 ,云南大学生命科学学院,省部共建生物资源保护与利用国家重点实验室,昆明 650091
,云南大学生命科学学院,省部共建生物资源保护与利用国家重点实验室,昆明 650091Mitogenome assembly strategies and software applications in the genome era
Weimin Kuang, Li Yu,State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, School of Life Sciences, Yunnan University, Kunming 650091, China通讯作者: 于黎,博士,研究员,研究方向:动物遗传与进化。E-mail:yuli@ynu.edu.cn
编委: 吴东东
收稿日期:2019-08-7修回日期:2019-09-25网络出版日期:2019-11-20
| 基金资助: |
Editorial board:
Received:2019-08-7Revised:2019-09-25Online:2019-11-20
| Fund supported: |
作者简介 About authors
匡卫民,博士,专业方向:遗传学。E-mail:

摘要
随着测序技术的不断发展,越来越多物种的全基因组数据被测定和广泛应用。在二代基因组数据爆发式增长的同时,除了核基因组数据,线粒体基因组数据也非常重要。高通量测序的全基因组序列中除了核基因组序列也包括线粒体基因组序列,如何从海量的全基因组数据中提取和拼装线粒体基因组序列并加以应用成为线粒体基因组在分子生物学、遗传学和医学等方面的研究方向之一。基于此,从全基因组数据中提取线粒体基因组序列的策略及相关的软件不断发展。根据从全基因组数据中锚定线粒体reads的方式和后续拼装策略的不同,可以分为有参考序列拼装方法和从头拼装方法,不同拼装策略及软件也表现出各自的优势和局限性。本文总结并比较了当前从全基因组数据中获得线粒体基因组数据的策略和软件应用,并对使用者在使用不同策略和相关软件方面给予建议,以期为线粒体基因组在生命科学的相关研究中提供方法上的参考。
关键词:
Abstract
With rapid advances in next-generation sequencing technologies, the genomes of many organisms have been sequenced and widely applied in different settings. Mitochondrial genome data is equally important and the high-throughput whole-genome data typically contain mitochondrial genome (mitogenome) sequences. How to extract and assemble the mitogenome from massive whole-genome sequencing (WGS) data remain a hot area in molecular biology, genetics and medicine. The cataloging and analysis of accumulating mitogenome data promotes the development of assembly strategies and corresponding software applications related to mitochondrial DNA from the WGS data. Mitogenome assembly strategies can be divided into mitogenome-reference strategy and de novo strategy. Each strategy has different advantages and limitations with respect to the difference of bait mitogenome-linked short reads from the WGS data and corresponding assembly strategy. In this review, we summarize and compare current mitogenome assembly strategies and the software applications available. We also provide suggestions related to use different assembly strategies and software applications, and the expected benefits and limitations of methods references in life science.
Keywords:
PDF (476KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
匡卫民, 于黎. 基因组时代线粒体基因组拼装策略及软件应用现状. 遗传[J], 2019, 41(11): 979-993 doi:10.16288/j.yczz.19-227
Weimin Kuang.
线粒体基因组(mitochondrial genome)作为一种特殊且容易获取的遗传标记,因具有高突变速率、无基因重组、高拷贝数和母系遗传等特点[1],被广泛应用在系统发育和生物地理研究[2,3,4,5]、群体遗传[6,7,8,9,10,11,12,13]、医学[14,15,16,17]和生态学研究[18,19,20]等领域。在早期的研究阶段,线粒体基因组序列的获取是首先通过长链链式反应(long range PCR, LR-PCR)和克隆PCR扩增,然后再通过引物步移(primer walking)桑格(Sanger)测序。这种方法准确性高,但通量低、耗时耗力和花费高。随着测序技术的发展,特别是新一代测序技术(next-generation sequencing, NGS)的发展及测序成本的快速下降,使得线粒体基因组序列的获取变得更为容易。目前,NGS及其衍生技术(如LR- PCR加NGS、RNA测序加缺口填补(gap filling)和直接鸟枪法测序[21,22,23]等)使得高通量测序成为普遍现象。相比传统的Sanger测序技术,NGS技术通量高、可以更快速且用更低的花费获得全基因组序列(whole- genome sequencing, WGS)、外显子序列和基因转录本[24]。新一代测序技术的基本原理是:测序平台对样本总DNA或分离纯化后的线粒体DNA随机打断成50~700 bp的单链DNA文库(DNA长短取决于文库构建平台),并将短片段的两端与测序接头序列连接起来,然后对产生的几百万条的DNA分子进行测序,高效、准确、快速地获得大量DNA序列,最后通过生物信息分析从海量的全基因组数据中获取线粒体基因组。近年来,以Pacific Biosciences (PacBio)和 Oxford Nanopore单分子测序技术为代表的第三代测序技术飞速发展,其测序过程无需进行DNA随机打碎和PCR扩增,并且读长增加到几十kb,甚至到100 kb,拼装后得到更高质量的全基因组序列。基因组技术的发展也促使线粒体序列数据爆发式地增加。因此,越来越多的研究者尝试采用多个不同的策略从WGS数据中获取线粒体基因组[23,25~39]。
在NGS时代如何高效分离和富集线粒体DNA而避免核DNA的污染是线粒体基因组测序及后续分析的关键,目前主要包括两种分离策略:(1)在NGS测序前,从总DNA中物理分离纯化线粒体DNA。这种策略先通过氯化铯密度梯度离心/差速离心或者试剂盒富集磁珠将核DNA和线粒体DNA分离[40,41],然后将分离纯化后的线粒体DNA进行文库构建和高通量测序。这样,通过在NGS测序前就将核DNA和线粒体DNA (或叶绿体DNA)分离,以保证获得的数据是来自于线粒体(或叶绿体)。该方法的优势在于避免了核DNA的污染,即线粒体序列转移到核基因的序列(nuclear mitochondrial pseudogenes, Numts[42])。但是,物理分离纯化的方法所用的试剂盒价格昂贵、操作比较繁琐和耗时耗力、对样品的质量和数量也都有一定的要求,因此目前仍然存在许多挑战[43,44],特别是在珍稀野生保护动物和古DNA (ancient DNA, aDNA)的研究领域则更为困难。(2)先进行PCR扩增,对扩增产物进行NGS测序。该策略是先用引物扩增出线粒体基因组目的片段,再将扩增产物直接上机进行NGS测序,无需构建DNA文库[45]。该方法的优势在于需要的起始DNA样本量少,特别适合小型昆虫和环境DNA研究领域,关键在于模板DNA的质量和PCR引物的特异性。
NGS数据被广泛应用在生命科学的很多领域,尤其是在进化生物学、群体遗传学等揭示物种的起源和扩散历史方面发挥了重要的作用。研究者们常常发现核基因数据和线粒体数据表现出不一致的谱系关系,特别是具有复杂的群体历史的类群(比如基因交流、遗传漂变、偏向性迁徙和祖先谱系分拣等)。可见,在分析NGS数据时,除了核基因组数据外,线粒体基因组数据也非常重要。然而,目前通过NGS方法获得的全基因组数据中即包括了线粒体基因组数据和核基因组数据。在全基因组数据中,虽然与核基因reads的测序深度相比,线粒体reads的测序深度是核基因的100~1000倍(细胞中存在几十到数百个拷贝)[46],但是线粒体基因组总的reads数量只占总WGS的reads很少一部分,而且常常受到核基因和叶绿体(绿色植物) reads的污染。因此,使用高效的生物信息工具和分析策略从海量的全基因组数据中快速准确地获得线粒体基因组reads并完整准确地进行后续线粒体基因组拼装就显得非常重要[36]。本文将总结当前常用的从WGS数据中获取线粒体基因组序列的拼装策略及相关软件,并对使用者在使用不同策略和相关软件方面给予建议。
1 有参考序列拼装策略及软件应用
有参考序列拼装策略需要选择近缘物种的线粒体基因组或部分片段作为参考序列从研究类群的全基因组数据中捕获线粒体reads。根据从WGS数据中捕获线粒体reads是否需要完整的线粒体基因组作为参考序列,目前常用的策略可以分为:(1)基于线粒体整个基因组的拼装策略;(2)基于线粒体片段的拼装策略[47,48](图1)。在数据分析流程上,首先使用全基因组比对工具(如BWA[49])将总reads映射(mapping)到线粒体参考序列上,根据序列的相似性捕获线粒体reads,然后再使用不同的序列延长策略对捕获到的线粒体reads进行序列延伸,直到延长到完整的线粒体基因组长度。1.1 基于线粒体基因组拼装策略及软件应用
基于线粒体基因组作为参考序列获取物种或群体的线粒体基因组序列的方法被广泛应用在系统发育和群体遗传学研究。如Ko等[50]将现存大熊猫的线粒体基因组作为参考序列,获取到一个2.2万年前大熊猫的线粒体基因组。其原理是根据同源比对的研究方法,将WGS数据映射到近缘物种的线粒体基因组上,再根据线粒体reads间相互重叠情况,从而完成序列的延长(图1)。这种方法较容易获取和参考基因组一致的序列(consensus sequence),并且准确性高,运算速度较快且不耗计算资源。图1
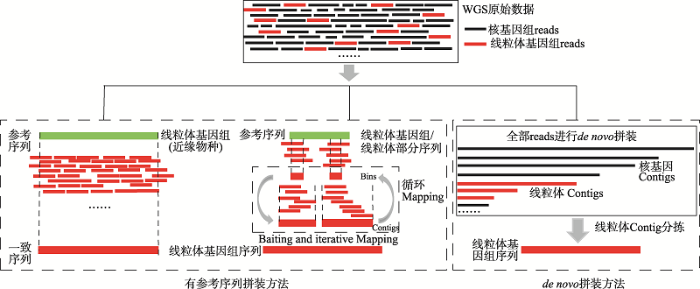 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1从全基因组测序数据中获得及拼装线粒体基因组策略
分析流程图根据参照文献[36,47,66]修改绘制。实线框代表全基因数组短reads序列;虚线框代表获取线粒体基因组序列的方法。
Fig. 1Strategies of mitogenome assembly from whole-genome sequencing data
Table 1
表1
表1线粒体基因组拼装软件信息
Table 1
| 软件名称 | 是否需要参考序列/ 参考序列类型 | 适用 物种 | 输入文件格式、 类型 | 变异 注释 | 结构可 视化 | 运行 环境 | 编程 语言 | 软件网址 |
|---|---|---|---|---|---|---|---|---|
| MIA | 是/自定义参考序列 | 任意 物种 | Fastq、SE reads和 PE reads | × | × | CUI | C/C++ | https://github.com/mpieva/mapping-iterative-assembler |
| MitoBamAnnotator | 是/ rCRS | 人 | Bam | √ | √ | Web | Java | http://bioinfo.bgu.ac.il/bsu/software/MITO-BAM |
| MitoSeek | 是/rCRS和hg19 | 人 | Bam | √ | × | GUI | Perl | |
| mtDNA- profiler | 是/rCRS | 人 | Fasta | × | √ | Web | Java | http://mtprofiler.yonsei.ac.kr |
| MITObim | 是/自定义参考序列 | 任意 物种 | Bam | × | × | CUI | Perl | |
| Mit-o-matic | 是/rCRS | 人 | Fastq、SE reads和 PE reads | √ | √ | Web/GUI | Java | |
| MToolBox | 是/rCRS和RSRS | 人 | Fastq/Bam/Sam、 SE reads和PE reads | √ | × | Web/CUI | Python | https://sourceforge.net/projects/mtoolbox |
| ARC | 是/自定义参考序列 | 任意 物种 | Fastq、SE reads和 PE reads | × | × | Web/CUI | Python | |
| Phy-Mer | 是/自定义参考序列 | 任意 物种 | Fasta/fastq/Bam、 SE reads和PE reads | × | √ | CUI | Python | https://github.com/danielnavarrogomez/phy-mer |
| mtDNA- Server | 是/rCRS和RSRS | 人 | Fastq/Bam/VCF、 SE reads和PE reads | √ | √ | Web | Java | |
| IOGA | 是/自定义参考序列 | 任意 物种 | Fastq、SE reads和 PE reads | × | × | CUI | Python | |
| NOVOPlasty | 是/自定义参考序列 | 任意 物种 | Fastq/fasta、SE reads 和PE reads | × | × | Web/CUI | Perl | https://github.com/ndierckx/NOVOPlasty |
| Norgal | 否 | 任意 物种 | Fastq、SE reads和 PE reads | × | × | CUI | Python/ Java | https://bitbucket.org/kosaidtu/norgal |
| Organelle- PBA | 是/自定义参考序列 | 任意 物种 | PacBio reads | × | × | CUI | Perl | https://github.com/aubombarely/Organelle_PBA |
| MitoSuite | 是/rCRS, RSRS, hg19, GRCh37和38 | 人 | Bam/Sam | √ | √ | GUI | Python | |
| ORG.Asm | 是/自定义参考序列 | 任意 物种 | Fastq、SE reads和 PE reads | × | × | CUI | Python | |
| MitoZ | 否 | 任意 物种 | Fastq、SE reads和 PE reads | √ | √ | CUI | Python | |
| GetOrganelle | 是/自定义参考序列 | 任意 物种 | Fastq、SE reads和 PE reads | × | × | CUI | Python | |
| Trimitomics | 是/自定义参考序列 | 任意 物种 | RNA-seq reads、 PE reads | × | × | Unknown | Unknown | Unknown |
新窗口打开|下载CSV
随着测序技术的发展,对数据分析能力的需求也在增加,特别是人类线粒体基因组研究领域,包括人类进化历史、人类线粒体疾病等方面的研究[51,52],推动了人类线粒体基因组的拼装和注释相关软件的发展(表1)。MIA是较早用于人类线粒体基因组拼装的软件,研究者对尼安德特古人类骨头提到的DNA进行高通量测序后,用现代人的线粒体基因组作为参考序列,使用该软件获取到尼安德特古人类的线粒体基因组[53]。随着人类线粒体基因组数据的不断累积和研究领域的不断扩大,对数据分析能力和软件的功能提出了新要求。一些网络或windows图形用户界面的软件被广泛使用,包括MitoBamAnnotator[54]、MitoSeek[55]、mtDNA-profiler[56]、mit-o-matic[57]、MToolBox[58]、Phy-Mer[59]、mtDNA-Server[60]和MitoSuite[61]等。这类软件支持多种输入文件格式,除了mtDNA-profiler和mit-o-matic外,其他软件都支持二进制的Bam格式文件。因此,这些软件可以直接读取不同软件的输出数据,加快了整个分析流程。值得注意的是,各种软件供用户选择的参考基因组数量有差异,如MitoBamAnnotator、mtDNA-profiler和mit-o-matic仅提供了1套人类基因组(rCRS),MitoSeek (rCRS, hg19)、mtDNA-Server (rCRS, RSRS)和MToolBox (rCRS, RSRS)提供了2套基因组数据,而MitoSuite提供了5套人类参考基因组(rCRS、RSRS、hg19、GRCh37和38)。使用Phy-Mer软件,用户可以自定义参考基因组序列。此外,通过MitoBamAnnotator、MitoSeek、MToolBox、mtDNA- Server、mit-o-matic和MitoSuite软件,用户可以设置相应参数(比如最小等位基因频率,MAF)来检测线粒体基因组的变异位点和异质性位点(heteroplasmic sites, 即线粒体基因组序列上同一个位置存在两种及两种以上的碱基类型,来源可能是外源污染,包括测序错误、特异性扩增,reads匹配错误等,也可能是内源线粒体异质体)。MitoBamAnnotator主要评估和预测线粒体异质性位点潜在的功能,但使用功能比较单一。MitoSeek 和MToolBox扩展了分析功能,包括线粒体拷贝数目、比对质量、结构变异检测等功能。MitoSeek还可以借助Circos[62]软件对检测出的变异进行可视化,包括基因结构变异(structural variations, SVs)和单核苷酸变异(single nucleotide polymorphism, SNPs)。MToolBox优势在于可以单次分析多个个体,并且将变异信息记录到VCF文件中,更容易被解析和注释。从用户操作运行方面比较,MitoSeek和MToolBox是一款基于Perl编程语言的Linux运算环境,并且需要加载多个独立的Perl模块和比对软件(BWA)以及变异检测软件(GATK[63]),对于非生物信息研究背景的用户安装和使用这类软件相对较困难。mtDNA-Server和mit-o-matic软件是网络用户图形分析工具,用户不需要复杂的安装过程,仅通过注册的邮箱后上传数据并进行分析,操作和数据分析相对简单,缺点是受输入文件大小的限制,特别是高测序深度的个体上传数据较缓慢。近期开发的MitoSuite软件扩展了更多实用功能,功能更强大,包括人类线粒体基因组的拼装、变异检测、疾病变异注释和功能预测、拷贝数目、质量检测和覆盖度的可视化等。MitoSuite相比于其他早期的软件,不需要安装其他复杂的计算模块,是图形化操作系统且能本地运行的一款容易操作的软件,可以直接从Bam文件中自动建立一致性序列后进行系统发育或群体遗传学的研究[61],所以对于人类线粒体基因组的研究领域,选择MitoSuite更具有优势。
