 ,2
,2Whole genome sequencing reveals the distribution of resistance and virulence genes of pathogenic Escherichia coli CCHTP from giant panda
Wenwen Deng1, Caiwu Li1, Siyue Zhao2, Rengui Li1, Yongguo He1, Daifu Wu1, Shengzhi Yang2, Yan Huang1, Hemin Zhang1, Likou Zou,2通讯作者: 邹立扣,博士,教授,博士生导师,研究方向:微生物资源利用、细菌耐药性。E-mail:zoulikou@sicau.edu.cn
编委: 谢建平
收稿日期:2019-09-17修回日期:2019-11-25网络出版日期:2019-12-20
| 基金资助: |
Editorial board:
Received:2019-09-17Revised:2019-11-25Online:2019-12-20
| Fund supported: |
作者简介 About authors
邓雯文,硕士研究生,研究方向:大熊猫分子遗传学。E-mail:dwenwen130@163.com。
李才武,硕士研究生,高级工程师,研究方向:大熊猫临床兽医。E-mail:83330019@qq.com;邓雯文和李才武并列第一作者。。

摘要
致病性大肠杆菌是引起动物泌尿系统感染的重要病原菌,本研究对泌尿生殖道感染出现潜血的大熊猫尿液中分离的一株致病性大肠杆菌(Escherichia coli CCHTP)进行全基因组测序,检测其中耐药基因和毒力因子的情况,同时对基因岛上耐药和毒力基因及其基因环境进行研究。研究发现,大肠杆菌CCHTP中存在多种类型的耐药基因,其中外排泵系统基因数量最多,包括mdfA、emrE和mdtN等介导多重耐药外排泵的基因。此外,该菌还携带166种毒力因子及563个相关毒力基因,其中属于黏附与侵袭类的毒力因子及相关基因数量最多。对19个基因岛分析发现,基因岛GIs011和GIs017中各有一段包含耐药和毒力基因的序列,两侧与可移动遗传元件(转座酶和插入序列)相连,这些结构可能介导耐药及毒力基因水平转移。本研究通过全基因组测序分析了大熊猫源致病性大肠杆菌中存在的耐药及毒力基因情况,对大熊猫相关疾病的科学治疗、合理用药有重要意义。
关键词:
Abstract
Pathogenic Escherichia coli (E. coli) is the most common pathogen causing urinary tract infection in animals. We investigated the antibiotic resistance and virulence genes of pathogenic E. coli CCHTP derived from urine with occult blood of the giant panda by whole genome sequencing. The flanking sequencing of resistance and virulence genes in genomic islands were also analyzed. Our results demonstrate that E. coli CCHTP contains different families of antibiotic resistance genes, most of which are efflux pump related genes, including multiple drug resistance efflux pump genes mdfA, emrE, and mdtN. A total of 166 virulence factors and 563 virulence genes were identified, and the most virulence factors and related genes are involved in host cell attachment and invasion processes. Furthermore, sequence analysis of 19 genomic islands revealed that antibiotic and virulence genes are associated with mobile genetic elements (transposon and insertion sequence) in GIs011 and GIs017. These structures can mediate horizontal transfer of antibiotic and virulence genes. Our work described the distribution of antibiotic resistance genes and virulence genes in E. coli CCHTP, which may provide an important guidance for treatment and rational drug use of E. coli CCHTP infection in the giant panda.
Keywords:
PDF (566KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
邓雯文, 李才武, 赵思越, 李仁贵, 何永果, 吴代福, 杨盛智, 黄炎, 张和民, 邹立扣. 大熊猫源致病大肠杆菌CCHTP全基因组测序及耐药和毒力基因分析. 遗传[J], 2019, 41(12): 1138-1147 doi:10.16288/j.yczz.19-243
Wenwen Deng.
大肠杆菌(Escherichia coli)通常存在于人和动物的胃肠道,属于条件性致病菌,在宿主健康的情况下,可起到保护宿主、防止病原菌定植的作用,但在特定情况下会产生致病性[1]。此外,致病性大肠杆菌还可在胃肠道以外,如尿道、血液中生存并引起严重的疾病,它们具有广泛的宿主谱,其防治仍是当今世界性的难题,对大熊猫感染性疾病的发生有重要影响[2,3]。
目前,用于治疗大熊猫细菌感染的药物主要为抗生素,抗生素通过直接杀死或抑制病原菌的生长来达到治疗效果,这种作用机理使细菌具有选择压力,并催生病原菌耐药性的产生[4]。同时,由于耐药基因的水平转移特性使耐药性可在不同细菌种属间进行传播,加速了耐药水平的提高。研究表明大熊猫源大肠杆菌已对不同大类抗生素,如四环素类、磺胺类和β-内酰胺类抗生素呈现出不同程度耐药[5]。此外,毒力因子是病原菌具有的能够使其在宿主环境中定植、繁殖和致病的基因产物[6]。大肠杆菌的致病性是由多种毒力相关因子共同决定,这些毒力因子相互协调,直接或间接参与与致病过程,进而引发宿主有害炎症反应。近年来随着基因组学的飞速发展,可从分子水平对毒力因子进行研究,揭示病原菌致病机理[7]。
目前关于大熊猫源大肠杆菌的研究主要集中在对大熊猫肠道内大肠杆菌的分离鉴定、耐药性检测等[3,5,8],但有关大熊猫肠外大肠杆菌相关的研究报道还较少。本实验通过对1株从泌尿生殖道感染出现潜血的大熊猫尿液中分离的大肠杆菌进行全基因测序,研究耐药及毒力因子情况,分析耐药及毒力相关基因的环境,可对大肠杆菌致病机理研究提供理论基础,为合理采用抗生素治疗大熊猫大肠杆菌感染提供依据。
1 材料与方法
1.1 菌株来源
2017年5月从中国大熊猫保护研究中心一只泌尿生殖道感染出现潜血的大熊猫尿液中分离得到的大肠杆菌。1.2 菌株纯化鉴定
大肠杆菌使用EMB培养基和TSA划线纯化,将纯化菌株腹腔注射小白鼠进行致病性实验[9],确认后按文献[1]的方法进行形态学和16S rDNA测序再次鉴定确认,鉴定结果为大肠杆菌(Escherichia coli CCHTP)。挑取单菌落,使用TSB培养基培养18~20 h,取1.8 mL过夜孵育的菌液到一个2 mL无菌离心管中,离心后吸掉上清培养基,获取大肠杆菌沉淀用于后续DNA提取。1.3 抗生素药敏试验
根据美国临床和实验室标准协会(Clinical and Laboratory Standards Institute, CLSL) 2019年发布的《抗菌药物敏感性试验执行标准》[10],采用K-B药敏纸片法,选择15种抗生素测定菌株的敏感性。利用无菌生理盐水稀释菌落制备菌悬浮液至0.5麦氏浓度,用无菌棉签蘸取菌液涂布于MHA琼脂平板上,镊子灭菌后分别夹取药敏纸片,按一定间隔贴在平板的不同区域,37℃恒温培养16~18 h。按CLSI标准判定耐药性。选择E. coli ATCC?25922, E. coli ATCC? 35218作为质控菌株。1.4 DNA提取
