑发育过程中表达,并被精确调控[3]。任何发育过程出现错误都有可能引起发育异常,从而导致脑发育疾病[4]。根据侧重点不同,脑发育疾病通常被划分为两类:一类是大脑整体结构出现明显异常,以形态改变为主要特征,即大脑皮层发育畸形(malformation of cortical development, MCD);另一类侧重大脑的功能障碍,以智力、言语、社交等高级认知和精神活动异常为指标,即神经精神疾病(neuropsychopathy)。大脑皮层发育畸形中的小颅畸形(microcephaly)和神经精神疾病中的孤独症谱系障碍(autism spectrum disorder, ASD)具有许多共同之处。比如,大多数小颅畸形和ASD患儿都智力低下,许多小颅畸形患儿有孤独症行为,>15%孤独症患儿有小颅畸形,许多小颅畸形基因突变可能与孤独症相关[5]。因此,本文主要以小颅畸形和ASD为例,对两类脑发育疾病的发病原因和机制展开介绍,以期为疾病的基础研究和治疗提供理论指导。
塞克尔综合征又称为小颅畸形侏儒Ⅰ型(microcephalic dwarfism type Ⅰ),与MCPH类似,也是一种常染色体隐性遗传病,表现为小颅畸形、智力缺陷、身材矮小,面部、牙齿和骨骼畸形。目前发现至少有9个基因(ATR、ATRIP、RBBP8、NIN、DNA2、CEP63、CEP152、CENPJ和CDK5RAP2,其中后4个基因与MCPH相关)的隐性突变能导致塞克尔综合征[7,42]。ATR是第一个被发现的塞克尔综合征基因,编码一种丝氨酸/苏氨酸蛋白激酶,主要参与DNA损伤应答的信号通路,对有丝分裂过程中DNA的正常复制起着重要作用[43]。除此之外,ATR还参与了细胞纤毛(cilia)的形成,敲除ATR能导致纤毛长度变短,对依赖纤毛的信号通路(如生长因子信号通路和Sonic hedgehog信号通路)的功能造成严重的影响[42]。因此,塞克尔综合征可能是一种由胚胎发育过程中依赖纤毛的信号通路功能异常引起的生长发育异常疾病。
1.2.2 小颅畸形骨发育不良先天性侏儒Ⅱ型(microcephalic osteodysplastic primordial dwarfism type Ⅱ, MOPD-Ⅱ)
ASD的成因复杂,有高度的遗传性。研究发现同卵双胞胎有一个儿童患有ASD,则另一个患ASD的概率高达36%~95%[50]。在过去的几十年中,外显子组测序和全基因组关联分析(genome-wide association study, GWAS)在鉴定ASD风险基因上发挥了重要的作用,包括新生突变(de novo mutations)、遗传变异(inherited variants)、拷贝数变异(copy number variants)和基因组结构变异(genomic structural variants)[5,55,56]。据估计,大约有1000个基因的突变可能与ASD相关[3,55]。这些基因往往在一个复杂的基因调控网络中共同发挥作用,而由单一基因突变引发的ASD比例不超过5%。
与ASD相关的基因种类繁多,同一个基因往往参与大脑发育的多个过程,在不同的信号途径和生物学过程中发挥作用,这也是为什么ASD常伴随着其他综合征发生的原因之一。2016年,Caitlin等[54]总结了ASD相关基因,根据它们的功能分成了细胞核信号传递(signaling to the nucleus)、局部调控(local regulation)、感受器(sensors)、兴奋性-抑制性协调(E:I coordination)、结构组成(structural)和发育(development)等6大类,并给出了每个基因的可信度评级。图4选取了可信度评级最高的44个基因,可以看到它们主要涉及两个方面的功能:一个是定位于细胞核,参与DNA和组蛋白的修饰、转录调控和染色质重塑过程;另一个是定位于突触,在神经递质受体和离子通道发挥正常功能和突触结构形成上起重要作用[64]。下面以定位于细胞核内的DNA结合蛋白基因CHD8、定位于突触后膜的支架蛋白基因SHANK3和细胞膜蛋白基因PTCHD1为例,介绍这些基因的突变导致ASD的可能机制。
A:44个高可信度的ASD基因的功能归类网络图。黄色方形:功能类别;灰色椭圆:基因;B:44个高可信度的ASD基因的亚细胞定位分布图。数字代表基因个数。基因引自Caitlin等[54]总结的“High Confidence”和“Strong Candidate”的ASD基因。基因的能类别信息来自KEGG数据库,亚细胞定位信息来自MGI数据库。 Fig. 4Molecular function and subcellular localization of ASD genes
CHD8 (chromodomain helicase DNA binding protein 8)是ASD基因组研究中最常见的相关基因之一[5,65~67]。CHD8属于染色质解旋酶DNA结合蛋白家族,是ATP依赖的染色质重塑因子,在染色质动态性、转录和细胞存活方面发挥重要作用[68]。CHD8参与调控Wnt-β-catenin信号通路。CHD8的单倍剂量不足能导致REST基因过度激活,从而抑制多种神经相关基因的表达[69]。在小鼠中敲降Chd8能导致神经元迁移延迟,神经元树突复杂性降低,同时小鼠表现出ASD表型[68,69]。除此之外,CHD8的单倍剂量不足或功能缺失还与发育延迟、智力障碍、肠胃障碍、睡眠障碍和巨头畸形等疾病相关[70]。
D'OrtenzioE, MatheronS, YazdanpanahY, de LamballerieX, HubertB, PiorkowskiG, MaquartM, DescampsD, DamondF, Leparc-Goffart I. Evidence of sexual transmission of Zika virus New Engl J Med, 2016,374(22):2195-2198. [本文引用: 1]
LiC, XuD, YeQ, HongS, JiangY, LiuX, ZhangN, ShiL, QinCF, XuZ . Zika virus disrupts neural progenitor development and leads to microcephaly in mice Cell Stem Cell, 2016,19(1):120-126. [本文引用: 2]
CalvetG, AguiarRS, MeloASO, SampaioSA, de FilippisI, FabriA, AraujoESM, de SequeiraPC, de Mendon?aMCL, de OliveiraL, TschoekeDA, SchragoCG, ThompsonFL, BrasilP, Dos SantosFB, NogueiraRMR, TanuriA, de FilippisAMB. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in brazil: a case study Lancet Infect Dis, 2016,16(6):653-660. [本文引用: 1]
