 ,1,2, 孙玉洁,1,2
,1,2, 孙玉洁,1,2DNMT3a mediates paclitaxel-induced abnormal expression of LINE-1 by increasing the intragenic methylation
Xinyuan Wang1,2, Yu Zhang1,2, Nan Yang1, He Cheng,1,2, Yujie Sun,1,2通讯作者: 孙玉洁,博士,教授,研究方向:增强子生物学功能、肿瘤耐药形成机制。E-mail:yujiesun@njmu.edu.cn;程禾,博士,讲师,研究方向:乳腺癌耐药形成机制。E-mail:chenghe@njmu.edu.cn
编委: 宋旭
收稿日期:2019-10-29修回日期:2019-12-27网络出版日期:2020-01-20
| 基金资助: |
Editorial board:
Received:2019-10-29Revised:2019-12-27Online:2020-01-20
| Fund supported: |
作者简介 About authors
王昕源,硕士,专业方向:LINE-1的异常激活机制。E-mail:wangxinyuan2727@gmail.com。

摘要
药物诱导的长散在重复序列LINE-1异常激活可促进细胞基因组不稳定,而基因组不稳定是促进肿瘤发生发展和耐药表型形成的重要因素。因此,探索LINE-1异常激活的分子机制具有重要的理论和临床意义。DNA甲基化是调控基因表达的重要方式,已知DNA甲基转移酶家族成员DNMT3a不仅能通过促进基因启动子甲基化抑制基因表达,还可通过增强基因内部甲基化上调基因表达。本实验室前期研究发现,将乳腺癌细胞暴露于化疗药物可诱导LINE-1异常高表达,但LINE-1启动子甲基化水平并无显著改变。本研究进一步探讨了在化疗药物压力下DNMT3a是否可通过增强LINE-1基因内部甲基化水平促进LINE-1在乳腺癌细胞中的异常高表达。ChIP实验和甲基分析结果显示,用化疗药物紫杉醇(PTX)处理乳腺癌细胞,不仅可以诱导DNMT3a表达,而且可以促进DNMT3a与LINE-1基因内部区域的结合,提升其基因内部甲基化水平, 进而上调LINE-1的表达水平。利用表达载体增加细胞内DNMT3a的表达水平,可显著上调LINE-1基因内部的甲基化及基因的表达水平,而下调DNMT3a的表达可有效抑制LINE-1表达。上述研究结果表明,DNMT3a介导的基因非启动子区甲基化在药物诱导的LINE-1异常激活中发挥重要作用,为认识LINE-1在乳腺癌化疗耐药性形成过程中异常激活的机制提供了新思路。
关键词:
Abstract
The activation of long interspersed nuclear element-1 (LINE-1) leads to genomic instability, which promotes carcinogenesis and drug resistant. Therefore, exploring the mechanism underlying LINE-1 abnormal activation has the theoretical and clinical significance. DNA methylation is an important way to regulate gene expression. DNMT3a, one member of the DNA methyltransferase family, not only inhibits gene expression by inducing promoter hypermethylation, but also activates gene expression by increasing the intragenic DNA methylation. Our previous studies found that the expression of LINE-1 did not increase significantly in the promoter methylation in breast cancer cells treated with paclitaxel (PTX), a first-line chemotherapeutic drug for breast cancer. Here we explored whether DMNMT3a could directly mediate the drug-induced activation of LINE-1 in breast cancer cells through increasing the LINE-1 intragenic methylation. Our ChIP experiments and methyl analysis showed that treatment of breast cancer cells with PTX not only induced DNMT3a expression, but also promoted the binding of DNMT3a to the inner region of the LINE-1 gene to increase its methylation, resulting in upregulation of LINE-1 expression. Using expression vectors or RNA interference to alter the DNMT3a expression levels in the cells significantly changed the intragenic methylation degree and LINE-1 expression. Moreover, down-regulation of DNMT3a expression effectively inhibited the expression of LINE-1. These results indicate that DNMT3a-mediated intragenic methylation plays an important role in drug-induced abnormal activation of LINE-1, which provides a new idea for understanding the mechanism of abnormal activation of Line-1 induced by chemotherapy drug stress in breast cancer cells.
Keywords:
PDF (1188KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王昕源, 张雨, 杨楠, 程禾, 孙玉洁. DNMT3a通过提升基因内部甲基化介导紫杉醇诱导的LINE-1异常表达. 遗传[J], 2020, 42(1): 100-111 doi:10.16288/j.yczz.19-258
Xinyuan Wang.
乳腺癌作为女性最常见的恶性肿瘤之一,与结肠癌、肺癌并列为全世界发病率最高的三大癌症[1]。在中国,乳腺癌已经成为女性患病率最高的癌症(17.1%),并且其发病率逐年上升[2]。化学药物治疗(简称化疗)是乳腺癌系统性治疗中不可缺少的一部分,然而癌细胞对化疗药物产生耐受性是阻碍化疗成功的主要原因之一[3,4,5]。
长散在重复序列LINE-1(long interspersed nuclear element-1, LINE-1)是一种能够自主发生转座的“逆转录转座元件”[6,7],约占人类基因组的18%[8,9]。完整的LINE-1主要包括2个开放阅读框,分别编码Orf-1和Orf-2两种蛋白。其中Orf-1具有RNA结合能力,Orf-2具有核酸内切酶和逆转录酶活性。Orf-1蛋白翻译后能够识别并结合编码他们的信使RNA (message RNA, mRNA),与Orf-2形成核糖核蛋白复合体(ribonucleoprotein complex, RNP)进入细胞核,Orf-2蛋白的核酸内切酶活性能够识别基因组DNA 上的目标序列(AATTT)并对其进行切割,其逆转录酶活性能够利用暴露的TTT为引物,以LINE-1的RNA为模板逆转录产生新的LINE-1 DNA并拷贝整合入基因组,这一过程被称为“靶点引导的逆转录过程”(target-site primed reverse transcription, TPRT)[10,11,12]。LINE-1的逆转录过程不仅可能会破坏基因或非编码功能区域的完整性,还可能会导致DNA双链断 裂,进而使得细胞基因组发生更多的非等位基因同源 性重组以及对染色质重组的无效性修复[10,13,14]。LINE-1所表达的两个蛋白,亦能单独发挥其自身功能。例如Orf-1能够单独发挥其RNA结合能力,参与mRNA和小核RNA (small nuclear RNA, snRNA)在内的多种RNA的活化[15,16]。而Orf-2也能直接对基因组DNA双链进行切割从而导致DNA双链断 裂[14,17]。而这些现象最终都会导致细胞基因组不稳定性的形成。
基因组不稳定性是一种以遗传改变频率升高为主要特点的突发性的细胞状态[18]。研究表明,基因组的不稳定会增进肿瘤异质性、促进肿瘤的克隆演变,从而使多种肿瘤产生原发性耐药或获得性耐药表型[19,20,21]。LINE-1在正常乳腺细胞中一般处于沉默状态,而在多种亚型的乳腺癌中均异常激活[22,23]。有研究表明多种癌症中异常激活的LINE-1会加剧细胞基因组不稳定性从而增加肿瘤细胞的多样性,促进肿瘤的发生发展,并且还会导致肿瘤耐药性的形成[10,11,20,24,25]。因此,深入探索LINE-1在乳腺癌中异常激活的机制,对于提升乳腺癌临床治疗效果,以及改善患者预后均具有极为重要的现实意义。
