 ,1,2
,1,2The effect of lncRNA TCONS_00815878 on differentiation of porcine skeletal muscle satellite cells
Ziying Huang1, Long Li1, Qianqian Li1, Xiangdong Liu1,2, Changchun Li,1,2通讯作者: 李长春,博士生导师,研究方向:猪重要经济性状的表观遗传学基础。E-mail:lichangchun@mail.hzau.edu.cn
编委: 李明洲
收稿日期:2019-05-21修回日期:2019-11-19网络出版日期:2019-12-20
| 基金资助: |
Editorial board:
Received:2019-05-21Revised:2019-11-19Online:2019-12-20
| Fund supported: |
作者简介 About authors
黄子莹,在读硕士研究生,专业方向:动物遗传育种大数据分析。E-mail:327620138@qq.com。

摘要
猪骨骼肌发育是一个复杂的生物学过程,其中骨骼肌卫星细胞分化是影响骨骼肌发育的重要环节。近年来发现长链非编码RNA (long non-coding RNA, lncRNA)在骨骼肌卫星细胞分化中具有重要作用。为探究lncRNA TCONS_00815878对猪骨骼肌卫星细胞分化的影响,本研究利用qRT-PCR技术检测出生7 d内大白仔猪6种组织(心脏、脾脏、肺脏、肾脏、背肌和腿肌)及从胚胎期到出生后5个不同时间点(35 d、45 d、55 d胚胎及产后第7 d和第200 d后腿肌肉组织) TCONS_00815878的表达情况;利用反义核苷酸(antisense oligonucleotides, ASO)在猪骨骼肌卫星细胞中敲低TCONS_00815878,检验分化标记基因MyoD、MyoG和MyHC表达情况;通过生物信息学分析预测TCONS_00815878靶基因,并利用DAVID软件在线预测其靶基因的功能与通路。结果表明:TCONS_00815878在猪心肌和腿肌中高表达;仔猪出生后7 d内,TCONS_00815878在猪肌肉组织中表达量不断升高,第7 d达到高峰;在猪骨骼肌卫星细胞增殖和分化过程中,TCONS_00815878在分化期表达量不断上升,且在分化30 h表达量达到峰值;敲低TCONS_00815878后,MyoD、MyoG和MyHC基因表达量降低,其中MyoD表达量显著下降(P<0.05)。此外,功能预测结果发现,其靶基因富集到糖酵解和丙酮酸代谢等与骨骼肌卫星细胞分化相关的多个生物学过程。本研究推测,lncRNA TCONS_00815878可能对猪骨骼肌卫星细胞的分化起促进作用。
关键词:
Abstract
Porcine skeletal muscle development is a complex biological process, and differentiation of skeletal muscle satellite cells is an important part of skeletal muscle development. In recent years, it has been found that lncRNA plays an important role in the differentiation of skeletal muscle satellite cells. Here we investigate the effect of lncRNA TCONS_00815878 on the differentiation of porcine skeletal muscle satellite cells. We first used qRT-PCR to detect the expression levels of TCONS_00815878 in six tissues (heart, spleen, lung, kidney, back muscles and leg muscles) of Yorkshire piglets within seven days of birth. At the same time, the expression levels of TCONS_00815878 at five different time points from the embryonic stage to the postnatal stage (35 d, 45 d, 55 d of embryos, and 7 d, 200 d of postpartum leg muscles) were examined. The expression of the differentiation marker genes MyoD, MyoG and MyHC was examined by knocking down TCONS_00815878 in porcine skeletal muscle satellite cells using antisense oligonucleotides (ASO). The target gene of TCONS_00815878 was predicted by bioinformatics analysis, and the function and pathway of its target gene were predicted online using DAVID software. The results showed that TCONS_00815878 had the highest expression level in pig myocardium and leg muscles. Within seven days after birth, TCONS_00815878 increased in the muscle tissue of pigs, and reached the peak of expression level on the 7th day. During the process of proliferation and differentiation of porcine skeletal muscle satellite cells, the expression level of TCONS_00815878 increased during the differentiation stage and peaked at 30 h of differentiation. After knocking down TCONS_00815878, the expression levels of MyoD, MyoG and MyHC were decreased, but the expression level of MyoD was significantly decreased (P<0.05). In addition, functional predictions revealed that the target gene of TCONS_00815878 is enriched in multiple biological processes, such as glycolysis and pyruvate metabolism, related to skeletal muscle satellite cell differentiation. This study speculates that lncRNA TCONS_00815878 may promote the differentiation of porcine skeletal muscle satellite cells.
Keywords:
PDF (872KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黄子莹, 李龙, 李倩倩, 刘向东, 李长春. lncRNA TCONS_00815878对猪骨骼肌卫星细胞分化的影响. 遗传[J], 2019, 41(12): 1119-1128 doi:10.16288/j.yczz.19-146
Ziying Huang.
猪(Sus scrofa)骨骼肌生长发育与其产肉量高低直接相关。此外,因与人具有相似的解剖学和生理学特征,研究猪骨骼肌的发育规律对人类相关研究也大有裨益[1]。骨骼肌发育是一个复杂且有序的过程,是各种遗传因素及调控因子共同作用的结果。骨骼肌卫星细胞是位于肌肉组织基膜和肌细胞膜之间的骨骼肌干细胞,在骨骼肌发育与再生过程中发挥着重要作用[2,3]。骨骼肌卫星细胞被激活后首先形成单核成肌细胞,增殖分化,随后融合成多核肌细胞,进一步相互连接形成肌纤维,最终并排形成肌肉组织[4,5]。
长链非编码RNA (long non-coding RNA, lncRNA)是一类长度大于200 nt的非编码RNA。研究证实,lncRNA在骨骼肌发育、肌细胞增殖与分化过程中发挥关键调控作用[6,7,8]。有关lncRNA参与骨骼肌发育的研究已有很多,但多集中对小鼠(Mus musculus)的研究[9,10]。近年来,随着生物信息学及生物检测技术的发展,已在猪上获得大量与肌肉发育潜在相关的lncRNA,但多数lncRNA作用还尚不明确[11]。TCONS_ 00815878是Tang等[12]对贵州小型猪9个组织和3个骨骼肌样本进行高通量测序鉴定得到的肌肉组织特异性lncRNA,位于15号染色体,全长1089 nt,包含2个外显子。为进一步探究TCONS_00815878在猪骨骼肌发育过程中的作用,本研究检测了出生7 d内大白仔猪6种组织及从胚胎期到出生后5个不同时间点TCONS_00815878的表达情况,进一步在猪骨骼肌卫星细胞中对TCONS_00815878进行敲低处理,检测其对猪骨骼肌卫星细胞分化产生的影响。最后通过生物信息学分析,预测TCONS_ 00815878的靶基因及其靶基因可能参与的生物学过程和通路。通过以上研究,推测TCONS_00815878可能对猪骨骼肌卫星细胞分化过程产生的影响,旨在为猪骨骼肌生长发育相关lncRNA的功能研究提供更有价值的参考。
1 材料与方法
1.1 猪骨骼肌卫星细胞分离和培养
出生7 d内大白仔猪(购自华中农业大学种猪场),取两后腿全部肌肉,置于装有20~30 mL PBS (美国GIBCO公司)的培养皿中,在PBS中漂洗3次去除肌肉表面筋膜,剪碎组织肉样至肉糜状;将肉糜移至T75培养瓶,吸净上清,向肌肉组织中加入2倍体积0.2% Ⅰ型胶原酶(美国GIBCO公司)工作液;37℃、1200 r/min水浴震荡消化2.5 h后加等体积终止培养基终止消化;过100目筛、200目筛及400目筛,2000 r/min离心10 min,除上清,加培养基重悬细胞,溶液移至10 cm培养皿;37℃、5% CO2贴壁培养2.5 h,将培养皿溶液上清移至新培养皿中继续培养;卫星细胞在37℃、5% CO2培养箱(美国Thermo Scientific公司)培养24 h,用PBS清洗2遍并更换培养基。1.2 样品采集