LiangQ, LuoZ, ZengJ, ChenW, FooSS, LeeSA, GeJ, WangS, GoldmanSA, ZlokovicBV, ZhaoZ, JungJU . Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy Cell Stem Cell, 2016,19(5):663-671. [本文引用: 1]
YuanL, HuangXY, LiuZY, ZhangF, ZhuXL, YuJY, JiX, XuYP, LiG, LiC, WangHJ, DengYQ, WuM, ChengML, YeQ, XieDY, LiXF, WangX, ShiW, HuB, ShiPY, XuZ, QinCF . A single mutation in the prM protein of Zika virus contributes to fetal microcephaly Science, 2017,358(6365):933-936. [本文引用: 2]
ZhangF, WangHJ, WangQ, LiuZY, YuanL, HuangXY, LiG, YeQ, YangH, ShiL, DengYQ, QinCF, XuZ . American strain of Zika virus causes more severe microcephaly than an old asian strain in neonatal mice E Bio Medicine, 2017,25:95-105. [本文引用: 1]
XuD, LiC, QinCF, XuZ . Update on the animal models and underlying mechanisms for ZIKV-Induced microcephaly Annu Rev Virol, 2019. [本文引用: 2]
LiC, DengYQ, WangS, MaF, AliyariR, HuangXY, ZhangNN, WatanabeM, DongHL, LiuP, LiXF, YeQ, TianM, HongS, FanJ, ZhaoH, LiL, VishlaghiN, ButhJE, AuC, LiuY, LuN, DuP, QinFX, ZhangB, GongD, DaiX, SunR, NovitchBG, XuZ, QinCF, ChengG . 25-Hydroxycholesterol protects host against Zika virus infection and its associated microcephaly in a mouse model Immunity, 2017,46(3):446-456. [本文引用: 1]
LiC, ZhuX, JiX, QuanquinN, DengYQ, TianM, AliyariR, ZuoX, YuanL, AfridiSK, LiXF, JungJU, Nielsen- SainesK, QinFX, QinCF, XuZ, ChengG . Chloroquine, a FDA-approved Drug, prevents zika virus infection and its associated congenital microcephaly in mice E Bio Medicine, 2017,24:189-194. [本文引用: 1]
WangS, HongS, DengYQ, YeQ, ZhaoLZ, ZhangFC, QinCF, XuZ . Transfer of convalescent serum to pregnant mice prevents Zika virus infection and microcephaly in offspring Cell Res, 2017,27(1):158-160. [本文引用: 1]
LiC, GaoF, YuL, WangR, JiangY, ShiX, YinC, TangX, ZhangF, XuZ, ZhangL . A single injection of human neutralizing antibody protects against Zika virus infection and microcephaly in developing mouse embryos Cell Rep, 2018,23(5):1424-1434. [本文引用: 1]
XuX, LeeJ, SternDF . Microcephalin is a DNA damage response protein involved in regulation of CHK1 and BRCA1 J Biol Chem, 2004,279(33):34091-34094. [本文引用: 2]
O'DriscollM, Ruiz-PerezVL, WoodsCG, JeggoPA, GoodshipJA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in seckel syndrome Nat Genet, 2003,33(4):497-501. [本文引用: 1]
BoberMB, JacksonAP . Microcephalic osteodysplastic primordial dwarfism, Type II: a clinical review Curr Osteoporos Rep, 2017,15(2):61-69. [本文引用: 1]
RauchA, ThielCT, SchindlerD, WickU, CrowYJ, EkiciAB, van EssenAJ, GoeckeTO, Al-GazaliL, ChrzanowskaKH, ZweierC, BrunnerHG, BeckerK, CurryCJ, DallapiccolaB, DevriendtK, D?rflerA, KinningE, MegarbaneA, MeineckeP, SempleRK, SprangerS, ToutainA, TrembathRC, VossE, WilsonL, HennekamR, de ZegherF, D?rrHG, ReisA. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism Science, 2008,319(5864):816-819.