DNA甲基化是由DNA甲基转移酶介导形成的表观遗传修饰方式之一,其功能与修饰区域在基因组上的相对位置密切相关。发生于启动子区域的甲基化一般与基因沉默相关[26],而发生于基因内部的甲基化被认为与基因表达激活和激活状态的维持相关[26]。DNA甲基转移酶家族(DNA methyltransferases, DNMTs)主要包括DNMT1、DNMT3a和DNMT3b。其中,DNMT1发挥“维持原有甲基化”的功能[27],而DNMT3a和DNMT3b则发挥“从头合成甲基化”(de nove methylation)的功能[28]。研究证明3种DNMTs均能作用于启动子甲基化[27],通过抑制转录因子在启动子区域的结合,抑制转录聚合体形成等方式抑制基因的表达。此外,DNMT3a还具有调控基因内部甲基化的功能,可通过直接提升基因内部甲基化水平,使得基因保持稳定持续的转录状态,从而促进基因的表达[28,29,30,31]。并且DNMT3a还能够在多种保守染色质区域上发挥其“从头合成甲基化”功能,建立新的甲基化位点,使得这些原本处于沉默状态的保守染色质区域重新激活[29,30,31]。但是目前为止,DNMT3a通过基因内部甲基化维持基因的转录或直接激活基因转录的潜在的机制还未有深入而详细的报道。
目前认为LINE-1启动子高水平甲基化修饰对于LINE-1转录具有显著抑制作用[32],但是关于基因内部甲基化能否调控LINE-1的转录依旧研究尚浅。本实验室前期研究结果也显示:当乳腺癌细胞暴露于化疗药物紫杉醇(paclitaxel, PTX)时,LINE-1表达水平的改变与其启动子甲基化水平变化并不存在显著相关性,这一结果引发了我们后续对LINE-1基因内部甲基化改变与LINE-1异常激活的关系的思考和探究。与处于LINE-1启动子区域的、密集存在并且形成CpG岛的CpG二核苷相比,LINE-1基因内部的CpG二核苷处于散在分布状态。但是在位于LINE-1转录起始位点之后,Orf-1开放阅读框内大约500 bp长度的序列上,却发现了相对于LINE-1基因其他区域更密集的CpG二核苷。因此,本研究旨在探索在化疗药物PTX作用下乳腺癌细胞中的DNMT3a能否通过提升LINE-1基因内部甲基化水平促进LINE-1的表达,进而提高乳腺癌细胞对化疗药物的耐受性。
1 材料与方法
1.1 细胞培养
人乳腺癌ZR-75-1细胞系、MCF-7细胞系、MDA-MB-468细胞系、MDA-MB-453细胞系、MDA-MB-231细胞系均购于美国ATCC公司。人乳腺癌细胞系MDA-MB-231培养于含有10%小牛血清(NCBS)、200 U/mL青霉素和100 U/mL链霉素的L15培养液中。人乳腺癌细胞系MDA-MB- 453、ZR-75-1和MDA-MB-468均培养于含有10%胎牛血清(FBS)、200 U/mL青霉素和100 U/mL链霉素的L15培养液中。人乳腺癌细胞系MCF-7培养与含有10%胎牛血清(FBS)、0.01 mg/mL胰岛素、200 U/mL青霉素和100 U/mL链霉素的MEM培养液中。上诉细胞均培养于37℃、5%CO2、饱和湿度的细胞培养箱中。所有实验均使用处于对数生长期的细胞。
1.2 脂质体介导的细胞转染
人乳腺癌细胞系以合适的密度(转染过表达质粒时细胞密度应为70%~90%,而转染si-RNA时细胞密度应为30%~50%)接种于相应的孔板中。接种16 h后,按使用说明书将适量的Lipofectamine 3000(Thermo Fisher,美国)均匀稀释于50倍体积的L15培养液中,之后将对应的DNMT3a过表达质粒或si-RNA (质粒和Lipo的质量体积比为2∶1,而si-RNA和Lipo的体积比也为2∶1)分别均匀稀释于50倍体积的L15培养液中(如果稀释的是质粒,则需要加入和Lipo相同体积的P3000,而如果稀释的是si-RNA则不用添加P3000),室温静置5 min。将稀释后的质粒或si-RNA和稀释后的Lipofectamine 3000混匀,室温孵育5 min。吸弃孔板中的原培养液,分别将合适体积的L15培养液加入每个培养皿。最后将孵育好的混合液分别加入对应的培养皿中,轻晃混匀。将培养皿放置于培养箱中静置培养4~6 h,之后更换细胞对应的完全培养液正常培养。转染后24 h、36 h、48 h后收样。1.3 总RNA提取和Real-time PCR
收集约1×106个细胞,使用Trizol法(Invitrogen,美国)提取细胞全RNA,采用微量紫外分光光度计测定RNA浓度以及纯度(D260/280在1.8~2.0之间为宜),500 ng RNA使用逆转录试剂盒(南京诺唯赞生物科技有限公司)逆转录为cDNA。以cDNA为模版,使用SYBR Green I Master Mix (TaKaRa, 日本)在Real-time PCR仪器上检测LINE-1 Orf-1和Orf-2 mRNA的相对表达量。反应条件为:95℃ 10 min,95℃ 10 s,60℃ 30 s,72℃ 20 s ;35个循环。采用2-ΔΔCt进行相对定量分析。所用引物序列见表1。Table 1
表1
表1Real-time PCR所用引物
Table 1
| 引物名称 | 引物序列(5′→3′) |
|---|---|
| L1-Orf-1 | F: CCCCAATCTAGCAAGG |
| R: AGAGATCCACTGTTAGT | |
| L1-Orf-2 | F: AAATGGTGCTGGGAAAACTG |
| R: GCCATTGCTTTTGGTGTTTT |
新窗口打开|下载CSV
1.4 Western blot分析
RIPA裂解法提取收集细胞总蛋白,采用BCA 法检测蛋白浓度。Western blot 法检测乳腺癌细胞系中DNMT3a蛋白表达量。SDS-PAGE进行蛋白分离,蛋白上样量为30 μg/孔,恒压电流,堆积胶80 V,分离胶120 V。湿转法进行蛋白转膜,冰浴中以100V恒压电转60 min。质量分数5%的脱脂奶粉室温封闭2 h。按照体积比1∶2000比例加入TBST稀释的DNMT3a抗体,4℃轻摇过夜。次日取出聚偏氟乙烯膜,TBST清洗3次,每次15 min。按照体积比1∶5000比例加入TBST稀释的二抗,室温轻摇2 h。取出二抗孵育的聚偏氟乙烯膜,TBST清洗3次,每次15 min。辣根过氧化物酶化学发光法显色。最后使用Image J软件对曝光所得的蛋白条带进行灰度分析。1.5 甲基化特异性PCR (methylation specific PCR, MSP)
收集约1×106个细胞,使用基因组DNA提取试剂盒(Axygen,美国)提取细胞基因组DNA,采用微量紫外分光光度计测定DNA浓度以及纯度。500 ng DNA使用重亚硫酸盐修饰试剂盒(ZYMO,美国)进行修饰。以修饰后的DNA作为模版,使用SYBR Primix Ex Taq II (TaKaRa,日本)在Real-time PCR仪器上检测LINE-1启动子区域和基因内部的甲基化修饰水平。LINE-1启动子区段对应的引物为LINE1- Promoter-MSP,而LINE-1基因内部两个区段S1和S2分别对应的引物为LINE1-GB1-MSP和LINE1- GB2-MSP。反应条件为:95℃ 10 min,95℃ 10 s,60℃ 30 s;40个循环。采用2-ΔΔCt进行相对定量分析。所用引物序列见表2。Table 2
表2
表2甲基化特异性PCR所用引物
Table 2
| 引物名称 | 引物序列(5′→3′) |
|---|---|
| LINE1- Promoter-MSP | F: GTCGAATAGGAATAGTTTCGG |
| R: ACTCCCTAACCCCTTACGCT | |
| LINE1-GB1- MSP | F: GTTTAGATTTAGGAAATATAGAGAAC |
| R: CTAACTTATAAAATTTCTACCGAA | |
| LINE1-GB2- MSP | F: GTTGGATGGAGAATGATTTTGAC |
| R: TTAATCACATCGACTCCTAAAAC | |
| Actin-MSP | F: TGGTGATGGAGGAGGTTTAGTAAG |
| R: AACCAATAAAACCTACTCCTCCCTTAA |
新窗口打开|下载CSV
1.6 染色质免疫共沉淀(chromatin immunoprecipitation, ChIP)
收集约1×107个细胞,使用ChIP试剂盒(Magna,美国)提取碎片化的基因组DNA片段,并将基因组DNA片段分装为3份。第一份基因组DNA片段和20 μL磁珠、3 μg DNMT3a抗体(Santa-Cruz,sc-20703,美国)混匀孵育过夜,第二份基因组DNA片段和20 μL磁珠、3 μL无免疫性IgG抗体混匀孵育过夜,第三份基因组DNA作为Input。孵育过后,磁珠将按照操作手册进行清洗并解交联得到DNA片段。使用解交联得到的DNA片段作为模版,在Real-time PCR仪器上检测LINE-1和DNMT3a是否存在相互作用,并且确定DNMT3a在LINE-1上的具体结合位置。引物LINE-1-SP对应的片段为LINE-1启动子中的片段SP,而引物LINE-1-SGB1所对应的片段为SGB1,引物LINE-1-SGB2所对应的片段为SGB2,以此类推。反应条件为:95℃ 10 min,95℃ 10 s,60℃ 30 s,72℃ 20 s ;35个循环。以Input的Ct值作为内参,采用2-ΔΔCt进行相对定量分析。所用引物序列见表3。Table 3
表3
表3染色质免疫共沉淀所用引物
Table 3
| 引物名称 | 引物序列(5′→3′) |
|---|---|
| LINE-1-SP | F: TCACTAGGGAGTGCCAGACAG |
| R: ATTTTCCAGGTGCGACCGTCA | |
| LINE-1-SGB1 | F: GTAGATAAAACCACAAAGATG |
| R: TTGACGAGCTGAGAGAAGAAG | |
| LINE-1-SGB2 | F: GGAACGCAGTTCCTCACCAGC |
| R: ATGTATAACTAGAATAACCAA | |
| LINE-1-SGB3 | F: GGCAAAGAAGTTGAAAACTTTG |
| R: TCAGCTCCATCAGCTCCTTTA | |
| LINE-1-SGB4 | F: AAGGAGCTGATGGAGCTGAAA |
| R: CTAAACTTCCCTTCTCGCTTCA | |
| LINE-1-SGB5 | F: CCGATGCGATCAACTGGAAGA |
| R: TAAACTTCCCTTCTCGCTTCA | |
| LINE-1-SGB6 | F: CCAAGAAATATGGGACTATGT |
| R: TAGATTGGGGAAGTTCTCCTG | |
| LINE-1-SGB7 | F: CAGGAGAACTTCCCCAATCTA |