取3头出生7 d内大白仔猪(购自华中农业大学种猪场),采集心脏、脾脏、肺脏、肾脏、背肌和后腿肌肉6种组织,用于检测TCONS_00815878在不同组织中的表达量。采集大白猪35 d、45 d、55 d胚胎及产后第7 d和第200 d的后腿肌肉组织,用于检测TCONS_ 00815878从猪胚胎期到出生后不同时间点的表达量。上述所有组织样品采集后,经液氮速冻,保存于-80℃备用。
收集增殖期12 h、24 h和分化期12 h、24 h的猪骨骼肌卫星细胞,检测增殖期Pax7、Myf5和分化期MyoD、MyHC和MyoG的表达量,以确定猪骨骼肌卫星细胞的增殖分化效果。
收集增殖期6 h、12 h、18 h、30 h、36 h和42 h及分化期0 h、6 h、12 h、24 h、30 h、36 h和42 h的猪骨骼肌卫星细胞,检测TCONS_00815878在猪骨骼肌卫星细胞增殖与分化不同时期的表达量。
收集诱导分化24 h的猪骨骼肌卫星细胞,分离其细胞质与细胞核RNA,用于检测TCONS_00815878在猪骨骼肌卫星细胞的细胞质和细胞核的表达量。
1.3 总RNA提取
组织总RNA提取:取-80℃冰箱保存的组织样品,从冻存管中取出适量迅速放入液氮预冷的研钵中,加入适当液氮后研磨,待研磨成粉后转移到1.5 mL无RNA酶的离心管中,按照RNA提取试剂盒(德国Epigentek公司)说明书提取上述组织样本中的总RNA,取1 μL样品于NanoDrop 2000 (美国Thermo Scientific公司)分光光度计检测浓度,A260/280值在1.8~2.0之间则表示RNA纯度较高。细胞总RNA提取:将六孔板内的猪骨骼肌卫星细胞弃掉培养基后,用PBS冲洗两次,每孔直接加入1 mL Trizol,用移液枪吹打数次,室温裂解5 min,然后吸入1.5 mL无RNA酶离心管,于-80℃冰箱储存,后续RNA提取步骤与组织样品RNA提取方法一致。
细胞核与细胞质RNA分离与提取:猪骨骼肌卫星细胞用预冷的PBS冲洗两次,2187 r/min弃上清液。将沉淀重悬于0.2 mL裂解缓冲液(50 mmol/L Tris-HCl pH 8.0、140 mmol/L NaCl、1.5 mmol/L MgCl2、0.5% IGEPAL、1 mmol/L DTT、1 U/μL RNase抑制剂)。将混合物冰上放置5 min,之后4℃、2187 r/min细胞悬液离心3 min。收集上清液到1.5 mL离心管(细胞质部分),14 000 r/min离心4 min。剩余沉淀用0.2 mL裂解液冲洗2次(细胞核部分)。最后用1 mL Trizol提取所有细胞核和细胞质中RNA。
1.4 引物及反义核苷酸(antisense oligonucleotides, ASO)片段的设计、合成
lncRNA TCONS_00815878序列参考文献[9];PFKM、ELOVL5、MBTPS1、FKBP10、MyoD、MyoG、MyHC、Myf5、Pax7和18S rRNA的mRNA序列由NCBI数据库获得。本研究采用Primer Premier 5软件设计引物。引物由生工生物工程(上海)股份有限公司合成。引物信息详见表1。通过网站(
1.5 cDNA合成和qRT-PCR扩增
参照SMARTerTM PCR cDNA Synthesis Kit试剂盒(日本TaKaRa公司)操作说明书反转录合成所有组织和猪骨骼肌卫星细胞中的cDNA,反转录产物于-20℃保存备用,以18S rRNA为内参,qRT-PCR检测TCONS_00815878表达量。反应总体系为10 μL:SYBR Green 5 μL,ddH2O 3.6 μL,引物各0.2 μL, cDNA 1 μL。反应条件:95℃ 2 min;95℃ 15 s, 56℃/60℃ 20 s,72℃ 15 s,40个循环;95℃ 5 s,58℃ 5 s,95℃ 50 s,1个循环。扩增结束后进行溶解曲线分析。1.6 TCONS_00815878靶基因及功能预测
根据参考文献[9]测序结果中的蛋白编码基因表达量数据,计算TCONS_00815878与蛋白编码基因之间的Pearson相关系数,预测TCONS_00815878靶基因。选择P<0.05、Pearon相关系数|r|>0.95的基因作为靶基因[13](附表1)。利用DAVID功能注释生物信息学芯片分析预测靶基因功能与通路,取结果P<0.05为有效功能预测结果。Table 1
表1
表1本文所用引物信息
Table 1
| 目的基因 | 引物序列( 5′→3′) | 复性温度(℃) | 产物大小(bp) |
|---|---|---|---|
| TCONS_00815878 | F: ACACCCTTTCCCAAAATCAA | 56 | 93 |
| R: GCAGACTGTCCAAATCTACCCT | |||
| PFKM | F: GAGAGCGTTTCCATGATGCTTC | 60 | 221 |
| R: AATCAAAGAGGGTGCCATCCAT | |||
| ELOVL5 | F: ATATGAAGATCATCCGCGTGCT | 60 | 59 |
| R: GTGATCTGGTGGTTGTTCTTGC | |||
| MBTPS1 | F: CCTCAACAGTGGTGGAATACGA | 60 | 179 |
| R: TTGGGATGATCTTCAAGCGTCA | |||
| MyoD | F: GGCTGCCCAAGGTGGAAATC | 60 | 170 |
| R: TGCGTCTGAGTCACCGCTGTAG | |||
| MyoG | F: ATGAGACATCCCCCTACTTCTACCA | 60 | 160 |
| R: GTCCCCAGCCCCTTATCTTCC | |||
| Pax7 | F: GGAGTACAAGAGGGAGAACCC | 60 | 122 |
| R: TTCTGAGCACGCGGCTAATC | |||
| Myf5 | F: GAATGCCATCCGCTACAT | 60 | 125 |
| R: AACTGCTGCTCTTTCTGG | |||
| MyHC | F: GTTCAGAGAAAGGCATCCCAAA | 60 | 135 |
| R: GAGAGTGACCGACACCACAAGTG | |||
| FKBP10 | F: ATGTGTCCTGGAGAGAGAAGGA | 60 | 209 |
| R: AATCAAAGAGGGTGCCATCCAT | |||
| 18S rRNA | F: TCCCGACGTGACTGCTC | 60 | 133 |
| R: GGTGACAGCGGGGTGG |
新窗口打开|下载CSV
1.7 lncRNA TCONS_00815878敲低实验及靶基因验证
1.7.1 猪骨骼肌卫星细胞转染及TCONS_ 00815878敲低实验在10 cm培养皿中培养猪骨骼肌卫星细胞,接种至六孔板,ASO敲低组(ASO)和对照组(NC)均设3个重复,待细胞密度达到80%左右进行转染实验,每孔ASO转染量为200 pmol。待细胞密度达到90%~ 100%更换分化培养基,分别于诱导分化后18 h、 30 h收取细胞,进行RNA提取。qRT-PCR检测TCONS_00815878及MyoD、MyoG和MyHC在不同分化时期表达量。
1.7.2 免疫荧光分析
将细胞用预冷的PBS洗涤2次,用预冷的4%多聚甲醛(广州赛国生物科技有限公司)室温静置固定15 min。PBS洗涤后,用预冷的0.25% TritonX-100 (广州赛国生物科技有限公司)室温静置处理10 min。PBS洗涤,封闭液4℃封闭1~2 h (封闭液:3%BSA, 0.3%T TritonX-100, 10%FBS,PBS)。PBS洗涤细胞,4℃孵育一抗过夜(抗体稀释液:3%BSA,0.3% TritonX-100, PBS)。之后37℃孵育二抗1 h。最后使用DAPI对细胞进行染色,荧光显微镜下观察荧光。
1.7.3 靶基因功能验证
使用敲低TCONS_00815878后的细胞样品。以18S rRNA为内参,qRT-PCR检测靶基因PFKM、ELOVL5、MBTPS1和FKBP10的表达量。反应总体系为10 μL:SYBR Green 5 μL,ddH2O 3.6 μL,引物各0.2 μL,cDNA 1 μL。反应条件:95℃ 2 min;95℃ 15 s,60℃ 20 s,72℃ 15 s,40个循环;95℃ 5 s,58℃ 5 s,95℃ 50 s,1个循环。扩增结束后进行溶解曲线分析。
1.8 统计分析
qRT-PCR扩增结果参照2-ΔΔCt方法[14]评估TCONS_00815878及PFKM、ELOVL5、MBTPS1、FKBP10、MyoD、MyoG、MyHC、Myf5和Pax7相对表达量,以18S rRNA作为内参基因,P<0.05表示为差异显著,P<0.01表示为差异极显著。每项检测设置3个生物学重复,每个生物学重复设置3个技术重复,由GraphPad Prism 6.0软件完成统计并作图。生物信息学分析结果由Excel软件统计,利用R软件分析作图。2 结果与分析
2.1 TCONS_00815878表达特征及核质定位
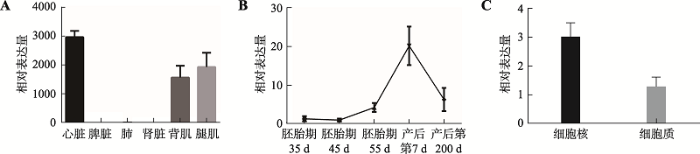
利用qRT-PCR检测出生7 d内大白仔猪心脏、脾脏、肺脏、肾脏、背肌和后腿肌肉6种组织中TCONS_00815878的表达量(图1A)。结果表明,TCONS_00815878在心脏、背肌和后腿肌肉中表达量较高,在其他组织中不表达。取35 d、45 d、55 d胚胎及产后第7 d和第200 d大白仔猪后腿肌肉组织,检测TCONS_00815878表达量(图1B)。结果表明,TCONS_00815878表达量在胚胎期35 d和45 d呈缓慢下降趋势,从45 d开始呈上升趋势,直至仔猪出生后第7 d达到峰值。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1TCONS_00815878表达特征及核质定位结果
A:lncRNA TCONS_00815878在猪不同组织中的表达量;B:TCONS_00815878从猪胚胎期到出生后不同时间点表达量;C:TCONS_00815878核质定位结果。
Fig. 1Expression characteristics and nuclear localization analysis results of TCONS_00815878
提取分化24 h猪骨骼肌卫星细胞中细胞质和细胞核RNA,qRT-PCR检测TCONS_00815878表达量(图1C)。结果表明,TCONS_00815878在细胞核中高表达,推测其可能在猪骨骼肌卫星细胞的细胞核中发挥作用。
2.2 TCONS_00815878对猪骨骼肌卫星细胞分化的影响
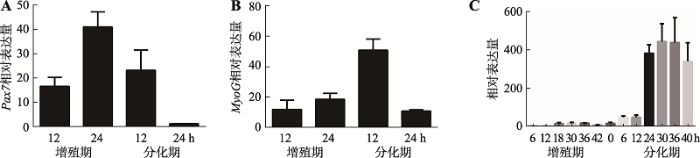
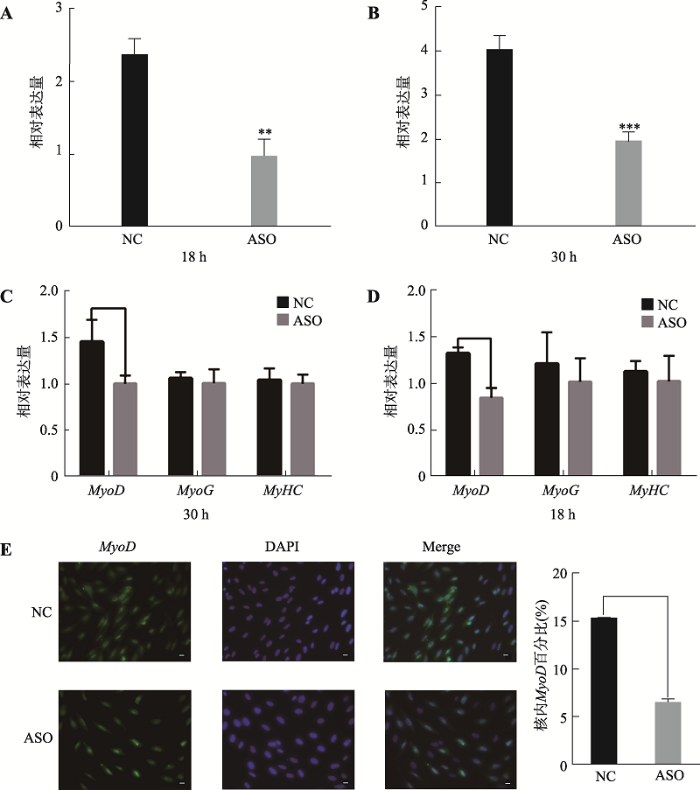