综上所述,使用上述方法及相关软件从全基因组数据中获取线粒体基因组序列,首先借助全基因组比对软件,包括常用的BWA和Bowtie/Bowtie2[64],将从总reads中捕获到线粒体基因组reads。这两种比对软件优势在于可以对reads错配或reads多处匹配进行筛选和过滤,通过后续的质控获取到纯净的线粒体reads。但是,无法区分Numts和线粒体拷贝数,从而影响线粒体异质性的检测。另外,这些方法及相关软件需要选择近缘物种的线粒体基因组参考序列,如果选择进化关系较远的物种的线粒体基因组作为参考序列,在全基因组比对的过程中可能会发生reads错配或者因序列分歧大导致部分区域比对不上而出现缺失数据(gap),从而影响到后续线粒体基因组拼装的准确性和完整性[38]。因此,选择合适物种的线粒体基因组作为参考序列是该方法和软件应用的关键。对于要研究的物种无法确定其近缘物种,或者是确定了其近缘物种但没有已有线粒体基因组数据的情况下,这个方法就有很大的局限性[36,39]。
1.2 基于线粒体片段拼装策略及软件应用
上述借助近缘物种的线粒体全基因组作为参考序列的拼装策略及相关的软件多数适用于人的线粒体基因组拼装、变异检测和变异注释等。随着越来越多其他物种的研究,线粒体基因组分析也被广泛应用在非模式物种的研究中[65]。仅用人的基因组作为参考序列的软件来获取和分析其他物种的线粒体基因组序列就表现出很大的局限性,因此迫切需要开发适用范围更广的线粒体基因组拼装软件。与总reads直接映射到线粒体基因组参考序列的拼装策略类似,但可以选择遗传关系较远或较近物种的线粒体基因组,甚至线粒体部分序列,来进行其它物种的线粒体基因组序列获取和拼装。该方法首先借助全基因组比对软件将过滤后的WGS数据映射到参考序列上,高覆盖度且连续的线粒体reads组成序列块(bins),这些单独的bins或者根据bins重叠情况连接成Contigs替换原先的参考序列,并作为下次映射的靶序列(baiting sequencing),依次反复将WGS数据映射到新生成的靶序列上延长序列,最后延长到完整的线粒体基因组长度(图1)。反复映射和替换靶序列可以避免参考序列和拼装方法的偏好性。拼装过程中需要调整Kmer值(拼装过程中reads打断成长度为K的一段固定核苷酸序列)大小,反复将WGS数据映射到靶序列上进行序列延长,因此需要消耗大量的计算资源,原始数据越大越消耗计算资源。如果选择遗传关系越远的物种或选择的靶序列越短,拼装时的序列延长则需要更多的循环次数,计算时间也就越长。Hahn等[66]开发的MITObim软件可以直接从WGS数据中拼装非模式物种的线粒体基因组,这个软件嵌入了MIRA和IMAGE计算模块。相比MIA,MITObim的准确性可以达到99.5%以上,在重复区域可以有效的填补gap,计算速度和内存消耗也占有优势,成为目前最广泛使用的线粒体基因组拼装软件。该软件不支持双端序列(paired-end reads, PE reads),支持Iontorrent、454和PacBio测序平台数据,而且建议原始数据reads数量不要超过20~40百万条。如果超出,建议从原始reads中随机抽取部分reads,这样就减少reads的数量,不过这样可能会影响拼装结果的准确性和完整性。当然,MITObim也无法解决线粒体基因组拼装中一些尤为复杂的问题,如Numts、复杂的无脊椎动物和植物的线粒体拼装等[67]。ARC[47]软件的拼装过程类似于MITObim软件,两者都可以选择亲缘关系较远的物种的线粒体基因组或者线粒体部分序列就可以得到完整的线粒体基因组序列,主要的差异在于序列延长方式。ARC是直接对bins进行拼装完成序列的延长,而MITObim则是反复将总reads往靶序列上映射完成延长序列。相比其他全基因组拼装软件,ARC不是将总reads进行从头拼装,而是先通过映射的方式对reads重叠的bins进行拼装,优势在于不耗内存,运行速度较快。此外,ARC基本上不受降解严重的DNA质量和低质量的reads的影响,特别是aDNA,而且运算速度比MITObim和传统的拼装方法快[47]。Li等[68]使用ARC软件对19个隐杆线虫(Caenorhabditis)物种进行线粒体基因组拼装,测试了不同测序平台(Roche、454、Illumina和Ion Torrent)对线粒体基因组拼装的影响,结果发现ARC软件对454平台的数据进行分析时会崩溃,可能的原因是序列长度范围大导致数据分析需要较大的计算资源。但是ARC拼装的完整性都要比MITObim好。然而,Dierckxsens等[47]用ARC软件对角胫叶甲属(Gonioctena Intermedia)进行线粒体基因组拼装,结果发现尽管ARC准确性高(99.99%),但不能将线粒体拼装到一条Contig上,完整性较差(覆盖到线粒体基因组的85.39%)。
Dierckxsens等[38]开发了NOVOPlasty软件,类似于SSAKE[69]和VCAKE[70]算法,将排序后的reads存放在哈希表中,以便reads的快速读取,因此运算速度较快。NOVOPlasty软件需要提供一条靶序列,可以是一条短read、一段编码基因序列,甚至是完整的线粒体基因组序列。值得注意的是,NOVOPlasty与ARC拼装策略不同的是,NOVOPlasty借助提供的靶序列从WGS数据中获取线粒体基因组的一条read,然后再对捕获到的read进行双向延伸。作者将NOVOPlasty与当前主流的拼装软件相比较,包括MITObim、MIRA、ARC、SOAPdenvo2和CLCbio,结果发现:除了ARC外,其余软件都将线粒体拼装在一条Contig。通过对NOVOPlasty拼装到的序列进行质量评估,没有发现缺失位点和不确定的碱基位点,表明准确性和完整性高。NOVOPlasty的计算速度最快、基因组覆盖度最高,CLCbio准确性同样也达到了100%,但是基因组的覆盖度不高(89.96%)。MIRA和ARC都体现最高的基因组覆盖度,但是准确性最低。增加测序覆盖度和reads的长度可以提高NOVOPlasty的完整性和准确性,特别是高重复和AT含量高的区域。NOVOPlasty运行不需要载入其他软件和模块,对于用户来说安装和操作比较简单[38]。
目前用于叶绿体基因组拼装软件同样适合线粒体基因组的拼装,包括IOGA[71]、GetOrganelle[72]和ORG.Asm[73]等。IOGA和GetOrganelle类似于MITObim 中的“Baiting and iterative 映射”分析流程。IOGA分析过程需要Bowtie2、SOAPdenovo2、SPAdes 3.0[37]和其他程序来捕获线粒体reads,拼装过程还需要调整拼装参数Kmer大小(范围为37~97),最后通过拼装似然评估(assembly likelihood estimation, ALE)从候选的Contigs序列里确定线粒体基因组[74]。这种方法适合降解程度较大的样品的线粒体基因组或叶绿体基因组拼装,比如博物馆样品等。与其他拼装软件比较,IOGA使用ALE检验来筛选拼装好的Contigs,最后通过最大似然值来判断最优的拼装
序列。GetOrganelle和IOGA数据分析流程非常相似。GetOrganelle嵌入了独立的Bowtie2、BLAST[75]和SPAdes 3.0分析模块,双端reads和单端reads (single- end reads,SE reads)均可以作为GetOrganelle的输入文件。GetOrganelle可以直接在SPAdes拼装的过程中进行reads错误矫正和错配过滤,保留高质量的reads作为后续分析,而IOGA和MITObim则需要用其他过滤软件提前进行低质量reads的过滤。IOGA和GetOrganelle拼装软件均嵌入SPAdes程序计算模块,在拼装过程中需要反复调试Kmer值的大小。选择合适的Kmer不仅能够保证线粒体Scaffolds或Contigs的完整性和准确性,还可以减少计算时间和运行内存[72]。
最近,随着单分子测序PacBio和Nanopore长片段测序技术的发展,一些复杂物种的全基因组序列被测序和应用,特别是多倍体物种和高重复的物种,显示了长片段测序技术的优势[27,76~80]。同时,已经开发出了一些适用于拼装PacBio和Nanopore长reads的软件,比如HGAP[81]、Falcon (
台可以得到更长的reads,但是仍然存在一定的碱基错误率,因此需要使用碱基矫正软件进行碱基矫正,比如Sprai。因PacBio和Nanopore测序平台不需要在建库的过程中进行DNA随机打断和扩增并且具有读长长特点,所以可以完整得将线粒体基因组一次性测通,有效避免了Numts的污染。但同时因为PacBio和Nanopore测序平台对样品DNA质量有极其严格的要求,要保证DNA的完整性,所以Organelle- PBA的使用也有局限性。
2 从头(de novo)拼装策略及软件应用
目前,世界上越来越多的物种的全基因组数据和线粒体基因组数据被公布,但也有绝大多数物种的基因组信息还未被测定,针对没有参考基因组序列的物种,从头拼装是一种快速和准确地获取遗传信息的策略,这种方法被广泛应用在DNA和RNA序列拼装。线粒体基因组的从头拼装与核基因组的拼装过程相似,首先从海量的全基因组数据中找到短reads的一致性序列,然后再根据不同长度的大片段文库进行Contigs的排序和连接,最后延长到Scaffolds水平。根据线粒体reads的来源不同,可以分为从全基因组数据中从头拼装线粒体基因组策略和从转录组数据中从头拼装线粒体基因组策略(图1)。2.1 从全基因组数据中从头拼装线粒体基因组策略及软件应用
从头拼装线粒体基因组方法不需要提供完整的线粒体基因组或线粒体部分序列作为参考序列。从头拼装首先将WGS的全部reads进行从头拼装[47,48],即将核基因和线粒体基因reads都分别拼装为长片段序列,然后依据线粒体基因组序列长度和高测序深度进行严格的Contigs过滤得到候选线粒体Contigs,最后反复将WGS数据映射到候选线粒体Contigs上,不断延长Contigs,直到延长到完整线粒体基因组长度(图1)。现有的软件有Norgal[36]和MitoZ[39]等。对于一些没有近缘物种线粒体基因组的物种,或者DNA降解严重的样品(比如aDNA序列),用有参考序列拼装方法就有很大的局限性。所以,对aDNA或者环境DNA首先进行NGS测序,再进行线粒体基因组从头拼装即是一个行之有效的策略。但是,这种方法常常要借助于全基因组或转录组拼装的软件和计算模块(包括SOAPdenovo2[90]、SPAdes[37]、Velvet[91]、BIGrat[92]、CLCbio (
传统的从头拼装软件,包括SOAPdenovo2、Newbler、SPAdes、Velvet、CLCbio、ALLPATHS[95]和Platanus[96]等,在全基因组序列拼装过程中,其线粒体Scaffolds或Contigs常常被过滤掉。从头拼装线粒体基因组则借助传统的从头拼装软件,在分析过程中考虑线粒体reads的高测序深度,而不是将其删除。目前已经有许多动植物的线粒体基因组用从头的拼装方法获得了完整的线粒体基因组序列。Lee等[97]对桔梗科的桔梗(Platycodon grandiflorus)和党参(Codonopsis lanceolata)进行了低覆盖度基因组测序并对线粒体基因组进行拼装。他们首先使用Celera、SOAPdenovo, SPAdes和CLCbio等4种全基因组拼装软件对全部reads 进行从头拼装,得到由核基因和线粒体组成的Contigs库,其次根据线粒体的Contigs和核基因组的Contigs平均测序深度的差异确定候选线粒体Contigs,再将WGS数据比对到候选线粒体Contigs上,如此循环完成Contig的延长,最后得到完整的线粒体基因组[97]。类似于这种拼装策略,Al-Nakeeb等[36]开发的Norgal软件,先使用MEGAHIT[98]拼装软件对NGS数据进行从头拼装,然后再将NGS数据重新映射到拼装好的Contig上,通过线粒体和核基因组的reads覆盖度来判断线粒体Contig(s)。他们通过与其他不同策略的线粒体基因组拼装软件比较发现,Norgal软件的准确性和NOVOPlasty软件相似,但是从运算速度上来比较,NOVOPlasty远比Norgal和MITObim要快,原因是Norgal需要调整不同Kmer大小对整个基因组进行拼装,然后再比对reads和计算核基因组reads的测序深度来判断拼装的可靠性[36]。
随着用户对数据分析的需求越来越大,要求简化及高效率的数据分析流程、功能全面和良好的用户体验的软件越来越成为迫切的需要。Meng等[39]开发的MitoZ软件可以“一键式”地对线粒体基因组进行拼装、注释和可视化。该软件包括了多种计算模块,包括原始数据的预处理、从头拼装、候选线粒体序列的富集和线粒体基因组的注释和可视化等功能。相比于其他软件,该软件能对低质量的reads、碱基大量缺失的reads和建库中PCR冗余的reads进行过滤,以保证后续分析数据的可靠性。MitoZ整合了SOAPdenovo-Trans的算法,从核基因组中的reads进行线粒体基因组的从头拼装,其原理是:根据线粒体基因组reads的平均测序深度远比核基因组的高,设置不同的Kmer参数来达到最佳的拼装效果。这个软件提供了两种拼装方式:快捷模式(quick model)和多Kmer模式。根据作者的建议尽可能使用多Kmer模式调整不同Kmer参数,以保证复杂线粒体基因组拼装的完整性和准确性。从拼装的基因数量和序列的总长度方面进行比较,MitoZ比有参考序列的拼装策略更具有优势,特别是对于物种间相似度很低的基因。此外,除了各类软件算法的差异,重复序列、AT含量和异质性率(异质性位点占总变异位点的数量)等也是影响线粒体基因组的拼装完整性和准确性的关键因素[39]。MitoZ对线粒体基因组的注释(Blast、Genewise、MiTFi和Infernal)以及可视化(Circos)功能集成了其他成熟的软件模块,因此间接地扩展了拼装软件的功能,也极大地简化了数据的分析过程。
2.2 从转录组数据中从头拼装线粒体基因组策略及软件应用
新一代测序技术的发展同时推动了转录组水平的研究,从转录组数据中获得基因组编码序列已经很成熟,而总的RNA转录本中包含大量的线粒体编码基因转录本,于是研究者开发了可以高效地从转录组数据中富集线粒体编码基因序列的一些软件。这些方法的原理是根据线粒体在细胞内多拷贝数的特征,线粒体编码基因mRNA的reads测序深度远比核基因组的编码基因reads高,具有高水平的基因表达量。Plese等[99]开发了Trimitomics软件能快速有效得从转录本reads里面对线粒体编码基因序列进行拼装。该软件的分析流程包括了NOVOPlasty、Bowtie2/Trinity和Velvet等3个独立拼装过程:(1)首先使用NOVOPlasty软件将全部的RNA reads进行从头拼装,根据Kmer大小范围(25、39、45和51)确定线粒体编码序列的完整性;(2)如果没有拼装到完整的线粒体编码序列或者拼装到部分序列,则先使用Trimmomatic 0.33[100]对原始RNA reads进行过滤,再用Bowtie2[64]软件将过滤后的reads 比对到近缘物种的线粒体基因组上,用Trinity[94,101]对mapped- read进行从头拼装;(3)使用Velvet软件对全部的转录本进行从头拼装,接着用BlastN软件[102]确定得到的线粒体Contigs。如果以上3种方法都没有拼装到完整的线粒体编码序列,那么再使用Geneious软件整合以上3种方法拼装的结果,再将整合的结果在NCBI数据库中进行同源性鉴定。作者通过对6个无脊椎动物进行线粒体编码基因的拼装,结果发现3种拼装过程都能够覆盖到97%以上的线粒体编码基因序列。从拼装完整性和准确性来评估NOVOPlasty、Bowtie2/Trinity和Velvet拼装过程的可靠性,结果发现3种拼装方法因物种差异而差异,如A.valida和P.dumerilii 这两种纽形动物,Bowtie2/Trinity拼装流程得到的线粒体编码序列的质量更好。而从运行时间、运行内存上比较,NOVOPlasty拼装流程更具有优势。值得注意的是,Trimitomics软件提供3种拼装流程,通过判断拼装结果的完整性来判断是否进行其他拼装流程。同时对于复杂物种的线粒体基因组,还可以整合3种拼装流程的结果,增加了可靠性。3 拼装策略及软件使用建议
当使用者在使用不同的线粒体基因组拼装软件时,首先要区分选择有参考线粒体序列拼装方法的软件还是从头拼装方法的软件。如果使用者要拼装的物种的遗传信息很清楚,可以选择有参考拼装方法的软件。如果要拼装的物种缺乏相关的遗传背景,特别是aDNA,建议选择从头拼装的策略。此外,用户选择不同的软件还需要注意以下几点:(1)了解各类软件的原理及适用性,特别是一些软件对基因组上高重复区有偏好性;(2)适用的物种,人或者非模式物种;(3)不同的软件依赖于不同的数据类型,首先需要区分数据是核基因组数据还是转录本数据,长片段还是短片段序列,单端reads还是双端reads等;(4)不同的软件对输入的文件格式有不同的要求;(5)根据使用者实际需要评估计算资源和操作系统选择不同的软件。影响线粒体基因组拼装的完整性和准确性的因素很多,包括基因组序列特征(比如重复元件,异质性)、测序深度和测序技术(reads长度和碱基错误率)都给序列拼装带来了挑战。此外,尽管基因组拼装算法和软件在不断地发展和优化,但在WGS数据中很难区分线粒体和核基因相似的reads,以及Numts污染[103]等问题,都会造成不同拼装软件在拼装结果上的冲突和后续研究分析结果的推断[104]。值得注意的是,有研究报道发现,不同的物种采用不同的拼装软件,拼装到的线粒体基因组的完整性(比如蛋白质编码区、rRNA和tRNA的数量)和准确性均有差异[105]。如果计算资源允许的情况下,应当选择多种拼装策略的软件进行线粒体基因组的拼装,而对于低覆盖区域或不同拼装软件间导致结果不一致的区域或gap,还需要Sanger测序进行验证[105]。本文共列举了19个从WGS数据中拼装线粒体基因组的软件(表1),多数软件的代码和软件包存储在GitHub,优势在于它是基于网站和云的服务,可以开源软件的代码,以及跟踪和控制对代码的更改。这些软件中有12个软件是命令行运行的方式(CUI),即可在Linux操作系统下完成,用户可以在参数设置文本文件或者命令行参数中设置软件运行参数。命令行运行方式的优点是可以跨平台进行大数据的计算,比如可以将任务提交到大型计算集群上进行计算,缺点是使用者必须要熟悉大量的计算机命令,而不是用鼠标操作就能实现。另外一种运行方式是网络(web server, Web)或windows图形用户界面运行(GUI),用户通过简单的鼠标操作就可以完成参数设置,非常适合对软件不熟悉或者生物信息研究的初****。
此外,本文列举的19个软件中,共有9个是用Python和Perl语言编写的(表1)。其他软件,如MIA使用的则是C/C++,而Norgal使用面向对象编程语言Java编写。这些编程语言具有可移植性、可扩展性和可嵌入性、具有丰富的库等特点。
4 结语与展望
新一代测序技术的不断发展使得越来越多物种的全基因组数据信息被公开和应用,这些数据包含线粒体DNA和核DNA。此外,即使在基因组时代,对线粒体基因组的研究仍然是不可缺少的,比如对于有复杂社会结构和与性别相关的扩散行为的物种的研究[13,106]等。这些研究都促进了线粒体基因组数据爆发式增长和拼装策略及相关软件的发展。线粒体基因组的拼装是非常复杂和快速发展的领域,包括获取线粒体基因组的技术和方法等都需要持续地改进和提高,好的拼装策略依赖于WGS数据集、计算能力和可获得的参考基因组。此外,成功获得一个高质量的线粒体基因组取决于许多因素,包括建库测序平台、基因组的结构特点(重复序列含量、GC含量等)[107]。数据类型也决定线粒体拼装的质量,如aDNA。最近测序技术和提取aDNA的发展推动了古基因组的研究,并利用生物信息学的手段从WGS数据中拼装古线粒体基因组序列。aDNA因长时间保存在土壤中或在博物馆中而导致DNA被降解成小的DNA片段,又加上发掘的aDNA的近缘物种的不确定性,因此为古线粒体基因组的拼装带来许多挑战。正如Meng等[39]指出,开发一款灵活性和高效率的软件,具有良好的用户体验的软件,使得用户能够把更多的时间和精力集中在生物学问题研究上,而不是如何获取线粒体基因组。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
.
