The concepts and research progress: from heritability to microbiability
Chaoliang Wen, Congjiao Sun, Ning Yang,National Engineering Laboratory for Animal Breeding, College of Animal Science andTechnology, China Agricultural University, Beijing100193, China
Supported by the National Natural Science Foundation of China No. (31930105) China Agriculture Research Systems No. (CARS-40) Programs for Changjiang Scholars and Innovative Research in Universities No.(IRT_15R62)
作者简介 About authors 文超良,博士研究生,专业方向:动物遗传育种与繁殖E-mail:clwen@cau.edu.cn。
Abstract Heritability, one of the central quantitative genetic parameters, is critically important to measure the genetic variation of traits, especially in the studies of the response to selection in evolutionary biology and agriculture, and the prediction of disease risks in medicine. The statistical model and method for estimating heritability have been continually developed and improved, since the genetic variance components was first proposed by Fisher in 1918. Recently, the term “microbiability” (m2), an analogous concept and estimated method to heritability, was introduced in gut microbiome research for evaluating the effect of entire microbiota on a host phenotype. In this review, we summarize the progress of statistical methods in the heritability estimation, as well as the current state of gut microbiome associations with the host genome, with a particular focus on the concept and estimated methods of microbiability. Our review will provide a reference for the future study of host phenotypic variation that can be inferred by the gut microbiota. Keywords:heritability;microbiability;association study;quantitative trait;variance component;relationship matrix
PDF (909KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 文超良, 孙从佼, 杨宁. 从遗传力到肠菌力:概念及研究进展[J]. 遗传, 2019, 41(11): 1023-1040 doi:10.16288/j.yczz.19-130 Chaoliang Wen, Congjiao Sun, Ning Yang. The concepts and research progress: from heritability to microbiability[J]. Hereditas(Beijing), 2019, 41(11): 1023-1040 doi:10.16288/j.yczz.19-130
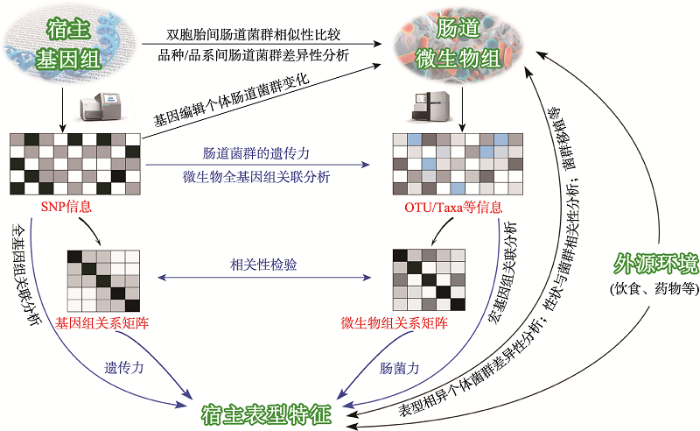
Fig. 1The interactions between host and gut microbiota and related research methods
1 宿主基因组与性状间的关系
1.1遗传力的定义
遗传力是一个基于数理统计、主要反映宿主数量性状遗传特征的重要参数,习惯上用h2表示,该符号最早是1920年美国遗传学家Wright[10]在豚鼠(Cavia porcellus)斑纹的通径分析(path analysis)中,用来表示宿主遗传因素对机体表型的决定程度。一般情况下指狭义遗传力(narrow sense heritability),即加性方差(即育种值方差)占据表型方差的比率,其生物统计学概念等同于加性效应对表型值的回归系数,或加性效应与表型值的相关系数的平方[11]。需要强调的是,世代更迭,只有基因通过配子进行传递,因此遗传力不能真实反映性状的传递能力,仅说明的是群体特定性状的变异情况[12]。
上述经典的遗传力估计方法虽然计算较为简单,但只能处理系谱关系简单、均衡的资料。对于系谱关系复杂(如多代和跨代)或者非均衡的群体,通过线性混合模型估计加性遗传方差和环境组分,再利用获得的方差组分计算遗传力则更为有效。1953年美国数量遗传学家Henderson发表了题为《Estimation of variance and covariance components》的论文[17],提出了3种适用于非均衡资料的方差组分估计方法,引起遗传学家和统计学家的广泛关注。为提高方差组分的估计准确性,各国的遗传育种工作者及对此感兴趣的统计学家进行了大量的探索,估计方法也随着新理论、新技术和新工具的不断涌现而更新和发展,如约束最大似然法(restricted maximum likelihood, REML)[18]、贝叶斯估计法(Bayesian method)[19]和Gibbs抽样法[20]等。此后,虽然新方法仍在不断出现,但是REML法和贝叶斯估计法已在全球范围内占据主导地位。
A:基于子代表型值对亲本中亲值的回归估计遗传力(根据参考文献[12]修改绘制);B:蛋鸡全同胞个体间的基因组关系系数分布(中间虚线为均值线,由于孟德尔抽样误差,全同胞个体间的亲缘系数可能偏离0.5);C:基于每条染色体上的SNP估计每条染色体所能解释目标性状的遗传方差。 Fig. 2The main source of information for heritability estimation