| R: CTGGCTGCCCTTAACATTT | |
| LINE-1-SGB8 | F: CAGATTCACCAAAGTTGAAATG |
| R: CCACTCTCTTCTGGCTTGTAG |
新窗口打开|下载CSV
1.7 统计学分析
实验数据以3次实验的平均值和标准误(mean± SEM)表示。组间比较用t检验,P<0.05时,为差异具有统计学意义(用*表示),P<0.01时为差异显著(用**表示)。2 结果与分析
2.1 PTX处理促进MDA-MB-231乳腺癌细胞系中DNMT3a蛋白和LINE-1 mRNA的 表达并提升LINE-1基因内部区域甲基化水平
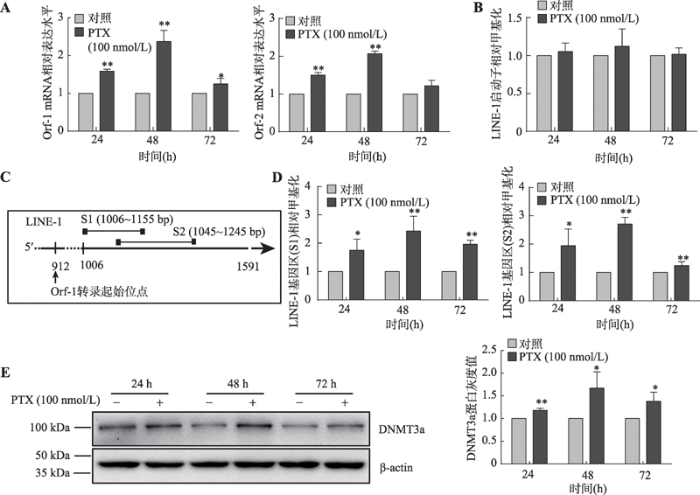
利用100 nmol/LPTX处理MDA-MB-231乳腺癌细胞后,结果发现细胞内LINE-1的mRNA表达水平显著升高(图1A)。为探究该现象与LINE-1启动子去甲基化是否存在相关性,本研究使用MSP检测LINE-1启动子甲基化水平,结果显示PTX处理前后LINE-1启动子甲基化水平并未发生显著性变化(P>0.05) (图1B)。由于LINE-1转录起始位点后,Orf-1开放阅读框中大约500 bp长度的区域内存在相对密集的CpG二核苷,为检测该区段的甲基化水平,设计了两对MSP引物(对应片段为S1和S2)(图1C),通过甲基化特异性PCR扩增这两个片段。
MSP实验结果显示:与未处理的MDA-MB-231细胞相比,经过PTX处理的细胞中片段S1和片段S2对应的LINE-1基因内部区段的甲基化水平显著上升(图1D),并且还伴随着细胞中DNMT3a蛋白表达上升(图1E)。
以上结果提示PTX处理引起的乳腺癌细胞中LINE-1mRNA表达水平的上升可能是通过提升LINE-1基因内部甲基化水平而非降低其启动子甲基化水平完成的,并且DNMT3a可能参与了该调节过程。
2.2 PTX诱导乳腺癌细胞中DNMT3a的表达并促进其与LINE-1基因内部区域的结合
为了证实上述推论,通过ChIP技术检测MDA- MB-231细胞中DNMT3a和LINE-1是否存在直接相互作用。实验选取LINE-1启动子区域和基因内部长度大约为800 bp的富含散在CpG二核苷的区域上分别设计引物,以探索DNMT3a在LINE-1的具体结合位置(图2A)。结果显示:在MDA-MB-231乳腺癌细胞中DNMT3a未与LINE-1启动子区域结合,而是结合在LINE-1基因内部(图2B),并且检测到的DNMT3a结合区域与检测LINE-1基因内部甲基化时引物LINE-1-GB1-MSP和LINE-1-GB2-MSP所对应的区域(片段S1和S2)相重合。使用100 nmol/L PTX处理细胞后,DNMT3a和LINE-1基因内部的结合增加(图2C)。
为进一步探究DNMT3a是否介导了PTX引起的LINE-1 mRNA表达水平上调,经PTX处理MDA- MB-231细胞后使用DNMT3asi-RNA干扰DNMT3a蛋白表达,并检测LINE-1的转录水平和其甲基化水平。结果表明:干扰DNMT3a蛋白的表达(图3A)可以部分逆转因PTX引起的LINE-1 mRNA上调(图3B)和基因内部甲基化水平的升高(图3C),且该过程中LINE-1启动子甲基化水平始终未发生显著性变化(P>0.05) (图3C)。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1PTX处理上调乳腺癌细胞中DNMT3a蛋白和LINE-1 mRNA表达并提升LINE-1基因内部而非启动子区的甲基化水平
A:Real-time PCR检测PTX处理后MDA-MB-231乳腺癌细胞中LINE-1的mRNA水平;B:MSP检测PTX处理后MDA-MB-231细胞中LINE-1启动子甲基化水平;C:利用甲基化特异性PCR扩增的LINE-1基因内部相应片段(S1和S2)位置示意图,片段位于LINE-1 Orf-1开放阅读框架中长度约500 bp富含CpG的区域;D:MSP检测PTX处理后MDA-MB-231细胞中LINE-1基因内部甲基化水平;E: Western blot检测PTX处理后MDA-MB-231细胞中DNMT3a蛋白的表达及相应的灰度分析结果。*: P<0.05表示有差异; **: P<0.01表示差异显著。
Fig. 1PTX treatment up-regulates the expression of DNMT3a proteins and LINE-1 mRNA in breast cancer cells and increases the methylation level of the LINE-1 gene body rather than the promoter region
以上结果表明:MDA-MB-231细胞在PTX刺激作用下能够通过提高DNMT3a蛋白的表达,使更多的DNMT3a蛋白和LINE-1基因区域结合,提升LINE-1基因内部甲基化水平。
2.3 DNMT3a通过提升LINE-1基因内部甲基化水平正向调控LINE-1的转录
为进一步探索乳腺癌细胞中DNMT3a与LINE-1 mRNA水平上调的关系,本研究检测了5种乳腺癌细胞系中DNMT3a蛋白的表达水平和LINE-1的mRNA水平,发现二者的表达呈现正相关性(图4,A和B)。随后选择在MDA-MB-231和MDA-MB-453两种乳腺癌细胞系中过表达DNMT3a蛋白,以探索LINE-1 mRNA表达水平否发生相应的上升。结果显示:在MDA-MB-231和MDA-MB-453两种乳腺癌细胞中过表达DNMT3a蛋白后(图5A),细胞中LINE-1 mRNA表达上调的同时(图5B),也促进了细胞内DNMT3a和LINE-1基因内部的结合(图5C)。与此同时两种乳腺癌细胞系中LINE-1启动子甲基化水平并未发生显著性变化(P>0.05),但是LINE-1基因内部甲基化水平却发生了显著提升(图5D)。
图2
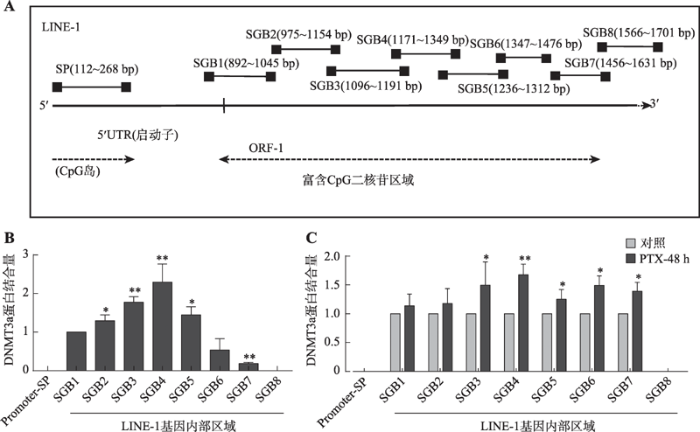 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2DNMT3a结合在LINE-1基因内部
A:LINE-1启动子区域和基因内部ChIP-PCR检测区段位置示意图;B:ChIP检测在MDA-MB-231乳腺癌细胞中DNMT3a蛋白与LINE-1启动子区和基因内部区域的结合情况;C:ChIP检测MDA-MB-231细胞经PTX处理后DNMT3a在LINE-1基因不同区段的结合情况。*: P<0.05表示有差异;**: P<0.01表示差异显著。
Fig. 2DNMT3a protein binds inside the LINE-1 gene body region
3 讨论
乳腺癌作为女性最为常见的恶性肿瘤,其发病率和死亡率多年以来一直高居女性肿瘤的首位,严重威胁着女性健康。随着医疗科技的发展,目前已经发展出多种治疗乳腺癌的方式,包括手术切除治疗、放疗、内分泌治疗、化疗以及生物靶向治疗。其中化疗仍是乳腺癌系统治疗的重要手段之一,但是肿瘤细胞耐药性的普遍产生已经成为了阻碍化疗成功的主要原因。因此研究探索乳腺癌耐药性产生机制对于进一步的推进化疗的有效性具有重要意义。长散在重复序列LINE-1的异常激活是影响细胞基因组不稳定性、增加细胞耐药性的因素之一。LINE-1在正常细胞中不表达而在多种肿瘤细胞(包括乳腺癌细胞)中异常激活[7]。有研究表明乳腺癌细胞中异常激活的LINE-1能够通过自身靶点引导的逆转录过程插入多种抑癌基因的基因区域[24],从而抑制这些基因的表达或直接破坏基因的完整性;或通过不同基因之间的同源重组概率、引起大片段的基因重复或缺失等途径使肿瘤细胞基因组不稳定性增加[14,15,16,17]。目前,人们对于乳腺癌耐药性形成的机制已经有了一定的研究,包括对药物造成的DNA损伤的精准修复[33]、全基因组甲基化水平紊乱推动耐药基因的异常表达[34]等。而基因组不稳定作为肿瘤的特征之一,也能通过增进肿瘤异质性、促进肿瘤的克隆演变[19,20,21]等方式促进肿瘤产生原发性耐药,或获得性耐药表型。因此研究LINE-1在乳腺癌如何被异常激活,对于了解乳腺癌细胞基因组不稳定性形成以及耐药性形成的原因具有重要的帮助作用。
多种肿瘤细胞中LINE-1的转录均受其启动子甲基化水平的负调控,但是本研究结果却发现MDA- MB-231乳腺癌细胞系在面对100 nmol/L PTX刺激时,LINE-1可以在维持启动子甲基化水平不变的情况下提高自身转录水平,与此同时LINE-1基因内部甲基化水平显著提升。与基因沉默相关联的启动子甲基化相比,基因内部甲基化常与多种基因的高表达相关联[35,36]。许多研究已经证明基因内部甲基化在多种生理学和疾病发生发展进程中通过转录调控、增强子活化、基因内部启动子沉默、反义链转录沉默等调控相应基因的表达和功能发挥[35,36,37]。因此这些结果提示在乳腺癌细胞面对PTX刺激时,细胞中LINE-1基因内部甲基化水平的升高可能导致LINE-1转录水平的提升。
图3
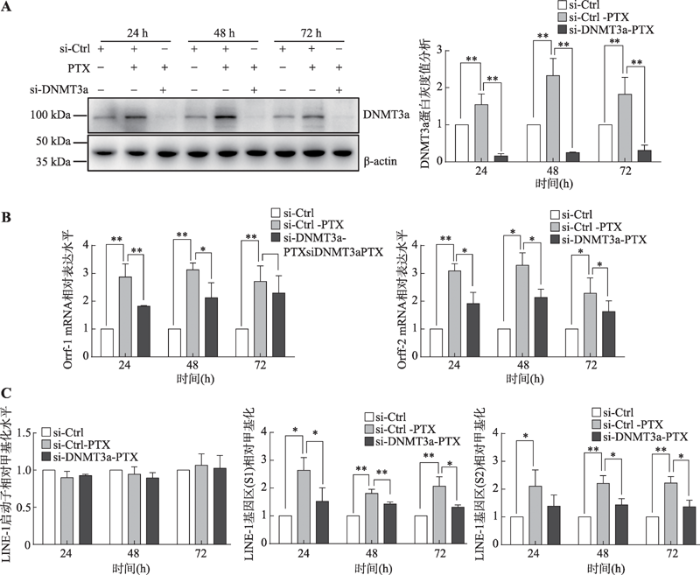 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3抑制DNMT3a蛋白的表达能部分逆转因PTX处理引起的LINE-1 mRNA表达水平的上升
A:Western blot检测MDA-MB-231乳腺癌细胞中si-DNMT3a对DNMT3a蛋白表达的抑制情况及相应的灰度分析结果;B:Real-time PCR检测抑制DNMT3a表达对PTX处理后LINE-1 mRNA表达情况的影响;C:MSP检测抑制DNMT3a表达对PTX处理后LINE-1启动子区和基因内部区段甲基化水平的影响。*:P<0.05表示有差异;**:P<0.01表示差异显著。
Fig. 3Inhibit the expression of DNMT3a could partly reverse the upregulation of LINE-1 mRNA induced by the PTX stimulation
在DNA甲基转移酶家族成员中,目前认为具有“从头合成甲基化”活性的DNMT3a和DNMT3b能够参与到基因内部甲基化的修饰调控过程中[30,31]。本研究结果显示:MDA-MB-231乳腺癌细胞中DNMT3a不仅能够和LINE-1基因内部区域发生直接的相互作用,并且在细胞面对PTX刺激时,细胞也能通过上调DNMT3a蛋白的表达从而提升LINE-1基因内部的甲基化水平,最终显著上调LINE-1的转录。且在两种DNMT3a和LINE-1均低表达的乳腺癌细胞(MDA-MB-231和MDA-MB-453)中直接过表达DNMT3a蛋白后发现,DNMT3a蛋白能够在不改变LINE-1在其启动子甲基化水平的同时,通过直接提升LINE-1基因内部甲基化水平从而正向调控LINE-1的转录。