收集增殖期12 h、24 h和分化期12 h、24 h猪骨骼肌卫星细胞,qRT-PCR检验骨骼肌细胞体外增殖及诱导分化效果。结果表明,增殖期标记基因Pax7和Myf5在猪骨骼肌卫星细胞增殖期呈上升趋势,至增值期24 h达到峰值,之后在分化期表达量不断下降(图2A;附图1A)。分化期标记基因MyoD和MyHC表达量在猪骨骼肌卫星细胞增殖期呈下降趋势,分化初期表达量上升,末期表达量下降。分化期标记基因MyoG在猪骨骼肌卫星细胞中的表达量,从增殖期到分化期12 h呈上升趋势,之后下降(图2B;附图1,B和C)。表明猪骨骼肌卫星细胞体外诱导分化成功。收集增殖期6 h、12 h、18 h、30 h、36 h和42 h及分化期6 h、12 h、24 h、30 h、36 h和42 h的猪骨骼肌卫星细胞,提取总RNA,反转录得到cDNA,qRT-PCR检测猪骨骼肌卫星细胞增殖期和分化期不同时间点TCONS_00815878的表达量。结果表明,在猪骨骼肌卫星细胞增殖期,TCONS_00815878表达量普遍较低,分化初期呈上升趋势,分化30 h达到峰值,说明TCONS_00815878可能作用于猪骨骼肌卫星细胞分化过程(图2C)。因此,本研究通过网站在线设计ASO片段,在分化期猪骨骼肌卫星细胞中敲低TCONS_00815878。检测结果表明,在分化期18 h和30 h的猪骨骼肌卫星卫星细胞中,ASO敲低组与对照组相比TCONS_00815878表达量显著降低(P<0.001),表明ASO片段具有明显敲低效果,可 用于后续TCONS_00815878表达调控分析(图3,A和B)。
在分化18 h和30 h收集细胞,qRT-PCR检测猪骨骼肌卫星细胞分化标记基因MyoD、MyoG和MyHC的表达量。结果表明,在分化18 h和30 h,MyoD基因表达量显著下降(P<0.05),MyoG和MyHC基因呈下降趋势,推测TCONS_00815878可能促进猪骨骼肌卫星细胞分化(图3,C和D)。通过免疫荧光检测敲低后细胞样品中MyoD基因表达量,发现MyoD基因表达量显著下降(P<0.01) (图3E)。以上结果初步说明TCONS_00815878可能影响猪骨骼肌卫星细胞的分化过程。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2猪骨骼肌卫星细胞分化效果及TCONS_00815878在猪骨骼肌卫星细胞不同时期表达量
A,B:增殖期Pax7基因与分化期MyoG基因的表达;C:TCONS_00815878在猪骨骼肌卫星细胞增殖和分化不同时间点的表达量。
Fig. 2Results of differentiation of porcine skeletal muscle satellite cells and TCONS_00815878 expression levels in porcine skeletal muscle satellite cells at different stages
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3TCONS_00815878对猪骨骼肌卫星细胞分化影响
A,B:诱导分化18 h和30 h后TCONS_00815878表达量;C,D:敲低TCONS_00815878分化18 h和30 h后分化标记基因的表达情况;E:敲低TCONS_00815878后免疫荧光图片(400?)及MyoD基因表达量。比例尺为20 μm。*表示P<0.05,差异显著;**表示P<0.01,***表示P<0.001,差异极显著;NC:正常对照。
Fig. 3Effect of TCONS_00815878 on differentiation of porcine skeletal muscle satellite cells
2.3 TCONS_00815878靶基因功能预测及验证
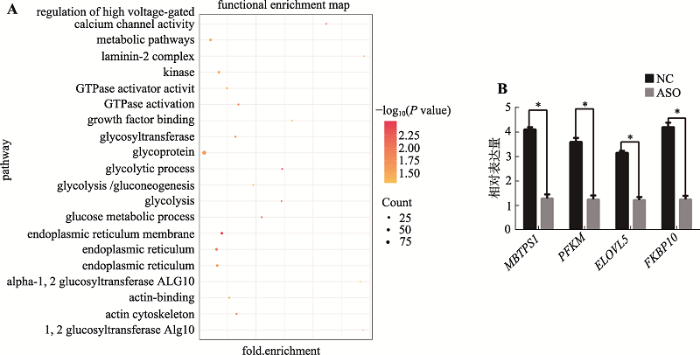
通过计算TCONS_00815878与其他蛋白编码基因的表达量相关性,预测得到TCONS_00815878的多个靶基因(附表1)。利用DAVID软件在线预测靶基因功能,推测TCONS_00815878可能通过调控其靶基因,参与生长因子合成、糖酵解和代谢等与骨骼肌发育有关的多个生物学过程(图4A,附表2)。其中PGK1、PFKM和GAPDH等基因参与糖酵解 代谢(glycolytic process,glycolysis和glycolysis/ gluconeogenesis)过程,ELOVL5、MBTPS1和FKBP10等基因参与生长因子结合(growth factor binding和GTPase activator activity)等过程。最后,本研究选取与TCONS_00815878表达呈正相关性的PFKM、ELOVL5、MBTPS1和FKBP10基因,利用qRT-PCR在敲低TCONS_00815878的细胞中检测其表达量。结果表明,MBTPS1、PFKM、ELOVL5和FKBP10基因表达量显著下降(P<0.05),验证了靶基因预测结果的准确性(图4B)。通过以上结果,提示TCONS_ 00815878可能通过调控其靶基因进而影响骨骼肌发育过程。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4TCONS_00815878靶基因功能预测及验证结果
A:GO富集分析和KEGG通路分析结果图;B:ASO敲低样品中靶基因表达量检测结果。*表示P<0.05,差异显著。
Fig. 4TCONS_00815878 target gene function prediction and verification results
3 讨论
运用高通量测序和生物信息学分析技术,目前已鉴定得到了大量的功能基因[15]。研究证实,人类基因组75%以上的基因被选择性地转录,但只有少部分被翻译,其余不具有任何编码能力的转录物被标注为非编码RNA(non-coding RNA, ncRNA)[16],其中长度大于200 nt的为lncRNA[17]。越来越多的研究表明,lncRNA参与多种细胞和组织的发育过程,如基因组印记、干细胞维持、胚胎发育及成肌过程等[18,19,20,21,22]。然而,目前lncRNA的功能研究,在猪上仍处于探索的初级阶段[23]。猪骨骼肌的发育主要取决于肌纤维的生成,胚胎期35~90 d的两次生长波以及出生后1~60 d肌纤维类型的转变,对骨骼肌最终的发育起关键作用[24,25]。本研究利用qRT-PCR检测大白猪胚胎期35~55 d及出生后7 d和200 d不同时间点后腿肌肉组织中TCONS_ 00815878表达量,结果推测该lncRNA可能在猪胚胎期和出生早期发挥作用。
猪骨骼肌卫星细胞是研究骨骼肌发育规律进行研究的理想材料[26],本研究利用猪骨骼肌卫星细胞进行TCONS_00815878核质定位实验,结果表明TCONS_00815878主要分布在猪骨骼肌卫星细胞的细胞核中。另外,在猪骨骼肌卫星细胞增殖与分化期不同时间点检测TCONS_00815878表达量,结果推测该基因主要在猪骨骼肌卫星细胞分化期发挥作用。在分化期猪骨骼肌卫星细胞中敲低TCONS_ 00815878,qRT-PCR和免疫荧光实验结果均表明,相较于对照组,敲低组中MyoD基因的表达量显著下降。以上结果推测TCONS_00815878可能促进猪骨骼肌卫星细胞的分化。
LncRNA作用范围广泛,调控机制复杂。根据lncRNA不同的作用模式分为顺式(cis)和反式(trans)两种调控模式,其中反式调控模式可以通过计算lncRNA与蛋白编码基因的表达量相关性预测lncRNA靶基因[27]。基于Tang等[12]分析得到的各基因表达量,本研究通过计算TCONS_00815878与其他蛋白编码基因的表达量相关性,找到其可能参与调控的多个靶基因。基因功能富集分析发现其靶基因富集到多个与肌肉发育相关的通路,其中PGK1、PFKM和GAPDH基因参与糖酵解代谢过程,ELOVL5、MBTPS1和FKBP10基因参与生长因子结合过程。已有研究表明,糖酵解过程可以通过促进成肌细胞增殖与分化来支持胚胎肌肉生长[28]。Mangano等[29]研究表明,ELOVL5基因可以通过促进多不饱和脂肪酸的合成间接促进肌肉的生长发育。而PGK1、PFKM和GAPDH基因作为糖酵解或丙酮酸代谢的关键基因,在肌肉生长发育中起促进作用[30]。为验证靶基因预测准确性,本研究利用qRT-PCR在敲低后细胞样品中检测与TCONS_00815878表达呈正相关性的PFKM、ELOVL5、MBTPS1和FKBP10基因表达量,发现PFKM、ELOVL5、MBTPS1和FKBP10基因表达量与对照组相比下降显著(P<0.05),由此推测TCONS_00815878可能通过调控其靶基因间接参与骨骼肌的发育。本研究初步探讨了lncRNA TCONS_ 00815878对猪骨骼肌卫星细胞分化可能起到的调控作用,为深入揭示lncRNA TCONS_00815878在猪骨骼肌卫星细胞分化和骨骼肌发育中的作用机制提供参考。
附录:
附表1、附表2和附图1见文章电子版Supplementary Table 1
附表1
附表1 靶基因列表
Supplementary Table 1
| 基因名称 | 基因名称 | 基因名称 | 基因名称 | 基因名称 | 基因名称 |
|---|---|---|---|---|---|
| ARFGAP3 | ADGRL4 | POLR3K | SLC25A26 | LAMC2 | ARHGEF10 |
| IFT27 | PGM1 | KCTD5 | MITF | INTS7 | PLXNA3 |
| PRPF40B | KIF2C | ADRA2B | GXYLT2 | LPGAT1 | REEP1 |
| IGFBP6 | SMOC2 | MAP4K4 | PPARG | HHAT | TECTA |
| NCKAP1L | CNKSR3 | TBC1D8 | GK5 | MGAT5 | SLC16A3 |
| STAT2 | NT5DC1 | CHST10 | P2RY14 | ADGRA2 | ADAMTS2 |
| BAZ2A | SNAP91 | ANKRD23 | SENP5 | HERC2 | ELOVL5 |
| RASSF8 | PSTPIP2 | SMYD1 | PARP14 | SCN3A | CES3 |
| KCNJ8 | CTIF | TMSB10 | CD47 | CALCRL | KDM4B |
| P3H3 | ZNF106 | DOK1 | OTC | TFPI | BBS9 |
| GAPDH | TYRO3 | DGUOK | ATP6AP2 | TMEFF2 | MPI |
| TNFRSF1A | RPAP1 | PREB | SLC9A7 | PLCL1 | PPIP5K2 |
| CRACR2A | ATP10A | NBAS | WDR45 | CARF | GREB1 |
| ITFG2 | ZNF516 | CD38 | SYNJ2 | PARD3B | PTPN6 |