DOI:10.1073/pnas.76.4.1967URLPMID:109836 [本文引用: 1]

Mitochondrial DNA was purified from four species of higher primates (Guinea baboon, rhesus macaque, guenon, and human) and digested with 11 restriction endonucleases. A cleavage map was constructed for the mitochondrial DNA of each species. Comparison of the maps, aligned with respect to the origin and direction of DNA replication, revealed that the species differ from one another at most of the cleavage sites. The degree of divergence in nucleotide sequence at these sites was calculated from the fraction of cleavage sites shared by each pair of species. By plotting the degree of divergence in mitochondrial DNA against time of divergence, the rate of base substitution could be calculated from the initial slope of the curve. The value obtained, 0.02 substitutions per base pair per million years, was compared with the value for single-copy nuclear DNA. The rate of evolution of the mitochondrial genome appears to exceed that of the single-copy fraction of the nuclear genome by a factor of about 10. This high rate may be due, in part, to an elevated rate of mutation in mitochondrial DNA. Because of the high rate of evolution, mitochondrial DNA is likely to be an extremely useful molecule to employ for high-resolution analysis of the evolutionary process.
.
DOI:10.1093/jhered/esw072URLPMID:28173059 [本文引用: 1]
The family Lepilemuridae includes 26 species of sportive lemurs, most of which were recently described. The cryptic morphological differences confounded taxonomy until recent molecular studies; however, some species’ boundaries remain uncertain. To better understand the genus Lepilemur, we analyzed 35 complete mitochondrial genomes representing all recognized 26 sportive lemur taxa and estimated divergence dates. With our dataset we recovered 25 reciprocally monophyletic lineages, as well as an admixed clade containing Lepilemur mittermeieri and Lepilemur dorsalis. Using modern distribution data, an ancestral area reconstruction and an ecological vicariance analysis were performed to trace the history of diversification and to test biogeographic hypotheses. We estimated the initial split between the eastern and western Lepilemur clades to have occurred in the Miocene. Divergence of most species occurred from the Pliocene to the Pleistocene. The biogeographic patterns recovered in this study were better addressed with a combinatorial approach including climate, watersheds, and rivers. Generally, current climate and watershed hypotheses performed better for western and eastern clades, while speciation of northern clades was not adequately supported using the ecological factors incorporated in this study. Thus, multiple mechanisms likely contributed to the speciation and distribution patterns in Lepilemur.
.
DOI:10.1073/pnas.0405785101URLPMID:15365171 [本文引用: 1]
The evolutionary history of the largest salamander family (Plethodontidae) is characterized by extreme morphological homoplasy. Analysis of the mechanisms generating such homoplasy requires an independent molecular phylogeny. To this end, we sequenced 24 complete mitochondrial genomes (22 plethodontids and two outgroup taxa), added data for three species from GenBank, and performed partitioned and unpartitioned Bayesian, maximum likelihood, and maximum parsimony phylogenetic analyses. We explored four dataset partitioning strategies to account for evolutionary process heterogeneity among genes and codon positions, all of which yielded increased model likelihoods and decreased numbers of supported nodes in the topologies (Bayesian posterior probability >0.95) relative to the unpartitioned analysis. Our phylogenetic analyses yielded congruent trees that contrast with the traditional morphology-based taxonomy; the monophyly of three of four major groups is rejected. Reanalysis of current hypotheses in light of these evolutionary relationships suggests that (i) a larval life history stage reevolved from a direct-developing ancestor multiple times; (ii) there is no phylogenetic support for the "Out of Appalachia" hypothesis of plethodontid origins; and (iii) novel scenarios must be reconstructed for the convergent evolution of projectile tongues, reduction in toe number, and specialization for defensive tail loss. Some of these scenarios imply morphological transformation series that proceed in the opposite direction than was previously thought. In addition, they suggest surprising evolutionary lability in traits previously interpreted to be conservative.
.
DOI:10.1073/pnas.0602325103URLPMID:16648252 [本文引用: 1]
We sequenced 15 complete mitochondrial genomes and performed comprehensive molecular phylogenetic analyses to study the origin and phylogeny of the Hynobiidae, an ancient lineage of living salamanders. Our phylogenetic analyses show that the Hynobiidae is a clade with well resolved relationships, and our results contrast with a morphology-based phylogenetic hypothesis. These salamanders have low vagility and are limited in their distribution primarily by deserts, mountains, and oceans. Our analysis suggests that the relationships among living hynobiids have been shaped primarily by geography. We show that four-toed species assigned to Batrachuperus do not form a monophyletic group, and those that occur in Afghanistan and Iran are transferred to the resurrected Paradactylodon. Convergent morphological characters in different hynobiid lineages are likely produced by similar environmental selective pressures. Clock-independent molecular dating suggests that hynobiids originated in the Middle Cretaceous [ approximately 110 million years ago (Mya)]. We propose an "out of North China" hypothesis for hynobiid origins and hypothesize an ancestral stream-adapted form. Given the particular distributional patterns and our molecular dating estimates, we hypothesize that: (i) the interior desertification from Mongolia to Western Asia began approximately 50 Mya; (ii) the Tibetan plateau (at least on the eastern fringe) experienced rapid uplift approximately 40 Mya and reached an altitude of at least 2,500 m; and (iii) the Ailao-Red River shear zone underwent the most intense orogenic movement approximately 24 Mya.
.
DOI:10.1016/j.ympev.2008.08.020URL [本文引用: 1]
Phylogenetic relationships of members of the salamander family Salamandridae were examined using complete mitochondrial genomes collected from 42 species representing all 20 salamandrid genera and five outgroup taxa. Weighted maximum parsimony, partitioned maximum likelihood, and partitioned Bayesian approaches all produce an identical, well-resolved phylogeny; most branches are strongly supported with greater than 90% bootstrap values and 1.0 Bayesian posterior probabilities. Our results support recent taxonomic changes in finding the traditional genera Mertensiella, Euproctus, and Triturus to be non-monophyletic species assemblages. We successfully resolved the current polytomy at the base of the salamandrid tree: the Italian newt genus Salamandrina is sister to all remaining salamandrids. Beyond Salamandrina, a clade comprising all remaining newts is separated from a clade containing the true salamanders. Among these newts, the branching orders of well-supported clades are: primitive newts (Echinotriton, Pleurodeles, and Tylototriton), New World newts (Notophthalmus-Taricha), Corsica-Sardinia newts (Euproctus), and modern European newts (Calotriton, Lissotriton, Mesotriton, Neurergus, Ommatotriton, and Triturus) plus modern Asian newts (Cynops, Pachytriton, and Paramesotriton).Two alternative sets of calibration points and two Bayesian dating methods (BEAST and MultiDivTime) were used to estimate timescales for salamandrid evolution. The estimation difference by dating methods is slight and we propose two sets of timescales based on different calibration choices. The two timescales suggest that the initial diversification of extant salamandrids took place in Europe about 97 or 69 Ma. North American salamandrids were derived from their European ancestors by dispersal through North Atlantic Land Bridges in the Late Cretaceous (∼69 Ma) or Middle Eocene (∼43 Ma). Ancestors of Asian salamandrids most probably dispersed to the eastern Asia from Europe, after withdrawal of the Turgai Sea (∼29 Ma).
.
DOI:10.1186/1471-2148-9-63URLPMID:19309521 [本文引用: 1]
Chad Basin, lying within the bidirectional corridor of African Sahel, is one of the most populated places in Sub-Saharan Africa today. The origin of its settlement appears connected with Holocene climatic ameliorations (aquatic resources) that started ~10,000 years before present (YBP). Although both Nilo-Saharan and Niger-Congo language families are encountered here, the most diversified group is the Chadic branch belonging to the Afro-Asiatic language phylum. In this article, we investigate the proposed ancient migration of Chadic pastoralists from Eastern Africa based on linguistic data and test for genetic traces of this migration in extant Chadic speaking populations.
.
DOI:10.1111/mec.12850URL [本文引用: 1]
Although the grey seal Halichoerus grypus is one of the most familiar and intensively studied of all pinniped species, its global population structure remains to be elucidated. Little is also known about how the species as a whole may have historically responded to climate-driven changes in habitat availability and anthropogenic exploitation. We therefore analysed samples from over 1500 individuals collected from 22 colonies spanning the Western and Eastern Atlantic and the Baltic Sea regions, represented by 350bp of the mitochondrial hypervariable region and up to nine microsatellites. Strong population structure was observed at both types of marker, and highly asymmetrical patterns of gene flow were also inferred, with the Orkney Islands being identified as a source of emigrants to other areas in the Eastern Atlantic. The Baltic and Eastern Atlantic regions were estimated to have diverged a little over 10000years ago, consistent with the last proposed isolation of the Baltic Sea. Approximate Bayesian computation also identified genetic signals consistent with postglacial population expansion across much of the species range, suggesting that grey seals are highly responsive to changes in habitat availability.
.
DOI:10.1016/j.ympev.2010.04.032URLPMID:20433932 [本文引用: 1]
Butterfly lizards of the genus Leiolepis (Agamidae) are widely distributed in coastal regions of Southeast Asia and South China, with the Reevese's Butterfly Lizard Leiolepis reevesii having a most northerly distribution that ranges from Vietnam to South China. To assess the genetic diversity within L. reevesii, and its population structure and evolutionary history, we sequenced 1004 bp of cytochrome b for 448 individuals collected from 28 localities covering almost the whole range of the lizard. One hundred and forty variable sites were observed, and 93 haplotypes were defined. We identified three genetically distinct clades, of which Clade A includes haplotypes mainly from southeastern Hainan, Clade B from Guangdong and northern Hainan, and Clade C from Vietnam and the other localities of China. Clade A was well distinguished and divergent from the other two. The Wuzhishan and Yinggeling mountain ranges were important barriers limiting gene exchange between populations on the both sides of the mountain series, whereas the Gulf of Tonkin and the Qiongzhou Strait were not. One plausible scenario to explain our genetic data is a historical dispersion of L. reevesii as proceeding from Vietnam to Hainan, followed by a second wave of dispersal from Hainan to Guangdong and Guangxi. Another equally plausible scenario is a historically widespread population that has been structured by vicariant factors such as the mountains in Hainan and sea level fluctuations.
.
DOI:10.1073/pnas.1102838108URLPMID:21709235 [本文引用: 1]
The Tasmanian devil (Sarcophilus harrisii) is threatened with extinction because of a contagious cancer known as Devil Facial Tumor Disease. The inability to mount an immune response and to reject these tumors might be caused by a lack of genetic diversity within a dwindling population. Here we report a whole-genome analysis of two animals originating from extreme northwest and southeast Tasmania, the maximal geographic spread, together with the genome from a tumor taken from one of them. A 3.3-Gb de novo assembly of the sequence data from two complementary next-generation sequencing platforms was used to identify 1 million polymorphic genomic positions, roughly one-quarter of the number observed between two genetically distant human genomes. Analysis of 14 complete mitochondrial genomes from current and museum specimens, as well as mitochondrial and nuclear SNP markers in 175 animals, suggests that the observed low genetic diversity in today's population preceded the Devil Facial Tumor Disease disease outbreak by at least 100 y. Using a genetically characterized breeding stock based on the genome sequence will enable preservation of the extant genetic diversity in future Tasmanian devil populations.
.
DOI:10.1046/j.1365-294x.2001.01235.xURLPMID:11348492 [本文引用: 1]
The dynamics and evolution of populations will critically depend on their spatial structure. Hence, a recent emphasis on one particular type of structure--the metapopulation concept of Levins--can only be justified by empirical assessment of spatial population structures in a wide range of organisms. This paper focuses on Aphodius fossor, a dung beetle specialized on cattle pastures. An agricultural database was used to locate nearly 50 000 local populations of A. fossor in Finland. Several independent methods were then used to quantify key processes in this vast population system. Allozyme markers and mitochondrial DNA (mtDNA) sequences were applied to examine genetic differentiation of local populations and to derive indirect estimates of gene flow. These estimates were compared to values expected on the basis of direct observations of dispersing individuals and assessments of local effective population size. Molecular markers revealed striking genetic homogeneity in A. fossor. Differentiation was only evident in mtDNA haplotype frequencies between the isolated Aland islands and the Finnish mainland. Thus, indirect estimates of gene flow agreed with direct observations that local effective population size in A. fossor is large (hundreds of individuals), and that in each generation, a substantial fraction (approximately one-fifth) of the individuals move between populations. Large local population size, extreme haplotype diversity and a high regional incidence of A. fossor all testify against recurrent population turnover. Taken together, these results provide strong evidence that the whole mainland population of A. fossor is better described as one large 'patchy population', with substantial movement between relatively persistent local populations, than as a classical metapopulation.
.
DOI:10.1186/1471-2148-12-248URLPMID:23259908 [本文引用: 1]
Marine fish, such as the Atlantic herring (Clupea harengus), often show a low degree of differentiation over large geographical regions. Despite strong environmental gradients (salinity and temperature) in the Baltic Sea, population genetic studies have shown little genetic differentiation among herring in this area, but some evidence for environmentally-induced selection has been uncovered. The mitochondrial genome is a likely target for selection in this system due to its functional role in metabolism.
.
DOI:10.1534/genetics.116.187369URLPMID:27474727 [本文引用: 1]
Recent genetic studies have established that the KhoeSan populations of southern Africa are distinct from all other African populations and have remained largely isolated during human prehistory until ~2000 years ago. Dozens of different KhoeSan groups exist, belonging to three different language families, but very little is known about their population history. We examine new genome-wide polymorphism data and whole mitochondrial genomes for >100 South Africans from the ≠Khomani San and Nama populations of the Northern Cape, analyzed in conjunction with 19 additional southern African populations. Our analyses reveal fine-scale population structure in and around the Kalahari Desert. Surprisingly, this structure does not always correspond to linguistic or subsistence categories as previously suggested, but rather reflects the role of geographic barriers and the ecology of the greater Kalahari Basin. Regardless of subsistence strategy, the indigenous Khoe-speaking Nama pastoralists and the N|u-speaking ≠Khomani (formerly hunter-gatherers) share ancestry with other Khoe-speaking forager populations that form a rim around the Kalahari Desert. We reconstruct earlier migration patterns and estimate that the southern Kalahari populations were among the last to experience gene flow from Bantu speakers, ~14 generations ago. We conclude that local adoption of pastoralism, at least by the Nama, appears to have been primarily a cultural process with limited genetic impact from eastern Africa.
