 ,东北师范大学生命科学学院,分子表观遗传学教育部重点实验室,长春 130024
,东北师范大学生命科学学院,分子表观遗传学教育部重点实验室,长春 130024Progresses in the plant 3D chromatin architecture
Qianli Dong, Jinbin Wang, Xiaochong Li, Lei Gong,Key Laboratory of Molecular Epigenetics of the Ministry of Education (MOE), Northeast Normal University, Changchun 130024, China通讯作者: 宫磊,博士,教授,研究方向:进化生物学,基因组学和表观基因组学。E-mail:gongl100@nenu.edu.cn
编委: 张飞雄
收稿日期:2019-10-29修回日期:2019-12-19网络出版日期:2020-01-20
| 基金资助: |
Editorial board:
Received:2019-10-29Revised:2019-12-19Online:2020-01-20
| Fund supported: |
作者简介 About authors
董芊里,博士研究生,专业方向:遗传学。E-mail:dongql043@nenu.edu.cn。
王金宾,博士研究生,专业方向:遗传学。E-mail:wangjb702@nenu.edu.cn。
李晓宠,博士研究生,专业方向:遗传学。E-mail:lixc800@nenu.edu.cn;董芊里、王金宾和李晓宠并列第一作者。

摘要
染色质在细胞核内的缠绕、折叠及其在细胞核内的空间排布是真核生物染色质构型的主要特征。在经典DNA探针荧光原位杂交显微观察的基础上,基于新一代测序技术的Hi-C及ChIA-PET染色质构型捕获技术已经被广泛应用于动物及植物细胞核染色质构型的研究中,并以新的角度定义了包括:染色体(质)域(chromosome territory)、A/B染色质区室(compartment A/B)、拓扑偶联结构域(topological associated domains, TADs)、染色质环(chromatin loops)等在内的多个更为精细的染色质构型。利用以上两种主流技术,越来越多的植物物种染色质构型特征被鉴定、分析和比较。本文系统分析和总结了近年来以植物细胞为模型的细胞核染色构型领域取得的重要成果,包括各级染色质构型特征的组成、建立机制和主要影响因素等。在此基础上,分析了目前研究植物染色质构型技术的瓶颈和突破性的技术进展,并对后续研究主要关注的问题和研究内容进行了展望,以期为相关领域的研究提供更多的理论参考和依据。
关键词:
Abstract
Chromatin architecture involves the patterns of chromatin coiling and packing as well as the mutual relative allocations of different chromatins. Besides the canonical microscopic observations, the chromatin architectural capture techniques, including the Hi-C and ChIA-PET, have been widely applied in characterization of chromatin architecture in various plant and animal model species, in which chromatin architectural features, such as the chromosome territory, compartment A/B, topological associated domains (TADs) and chromatin loops, were defined. As for the studies in plant species, replying on the two techniques above (with differences in experimental techniques and data structures), scientists have compared the variation of specific chromatin architecture features across species and/or in different cell types of the same plant species, besides detailed analyses in each individual model. Here, we mainly review the recent progresses in studies of plant chromatin architectures, in which their composition, establishing mechanism and effective factors were described and discussed. We also propose the main technical bottlenecks, describe the breaking-through progresses, and anticipate future research directions, which may offer more theoretical references for related researches in the field.
Keywords:
PDF (774KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
董芊里, 王金宾, 李晓宠, 宫磊. 植物三维染色质构型研究进展. 遗传[J], 2020, 42(1): 73-86 doi:10.16288/j.yczz.19-326
Qianli Dong.