| BID | SERPINB2 | KLHL2 | PGK1 | BARD1 | PXDC1 |
| SLC22A23 | DPP8 | GUCY1B1 | ZMAT1 | CYP27A1 | KDSR |
| SERPINB1 | ARF6 | SLC4A4 | ELF4 | PRKAG3 | ST6GAL1 |
| JARID2 | KANK1 | ARHGAP10 | ENOX2 | GPC1 | NOA1 |
| PHF1 | NPR2 | ANXA5 | FMR1 | HDLBP | IGFBP5 |
| VEGFA | TMEM8B | STPG2 | TMEM185A | SNED1 | SRPK2 |
| ZXDC | ZCCHC7 | MAPK10 | RENBP | NEB | SLC25A40 |
| TMEM266 | FKTN | PAQR3 | NADSYN1 | CAV2 | SCAP |
| LINGO1 | OLFML2A | FGF9 | BRMS1 | DBNL | ACSM5 |
| EDC3 | DNM1 | EBPL | EHBP1L1 | ZNF282 | PADI2 |
| AKAP5 | EXD3 | PHF11 | MAP4K2 | CDK3 | DCUN1D2 |
| GALNT16 | FBXL6 | MPHOSPH8 | MARK2 | GALK1 | CARHSP1 |
| IFT43 | MYC | DNAH10 | DTX4 | TANC2 | B4GALT2 |
| VASH1 | NCOA2 | TMEM120B | KBTBD4 | FKBP10 | CTC1 |
| IRF2BPL | SCYL3 | PPTC7 | ABTB2 | TBKBP1 | LMAN2L |
| SETD3 | F5 | ACAD10 | LMO2 | TSPOAP1 | CASP10 |
| WDR20 | PBX1 | HSPB8 | MPPED2 | ABR | STK36 |
| MBTPS1 | CD84 | NOS1 | TEAD1 | ANKFY1 | BDKRB2 |
| THAP11 | SLAMF8 | MIF | SCAMP4 | SMTNL2 | MED31 |
| SLC9A5 | SYT11 | LZTR1 | ANKRD24 | ENO3 | CENPH |
| YIF1B | CERS2 | DGCR2 | CHAF1A | BCL6B | PANK4 |
| ARHGEF1 | FMO5 | SIPA1L2 | PNPLA6 | MYH8 | ESYT3 |
| IRF2BP1 | ZNF697 | ARID5B | ZNF608 | SLC5A10 | OBSL1 |
| PLEKHA4 | TTF2 | SPOCK2 | PDLIM4 | ARHGAP23 | CASP8 |
| BCAT2 | CTTNBP2NL | CEP55 | IL5 | PFKM | FLYWCH1 |
| BAX | ARHGAP29 | TBC1D12 | TRPC7 | OXTR | VPS37C |
| CACNG6 | GLMN | SORBS1 | FZD4 | KCNA5 | PARVA |
| PUSL1 | TGFBR3 | ZDHHC6 | ARHGAP42 | CEP19 | PHACTR4 |
| MMEL1 | GBP2 | EIF3A | HINFP | DUS2 | RASIP1 |
| CLCN6 | HSPA12B | PPP1R12B | SORL1 | DIS3L2 | OLFM1 |
| NPPA | TTI1 | RPP25L | NTM | HS6ST1 | CCDC112 |
| VPS13D | KCNB1 | CACNB2 | PRELP | STK38L | HS1BP3 |
| DHRS3 | GUSB | CREM | SRI | WDR73 | AHR |
| FHAD1 | CTF1 | TRAK1 | CALCR | IL20RB | TBC1D23 |
| HMGN2 | SETD1A | PTH1R | CROT | CALM3 | LYSMD4 |
| EPB41 | REXO5 | TEX264 | PHTF2 | WDR4 | PDIK1L |
| CLSPN | ZNF174 | NISCH | LAMB1 | NFIA | |
| MOCOS | RHBDF1 | GLT8D1 | KIAA1614 | TIA1 |
新窗口打开|下载CSV
附图1
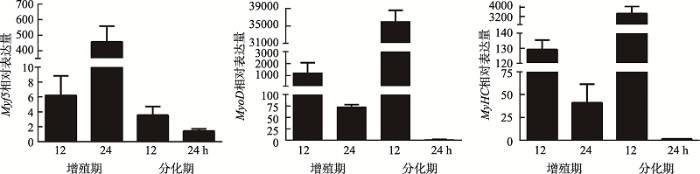 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图1猪骨骼肌卫星细胞增殖期和分化期标记基因的表达
A:Myf5基因表达量;B:MyoD基因表达量;C:MyHC基因表达量。
Supplementary Fig. 1Expression level of marker genes in proliferative and differentiation phases of porcine skeletal muscle satellite cells
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/nature11622URL [本文引用: 1]

For 10,000 years pigs and humans have shared a close and complex relationship. From domestication to modern breeding practices, humans have shaped the genomes of domestic pigs. Here we present the assembly and analysis of the genome sequence of a female domestic Duroc pig (Sus scrofa) and a comparison with the genomes of wild and domestic pigs from Europe and Asia. Wild pigs emerged in South East Asia and subsequently spread across Eurasia. Our results reveal a deep phylogenetic split between European and Asian wild boars similar to 1 million years ago, and a selective sweep analysis indicates selection on genes involved in RNA processing and regulation. Genes associated with immune response and olfaction exhibit fast evolution. Pigs have the largest repertoire of functional olfactory receptor genes, reflecting the importance of smell in this scavenging animal. The pig genome sequence provides an important resource for further improvements of this important livestock species, and our identification of many putative disease-causing variants extends the potential of the pig as a biomedical model.
DOI:10.1038/nrm.2016.7URLPMID:26956195 [本文引用: 1]
Satellite cells are adult myogenic stem cells that repair damaged muscle. The enduring capacity for muscle regeneration requires efficient satellite cell expansion after injury, their differentiation to produce myoblasts that can reconstitute damaged fibres and their self-renewal to replenish the muscle stem cell pool for subsequent rounds of injury and repair. Emerging studies indicate that misregulation of satellite cell fate and function can contribute to age-associated muscle dysfunction and influence the severity of muscle diseases, including Duchenne muscular dystrophy (DMD). It has also become apparent that satellite cell fate during muscle regeneration and ageing, and in the context of DMD, is governed by an intricate network of intrinsic and extrinsic regulators. Targeted manipulation of this network may offer unique opportunities for muscle regenerative medicine.
DOI:10.1038/nature07384URLPMID:18806774 [本文引用: 1]