.
DOI:10.1093/molbev/msy220URLPMID:30481341 [本文引用: 2]
The origin and population history of the endangered golden snub-nosed monkey (Rhinopithecus roxellana) remain largely unavailable and/or controversial. We here integrate analyses of multiple genomic markers, including mitochondrial (mt) genomes, Y-chromosomes, and autosomes of 54 golden monkey individuals from all three geographic populations (SG, QL, and SNJ). Our results reveal contrasting population structures. Mt analyses suggest a division of golden monkeys into five lineages: one in SNJ, two in SG, and two in QL. One of the SG lineages (a mixed SG/QL lineage) is basal to all other lineages. In contrast, autosomal analyses place SNJ as the most basal lineage and identify one QL and three SG lineages. Notably, Y-chromosome analyses bear features similar to mt analyses in placing the SG/QL-mixed lineage as the first diverging lineage and dividing SG into two lineages, while resembling autosomal analyses in identifying one QL lineage. We further find bidirectional gene flow among all three populations at autosomal loci, while asymmetric gene flow is suggested at mt genomes and Y-chromosomes. We propose that different population structures and gene flow scenarios are the result of sex-linked differences in the dispersal pattern of R. roxellana. Moreover, our demographic simulation analyses support an origin hypothesis suggesting that the ancestral R. roxellana population was once widespread and then divided into SNJ and non-SNJ (SG and QL) populations. This differs from previous mt-based "mono-origin (SG is the source population)" and "multiorigin (SG is a fusion of QL and SNJ)" hypotheses. We provide a detailed and refined scenario for the origin and population history of this endangered primate species, which has a broader significance for Chinese biogeography. In addition, this study highlights the importance to investigate multiple genomic markers with different modes of inheritance to trace the complete evolutionary history of a species, especially for those exhibiting differential or mixed patterns of sex dispersal.
.
DOI:10.1038/s41467-018-07841-3URLPMID:30604764 [本文引用: 1]
Molecular mechanisms driving disease course and response to therapy in ulcerative colitis (UC) are not well understood. Here, we use RNAseq to define pre-treatment rectal gene expression, and fecal microbiota profiles, in 206 pediatric UC patients receiving standardised therapy. We validate our key findings in adult and paediatric UC cohorts of 408 participants. We observe a marked suppression of mitochondrial genes and function across cohorts in active UC, and that increasing disease severity is notable for enrichment of adenoma/adenocarcinoma and innate immune genes. A subset of severity genes improves prediction of corticosteroid-induced remission in the discovery cohort; this gene signature is also associated with response to anti-TNFα and anti-α4β7 integrin in adults. The severity and therapeutic response gene signatures were in turn associated with shifts in microbes previously implicated in mucosal homeostasis. Our data provide insights into UC pathogenesis, and may prioritise future therapies for nonresponders to current approaches.
.
DOI:10.1002/stem.2637URLPMID:28544378 [本文引用: 1]
High attrition rates and loss of capital plague the drug discovery process. This is particularly evident for mitochondrial disease that typically involves neurological manifestations and is caused by nuclear or mitochondrial DNA defects. This group of heterogeneous disorders is difficult to target because of the variability of the symptoms among individual patients and the lack of viable modeling systems. The use of induced pluripotent stem cells (iPSCs) might significantly improve the search for effective therapies for mitochondrial disease. iPSCs can be used to generate patient-specific neural cell models in which innovative compounds can be identified or validated. Here we discuss the promises and challenges of iPSC-based drug discovery for mitochondrial disease with a specific focus on neurological conditions. We anticipate that a proper use of the potent iPSC technology will provide critical support for the development of innovative therapies against these untreatable and detrimental disorders. Stem Cells 2017;35:1655-1662.
.
DOI:10.3109/07853899708999341URLPMID:9240629 [本文引用: 1]
Mitochondrial diseases are a group of disorders characterized by morphological or functional defects of the mitochondria, the organelles producing most of our cellular energy. As the only extranuclear site carrying genetic information, the mitochondria add an important chapter into the inheritance patterns of genetic diseases. Mitochondrial DNA (mtDNA) is exclusively maternally inherited in humans, but a mitochondrial disorder may follow either maternal or Mendelian inheritance, depending on the site of the primary gene defect. After the initial finding of mtDNA mutations in rare ocular myopathies in 1988, an explosion in the amount of information on mitochondrial diseases has occurred. Because the mitochondria produce energy in all the tissues, symptoms resulting from mtDNA mutations may originate from any organ system, and the clinical spectrum of mitochondrial diseases has expanded to virtually all branches of medicine. Subgroups of several common diseases, such as diabetes, deafness and inherited cardiomyopathies, have been found to be caused by mtDNA mutations, and some mtDNA defects have been suggested to modify the outcome of diseases primarily caused by other factors, such as Parkinson's or Alzheimer's disease. Although no breakthroughs in the therapeutic trials on the devastating mitochondrial diseases have so far been achieved, detection of mtDNA mutations offers an accurate diagnosis and is a prerequisite for genetic counselling, being now accessible to most clinicians.
.
URLPMID:17713117 [本文引用: 1]
Parkinson's disease (PD) is the second most common neurodegenerative disorder in the world. The occurrence of PD is largely sporadic, while several families with Mendelian segregation of PD have been reported. PD is thought to be caused by mitochondrial dysfunction, oxidative stress and inflammation based on multiple genetic and environmental factors, resulting in the apoptosis of dopaminergic cells. Six causal genes for Mendelian inherited PD have been identified to date, which indicate the importance of the ubiquitin-proteasome pathway in the molecular pathogenesis of dopaminergic cell death. Recent studies have also indicated the involvement of genetic factors in the pathogenesis of sporadic PD. Many association studies on candidate genes have examined the relationship between PD and polymorphisms; We identified a-synuclein as a definite susceptibility gene for sporadic PD. Since 2001, significant linkage to several loci have been reported in samples of affected sibling pairs. With the recent advances in human genome analyses, genome-wide association studies by SNP chip are being performed to identify susceptibility genes and to establish tailor-made medicine for PD.
.
DOI:10.1126/science.1101156URLPMID:15459382 [本文引用: 1]
Diatoms are unicellular algae with plastids acquired by secondary endosymbiosis. They are responsible for approximately 20% of global carbon fixation. We report the 34 million-base pair draft nuclear genome of the marine diatom Thalassiosira pseudonana and its 129 thousand-base pair plastid and 44 thousand-base pair mitochondrial genomes. Sequence and optical restriction mapping revealed 24 diploid nuclear chromosomes. We identified novel genes for silicic acid transport and formation of silica-based cell walls, high-affinity iron uptake, biosynthetic enzymes for several types of polyunsaturated fatty acids, use of a range of nitrogenous compounds, and a complete urea cycle, all attributes that allow diatoms to prosper in aquatic environments.
.
DOI:10.1073/pnas.1621504114URLPMID:28716927 [本文引用: 1]
DNA sequencing brings another dimension to exploration of biodiversity, and large-scale mitochondrial DNA cytochrome oxidase I barcoding has exposed many potential new cryptic species. Here, we add complete nuclear genome sequencing to DNA barcoding, ecological distribution, natural history, and subtleties of adult color pattern and size to show that a widespread neotropical skipper butterfly known as Udranomia kikkawai (Weeks) comprises three different species in Costa Rica. Full-length barcodes obtained from all three century-old Venezuelan syntypes of U. kikkawai show that it is a rainforest species occurring from Costa Rica to Brazil. The two new species are Udranomia sallydaleyae Burns, a dry forest denizen occurring from Costa Rica to Mexico, and Udranomia tomdaleyi Burns, which occupies the junction between the rainforest and dry forest and currently is known only from Costa Rica. Whereas the three species are cryptic, differing but slightly in appearance, their complete nuclear genomes totaling 15 million aligned positions reveal significant differences consistent with their 0.00065-Mbp (million base pair) mitochondrial barcodes and their ecological diversification. DNA barcoding of tropical insects reared by a massive inventory suggests that the presence of cryptic species is a widespread phenomenon and that further studies will substantially increase current estimates of insect species richness.
.
DOI:10.1016/j.ijpara.2007.04.014URL [本文引用: 1]
Abstract
An increasing number of complete sequences of mitochondrial (mt) genomes provides the opportunity to optimise the choice of molecular markers for phylogenetic and ecological studies. This is particularly the case where mt genomes from closely related taxa have been sequenced; e.g., within Schistosoma. These blood flukes include species that are the causative agents of schistosomiasis, where there has been a need to optimise markers for species and strain recognition. For many phylogenetic and population genetic studies, the choice of nucleotide sequences depends primarily on suitable PCR primers. Complete mt genomes allow individual gene or other mt markers to be assessed relative to one another for potential information content, prior to broad-scale sampling. We assess the phylogenetic utility of individual genes and identify regions that contain the greatest interspecific variation for molecular ecological and diagnostic markers. We show that variable characters are not randomly distributed along the genome and there is a positive correlation between polymorphism and divergence. The mt genomes of African and Asian schistosomes were compared with the available intraspecific dataset of Schistosoma mansoni through sliding window analyses, in order to assess whether the observed polymorphism was at a level predicted from interspecific comparisons. We found a positive correlation except for the two genes (cox1 and nad1) adjoining the putative control region in S. mansoni. The genes nad1, nad4, nad5, cox1 and cox3 resolved phylogenies that were consistent with a benchmark phylogeny and in general, longer genes performed better in phylogenetic reconstruction. Considering the information content of entire mt genome sequences, partial cox1 would not be the ideal marker for either species identification (barcoding) or population studies with Schistosoma species. Instead, we suggest the use of cox3 and nad5 for both phylogenetic and population studies. Five primer pairs designed against Schistosoma mekongi and Schistosoma malayensis were tested successfully against Schistosoma japonicum. In combination, these fragments encompass 20–27% of the variation amongst the genomes (average total length ∼14,000 bp), thus providing an efficient means of encapsulating the greatest amount of variation within the shortest sequence. Comparative mitogenomics provides the basis of a rational approach to molecular marker selection and optimisation..
DOI:10.1038/nprot.2007.358URLPMID:17947975 [本文引用: 1]
Exploring mitochondrial (mt) genomes has significant implications for various fundamental research areas, including mt biochemistry and physiology, and, importantly, such genomes provide a rich source of markers for population genetics and systematic studies. Although some progress has been made, there is a paucity of information on mt genomes for many metazoan organisms, particularly invertebrates such as parasitic helminths, which relates mainly to the technical limitations associated with sequencing from tiny amounts of material. In this article, we describe a practical long PCR approach for the amplification and subsequent sequencing of the entire mt genome from individual helminths, which overcomes these limitations. The protocol includes the isolation of genomic DNA, long PCR amplification, electrophoresis and sequencing, and takes approximately 1-3 weeks to carry out. The present user-friendly, cost-effective approach has demonstrated utility to the study of a range of parasites, and has the potential to be applied to a wide range of organisms.
.
DOI:10.1016/j.ympev.2010.06.009URLPMID:20601014 [本文引用: 1]
Classically, the mitochondrial genome is sequenced by a series of amplicons using conserved PCR primers. Here we show how shot-gun transcriptome sequencing can be used to obtain the complete set of protein-coding genes from the mtDNA of four passerine bird species. With these sequences, we address the still unresolved basal Passerida relationships (Aves: Passeriformes). Our analysis suggests a new hypothesis for the basal relationships of Passerida, namely a clade grouping Sylvioidea and Passeroidea, with Paridae and Muscicapidae as successive sister groups to this clade. This study demonstrates the usefulness of next-generation sequencing transcriptome sequencing for obtaining new mtDNA genomes.
.
DOI:10.1093/nar/gkq807URLPMID:20876691 [本文引用: 2]
Mitochondrial genome sequences are important markers for phylogenetics but taxon sampling remains sporadic because of the great effort and cost required to acquire full-length sequences. Here, we demonstrate a simple, cost-effective way to sequence the full complement of protein coding mitochondrial genes from pooled samples using the 454/Roche platform. Multiplexing was achieved without the need for expensive indexing tags ('barcodes'). The method was trialled with a set of long-range polymerase chain reaction (PCR) fragments from 30 species of Coleoptera (beetles) sequenced in a 1/16th sector of a sequencing plate. Long contigs were produced from the pooled sequences with sequencing depths ranging from ~10 to 100× per contig. Species identity of individual contigs was established via three 'bait' sequences matching disparate parts of the mitochondrial genome obtained by conventional PCR and Sanger sequencing. This proved that assembly of contigs from the sequencing pool was correct. Our study produced sequences for 21 nearly complete and seven partial sets of protein coding mitochondrial genes. Combined with existing sequences for 25 taxa, an improved estimate of basal relationships in Coleoptera was obtained. The procedure could be employed routinely for mitochondrial genome sequencing at the species level, to provide improved species 'barcodes' that currently use the cox1 gene only.
.
DOI:10.1038/nrg2626URLPMID:19997069 [本文引用: 1]
Demand has never been greater for revolutionary technologies that deliver fast, inexpensive and accurate genome information. This challenge has catalysed the development of next-generation sequencing (NGS) technologies. The inexpensive production of large volumes of sequence data is the primary advantage over conventional methods. Here, I present a technical review of template preparation, sequencing and imaging, genome alignment and assembly approaches, and recent advances in current and near-term commercially available NGS instruments. I also outline the broad range of applications for NGS technologies, in addition to providing guidelines for platform selection to address biological questions of interest.
.
DOI:10.1111/1755-0998.12365URLPMID:25545584 [本文引用: 2]
Use of complete mitochondrial genomes (mitogenomes) can greatly increase the resolution achievable in phylogeographic and historical demographic studies. Using next-generation sequencing methods, it is now feasible to efficiently sequence mitogenomes of large numbers of individuals once a reference mitogenome is available. However, assembling the initial mitogenomes of nonmodel organisms can present challenges, for example, in birds, where mtDNA is often subject to gene rearrangements and duplications. We developed a workflow based on Illumina paired-end, whole-genome shotgun sequencing, which we used to generate complete 19-kilobase mitogenomes for each of three species of North Pacific albatross, a group of birds known to carry a tandem duplication. Although this duplication had been described previously, our procedure did not depend on this prior knowledge, nor did it require a closely related reference mitogenome (e.g. a mammalian mitogenome was sufficient). We employed an iterative process including de novo assembly, reference-guided assembly and gap closing, which enabled us to detect duplications, determine gene order and identify sequence for primer positioning to resolve any mitogenome ambiguity (via minimal targeted Sanger sequencing). We present full mtDNA annotations, including 22 tRNAs, 2 rRNAs, 13 protein-coding genes, a control region and a duplicated feature for all three species. Pairwise comparisons supported previous hypotheses regarding the phylogenetic relationships within this group and occurrence of a shared tandem duplication. The resulting mitogenome sequences will enable rapid, high-throughput NGS mitogenome sequencing of North Pacific albatrosses via direct reference-guided assembly. Moreover, our approach to assembling mitogenomes should be applicable to any taxon.
.
DOI:10.1038/srep31533URLPMID:27530092 [本文引用: 1]
Sugarcane accounts for a large portion of the worlds sugar production. Modern commercial cultivars are complex hybrids of S. officinarum and several other Saccharum species. Historical records identify New Guinea as the origin of S. officinarum and that a small number of plants originating from there were used to generate all modern commercial cultivars. The mitochondrial genome can be a useful way to identify the maternal origin of commercial cultivars. We have used the PacBio RSII to sequence and assemble the mitochondrial genome of a South East Asian commercial cultivar, known as Khon Kaen 3. The long read length of this sequencing technology allowed for the mitochondrial genome to be assembled into two distinct circular chromosomes with all repeat sequences spanned by individual reads. Comparison of five commercial hybrids, two S. officinarum and one S. spontaneum to our assembly reveals no structural rearrangements between our assembly, the commercial hybrids and an S. officinarum from New Guinea. The S. spontaneum, from India, and one sample of S. officinarum (unknown origin) are substantially rearranged and have a large number of homozygous variants. This supports the record that S. officinarum plants from New Guinea are the maternal source of all modern commercial hybrids.
.
DOI:10.1093/gbe/evy179URLPMID:30137422 [本文引用: 2]
Reconstructions of vascular plant mitochondrial genomes (mt-genomes) are notoriously complicated by rampant recombination that has resulted in comparatively few plant mt-genomes being available. The dearth of plant mitochondrial resources has limited our understanding of mt-genome structural diversity, complex patterns of RNA editing, and the origins of novel mt-genome elements. Here, we use an efficient long read (PacBio) iterative assembly pipeline to generate mt-genome assemblies for Leucaena trichandra (Leguminosae: Caesalpinioideae: mimosoid clade), providing the first assessment of non-papilionoid legume mt-genome content and structure to date. The efficiency of the assembly approach facilitated the exploration of alternative structures that are common place among plant mitochondrial genomes. A compact version (729 kbp) of the recovered assemblies was used to investigate sources of mt-genome size variation among legumes and mt-genome sequence similarity to the legume associated root holoparasite Lophophytum. The genome and an associated suite of transcriptome data from select species of Leucaena permitted an in-depth exploration of RNA editing in a diverse clade of closely related species that includes hybrid lineages. RNA editing in the allotetraploid, Leucaena leucocephala, is consistent with co-option of nearly equal maternal and paternal C-to-U edit components, generating novel combinations of RNA edited sites. A preliminary investigation of L. leucocephala C-to-U edit frequencies identified the potential for a hybrid to generate unique pools of alleles from parental variation through edit frequencies shared with one parental lineage, those intermediate between parents, and transgressive patterns.