根据UltraClean?Microbial DNA Isolation Kit试剂盒手册,提取细菌DNA。取60 ng DNA样品1.0%琼脂糖凝胶电泳,在80V电压电泳30 min,检测DNA浓度。1.5 全基因组测序
将DNA送往北京诺禾致源生物信息科技有限公司(Novogene)进行3代Pacbio平台测序。经电泳检测合格的DNA样品用Covaris g-TUBE打断成构建文库所需大小的目的片段,经DNA损伤修复及末端修复后,使用DNA黏合酶将发卡型接头连接在DNA片段两端,使用AMpure PB磁珠对DNA片段进行纯化选择,构建SMRT Bell文库。纯化后的片段经buffer回溶后,使用BluePipin片段筛选特定大小的片段,并使用AMpure PB磁珠对DNA片段进行纯化。构建好的文库经Qubit浓度定量,并利用Agilent 2100检测插入片段大小,随后用PacBio平台进行测序。1.6 数据处理
对原始数据进行过滤处理,得到有效数据(clean data)。使用SMRT Link v5.0.1软件对reads进行组装[11,12],得到初步组装结果,把reads比对到组装序列上,统计测序深度的分布情况。将得到的初步组装结果进行比对分析,并将染色体序列组装成为一个环状基因组,即最终的0 gap完成图序列。使用GeneMarkS软件(2 结果与分析
2.1 药敏实验结果
药敏实验结果显示,获得的大肠杆菌CCHTP对阿莫西林、氨苄西林、阿莫西林/克拉维酸、头孢克洛、头孢曲松、头孢噻肟、庆大霉素、红霉素、阿奇霉素、氧氟沙星、诺氟沙星、恩诺沙星和四环素耐药,对氨曲南和卡那霉素敏感(附表1)。2.2 大肠杆菌CCHTP全基因组测序结果
大肠杆菌CCHTP经PacBio测序平台进行全基因组测序后,通过对所测序列进行组装,最终获得1个contig,组装的contigs总长度为5120 663 bp,N50为5120 663 bp。通过组装获得一个长度为5106 047 bp的环状染色体序列(图1),鸟嘌呤-胞嘧啶(GC)含量为50.49%对基因组编码基因预测,注释得到的基因总数为4896,注释得到的基因总长度为4470 000 bp。对基因组非编码RNA (ncRNA)预测,得到61个sRNA、22个rRNA (包括5S、16S、23S)和89个tRNA。图1
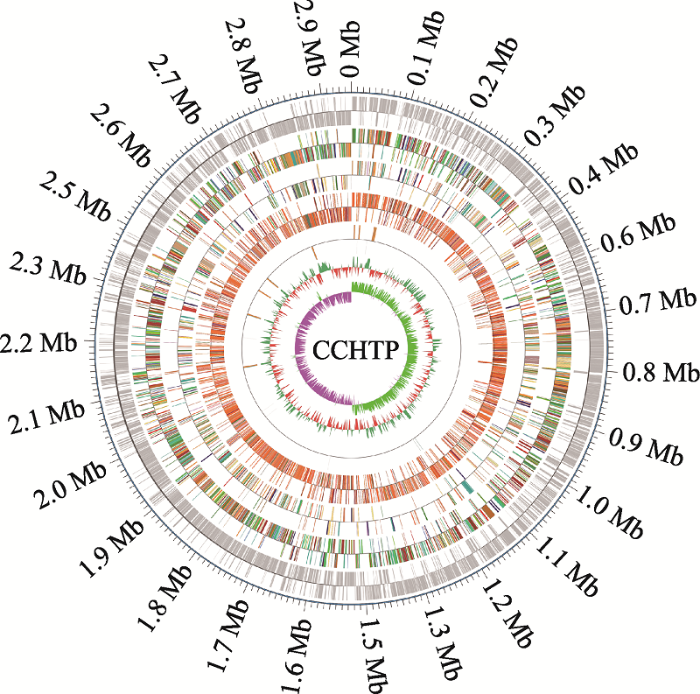 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1大肠杆菌CCHTP基因组图谱
从外圈至内圈:1、2圈为编码基因;3、4圈为eggNOG;4、5圈为KEGG;7、8圈为GO;9、10圈为ncRNA;11、12圈为基因组GC含量;13、14圈为基因组GC skew值分布。
Fig. 1The chromosome genome of E. coli CCHTP
经预测,共有4431个基因具有COG功能分类,其中信息储存和处理基因共745个,细胞过程和信号转导基因共1194个,代谢类基因共1945个,功能未知基因547个。菌株共有13 862个基因在GO数据库3大分支数据库中被注释到。共有43种功能注释结果,其中细胞学组件类有12个分支,共3360个注释结果,生物学途径类有23个分支,共7432基因注释结果,分子功能类存在10个分支共4107个相关注释结果。KEGG分析显示,共1544 个基因富集在 192条代谢通路中,其中涉及基因最多的通路主要有ABC转运蛋白代谢通路(ko02010) (200个基因)、双组分调节系统代谢通(ko02060) (142个基因)和嘌呤代谢通路(ko00230) (83个基因)。
2.3 大肠杆菌CCHTP中抗生素耐药基因分析
通过对染色体基因序列进行比对,发现大肠杆菌携带了多种类型的抗生素耐药基因,包括14大类161个抗生素耐药基因。从表1中可知,外排泵系统基因数量最多,主要包括macB (13个)、adeL (6个)、patA (6个)、evgS (5个)等115个耐药基因。其次为介导糖肽类抗生素(万古霉素)耐药的vanTG/ TC/RI等10个耐药基因、介导多肽类抗生素(多粘菌素)耐药的mcr-3、PmrF/E/C等9个耐药基因和介导四环素类抗生素(四环素)耐药的tetT/B(60)/A(48)/ 34。此外,少量介导喹诺酮类、磺胺类和β-内酰胺类等抗生素耐药的基因也在基因组中识别到。Table 1
表1
表1大肠杆菌CCHTP中耐药基因分析结果
Table 1
| 耐抗生素大类 | 耐药基因 | 基因数量 |
|---|---|---|
| 外排泵 | macB、adeL、patA、evgS、mdtF、 patB、sav1866和TaeA等 | 115 |
| 糖肽类 | vanTG、vanTC、vanRI、vanRF、vanRE、 vanRB、vanHD、vanG、baeS和baeR | 12 |
| 多肽类 | pmrF、pmrE、pmrC、mcr-3、 basS、arnA和arlS | 9 |
| 四环素类 | tetT、tetB(60)、tetA(48)和tet34 | 7 |
| 喹诺酮类 | gyrB、gyrA和mfd | 4 |
| 抗结核药 | ndh、katG和kasA | 3 |
| 磺胺类 | sul3和leuO | 2 |
| β-内酰胺类 | mecC和blaCMY-63 | 2 |
| 多磷类 | glpT和murA | 2 |
| 大环内酯类 | chrB | 1 |
| 环脂肽类 | cls | 1 |
| 肽类抗生素 | bacA | 1 |
| 林可酰胺类 | clbB | 1 |
| 利福霉素类 | rphB | 1 |
新窗口打开|下载CSV
2.4 大肠杆菌CCHTP中毒力因子分析
经VFDB数据库比对,发现大肠杆菌CCHTP共携带了166种毒力因子及563个相关毒力基因。按毒力因子大类(黏附与侵袭、分泌系统、铁转运和细菌毒素)来分析,发现属于黏附与侵袭类的毒力因子及相关基因数量最多,共有47种毒力因子及239个相关毒力基因,毒力基因数量前15的黏附与侵袭类毒力因子如图2所示,主要黏附因子包括5种菌毛类:Type I fimbriae (Ⅰ型菌毛)、Type Ⅳ pili (Ⅳ型菌毛)、Pix pilus (Pix菌毛)、Pap pilus (盂肾炎相关菌毛/P菌毛)和E.coli common pilus (大肠杆菌共生菌毛)和3种鞭毛类:Peritrichous flagella (周生鞭毛)、S fimbriae (S鞭毛)和P fimbriae (P鞭毛)。研究发现Peritrichous flagella相关毒力基因数量最多,包括filZ/Y/T/S、flhA/B/C/D、flgN/M/L等51个毒力基因,其次为LOS (脂寡糖)基因族中的rfaF/E/D、lpxK/H/ D/C等25个基因,Capsule (荚膜)基因族中的rmlA、oppF、lipA等15个基因和TypeⅠ fimbriae基因族中的fimA/B/C等12个基因。图2
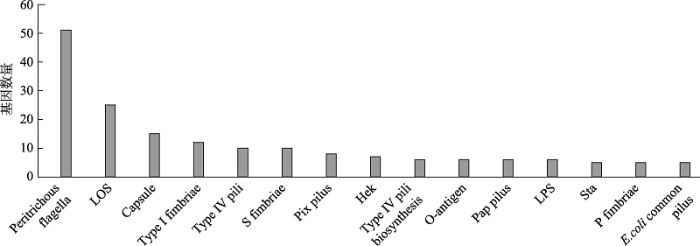 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2黏附与侵袭类主要毒力因子
Fig. 2Main virulence factors related to host cell attachment and invasion processes
分泌系统共发现16种毒力因子及76个相关毒力基因(图3)。按照分泌系统类型来看,共识别到5类,属于Ⅱ型分泌系统的毒力因子类型最多,包括gsp、T2SS、out、pul、GspA和exe,其次为Ⅵ型分泌系统,包括ACE T6SS、SCI-I T6SS、CTS2和T6SS。其余3类分泌系统各识别到两种毒力因子,分别为Trehalose-recycling ABC transporter和ABC transporter (Ⅰ型分泌系统)、T3SS和IcsP (SopA) (Ⅲ型分泌系统)、CTS2和T6SS (Ⅵ型分泌系统)。与ACE T6SS相关的毒力基因数量最多,主要为假定蛋白编码基因aec17/18/22/23/26等19个基因,其次为SCI-I T6SS基因族中的c3402、ECP_2814和LF82_ 443等17个基因。
图3