1996年,美国斯坦福医学院Risch等[42]发现复杂疾病遗传学研究中的关联分析比连锁分析具有更高的检测效力,随即提出全基因关联分析(genome-wide association study, GWAS)的概念:在整个基因组范围内筛选与目标性状相关联的分子标记。基于测序和基因分型技术,2005年Klein等[43]首次报道了一项有关年龄相关性视网膜黄斑变性的GWAS研究。之后,一系列与人类复杂疾病等相关表型的GWAS研究被陆续报道[44,45,46],并且GWAS在畜禽各种重要经济性状及复杂疾病抗性的遗传研究中也得到了广泛应用[47,48,49]。GWAS研究为揭示机体复杂性状的遗传奥秘开辟了新的渠道,筛选出大量与目标性状关联的主效和微效基因,并被纳入临床应用[50]和遗传选育[51]。
2宿主基因组与肠道菌群的关系
动物表型变异除了受宿主自身遗传因素的影响,肠道内栖息的数量庞大而复杂的微生物及其代谢产物的作用亦不能小觑,它们通过肠-肝[52]、肠-脑[53]等调控轴与宿主免疫疾病[54,55,56]、营养代谢[57,58,59]和机体行为[60,61,62]等诸多方面密切联系。肠道菌群究竟是先天遗传因素主导,还是后天环境因素驱动?这一直是肠道微生物研究领域的焦点之一。国内外****采用各种研究手段和方法,持续追问“nature or nurture?”
2010年美国内布拉斯加大学Beason等[5]提出将肠道微生物作为宿主的复杂数量性状进行研究。此后,诸多****将肠道菌群丰度、α和β多样性以及微生物基因功能丰度等视为数量性状(稀有的分类群视为二分类性状),估计它们的遗传力,辨别受宿主遗传因素调控的微生物群落,并进一步通过微生物全基因组关联分析(microbial genome-wide association study, mGWAS)挖掘导致肠道菌群可遗传性的宿主遗传变异。
A:宿主自身基因型矩阵和肠道菌群丰度矩阵,部分菌群受到宿主基因型的影响;B:利用A图中的两个矩阵构建的基因组关系矩阵和微生物相似矩阵;C:两两个体间基因组关系系数与Bray-Curtis相异度的散点图,两者相关性不显著。 Fig. 3The association between host genetics and gut microbiota
Fig. 4The diagram of microbial relationship matrix construction using high-throughput sequencing technologies
3.3 宏基因组关联分析
为了能够高分辨率的研究肠道微生物组与宿主表型的关联,Qin等[121]借鉴GWAS研究,于2012年提出了宏基因组关联分析(metagenome-wide association study, MWAS)的概念和方法。该研究对345个中国参与者的粪便微生物进行了两阶段MWAS,共鉴定出约6万个与Ⅱ型糖尿病相关的分子标记,并从菌种、功能以及微生态群落详尽展示了肠道微生物与宿主表型的关联特征。
RossEM, MoatePJ, MarettLC, CocksBG, HayesBJ . Metagenomic predictions: from microbiome to complex health and environmental phenotypes in humans and cattle PLoS One, 2013,8(9):e73056. [本文引用: 4]
GoodrichJK, WatersJL, PooleAC, SutterJL, KorenO, BlekhmanR, BeaumontM, Van TreurenW, KnightR, BellJT, SpectorTD, ClarkAG, LeyRE . Human genetics shape the gut microbiome Cell, 2014,159(4):789-799. [本文引用: 2]
BlekhmanR, GoodrichJK, HuangK, SunQ, BukowskiR, BellJT, SpectorTD, KeinanA, LeyRE, GeversD, ClarkAG . Host genetic variation impacts microbiome composition across human body sites Genome Biol, 2015,16(1):191. [本文引用: 3]
WrightS . The relative importance of heredity and environment in determining the piebald pattern of guinea-pigs Proc Natl Acad Sci USA, 1920,6(6):320-332. [本文引用: 1]
RobertsonFW . Studies in quantitative inheritance XI. Genetic and environmental correlation between body size and egg production in Drosophila Melanogaster J Genet, 1957,55(3):428. [本文引用: 1]
FredeenHT, JonssonP . Genic variance and covariance in Danish Landrace swine as evaluated under a system of individual feeding of progeny test group J Anim Breed Genet, 1957,70(4):348-363. [本文引用: 1]
HendersonCR . Estimation of variance and covariance components Biometrics, 1953,(9):226-252. [本文引用: 1]
PattersonHD, ThompsonR . Recovery of inter-block information when block sizes are unequal Biometrika, 1971,58(3):545-554. [本文引用: 1]
GianolaD, FernandoRL . Bayesian methods in animal breeding theory J Anim Sci, 1986,63(1):217-244. [本文引用: 1]
GemanS, GemanD . Stochastic relaxation, gibbs distributions, and the bayesian restoration of images IEEE Trans Pattern Anal Mach Intell, 1984,6(6):721-741. [本文引用: 1]
HillWG . Applications of population genetics to animal breeding, from Wright, Fisher and Lush to genomic prediction Genetics, 2014,196(1):1-16. [本文引用: 1]
ThompsonEA . The estimation of pairwise relationships Ann Hum Genet, 1975,39(2):173-188. [本文引用: 1]
LynchM . Estimation of relatedness by DNA fingerprinting Mol Biol Evol, 1988,5(5):584-599. [本文引用: 1]
RitlandK . Marker-based method for inferences about quantitative inheritance in natural populations Evolution, 1996,50(3):1062-1073. [本文引用: 1]