染色质(chromatin)是由基因组DNA与组蛋白等结构性蛋白通过不同层级的缠绕而形成[1]。作为真核生物遗传物质的载体,染色质需要填充在微米级的狭小细胞核中。其缠绕、折叠及染色质在细胞核空间内的不同排布模式,组成了染色质空间构型(chromatin architectures)研究的主要内容。基于光学显微镜观察的DNA探针荧光原位杂交(fluorescence in situ hybridization, FISH),是研究染色质构型的经典研究方法,该方法能够实现通过肉眼观察细胞内整体或特定区段的染色质空间构型[2,3]。但受限于技术本身的局限(DNA探针杂交的灵敏度等因素),FISH技术能够检测到的染色质构型的分辨率有限,且其不能量化染色质区段的空间距离。受益于近年来新一代测序和显微成像技术的发展,通过体外测序和体内原位观察的方法实现了染色质构型的精细定量和分析,也使得基于生物信息学的染色质构型研究成为目前的热点研究领域之一[4,5,6,7]。
目前,染色质构型研究的相关研究成果还主要集中在哺乳动物细胞系或其他动物模型中[8],鉴于植物和动物在细胞核结构和基因组组成等方面的差异,植物细胞中的染色质构型的模式、特征(不同植物组织和不同类型细胞中的保守性和特异性)以及形成机制等还亟需系统探讨[9,10]。本文在简要回顾近年来染色质构型研究技术发展的基础上,系统分析和总结了以植物细胞为模型的细胞核染色质构型领域取得的重要成果,阐述了植物染色质构型研究领域内目前的技术瓶颈,并探讨了后续研究中主要关注的问题和发展前景,以期为相关领域的研究提供更多的理论参考和依据。
1 染色质构型研究技术
目前,三维基因组研究领域内最为主流的染色质构型研究技术,包括:断层扫描电镜成像(chromosome electron microscopy tomography)、高通量染色质构象捕获(high-throughput chromosome conformation capture, Hi-C)以及末端配对标签测序(paired- end tag sequencing, ChIA-PET)[11,12]。其中,电镜成像虽然可以在极高的分辨率下直接解析染色质结构,但由于其检测到的染色质互作区段有限,因此本文未将其列为介绍的内容,主要对Hi-C和ChIA-PET两种染色质构型分析技术进行详细介绍。1.1 Hi-C技术原理
2002年,Dekker等[13]开发了染色质构象捕获技术(chromosome conformation capture, 3C)。3C技术首先是用甲醛瞬时固定细胞核染色质,用限制性内切酶酶切消化染色质—蛋白质交联物。随后,在DNA浓度极低的条件下,使用高浓度连接酶连接消化物,使得交联在一起的DNA片段粘性末端在交联片段之间重新连接。最后,使用蛋白酶消化交联物进而释放出结合的蛋白质,并根据实验目的的不同,用推测可能有互作的目的片段的引物进行普通PCR或定量PCR扩增,最终确定其是否存在显著的相互作用。该技术是一种“一对一”(One vs. One)的检测染色质相互作用的技术,即对于基因组上两个已知的目的染色质区段,利用3C技术可以得知这两个片段的相互作用情况。以3C技术为基础,2009年,Lieberman-Aiden等[11]将3C技术与新一代测序技术(next-generation sequencing, NGS)技术结合,研发 出了高通量染色质构象捕获方法,即Hi-C (high- throughput chromosome conformation capture)技术(图1A)。