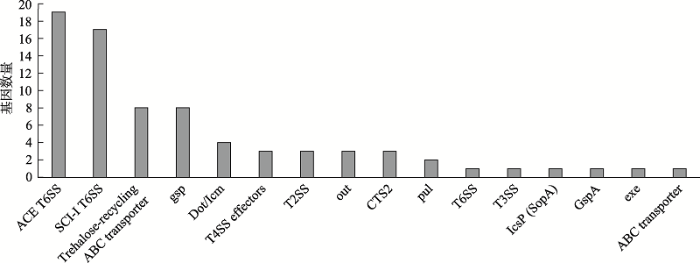 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3分泌系统类主要毒力因子
Fig. 3Main virulence factors of bacterial secretion systems
本研究共识别12种毒力因子及59个相关毒力基因属于铁转运系统。如图4所示,与毒力因子Enterobactin (肠细菌素)和血红素合成(Heme biosynthesis)相关的基因数量最多。其次为Yersiniabactin siderophore (耶尔森菌素合成)、Yersiniabactin (耶尔森菌素)及Salmochelin (沙门菌素)。
图4
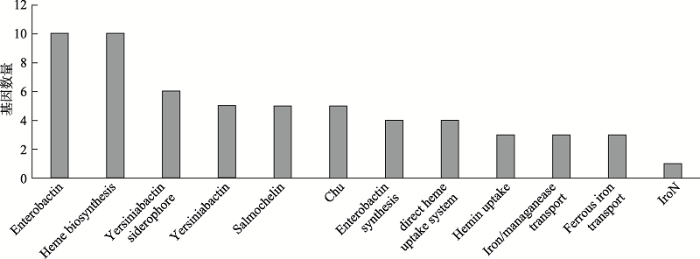 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4铁转运主要毒力因子
Fig. 4Main virulence factors of iron-acquisition systems
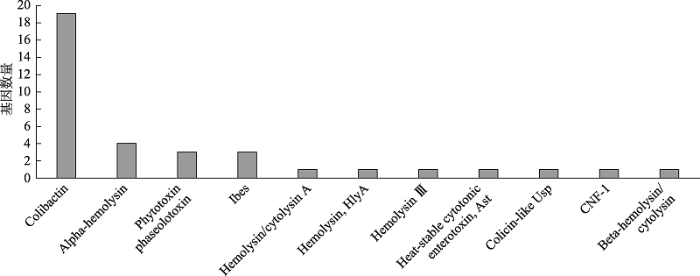
如图5,通过比对得到细菌毒素类毒力因子11种,毒力基因36个。毒力因子主要包括5种溶血素:Alpha-hemolysin (α溶血素)、cytolysin A (溶血素A)、Hemolysin, HlyA (HlyA溶血素)、Hemolysin Ⅲ (Ⅲ型溶血素)和Beta-hemolysin (β溶血素)。研究发现Colibactin (聚酮肽基因毒素)相关毒力基因数量最多, 包括clbR/Q/P/O/N等19个基因。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5细菌毒素主要毒力因子
Fig. 5Main virulence factors of bacterial toxins
2.5 大肠杆菌E. coli CCHTP中耐药和毒力基因环境分析
在染色体序列共发现19个基因岛,总长在5187~74 031 bp之间,其中发现两个基因岛GIs011和GIs017上各有一段含可移动遗传元件(插入序列)、毒力因子和耐药基因的序列,基因环境绘图如图6所示,在基因岛GIs011上发现一段序列包含外排泵基因调控因子(emrR)和四环素类耐药基因(tetA(60)),F1C菌毛(focY/H)和S菌毛(sfaG/F/E/D/A/B/C) 相关基因,在这些基因两侧存在可移动遗传元件IS630、转座酶基因及IS3fA。GIs017上也发现在一段包含外排泵基因cpxR、α-溶血素基因hlyA/B/C/D及细菌毒素CNF-1,两侧分别与转座酶基因、插入序列IS3fB以及IS66相连,大肠杆菌中的耐药或毒力基因可由可移动遗传元件介导传播。图6
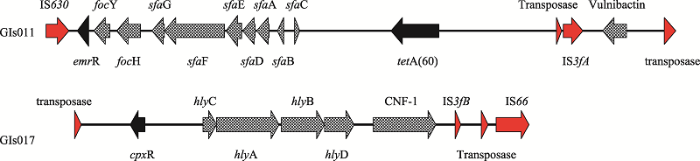 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6抗生素耐药基因和毒力基因环境
Fig. 6Genetic environment of antibiotic resistance and virulence genes
此外,对多肽类耐药基因mcr-3及其侧翼环境进行分析(图7),发现基因mcr-3一侧与假定抗性蛋白(putative resistance protein, PRP),并具有假定蛋白(hypothetical protein, HP) YiaG。另一侧与假定蛋白相连,并发现Capsule类毒力基因oppF与ABC转运器和假定蛋白相连,另外在基因mcr-3周围序列中还发现chu类毒力基因chuA和chuT以及外排泵基因adeL、gadX、gadW和mdtE。
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7mcr-3基因环境
PRP: putative resistance protein,假定抗性蛋白;HP: hypothetical protein,假定蛋白。
Fig. 7Genetic environment of mcr-3
3 讨论
许多已知的病原菌都能引起尿路感染,如白色念珠菌(Candida albicans)、肺炎克雷伯氏菌(Klebsiella pneumoniae)、铜绿假单胞菌(Pseudomonas aeruginosa)、奇异变形杆菌(Proteus mirabilis)、金黄色葡萄球菌(Staphylococcus saprophyticus)和大肠杆菌等[15,16]。然而,目前对大熊猫泌尿生殖道感染的研究较少,仅发现肺炎克雷伯氏菌可引起大熊猫的泌尿生殖道血尿症出现[17]。本研究从一只泌尿生殖道感染出现潜血的大熊猫尿液中分离出大肠杆菌CCHTP。通过全基因组测序,发现该菌携带多种类型的抗生素耐药基因,并以外排泵基因为主,表明外排泵系统可能为大肠杆菌CCHTP产生耐药性的主要因素。此外,本研究还发现mdfA、emrE、mdtK和hmrM等多重耐药外排泵系统基因。目前已有大量研究表明多重耐药外排泵能够介导对抗生素、消毒剂、洗涤剂和染料等有毒化合物的内在抗性[18],阻止药物在细菌体内积聚,是细菌多重耐药性产生的主要机制之一[19]。其中,mdfA外排泵能够外排环丙沙星,卡那霉素、新霉素及季铵盐类消毒剂等[20,21],mdtK基因可表达细菌对诺氟沙星、阿霉素及吖啶黄的外排,emrE编码的外排泵能外排四环素、红霉素、结晶紫及染色剂溴化乙锭[22]。此外,研究检测到四环素类、磺胺类和β-内酰胺类耐药基因,这与部分****在大熊猫粪便源大肠杆菌检测到耐药基因结果相似[5,23]。四环素类抗生素广泛用于动物疾病治疗 中,目前已发现40多种不同类型的四环素耐药基 因,主要介导3种耐药机制,分别为外排泵机制、核糖体保护机制和酶降解机制[24],其中基因tetA和tetB可编码主要易化子超家族(MFS)外排泵蛋白将四环素药物泵出细胞外,降低细胞内药物浓度,四环耐药基因tetT通过编码核糖体保护机制,使耐药细菌可以产生核糖体保护蛋白与核糖体结合引起构型改变,从而弱化了对二甲胺四环素和强力霉素的抑制作用,导致耐药性的产生[25]。研究还发现多粘菌素耐药基因mcr-3,这种新型基因在2017年被Yin等[26]首次发现于大肠杆菌的IncHI2质粒中,其在大肠杆菌中的表达可有效阻止多粘菌素所引发的活性氧形成,并一定程度地对宿主细菌造成一定的代谢压力。研究表明基因mcr-3的氨基酸序列与前期发现的多粘菌素基因mcr-1和mcr-2差异较大,仅有32.5%和31.7%的一致性[27],具有多种亚型(如mcr-3.3、mcr-3.5和mcr-3.7),宿主范围广,主要发现于大肠杆菌、肺炎克雷伯氏菌和鼠伤寒沙门氏菌中[28]。基因mcr-3可由染色体和质粒介导,通过质粒、插入序列、转座子等方式介导传播[27,29]。Ling等[29]对气单胞菌中有染色体介导的mcr-3.3基因环境进行分析,与本实验中mcr-3基因环境对比发现具有部分相同的序列结构,基因两侧均有假定蛋白,且其中一侧与假定蛋白相连。但仍存在差异,表现为基因mcr-3.3周围序列中发现插入序列遗传标记ISAsI。此外,mcr-3基因可与其他耐药基因共存[28,30,31],Liu等[27]发现由mcr-3位于IncP质粒中的TnAs3转座子上,并与IncHI2质粒携带的基因mcr-1和IncX3质粒携带的基因blaNDM-5共存于一株大肠杆菌中,引起多重耐药,进一步增强宿主细菌的耐药性。近年来随着人们对于各种毒力因子的逐步了解,一些毒力因子的致病机理也逐渐被揭示出来。通过与VFDB数据库比对,发现大肠杆菌E. coli CCHTP携带了大量毒力因子及相关毒力基因。例如属于黏附与侵袭类毒力因子的Ⅰ型菌毛,其普遍存在于大肠杆菌中,主要在大肠杆菌致病的第一阶段起作用[32,33]。Vizcarra等[33]研究显示Ⅰ型菌毛还能产生屏蔽效应增强细胞内细菌对抗生素的抵抗能力,造成慢性感染发生,表明Ⅰ型菌毛对细菌抗生素耐药性的产生有一定影响。研究检测到了fimA/B/C/D/H等12个编码Ⅰ型菌毛的基因,其中fimC和fimD主要在协调菌毛装配及细菌致病过程中起作用,fimH介导 粘附作用[32]。此外,Ⅰ型菌毛和P菌毛都能介导大肠杆菌黏附于上尿道,也是尿道感染发生的重要因素[33,34]。此外,细菌毒素类包含多种溶血素,其中α溶血素毒力基因hlyA具有广谱杀细胞作用,可作用于红细胞、肾脏上皮细胞和免疫细胞,诱导细胞凋亡[35]。Reijer等[36]研究表明溶血素在金黄色葡萄球菌生物膜形成等致病过程中也起着重要作用,增加了金葡菌的危害性。编码毒素Colibactin相关毒力的基因数量最多,该毒素可导致慢性有丝分裂、染色体畸变和基因突变频率增加,能够引发肿瘤[37]。本研究通过对基因岛分析发现,大量抗生素耐药及毒力基因两侧存在插入序列。 研究表明插入序列是染色体特殊组成部分,可携带耐药基因转移,同时插入序列可通过自身携带的转座子提高耐药基因表达量从而提高细菌耐药程度[38,39]。因此,研究结果提示大肠杆菌中耐药或毒力基因可能通过可移动遗传元件介导传播。