RitlandK, RitlandC . Inferences about quantitative inheritance based on natural population structure in the yellow monkeyflower, Mimulus guttatus Evolution, 1996,50(3):1074-1082. [本文引用: 1]
MousseauTA, RitlandK, HeathDD . A novel method for estimating heritability using molecular markers Heredity, 1998,80(2):218-224. [本文引用: 1]
ThomasSC, ColtmanDW, PembertonJM . The use of marker-based relationship information to estimate the heritability of body weight in a natural population: a cautionary tale J Evolution Biol, 2002,15(1):92-99. [本文引用: 1]
VisscherPM, MedlandSE, FerreiraMAR, MorleyKI, ZhuG, CornesBK, MontgomeryGW, MartinNG . Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings PLoS Genet, 2006,2(3):e41. [本文引用: 1]
YangJ, BenyaminB, McEvoy BP, GordonS, HendersAK, NyholtDR, MaddenPA, HeathAC, MartinNG, MontgomeryGW, GoddardME, VisscherPM . Common SNPs explain a large proportion of the heritability for human height Nat Genet, 2010,42(7):565-569. [本文引用: 1]
YangJ, ZengJ, GoddardME, WrayNR, VisscherPM . Concepts, estimation and interpretation of SNP-based heritability Nat Genet, 2017,49(9):1304-1310. [本文引用: 1]
BenjaminDJ, Cesarini D, vander Loos MJHM, DawesCT, KoellingerPD, MagnussonPKE, ChabrisCF, ConleyD, LaibsonD, JohannessonM, VisscherPM . The genetic architecture of economic and political preferences Proc Natl Acad Sci USA, 2012,109(21):8026-8031. [本文引用: 1]
YangJ, LeeSH, GoddardME, VisscherPM . GCTA: a tool for genome-wide complex trait analysis Am J Hum Genet, 2011,88(1):76-82. [本文引用: 1]
YangJ, ManolioTA, PasqualeLR, BoerwinkleE, CaporasoN, Cunningham JM, de Andrade M, FeenstraB, FeingoldE, HayesMG, HillWG, LandiMT, AlonsoA, LettreG, LinP, LingH, LoweW, MathiasRA, MelbyeM, PughE, CornelisMC, WeirBS, GoddardME, VisscherPM . Genome partitioning of genetic variation for complex traits using common SNPs Nat Genet, 2011,43(6):519-525. [本文引用: 1]
DaviesG, TenesaA, PaytonA, YangJ, HarrisSE, LiewaldD, KeXY, Le HellardS, ChristoforouA, LucianoM, McGhee K, LopezL, GowAJ, CorleyJ, RedmondP, FoxHC, HaggartyP, WhalleyLJ, McNeillG, GoddardME, EspesethT, LundervoldAJ, ReinvangI, PicklesA, SteenVM, OllierW, PorteousDJ, HoranM, StarrJM, PendletonN, VisscherPM, DearyIJ . Genome-wide association studies establish that human intelligence is highly heritable and polygenic Mol Psychiatry, 2011,16(10):996-1005. [本文引用: 1]
VinkhuyzenAAE, PedersenNL, YangJ, LeeSH, MagnussonPKE, IaconoWG, McGue M, MaddenPAF, HeathAC, LucianoM, PaytonA, HoranM, OllierW, PendletonN, DearyIJ, MontgomeryGW, MartinNG, VisscherPM, WrayNR, . Common SNPs explain some of the variation in the personality dimensions of neuroticism and extraversion Transl Psychiatry, 2012,2(4):e102. [本文引用: 1]
LohP, BhatiaG, GusevA, FinucaneHK, Bulik-SullivanBK, Pollack SJ, deCandia TR, LeeSH, WrayNR, KendlerKS, O'DonovanMC, NealeBM, PattersonN, PriceAL . Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis Nat Genet, 2015,47(12):1385-1392. [本文引用: 1]
YuanJ, SunC, DouT, YiG, QuL, QuL, WangK, YangN . Identification of promising mutants associated with egg production traits revealed by genome-wide association study PLoS One, 2015,10(10):e0140615. [本文引用: 1]
MathewB, LéonJ, Sillanp?? MJ . A novel linkage-disequilibrium corrected genomic relationship matrix for SNP-heritability estimation and genomic prediction Heredity, 2018,120(4):356-368. [本文引用: 1]