Adult muscle satellite cells have a principal role in postnatal skeletal muscle growth and regeneration. Satellite cells reside as quiescent cells underneath the basal lamina that surrounds muscle fibres and respond to damage by giving rise to transient amplifying cells (progenitors) and myoblasts that fuse with myofibres. Recent experiments showed that, in contrast to cultured myoblasts, satellite cells freshly isolated or satellite cells derived from the transplantation of one intact myofibre contribute robustly to muscle repair. However, because satellite cells are known to be heterogeneous, clonal analysis is required to demonstrate stem cell function. Here we show that when a single luciferase-expressing muscle stem cell is transplanted into the muscle of mice it is capable of extensive proliferation, contributes to muscle fibres, and Pax7(+)luciferase(+) mononucleated cells can be readily re-isolated, providing evidence of muscle stem cell self-renewal. In addition, we show using in vivo bioluminescence imaging that the dynamics of muscle stem cell behaviour during muscle repair can be followed in a manner not possible using traditional retrospective histological analyses. By imaging luciferase activity, real-time quantitative and kinetic analyses show that donor-derived muscle stem cells proliferate and engraft rapidly after injection until homeostasis is reached. On injury, donor-derived mononucleated cells generate massive waves of cell proliferation. Together, these results show that the progeny of a single luciferase-expressing muscle stem cell can both self-renew and differentiate after transplantation in mice, providing new evidence at the clonal level that self-renewal is an autonomous property of a single adult muscle stem cell.
DOI:10.2527/jas1979.491115xURLPMID:500507 [本文引用: 1]
DOI:10.1016/j.gde.2006.08.008URL [本文引用: 1]
Continuing research on the onset of skeletal myogenesis in the somite is providing new insights into the behaviour of early myogenic progenitor cells and how signalling molecules affect cell fate decisions, in addition to subsequent muscle growth. Genetic manipulations have revealed new regulatory aspects, including the role of Six transcription factors and the CXCR4 cytokine receptor during embryonic myogenesis. An important recent development is the identification of a novel population of somite-derived cells that make a major contribution to muscle growth. These cells, which are characterised by the expression of Pax3 and Pax7, also give rise to the satellite cells of postnatal muscle. The relationship between Pax and Myogenic regulatory factors has been explored. Furthermore, Pax7 is now shown to be required for the maintenance of satellite cells. New approaches that permit the grafting of purified satellite cells demonstrate their capacity for efficient muscle repair and for self-renewal. Regeneration in amphibians is now also shown to involve Pax-positive progenitor cells.
DOI:10.1007/978-981-10-5203-3_10URLPMID:28815544 [本文引用: 1]
It is estimated that more than 90% of the mammalian genome is transcribed as non-coding RNAs. Recent evidences have established that these non-coding transcripts are not junk or just transcriptional noise, but they do serve important biological purpose. One of the rapidly expanding fields of this class of transcripts is the regulatory lncRNAs, which had been a major challenge in terms of their molecular functions and mechanisms of action. The emergence of high-throughput technologies and the development in various conventional approaches have led to the expansion of the lncRNA world. The combination of multidisciplinary approaches has proven to be essential to unravel the complexity of their regulatory networks and helped establish the importance of their existence. Here, we review the current methodologies available for discovering and investigating functions of long non-coding RNAs (lncRNAs) and focus on the powerful technological advancement available to specifically address their functional importance.
DOI:10.3724/SP.J.1005.2014.0456URL [本文引用: 1]
With the completion of Human Genome Project (HGP), it was revealed that among the 3 billion base pairs in human genome, only 1.5% of them encodes proteins. The remaining 98.5% of the sequence does not encode any protein, and was once regarded as accumulated “junk sequences” during evolution. However, in the subsequently initiated ENCODE project, it was unexpectedly found that about 75% of the human genome was transcribed into RNAs. Seventy-four percent of them are non-protein-coding RNAs (non-coding RNAs, ncRNAs). In this RNA category, most of the transcripts are longer than 200 nucleotides and thus named as “long non-coding RNAs (lncRNAs) ”. ncRNAs regulate gene expression at the transcriptional and post-transcriptional levels, function in fundamental biological processes including cell differentiation and organ development, and are closely associated with many human diseases. In this paper, we review the recent progress in the discovery, classification, expression, and function study of lncRNAs, as well as their roles in the pathogenesis of hu-man diseases.
