Abstract β-thalassemia (β-thal) is a fatal and disabling inherited blood disorder with diverse phenotypes. The same or similar genotype of β-thal can manifest variable clinical severities. It is the hotspot and emphasis in the field of hematopathy and genetic diseases to explore genetic modifiers that influence the phenotype of β-thal. This review illustrates the deteriorating and amelioratig modifiers from two aspects: genotypes of α-globin and quantitative trait locus of fetal hemoglobin (Hb F). Variations of transcription factors which reactive the γ-globin gene expression and β-globin cluster cis-acting elements were introduced emphatically. Finally, clinical applications and future development prospects of β-thal genetic modifiers are introduced by examples. Keywords:β-thalassemia;fetal hemoglobin;genetic modifier effects;gene editing
PDF (427KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 张倩倩, 商璇, 林宛颖, 徐湘民. 影响β-地中海贫血表型的遗传修饰作用[J]. 遗传, 2019, 41(8): 669-676 doi:10.16288/j.yczz.19-131 Qianqian Zhang, Xuan Shang, Wanying Lin, Xiangmin Xu. Effect of genetic modifiers on the clinical severity of β-thalassemia[J]. Hereditas(Beijing), 2019, 41(8): 669-676 doi:10.16288/j.yczz.19-131
ShangX, XuX . Update in the genetics of thalassemia: What clinicians need to know Best Pract Res Cl Ob, 2017,9:3-15. [本文引用: 1]
MettanandaS, HiggsDR . Molecular basis and genetic modifiers of thalassemia Hematol Oncol Clin N, 2018,32(2):177-191. [本文引用: 1]
ClarkB, ShooterC, SmithF, BrawandD, SteedmanL, OakleyM, RushtonP, RooksH, WangX, DrousiotouA, KyrriA, HadjigavrielM, WillA, FisherC, HiggsDR, PhylipsenM, HarteveldC, KleanthousM, TheinSL . Beta thalassaemia intermedia due to co-inheritance of three unique alpha globin cluster duplications characterised by next generation sequencing analysis Br J Haematol, 2018,180(1):160-164. [本文引用: 1]
GrandchampB, HetetG, KannengiesserC, OudinC, BeaumontC, Rodrigues-FerreiraS, AmsonR, TelermanA, NielsenP, KohneE, BalserC, HeimpelH . A novel type of congenital hypochromic anemia associated with a nonsense mutation in the STEAP3/TSAP6 gene Blood, 2011,118(25):6660-6666. [本文引用: 1]
LiuD, YiS, ZhangX, FangP, ZhengC, LinL, CaiR, YeY, ZhouY, LiangY, ChengF, ZhangX, ZhouW, MohandasN, AnX, XuX . Human STEAP3 mutations with no phenotypic red cell changes Blood, 2016,127(8):1067-1071. [本文引用: 1]
RagabSM, BadrEA, IbrahimAS . Evaluation of glutathione-S-Transferase P1 polymorphism and its relation to bone mineral density in egyptian children and adolescents with Beta-thalassemia Major Mediterr J Hematol Infect Dis, 2016,8(1):e2016004. [本文引用: 1]
MokhtarGM, SherifEM, HabeebNM, AbdelmaksoudAA, El-GhorouryEA, IbrahimAS, HamedEM . Glutathione S-transferase gene polymorphism: relation to cardiac iron overload in egyptian patients with beta thalassemia major Hematology, 2015,21(1):46-53. [本文引用: 1]
MettanandaS, GibbonsRJ, HiggsDR . α-Globin as a molecular target in the treatment of β-thalassemia Blood, 2015,125(24):3694-701. [本文引用: 1]
UdaM, GalanelloR, SannaS, LettreG, SankaranVG, ChenW, UsalaG, BusoneroF, MaschioA, AlbaiG, PirasMG, SestuN, LaiS, DeiM, MulasA, CrisponiL, NaitzaS, AsunisI, DeianaM, NagarajaR, PerseuL, SattaS, CipollinaMD, SollainoC, MoiP, HirschhornJN, OrkinSH, AbecasisGR, SchlessingerD, CaoA . Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia Proc Natl Acad Sci USA, 2008,105(5):1620-1625. [本文引用: 1]