McCoyAM, NortonEM, KemperAM, BeesonSK, MickelsonJR, McCueME . SNP-based heritability and genetic architecture of tarsal osteochondrosis in North American Standardbred horses Anim Genet, 2019,50(1):78-81. [本文引用: 1]
RischN, MerikangasK . The future of genetic studies of complex human diseases Science, 1996,273(5281):1516-1517. [本文引用: 1]
ZhangT, WangWH, ZhangGX, WangJY, XueQ, GuYP . A genome-wide association study on body weight traits of Jinghai yellow chicken, Hereditas(Beijing), 2015,37(8):811-820. [本文引用: 1]
SunCJ, QuL, YiGQ, YuanJW, DuanZY, ShenMM, QuLJ, XuGY, WangKH, YangN . Genome-wide association study revealed a promising region and candidate genes for eggshell quality in an F2 resource population BMC Genomics, 2015,16(1):565. [本文引用: 1]
ManolioTA . Bringing genome-wide association findings into clinical use Nat Rev Genet, 2013,14(8):549-558. [本文引用: 1]
LopesMS, Bovenhuis H, vanSon M, Nordb??, GrindflekEH, KnolEF, BastiaansenJWM . Using markers with large effect in genetic and genomic predictions J Anim Sci, 2017,95(1):59-71. [本文引用: 1]
TripathiA, DebeliusJ, Brenner DA, KarinM, LoombaR, SchnablB, KnightR . The gut-liver axis and the intersection with the microbiome Nat Rev Gastroenterol Hepatol, 2018,15(7):397-411. [本文引用: 1]
ClementeJC, UrsellLK, ParfreyLW, KnightR . The impact of the gut microbiota on human health: an integrative view Cell, 2012,148(6):1258-1270. [本文引用: 1]
GensollenT, IyerSS, KasperDL, BlumbergRS . How colonization by microbiota in early life shapes the immune system Science, 2016,352(6285):539-544. [本文引用: 1]
SannaS, van ZuydamNR, MahajanA, KurilshikovA, Vich VilaA, V?saU, MujagicZ, MascleeAAM, JonkersDMAE, OostingM, JoostenLAB, NeteaMG, FrankeL, ZhernakovaA, FuJ, WijmengaC, McCarthyMI . Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases Nat Genet, 2019,51(4):600-605. [本文引用: 1]
TurnbaughPJ, LeyRE, MahowaldMA, MagriniV, MardisER, GordonJI . An obesity-associated gut microbiome with increased capacity for energy harvest Nature, 2006,444(7122):1027-1131. [本文引用: 1]
Van de MerweJP, StegemanJH, HazenbergMP . The resident faecal flora is determined by genetic characterristics of the host. Implications for Crohn's disease? Antonie Van Leeuwenhoek, 1983,49(2):119-124. [本文引用: 1]
ZoetendalEG, AkkermansADL, Akkermans-van Vliet WM, de VisserJAGM, de VosWM . The host genotype affects the bacterial community in the human gastrointestinal tract Microb Ecol Health Dis, 2001,13(3):129-134. [本文引用: 1]
StewartJA, ChadwickVS, MurrayA . Investigations into the influence of host genetics on the predominant eubacteria in the faecal microflora of children J Med Microbiol, 2005,54(12):1239-1242. [本文引用: 1]
TurnbaughPJ, HamadyM, YatsunenkoT, CantarelBL, DuncanA, LeyRE, SoginML, JonesWJ, RoeBA, AffourtitJP, EgholmM, HenrissatB, HeathAC, KnightR, GordonJI . A core gut microbiome in obese and lean twins Nature, 2009,457(7228):480-484. [本文引用: 1]
YatsunenkoT, ReyFE, ManaryMJ, TrehanI, Dominguez-BelloMG, ContrerasM, MagrisM, HidalgoG, BaldassanoRN, AnokhinAP, HeathAC, WarnerB, ReederJ, KuczynskiJ, CaporasoJG, LozuponeCA, LauberC, ClementeJC, KnightsD, KnightR, GordonJI . Human gut microbiome viewed across age and geography Nature, 2012,486(7402):222-227. [本文引用: 2]