分泌系统和铁转运系统虽未直接涉及致病过程,但对细菌致病性的产生也至关重要。目前在革兰氏阴性菌中已经发现并命名9种不同类型的分泌系统(Ⅰ~Ⅸ型),致病菌借助分泌系统将特异蛋白直接注入宿主细胞内,可导致细菌的定殖和感染[40]。在本研究共识别到5类(Ⅰ~Ⅳ型),其中Ⅲ型分泌系统广泛存在于革兰氏阴性致病菌,如耶尔森氏菌属、沙门氏菌属、大肠杆菌、副溶血性弧菌等中[7,41]。沙门菌素和肠杆菌素是肠杆菌科病原菌中常见的铁载体,其中肠杆菌素主要通过于血浆转铁蛋白中的铁结合来促进细菌生长而引起感染,与其他已知的铁载体蛋白相比具有最高的铁亲和力[42]。
综上所述,本研究通过全基因组测序技术首次对泌尿生殖道感染出现潜血的大熊猫尿液中分离的大肠杆菌进行检测,主要研究了其中毒力因子及耐药基因情况,可较为全面地预测大肠杆菌中耐药基因、耐药谱、毒力因子及相关毒力基因等重要致病特性。研究结果可为圈养大熊猫临床治疗和合理用药提供指导,为后续开展圈养大熊猫源病原菌中毒力因子和耐药基因研究提供依据。
附录:
附表1详见文章电子版www.chinagene.cn。Supplementary Table 1
附录:
附录:大肠杆菌CCHTP耐药情况
Supplementary Table 1
| 类别 | 抗生素 | 敏感(S) | 耐药(R) |
|---|---|---|---|
| 四环素类 | 四环素 | R | |
| β内酰胺抑制剂 | 阿莫西林/克拉维酸 | S | |
| 单环内酰胺类 | 氨曲南 | S | |
| 氨基糖苷类 | 卡那霉素 | R | |
| 庆大霉素 | R | ||
| 大环内酯类 | 红霉素 | R | |
| 阿奇霉素 | R | ||
| 喹诺酮类 | 诺氟沙星 | R | |
| 氧氟沙星 | R | ||
| 恩诺沙星 | R | ||
| β-内酰胺类 | 阿莫西林 | R | |
| 氨苄西林 | R | ||
| 头孢克洛 | R | ||
| 头孢噻肟 | R | ||
| 头孢曲松 | R |
新窗口打开|下载CSV
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 2]
DOI:10.1128/JB.06375-11URL [本文引用: 1]

Escherichia coli strains that cause disease outside the intestine are known as extraintestinal pathogenic E. coli (ExPEC) and include pathogens of humans and animals. Previously, the genome of avian-pathogenic E. coil (APEC) O1:K1:H7 strain O1, from ST95, was sequenced and compared to those of several other E. coli strains, identifying 43 genomic islands. Here, the genomic islands of APEC O1 were compared to those of other sequenced E. coli strains, and the distribution of 81 genes belonging to 12 APEC O1 genomic islands among 828 human and avian ExPEC and commensal E. coli isolates was determined. Multiple islands were highly prevalent among isolates belonging to the O1 and 018 serogroups within phylogenetic group B2, which are implicated in human neonatal meningitis. Because of the extensive genomic similarities between APEC O1 and other human ExPEC strains belonging to the ST95 phylogenetic lineage, its ability to cause disease in a rat model of sepsis and meningitis was assessed. Unlike other ST95 lineage strains, APEC O1 was unable to cause bacteremia or meningitis in the neonatal rat model and was significantly less virulent than uropathogenic E. con (UPEC) CFT073 in a mouse sepsis model, despite carrying multiple neonatal meningitis E. coli (NMEC) virulence factors and belonging to the ST95 phylogenetic lineage. These results suggest that host adaptation or genome modifications have occurred either in APEC O1 or in highly virulent ExPEC isolates, resulting in differences in pathogenicity. Overall, the genomic islands examined provide targets for further discrimination of the different ExPEC subpathotypes, serogroups, phylogenetic types, and sequence types.
[本文引用: 2]
[本文引用: 2]
DOI:10.1360/972013-351URL [本文引用: 1]
DOI:10.1360/972013-351URL [本文引用: 1]
DOI:10.1111/jam.12820URLPMID:25846200 [本文引用: 3]
The study aims to demonstrate the antimicrobial and disinfectant resistance phenotypes and genotypes of Escherichia coli isolates obtained from giant pandas (Ailuropoda melanoleuca).
DOI:10.1186/cc7091URLPMID:19014410 [本文引用: 1]