图4
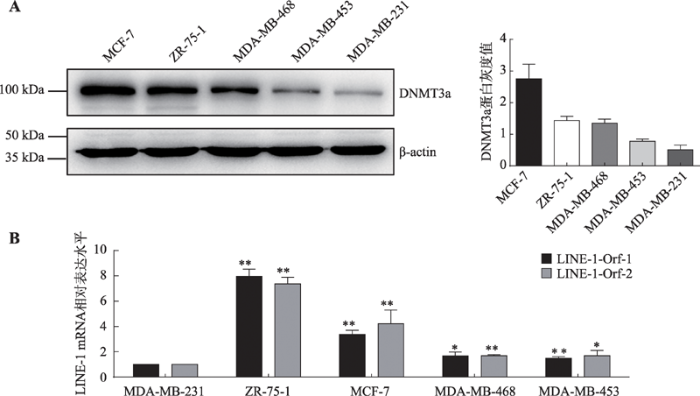 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4各个乳腺癌细胞系中DNMT3a蛋白表达和LINE-1 mRNA水平
A:Western blot检测5种乳腺癌细胞系中DNMT3a蛋白的表达水平;B:Real-time PCR检测5种乳腺癌细胞系中LINE-1 mRNA的表达水平。*:P<0.05表示有差异;**:P<0.01表示差异显著。
Fig. 4The expression of LINE-1 mRNA and DNMT3a protein levels in five breast cancer cells
但是基因内部甲基化上调LINE-1转录的机制还有待进一步的研究。有研究表明基因内部甲基化可能通过稳定转录单位(transcribed unit)的有序结构,使得RNA聚合酶能够更加顺利的在转录单位中延伸,提升转录单位获得参与转录的几率,从而正向调控基因的转录[36]。此外,很多研究结果显示DNA甲基化的改变能够影响相应区域内组蛋白甲基化的形成,并且反之亦然[38,39,40]。某些处于激活状态的基因,其启动子一般不存在H3K36me3而基因内部区域常带有高水平的H3K36me3。并且这样的基因启动子区一般不存在甲基化或处于低甲基化水平,而基因内部区域常常表现为高甲基化[41,42]。这表明H3K36me3和基因内部甲基化的出现呈现一致性。并且因为DNMT3a能够作用于基因内部甲基化的同时,其蛋白中的PWWP结构域与H3K36me3之间又存在直接相互作用[43],而H3K36me3作为转录激活性的组蛋白标志物,能够促进基因的转录。所以DNMT3a可能通过促进基因内部甲基化的形成从而招募转录激活性的H3K36me3组蛋白从而促进基因的转录[44]。而这些仍需要进一步的探索和研究。
在乳腺癌细胞中DNMT3a蛋白的表达可以作为LINE-1异常激活的因素之一,并且LINE-1的转录在受其启动子甲基化水平负调控的同时,也会受到其基因内部甲基化水平的正向调控。虽然目前的研究对于DNMT3a和乳腺癌耐药性的相关研究还比较少,但本研究结果显示,化疗药物作用后DNMT3a在乳腺癌组织中的表达上调,且正向调控LINE-1的转录,促进乳腺癌耐药表型的形成。这提示在乳腺癌的化疗过程中,在以PTX为主的化疗方案中同时抑制DNMT3a的表达可能会取得更好的化疗效果。但这一推测仍需要进一步的研究证实。
综上所述,本研究发现在乳腺癌细胞系中DNMT3a能够直接结合到LINE-1基因内部,通过提升LINE-1基因内部的散在分布的CpG二核苷的甲基化水平,从而正向调控LINE-1 mRNA的表达水平。并且在细胞面对化疗药物刺激时,DNMT3a也能通过相同的机制提升LINE-1 mRNA的表达水平。本研究结果为探索LINE-1在乳腺癌化疗耐药过程中异常激活的原因提供了一定的新思路。
图5
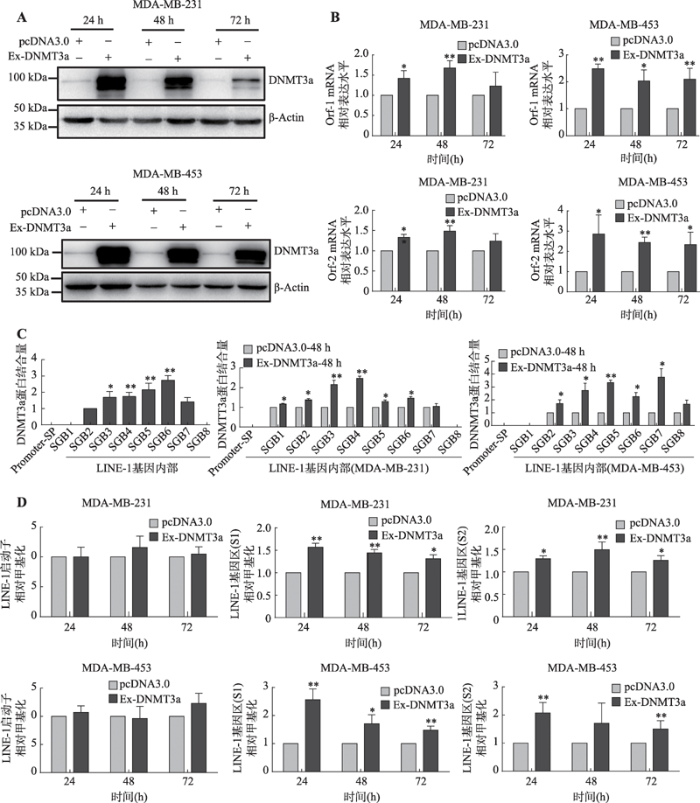 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5DNMT3a在乳腺癌细胞系中通过提升LINE-1基因内部甲基化水平从而正向调控LINE-1的转录
A:Western blot检测MDA-MB-231和MDA-MB-453乳腺癌细胞中转染DNMT3a过表达质粒后DNMT3a蛋白的表达情况;B:Real-time PCR检测在MDA-MB-231和MDA-MB-453乳腺癌细胞中过表达DNMT3a蛋白后LINE-1 mRNA的表达水平;C:ChIP检测过表达DNMT3a对其与LINE-1基因启动子区和基因内部区域结合情况的影响;D:MSP检测过表达DNMT3a对LINE-1启动子和基因内部甲基化水平的影响。*:P<0.05表示有差异;**:P<0.01表示差异显著。
Fig. 5DNMT3a up-regulates the transcription of LINE-1 in breast cancer cells by enhancing the methylation of the LINE-1 gene body region
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/S1015-9584(10)60017-6URL [本文引用: 1]

Objective
Age is a known risk factor for breast cancer behaviour. We studied the relationship of age with clinical characteristics, tumour pathology, therapeutic options and outcome in an affluent Asian population.Methods
From 2003 to 2008, data on newly diagnosed breast carcinoma patients under the care of the multidisciplinary breast cancer team based at a private hospital in Hong Kong were collected prospectively. Patients were divided into three groups: age < 40 years (group I), 41–69 years (group II), and ≥ 70 years (group III).Results
There were 2,079 patients: 334 in group I, 1,538 in group II and 148 in group III. The clinical presentation and tumour stages were similar. Younger patients had higher tumour grading (p = 0.000) and more lymphovascular permeation (p = 0.011). For older patients, combination therapy was employed less frequently (p < 0.0005), and more radical resection with less reconstructive procedures were performed (p = 0.000). The 3-year disease-free survival was 97.8% and there was no difference between the three groups.Conclusion
Although breast cancer in younger Chinese patients was more aggressive pathologically, the differences between clinical presentation, tumour staging and survival were similar. Treatment strategies should follow the clinical condition of the patient rather than age alone.DOI:10.1002/ijc.25516URLPMID:21351269 [本文引用: 1]
Estimates of the worldwide incidence and mortality from 27 cancers in 2008 have been prepared for 182 countries as part of the GLOBOCAN series published by the International Agency for Research on Cancer. In this article, we present the results for 20 world regions, summarizing the global patterns for the eight most common cancers. Overall, an estimated 12.7 million new cancer cases and 7.6 million cancer deaths occur in 2008, with 56% of new cancer cases and 63% of the cancer deaths occurring in the less developed regions of the world. The most commonly diagnosed cancers worldwide are lung (1.61 million, 12.7% of the total), breast (1.38 million, 10.9%) and colorectal cancers (1.23 million, 9.7%). The most common causes of cancer death are lung cancer (1.38 million, 18.2% of the total), stomach cancer (738,000 deaths, 9.7%) and liver cancer (696,000 deaths, 9.2%). Cancer is neither rare anywhere in the world, nor mainly confined to high-resource countries. Striking differences in the patterns of cancer from region to region are observed.