CaporasoJG, LauberCL, CostelloEK, Berg-LyonsD, GonzalezA, StombaughJ, KnightsD, GajerP, RavelJ, FiererN, GordonJI, KnightR . Moving pictures of the human microbiome Genome Biol, 2011,12(5):R50. [本文引用: 1]
XieH, GuoR, ZhongH, FengQ, LanZ, QinB, WardKJ, JacksonMA, XiaY, ChenX, ChenB, XiaH, XuC, LiF, XuX, Al-AamaJY, YangH, WangJ, KristiansenK, WangJ, StevesCJ, BellJT, LiJ, SpectorTD, JiaH . Shotgun metagenomics of 250 adult twins reveals genetic and environmental impacts on the gut microbiome Cell Syst, 2016,3(6):572-584. [本文引用: 1]
FriswellMK, GikaH, StratfordIJ, TheodoridisG, TelferB, WilsonID, McBain AJ. Site and strain-specific variation in gut microbiota profiles and metabolism in experimental mice PLoS One, 2010,5(1):e8584. [本文引用: 1]
DingJ, ZhaoL, WangL, ZhaoW, ZhaiZ, LengL, WangY, HeC, ZhangY, ZhangH, LiH, MengH . Divergent selection-induced obesity alters the composition and functional pathways of chicken gut microbiota Genet Sel Evol, 2016,48(1):93. [本文引用: 1]
YangL, LiuS, DingJ, DaiR, HeC, XuK, HonakerCF, ZhangY, SiegelP, MengH . Gut microbiota co-microevolution with selection for host humoral immunity Front Microbiol, 2017,8:1243. [本文引用: 1]
PanditRJ, HinsuAT, PatelNV, KoringaPG, JakhesaraSJ, ThakkarJR, ShahTM, LimonG, PsifidiA, GuitianJ, HumeDA, TomleyFM, RankDN, RamanM, TirumurugaanKG, BlakeDP, JoshiCG . Microbial diversity and community composition of caecal microbiota in commercial and indigenous Indian chickens determined using 16s rDNA amplicon sequencing Microbiome, 2018,6(1):115. [本文引用: 1]
XiaoY, KongF, XiangY, ZhouW, WangJ, YangH, ZhangG, ZhaoJ . Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs Sci Rep, 2018,8(1):5985. [本文引用: 1]
KingEE, SmithRP, St-PierreB, WrightAG . Differences in the rumen methanogen populations of lactating Jersey and Holstein dairy cows under the same diet regimen Appl Environ Microbiol, 2011,77(16):5682-5687. [本文引用: 1]
Gonzalez-RecioO, ZubiriaI, García-RodríguezA, HurtadoA, AtxaerandioR . Short communication: Signs of host genetic regulation in the microbiome composition in 2 dairy breeds: Holstein and Brown Swiss J Dairy Sci, 2018,101(3):2285-2292. [本文引用: 1]
LiFY, HitchTCA, ChenYH, CreeveyCJ, GuanLL . Comparative metagenomic and metatranscriptomic analyses reveal the breed effect on the rumen microbiome and its associations with feed efficiency in beef cattle Microbiome, 2019,7(1):6. [本文引用: 1]
ShiPJ, MengK, ZhouZG, WangYR, DiaoQY, YaoB . The host species affects the microbial community in the goat rumen Lett Appl Microbiol, 2008,46(1):132-135. [本文引用: 1]
DouglasJL, WorganHJ, EastonGL, PoretL, WolfBT, EdwardsA, DaviesE, RossD, McEwanNR. Microbial diversity in the digestive tract of two different breeds of sheep J Appl Microbiol, 2016,120(5):1382-1389. [本文引用: 1]
GoodrichJK, DavenportER, WatersJL, ClarkAG, LeyRE . Cross-species comparisons of host genetic associations with the microbiome Science, 2016,352(6285):532-535. [本文引用: 1]
TurpinW, Espin-GarciaO, XuW, SilverbergMS, KevansD, SmithMI, GuttmanDS, GriffithsA, PanaccioneR, OtleyA, XuL, ShestopaloffK, Moreno-HagelsiebG, PatersonAD, CroitoruK . Association of host genome with intestinal microbial composition in a large healthy cohort Nat Genet, 2016,48(11):1413-1417. [本文引用: 3]