.
DOI:10.3390/genes9110547URLPMID:30424578 [本文引用: 1]
Diversity in structure and organization is one of the main features of angiosperm mitochondrial genomes (mitogenomes). The ultra-long reads of Oxford Nanopore Technology (ONT) provide an opportunity to obtain a complete mitogenome and investigate the structural variation in unprecedented detail. In this study, we compared mitogenome assembly methods using Illumina and/or ONT sequencing data and obtained the complete mitogenome (208 kb) of Chrysanthemum nankingense based on the hybrid assembly method. The mitogenome encoded 19 transfer RNA genes, three ribosomal RNA genes, and 34 protein-coding genes with 21 group II introns disrupting eight intron-contained genes. A total of seven medium repeats were related to homologous recombination at different frequencies as supported by the long ONT reads. Subsequently, we investigated the variations in gene content and constitution of 28 near-complete mitogenomes from Asteraceae. A total of six protein-coding genes were missing in all Asteraceae mitogenomes, while four other genes were not detected in some lineages. The core fragments (~88 kb) of the Asteraceae mitogenomes had a higher GC content (~46.7%) than the variable and specific fragments. The phylogenetic topology based on the core fragments of the Asteraceae mitogenomes was highly consistent with the topologies obtained from the corresponding plastid datasets. Our results highlighted the advantages of the complete assembly of the C. nankingense mitogenome and the investigation of its structural variation based on ONT sequencing data. Moreover, the method based on local collinear blocks of the mitogenomes could achieve the alignment of highly rearrangeable and variable plant mitogenomes as well as construct a robust phylogenetic topology.
.
DOI:10.1016/j.margen.2019.02.002URLPMID:30928201 [本文引用: 1]
Despite recent advances in sequencing technology, a complete mitogenome assembly is still unavailable for the gecarcinid land crabs that include the iconic Christmas Island red crab (Gecarcoidea natalis) which is known for its high population density, annual mass breeding migration and ecological significance in maintaining rainforest structure. Using sequences generated from Nanopore and Illumina platforms, we assembled the complete mitogenome for G. natalis, the first for the genus and only second for the family Gecarcinidae. Nine Nanopore long reads representing 0.15% of the sequencing output from an overnight MinION Nanopore run were aligned to the mitogenome. Two of them were >10?kb and combined are sufficient to span the entire G. natalis mitogenome. The use of Illumina genome skimming data only resulted in a fragmented assembly that can be attributed to low to zero sequencing coverage in multiple high AT-regions including the mitochondrial protein-coding genes (NAD4 and NAD5), 16S ribosomal rRNA and non-coding control region. Supplementing the mitogenome assembly with previously acquired transcriptome dataset containing high abundance of mitochondrial transcripts improved mitogenome sequence coverage and assembly reliability. We then inferred the phylogeny of the Eubrachyura using Maximum Likelihood and Bayesian approaches, confirming the phylogenetic placement of G. natalis within the family Gecarcinidae based on whole mitogenome alignment. Given the substantial impact of AT-content on mitogenome assembly and the value of complete mitogenomes in phylogenetic and comparative studies, we recommend that future mitogenome sequencing projects consider generating a modest amount of Nanopore long reads to facilitate the closing of problematic and fragmented mitogenome assemblies.
.
DOI:10.1038/s41598-018-36693-6URLPMID:30655548
Quinoa has recently gained international attention because of its nutritious seeds, prompting the expansion of its cultivation into new areas in which it was not originally selected as a crop. Improving quinoa production in these areas will benefit from the introduction of advantageous traits from free-living relatives that are native to these, or similar, environments. As part of an ongoing effort to characterize the primary and secondary germplasm pools for quinoa, we report the complete mitochondrial and chloroplast genome sequences of quinoa accession PI 614886 and the identification of sequence variants in additional accessions from quinoa and related species. This is the first reported mitochondrial genome assembly in the genus Chenopodium. Inference of phylogenetic relationships among Chenopodium species based on mitochondrial and chloroplast variants supports the hypotheses that 1) the A-genome ancestor was the cytoplasmic donor in the original tetraploidization event, and 2) highland and coastal quinoas were independently domesticated.
.
DOI:10.1371/journal.pntd.0004384URLPMID:26872064
The scabies mite, Sarcoptes scabiei, is an obligate parasite of the skin that infects humans and other animal species, causing scabies, a contagious disease characterized by extreme itching. Scabies infections are a major health problem, particularly in remote Indigenous communities in Australia, where co-infection of epidermal scabies lesions by Group A Streptococci or Staphylococcus aureus is thought to be responsible for the high rate of rheumatic heart disease and chronic kidney disease. We collected and separately sequenced mite DNA from several pools of thousands of whole mites from a porcine model of scabies (S. scabiei var. suis) and two human patients (S. scabiei var. hominis) living in different regions of northern Australia. Our sequencing samples the mite and its metagenome, including the mite gut flora and the wound micro-environment. Here, we describe the mitochondrial genome of the scabies mite. We developed a new de novo assembly pipeline based on a bait-and-reassemble strategy, which produced a 14 kilobase mitochondrial genome sequence assembly. We also annotated 35 genes and have compared these to other Acari mites. We identified single nucleotide polymorphisms (SNPs) and used these to infer the presence of six haplogroups in our samples, Remarkably, these fall into two closely-related clades with one clade including both human and pig varieties. This supports earlier findings that only limited genetic differences may separate some human and animal varieties, and raises the possibility of cross-host infections. Finally, we used these mitochondrial haplotypes to show that the genetic diversity of individual infections is typically small with 1-3 distinct haplotypes per infestation.
.
DOI:10.1007/s10709-018-0018-yURLPMID:29748765
Up to date, the scarcity of publicly available complete mitochondrial sequences for European wild pigs hampers deeper understanding about the genetic changes following domestication. Here, we have assembled 26 de novo mtDNA sequences of European wild boars from next generation sequencing (NGS) data and downloaded 174 complete mtDNA sequences to assess the genetic relationship, nucleotide diversity, and selection. The Bayesian consensus tree reveals the clear divergence between the European and Asian clade and a very small portion (10 out of 200 samples) of maternal introgression. The overall nucleotides diversities of the mtDNA sequences have been reduced following domestication. Interestingly, the selection efficiencies in both European and Asian domestic pigs are reduced, probably caused by changes in both selection constraints and maternal population size following domestication. This study suggests that de novo assembled mitogenomes can be a great boon to uncover the genetic turnover following domestication. Further investigation is warranted to include more samples from the ever-increasing amounts of NGS data to help us to better understand the process of domestication.
.
DOI:10.3897/zookeys.793.28977URLPMID:30405308
Lack of mitochondrial genome data of Scleractinia is hampering progress across genetic, systematic, phylogenetic, and evolutionary studies concerning this taxon. Therefore, in this study, the complete mitogenome sequence of the stony coral Echinophylliaaspera (Ellis & Solander, 1786), has been decoded for the first time by next generation sequencing and genome assembly. The assembled mitogenome is 17,697 bp in length, containing 13 protein coding genes (PCGs), two transfer RNAs and two ribosomal RNAs. It has the same gene content and gene arrangement as in other Scleractinia. All genes are encoded on the same strand. Most of the PCGs use ATG as the start codon except for ND2, which uses ATT as the start codon. The A+T content of the mitochondrial genome is 65.92% (25.35% A, 40.57% T, 20.65% G, and 13.43% for C). Bayesian and maximum likelihood phylogenetic analysis have been performed using PCGs, and the result shows that E.aspera clustered closely with Sclerophylliamaxima (Sheppard & Salm, 1988), both of which belong to Lobophylliidae, when compared with species belonging to Merulinidae and other scleractinian taxa used as outgroups. The complete mitogenome of E.aspera provides essential and important DNA molecular data for further phylogenetic and evolutionary analyses of corals.
.
DOI:10.3109/19401736.2015.1101565URLPMID:26641940
The complete mitochondrial genome of Salmo trutta fario, commonly known as brown trout, was sequenced using NGS technology. The mitochondrial genome size was determined to be 16 677?bp and composed of 13 protein-coding gene (PCG), 22 tRNAs, 2 rRNA genes, and 1 putative control region. The overall mitogenome composition of S. trutta fario is A: 28.13%, G: 16.44%, C: 29.47%, and T: 25.96% with A?+?T content of 54.09% and G?+?C content of 45.91%. The gene arrangement and the order are similar to other vertebrates. The phylogenetic tree constructed using 42 complete mitogenomes of Salmonidae fishes confirmed the position of the present species under the genus Salmo of subfamily Salmoninae. NGS platform was proved to be a rapid and time-saving technology to reveal complete mitogenomes.
.
DOI:10.1371/journal.pone.0202485URLPMID:30114217
Sophora japonica L. (Faboideae, Leguminosae) is an important traditional Chinese herb with a long history of cultivation. Its flower buds and fruits contain abundant flavonoids, and therefore, the plants are cultivated for the industrial extraction of rutin. Here, we determined the complete nucleotide sequence of the mitochondrial genome of S. japonica 'JinhuaiJ2', the most widely planted variety in Guangxi region of China. The total length of the mtDNA sequence is 484,916 bp, with a GC content of 45.4%. Sophora japonica mtDNA harbors 32 known protein-coding genes, 17 tRNA genes, and three rRNA genes with 17 cis-spliced and five trans-spliced introns disrupting eight protein-coding genes. The gene coding and intron regions, and intergenic spacers account for 7.5%, 5.8% and 86.7% of the genome, respectively. The gene profile of S. japonica mitogenome differs from that of the other Faboideae species by only one or two gene gains or losses. Four of the 17 cis-spliced introns showed distinct length variations in the Faboideae, which could be attributed to the homologous recombination of the short repeats measuring a few bases located precisely at the edges of the putative deletions. This reflects the importance of small repeats in the sequence evolution in Faboideae mitogenomes. Repeated sequences of S. japonica mitogenome are mainly composed of small repeats, with only 20 medium-sized repeats, and one large repeat, adding up to 4% of its mitogenome length. Among the 25 pseudogene fragments detected in the intergenic spacer regions, the two largest ones and their corresponding functional gene copies located in two different sets of medium-sized repeats, point to their origins from homologous recombinations. As we further observed the recombined reads associated with the longest repeats of 2,160 bp with the PacBio long read data set of just 15 × in depth, repeat mediated homologous recombinations may play important role in the mitogenomic evolution of S. japonica. Our study provides insightful knowledge to the genetic background of this important herb species and the mitogenomic evolution in the Faboideae species.
.
DOI:10.1186/s12859-017-1927-yURLPMID:29162031 [本文引用: 6]
Whole-genome sequencing (WGS) projects provide short read nucleotide sequences from nuclear and possibly organelle DNA depending on the source of origin. Mitochondrial DNA is present in animals and fungi, while plants contain DNA from both mitochondria and chloroplasts. Current techniques for separating organelle reads from nuclear reads in WGS data require full reference or partial seed sequences for assembling.
.
DOI:10.1089/cmb.2012.0021URL [本文引用: 2]
The lion's share of bacteria in various environments cannot be cloned in the laboratory and thus cannot be sequenced using existing technologies. A major goal of single-cell genomics is to complement gene-centric metagenomic data with whole-genome assemblies of uncultivated organisms. Assembly of single-cell data is challenging because of highly non-uniform read coverage as well as elevated levels of sequencing errors and chimeric reads. We describe SPAdes, a new assembler for both single-cell and standard (multicell) assembly, and demonstrate that it improves on the recently released E+V-SC assembler (specialized for single-cell data) and on popular assemblers Velvet and SoapDeNovo (for multicell data). SPAdes generates single-cell assemblies, providing information about genomes of un-cultivatable bacteria that vastly exceeds what may be obtained via traditional metagenomics studies. SPAdes is available online (http://bioinf.spbau.ru/spades). It is distributed as open source software.
.
DOI:10.1093/nar/gkw955URLPMID:28204566 [本文引用: 3]
The evolution in next-generation sequencing (NGS) technology has led to the development of many different assembly algorithms, but few of them focus on assembling the organelle genomes. These genomes are used in phylogenetic studies, food identification and are the most deposited eukaryotic genomes in GenBank. Producing organelle genome assembly from whole genome sequencing (WGS) data would be the most accurate and least laborious approach, but a tool specifically designed for this task is lacking. We developed a seed-and-extend algorithm that assembles organelle genomes from whole genome sequencing (WGS) data, starting from a related or distant single seed sequence. The algorithm has been tested on several new (Gonioctena intermedia and Avicennia marina) and public (Arabidopsis thaliana and Oryza sativa) whole genome Illumina data sets where it outperforms known assemblers in assembly accuracy and coverage. In our benchmark, NOVOPlasty assembled all tested circular genomes in less than 30 min with a maximum memory requirement of 16 GB and an accuracy over 99.99%. In conclusion, NOVOPlasty is the sole de novo assembler that provides a fast and straightforward extraction of the extranuclear genomes from WGS data in one circular high quality contig. The software is open source and can be downloaded at https://github.com/ndierckx/NOVOPlasty.
.
DOI:10.1093/nar/gkz173URLPMID:30864657 [本文引用: 6]
Mitochondrial genome (mitogenome) plays important roles in evolutionary and ecological studies. It becomes routine to utilize multiple genes on mitogenome or the entire mitogenomes to investigate phylogeny and biodiversity of focal groups with the onset of High Throughput Sequencing?(HTS)?technologies. We developed a mitogenome toolkit MitoZ, consisting of independent modules of de novo assembly, findMitoScaf (find Mitochondrial Scaffolds), annotation and visualization, that can generate mitogenome assembly together with annotation and visualization results from HTS raw reads. We evaluated its performance using a total of 50 samples of which mitogenomes are publicly available. The results showed that MitoZ can recover more full-length mitogenomes with higher accuracy compared to the other available mitogenome assemblers. Overall, MitoZ provides a one-click solution to construct the annotated mitogenome from HTS raw data and will facilitate large scale ecological and evolutionary studies. MitoZ is free open source software distributed under GPLv3 license and available at https://github.com/linzhi2013/MitoZ.
.
DOI:10.1385/0-89603-319-8:109URLPMID:8924972 [本文引用: 1]
.
DOI:10.1101/gr.186668.114URLPMID:26518481 [本文引用: 1]
Inter-species hybridization has been recently recognized as potentially common in wild animals, but the extent to which it shapes modern genomes is still poorly understood. Distinguishing historical hybridization events from other processes leading to phylogenetic discordance among different markers requires a well-resolved species tree that considers all modes of inheritance and overcomes systematic problems due to rapid lineage diversification by sampling large genomic character sets. Here, we assessed genome-wide phylogenetic variation across a diverse mammalian family, Felidae (cats). We combined genotypes from a genome-wide SNP array with additional autosomal, X- and Y-linked variants to sample ~150 kb of nuclear sequence, in addition to complete mitochondrial genomes generated using light-coverage Illumina sequencing. We present the first robust felid time tree that accounts for unique maternal, paternal, and biparental evolutionary histories. Signatures of phylogenetic discordance were abundant in the genomes of modern cats, in many cases indicating hybridization as the most likely cause. Comparison of big cat whole-genome sequences revealed a substantial reduction of X-linked divergence times across several large recombination cold spots, which were highly enriched for signatures of selection-driven post-divergence hybridization between the ancestors of the snow leopard and lion lineages. These results highlight the mosaic origin of modern felid genomes and the influence of sex chromosomes and sex-biased dispersal in post-speciation gene flow. A complete resolution of the tree of life will require comprehensive genomic sampling of biparental and sex-limited genetic variation to identify and control for phylogenetic conflict caused by ancient admixture and sex-biased differences in genomic transmission.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
.
DOI:10.1186/s41065-017-0028-2URLPMID:28396619 [本文引用: 1]
With the increasing capacity of present-day next-generation sequencers the field of mitogenomics is rapidly changing. Enrichment of the mitochondrial fraction, is no longer necessary for obtaining mitogenomic data. Despite the benefits, shotgun sequencing approaches also have disadvantages. They do not guarantee obtaining the complete mitogenome, generally require larger amounts of input DNA and coverage is low compared to sequencing with enrichment strategies. If the mitogenome could be amplified in a single amplification, additional time and costs for sample preparation might outweigh these disadvantages.
.