Bacterial pathogens possess an array of specific mechanisms that confer virulence and the capacity to avoid host defence mechanisms. Mechanisms of virulence are often mediated by the subversion of normal aspects of host biology. In this way the pathogen modifies host function so as to promote the pathogen's survival or proliferation. Such subversion is often mediated by the specific interaction of bacterial effector molecules with host encoded proteins and other molecules. The importance of these mechanisms for bacterial pathogens that cause infections leading to severe community-acquired infections is well established. In contrast, the importance of specialised mechanisms of virulence in the genesis of nosocomial bacterial infections, which occur in the context of local or systemic defects in host immune defences, is less well established. Specific mechanisms of bacterial resistance to host immunity might represent targets for therapeutic intervention. The clinical utility of such an approach for either prevention or treatment of bacterial infection, however, has not been determined.
[本文引用: 2]
[本文引用: 2]
DOI:10.1016/j.micpath.2018.01.034URLPMID:29414607 [本文引用: 1]
Escherichia coli (E. coli) is considered as a common opportunistic pathogen, which causes seriously intestinal infections to giant pandas (Ailuropoda melanoleuca) and other animals. The aim of this investigation was to characterize the antimicrobial resistance and integron gene cassettes in E. coli isolated from the faeces of giant pandas in China. A total of 89 E. coli were isolated, after diagnosis of isolates and genomes were extracted. All the isolates were screened for the presence of related drug-resistance genes and integron gene cassettes through the Polymerase Chain Reaction (PCR) and sequencing. In addition, antimicrobial resistance testing was performed according to the standard disk diffusion method (CLSI 2013). The results demonstrated that all the isolates were multi-drug resistance (MDR). High resistance proportions of the E. coli isolates were to streptomycin (93%), cefazolin (90%), amikacin (75%), tetracycline (65%), ampicillin (62%), cefotaxime and trimethoprim-sulfamethoxazole (54%, each). With respect to the various resistance genes; blaCTX-M, sul1, ant (3')-Ia, tetA, qnrB, tetE, floR, aac (6')-Ib, sul2, rmtA, cmlA, rmtB and tetC were identified with the respective frequencies of 44%, 45%, 38%, 37%, 35%, 27%, 26%, 20%, 18%, 15%, 10%, 7% and 4%. None of the isolates was positive for qnrA and cfr genes. Moreover, a further investigation of integron revealed that the emergence of class 1 and 2 integrons were in 47% and 8% isolates, respectively. While class 3 integron was not screened. Six types of containing in class 1 integron specific gene cassettes (dfrA12-orfF-aadA2, dfrA17-aadA5, aadA1, aadA5, dfrA1 and dfrA7) were identified. However, only one gene cassette (dfrA1-sat2-aadA1) was detected in class 2 integron. These finding emphasize that a high level of E. coli isolates harbored antibiotics resistance and integron gene cassettes, which may bring so many potential threats to the health of giant pandas.
DOI:10.3969/cjz.j.issn.1002-2694.2015.02.008URL [本文引用: 1]
To investigate the serotypes and virulence genes of extraintestinal pathogenic Escherichia coli (ExPEC) in Qinhuangdao City, China, a total of 30 tissue samples were collected from pigs during 2013 to 2014 and 8 isolates of ExPEC were isolated which were identified by colony morphology, culture characteristics identification, and biochemical observation. The O serogroups were identified by agglutination with specific antisera. All strains were screened for virulence genes by PCR. Three O serogroups were identified for 5 E. coli isolates, of which O53, O93 and O157 were dominant serogroups. In addition, the virulence genes were detected by PCR and the results showed that ler, iutA, irp2, fyuA, and astA were positive in the 8 isolates by 100%, 75.0%, 50.0%, 50.0%, and 25.0%, respectively. It is concluded that O53, O93 and O157 are dominant serogroups associate with the disease of swine. Isolates carrying pathogenicity islands gene irp2, fyuA and ler were proved to be pathogenic to mice.