LimMY, YouHJ, YoonHS, KwonB, LeeJY, LeeS, SongY, LeeK, SungJ, KoG . The effect of heritability and host genetics on the gut microbiota and metabolic syndrome Gut, 2017,66(6):1031-1038. [本文引用: 2]
GoodrichJK, DavenportER, BeaumontM, JacksonMA, KnightR, OberC, SpectorTD, BellJT, ClarkAG, LeyRE . Genetic determinants of the gut microbiome in UK twins Cell Host Microbe, 2016,19(5):731-743. [本文引用: 5]
ZhaoL, WangG, SiegelP, HeC, WangH, ZhaoW, ZhaiZ, TianF, ZhaoJ, ZhangH, SunZ, ChenW, ZhangY, MengH . Quantitative genetic background of the host influences gut microbiomes in chickens Sci Rep, 2013,3(1):1163. [本文引用: 1]
MengH, ZhangY, ZhaoL, ZhaoWJ, HeC, Honaker CF, ZhaiZX, SunZK, SiegelPB . Body weight selection affects quantitative genetic correlated responses in gut microbiota PLoS One, 2014,9(3):e89862. [本文引用: 1]
WenCL, YanW, SunCJ, JiCJ, ZhouQQ, ZhangDX, ZhengJX, YangN . The gut microbiota is largely independent of host genetics in regulating fat deposition in chickens ISME J, 2019,13(6):1422-1436. [本文引用: 6]
SassonG, Ben-ShabatSK, SeroussiE, Doron-FaigenboimA, ShterzerN, YaacobyS, BergMM, WhiteBA, HalperinE, MizrahiI . Heritable bovine rumen bacteria are phylogenetically related and correlated with the cow's capacity To harvest energy from its feed MBio, 2017,8(4):e00703-17. [本文引用: 1]
DiffordGF, PlichtaDR, L?vendahlP, LassenJ, NoelSJ, H?jbergO, WrightAG, ZhuZ, KristensenL, NielsenHB, GuldbrandtsenB, SahanaG . Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows PLoS Genet, 2018,14(10):e1007580. [本文引用: 3]
LiFY, LiCX, ChenYY, LiuJH, ZhangCY, IrvingB, FitzsimmonsC, PlastowG, GuanLL . Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle Microbiome, 2019,7(1):92. [本文引用: 2]
ChenCY, HuangXC, FangSM, YangH, HeMZ, ZhaoYZ, HuangLS . Contribution of host genetics to the variation of microbial composition of cecum lumen and feces in pigs Front Microbiol, 2018,9:2626. [本文引用: 2]
Camarinha-SilvaA, MaushammerM, WellmannR, VitalM, PreussS, BennewitzJ . Host genome influence on gut microbial composition and microbial prediction of complex traits in pigs Genetics, 2017,206(3):1637-1644. [本文引用: 3]
LuD, TiezziF, SchillebeeckxC, McNulty NP, SchwabC, ShullC, MalteccaC . Host contributes to longitudinal diversity of fecal microbiota in swine selected for lean growth Microbiome, 2018,6(1):4. [本文引用: 1]
OrgE, ParksBW, JooJW, EmertB, SchwartzmanW, KangEY, MehrabianM, PanC, KnightR, GunsalusR, DrakeTA, EskinE, LusisAJ . Genetic and environmental control of host-gut microbiota interactions Genome Res, 2015,25(10):1558-1569. [本文引用: 2]
O’ConnorA, QuizonPM, AlbrightJE, LinFT, BennettBJ . Responsiveness of cardiometabolic-related microbiota to diet is influenced by host genetics Mamm Genome, 2014,25(11-12):583-599. [本文引用: 1]
BonderMJ, KurilshikovA, TigchelaarEF, MujagicZ, ImhannF, VilaAV, DeelenP, VatanenT, SchirmerM, SmeekensSP, ZhernakovaDV, JankipersadsingSA, JaegerM, OostingM, CenitMC, MascleeAAM, SwertzMA, LiY, KumarV, JoostenL, HarmsenH, WeersmaRK, FrankeL, HofkerMH, XavierRJ, JonkersD, NeteaMG, WijmengaC, FuJ, ZhernakovaA . The effect of host genetics on the gut microbiome Nat Genet, 2016,48(11):1407-1412. [本文引用: 2]