DOI:10.1016/j.fsigen.2014.06.001URLPMID:24973578 [本文引用: 1]
Mitochondrial DNA typing in forensic genetics has been performed traditionally using Sanger-type sequencing. Consequently sequencing of a relatively-large target such as the mitochondrial genome (mtGenome) is laborious and time consuming. Thus, sequencing typically focuses on the control region due to its high concentration of variation. Massively parallel sequencing (MPS) has become more accessible in recent years allowing for high-throughput processing of large target areas. In this study, Nextera(?) XT DNA Sample Preparation Kit and the Illumina MiSeq? were utilized to generate quality whole genome mitochondrial haplotypes from 283 individuals in a both cost-effective and rapid manner. Results showed that haplotypes can be generated at a high depth of coverage with limited strand bias. The distribution of variants across the mitochondrial genome was described and demonstrated greater variation within the coding region than the non-coding region. Haplotype and haplogroup diversity were described with respect to whole mtGenome and HVI/HVII. An overall increase in haplotype or genetic diversity and random match probability, as well as better haplogroup assignment demonstrates that MPS of the mtGenome using the Illumina MiSeq system is a viable and reliable methodology.
.
[本文引用: 6]
.
DOI:10.1111/1755-0998.12492URLPMID:26607054 [本文引用: 2]
Next-generation sequencing continues to revolutionize biodiversity studies by generating unprecedented amounts of DNA sequence data for comparative genomic analysis. However, these data are produced as millions or billions of short reads of variable quality that cannot be directly applied in comparative analyses, creating a demand for methods to facilitate assembly. We optimized an in silico strategy to efficiently reconstruct high-quality mitochondrial genomes directly from genomic reads. We tested this strategy using sequences from five species of frogs: Hylodes meridionalis (Hylodidae), Hyloxalus yasuni (Dendrobatidae), Pristimantis fenestratus (Craugastoridae), and Melanophryniscus simplex and Rhinella sp. (Bufonidae). These are the first mitogenomes published for these species, the genera Hylodes, Hyloxalus, Pristimantis, Melanophryniscus and Rhinella, and the families Craugastoridae and Hylodidae. Sequences were generated using only half of one lane of a standard Illumina HiqSeq 2000 flow cell, resulting in fewer than eight million reads. We analysed the reads of Hylodes meridionalis using three different assembly strategies: (1) reference-based (using bowtie2); (2) de novo (using abyss, soapdenovo2 and velvet); and (3) baiting and iterative mapping (using mira and mitobim). Mitogenomes were assembled exclusively with strategy 3, which we employed to assemble the remaining mitogenomes. Annotations were performed with mitos and confirmed by comparison with published amphibian mitochondria. In most cases, we recovered all 13 coding genes, 22 tRNAs, and two ribosomal subunit genes, with minor gene rearrangements. Our results show that few raw reads can be sufficient to generate high-quality scaffolds, making any Illumina machine run using genomic multiplex libraries a potential source of data for organelle assemblies as by-catch.
.
DOI:10.1093/bioinformatics/btp324URLPMID:19451168 [本文引用: 1]
The enormous amount of short reads generated by the new DNA sequencing technologies call for the development of fast and accurate read alignment programs. A first generation of hash table-based methods has been developed, including MAQ, which is accurate, feature rich and fast enough to align short reads from a single individual. However, MAQ does not support gapped alignment for single-end reads, which makes it unsuitable for alignment of longer reads where indels may occur frequently. The speed of MAQ is also a concern when the alignment is scaled up to the resequencing of hundreds of individuals.
.
DOI:10.1016/j.cub.2018.05.008URLPMID:29920259 [本文引用: 1]
Present-day giant pandas (Ailuropoda melanoleuca) are estimated to have diverged from their closest relatives, all other bears, ~20 million years ago, based on molecular data [1]. With fewer than 2,500 individuals living today [2], it is unclear how well genetic data from extant and historical giant pandas [3] reflect the past [3]. To date, there has been no complete mitochondrial DNA (mtDNA) sequenced from an ancient giant panda. Here, we use ancient DNA capture techniques [4] to sequence the complete mitochondrial genome of a ~22,000-year-old giant panda specimen (radiocarbon date of 21,910-21,495 cal BP with ± 2σ at 95.4% probability; Lab.no Beta-473743) from the Cizhutuo Cave, in Leye County, Guangxi Province, China (Figure 1A). Its date and location in Guangxi, where no wild giant pandas live today, as well as the difficulty of DNA preservation in a hot and humid region, place it as a unique specimen to learn about ancient giant pandas from the last glacial maximum. We find that the mtDNA lineage of the Cizhutuo panda coalesced with present-day pandas ~183 thousand years ago (kya, 95% HPD, 227-144 kya), earlier than the time to the most recent common ancestor (TMRCA) of mtDNA lineages shared by present-day pandas (~72 kya, 95% HPD, 94-55 kya, Supplemental Information). Furthermore, the Cizhutuo panda possessed 18 non-synonymous mutations across six mitochondrial genes. Our results show that the Cizhutuo mtDNA lineage underwent a distinct history from that of present-day populations.
.
DOI:10.1038/nrg1606URLPMID:15861210 [本文引用: 1]
The human mitochondrial genome is extremely small compared with the nuclear genome, and mitochondrial genetics presents unique clinical and experimental challenges. Despite the diminutive size of the mitochondrial genome, mitochondrial DNA (mtDNA) mutations are an important cause of inherited disease. Recent years have witnessed considerable progress in understanding basic mitochondrial genetics and the relationship between inherited mutations and disease phenotypes, and in identifying acquired mtDNA mutations in both ageing and cancer. However, many challenges remain, including the prevention and treatment of these diseases. This review explores the advances that have been made and the areas in which future progress is likely.
.
DOI:10.1016/j.tig.2006.04.001URLPMID:16678300 [本文引用: 1]
Human mitochondrial DNA (mtDNA) studies have entered a new phase since the blossoming of complete genome analyses. Sequencing complete mtDNAs is more expensive and more labour intensive than restriction analysis or simply sequencing the control region of the molecule. But the efforts are paying off, as the phylogenetic resolution of the mtDNA tree has been greatly improved, and, in turn, phylogeographic interpretations can be given correspondingly greater precision in terms of the timing and direction of human dispersals. Therefore, despite mtDNA being only a fraction of our total genome, the deciphering of its evolution is profoundly changing our perception about how modern humans spread across our planet. Here we illustrate the phylogeographic approach with two case studies: the initial dispersal out of Africa, and the colonization of Europe.
.
DOI:10.1016/j.cell.2008.06.021URLPMID:18692465 [本文引用: 1]
A complete mitochondrial (mt) genome sequence was reconstructed from a 38,000 year-old Neandertal individual with 8341 mtDNA sequences identified among 4.8 Gb of DNA generated from approximately 0.3 g of bone. Analysis of the assembled sequence unequivocally establishes that the Neandertal mtDNA falls outside the variation of extant human mtDNAs, and allows an estimate of the divergence date between the two mtDNA lineages of 660,000 +/- 140,000 years. Of the 13 proteins encoded in the mtDNA, subunit 2 of cytochrome c oxidase of the mitochondrial electron transport chain has experienced the largest number of amino acid substitutions in human ancestors since the separation from Neandertals. There is evidence that purifying selection in the Neandertal mtDNA was reduced compared with other primate lineages, suggesting that the effective population size of Neandertals was small.
.
DOI:10.1016/j.mito.2011.08.005URL [本文引用: 1]
The use of Next-Generation Sequencing of mitochondrial DNA is becoming widespread in biological and clinical research. This, in turn, creates a need for a convenient tool that detects and analyzes heteroplasmy. Here we present MitoBamAnnotator, a user friendly web-based tool that allows maximum flexibility and control in heteroplasmy research. MitoBamAnnotator provides the user with a comprehensively annotated overview of mitochondrial genetic variation, allowing for an in-depth analysis with no prior knowledge in programming. (C) 2011 Elsevier B.V. and Mitochondria Research Society.
.
DOI:10.1093/bioinformatics/btt118URL [本文引用: 1]
Motivation: Exome capture kits have capture efficiencies that range from 40 to 60%. A significant amount of off-target reads are from the mitochondrial genome. These unintentionally sequenced mitochondrial reads provide unique opportunities to study the mitochondria genome.
Results: MitoSeek is an open-source software tool that can reliably and easily extract mitochondrial genome information from exome and whole genome sequencing data. MitoSeek evaluates mitochondrial genome alignment quality, estimates relative mitochondrial copy numbers and detects heteroplasmy, somatic mutation and structural variants of the mitochondrial genome. MitoSeek can be set up to run in parallel or serial on large exome sequencing datasets.
.
DOI:10.1111/1556-4029.12139URLPMID:23682804 [本文引用: 1]
Mitochondrial DNA (mtDNA) is a valuable tool in the fields of forensic, population, and medical genetics. However, recording and comparing mtDNA control region or entire genome sequences would be difficult if researchers are not familiar with mtDNA nomenclature conventions. Therefore, mtDNAprofiler, a Web application, was designed for the analysis and comparison of mtDNA sequences in a string format or as a list of mtDNA single-nucleotide polymorphisms (mtSNPs). mtDNAprofiler which comprises four mtDNA sequence-analysis tools (mtDNA nomenclature, mtDNA assembly, mtSNP conversion, and mtSNP concordance-check) supports not only the accurate analysis of mtDNA sequences via an automated nomenclature function, but also consistent management of mtSNP data via direct comparison and validity-check functions. Since mtDNAprofiler consists of four tools that are associated with key steps of mtDNA sequence analysis, mtDNAprofiler will be helpful for researchers working with mtDNA. mtDNAprofiler is freely available at http://mtprofiler.yonsei.ac.kr.
.
DOI:10.1002/humu.22767URLPMID:25677119 [本文引用: 1]
The human mitochondrial genome has been reported to have a very high mutation rate as compared with the nuclear genome. A large number of mitochondrial mutations show significant phenotypic association and are involved in a broad spectrum of diseases. In recent years, there has been a remarkable progress in the understanding of mitochondrial genetics. The availability of next-generation sequencing (NGS) technologies have not only reduced sequencing cost by orders of magnitude but has also provided us good quality mitochondrial genome sequences with high coverage, thereby enabling decoding of a number of human mitochondrial diseases. In this study, we report a computational and experimental pipeline to decipher the human mitochondrial DNA variations and examine them for their clinical correlation. As a proof of principle, we also present a clinical study of a patient with Leigh disease and confirmed maternal inheritance of the causative allele. The pipeline is made available as a user-friendly online tool to annotate variants and find haplogroup, disease association, and heteroplasmic sites. The "mit-o-matic" computational pipeline represents a comprehensive cloud-based tool for clinical evaluation of mitochondrial genomic variations from NGS datasets. The tool is freely available at http://genome.igib.res.in/mitomatic/.
.
DOI:10.1093/bioinformatics/btu483URL [本文引用: 1]
Motivation: The increasing availability of mitochondria-targeted and off-target sequencing data in whole-exome and whole-genome sequencing studies (WXS and WGS) has risen the demand of effective pipelines to accurately measure heteroplasmy and to easily recognize the most functionally important mitochondrial variants among a huge number of candidates. To this purpose, we developed MToolBox, a highly automated pipeline to reconstruct and analyze human mitochondrial DNA from high-throughput sequencing data.
Results: MToolBox implements an effective computational strategy for mitochondrial genomes assembling and haplogroup assignment also including a prioritization analysis of detected variants. MToolBox provides a Variant Call Format file featuring, for the first time, allele-specific heteroplasmy and annotation files with prioritized variants. MToolBox was tested on simulated samples and applied on 1000 Genomes WXS datasets.
.
DOI:10.1093/bioinformatics/btu825URLPMID:25505086 [本文引用: 1]
All current mitochondrial haplogroup classification tools require variants to be detected from an alignment with the reference sequence and to be properly named according to the canonical nomenclature standards for describing mitochondrial variants, before they can be compared with the haplogroup determining polymorphisms. With the emergence of high-throughput sequencing technologies and hence greater availability of mitochondrial genome sequences, there is a strong need for an automated haplogroup classification tool that is alignment-free and agnostic to reference sequence.
.
DOI:10.1093/nar/gkw247URLPMID:27084948 [本文引用: 1]
Next generation sequencing (NGS) allows investigating mitochondrial DNA (mtDNA) characteristics such as heteroplasmy (i.e. intra-individual sequence variation) to a higher level of detail. While several pipelines for analyzing heteroplasmies exist, issues in usability, accuracy of results and interpreting final data limit their usage. Here we present mtDNA-Server, a scalable web server for the analysis of mtDNA studies of any size with a special focus on usability as well as reliable identification and quantification of heteroplasmic variants. The mtDNA-Server workflow includes parallel read alignment, heteroplasmy detection, artefact or contamination identification, variant annotation as well as several quality control metrics, often neglected in current mtDNA NGS studies. All computational steps are parallelized with Hadoop MapReduce and executed graphically with Cloudgene. We validated the underlying heteroplasmy and contamination detection model by generating four artificial sample mix-ups on two different NGS devices. Our evaluation data shows that mtDNA-Server detects heteroplasmies and artificial recombinations down to the 1% level with perfect specificity and outperforms existing approaches regarding sensitivity. mtDNA-Server is currently able to analyze the 1000G Phase 3 data (n = 2,504) in less than 5 h and is freely accessible at https://mtdna-server.uibk.ac.at.
.
DOI:10.7717/peerj.3406URLPMID:28584729 [本文引用: 2]
Recent rapid advances in high-throughput, next-generation sequencing (NGS) technologies have promoted mitochondrial genome studies in the fields of human evolution, medical genetics, and forensic casework. However, scientists unfamiliar with computer programming often find it difficult to handle the massive volumes of data that are generated by NGS. To address this limitation, we developed MitoSuite, a user-friendly graphical tool for analysis of data from high-throughput sequencing of the human mitochondrial genome. MitoSuite generates a visual report on NGS data with simple mouse operations. Moreover, it analyzes high-coverage sequencing data but runs on a stand-alone computer, without the need for file upload. Therefore, MitoSuite offers outstanding usability for handling massive NGS data, and is ideal for evolutionary, clinical, and forensic studies on the human mitochondrial genome variations. It is freely available for download from the website https://mitosuite.com.
.
DOI:10.1101/gr.092759.109URLPMID:19541911 [本文引用: 1]
We created a visualization tool called Circos to facilitate the identification and analysis of similarities and differences arising from comparisons of genomes. Our tool is effective in displaying variation in genome structure and, generally, any other kind of positional relationships between genomic intervals. Such data are routinely produced by sequence alignments, hybridization arrays, genome mapping, and genotyping studies. Circos uses a circular ideogram layout to facilitate the display of relationships between pairs of positions by the use of ribbons, which encode the position, size, and orientation of related genomic elements. Circos is capable of displaying data as scatter, line, and histogram plots, heat maps, tiles, connectors, and text. Bitmap or vector images can be created from GFF-style data inputs and hierarchical configuration files, which can be easily generated by automated tools, making Circos suitable for rapid deployment in data analysis and reporting pipelines.
.
DOI:10.1101/gr.107524.110URLPMID:20644199 [本文引用: 1]
Next-generation DNA sequencing (NGS) projects, such as the 1000 Genomes Project, are already revolutionizing our understanding of genetic variation among individuals. However, the massive data sets generated by NGS--the 1000 Genome pilot alone includes nearly five terabases--make writing feature-rich, efficient, and robust analysis tools difficult for even computationally sophisticated individuals. Indeed, many professionals are limited in the scope and the ease with which they can answer scientific questions by the complexity of accessing and manipulating the data produced by these machines. Here, we discuss our Genome Analysis Toolkit (GATK), a structured programming framework designed to ease the development of efficient and robust analysis tools for next-generation DNA sequencers using the functional programming philosophy of MapReduce. The GATK provides a small but rich set of data access patterns that encompass the majority of analysis tool needs. Separating specific analysis calculations from common data management infrastructure enables us to optimize the GATK framework for correctness, stability, and CPU and memory efficiency and to enable distributed and shared memory parallelization. We highlight the capabilities of the GATK by describing the implementation and application of robust, scale-tolerant tools like coverage calculators and single nucleotide polymorphism (SNP) calling. We conclude that the GATK programming framework enables developers and analysts to quickly and easily write efficient and robust NGS tools, many of which have already been incorporated into large-scale sequencing projects like the 1000 Genomes Project and The Cancer Genome Atlas.
.
DOI:10.1038/NMETH.1923URL [本文引用: 2]
As the rate of sequencing increases, greater throughput is demanded from read aligners. The full-text minute index is often used to make alignment very fast and memory-efficient, but the approach is ill-suited to finding longer, gapped alignments. Bowtie 2 combines the strengths of the full-text minute index with the flexibility and speed of hardware-accelerated dynamic programming algorithms to achieve a combination of high speed, sensitivity and accuracy.
.
DOI:10.1016/j.tig.2007.12.006URL [本文引用: 1]
New DNA sequencing technologies can sequence up to one billion bases in a single day at low cost, putting large-scale sequencing within the reach of many scientists. Many researchers are forging ahead with projects to sequence a range of species using the new technologies. However, these new technologies produce read lengths as short as 35–40 nucleotides, posing challenges for genome assembly and annotation. Here we review the challenges and describe some of the bioinformatics systems that are being proposed to solve them. We specifically address issues arising from using these technologies in assembly projects, both de novo and for resequencing purposes, as well as efforts to improve genome annotation in the fragmented assemblies produced by short read lengths.
.