GriffithE, WalkerS, MartinCA, VagnarelliP, StiffT, VernayB, Al SannaN, SaggarA, HamelB, EarnshawWC, JeggoPA, JacksonAP, O'DriscollM. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling Nat Genet, 2008,40(2):232-236. [本文引用: 1]
KimJ, KimJ, RheeK. PCNT is critical for the association and conversion of centrioles to centrosomes during mitosis J Cell Sci, 2019,132(6): jcs225789. [本文引用: 2]
KimJ, LeeK, RheeK . PLK1 regulation of PCNT cleavage ensures fidelity of centriole separation during mitotic exit Nat Commun, 2015,6:10076. [本文引用: 1]
BhatS, AcharyaUR, AdeliH, BairyGM, AdeliA . Autism: cause factors, early diagnosis and therapies Rev Neurosci, 2014,25(6):841-850. [本文引用: 6]
BaioJ, WigginsL, ChristensenDL, MaennerMJ, DanielsJ, WarrenZ, Kurzius-SpencerM, ZahorodnyW, Robinson RosenbergC, WhiteT, DurkinMS, ImmP, NikolaouL, Yeargin-AllsoppM, LeeLC, HarringtonR, LopezM, FitzgeraldRT, HewittA, PettygroveS, ConstantinoJN, VehornA, ShenoudaJ, Hall-LandeJ, van Naarden BraunK, DowlingNF. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 Sites, United States, 2014 MMWR Surveill Summ, 2018,67(6):1-23. [本文引用: 2]
BelmonteMK, AllenG, Beckel-MitchenerA, BoulangerLM, CarperRA, WebbSJ . Autism and abnormal development of brain connectivity J Neurosci, 2004,24(42):9228-9231. [本文引用: 1]
JesteSS, GeschwindDH . Disentangling the heterogeneity of autism spectrum disorder through genetic findings Nat Rev Neurol, 2014,10(2):74-81. [本文引用: 1]
WernerE, DawsonG, MunsonJ, OsterlingJ . Variation in early developmental course in autism and its relation with behavioral outcome at 3-4 years of age J Autism Dev Disord, 2005,35(3):337-350. [本文引用: 1]
MullinsC, FishellG, TsienRW . Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops Neuron, 2016,89(6):1131-1156. [本文引用: 6]
ParkHR, LeeJM, MoonHE, LeeDS, KimBN, KimJ, KimDG, PaekSH . A short review on the current understanding of autism spectrum disorders Exp Neurobiol, 2016,25(1):1-13. [本文引用: 1]
GilmanSR, IossifovI, LevyD, RonemusM, WiglerM, VitkupD . Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses Neuron, 2011,70(5):898-907. [本文引用: 1]
BaribeauDA, AnagnostouE . A comparison of neuroimaging findings in childhood onset schizophrenia and autism spectrum disorder: a review of the literature Front Psychiatry, 2013,4:175. [本文引用: 1]