WangJ, ThingholmLB, Skiecevi?ien?J, RauschP, KummenM, HovJR, DegenhardtF, HeinsenFA, RühlemannMC, SzymczakS, HolmK, EskoT, SunJ, Pricop-JeckstadtM, Al-DuryS, BohovP, BethuneJ, SommerF, EllinghausD, BergeRK, HübenthalM, KochM, SchwarzK, RimbachG, HübbeP, PanW, Sheibani-TezerjiR, H?slerR, RosenstielP, D'Amato M, Cloppenborg-SchmidtK, KünzelS, LaudesM, MarschallH, LiebW, N?thlingsU, KarlsenTH, BainesJF, FrankeA . Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota Nat Genet, 2016,48(11):1396-1406. [本文引用: 3]
KoldeR, FranzosaEA, RahnavardG, HallAB, VlamakisH, StevensC, DalyMJ, XavierRJ, HuttenhowerC . Host genetic variation and its microbiome interactions within the Human Microbiome Project Genome Med, 2018,10(1):6. [本文引用: 2]
DavenportER, CusanovichDA, MicheliniK, BarreiroLB, OberC, GiladY . Genome-wide association studies of the human gut microbiota PLoS One, 2015,10(11):e0140301. [本文引用: 1]
BeaumontM, GoodrichJK, JacksonMA, YetI, DavenportER, Vieira-SilvaS, DebeliusJ, PallisterT, ManginoM, RaesJ, KnightR, ClarkAG, LeyRE, SpectorTD, BellJT . Heritable components of the human fecal microbiome are associated with visceral fat Genome Biol, 2016,17(1):189. [本文引用: 1]
SuzukiTA, Phifer-RixeyM, MackKL, SheehanMJ, LinD, BiK, NachmanMW . Host genetic determinants of the gut microbiota of wild mice Mol Ecol, 2019,28(13):3197-3207. [本文引用: 2]
Crespo-PiazueloD, Migura-GarciaL, EstelléJ, Criado-MesasL, RevillaM, CastellóA, Mu?ozM, García-CascoJM, FernándezAI, BallesterM, FolchJM . Association between the pig genome and its gut microbiota composition Sci Rep, 2019,9(1):8791. [本文引用: 1]
Vijay-KumarM, AitkenJD, CarvalhoFA, CullenderTC, MwangiS, SrinivasanS, SitaramanSV, KnightR, LeyRE, GewirtzAT . Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5 Science, 2010,328(5975):228-231. [本文引用: 1]
MassacciFR, ClarkA, RuetA, LansadeL, CostaM, MachN . Inter-breed diversity and temporal dynamics of the faecal microbiota in healthy horses J Anim Breed Genet, 2019, doi: 10.1111/jbg.12441. [本文引用: 1]
van OpstalEJ, BordensteinSR . MICROBIOME. Rethinking heritability of the microbiome Science, 2015,349(6253):1172-1173. [本文引用: 1]
WeissbrodO, RothschildD, BarkanE, SegalE . Host genetics and microbiome associations through the lens of genome wide association studies Curr Opin Microbiol, 2018,44:9-19. [本文引用: 1]
DiffordGF, LassenJ, L?vendahlP. Genes and microbes, the next step in dairy cattle breeding. In: EAAP-67th Annual Meeting, Belfast, United Kingdom, 2016,285. [本文引用: 1]
EspeyMG . Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota Free Radic Biol Med, 2013,55:130-140. [本文引用: 1]
O'MayGA, Reynolds N, SmithAR, KennedyA, MacfarlaneGT . Effect of pH and antibiotics on microbial overgrowth in the stomachs and duodena of patients undergoing percutaneous endoscopic gastrostomy feeding J Clin Microbiol, 2005,43(7):3059-3065. [本文引用: 1]
TropiniC, EarleKA, HuangKC, SonnenburgJL . The gut microbiome: connecting spatial organization to function Cell Host Microbe, 2017,21(4):433-442. [本文引用: 1]
FuJY, BonderMJ, CenitMC, TigchelaarEF, MaatmanA, DekensJAM, BrandsmaE, MarczynskaJ, ImhannF, WeersmaRK, FrankeL, PoonTW, XavierRJ, GeversD, HofkerMH, WijmengaC, ZhernakovaA . The gut microbiome contributes to a substantial proportion of the variation in blood lipids Circ Res, 2015,117(9):817-824. [本文引用: 1]
TalmudPJ, HingoraniAD, CooperJA, MarmotMG, BrunnerEJ, KumariM, Kivim?kiM, HumphriesSE . Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study BMJ, 2010,340:b4838. [本文引用: 1]