虽然Hi-C技术脱胎于3C技术,但整个操作过程相对于3C技术更加的冗长和繁琐,针对此类问题,赵志虎等对Hi-C实验流程中的关键步骤进行了优化和改进,促进了Hi-C技术进一步的广泛应用[14]。Hi-C技术分析步骤主要包括:(1)甲醛交联:使用甲醛瞬间固定细胞核染色质。使得细胞内染色质交联,生成DNA-蛋白质复合物或蛋白质-蛋白质复合物;(2)酶切:使用限制性内切酶消化染色质,通常使用六碱基内切酶(6-cutter),如Hind Ⅲ、Bgl Ⅱ、或EcoR Ⅰ等,或四碱基内切酶(4-cutter),如Mbo Ⅰ、Dpn Ⅱ或Aci Ⅰ等;(3)生物素标记:用生物素标记的核苷酸片段,连接至酶切时产生的粘性末端;(4)片段末端链接:利用DNA连接酶,将不同的切口末端连接起来,形成环状嵌合分子;(5)剪切并纯化:利用机械力打断环状嵌合分子,产生小片段DNA。并利用亲和素特异性结合生物素的原理,将携带生物素核苷酸的DNA片段富集;(6)文库构建及NGS测序:将携带生物素核苷酸的DNA片段两端添加测序接头,构建Hi-C测序文库,利用Illumina NGS测序平台进行高通量测序,并将测序结果转化为相应大小的染色质区段之间的相互作用矩阵和热图。1.2 ChIA-PET技术原理
2009年,Fullwood等[12]开发了染色质远程交互测序技术(chromatin interaction analysis by paired- end tag sequencing, ChIA-PET)。ChIA-PET是基于免疫共沉淀(chromatin immunoprecipitation, ChIP)、染色质邻近连接(chromatin proximity ligation)、配对末端标签(paired-end tags)以及新一代测序技术,所开发出的一种分析全基因组范围内远程染色质相互作用的新技术。与Hi-C技术相比,ChIA-PET在甲醛交联固定后,优先进行ChIP富集;随后的操作中不同于Hi-C实验中使用的限制性内切酶消化,ChIA- PET则使用超声破碎进行物理打断。后续的操作步骤基本与Hi-C技术相似,即连接生物素并连接、纯化以及建库并测序(图1B)。图1
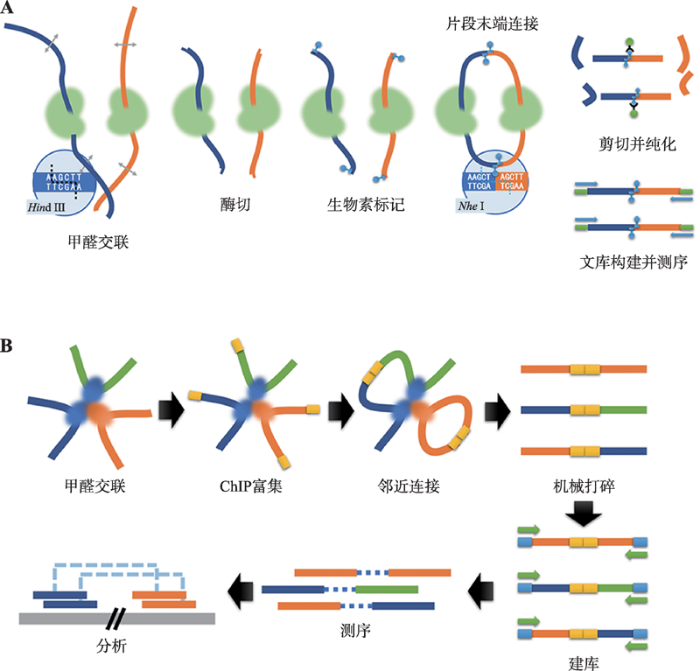 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1Hi-C及ChIA-PET技术流程示意图
A:Hi-C技术流程示意图;B:ChIA-PET技术流程示意图。
Fig. 1Workflows of Hi-C and ChIA-PET
1.3 Hi-C与ChIA-PET技术的特征和应用
鉴于Hi-C技术与ChIA-PET技术在原理层面既存在相似又有不同,因此两种方法有着各自的优点与局限。有关两种技术的特征和应用范围的具体比较,详见表1。Table 1
表1
表1Hi-C与ChIA-PET技术的特征和应用范围的比较
Table 1
| 技术 | 互作方式 | 覆盖范围 | 检测方法 | 技术优势 | 技术局限 | 研究应用 |
|---|---|---|---|---|---|---|