DOI:10.1093/nar/gkt371URLPMID:23661685 [本文引用: 2]
We present an in silico approach for the reconstruction of complete mitochondrial genomes of non-model organisms directly from next-generation sequencing (NGS) data-mitochondrial baiting and iterative mapping (MITObim). The method is straightforward even if only (i) distantly related mitochondrial genomes or (ii) mitochondrial barcode sequences are available as starting-reference sequences or seeds, respectively. We demonstrate the efficiency of the approach in case studies using real NGS data sets of the two monogenean ectoparasites species Gyrodactylus thymalli and Gyrodactylus derjavinoides including their respective teleost hosts European grayling (Thymallus thymallus) and Rainbow trout (Oncorhynchus mykiss). MITObim appeared superior to existing tools in terms of accuracy, runtime and memory requirements and fully automatically recovered mitochondrial genomes exceeding 99.5% accuracy from total genomic DNA derived NGS data sets in <24 h using a standard desktop computer. The approach overcomes the limitations of traditional strategies for obtaining mitochondrial genomes for species with little or no mitochondrial sequence information at hand and represents a fast and highly efficient in silico alternative to laborious conventional strategies relying on initial long-range PCR. We furthermore demonstrate the applicability of MITObim for metagenomic/pooled data sets using simulated data. MITObim is an easy to use tool even for biologists with modest bioinformatics experience. The software is made available as open source pipeline under the MIT license at https://github.com/chrishah/MITObim.
.
DOI:10.1007/978-1-4939-9176-1_18URLPMID:30875055 [本文引用: 1]
Recent methodological advances have transformed the field of ancient DNA (aDNA). Basic bioinformatics skills are becoming essential requirements to process and analyze the sheer amounts of data generated by current aDNA studies and in biomedical research in general. This chapter is intended as a practical guide to the assembly of ancient mitochondrial genomes, directly from genomic DNA-derived next-generation sequencing (NGS) data, specifically in the absence of closely related reference genomes. In a hands-on tutorial suitable for readers with little to no prior bioinformatics experience, we reconstruct the mitochondrial genome of a woolly mammoth deposited ~45,000?years ago. We introduce key software tools and outline general strategies for mitogenome assembly, including the critical quality assessment of assembly results without a reference genome.
.
DOI:10.1093/dnares/dsy026URLPMID:30085012 [本文引用: 1]
Mitochondrial genome (mtDNA) carries not only well-conserved protein coding, tRNA and rRNA genes, but also highly variable non-coding regions (NCRs). However, the NCRs show poor conservation across species, making their function and evolution elusive. Identification and functional characterization of NCRs across species would be critical for addressing these questions. To this end, we devised a computational pipeline and performed de novo assembly and annotation of mtDNA from 19 Caenorhabditis species using next-generation sequencing (NGS) data. The mtDNAs for 14 out of the 19 species are reported for the first time. Comparison of the 19 genomes reveals species-specific sampling of partial displacement-loop (D-loop) sequence as a novel NCR inserted into a unique tRNA cluster, suggesting an important role of the D-loop and the tRNA cluster in shaping NCR evolution. Intriguingly, RNA-Seq analysis suggests that a novel NCR resulting from a recent duplication of NADH dehydrogenase subunit 5 (ND5) could be utilized as a 3' UTR for up-regulation of its upstream gene. The expression analysis shows a species- and sex-specific expression of mitochondrial genes encoded by mtDNA and nucleus, respectively. Our analyses provide important insights into the function and evolution of mitochondrial NCRs and pave the way for further studying the function and evolution of mitochondrial genome.
.
DOI:10.1093/bioinformatics/btl629URLPMID:17158514 [本文引用: 1]
Novel DNA sequencing technologies with the potential for up to three orders magnitude more sequence throughput than conventional Sanger sequencing are emerging. The instrument now available from Solexa Ltd, produces millions of short DNA sequences of 25 nt each. Due to ubiquitous repeats in large genomes and the inability of short sequences to uniquely and unambiguously characterize them, the short read length limits applicability for de novo sequencing. However, given the sequencing depth and the throughput of this instrument, stringent assembly of highly identical sequences can be achieved. We describe SSAKE, a tool for aggressively assembling millions of short nucleotide sequences by progressively searching through a prefix tree for the longest possible overlap between any two sequences. SSAKE is designed to help leverage the information from short sequence reads by stringently assembling them into contiguous sequences that can be used to characterize novel sequencing targets.
.
DOI:10.1093/bioinformatics/btm451URLPMID:17893086 [本文引用: 1]
Inexpensive de novo genome sequencing, particularly in organisms with small genomes, is now possible using several new sequencing technologies. Some of these technologies such as that from Illumina's Solexa Sequencing, produce high genomic coverage by generating a very large number of small reads ( approximately 30 bp). While prior work shows that partial assembly can be performed by k-mer extension in error-free reads, this algorithm is unsuccessful with the sequencing error rates found in practice. We present VCAKE (Verified Consensus Assembly by K-mer Extension), a modification of simple k-mer extension that overcomes error by using high depth coverage. Though it is a simple modification of a previous approach, we show significant improvements in assembly results on simulated and experimental datasets that include error.
.
[本文引用: 1]
.
[本文引用: 2]
.
DOI:10.1111/mec.13549URLPMID:26821259 [本文引用: 1]
DNA barcoding has had a major impact on biodiversity science. The elegant simplicity of establishing massive scale databases for a few barcode loci is continuing to change our understanding of species diversity patterns, and continues to enhance human abilities to distinguish among species. Capitalizing on the developments of next generation sequencing technologies and decreasing costs of genome sequencing, there is now the opportunity for the DNA barcoding concept to be extended to new kinds of genomic data. We illustrate the benefits and capacity to do this, and also note the constraints and barriers to overcome before it is truly scalable. We advocate a twin track approach: (i) continuation and acceleration of global efforts to build the DNA barcode reference library of life on earth using standard DNA barcodes and (ii) active development and application of extended DNA barcodes using genome skimming to augment the standard barcoding approach.
.
DOI:10.1093/bioinformatics/bts723URL [本文引用: 1]
Motivation: Researchers need general purpose methods for objectively evaluating the accuracy of single and metagenome assemblies and for automatically detecting any errors they may contain. Current methods do not fully meet this need because they require a reference, only consider one of the many aspects of assembly quality or lack statistical justification, and none are designed to evaluate metagenome assemblies.
Results: In this article, we present an Assembly Likelihood Evaluation (ALE) framework that overcomes these limitations, systematically evaluating the accuracy of an assembly in a reference-independent manner using rigorous statistical methods. This framework is comprehensive, and integrates read quality, mate pair orientation and insert length (for paired-end reads), sequencing coverage, read alignment and k-mer frequency. ALE pinpoints synthetic errors in both single and metagenomic assemblies, including single-base errors, insertions/deletions, genome rearrangements and chimeric assemblies presented in metagenomes. At the genome level with real-world data, ALE identifies three large misassemblies from the Spirochaeta smaragdinae finished genome, which were all independently validated by Pacific Biosciences sequencing. At the single-base level with Illumina data, ALE recovers 215 of 222 (97%) single nucleotide variants in a training set from a GC-rich Rhodobacter sphaeroides genome. Using real Pacific Biosciences data, ALE identifies 12 of 12 synthetic errors in a Lambda Phage genome, surpassing even Pacific Biosciences' own variant caller, EviCons. In summary, the ALE framework provides a comprehensive, reference-independent and statistically rigorous measure of single genome and metagenome assembly accuracy, which can be used to identify misassemblies or to optimize the assembly process.
.
DOI:10.1186/1471-2105-10-421URLPMID:20003500 [本文引用: 1]
Sequence similarity searching is a very important bioinformatics task. While Basic Local Alignment Search Tool (BLAST) outperforms exact methods through its use of heuristics, the speed of the current BLAST software is suboptimal for very long queries or database sequences. There are also some shortcomings in the user-interface of the current command-line applications.
.
DOI:10.1093/gigascience/gix007URLPMID:28327976 [本文引用: 1]
Environmental metagenomic analysis is typically accomplished by assigning taxonomy and/or function from whole genome sequencing or 16S amplicon sequences. Both of these approaches are limited, however, by read length, among other technical and biological factors. A nanopore-based sequencing platform, MinION?, produces reads that are ≥1 × 104 bp in length, potentially providing for more precise assignment, thereby alleviating some of the limitations inherent in determining metagenome composition from short reads. We tested the ability of sequence data produced by MinION (R7.3 flow cells) to correctly assign taxonomy in single bacterial species runs and in three types of low-complexity synthetic communities: a mixture of DNA using equal mass from four species, a community with one relatively rare (1%) and three abundant (33% each) components, and a mixture of genomic DNA from 20 bacterial strains of staggered representation. Taxonomic composition of the low-complexity communities was assessed by analyzing the MinION sequence data with three different bioinformatic approaches: Kraken, MG-RAST, and One Codex. Results: Long read sequences generated from libraries prepared from single strains using the version 5 kit and chemistry, run on the original MinION device, yielded as few as 224 to as many as 3497 bidirectional high-quality (2D) reads with an average overall study length of 6000 bp. For the single-strain analyses, assignment of reads to the correct genus by different methods ranged from 53.1% to 99.5%, assignment to the correct species ranged from 23.9% to 99.5%, and the majority of misassigned reads were to closely related organisms. A synthetic metagenome sequenced with the same setup yielded 714 high quality 2D reads of approximately 5500 bp that were up to 98% correctly assigned to the species level. Synthetic metagenome MinION libraries generated using version 6 kit and chemistry yielded from 899 to 3497 2D reads with lengths averaging 5700 bp with up to 98% assignment accuracy at the species level. The observed community proportions for “equal” and “rare” synthetic libraries were close to the known proportions, deviating from 0.1% to 10% across all tests. For a 20-species mock community with staggered contributions, a sequencing run detected all but 3 species (each included at <0.05% of DNA in the total mixture), 91% of reads were assigned to the correct species, 93% of reads were assigned to the correct genus, and >99% of reads were assigned to the correct family. Conclusions: At the current level of output and sequence quality (just under 4 × 103 2D reads for a synthetic metagenome), MinION sequencing followed by Kraken or One Codex analysis has the potential to provide rapid and accurate metagenomic analysis where the consortium is comprised of a limited number of taxa. Important considerations noted in this study included: high sensitivity of the MinION platform to the quality of input DNA, high variability of sequencing results across libraries and flow cells, and relatively small numbers of 2D reads per analysis limit. Together, these limited detection of very rare components of the microbial consortia, and would likely limit the utility of MinION for the sequencing of high-complexity metagenomic communities where thousands of taxa are expected. Furthermore, the limitations of the currently available data analysis tools suggest there is considerable room for improvement in the analytical approaches for the characterization of microbial communities using long reads. Nevertheless, the fact that the accurate taxonomic assignment of high-quality reads generated by MinION is approaching 99.5% and, in most cases, the inferred community structure mirrors the known proportions of a synthetic mixture warrants further exploration of practical application to environmental metagenomics as the platform continues to develop and improve. With further improvement in sequence throughput and error rate reduction, this platform shows great promise for precise real-time analysis of the composition and structure of more complex microbial communities.
.
DOI:10.1038/ncomms14515URLPMID:28218240
Third generation sequencing technologies provide the opportunity to improve genome assemblies by generating long reads spanning most repeat sequences. However, current analysis methods require substantial amounts of sequence data and computational resources to overcome the high error rates. Furthermore, they can only perform analysis after sequencing has completed, resulting in either over-sequencing, or in a low quality assembly due to under-sequencing. Here we present npScarf, which can scaffold and complete short read assemblies while the long read sequencing run is in progress. It reports assembly metrics in real-time so the sequencing run can be terminated once an assembly of sufficient quality is obtained. In assembling four bacterial and one eukaryotic genomes, we show that npScarf can construct more complete and accurate assemblies while requiring less sequencing data and computational resources than existing methods. Our approach offers a time- and resource-effective strategy for completing short read assemblies.
.
DOI:10.1038/s41467-018-07271-1URLPMID:30451840
Long-read sequencing technologies have greatly facilitated assemblies of large eukaryotic genomes. In this paper, Oxford Nanopore sequences generated on a MinION sequencer are combined with Bionano Genomics Direct Label and Stain (DLS) optical maps to generate a chromosome-scale de novo assembly of the repeat-rich Sorghum bicolor Tx430 genome. The final assembly consists of 29 scaffolds, encompassing in most cases entire chromosome arms. It has a scaffold N50 of 33.28 Mbps and covers 90% of the expected genome length. A sequence accuracy of 99.85% is obtained after aligning the assembly against Illumina Tx430 data and 99.6% of the 34,211 public gene models align to the assembly. Comparisons of Tx430 and BTx623 DLS maps against the public BTx623 v3.0.1 genome assembly suggest substantial discrepancies whose origin remains to be determined. In summary, this study demonstrates that informative assemblies of complex plant genomes can be generated by combining nanopore sequencing with DLS optical maps.
.
DOI:10.1101/gr.191395.115URLPMID:26447147
Monitoring the progress of DNA molecules through a membrane pore has been postulated as a method for sequencing DNA for several decades. Recently, a nanopore-based sequencing instrument, the Oxford Nanopore MinION, has become available, and we used this for sequencing the Saccharomyces cerevisiae genome. To make use of these data, we developed a novel open-source hybrid error correction algorithm Nanocorr specifically for Oxford Nanopore reads, because existing packages were incapable of assembling the long read lengths (5-50 kbp) at such high error rates (between ~5% and 40% error). With this new method, we were able to perform a hybrid error correction of the nanopore reads using complementary MiSeq data and produce a de novo assembly that is highly contiguous and accurate: The contig N50 length is more than ten times greater than an Illumina-only assembly (678 kb versus 59.9 kbp) and has >99.88% consensus identity when compared to the reference. Furthermore, the assembly with the long nanopore reads presents a much more complete representation of the features of the genome and correctly assembles gene cassettes, rRNAs, transposable elements, and other genomic features that were almost entirely absent in the Illumina-only assembly.
.
DOI:10.1371/journal.pone.0197393URLPMID:29795601 [本文引用: 1]
Actinidia arguta is the most basal species in a phylogenetically and economically important genus in the family Actinidiaceae. To better understand the molecular basis of the Actinidia arguta chloroplast (cp), we sequenced the complete cp genome from A. arguta using Illumina and PacBio RS II sequencing technologies. The cp genome from A. arguta was 157,611 bp in length and composed of a pair of 24,232 bp inverted repeats (IRs) separated by a 20,463 bp small single copy region (SSC) and an 88,684 bp large single copy region (LSC). Overall, the cp genome contained 113 unique genes. The cp genomes from A. arguta and three other Actinidia species from GenBank were subjected to a comparative analysis. Indel mutation events and high frequencies of base substitution were identified, and the accD and ycf2 genes showed a high degree of variation within Actinidia. Forty-seven simple sequence repeats (SSRs) and 155 repetitive structures were identified, further demonstrating the rapid evolution in Actinidia. The cp genome analysis and the identification of variable loci provide vital information for understanding the evolution and function of the chloroplast and for characterizing Actinidia population genetics.
.
DOI:10.1038/nmeth.2474URLPMID:23644548 [本文引用: 1]
We present a hierarchical genome-assembly process (HGAP) for high-quality de novo microbial genome assemblies using only a single, long-insert shotgun DNA library in conjunction with Single Molecule, Real-Time (SMRT) DNA sequencing. Our method uses the longest reads as seeds to recruit all other reads for construction of highly accurate preassembled reads through a directed acyclic graph-based consensus procedure, which we follow with assembly using off-the-shelf long-read assemblers. In contrast to hybrid approaches, HGAP does not require highly accurate raw reads for error correction. We demonstrate efficient genome assembly for several microorganisms using as few as three SMRT Cell zero-mode waveguide arrays of sequencing and for BACs using just one SMRT Cell. Long repeat regions can be successfully resolved with this workflow. We also describe a consensus algorithm that incorporates SMRT sequencing primary quality values to produce de novo genome sequence exceeding 99.999% accuracy.
.
DOI:10.1101/gr.215087.116URLPMID:28298431 [本文引用: 1]
Long-read single-molecule sequencing has revolutionized de novo genome assembly and enabled the automated reconstruction of reference-quality genomes. However, given the relatively high error rates of such technologies, efficient and accurate assembly of large repeats and closely related haplotypes remains challenging. We address these issues with Canu, a successor of Celera Assembler that is specifically designed for noisy single-molecule sequences. Canu introduces support for nanopore sequencing, halves depth-of-coverage requirements, and improves assembly continuity while simultaneously reducing runtime by an order of magnitude on large genomes versus Celera Assembler 8.2. These advances result from new overlapping and assembly algorithms, including an adaptive overlapping strategy based on tf-idf weighted MinHash and a sparse assembly graph construction that avoids collapsing diverged repeats and haplotypes. We demonstrate that Canu can reliably assemble complete microbial genomes and near-complete eukaryotic chromosomes using either Pacific Biosciences (PacBio) or Oxford Nanopore technologies and achieves a contig NG50 of >21 Mbp on both human and Drosophila melanogaster PacBio data sets. For assembly structures that cannot be linearly represented, Canu provides graph-based assembly outputs in graphical fragment assembly (GFA) format for analysis or integration with complementary phasing and scaffolding techniques. The combination of such highly resolved assembly graphs with long-range scaffolding information promises the complete and automated assembly of complex genomes.
.