DOI:10.3969/cjz.j.issn.1002-2694.2015.02.008URL [本文引用: 1]
To investigate the serotypes and virulence genes of extraintestinal pathogenic Escherichia coli (ExPEC) in Qinhuangdao City, China, a total of 30 tissue samples were collected from pigs during 2013 to 2014 and 8 isolates of ExPEC were isolated which were identified by colony morphology, culture characteristics identification, and biochemical observation. The O serogroups were identified by agglutination with specific antisera. All strains were screened for virulence genes by PCR. Three O serogroups were identified for 5 E. coli isolates, of which O53, O93 and O157 were dominant serogroups. In addition, the virulence genes were detected by PCR and the results showed that ler, iutA, irp2, fyuA, and astA were positive in the 8 isolates by 100%, 75.0%, 50.0%, 50.0%, and 25.0%, respectively. It is concluded that O53, O93 and O157 are dominant serogroups associate with the disease of swine. Isolates carrying pathogenicity islands gene irp2, fyuA and ler were proved to be pathogenic to mice.
DOI:10.1007/s10096-019-03735-4URLPMID:31720945 [本文引用: 1]
The disc diffusion test is used for antimicrobial susceptibility testing worldwide. In this study, the performance of both Bio-Rad? antibiotic discs (as compared with Oxoid? discs) and the ADAGIO? automated system for the reading of disc diffusion test results was evaluated with American Type Culture Collection (ATCC) quality control (QC) and wild strains of bacteria. Inhibition zones of both disc brands were read manually and through use of the ADAGIO? system. Categorized interpretation of the results for each strain and antibiotic combination was summarized according to the Clinical Laboratory Standards Institute MS-100 (2017 update) manual and ADAGIO? readings. Eight ATCC QC strains and 120 different wild strains were evaluated, to give a total of 1226 antibiotic/bacteria combinations and 2486 manual readings. One major error and four minor errors (0.08% and 0.34%, respectively) were detected via manual readings of the Bio-Rad? discs as compared with the Oxoid? discs. For the same number of antibiotic/bacteria combinations, five minor errors and one major error (0.42% and 0.08%, respectively) were detected with the Bio-Rad? discs read by the ADAGIO? system. In addition, the number of times the automatic reading needed manual edition with Bio-Rad? discs was statistically lower than it did with Oxoid? discs (3.7% vs. 5.7%, p < 0.05). These findings support the hypothesis that Bio-Rad discs are not inferior to Oxoid? discs, and the performance of the ADAGIO? system is comparable to that of manual readings with both disc brands.
DOI:10.1101/gr.097261.109URLPMID:20019144 [本文引用: 1]
Next-generation massively parallel DNA sequencing technologies provide ultrahigh throughput at a substantially lower unit data cost; however, the data are very short read length sequences, making de novo assembly extremely challenging. Here, we describe a novel method for de novo assembly of large genomes from short read sequences. We successfully assembled both the Asian and African human genome sequences, achieving an N50 contig size of 7.4 and 5.9 kilobases (kb) and scaffold of 446.3 and 61.9 kb, respectively. The development of this de novo short read assembly method creates new opportunities for building reference sequences and carrying out accurate analyses of unexplored genomes in a cost-effective way.
DOI:10.1093/bioinformatics/btn025URLPMID:18227114 [本文引用: 1]
We have developed a program SOAP for efficient gapped and ungapped alignment of short oligonucleotides onto reference sequences. The program is designed to handle the huge amounts of short reads generated by parallel sequencing using the new generation Illumina-Solexa sequencing technology. SOAP is compatible with numerous applications, including single-read or pair-end resequencing, small RNA discovery and mRNA tag sequence mapping. SOAP is a command-driven program, which supports multi-threaded parallel computing, and has a batch module for multiple query sets.
DOI:10.1038/srep08365URLPMID:25666585 [本文引用: 1]
The RAST (Rapid Annotation using Subsystem Technology) annotation engine was built in 2008 to annotate bacterial and archaeal genomes. It works by offering a standard software pipeline for identifying genomic features (i.e., protein-encoding genes and RNA) and annotating their functions. Recently, in order to make RAST a more useful research tool and to keep pace with advancements in bioinformatics, it has become desirable to build a version of RAST that is both customizable and extensible. In this paper, we describe the RAST tool kit (RASTtk), a modular version of RAST that enables researchers to build custom annotation pipelines. RASTtk offers a choice of software for identifying and annotating genomic features as well as the ability to add custom features to an annotation job. RASTtk also accommodates the batch submission of genomes and the ability to customize annotation protocols for batch submissions. This is the first major software restructuring of RAST since its inception.
DOI:10.1186/s13099-017-0213-xURLPMID:29201148 [本文引用: 1]
The aim of this study was to generate a reference set of Salmonella enterica genomes isolated from wildlife from the United States and to determine the antimicrobial resistance and virulence gene profile of the isolates from the genome sequence data. We sequenced the whole genomes of 103 Salmonella isolates sampled between 1988 and 2003 from wildlife and exotic pet cases that were submitted to the Oklahoma Animal Disease Diagnostic Laboratory, Stillwater, Oklahoma. Among 103 isolates, 50.48% were from wild birds, 0.9% was from fish, 24.27% each were from reptiles and mammals. 50.48% isolates showed resistance to at least one antibiotic. Resistance against the aminoglycoside streptomycin was most common while 9 isolates were found to be multi-drug resistant having resistance against more than three antibiotics. Determination of virulence gene profile revealed that the genes belonging to csg operons, the fim genes that encode for type 1 fimbriae and the genes belonging to type III secretion system were predominant among the isolates. The universal presence of fimbrial genes and the genes encoded by pathogenicity islands 1-2 among the isolates we report here indicates that these isolates could potentially cause disease in humans. Therefore, the genomes we report here could be a valuable reference point for future traceback investigations when wildlife is considered to be the potential source of human Salmonellosis.
DOI:10.1186/1471-2334-13-19URLPMID:23327474 [本文引用: 1]
Urinary tract infection (UTI) is one of the most common infectious diseases at the community level. In order to assess the adequacy of the empirical therapy, the prevalence and the resistance pattern of the main bacteria responsible for UTI in the community (in Aveiro, Portugal) was evaluated throughout a ten-year period.
DOI:10.1371/journal.pone.0004752URLPMID:19270734 [本文引用: 1]
Type II secretion systems (T2SS) and the evolutionarily related type IV pili (T4P) are important virulence determinants in many Gram-negative bacterial pathogens. However, the roles of T2SS and T4P in the virulence of extraintestinal pathogenic Escherichia coli have not been determined.