DOI:10.3724/SP.J.1005.2014.0456URL [本文引用: 1]
With the completion of Human Genome Project (HGP), it was revealed that among the 3 billion base pairs in human genome, only 1.5% of them encodes proteins. The remaining 98.5% of the sequence does not encode any protein, and was once regarded as accumulated “junk sequences” during evolution. However, in the subsequently initiated ENCODE project, it was unexpectedly found that about 75% of the human genome was transcribed into RNAs. Seventy-four percent of them are non-protein-coding RNAs (non-coding RNAs, ncRNAs). In this RNA category, most of the transcripts are longer than 200 nucleotides and thus named as “long non-coding RNAs (lncRNAs) ”. ncRNAs regulate gene expression at the transcriptional and post-transcriptional levels, function in fundamental biological processes including cell differentiation and organ development, and are closely associated with many human diseases. In this paper, we review the recent progress in the discovery, classification, expression, and function study of lncRNAs, as well as their roles in the pathogenesis of hu-man diseases.
DOI:10.16288/j.yczz.17-120URLPMID:29254923 [本文引用: 1]
Long non-coding RNAs (lncRNAs) are important transcripts that are more than 200 nucleotides in length, and distribute extensively in animal and plant genomes. Accumulated studies demonstrate that lncRNAs play critical roles in biological processes related to embryogenesis, muscle development, lipid deposition and immune responses. They assist protein complexes in translocating to appropriate locations and participate in regulating gene activation and inactivation. Recently, rapid progress of lncRNA research is emerging, largely due to molecular biological technologies and information developed in the human genome project and the Encyclopedia of DNA Elements (ENCODE) project. For example, a dwarf open reading frame (DWORF) encoded by an annotated lncRNA was reported to activate the SERCA pump. Moreover, small regulatory polypeptide of amino acid response (SPAR) encoded by lncRNA LINC00961 was found to regulate muscle regeneration. These new results have revealed a novel model that lncRNA regulates biological processes using its small peptide product. In this review, we summarize the characteristics, databases, biological functions and molecular regulatory models, as well as research interests of lncRNAs in the future.
DOI:10.16288/j.yczz.17-120URLPMID:29254923 [本文引用: 1]
Long non-coding RNAs (lncRNAs) are important transcripts that are more than 200 nucleotides in length, and distribute extensively in animal and plant genomes. Accumulated studies demonstrate that lncRNAs play critical roles in biological processes related to embryogenesis, muscle development, lipid deposition and immune responses. They assist protein complexes in translocating to appropriate locations and participate in regulating gene activation and inactivation. Recently, rapid progress of lncRNA research is emerging, largely due to molecular biological technologies and information developed in the human genome project and the Encyclopedia of DNA Elements (ENCODE) project. For example, a dwarf open reading frame (DWORF) encoded by an annotated lncRNA was reported to activate the SERCA pump. Moreover, small regulatory polypeptide of amino acid response (SPAR) encoded by lncRNA LINC00961 was found to regulate muscle regeneration. These new results have revealed a novel model that lncRNA regulates biological processes using its small peptide product. In this review, we summarize the characteristics, databases, biological functions and molecular regulatory models, as well as research interests of lncRNAs in the future.
DOI:10.1111/age.12493URLPMID:27615279 [本文引用: 2]
Genome-wide association studies in livestock based on high-resolution genotyping and sequencing have revealed that the majority of signals associated with complex phenotypic traits are located outside of annotated protein-coding regions in the genome. The approaches of next-generation sequencing applied to whole transcriptome and chromatin profiles have provided information about existing genome-wide transcriptional activity and have revealed that the genomes are templates for thousands of long noncoding transcripts (lncRNAs). Despite their lack of coding capacity, many lncRNAs have been found to play functional roles in a variety of biological processes, which is adding a novel regulatory network to the complex structural organization and function of the genome. Here, we summarize main features of lncRNAs, provide an overview about computational tools and pipelines used for identification of lncRNAs from whole transcriptome datasets and review the current state of knowledge about lncRNAs in livestock species. Although lncRNAs are increasingly emerging as an integral component of the regulatory information encoded in the genome, the complexity of the transcriptomes in domesticated animals is inadequately characterized in comparison to human and mouse. Progress in elucidating whole transcriptomes of livestock species, including identification, functional annotation and characterization of lncRNAs, will be essential for a better understanding of basic biological processes associated with developmental, metabolic and immunological regulation and adaptation and phenotypic variation of complex traits in domesticated animals.
DOI:10.16288/j.yczz.18-053URLPMID:30369479 [本文引用: 1]
The long non-coding RNAs (lncRNAs) are a type of RNAs with more than 200nt in length and without any long open reading frame, but often have mRNA structural features. They can regulate the expression of target genes in different manners at the transcriptional and post-transcriptional levels. In recent years, various studies demonstrated that lncRNAs play crucial roles in adipogenesis. The long non-coding RNA, lnc-RAP3, located on the mouse chromosome 17, possesses a significantly differential expression pattern during mouse adipocyte differentiation; but its specific biological function (s) remains unclear. To investigate the effect of lnc-RAP3 on adipogenesis in the mouse 3T3-L1 preadipocytes, we first constructed a eukaryotic expression vector pcDNA3.1-RAP3. pcDNA3.1-RAP3 and synthetic RAP3-siRNAs were transfected individually into 3T3-L1 preadipocytes by Lipofectamine TM 2000, thereby over-expressing and knocking-down lnc-RAP3 expression, respectively. The transfected preadipocytes were induced to undergo adipogenic differentiation. Oil Red O staining and qRT-PCR were used to detect the effects of lnc-RAP3 overexpression/knockdown on 3T3-L1 preadipocyte differentiation. The results showed that overexpression of lnc-RAP3 led to a notable decrease in lipid accumulation (P<0.05) and remarkably reduced the mRNA expression levels of C/EBPα, Glut4, PPARγ, LPL and FAS on day 0, day 2, and day 4 post differentiation (P<0.05, P<0.01). In contrast, quantitative analysis of Oil Red O on day 4 of differentiation revealed that inhibition of lnc-RAP3 increased the formation of neutral lipid droplets (P<0.05). In addition, silencing lnc-RAP3 also significantly increased the mRNA expression of PPARγ, LPL, C/EBPα, FAS and Glut4 on day 0 and day 2 post differentiation (P<0.05, P<0.01). Our study suggests that lnc-RAP3 might suppress 3T3-L1 preadipocyte differentiation by affecting the expression of the genes involved in adipogenic differentiation.
DOI:10.16288/j.yczz.18-053URLPMID:30369479 [本文引用: 1]
The long non-coding RNAs (lncRNAs) are a type of RNAs with more than 200nt in length and without any long open reading frame, but often have mRNA structural features. They can regulate the expression of target genes in different manners at the transcriptional and post-transcriptional levels. In recent years, various studies demonstrated that lncRNAs play crucial roles in adipogenesis. The long non-coding RNA, lnc-RAP3, located on the mouse chromosome 17, possesses a significantly differential expression pattern during mouse adipocyte differentiation; but its specific biological function (s) remains unclear. To investigate the effect of lnc-RAP3 on adipogenesis in the mouse 3T3-L1 preadipocytes, we first constructed a eukaryotic expression vector pcDNA3.1-RAP3. pcDNA3.1-RAP3 and synthetic RAP3-siRNAs were transfected individually into 3T3-L1 preadipocytes by Lipofectamine TM 2000, thereby over-expressing and knocking-down lnc-RAP3 expression, respectively. The transfected preadipocytes were induced to undergo adipogenic differentiation. Oil Red O staining and qRT-PCR were used to detect the effects of lnc-RAP3 overexpression/knockdown on 3T3-L1 preadipocyte differentiation. The results showed that overexpression of lnc-RAP3 led to a notable decrease in lipid accumulation (P<0.05) and remarkably reduced the mRNA expression levels of C/EBPα, Glut4, PPARγ, LPL and FAS on day 0, day 2, and day 4 post differentiation (P<0.05, P<0.01). In contrast, quantitative analysis of Oil Red O on day 4 of differentiation revealed that inhibition of lnc-RAP3 increased the formation of neutral lipid droplets (P<0.05). In addition, silencing lnc-RAP3 also significantly increased the mRNA expression of PPARγ, LPL, C/EBPα, FAS and Glut4 on day 0 and day 2 post differentiation (P<0.05, P<0.01). Our study suggests that lnc-RAP3 might suppress 3T3-L1 preadipocyte differentiation by affecting the expression of the genes involved in adipogenic differentiation.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/srep43166URLPMID:28233874 [本文引用: 2]
Despite modest sequence conservation and rapid evolution, long non-coding RNAs (lncRNAs) appear to be conserved in expression pattern and function. However, analysis of lncRNAs across tissues and developmental stages remains largely uncharacterized in mammals. Here, we systematically investigated the lncRNAs of the Guizhou miniature pig (Sus scrofa), which was widely used as biomedical model. We performed RNA sequencing across 9 organs and 3 developmental skeletal muscle, and developed a filtering pipeline to identify 10,813 lncRNAs (9,075 novel). Conservation patterns analysis revealed that 57% of pig lncRNAs showed homology to humans and mice based on genome alignment. 5,455 lncRNAs exhibited typical hallmarks of regulatory molecules, such as high spatio-temporal specificity. Notably, conserved lncRNAs exhibited higher tissue specificity than pig-specific lncRNAs and were significantly enriched in testis and ovary. Weighted co-expression network analysis revealed a set of conserved lncRNAs that are likely involved in postnatal muscle development. Based on the high degree of similarity in the structure, organization, and dynamic expression of pig lncRNAs compared with human and mouse lncRNAs, we propose that these lncRNAs play an important role in organ physiology and development in mammals. Our results provide a resource for studying animal evolution, morphological complexity, breeding, and biomedical research.