DOI:10.1016/j.mce.2015.09.035URLPMID:26455641 [本文引用: 1]
Tumors that express detectable levels of the product of the ESR1 gene (estrogen receptor-α; ERα) represent the single largest molecular subtype of breast cancer. More women eventually die from ERα+ breast cancer than from either HER2+ disease (almost half of which also express ERα) and/or from triple negative breast cancer (ERα-negative, progesterone receptor-negative, and HER2-negative). Antiestrogens and aromatase inhibitors are largely indistinguishable from each other in their abilities to improve overall survival and almost 50% of ERα+ breast cancers will eventually fail one or more of these endocrine interventions. The precise reasons why these therapies fail in ERα+ breast cancer remain largely unknown. Pharmacogenetic explanations for Tamoxifen resistance are controversial. The role of ERα mutations in endocrine resistance remains unclear. Targeting the growth factors and oncogenes most strongly correlated with endocrine resistance has proven mostly disappointing in their abilities to improve overall survival substantially, particularly in the metastatic setting. Nonetheless, there are new concepts in endocrine resistance that integrate molecular signaling, cellular metabolism, and stress responses including endoplasmic reticulum stress and the unfolded protein response (UPR) that provide novel insights and suggest innovative therapeutic targets. Encouraging evidence that drug combinations with CDK4/CDK6 inhibitors can extend recurrence free survival may yet translate to improvements in overall survival. Whether the improvements seen with immunotherapy in other cancers can be achieved in breast cancer remains to be determined, particularly for ERα+ breast cancers. This review explores the basic mechanisms of resistance to endocrine therapies, concluding with some new insights from systems biology approaches further implicating autophagy and the UPR in detail, and a brief discussion of exciting new avenues and future prospects.
DOI:10.1073/pnas.93.12.5925URLPMID:8650195 [本文引用: 1]
We have cloned a novel member of the nuclear receptor superfamily. The cDNA of clone 29 was isolated from a rat prostate cDNA library and it encodes a protein of 485 amino acid residues with a calculated molecular weight of 54.2 kDa. Clone 29 protein is unique in that it is highly homologous to the rat estrogen receptor (ER) protein, particularly in the DNA-binding domain (95%) and in the C-terminal ligand-binding domain (55%). Expression of clone 29 in rat tissues was investigated by in situ hybridization and prominent expression was found in prostate and ovary. In the prostate clone 29 is expressed in the epithelial cells of the secretory alveoli, whereas in the ovary the granuloma cells in primary, secondary, and mature follicles showed expression of clone 29. Saturation ligand-binding analysis of in vitro synthesized clone 29 protein revealed a single binding component for 17beta-estradiol (E2) with high affinity (Kd= 0.6 nM). In ligand-competition experiments the binding affinity decreased in the order E2 &gt; diethylstilbestrol &gt; estriol &gt; estrone &gt; 5alpha-androstane-3beta,17beta-diol &gt; testosterone = progesterone = corticosterone = 5alpha-androstane-3alpha,17beta-diol. In cotransfection experiments of Chinese hamster ovary cells with a clone 29 expression vector and an estrogen-regulated reporter gene, maximal stimulation (about 3-fold) of reporter gene activity was found during incubation with 10 nM of E2. Neither progesterone, testosterone, dexamethasone, thyroid hormone, all-trans-retinoic acid, nor 5alpha-androstane-3alpha,I7beta-diol could stimulate reporter gene activity, whereas estrone and 5alpha-androstane-3beta,17beta-diol did. We conclude that clone 29 cDNA encodes a novel rat ER, which we suggest be named rat ERbeta to distinguish it from the previously cloned ER (ERalpha) from rat uterus.
DOI:10.1097/COC.0b013e3181931049URLPMID:19675449 [本文引用: 1]
Resistance to chemotherapeutic agents is a significant obstacle to the effective treatment of metastatic breast cancer (MBC). Anthracycline- and taxane-based regimens are active as first-line treatment for MBC; however, MBC often progresses because of primary or acquired resistance to anthracyclines and taxanes. There are few options for the treatment of patients with anthracycline- and taxane-resistant or taxane-refractory MBC. This article reviews several single agents that have demonstrated activity as treatment for patients with MBC who progress during, or rapidly following, treatment with anthracyclines and taxanes. Results from clinical trials evaluating agents such as ixabepilone, albumin-bound paclitaxel, capecitabine, vinorelbine, pemetrexed, and irinotecan are presented. Single-agent capecitabine is approved for the treatment of patients after failure of anthracyclines and taxanes. Ixabepilone has demonstrated efficacy in patients with MBC resistant to multiple chemotherapeutic agents and is the only agent approved by the Food and Drug Administration as monotherapy for anthracycline-, taxane-, and capecitabine-resistant MBC. Improved treatment strategies and further evaluation of newer agents may reduce the current burden of treatment-resistant or treatment-refractory MBC.
DOI:10.1186/s13100-015-0041-9URLPMID:26045719 [本文引用: 1]
Repbase Update (RU) is a database of representative repeat sequences in eukaryotic genomes. Since its first development as a database of human repetitive sequences in 1992, RU has been serving as a well-curated reference database fundamental for almost all eukaryotic genome sequence analyses. Here, we introduce recent updates of RU, focusing on technical issues concerning the submission and updating of Repbase entries and will give short examples of using RU data. RU sincerely invites a broader submission of repeat sequences from the research community.
DOI:10.1101/gr.5558208URLPMID:18256243 [本文引用: 2]
Transposable elements (TEs) have shared an exceptionally long coexistence with their host organisms and have come to occupy a significant fraction of eukaryotic genomes. The bulk of the expansion occurring within mammalian genomes has arisen from the activity of type I retrotransposons, which amplify in a &quot;copy-and-paste&quot; fashion through an RNA intermediate. For better or worse, the sequences of these retrotransposons are now wedded to the genomes of their mammalian hosts. Although there are several reported instances of the positive contribution of mobile elements to their host genomes, these discoveries have occurred alongside growing evidence of the role of TEs in human disease and genetic instability. Here we examine, with a particular emphasis on human retrotransposon activity, several newly discovered aspects of mammalian retrotransposon biology. We consider their potential impact on host biology as well as their ultimate implications for the nature of the TE-host relationship.
DOI:10.1146/annurev-genom-082509-141802URLPMID:21801021 [本文引用: 1]
The completion of the human genome reference sequence ushered in a new era for the study and discovery of human transposable elements. It now is undeniable that transposable elements, historically dismissed as junk DNA, have had an instrumental role in sculpting the structure and function of our genomes. In particular, long interspersed element-1 (LINE-1 or L1) and short interspersed elements (SINEs) continue to affect our genome, and their movement can lead to sporadic cases of disease. Here, we briefly review the types of transposable elements present in the human genome and their mechanisms of mobility. We next highlight how advances in DNA sequencing and genomic technologies have enabled the discovery of novel retrotransposons in individual genomes. Finally, we discuss how L1-mediated retrotransposition events impact human genomes.
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/1475-2867-6-13URLPMID:16670018 [本文引用: 3]
Long interspersed nuclear elements (LINEs), Alu and endogenous retroviruses (ERVs) make up some 45% of human DNA. LINE-1 also called L1, is the most common family of non-LTR retrotransposons in the human genome and comprises about 17% of the genome. L1 elements require the integration into chromosomal target sites using L1-encoded endonuclease which creates staggering DNA breaks allowing the newly transposed L1 copies to integrate into the genome. L1 expression and retrotransposition in cancer cells might cause transcriptional deregulation, insertional mutations, DNA breaks, and an increased frequency of recombinations, contributing to genome instability. There is however little evidence on the mechanism of L1-induced genetic instability and its impact on cancer cell growth and proliferation.
DOI:10.1016/s0092-8674(00)81997-2URLPMID:8945517 [本文引用: 2]
Human L1 elements are highly abundant poly(A) (non-LTR) retrotransposons whose second open reading frame (ORF2) encodes a reverse transcriptase (RT). We have identified an endonuclease (EN) domain at the L1 ORF2 N-terminus that is highly conserved among poly(A) retrotransposons and resembles the apurinic/apyrimidinic (AP) endonucleases. Purified L1 EN protein (L1 ENp) makes 5'-PO4, 3'-OH nicks in supercoiled plasmids, shows no preference for AP sites, and preferentially cleaves sequences resembling L1 in vivo target sequences. Mutations in conserved amino acid residues of L1 EN abolish its nicking activity and eliminate L1 retrotransposition. We propose that L1 EN cleaves the target site for L1 insertion and primes reverse transcription.
DOI:10.1016/s0378-1119(98)00252-2URLPMID:9666081 [本文引用: 1]
Long interspersed elements, or LINEs, are retrotransposons that move via an RNA intermediate. In mice, one polymorphic variant of L1 has amplified relatively recently, giving rise to the A-type subfamily in species belonging to the genus and subgenus Mus. Retrotransposition of LINE-1 (L1) requires the function of the L1-encoded reverse transcriptase that is produced from open reading frame 2 (ORF2). Here, we employ a convenient yeast genetic assay to determine the reverse transcriptase activity of the ORF2 obtained from three A-type L1 elements: one, a cDNA from the RNA in ribonucleoprotein particles; another with a purported inactivating mutation; and the third, a hypothetical ancestral construct. Because there are no examples of A-type elements that have transposed recently to inactivate a gene, this assay is the first step towards demonstrating the functional capability of mouse A-type LINE-1 elements. One of the three elements was believed to have been inactivated during evolution by the substitution of leucine for a highly conserved phenylalanine or tryptophan residue among known reverse transcriptases. This mutation did not inactivate the L1 reverse transcriptase in the yeast assay; thus, all three of the elements tested encoded reverse transcriptase activity. We further examined the minimal reverse transcriptase domain within ORF2 by creating a series of deletions. The results demonstrate that removal of the L1 endonuclease domain from the N-terminal region of ORF2 does not affect reverse transcriptase activity as determined by this assay, and that approximately half of the ORF2 coding sequence from mouse A-type L1 elements is required for functional reverse transcriptase.