[本文引用: 1]
[本文引用: 1]
DOI:10.1128/AAC.45.4.1126-1136.2001URLPMID:11257026 [本文引用: 1]
The contribution of seven known and nine predicted genes or operons associated with multidrug resistance to the susceptibility of Escherichia coli W3110 was assessed for 20 different classes of antimicrobial compounds that include antibiotics, antiseptics, detergents, and dyes. Strains were constructed with deletions for genes in the major facilitator superfamily, the resistance nodulation-cell division family, the small multidrug resistance family, the ATP-binding cassette family, and outer membrane factors. The agar dilution MICs of 35 compounds were determined for strains with deletions for multidrug resistance (MDR) pumps. Deletions in acrAB or tolC resulted in increased susceptibilities to the majority of compounds tested. The remaining MDR pump gene deletions resulted in increased susceptibilities to far fewer compounds. The results identify which MDR pumps contribute to intrinsic resistance under the conditions tested and supply practical information useful for designing sensitive assay strains for cell-based screening of antibacterial compounds.
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/jac/dku197URLPMID:24908046 [本文引用: 1]
To examine the distribution of all genes known to be responsible for resistance to quaternary ammonium compounds (QACs), and their association with resistance to QACs and other antimicrobials, in Escherichia coli recovered from retail meats.
DOI:10.1128/jb.179.7.2274-2280.1997URLPMID:9079913 [本文引用: 1]
Multidrug resistance (MDR) translocators recently identified in bacteria constitute an excellent model system for studying the MDR phenomenon and its clinical relevance. Here we describe the identification and characterization of an unusual MDR gene (mdfA) from Escherichia coli. mdfA encodes a putative membrane protein (MdfA) of 410 amino acid residues which belongs to the major facilitator superfamily of transport proteins. Cells expressing MdfA from a multicopy plasmid are substantially more resistant to a diverse group of cationic or zwitterionic lipophilic compounds such as ethidium bromide, tetraphenylphosphonium, rhodamine, daunomycin, benzalkonium, rifampin, tetracycline, and puromycin. Surprisingly, however, MdfA also confers resistance to chemically unrelated, clinically important antibiotics such as chloramphenicol, erythromycin, and certain aminoglycosides and fluoroquinolones. Transport experiments with an E. coli strain lacking F1-F0 proton ATPase activity indicate that MdfA is a multidrug transporter that is driven by the proton electrochemical gradient.
DOI:10.1073/pnas.0400137101URL [本文引用: 1]
DOI:10.1016/j.micpath.2018.01.034URLPMID:29414607 [本文引用: 1]
Escherichia coli (E. coli) is considered as a common opportunistic pathogen, which causes seriously intestinal infections to giant pandas (Ailuropoda melanoleuca) and other animals. The aim of this investigation was to characterize the antimicrobial resistance and integron gene cassettes in E. coli isolated from the faeces of giant pandas in China. A total of 89 E. coli were isolated, after diagnosis of isolates and genomes were extracted. All the isolates were screened for the presence of related drug-resistance genes and integron gene cassettes through the Polymerase Chain Reaction (PCR) and sequencing. In addition, antimicrobial resistance testing was performed according to the standard disk diffusion method (CLSI 2013). The results demonstrated that all the isolates were multi-drug resistance (MDR). High resistance proportions of the E. coli isolates were to streptomycin (93%), cefazolin (90%), amikacin (75%), tetracycline (65%), ampicillin (62%), cefotaxime and trimethoprim-sulfamethoxazole (54%, each). With respect to the various resistance genes; blaCTX-M, sul1, ant (3')-Ia, tetA, qnrB, tetE, floR, aac (6')-Ib, sul2, rmtA, cmlA, rmtB and tetC were identified with the respective frequencies of 44%, 45%, 38%, 37%, 35%, 27%, 26%, 20%, 18%, 15%, 10%, 7% and 4%. None of the isolates was positive for qnrA and cfr genes. Moreover, a further investigation of integron revealed that the emergence of class 1 and 2 integrons were in 47% and 8% isolates, respectively. While class 3 integron was not screened. Six types of containing in class 1 integron specific gene cassettes (dfrA12-orfF-aadA2, dfrA17-aadA5, aadA1, aadA5, dfrA1 and dfrA7) were identified. However, only one gene cassette (dfrA1-sat2-aadA1) was detected in class 2 integron. These finding emphasize that a high level of E. coli isolates harbored antibiotics resistance and integron gene cassettes, which may bring so many potential threats to the health of giant pandas.
DOI:10.1186/1746-6148-10-155URLPMID:25015125 [本文引用: 1]
In Escherichia coli the genes involved in the acquisition of tetracycline resistance are mainly tet(A) and tet(B). In addition, tet(M) is the most common tetracycline resistance determinant in enterococci and it is associated with conjugative transposons and plasmids. Although tet(M) has been identified in E. coli, to our knowledge, there are no previous reports studying the linkage of the tet(M) gene in E. coli to different mobile genetic elements. The aim of this study was to determine the occurrence of tet(A), tet(B), and tet(M) genes in doxycycline-resistant E. coli isolates from pigs, as well as the detection of mobile genetic elements linked to tet(M) in E. coli and its possible transfer from enterococci.
DOI:10.1515/hsz-2013-0292URLPMID:24497223 [本文引用: 1]
The ribosome and protein synthesis are major targets within the cell for inhibition by antibiotics, such as the tetracyclines. The tetracycline family of antibiotics represent a large and diverse group of compounds, ranging from the naturally produced chlortetracycline, introduced into medical usage in the 1940s, to second and third generation semi-synthetic derivatives of tetracycline, such as doxycycline, minocycline and more recently the glycylcycline tigecycline. Here we describe the mode of interaction of tetracyclines with the ribosome and mechanism of action of this class of antibiotics to inhibit translation. Additionally, we provide an overview of the diverse mechanisms by which bacteria obtain resistance to tetracyclines, ranging from efflux, drug modification, target mutation and the employment of specialized ribosome protection proteins.
DOI:10.1128/mBio.00543-17URLPMID:28655818 [本文引用: 1]
The mobile colistin resistance gene mcr-1 has attracted global attention, as it heralds the breach of polymyxins, one of the last-resort antibiotics for the treatment of severe clinical infections caused by multidrug-resistant Gram-negative bacteria. To date, six slightly different variants of mcr-1, and a second mobile colistin resistance gene, mcr-2, have been reported or annotated in the GenBank database. Here, we characterized a third mobile colistin resistance gene, mcr-3 The gene coexisted with 18 additional resistance determinants in the 261-kb IncHI2-type plasmid pWJ1 from porcine Escherichia colimcr-3 showed 45.0% and 47.0% nucleotide sequence identity to mcr-1 and mcr-2, respectively, while the deduced amino acid sequence of MCR-3 showed 99.8 to 100% and 75.6 to 94.8% identity to phosphoethanolamine transferases found in other Enterobacteriaceae species and in 10 Aeromonas species, respectively. pWJ1 was mobilized to an E.?coli recipient by conjugation and contained a plasmid backbone similar to those of other mcr-1-carrying plasmids, such as pHNSHP45-2 from the original mcr-1-harboring E.?coli strain. Moreover, a truncated transposon element, TnAs2, which was characterized only in Aeromonas salmonicida, was located upstream of mcr-3 in pWJ1. This ΔTnAs2-mcr-3 element was also identified in a shotgun genome sequence of a porcine E.?coli isolate from Malaysia, a human Klebsiella pneumoniae isolate from Thailand, and a human Salmonella enterica serovar Typhimurium isolate from the United States. These results suggest the likelihood of a wide dissemination of the novel mobile colistin resistance gene mcr-3 among Enterobacteriaceae and aeromonads; the latter may act as a potential reservoir for mcr-3IMPORTANCE The emergence of the plasmid-mediated colistin resistance gene mcr-1 has attracted substantial attention worldwide. Here, we examined a colistin-resistant Escherichia coli isolate that was negative for both mcr-1 and mcr-2 and discovered a novel mobile colistin resistance gene, mcr-3 The amino acid sequence of MCR-3 aligned closely with phosphoethanolamine transferases from Enterobacteriaceae and Aeromonas species originating from both clinical infections and environmental samples collected in 12 countries on four continents. Due to the ubiquitous profile of aeromonads in the environment and the potential transfer of mcr-3 between Enterobacteriaceae and Aeromonas species, the wide spread of mcr-3 may be largely underestimated. As colistin has been and still is widely used in veterinary medicine and used at increasing frequencies in human medicine, the continuous monitoring of mobile colistin resistance determinants in colistin-resistant Gram-negative bacteria is imperative for understanding and tackling the dissemination of mcr genes in both the agricultural and health care sectors.