LiuD, ZhangX, YuL, CaiR, MaX, ZhengC, ZhouY, LiuQ, WeiX, LinL, YanT, HuangJ, MohandasN, AnX, XuX . KLF1 mutations are relatively more common in a thalassemia endemic region and ameliorate the severity of β-thalassemia Blood, 2014,124(5):803-811. [本文引用: 2]
RujitoL, BasalamahM, SiswandariW, SetyonoJ, WulandariG, MulatsihS, SofroAS, SadewaAH, SutaryoS . Modifying effect of XmnI, BCL11A, and HBS1L-MYB on clinical appearances: a study on β-thalassemia and hemoglobin E/β-thalassemia patients in indonesia Hematol Oncol Stem Cell Ther, 2016,9(2):55-63. [本文引用: 1]
LiuRR, WangMY, LaiYR . Analysis of Gγ-158(C→T) polymorphism in hemoglobin E/β-thalassemia major in Southern China J Hematol Oncol, 2010,3:29. [本文引用: 1]
MtatiroSN, MakaniJ, MmbandoB, TheinSL, MenzelS, CoxSE . Genetic variants at HbF-modifier loci moderate anemia and leukocytosis in sickle cell disease in tanzania Am J Hematol, 2015,90(1):E1-4. [本文引用: 1]
MartynGE, WienertB, KuritaR, NakamuraY, QuinlanKGR, CrossleyM . A natural regulatory mutation in the proximal promoter elevates fetal globin expression by creating a de novo GATA1 site Blood, 2019,133(8):852-856. [本文引用: 1]
JolyP, LacanP, GarciaC, MeleyR, PondarréC, FrancinaA . A novel deletion/insertion caused by a replication error in the β-globin gene locus control region Hemoglobin, 2011,35(4):316-322. [本文引用: 1]
KimYW, KimA . Deletion of transcription factor binding motifs using the CRISPR/spCas9 system in the β-globin LCR Bioscience Rep, 2017, 37(4): BSR20170976. [本文引用: 1]
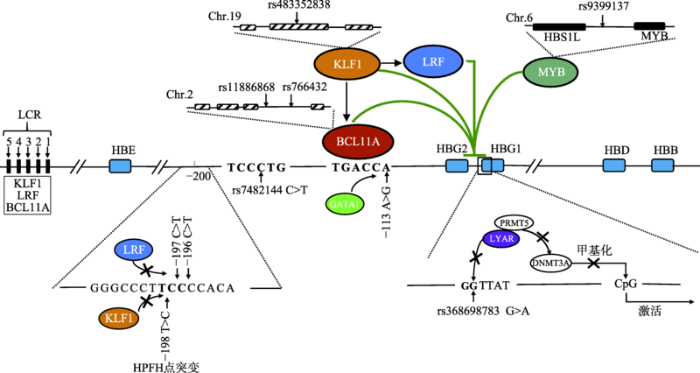
JuJY, ZhaoQ . Regulation of γ-globin gene expression and its clinical applications Hereditas(Beijing), 2018,40(6):429-444. [本文引用: 2]
LeyTJ, AnagnouNP, NoguchiCT, SchechterAN, DeSimoneJ, HellerP, NienhuisAW . DNA methylation and globin gene expression in patients treated with 5-azacytidine Prog Clin Biol Res, 1983,134:457-474. [本文引用: 1]
ChenD, ZuoY, ZhangX, YeY, BaoX, HuangH, TepakhanW, WangL, JuJ, ChenG, ZhengM, LiuD, HuangS, ZongL, LiC, ChenY, ZhengC, ShiL, ZhaoQ, WuQ, FucharoenS, ZhaoC, XuX . A genetic variant ameliorates β-Thalassemia severity by epigenetic-mediated elevation of human fetal hemoglobin expression Am J Hum Genet, 2017,101(1):130-138. [本文引用: 1]
MettanandaS, FisherCA, Sloane-StanleyJA, TaylorS, OppermannU, GibbonsRJ, HiggsDR . Selective silencing of α-globin by the histone demethylase inhibitor IOX1: a potentially new pathway for treatment of β-thalassemia Haematologica, 2017,102(3):e80-e84. [本文引用: 1]
DaiY, ChenT, IjazH, ChoEH, SteinbergMH . SIRT1 activates the expression of fetal hemoglobin genes Am J Hematol, 2017,92(11):1177-1186. [本文引用: 1]
DanjouF, FrancavillaM, AnniF, SattaS, DemartisFR, PerseuL, MancaM, SollainoMC, ManunzaL, MereuE, MarcedduG, PissardS, JolyP, ThuretI, OrigaR, BorgJ, ForniGL, PigaA, LaiME, BadensC, MoiP, GalanelloR . A genetic score for the prediction of beta-thalassemia severity Haematologica, 2015,100(4):452-457. [本文引用: 1]
 ,南方医科大学基础医学院医学遗传学教研室,广州 510800
,南方医科大学基础医学院医学遗传学教研室,广州 510800
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT

{kind=link}
{kind=link}