NiuDY, YanWL . The application of genetic risk score in genetic studies of complex human diseases Hereditas(Beijing), 2015,37(12):1204-1210. [本文引用: 1]
WangJ, JiaH . Metagenome-wide association studies: fine-mining the microbiome Nat Rev Microbiol, 2016,14(8):508-522. [本文引用: 1]
KarlssonFH, TremaroliV, NookaewI, Bergstr?mG, BehreCJ, FagerbergB, NielsenJ, B?ckhedF . Gut metagenome in European women with normal, impaired and diabetic glucose control Nature, 2013,498(7452):99-103. [本文引用: 1]
NielsenHB, AlmeidaM, JunckerA, RasmussenS, LiJ, SunagawaS, PlichtaDR, GautierL, PedersenAG, Le ChatelierE, PelletierE, BondeI, NielsenT, ManichanhC, ArumugamM, BattoJM, Quintanilha Dos SantosMB, BlomN, BorruelN, BurgdorfKS, BoumezbeurF, CasellasF, DoréJ, DworzynskiP, GuarnerF, HansenT, HildebrandF, KaasRS, KennedyS, KristiansenK, KultimaJR, LéonardP, LevenezF, LundO, MoumenB, Le Paslier D, PonsN, PedersenO, PriftiE, QinJ, RaesJ, S?rensenS, TapJ, TimsS, UsseryDW, YamadaT, JametA, MérieuxA, CultroneA, TorrejonA, QuinquisB, BrechotC, DelormeC, M'RiniC, de Vos WM, MaguinE, VarelaE, GuedonE, GwenF, HaimetF, ArtiguenaveF, VandemeulebrouckG, DenariazG, KhaciG, BlottièreH, KnolJ, WeissenbachJ, Hylckama Vlieg VJE, TorbenJ, ParkhillJ, TurnerK, van de Guchte M, AntolinM, RescignoM, KleerebezemM, DerrienM, GalleronN, SanchezN, GrarupN, VeigaP, OozeerR, DervynR, LayecS, BrulsT, WinogradskiY, ErwinGZ, RenaultD, Sicheritz-PontenT, BorkP, WangJ, BrunakS, EhrlichSD, MetaHITC . Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes Nat Biotechnol, 2014,32(8):822-828. [本文引用: 2]
Le ChatelierE, NielsenT, QinJ, PriftiE, HildebrandF, FalonyG, AlmeidaM, ArumugamM, BattoJM, KennedyS, LeonardP, LiJ, BurgdorfK, GrarupN, J?rgensenT, BrandslundI, NielsenHB, JunckerAS, BertalanM, LevenezF, PonsN, RasmussenS, SunagawaS, TapJ, TimsS, ZoetendalEG, BrunakS, ClémentK, DoréJ, KleerebezemM, KristiansenK, RenaultP, Sicheritz-PontenT, de VosWM, ZuckerJD, RaesJ, HansenT, BorkP, WangJ, EhrlichSD, PedersenO . Richness of human gut microbiome correlates with metabolic markers Nature, 2013,500(7464):541-546. [本文引用: 1]
ZellerG, TapJ, VoigtAY, SunagawaS, KultimaJR, CosteaPI, AmiotA, B?hmJ, BrunettiF, HabermannN, HercogR, KochM, LucianiA, MendeDR, SchneiderMA, Schrotz-KingP, TournigandC, Tran Van NhieuJ, YamadaT, ZimmermannJ, BenesV, KloorM, UlrichCM, von Knebel DoeberitzM, SobhaniI, BorkP . Potential of fecal microbiota for early-stage detection of colorectal cancer Mol Syst Biol, 2014,10(11):766. [本文引用: 1]
ZhangX, ZhangD, JiaH, FengQ, WangD, LiangD, WuX, LiJ, TangL, LiY, LanZ, ChenB, LiY, ZhongH, XieH, JieZ, ChenW, TangS, XuX, WangX, CaiX, LiuS, XiaY, LiJ, QiaoX, Al-AamaJ Y, ChenH, WangL, WuQ, ZhangF, ZhengW, LiY, ZhangM, LuoG, XueW, XiaoL, LiJ, ChenW, XuX, YinY, YangH, WangJ, KristiansenK, LiuL, LiT, HuangQ, LiY, WangJ . The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment Nat Med, 2015,21(8):895-905. [本文引用: 1]
 ,中国农业大学动物科技学院,畜禽国家育种工程实验室,北京 100193
,中国农业大学动物科技学院,畜禽国家育种工程实验室,北京 100193
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT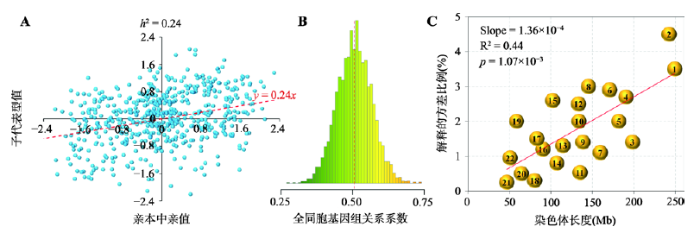 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT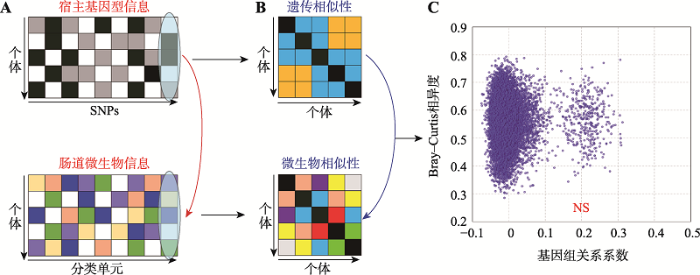 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT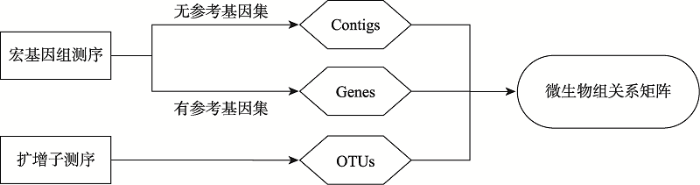 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}