DOI:10.1128/AAC.01757-17URLPMID:28971871 [本文引用: 3]
A colistin- and carbapenem-resistant Escherichia coli clinical isolate was found to carry two plasmid-borne colistin-resistant genes, mcr-1 and the newly identified mcr-3, and a carbapenemase gene, blaNDM-5mcr-3 is a new variant (mcr-3.5) in the isolate and encodes three amino acid substitutions compared with the original MCR-3. mcr-3 was carried by a TnAs3-like transposon on a self-transmissible IncP plasmid in the isolate, highlighting that mcr-3 may have widely spread.
[本文引用: 2]
DOI:10.1128/AAC.01272-17URLPMID:28848017 [本文引用: 2]
Two adjacent colistin resistance gene variants, termed mcr-3.3 and mcr-3-like, were identified in the chromosome of an Aeromonas veronii isolate obtained from retail chicken meat. The variants showed 95.20% and 84.19% nucleotide sequence identity, respectively, to mcr-3 from porcine Escherichia coli Functional cloning indicated that only mcr-3.3 conferred polymyxin resistance in both E. coli and Aeromonas salmonicida The mcr-3.3-mcr-3-like segment was also observed in other Aeromonas species, including A. media, A. caviae, and A. hydrophila.
DOI:10.1016/j.ijantimicag.2018.07.007URLPMID:30009959 [本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1038/srep18109URLPMID:26728082 [本文引用: 3]
To survive antibiotics, bacteria use two different strategies: counteracting antibiotic effects by expression of resistance genes or evading their effects e.g. by persisting inside host cells. Since bacterial adhesins provide access to the shielded, intracellular niche and the adhesin type 1 fimbriae increases bacterial survival chances inside macrophages, we asked if fimbriae also influenced survival by antibiotic evasion. Combined gentamicin survival assays, flow cytometry, single cell microscopy and kinetic modeling of dose response curves showed that type 1 fimbriae increased the adhesion and internalization by macrophages. This was caused by strongly decreased off-rates and affected the number of intracellular bacteria but not the macrophage viability and morphology. Fimbriae thus promote antibiotic evasion which is particularly relevant in the context of chronic infections.
[本文引用: 1]
[本文引用: 1]
DOI:10.1046/j.1365-2958.2003.03391.xURLPMID:12622819 [本文引用: 1]
Examination of 55 clinical isolates of uropathogenic Escherichia coli producing the CNF1 toxin demonstrated that the cnf1 gene is systematically associated with a hly operon via a highly conserved hlyD-cnf1 intergenic region (igs, 943 bp) as shown in the J96 UPEC strain. We examined if this association could reflect a co-regulation of the production of these toxins. Translation of cnf1 from an immediately upstream promoter has been shown to be controlled by means of an anti-Shine-Dalgarno sequence present in the cnf1 coding sequence [fold-back inhibition (cnf1 fbi)]. The cnf1 fbi was not regulated by elements present in the igs. An RNA covering the full hlyD sequence, the igs and extending on the cnf1 gene, was then detected in the J96 strain. This RNA could be part of a HlyCABD mRNA. Transcription of the haemolysin operon requires RfaH antitermination activity. Inactivation of rfaH in J96 resulted in a 100-fold reduction of the CNF1 content of bacteria. The production of CNF1 from a plasmidic igscnf1 DNA was not sensitive to RfaH, indicating that this factor acted on cnf1 transcription via the hly promoter. This way the cnf1 fbi mechanism might be overcome by transcription of cnf1 from the haemolysin promoter and antitermination by RfaH. This constitutes a novel system of bacterial virulence factors co-regulation.
DOI:10.1371/journal.pone.0145722URLPMID:26741798 [本文引用: 1]
The ability of Staphylococcus aureus to successfully colonize (a)biotic surfaces may be explained by biofilm formation and the actions of virulence factors. The aim of the present study was to establish the presence of 52 proteins, including virulence factors such as alpha-toxin, during biofilm formation of five different (methicillin resistant) S. aureus strains on Leiden human epidermal models (LEMs) and polystyrene surfaces (PS) using a competitive Luminex-based assay.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00239-004-0046-3URL [本文引用: 1]
Prokaryotes contain short DNA repeats known as CRISPR, recognizable by the regular spacing existing between the recurring units. They represent the most widely distributed family of repeats among prokaryotic genomes, suggesting a biological function. The origin of the intervening sequences, at present unknown, could provide clues about their biological activities. Here we show that CRISPR spacers derive from preexisting sequences, either chromosomal or within transmissible genetic elements such as bacteriophages and conjugative plasmids. Remarkably, these extrachromosomal elements fail to infect the specific spacer-carrier strain, implying a relationship between CRISPR and immunity against targeted DNA. Bacteriophages and conjugative plasmids are involved in prokaryotic population control, evolution, and pathogenicity. All these biological traits could be influenced by the presence of specific spacers. CRISPR loci can be visualized as mosaics of a repeated unit, separated by sequences at some time present elsewhere in the cell.
[本文引用: 1]
[本文引用: 1]
DOI:10.13343/j.cnki.wsxb.20180270URL [本文引用: 1]
Specialized substrates are injected into a host cell through secretion system, to destroy various signal pathways, which is an effective way for bacterial colonization and infection. Vibrio parahaemolyticus being an important foodborne pathogen, its Type Ⅲ Secretion System (T3SS) and Type VI Secretion System (T6SS) are important virulence factors to the hosts. In this review, we summarize the specific roles of T3SS and T6SS effectors in pathogenicity and the related regulatory mechanisms. This paper provides a reference to further understand the diseases caused by Vibrio parahaemolyticus, to investigate its pathogenesis and find the pathogenic targets.
DOI:10.13343/j.cnki.wsxb.20180270URL [本文引用: 1]
Specialized substrates are injected into a host cell through secretion system, to destroy various signal pathways, which is an effective way for bacterial colonization and infection. Vibrio parahaemolyticus being an important foodborne pathogen, its Type Ⅲ Secretion System (T3SS) and Type VI Secretion System (T6SS) are important virulence factors to the hosts. In this review, we summarize the specific roles of T3SS and T6SS effectors in pathogenicity and the related regulatory mechanisms. This paper provides a reference to further understand the diseases caused by Vibrio parahaemolyticus, to investigate its pathogenesis and find the pathogenic targets.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}