| Hi-C | 全部互作 (all vs. all) | 全基因 组范围 | 新一代 测序技术 | 可以获得较高分辨率的互作矩阵 | 需大量细胞进行建库,以产出较高分辨率Hi-C矩阵。基于由此得到的高分辨率Hi-C矩阵,所反映的染色体(质)特征为大量组织样本中大量细胞的平均值;使用限制性内切酶对染色体进行剪切时,可能会由于酶的剪切偏好性,对互作结果产生一定程度的影响 | 可产出较高分辨率互作矩阵,用以分析目前已知的各级染色体(质)构型(compartment、TAD、chromatin loop等);Hi-C技术亦可用于辅助基因组拼接 |
| scHi-C | 可以获得单个细胞的互作热图,观察单个细胞内的染色体(质)特征 | 单个细胞中DNA含量较少,无法绘制较高分辨率的Hi-C热图,所以无法分析需要较高分辨率下可以分析的TAD结构或chromatin loop结构;使用限制性内切酶对染色体进行剪切时,可能会由于酶的剪切偏好性,对互作结果产生一定程度的影响。 | 可研究单个细胞内的染色体(质)构型,可以更精准的阐述染色体(质)构型特征 | |||
| ChIA-PET | 特定蛋白介导的全部互作(many vs. all) | 可以构建已知转录因子介导的染色体(质)互作网络;由于使用超声的方法进行机械破碎,不会由于限制性内切酶的剪切偏好性对互作结果产生影响;具有更高的分辨率(100 bp水平)[15] | 由于ChIA-PET实验优先使用特异性的蛋白抗体,其选择性捕获与目标蛋白交联在一起的DNA片段,可能会忽略其他的互作 | 主要包含靶蛋白结合位点和由靶蛋白介导的结合位点之间的染色质相互作用信息 |
新窗口打开|下载CSV
2 植物不同等级的染色质构型特征
作为基于概率模型分析染色质构型的主流技术,Hi-C和ChIA-PET已经被广泛应用在不同植物的染色质构型特征的鉴定、功能分析以及相互关系的研究中(表2)。按照目前业内广泛接受的标准(包含染色质区段大小的尺度和功能特征),植物染色质构型涵盖的等级主要包括:染色体(质)域(chromosome territory, CT)、A/B染色质区室(compartment A/B)、拓扑偶联结构域(topological associated domains, TADs)、染色质环(chromatin loops)以及IHIs/ KEEs (interactive heterochromatic islands/KNOT ENGAGED ELEMENTs)[5,16](图2)。接下来将从植物物种、材料性质、研究方法(Hi-C或ChIA-PET)、组成、建立机制或者主要的影响因素等方面,详细说明植物不同等级染色质构型的主要特征。Table 2
表2
表2植物不同等级的染色质构型特征汇总
Table 2
| 物种 | 技术 | 染色体 (质)域 | A/B染色质 区室 | 拓扑偶联 结构域 | 染色质环 | IHIs/KEEs | 参考文献 |
|---|---|---|---|---|---|---|---|
| 深山南芥 (Arabidopsis lyrata) | Hi-C | √ | [17] | ||||
| 拟南芥 (Arabidopsis thaliana) | Hi-C | √ | √ | √ | [18] | ||
| Hi-C | √ | √ | √ | [19] | |||
| Hi-C | √ | √ | √ | √ | [20] | ||
| 四倍体拟南芥 (Arabidopsis thaliana (4×Columbia)) | Hi-C | √ | √ | √ | [21] | ||
| 甘蓝 (Brassica oleracea) | Hi-C | √ | √ | √ | √ | [22] | |