DOI:10.1093/nar/gkj522URLPMID:16507671 [本文引用: 1]
Long Interspersed Elements (LINE-1s, L1s) are the most active mobile elements in the human genome and account for a significant fraction of its mass. The propagation of L1 in the human genome requires disruption and repair of DNA at the site of integration. As Barbara McClintock first hypothesized, genotoxic stress may contribute to the mobilization of transposable elements, and conversely, element mobility may contribute to genotoxic stress. We tested the ability of genotoxic agents to increase L1 retrotransposition in a cultured cell assay. We observed that cells exposed to gamma radiation exhibited increased levels of L1 retrotransposition. The L1 retrotransposition frequency was proportional to the number of phosphorylated H2AX foci, an indicator of genotoxic stress. To explore the role of the L1 endonuclease in this context, endonuclease-deficient tagged L1 constructs were produced and tested for their activity in irradiated cells. The activity of the endonuclease-deficient L1 was very low in irradiated cells, suggesting that most L1 insertions in irradiated cells still use the L1 endonuclease. Consistent with this interpretation, DNA sequences that flank L1 insertions in irradiated cells harbored target site duplications. These results suggest that increased L1 retrotransposition in irradiated cells is endonuclease dependent. The mobilization of L1 in irradiated cells potentially contributes to genomic instability and could be a driving force for secondary mutations in patients undergoing radiation therapy.
.
DOI:10.1016/j.jmb.2006.01.089URLPMID:16490214 [本文引用: 3]
Long interspersed element-1 (L1) is an autonomous retroelement that is active in the human genome. The proposed mechanism of insertion for L1 suggests that cleavage of both strands of genomic DNA is required. We demonstrate that L1 expression leads to a high level of double-strand break (DSB) formation in DNA using immunolocalization of gamma-H2AX foci and the COMET assay. Similar to its role in mediating DSB repair in response to radiation, ATM is required for L1-induced gamma-H2AX foci and for L1 retrotransposition. This is the first characterization of a DNA repair response from expression of a non-long terminal repeat (non-LTR) retrotransposon in mammalian cells as well as the first demonstration that a host DNA repair gene is required for successful integration. Notably, the number of L1-induced DSBs is greater than the predicted numbers of successful insertions, suggesting a significant degree of inefficiency during the integration process. This result suggests that the endonuclease activity of endogenously expressed L1 elements could contribute to DSB formation in germ-line and somatic tissues.
DOI:10.1101/gr.5870107URLPMID:17416749 [本文引用: 2]
Long Interspersed Element-1 (LINE-1 or L1) sequences comprise approximately 17% of human DNA and ongoing L1 retrotransposition continues to impact genome evolution. The L1-encoded proteins also can mobilize other cellular RNAs (e.g., Alu retrotransposons, SVA retrotransposons, and U6 snRNAs), which comprise approximately 13% of human DNA. Here, we demonstrate that the trans-mediated mobilization of non-L1 RNAs can occur by either template choice or template-switching mechanisms. Remarkably, these mechanisms are not mutually exclusive, as both processes can operate sequentially on the same RNA template. Finally, we provide evidence that efficient U6 snRNA retrotransposition requires both ORF1p and ORF2p, providing indirect evidence for the action of ORF1p in U6 snRNA retrotransposition. Thus, we propose that the LINE-1-encoded reverse transcriptase can mediate the retrotransposition of non-L1 RNAs by distinct mechanisms.
DOI:10.1128/MCB.21.4.1429-1439.2001URLPMID:11158327 [本文引用: 2]
Long interspersed nuclear elements (LINEs or L1s) comprise approximately 17% of human DNA; however, only about 60 of the approximately 400,000 L1s are mobile. Using a retrotransposition assay in cultured human cells, we demonstrate that L1-encoded proteins predominantly mobilize the RNA that encodes them. At much lower levels, L1-encoded proteins can act in trans to promote retrotransposition of mutant L1s and other cellular mRNAs, creating processed pseudogenes. Mutant L1 RNAs are mobilized at 0.2 to 0.9% of the retrotransposition frequency of wild-type L1s, whereas cellular RNAs are mobilized at much lower frequencies (ca. 0.01 to 0.05% of wild-type levels). Thus, we conclude that L1-encoded proteins demonstrate a profound cis preference for their encoding RNA. This mechanism could enable L1 to remain retrotransposition competent in the presence of the overwhelming number of nonfunctional L1s present in human DNA.
DOI:10.1186/s13100-016-0064-xURLPMID:27099633 [本文引用: 2]
Approximately 17 % of the human genome is comprised of the Long INterspersed Element-1 (LINE-1 or L1) retrotransposon, the only currently active autonomous family of retroelements. Though L1 elements have helped to shape mammalian genome evolution over millions of years, L1 activity can also be mutagenic and result in human disease. L1 expression has the potential to contribute to genomic instability via retrotransposition and DNA double-strand breaks (DSBs). Additionally, L1 is responsible for structural genomic variations induced by other transposable elements such as Alu and SVA, which rely on the L1 ORF2 protein for their propagation. Most of the genomic damage associated with L1 activity originates with the endonuclease domain of the ORF2 protein, which nicks the DNA in preparation for target-primed reverse transcription.
DOI:10.1038/890URLPMID:9662389 [本文引用: 1]
URLPMID:12801837 [本文引用: 2]
The p210 bcr-abl fusion protein has a key role in the pathogenesis of chronic myeloid leukemia (CML). However, its influence on disease progression to blast crisis is marginal and mostly due to its effect of impairing the genomic stability of clonal myeloid progenitors through pathways still largely unknown.
DOI:10.1038/nature10762URL [本文引用: 3]
Cancers evolve by a reiterative process of clonal expansion, genetic diversification and clonal selection within the adaptive landscapes of tissue ecosystems. The dynamics are complex, with highly variable patterns of genetic diversity and resulting clonal architecture. Therapeutic intervention may destroy cancer clones and erode their habitats, but it can also inadvertently provide a potent selective pressure for the expansion of resistant variants. The inherently Darwinian character of cancer is the primary reason for this therapeutic failure, but it may also hold the key to more effective control.
DOI:10.1586/era.12.85URLPMID:23030222 [本文引用: 2]
There is burgeoning evidence to suggest that tumor evolution follows the laws of Darwinian evolution, whereby individual tumor cell clones harbor private genetic aberrations in addition to the founder mutations, and that these distinct populations of cancer cells interact in competitive and mutualistic manners. The combined effect of genetic and epigenetic instability, and differential selective pressures according to the microenvironment and therapeutic interventions, create many different evolutionary routes such that intratumor heterogeneity is inevitable. Numerous cytogenetic, comparative genomic hybridization and, more recently, massively parallel sequencing studies have generated indisputable evidence of this phenomenon. The impact of intratumor heterogeneity on response and resistance to therapy is beginning to be understood; this information may prove crucial for the potentials of personalized medicine to be realized. In this review, the evidence of intratumor heterogeneity in breast cancer, its potential causes and implications for the clinical management of breast cancer patients are discussed.
DOI:10.1007/s10549-012-2246-7URLPMID:23053642 [本文引用: 1]
Long interspersed nuclear element 1 (L1) belongs to a family of retrotransposons. Expression of the normally repressed L1 retrotransposons has been shown to induce genome instability by creating DNA double-stranded breaks and chromosomal rearrangements through the process of retrotransposition. At present, little is known about the expression of L1-encoded ORF1p and ORF2p which are indispensable for its retrotransposition activity. Given its potentially harmful effects on the genome, we investigated the implications of both ORF1p and ORF2p expression and their subcellular localization in a range of breast cancer cell lines and breast tumor tissues including 15 normal breast tissues, 25 fibroadenomas, 25 ductal carcinomas in situ (DCIS), and 95 invasive cancers. Clinicopathologic parameters and survival outcomes were investigated in association with the cytoplasmic and nuclear expression of ORF1p and ORF2p using univariate and multivariate analysis. High cytoplasmic expression of ORF1p and ORF2p was seen in DCIS tumors, but they were not related with survival outcome. The majority of invasive cancers were found to express both ORF1p and ORF2p in the cytoplasm, while nuclear expression was also seen in a subclass of those invasive cancers in the range of 28-31?%. Tumors with high nuclear expression of ORF1p and ORF2p were more significantly associated with lymph node metastasis (p?=?0.001) and the worst patient survival (p?&lt;?0.0001) than those with cytoplasmic expression. This is the first study examining the effects of both ORF1p and ORF2p expression in breast cancer tissues. Our observation shows altered expression patterns of ORF1p and ORF2p within invasive cancers, which are related to differences in overall patient survival. The differing patterns of both cytoplasmic and nuclear ORF1p and ORF2p expression indicate that further studies of the biology and function of L1 retrotransposons are required in breast cancer.
DOI:10.1371/journal.pone.0100429URLPMID:24971511 [本文引用: 1]
The changes in DNA methylation status in cancer cells are characterized by hypermethylation of promoter CpG islands and diffuse genomic hypomethylation. Alu and long interspersed nucleotide element-1 (LINE-1) are non-coding genomic repetitive sequences and methylation of these elements can be used as a surrogate marker for genome-wide methylation status. This study was designed to evaluate the changes of Alu and LINE-1 hypomethylation during breast cancer progression from normal to pre-invasive lesions and invasive breast cancer (IBC), and their relationship with characteristics of IBC. We analyzed the methylation status of Alu and LINE-1 in 145 cases of breast samples including normal breast tissue, atypical ductal hyperplasia/flat epithelial atypia (ADH/FEA), ductal carcinoma in situ (DCIS) and IBC, and another set of 129 cases of IBC by pyrosequencing. Alu methylation showed no significant changes during multistep progression of breast cancer, although it tended to decrease during the transition from DCIS to IBC. In contrast, LINE-1 methylation significantly decreased from normal to ADH/FEA, while it was similar in ADH/FEA, DCIS and IBC. In IBC, Alu hypomethylation correlated with negative estrogen receptor (ER) status, and LINE-1 hypomethylation was associated with negative ER status, ERBB2 (HER2) amplification and p53 overexpression. Alu and LINE-1 methylation status was significantly different between breast cancer subtypes, and the HER2 enriched subtype had lowest methylation levels. In survival analyses, low Alu methylation status tended to be associated with poor disease-free survival of the patients. Our findings suggest that LINE-1 hypomethylation is an early event and Alu hypomethylation is probably a late event during breast cancer progression, and prominent hypomethylation of Alu and LINE-1 in HER2 enriched subtype may be related to chromosomal instability of this specific subtype.