HuangMX, LiuXH, ZhangZJ, ChenC, WangD, HouX, ChenH, XiaK . Functional connection between the stereotyped behavior and the motor front area in children with autism Br J Neurosurg, 2018: 1-4. [本文引用: 1]
TyszkaJM, KennedyDP, PaulLK, AdolphsR . Largely typical patterns of resting-state functional connectivity in high-functioning adults with autism Cereb Cortex, 2014,24(7):1894-1905. [本文引用: 1]
HeQ, DuanY, KarschK, MilesJ . Detecting corpus callosum abnormalities in autism based on anatomical landmarks Psychiatry Res, 2010,183(2):126-132. [本文引用: 1]
BoothR, WallaceGL, HappéF . Connectivity and the corpus callosum in autism spectrum conditions: insights from comparison of autism and callosal agenesis Prog Brain Res, 2011,189:303-317. [本文引用: 1]
ZhaoH, ZhangYC, ZhangYQ . Recent progresses in molecular genetics of autism spectrum disorders Hereditas (Beijing), 2015,37(9):845-854. [本文引用: 1]
YasinH, GibsonWT, LangloisS, StoweRM, TsangES, LeeL, PoonJ, TranG, TysonC, WongCK, MarraMA, FriedmanJM, ZahirFR . A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8 J Hum Genet, 2019,64(4):271-280. [本文引用: 1]
DurandCM, BetancurC, BoeckersTM, BockmannJ, ChasteP, FauchereauF, NygrenG, RastamM, GillbergIC, Anckars?terH, SponheimE, Goubran-BotrosH, DelormeR, ChabaneN, Mouren-SimeoniMC, de MasP, BiethE, RogéB, HéronD, BurglenL, GillbergC, LeboyerM, BourgeronT. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders Nat Genet, 2007,39(1):25-27. [本文引用: 1]
JiangYH, EhlersMD . Modeling autism by SHANK gene mutations in mice Neuron, 2013,78(1):8-27. [本文引用: 1]
ZhaoH, TuZ, XuH, YanS, YanH, ZhengY, YangW, ZhengJ, LiZ, TianR, LuY, GuoX, JiangYH, LiXJ, ZhangYQ . Altered neurogenesis and disrupted expression of synaptic proteins in prefrontal cortex of SHANK3- deficient non-human primate Cell Res, 2017,27(10):1293-1297. [本文引用: 2]
LiY, JiaX, WuH, XunG, OuJ, ZhangQ, LiH, BaiT, HuZ, ZouX, XiaK, GuoH . Genotype and phenotype correlations for SHANK3 de novo mutations in neurodevelopmental disorders Am J Med Genet A, 2018,176(12):2668-2676. [本文引用: 2]
ToraD, GomezAM, MichaudJF, YamPT, CharronF, ScheiffeleP . Cellular functions of the autism risk factor PTCHD1 in mice J Neurosci, 2017,37(49):11993-12005. [本文引用: 2]
GauSS, LiaoHM, HongCC, ChienWH, ChenCH . Identification of two inherited copy number variants in a male with autism supports two-hit and compound heterozygosity models of autism Am J Med Genet B Neuropsychiatr Genet, 2012,159B(6):710-717.