DOI:10.1093/gbe/evu113URLPMID:24891613 [本文引用: 1]
Thousands of long intergenic noncoding RNAs (lincRNAs) have been identified in the human and mouse genomes, some of which play important roles in fundamental biological processes. The pig is an important domesticated animal, however, pig lincRNAs remain poorly characterized and it is unknown if they were involved in the domestication of the pig. Here, we used available RNA-seq resources derived from 93 samples and expressed sequence tag data sets, and identified 6,621 lincRNA transcripts from 4,515 gene loci. Among the identified lincRNAs, some lincRNA genes exhibit synteny and sequence conservation, including linc-sscg2561, whose gene neighbor Dnmt3a is associated with emotional behaviors. Both linc-sscg2561 and Dnmt3a show differential expression in the frontal cortex between domesticated pigs and wild boars, suggesting a possible role in pig domestication. This study provides the first comprehensive genome-wide analysis of pig lincRNAs.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s11427-015-0349-4URLPMID:27294835 [本文引用: 1]
Bioinformatics methods for various RNA-seq data analyses are in fast evolution with the improvement of sequencing technologies. However, many challenges still exist in how to efficiently process the RNA-seq data to obtain accurate and comprehensive results. Here we reviewed the strategies for improving diverse transcriptomic studies and the annotation of genetic variants based on RNA-seq data. Mapping RNA-seq reads to the genome and transcriptome represent two distinct methods for quantifying the expression of genes/transcripts. Besides the known genes annotated in current databases, many novel genes/transcripts (especially those long noncoding RNAs) still can be identified on the reference genome using RNA-seq. Moreover, owing to the incompleteness of current reference genomes, some novel genes are missing from them. Genome- guided and de novo transcriptome reconstruction are two effective and complementary strategies for identifying those novel genes/transcripts on or beyond the reference genome. In addition, integrating the genes of distinct databases to conduct transcriptomics and genetics studies can improve the results of corresponding analyses.
DOI:10.1038/nature11233URL [本文引用: 1]
Eukaryotic cells make many types of primary and processed RNAs that are found either in specific subcellular compartments or throughout the cells. A complete catalogue of these RNAs is not yet available and their characteristic subcellular localizations are also poorly understood. Because RNA represents the direct output of the genetic information encoded by genomes and a significant proportion of a cell's regulatory capabilities are focused on its synthesis, processing, transport, modification and translation, the generation of such a catalogue is crucial for understanding genome function. Here we report evidence that three-quarters of the human genome is capable of being transcribed, as well as observations about the range and levels of expression, localization, processing fates, regulatory regions and modifications of almost all currently annotated and thousands of previously unannotated RNAs. These observations, taken together, prompt a redefinition of the concept of a gene.
DOI:10.1007/s10142-016-0524-xURLPMID:27681237 [本文引用: 1]
Long intergenic non-coding RNAs (lincRNAs) are defined as RNA transcripts that are longer than 200 nucleotides. By definition, these RNAs must not have open reading frames that encode proteins. Many of these transcripts are encoded by RNA polymerase II, are spliced, and are poly-adenylated. This final fact indicates that there is a trove of information about lincRNAs in databases such as the Gene Expression Omnibus (GEO), which is a repository for RNAseq and microarray data. Recent experiments indicate that there are upwards of 15,000 lincRNAs encoded by the human genome. The term &quot;intergenic&quot; refers to the identification of these transcripts from regions of the genome that do not contain protein-encoding genes. These regions coincide with what was once labeled as the &quot;junk DNA&quot; portions of our genomes, which, upon careful examination by whole genome RNA sequencing experiments, clearly encode RNA transcripts. LincRNAs also contain promoter- or enhancer-associated RNAs that are gene proximal and can be either in the sense or antisense orientation, relative to the protein-coding gene with which they are associated. In this review, we describe the functions of lincRNAs playing roles in biological processes such as gene expression control, scaffold formation, and epigenetic control.
DOI:10.1016/j.tig.2017.08.002URLPMID:28870653 [本文引用: 1]
The past decade has seen a major increase in the study of noncoding RNAs (ncRNAs). However, there remains a great deal of confusion and debate over the levels of functionality and mechanisms of action of the majority of these new transcripts. This Opinion article addresses several of these issues, focusing particularly on long ncRNAs (lncRNAs). We reemphasize the unique abilities of RNAs to form myriad structures as well as to interact with other RNAs, DNA, and proteins, which provide them with unique and powerful abilities. One of these, the ability to interact sequence specifically with DNA, has been largely overlooked. Accumulating evidence suggests that evolution has taken advantage of RNA's properties via the rapid acquisition of new noncoding genes in testes, with subsequent gains of function in other tissues. This amplification process appears to be one of the major forces driving metazoan evolution and diversity.
DOI:10.1016/j.canlet.2017.12.015URLPMID:29253523 [本文引用: 1]
Skeletal muscle myogenesis during development and the injury induced regeneration contribute to the formation and maintenance of muscle tissue. Emerging studies have demonstrated that long non-coding RNAs (lncRNAs) participate in the regulation of gene expression during skeletal myogenesis and their aberrant expression is associated with several muscular diseases. In this review, we summarize recent studies of lncRNAs in the regulation of myogenesis and muscle diseases with mechanistic characterization. These findings have greatly enhanced our understanding of gene regulatory mechanisms governing muscle formation and regeneration, which will eventually lead to novel therapeutics against various muscle diseases.
DOI:10.1016/j.stem.2014.05.014URL [本文引用: 1]
In recent years, long noncoding RNAs (IncRNAs) have emerged as an important class of regulators of gene expression. IncRNAs exhibit several distinctive features that confer unique regulatory functions, including exquisite cell-and tissue-specific expression and the capacity to transduce higher-order spatial information. Here we review evidence showing that lncRNAs exert critical functions in adult tissue stem cells, including skin, brain, and muscle, as well as in developmental patterning and pluripotency. We highlight new approaches for ascribing IncRNA functions and discuss mammalian dosage compensation as a classic example of an lncRNA network coupled to stem cell differentiation.
DOI:10.16288/j.yczz.17-193URLPMID:29258985 [本文引用: 1]
As one of the first identified long non-coding RNAs (lncRNAs), H19 plays a wide range of roles in vivo, including not only as a tumor suppressor and oncogene involved in disease process, but also as a regulator of growth and development of multiple tissues in mammalian embryos. The function of H19 in muscles (both skeletal and cardiac muscle) draws widespread attention due to the following two reasons. On one hand, H19 promotes myogenic differentiation and myogenesis of skeletal muscle satellite cells (SMSCs) via regulating Igf2 in cis. On the other hand, H19 also modulates the target genes in trans, including sponging let-7, miR-106 or miR-29 to mediate myocyte glucose uptake, cardiomyocyte proliferation and tendon repair, as well as promote embryonic development and muscle regeneration through binding to MBD1 as a chromatin modifier. In this review, we summarize the role of H19 in mammalian muscles, which will provide a reference for further research to unveil the molecular mechanism of muscle growth and development.