DOI:10.1186/1471-2164-15-699URLPMID:25142801 [本文引用: 1]
The availability of diverse second- and third-generation sequencing technologies enables the rapid determination of the sequences of bacterial genomes. However, identifying the sequencing technology most suitable for producing a finished genome with multiple chromosomes remains a challenge. We evaluated the abilities of the following three second-generation sequencers: Roche 454 GS Junior (GS Jr), Life Technologies Ion PGM (Ion PGM), and Illumina MiSeq (MiSeq) and a third-generation sequencer, the Pacific Biosciences RS sequencer (PacBio), by sequencing and assembling the genome of Vibrio parahaemolyticus, which consists of a 5-Mb genome comprising two circular chromosomes.
.
DOI:10.1186/s12864-016-3412-9URLPMID:28061749 [本文引用: 1]
The development of long-read sequencing technologies, such as single-molecule real-time (SMRT) sequencing by PacBio, has produced a revolution in the sequencing of small genomes. Sequencing organelle genomes using PacBio long-read data is a cost effective, straightforward approach. Nevertheless, the availability of simple-to-use software to perform the assembly from raw reads is limited at present.
.
DOI:10.1186/1471-2105-13-238URLPMID:22988817 [本文引用: 1]
Recent methods have been developed to perform high-throughput sequencing of DNA by Single Molecule Sequencing (SMS). While Next-Generation sequencing methods may produce reads up to several hundred bases long, SMS sequencing produces reads up to tens of kilobases long. Existing alignment methods are either too inefficient for high-throughput datasets, or not sensitive enough to align SMS reads, which have a higher error rate than Next-Generation sequencing.
.
DOI:10.1093/bioinformatics/btp352URLPMID:19505943 [本文引用: 1]
The Sequence Alignment/Map (SAM) format is a generic alignment format for storing read alignments against reference sequences, supporting short and long reads (up to 128 Mbp) produced by different sequencing platforms. It is flexible in style, compact in size, efficient in random access and is the format in which alignments from the 1000 Genomes Project are released. SAMtools implements various utilities for post-processing alignments in the SAM format, such as indexing, variant caller and alignment viewer, and thus provides universal tools for processing read alignments.
.
DOI:10.1093/nar/gkh435URLPMID:15215342 [本文引用: 1]
Basic Local Alignment Search Tool (BLAST) is one of the most heavily used sequence analysis tools available in the public domain. There is now a wide choice of BLAST algorithms that can be used to search many different sequence databases via the BLAST web pages (http://www.ncbi.nlm.nih.gov/BLAST/). All the algorithm-database combinations can be executed with default parameters or with customized settings, and the results can be viewed in a variety of ways. A new online resource, the BLAST Program Selection Guide, has been created to assist in the definition of search strategies. This article discusses optimal search strategies and highlights some BLAST features that can make your searches more powerful.
.
DOI:10.1186/1471-2105-15-211URLPMID:24950923 [本文引用: 1]
The recent introduction of the Pacific Biosciences RS single molecule sequencing technology has opened new doors to scaffolding genome assemblies in a cost-effective manner. The long read sequence information is promised to enhance the quality of incomplete and inaccurate draft assemblies constructed from Next Generation Sequencing (NGS) data.
.
DOI:10.1093/bioinformatics/btq033URLPMID:20110278 [本文引用: 1]
Testing for correlations between different sets of genomic features is a fundamental task in genomics research. However, searching for overlaps between features with existing web-based methods is complicated by the massive datasets that are routinely produced with current sequencing technologies. Fast and flexible tools are therefore required to ask complex questions of these data in an efficient manner.
.
DOI:10.1186/2047-217X-1-18URLPMID:23587118 [本文引用: 1]
There is a rapidly increasing amount of de novo genome assembly using next-generation sequencing (NGS) short reads; however, several big challenges remain to be overcome in order for this to be efficient and accurate. SOAPdenovo has been successfully applied to assemble many published genomes, but it still needs improvement in continuity, accuracy and coverage, especially in repeat regions.
.
DOI:10.1101/gr.074492.107URLPMID:18349386 [本文引用: 1]
We have developed a new set of algorithms, collectively called "Velvet," to manipulate de Bruijn graphs for genomic sequence assembly. A de Bruijn graph is a compact representation based on short words (k-mers) that is ideal for high coverage, very short read (25-50 bp) data sets. Applying Velvet to very short reads and paired-ends information only, one can produce contigs of significant length, up to 50-kb N50 length in simulations of prokaryotic data and 3-kb N50 on simulated mammalian BACs. When applied to real Solexa data sets without read pairs, Velvet generated contigs of approximately 8 kb in a prokaryote and 2 kb in a mammalian BAC, in close agreement with our simulated results without read-pair information. Velvet represents a new approach to assembly that can leverage very short reads in combination with read pairs to produce useful assemblies.
.
DOI:10.1186/1756-0500-5-567URLPMID:23069129 [本文引用: 1]
As more and more reference genome sequences are assembled, it becomes practical to assemble individual genomes from large amount of raw read data based on a reference sequence. However, most available assembly tools are designed for de-novo genome assembly. There is one commercial tool box (Newbler) developed for re-sequencing projects based on the Roche 454 sequencing platform. However, the genome with large repeat regions cannot be well assembled in Newbler.
.
DOI:10.1093/bioinformatics/btu077URL [本文引用: 1]
Motivation: Transcriptome sequencing has long been the favored method for quickly and inexpensively obtaining a large number of gene sequences from an organism with no reference genome. Owing to the rapid increase in throughputs and decrease in costs of next- generation sequencing, RNA-Seq in particular has become the method of choice. However, the very short reads (e.g. 2 x 90 bp paired ends) from next generation sequencing makes de novo assembly to recover complete or full-length transcript sequences an algorithmic challenge.
Results: Here, we present SOAPdenovo-Trans, a de novo transcriptome assembler designed specifically for RNA-Seq. We evaluated its performance on transcriptome datasets from rice and mouse. Using as our benchmarks the known transcripts from these well-annotated genomes (sequenced a decade ago), we assessed how SOAPdenovo-Trans and two other popular transcriptome assemblers handled such practical issues as alternative splicing and variable expression levels. Our conclusion is that SOAPdenovo-Trans provides higher contiguity, lower redundancy and faster execution.
.
DOI:10.1038/nprot.2013.084URLPMID:23845962 [本文引用: 2]
De novo assembly of RNA-seq data enables researchers to study transcriptomes without the need for a genome sequence; this approach can be usefully applied, for instance, in research on 'non-model organisms' of ecological and evolutionary importance, cancer samples or the microbiome. In this protocol we describe the use of the Trinity platform for de novo transcriptome assembly from RNA-seq data in non-model organisms. We also present Trinity-supported companion utilities for downstream applications, including RSEM for transcript abundance estimation, R/Bioconductor packages for identifying differentially expressed transcripts across samples and approaches to identify protein-coding genes. In the procedure, we provide a workflow for genome-independent transcriptome analysis leveraging the Trinity platform. The software, documentation and demonstrations are freely available from http://trinityrnaseq.sourceforge.net. The run time of this protocol is highly dependent on the size and complexity of data to be analyzed. The example data set analyzed in the procedure detailed herein can be processed in less than 5 h.
.
DOI:10.1186/gb-2009-10-10-r103URLPMID:19796385 [本文引用: 1]
We demonstrate that genome sequences approaching finished quality can be generated from short paired reads. Using 36 base (fragment) and 26 base (jumping) reads from five microbial genomes of varied GC composition and sizes up to 40 Mb, ALLPATHS2 generated assemblies with long, accurate contigs and scaffolds. Velvet and EULER-SR were less accurate. For example, for Escherichia coli, the fraction of 10-kb stretches that were perfect was 99.8% (ALLPATHS2), 68.7% (Velvet), and 42.1% (EULER-SR).
.
DOI:10.1038/s41467-019-09575-2URLPMID:30979905 [本文引用: 1]
The ultimate goal for diploid genome determination is to completely decode homologous chromosomes independently, and several phasing programs from consensus sequences have been developed. These methods work well for lowly heterozygous genomes, but the manifold species have high heterozygosity. Additionally, there are highly divergent regions (HDRs), where the haplotype sequences differ considerably. Because HDRs are likely to direct various interesting biological phenomena, many genomic analysis targets fall within these regions. However, they cannot be accessed by existing phasing methods, and we have to adopt costly traditional methods. Here, we develop a de novo haplotype assembler, Platanus-allee ( http://platanus.bio.titech.ac.jp/platanus2 ), which initially constructs each haplotype sequence and then untangles the assembly graphs utilizing sequence links and synteny information. A comprehensive benchmark analysis reveals that Platanus-allee exhibits high recall and precision, particularly for HDRs. Using this approach, previously unknown HDRs are detected in the human genome, which may uncover novel aspects of genome variability.
.
DOI:10.3390/genes9080383URLPMID:30061537 [本文引用: 2]
Platycodongrandiflorus (balloon flower) and Codonopsislanceolata (bonnet bellflower) are important herbs used in Asian traditional medicine, and both belong to the botanical family Campanulaceae. In this study, we designed and implemented a de novo DNA sequencing and assembly strategy to map the complete mitochondrial genomes of the first two members of the Campanulaceae using low-coverage Illumina DNA sequencing data. We produced a total of 28.9 Gb of paired-end sequencing data from the genomic DNA of P.grandiflorus (20.9 Gb) and C.lanceolata (8.0 Gb). The assembled mitochondrial genome of P.grandiflorus was found to consist of two circular chromosomes; the master circle contains 56 genes, and the minor circle contains 42 genes. The C.lanceolata mitochondrial genome consists of a single circle harboring 54 genes. Using a comparative genome structure and a pattern of repeated sequences, we show that the P.grandiflorus minor circle resulted from a recombination event involving the direct repeats of the master circle. Our dataset will be useful for comparative genomics and for evolutionary studies, and will facilitate further biological and phylogenetic characterization of species in the Campanulaceae.
.
DOI:10.1093/bioinformatics/btv033URLPMID:25609793 [本文引用: 1]
MEGAHIT is a NGS de novo assembler for assembling large and complex metagenomics data in a time- and cost-efficient manner. It finished assembling a soil metagenomics dataset with 252?Gbps in 44.1 and 99.6?h on a single computing node with and without a graphics processing unit, respectively. MEGAHIT assembles the data as a whole, i.e. no pre-processing like partitioning and normalization was needed. When compared with previous methods on assembling the soil data, MEGAHIT generated a three-time larger assembly, with longer contig N50 and average contig length; furthermore, 55.8% of the reads were aligned to the assembly, giving a fourfold improvement.
.
[本文引用: 1]
.
DOI:10.1093/bioinformatics/btu170URL [本文引用: 1]
Motivation: Although many next-generation sequencing (NGS) read preprocessing tools already existed, we could not find any tool or combination of tools that met our requirements in terms of flexibility, correct handling of paired-end data and high performance. We have developed Trimmomatic as a more flexible and efficient preprocessing tool, which could correctly handle paired-end data.
Results: The value of NGS read preprocessing is demonstrated for both reference-based and reference-free tasks. Trimmomatic is shown to produce output that is at least competitive with, and in many cases superior to, that produced by other tools, in all scenarios tested.
.
DOI:10.1038/nbt.1883URLPMID:21572440 [本文引用: 1]
Massively parallel sequencing of cDNA has enabled deep and efficient probing of transcriptomes. Current approaches for transcript reconstruction from such data often rely on aligning reads to a reference genome, and are thus unsuitable for samples with a partial or missing reference genome. Here we present the Trinity method for de novo assembly of full-length transcripts and evaluate it on samples from fission yeast, mouse and whitefly, whose reference genome is not yet available. By efficiently constructing and analyzing sets of de Bruijn graphs, Trinity fully reconstructs a large fraction of transcripts, including alternatively spliced isoforms and transcripts from recently duplicated genes. Compared with other de novo transcriptome assemblers, Trinity recovers more full-length transcripts across a broad range of expression levels, with a sensitivity similar to methods that rely on genome alignments. Our approach provides a unified solution for transcriptome reconstruction in any sample, especially in the absence of a reference genome.
.
DOI:10.1016/S0022-2836(05)80360-2URLPMID:2231712 [本文引用: 1]
A new approach to rapid sequence comparison, basic local alignment search tool (BLAST), directly approximates alignments that optimize a measure of local similarity, the maximal segment pair (MSP) score. Recent mathematical results on the stochastic properties of MSP scores allow an analysis of the performance of this method as well as the statistical significance of alignments it generates. The basic algorithm is simple and robust; it can be implemented in a number of ways and applied in a variety of contexts including straightforward DNA and protein sequence database searches, motif searches, gene identification searches, and in the analysis of multiple regions of similarity in long DNA sequences. In addition to its flexibility and tractability to mathematical analysis, BLAST is an order of magnitude faster than existing sequence comparison tools of comparable sensitivity.
.
DOI:10.1093/nar/gks499URLPMID:22649055 [本文引用: 1]
Enriching target sequences in sequencing libraries via capture hybridization to bait/probes is an efficient means of leveraging the capabilities of next-generation sequencing for obtaining sequence data from target regions of interest. However, homologous sequences from non-target regions may also be enriched by such methods. Here we investigate the fidelity of capture enrichment for complete mitochondrial DNA (mtDNA) genome sequencing by analyzing sequence data for nuclear copies of mtDNA (NUMTs). Using capture-enriched sequencing data from a mitochondria-free cell line and the parental cell line, and from samples previously sequenced from long-range PCR products, we demonstrate that NUMT alleles are indeed present in capture-enriched sequence data, but at low enough levels to not influence calling the authentic mtDNA genome sequence. However, distinguishing NUMT alleles from true low-level mutations (e.g. heteroplasmy) is more challenging. We develop here a computational method to distinguish NUMT alleles from heteroplasmies, using sequence data from artificial mixtures to optimize the method.
[本文引用: 1]
[本文引用: 1]
.
DOI:10.1371/journal.pone.0179971URLPMID:28662089 [本文引用: 2]
Mitogenome sequences are highly desired because they are used in several biological disciplines. Their elucidation has been facilitated through the development of massive parallel sequencing, accelerating their deposition in public databases. However, sequencing, assembly and annotation methods might induce variability in their quality, raising concerns about the accuracy of the sequences that have been deposited in public databases. In this work we show that different sequencing methods (number of species pooled in a library, insert size and platform) and assembly and annotation methods generated variable completeness and similarity of the resulting mitogenome sequences, using three species of predaceous ladybird beetles as models. The identity of the sequences varied considerably depending on the method used and ranged from 38.19 to 90.1% for Cycloneda sanguinea, 72.85 to 91.06% for Harmonia axyridis and 41.15 to 93.60% for Hippodamia convergens. Dissimilarities were frequently found in the non-coding A+T rich region, but were also common in coding regions, and were not associated with low coverage. Mitogenome completeness and sequence identity were affected by the sequencing and assembly/annotation methods, and high within-species variation was also found for other mitogenome depositions in GenBank. This indicates a need for methods to confirm sequence accuracy, and guidelines for verifying mitogenomes should be discussed and developed by the scientific community.
.
DOI:10.1111/j.1365-294X.2012.05612.xURL [本文引用: 1]
Genetic studies of waterfowl (Anatidae) have observed the full spectrum of mitochondrial (mt) DNA population divergence, from apparent panmixia to deep, reciprocally monophyletic lineages. Yet, these studies often found weak or no nuclear (nu) DNA structure, which was often attributed to male-biased gene flow, a common behaviour within this family. An alternative explanation for this conflict is that the smaller effective population size and faster sorting rate of mtDNA relative to nuDNA lead to different signals of population structure. We tested these alternatives by sequencing 12 nuDNA introns for a Holarctic pair of waterfowl subspecies, the European goosander (Mergus merganser merganser) and the North American common merganser (M. m. americanus), which exhibit strong population structure in mtDNA. We inferred effective population sizes, gene flow and divergence times from published mtDNA sequences and simulated expected differentiation for nuDNA based on those histories. Between Europe and North America, nuDNA Phi(ST) was 3.4-fold lower than mtDNA Phi(ST), a result consistent with differences in sorting rates. However, despite geographically structured and monophyletic mtDNA lineages within continents, nuDNA Phi(ST) values were generally zero and significantly lower than predicted. This between- and within-continent contrast held when comparing mtDNA and nuDNA among published studies of ducks. Thus, male-mediated gene flow is a better explanation than slower sorting rates for limited nuDNA differentiation within continents, which is also supported by nonmolecular data. This study illustrates the value of quantitatively testing discrepancies between mtDNA and nuDNA to reject the null hypothesis that conflict simply reflects different sorting rates.
.
DOI:10.1186/1471-2164-15-467URLPMID:24923674 [本文引用: 1]
Genome and transcriptome sequencing applications that rely on variation in sequence depth can be negatively affected if there are systematic biases in coverage. We have investigated patterns of local variation in sequencing coverage by utilising ultra-deep sequencing (>100,000X) of mtDNA obtained during sequencing of two vertebrate genomes, wolverine (Gulo gulo) and collared flycatcher (Ficedula albicollis). With such extreme depth, stochastic variation in coverage should be negligible, which allows us to provide a very detailed, fine-scale picture of sequence dependent coverage variation and sequencing error rates.

{kind=link}
{kind=link}