DOI:10.3748/wjg.v19.i7.1068URLPMID:23466962 [本文引用: 2]
To clarify the specific roles and mechanisms of long interspersed nuclear element-1 ORF-1 protein [human long interspersed nuclear element-1 (LINE-1), ORF-1p] in chemotherapeutic drug resistance and cell proliferation regulation in hepatocellular carcinoma (HCC) cells.
DOI:10.2147/CMAR.S176088URLPMID:30349375 [本文引用: 1]
LINE-1 ORF-1p is encoded by the human pro-oncogene LINE-1. Our previous work showed that LINE-1 ORF-1p could enhance the resistance of hepatocellular carcinoma (HCC) cells to antitumor agents. However, the mechanisms involved in LINE-1 ORF-1p-mediated drug resistance remain largely unknown.
DOI:10.1038/nrg2341URLPMID:18463664 [本文引用: 2]
The genomes of many animals, plants and fungi are tagged by methylation of DNA cytosine. To understand the biological significance of this epigenetic mark it is essential to know where in the genome it is located. New techniques are making it easier to map DNA methylation patterns on a large scale and the results have already provided surprises. In particular, the conventional view that DNA methylation functions predominantly to irreversibly silence transcription is being challenged. Not only is promoter methylation often highly dynamic during development, but many organisms also seem to target DNA methylation specifically to the bodies of active genes.
DOI:10.1007/978-1-4419-9967-2_1URLPMID:22956494 [本文引用: 2]
The maintenance DNA methyltransferase (DNMT) 1 and the de novo methyltransferases DNMT3A and DNMT3B are all essential for mammalian development. DNA methylation, catalyzed by the DNMTs, plays an important role in maintaining genome stability. Aberrant expression of DNMTs and disruption of DNA methylation patterns are closely associated with many forms of cancer, although the exact mechanisms underlying this link remain elusive. DNA damage repair systems have evolved to act as a genome-wide surveillance mechanism to maintain chromosome integrity by recognizing and repairing both exogenous and endogenous DNA insults. Impairment of these systems gives rise to mutations and directly contributes to tumorigenesis. Evidence is mounting for a direct link between DNMTs, DNA methylation, and DNA damage repair systems, which provide new insight into the development of cancer. Like tumor suppressor genes, an array of DNA repair genes frequently sustain promoter hypermethylation in a variety of tumors. In addition, DNMT1, but not the DNMT3s, appear to function coordinately with DNA damage repair pathways to protect cells from sustaining mutagenic events, which is very likely through a DNA methylation-independent mechanism. This chapter is focused on reviewing the links between DNA methylation and the DNA damage response.
DOI:10.1038/nrc3895URLPMID:25693834 [本文引用: 2]
DNA methylation patterns are disrupted in various malignancies, suggesting a role in the development of cancer, but genetic aberrations directly linking the DNA methylation machinery to malignancies were rarely observed, so this association remained largely correlative. Recently, however, mutations in the gene encoding DNA methyltransferase 3A (DNMT3A) were reported in patients with acute myeloid leukaemia (AML), and subsequently in patients with various other haematological malignancies, pointing to DNMT3A as a critically important new tumour suppressor. Here, we review the clinical findings related to DNMT3A, tie these data to insights from basic science studies conducted over the past 20 years and present a roadmap for future research that should advance the agenda for new therapeutic strategies.
DOI:10.1038/ng.2836URLPMID:24270360 [本文引用: 2]
Gains and losses in DNA methylation are prominent features of mammalian cell types. To gain insight into the mechanisms that promote shifts in DNA methylation and contribute to changes in cell fate, including malignant transformation, we performed genome-wide mapping of 5-methylcytosine and 5-hydroxymethylcytosine in purified mouse hematopoietic stem cells. We discovered extended regions of low methylation (canyons) that span conserved domains frequently containing transcription factors and are distinct from CpG islands and shores. About half of the genes in these methylation canyons are coated with repressive histone marks, whereas the remainder are covered by activating histone marks and are highly expressed in hematopoietic stem cells (HSCs). Canyon borders are demarked by 5-hydroxymethylcytosine and become eroded in the absence of DNA methyltransferase 3a (Dnmt3a). Genes dysregulated in human leukemias are enriched for canyon-associated genes. The new epigenetic landscape we describe may provide a mechanism for the regulation of hematopoiesis and may contribute to leukemia development.
DOI:10.1126/science.1190485URLPMID:20651149 [本文引用: 3]
DNA methylation at proximal promoters facilitates lineage restriction by silencing cell type-specific genes. However, euchromatic DNA methylation frequently occurs in regions outside promoters. The functions of such nonproximal promoter DNA methylation are unclear. Here we show that the de novo DNA methyltransferase Dnmt3a is expressed in postnatal neural stem cells (NSCs) and is required for neurogenesis. Genome-wide analysis of postnatal NSCs indicates that Dnmt3a occupies and methylates intergenic regions and gene bodies flanking proximal promoters of a large cohort of transcriptionally permissive genes, many of which encode regulators of neurogenesis. Surprisingly, Dnmt3a-dependent nonproximal promoter methylation promotes expression of these neurogenic genes by functionally antagonizing Polycomb repression. Thus, nonpromoter DNA methylation by Dnmt3a may be used for maintaining active chromatin states of genes critical for development.
DOI:10.1016/j.ccr.2014.07.028URL [本文引用: 3]
DNA methylation in promoters is well known to silence genes and is the presumed therapeutic target of methylation inhibitors. Gene body methylation is positively correlated with expression, yet its function is unknown. We show that 5-aza-2'-deoxycytidine treatment not only reactivates genes but decreases the overexpression of genes, many of which are involved in metabolic processes regulated by c-MYC. Downregulation is caused by DNA demethylation of the gene bodies and restoration of high levels of expression requires remethylation by DNMT3B. Gene body methylation may, therefore, be an unexpected therapeutic target for DNA methylation inhibitors, resulting in the normalization of gene overexpression induced during carcinogenesis. Our results provide direct evidence for a causal relationship between gene body methylation and transcription.
DOI:10.1016/s0168-9525(97)01181-5URLPMID:9260521 [本文引用: 1]
Most of the 5-methylcytosine in mammalian DNA resides in transposons, which are specialized intragenomic parasites that represent at least 35% of the genome. Transposon promoters are inactive when methylated and, over time, C--&gt;T transition mutations at methylated sites destroy many transposons. Apart from that subset of genes subject to X inactivation and genomic imprinting, no cellular gene in a non-expressing tissue has been proven to be methylated in a pattern that prevents transcription. It has become increasingly difficult to hold that reversible promoter methylation is commonly involved in developmental gene control; instead, suppression of parasitic sequence elements appears to be the primary function of cytosine methylation, with crucial secondary roles in allele-specific gene expression as seen in X inactivation and genomic imprinting.
DOI:10.1038/nature18325URLPMID:27443740 [本文引用: 1]
Cells deficient in the Brca1 and Brca2 genes have reduced capacity to repair DNA double-strand breaks by homologous recombination and consequently are hypersensitive to DNA-damaging agents, including cisplatin and poly(ADP-ribose) polymerase (PARP) inhibitors. Here we show that loss of the MLL3/4 complex protein, PTIP, protects Brca1/2-deficient cells from DNA damage and rescues the lethality of Brca2-deficient embryonic stem cells. However, PTIP deficiency does not restore homologous recombination activity at double-strand breaks. Instead, its absence inhibits the recruitment of the MRE11 nuclease to stalled replication forks, which in turn protects nascent DNA strands from extensive degradation. More generally, acquisition of PARP inhibitors and cisplatin resistance is associated with replication fork protection in Brca2-deficient tumour cells that do not develop Brca2 reversion mutations. Disruption of multiple proteins, including PARP1 and CHD4, leads to the same end point of replication fork protection, highlighting the complexities by which tumour cells evade chemotherapeutic interventions and acquire drug resistance.
DOI:10.3892/mmr.2014.2276URL [本文引用: 1]
Bone morphogenetic protein 2 (BMP2) is a growth factor that is involved in the development and progression of various types of cancer. However, the epigenetic regulation of the expression of BMP2 and the association between BMP2 expression and drug resistance in breast cancer remains to be elucidated. The present study reported that the expression of BMP2 was significantly decreased in primary breast cancer samples and the MCF-7/ADR breast cancer mulitdrug resistance cell line, which was closely associated with its promoter DNA methylation status. The expression of BMP2 in MCF-7/ADR cells markedly increased when treated with 5-Aza-2'-deoxycytidine. Knockdown of BMP2 by specific small interfering RNA enhanced the chemoresistance of the MCF-7 breast cancer cell line. These findings indicated that epigenetic silencing of BMP2 in breast cancer may be involved in breast cancer progression and drug resistance, and provided a novel prognostic marker and therapeutic strategy for breast cancer.
DOI:10.1038/ng.2443URLPMID:23064414 [本文引用: 2]
We have extensively characterized the DNA methylomes of 139 patients with chronic lymphocytic leukemia (CLL) with mutated or unmutated IGHV and of several mature B-cell subpopulations through the use of whole-genome bisulfite sequencing and high-density microarrays. The two molecular subtypes of CLL have differing DNA methylomes that seem to represent epigenetic imprints from distinct normal B-cell subpopulations. DNA hypomethylation in the gene body, targeting mostly enhancer sites, was the most frequent difference between naive and memory B cells and between the two molecular subtypes of CLL and normal B cells. Although DNA methylation and gene expression were poorly correlated, we identified gene-body CpG dinucleotides whose methylation was positively or negatively associated with expression. We have also recognized a DNA methylation signature that distinguishes new clinico-biological subtypes of CLL. We propose an epigenomic scenario in which differential methylation in the gene body may have functional and clinical implications in leukemogenesis.