DOI:10.16288/j.yczz.17-193URLPMID:29258985 [本文引用: 1]
As one of the first identified long non-coding RNAs (lncRNAs), H19 plays a wide range of roles in vivo, including not only as a tumor suppressor and oncogene involved in disease process, but also as a regulator of growth and development of multiple tissues in mammalian embryos. The function of H19 in muscles (both skeletal and cardiac muscle) draws widespread attention due to the following two reasons. On one hand, H19 promotes myogenic differentiation and myogenesis of skeletal muscle satellite cells (SMSCs) via regulating Igf2 in cis. On the other hand, H19 also modulates the target genes in trans, including sponging let-7, miR-106 or miR-29 to mediate myocyte glucose uptake, cardiomyocyte proliferation and tendon repair, as well as promote embryonic development and muscle regeneration through binding to MBD1 as a chromatin modifier. In this review, we summarize the role of H19 in mammalian muscles, which will provide a reference for further research to unveil the molecular mechanism of muscle growth and development.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1051/rnd:2002035URLPMID:12537254 [本文引用: 1]
In farm animals (bovine, ovine, swine, rabbit and poultry), muscle fibre characteristics play a key role in meat quality. The present review summarises the knowledge on muscle fibre characteristics and ontogenesis in these species. Myofibre ontogenesis begins very early during embryonic life, with the appearance of two or three successive waves of myoblasts which constitute the origin of the different types of muscle fibres. In small animals (rodents and poultry), a primary and a secondary generation of fibres arise respectively during the embryonic and foetal stages of development. In the largest species (bovines, sheep, pigs) a third generation arises in the late foetal or early postnatal period. Following these two or three waves of myogenesis, the total number of fibres is fixed. This occurs during foetal life (bovines, ovines, pigs and poultry) or during the first postnatal month in rabbits. Contractile and metabolic differentiation proceed by steps in parallel to myogenesis and are partially linked to each other. In bovines and ovines, the main events occur during foetal life, whereas they occur soon after birth in the pig, poultry and rabbit, but some plasticity remains later in life in all species. This comparative survey shows that the cellular processes of differentiation are comparable between species, while their timing is usually species specific.
DOI:10.1016/0309-1740(86)90026-4URLPMID:22054929 [本文引用: 1]
Post-natal changes in histoenzymatic and enzymatic characteristics were studied in porcine longissimus, psoas major and tibialis cranialis muscles. The experiment was carried out on thirty-five female large White pigs: fifteen were slaughtered between birth and 120 kg body weight (BW) and twenty at the market liveweight of 100 kg. The relative growth of each of the three muscles in relation to BW appeared to be monophasic, the allometric growth rates differed significantly between muscles: 1·21, 1·08 and 0·97, respectively. Myofibers were classified according to their contractile and metabolic properties as βR, αR and αW fibers. In the three muscles, the percentage of β fibers increased from birth up to 2 months of age and little change occurred thereafter. Glycolytic fibers were detectable only from about one month onwards. Their percentage then increased rapidly up to 30-50 kg BW to reach 60%, 30% and 12% in longissimus, psoas major and tibialis cranialis muscles, respectively. Thereafter, these values only changed slightly. This histoenzymatic differentiation led to the typical type grouping found in pigs. Throughout the period studied, the cross-sectional area of the myofibers increased with some differences according to fiber type and muscle. Lactate dehydrogenase (LDH) specific activity was similar in the three muscles at birth and then increased rapidly up to 4-5kg BW (15-20 days of age), although to a greater extent in the future glycolytic muscle (longissimus) This increase then abruptly slowed down. The period between 15 and 20 days of age corresponds to a critical stage already known in the rearing of pigs, i.e. growth rate, body composition and food intake. Changes in isocitrate dehydrogenase (ICDH) specific activity were much slighter and differed according to the muscle. There were close correlations between percentages of αW fibers and enzymatic kinetic measurements from one month onwards.
DOI:10.1006/excr.2002.5653URLPMID:12441128 [本文引用: 1]
The satellite cell compartment provides skeletal muscle with a remarkable capacity for regeneration. Here, we have used isolated myofibers to investigate the activation and proliferative potential of satellite cells. We have previously shown that satellite cells are heterogeneous: the majority express Myf5 and M-cadherin protein, presumably reflecting commitment to myogenesis, while a minority is negative for both. Although MyoD is rarely detected in quiescent satellite cells, over 98% of satellite cells contain MyoD within 24 h of stimulation. Significantly, MyoD is only observed in cells that are already expressing Myf5. In contrast, a minority population does not activate by the criteria of Myf5 or MyoD expression. Following the synchronous activation of the myogenic regulatory factor+ve satellite cells, their daughter myoblasts proliferate with a doubling time of approximately 17 h, irrespective of the fiber type (type I, IIa, or IIb) from which they originate. Although fast myofibers have fewer associated satellite cells than slow, and accordingly produce fewer myoblasts, each myofiber phenotype is associated with a complement of satellite cells that has sufficient proliferative potential to fully regenerate the parent myofiber within 4 days. This time course is similar to that observed in vivo following acute injury and indicates that cells other than satellite cells are not required for complete myofiber regeneration.
DOI:10.1093/nar/gkq1348URLPMID:21247874 [本文引用: 1]
Although accumulating evidence has provided insight into the various functions of long-non-coding RNAs (lncRNAs), the exact functions of the majority of such transcripts are still unknown. Here, we report the first computational annotation of lncRNA functions based on public microarray expression profiles. A coding-non-coding gene co-expression (CNC) network was constructed from re-annotated Affymetrix Mouse Genome Array data. Probable functions for altogether 340 lncRNAs were predicted based on topological or other network characteristics, such as module sharing, association with network hubs and combinations of co-expression and genomic adjacency. The functions annotated to the lncRNAs mainly involve organ or tissue development (e.g. neuron, eye and muscle development), cellular transport (e.g. neuronal transport and sodium ion, acid or lipid transport) or metabolic processes (e.g. involving macromolecules, phosphocreatine and tyrosine).
DOI:10.1073/pnas.0330960100URLPMID:12677001 [本文引用: 1]
The nemaline myopathies (NMs) are a clinically and genetically heterogeneous group of disorders characterized by nemaline rods and skeletal muscle weakness. Mutations in five sarcomeric thin filament genes have been identified. However, the molecular consequences of these mutations are unknown. Using Affymetrix oligonucleotide microarrays, we have analyzed the expression patterns of &gt;21,000 genes and expressed sequence tags in skeletal muscles of 12 NM patients and 21 controls. Multiple complementary approaches were used for data analysis, including geometric fold analysis, two-tailed unequal variance t test, hierarchical clustering, relevance network, and nearest-neighbor analysis. We report the identification of high satellite cell populations in NM and the significant down-regulation of transcripts for key enzymes of glucose and glycogen metabolism as well as a possible regulator of fatty acid metabolism, UCP3. Interestingly, transcript level changes of multiple genes suggest possible changes in Ca(2+) homeostasis. The increased expression of multiple structural proteins was consistent with increased fibrosis. This comprehensive study of downstream molecular consequences of NM gene mutations provides insights in the cellular events leading to the NM phenotype.
DOI:10.1007/s11914-013-0149-0URLPMID:23857286 [本文引用: 1]
Age-related bone and muscle loss are major public health problems. Investigational therapies to reduce these losses include anti-inflammatory dietary supplementations, such as polyunsaturated fatty acids (PUFA). Surprisingly, this topic has received little attention in the osteoporosis community. Recent research highlights the role of PUFA in inflammatory regulation of bone remodeling via cellular pathways. Emerging research suggests significant roles for PUFA in reducing bone and muscle loss with aging; however, findings are conflicted for PUFA and fracture risk. Limited studies suggest a relation between higher omega-3 FA and better muscle/bone in older adults. This review highlights new research since 2008 and synthesizes our current understanding of PUFA in relation to bone and muscle. Across study designs, evidence indicates that PUFA has positive effects upon bone. As data are sparse, future clinical trials and prospective studies are important to determine the long term benefits of PUFA supplementation upon bone and muscle outcomes.
DOI:10.4239/wjd.v6.i4.626URLPMID:25987960 [本文引用: 1]
Under normal metabolic conditions insulin stimulates microvascular perfusion (capillary recruitment) of skeletal muscle and subcutaneous adipose tissue and thus increases blood flow mainly after meal ingestion or physical exercise. This helps the delivery of insulin itself but also that of substrates and of other signalling molecules to multiple tissues beds and facilitates glucose disposal and lipid kinetics. This effect is impaired in insulin resistance and type 2 diabetes early in the development of metabolic dysregulation and reflects early-onset endothelial dysfunction. Failure of insulin to increase muscle and adipose tissue blood flow results in decreased glucose handling. In fat depots, a blunted postprandial blood flow response will result in an insufficient suppression of lipolysis and an increased spill over of fatty acids in the circulation, leading to a more pronounced insulin resistant state in skeletal muscle. This defect in blood flow response is apparent even in the prediabetic state, implying that it is a facet of insulin resistance and exists long before overt hyperglycaemia develops. The following review intends to summarize the contribution of blood flow impairment to the development of the atherogenic dysglycemia and dyslipidaemia.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}