| 芜菁 (Brassica rapa) | Hi-C | √ | √ | √ | √ | [22] | |
| 树棉 (Gossypium arboreum) | Hi-C | √ | √ | √ | [23] | ||
| 海岛棉 (Gossypium barbadense) | Hi-C | √ | √ | √ | [23] | ||
| 陆地棉 (Gossypium hirsutum) | Hi-C | √ | [24] | ||||
| 雷蒙德氏棉 (Gossypium raimondii) | Hi-C | √ | √ | √ | [23] | ||
| 大麦 (Hordeum vulgare) | Hi-C | √ | [25] | ||||
| 水稻 明恢63 (Oryza Sativa L. spp. indica (Minghui63)) | Hi-C | √ | √ | √ | [26] | ||
| 水稻 日本晴 (Oryza sativa L. spp. Japonica) | Hi-C | √ | √ | √ | √ | √ | [27] |
| Hi-C | √ | √ | √ | √ | [28] | ||
| scHi-C | √ | √ | [29] | ||||
| ChIA-PET | √ | √ | √ | [15] | |||
| 粟 (Setaria italic) | Hi-C | √ | √ | √ | √ | [28] | |
| 番茄 (Solanum lycopersicum) | Hi-C | √ | √ | √ | √ | [28] | |
| 高粱 (Sorghum bicolor) | Hi-C | √ | √ | √ | √ | [28] | |
| 玉米 (Zea mays) | Hi-C | √ | √ | √ | √ | [28] | |
| ChIA-PET | √ | [30] |
新窗口打开|下载CSV
2.1 染色体(质)域CT
在细胞分裂间期(多数细胞主要处于的时期),虽然植物染色质主要呈现较为松散的状态,但在整个细胞核空间内仍呈现非随机的排布方式。每条染色质占有自己特有的一个细胞核空间CT。CT的概念,最早是由Rabl在1885年观察火蜥蜴(Salamander)细胞分裂的过程中提出;而后在1909年,由Boveri[31]对CT概念进行了进一步的凝练;在20世纪80年代,在人的细胞中利用染色体特异性的DNA探针完成的FISH观察,最终确认了CT的存在[1,32]。而植物CT的存在,首先是在拟南芥(Arabidopsis thaliana)中,利用细菌人工染色体(bacterial artificial chromosome)片段标记探针后,由FISH观察而确认的[1,2]。利用相似的FISH观察方法,通过特异性标记着丝粒、端粒以及其他染色体区段的BAC文库探针,不同植物物种中的CT排布特征被详细分析和归纳为不同的染色质构象(图3,A和B分别展示的Rabl和Rosette构象)。图2
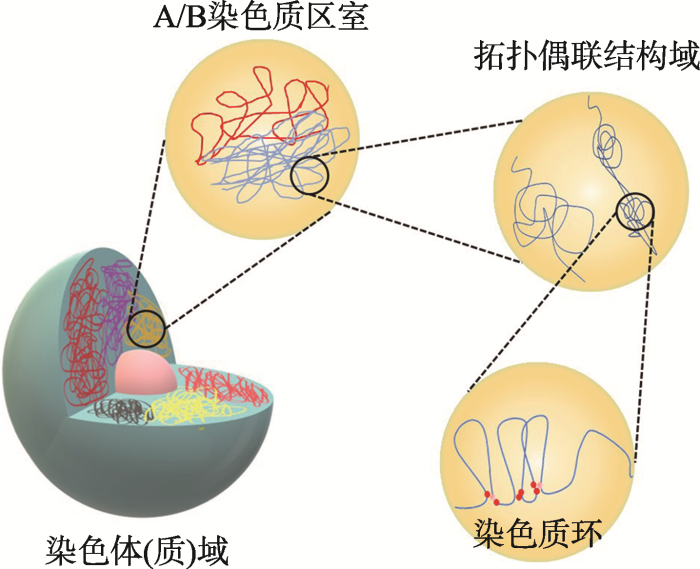 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2植物中染色质层次结构示意图
单个染色体在核中占据染色体(质)域(chromosome territories)的一个子空间。染色体(质)域可以进一步划分为不同的A/B染色质区室(compartment A/B)。拓扑偶联结构域内(TADs)的基因组区域显示出更强的相互作用,而它们与拓扑偶联结构域之外的相邻区域相互作用则相当有限。TADs内调节元件与其靶基因座连接形成的染色质环[10]。