DOI:10.1038/nature09165URLPMID:20613842 [本文引用: 3]
Although it is known that the methylation of DNA in 5' promoters suppresses gene expression, the role of DNA methylation in gene bodies is unclear. In mammals, tissue- and cell type-specific methylation is present in a small percentage of 5' CpG island (CGI) promoters, whereas a far greater proportion occurs across gene bodies, coinciding with highly conserved sequences. Tissue-specific intragenic methylation might reduce, or, paradoxically, enhance transcription elongation efficiency. Capped analysis of gene expression (CAGE) experiments also indicate that transcription commonly initiates within and between genes. To investigate the role of intragenic methylation, we generated a map of DNA methylation from the human brain encompassing 24.7 million of the 28 million CpG sites. From the dense, high-resolution coverage of CpG islands, the majority of methylated CpG islands were shown to be in intragenic and intergenic regions, whereas less than 3% of CpG islands in 5' promoters were methylated. The CpG islands in all three locations overlapped with RNA markers of transcription initiation, and unmethylated CpG islands also overlapped significantly with trimethylation of H3K4, a histone modification enriched at promoters. The general and CpG-island-specific patterns of methylation are conserved in mouse tissues. An in-depth investigation of the human SHANK3 locus and its mouse homologue demonstrated that this tissue-specific DNA methylation regulates intragenic promoter activity in vitro and in vivo. These methylation-regulated, alternative transcripts are expressed in a tissue- and cell type-specific manner, and are expressed differentially within a single cell type from distinct brain regions. These results support a major role for intragenic methylation in regulating cell context-specific alternative promoters in gene bodies.
DOI:10.1101/gr.147942.112URL [本文引用: 1]
As studies of DNA methylation increase in scope, it has become evident that methylation has a complex relationship with gene expression, plays an important role in defining cell types, and is disrupted in many diseases. We describe large-scale single-base resolution DNA methylation profiling on a diverse collection of 82 human cell lines and tissues using reduced representation bisulfite sequencing (RRBS). Analysis integrating RNA-seq and ChIP-seq data illuminates the functional role of this dynamic mark. Loci that are hypermethylated across cancer types are enriched for sites bound by NANOG in embryonic stem cells, which supports and expands the model of a stem/progenitor cell signature in cancer. CpGs that are hypomethylated across cancer types are concentrated in megabase-scale domains that occur near the telomeres and centromeres of chromosomes, are depleted of genes, and are enriched for cancer-specific EZH2 binding and H3K27me3 (repressive chromatin). In noncancer samples, there are cell-type specific methylation signatures preserved in primary cell lines and tissues as well as methylation differences induced by cell culture. The relationship between methylation and expression is context-dependent, and we find that CpG-rich enhancers bound by EP300 in the bodies of expressed genes are unmethylated despite the dense gene-body methylation surrounding them. Non-CpG cytosine methylation occurs in human somatic tissue, is particularly prevalent in brain tissue, and is reproducible across many individuals. This study provides an atlas of DNA methylation across diverse and well-characterized samples and enables new discoveries about DNA methylation and its role in gene regulation and disease.
DOI:10.1038/nature05864URLPMID:17554311 [本文引用: 1]
Epigenetic regulation involves reversible changes in DNA methylation and/or histone modification patterns. Short interfering RNAs (siRNAs) can direct DNA methylation and heterochromatic histone modifications, causing sequence-specific transcriptional gene silencing. In animals and yeast, histone H2B is known to be monoubiquitinated, and this regulates the methylation of histone H3 (refs 10, 11). However, the relationship between histone ubiquitination and DNA methylation has not been investigated. Here we show that mutations in an Arabidopsis deubiquitination enzyme, SUP32/UBP26, decrease the dimethylation on lysine 9 of H3, suppress siRNA-directed methylation of DNA and release heterochromatic silencing of transgenes as well as transposons. We found that Arabidopsis histone H2B is monoubiquitinated at lysine 143 and that the levels of ubiquitinated H2B and trimethyl H3 at lysine 4 increase in sup32 mutant plants. SUP32/UBP26 can deubiquitinate H2B, and chromatin immunoprecipitation assays suggest an association between H2B ubiquitination and release of silencing. These data suggest that H2B deubiquitination by SUP32/UBP26 is required for heterochromatic histone H3 methylation and DNA methylation.
DOI:10.1242/dev.033902URLPMID:19820181 [本文引用: 1]
Polycomb group (PcG) protein complexes dynamically define cellular identity through the regulation of key developmental genes. Important advances in the PcG field have come from genome-wide mapping studies in a variety of tissues and cell types that have analyzed PcG protein complexes, their associated histone marks and putative mechanisms of PcG protein recruitment. We review how these analyses have contributed to our understanding of PcG protein complex targeting to chromatin and consider the importance of diverse PcG protein complex composition for gene regulation. Finally, we focus on the dynamics of PcG protein complex action during cell fate transitions and on the implications of histone modifications for cell lineage commitment.
DOI:10.1016/j.stem.2010.03.018URLPMID:20452322 [本文引用: 1]
Human embryonic stem cells (hESCs) share an identical genome with lineage-committed cells, yet possess the remarkable properties of self-renewal and pluripotency. The diverse cellular properties in different cells have been attributed to their distinct epigenomes, but how much epigenomes differ remains unclear. Here, we report that epigenomic landscapes in hESCs and lineage-committed cells are drastically different. By comparing the chromatin-modification profiles and DNA methylomes in hESCs and primary fibroblasts, we find that nearly one-third of the genome differs in chromatin structure. Most changes arise from dramatic redistributions of repressive H3K9me3 and H3K27me3 marks, which form blocks that significantly expand in fibroblasts. A large number of potential regulatory sequences also exhibit a high degree of dynamics in chromatin modifications and DNA methylation. Additionally, we observe novel, context-dependent relationships between DNA methylation and chromatin modifications. Our results provide new insights into epigenetic mechanisms underlying properties of pluripotency and cell fate commitment.
DOI:10.1038/nrm3274URLPMID:22266761 [本文引用: 1]
Histone side chains are post-translationally modified at multiple sites, including at Lys36 on histone H3 (H3K36). Several enzymes from yeast and humans, including the methyltransferases SET domain-containing 2 (Set2) and nuclear receptor SET domain-containing 1 (NSD1), respectively, alter the methylation status of H3K36, and significant progress has been made in understanding how they affect chromatin structure and function. Although H3K36 methylation is most commonly associated with the transcription of active euchromatin, it has also been implicated in diverse processes, including alternative splicing, dosage compensation and transcriptional repression, as well as DNA repair and recombination. Disrupted placement of methylated H3K36 within the chromatin landscape can lead to a range of human diseases, underscoring the importance of this modification.
DOI:10.1038/nature11326URL [本文引用: 1]
Set2-mediated methylation of histone H3 at Lys 36 (H3K36me) is a co-transcriptional event that is necessary for the activation of the Rpd3S histone deacetylase complex, thereby maintaining the coding region of genes in a hypoacetylated state(1,2). In the absence of Set2, H3K36 or Rpd3S acetylated histones accumulate on open reading frames (ORFs), leading to transcription initiation from cryptic promoters within ORFs(1,3). Although the co-transcriptional deacetylation pathway is well characterized, the factors responsible for acetylation are as yet unknown. Here we show that, in yeast, co-transcriptional acetylation is achieved in part by histone exchange over ORFs. In addition to its function of targeting and activating the Rpd3S complex, H3K36 methylation suppresses the interaction of H3 with histone chaperones, histone exchange over coding regions and the incorporation of new acetylated histones. Thus, Set2 functions both to suppress the incorporation of acetylated histones and to signal for the deacetylation of these histones in transcribed genes. By suppressing spurious cryptic transcripts from initiating within ORFs, this pathway is essential to maintain the accuracy of transcription by RNA polymerase II.
DOI:10.4161/epi.28324URL [本文引用: 1]
DNA methylation, one of the best-characterized epigenetic modifications, plays essential roles in development, aging and diseases. The de novo DNA methyltransferase DNMT3A is responsible for the establishment of de novo genomic DNA methylation patterns and, as such, involved in normal development as well as in many diseases including cancer. In recent years, our understanding of this important protein has made significant progress, which was facilitated by stunning development in the analysis of the DNA methylome of multiple organs and cell types. In this review, recent developments in the characterization of DNMT3A were discussed with special emphasis on the roles of DNMT3A in development and cancer.
DOI:10.3724/SP.J.1005.2014.0403URL [本文引用: 1]
DNA methylation plays important roles in cell differentiation, embryonic development, host adaptations to environmental factors, and pathogenesis through regulation of gene transcription and imprinting, X-inactivation, and defense of foreign genetic material invasion, is currently one of the hottest research fields on epigenetics. In the past few years, a number of important findings on DNA methylation have been achieved. These findings include discovery of TETs-catalyzed cytosine hydroxymethylation and its functions in the early embryonic development; the relationship between active and passive DNA demethylation; establishment and maintenance of DNA methylation patterns and their associations with histone modifications, chromatin configuration, polycomb group proteins and non-coding RNA bindings. DNA methylation has become a new potential biomarker and therapy target.
DOI:10.3724/SP.J.1005.2014.0403URL [本文引用: 1]
DNA methylation plays important roles in cell differentiation, embryonic development, host adaptations to environmental factors, and pathogenesis through regulation of gene transcription and imprinting, X-inactivation, and defense of foreign genetic material invasion, is currently one of the hottest research fields on epigenetics. In the past few years, a number of important findings on DNA methylation have been achieved. These findings include discovery of TETs-catalyzed cytosine hydroxymethylation and its functions in the early embryonic development; the relationship between active and passive DNA demethylation; establishment and maintenance of DNA methylation patterns and their associations with histone modifications, chromatin configuration, polycomb group proteins and non-coding RNA bindings. DNA methylation has become a new potential biomarker and therapy target.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}