WangS, TanN, ZhuX, YaoM, WangY, ZhangX, XuZ . Sh3rf2 haploinsufficiency leads to unilateral neuronal development deficits and autistic-like behaviors in mice Cell Rep, 2018,25(11):2963-2971. [本文引用: 1]
GoldWA, KrishnarajyR, EllawayC, ChristodoulouJ . Rett syndrome: a genetic update and clinical review focusing on comorbidities ACS Chem Neurosci, 2018,9(2):167-176. [本文引用: 3]
ZhaiW, HuHX, LeL, ZhuangFF, WangKZ, ZhaoY, WangK, LiuXM, SunDA, WangXY, KuangSH, HuKP . Generation and analysis of the Rett syndrome-associated MeCP2- null rat model Hereditas (Beijing), 2016,38(11):1004-1011. [本文引用: 1]
SharmaK, SinghJ, PillaiPP . MeCP2 differentially regulate the myelin MBP and PLP protein expression in oligodendrocytes and C6 glioma J Mol Neurosci, 2018,65(3):343-350. [本文引用: 1]
ChengTL, WangZ, LiaoQ, ZhuY, ZhouWH, XuW, QiuZ . MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex Dev Cell, 2014,28(5):547-560. [本文引用: 1]
YangWX, PanH . Regulation mechanism and research progress of MeCP2 in Rett syndrome Hereditas (Beijing), 2014,36(7):625-630. Magsci [本文引用: 1] Rett综合征(Rett syndrome, RTT)是一种X连锁的神经发育障碍性遗传病, 是导致女性严重智力障碍的主要原因之一。编码甲基化CpG结合蛋白2(Methyl-CpG-binding protein 2, MeCP2)基因突变是RTT主要的遗传病理学改变, MeCP2作为转录抑制因子调控基因表达。在RTT发病机制中, 由于缺乏MeCP2与甲基化DNA的正确结合, 阻碍了它对下游靶基因表达的正常调控, 最终导致脑功能障碍。目前, 对MeCP2在脑发育过程中的作用以及如何导致RTT的发生, 其机制尚不清楚。文章从MECP2基因和MeCP2蛋白两个方面, 对基因结构、蛋白质功能以及在分子水平上的调控机制进行了综述, 以期为RTT的发病机制研究提供新思路。 杨文旭, 潘虹 . MeCP2在Rett综合征中的调控机制 遗传, 2014,36(7):625-630. Magsci [本文引用: 1] Rett综合征(Rett syndrome, RTT)是一种X连锁的神经发育障碍性遗传病, 是导致女性严重智力障碍的主要原因之一。编码甲基化CpG结合蛋白2(Methyl-CpG-binding protein 2, MeCP2)基因突变是RTT主要的遗传病理学改变, MeCP2作为转录抑制因子调控基因表达。在RTT发病机制中, 由于缺乏MeCP2与甲基化DNA的正确结合, 阻碍了它对下游靶基因表达的正常调控, 最终导致脑功能障碍。目前, 对MeCP2在脑发育过程中的作用以及如何导致RTT的发生, 其机制尚不清楚。文章从MECP2基因和MeCP2蛋白两个方面, 对基因结构、蛋白质功能以及在分子水平上的调控机制进行了综述, 以期为RTT的发病机制研究提供新思路。
LiEH, ZhaoX, ZhangC, LiuW . Fragile X mental retardation protein participates in non-coding RNA pathways Hereditas (Beijing), 2018,40(2):87-94. [本文引用: 2]
 ,中国科学院遗传与发育生物学研究所发育生物学研究中心,北京 100101
,中国科学院遗传与发育生物学研究所发育生物学研究中心,北京 100101
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT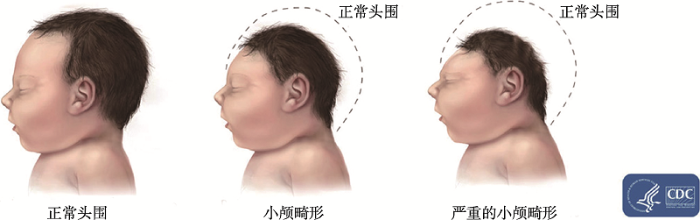 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT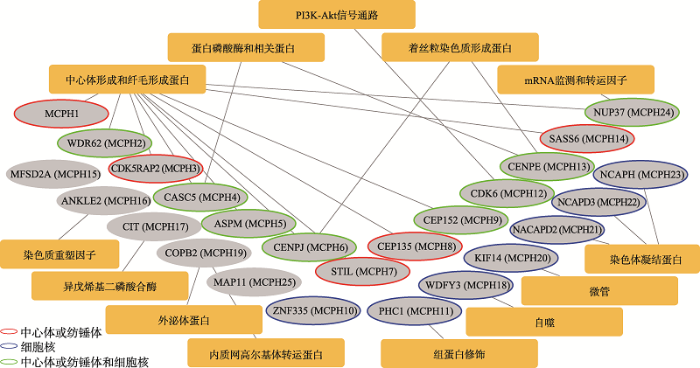 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT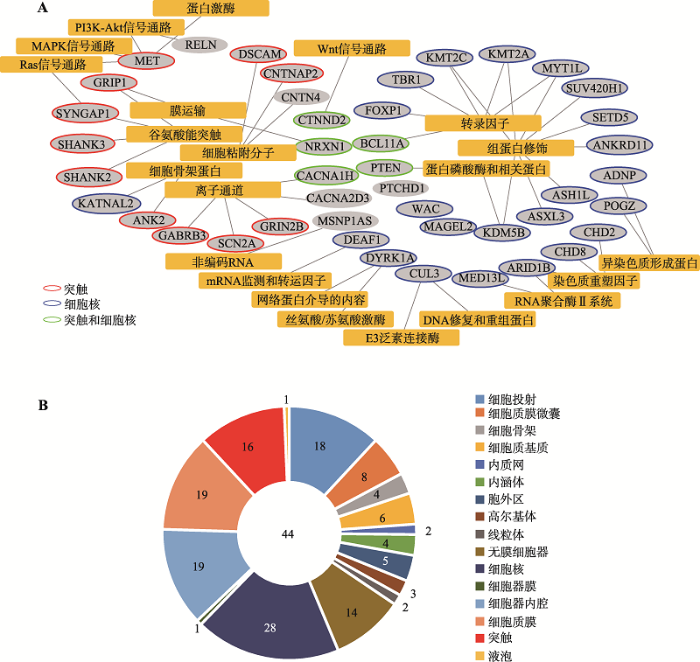 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}