Fig. 2Schematic representation of hierarchical chromatin organization in plants
利用Hi-C数据结合后期3D模型重构的方法(chromatin 3D remodeling),2009年Lieberman-Aiden等[11]首次应用成功绘制出了1M分辨率下人类淋巴母细胞(GM06990)的三维染色体结构,并验证了之前通过FISH技术发现的CTs的存在。利用已有的高分辨率Hi-C数据,本文也重构了拟南芥混合细胞类型平均呈现的3D染色质构象(图4)。与经典的FISH研究结果一致[1,33,34],拟南芥间期细胞主要呈现(混合细胞平均呈现出的相似特征)莲座状构象(Rosette configuration):端粒在核仁区聚集,着丝粒围绕在外围(图3B,图4)。这一构象特征与Hi-C互作热图显示的互作状态一致。前期FISH研究发现,拥有较大基因组的物种,如:玉米(Zea mays)、大麦(Hordeum vulgare)等,其间期细胞的染色质主要呈现Rabl构象(Rabl configuration):端粒和着丝粒分处于细胞核的两级,不同染色质的着丝粒左右的染色质臂之间紧密贴合(图3A)。近期玉米叶肉细胞(单一细胞类型)的Hi-C互作热图显示,其不同染色质的着丝粒和端粒之间的确呈现高强度的互作,而且不同染色质的着丝粒左右的染色质臂之间也一致性的存在染色质内及染色质之间的紧密互作。
图3
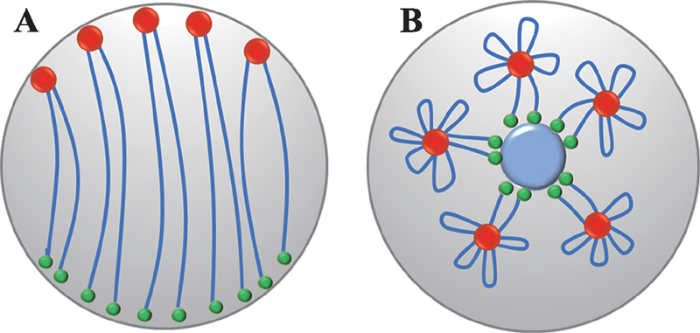 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Rabl及Rosette构象的构象展示
A:Rabl构象示意图;B:Rosette构象示意图。红色圆圈:近着丝粒区段;绿色圆圈:端粒区段;蓝色线段:染色体臂;蓝色圆圈:核仁。
Fig. 3Cartoon illustration of Rabl and rosette configurations
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4拟南芥Hi-C数据进行的染色体三维模拟结构图
基于Liu等[35]已发表的拟南芥Hi-C数据,进行染色体三维结构模拟。A:拟南芥5条染色体三维结构图;B:红色部分为着丝粒区域,绿色球体为端粒区域。可以看到基于Hi-C数据模拟出的染色体三维结构图与Rosette结构一致。
Fig. 4Chromosome three-dimensional simulation structure diagram based on Arabidopsis thaliana Hi-C data
然而,考虑到相同物种的不同细胞类型可能拥有不同的CT特征(呈现不同染色质构象),混合细胞类型的Hi-C测序数据不能够准确反映特定细胞呈现的CT染色质构象特征,故而后者需要更为精准的单细胞Hi-C技术(scHi-C)。近期,Zhou等[29]对水稻卵细胞、精子细胞、单细胞合子和芽叶肉细胞,进行了scHi-C测序和分析。通过3D模型重构,发现水稻卵子和精子细胞的染色质结构与叶肉细胞的染色质结构相当,并且在受精后得以重组。类似的scHi-C技术为在植物系统中更为精细的研究包括CT染色质构象在内的各层级染色质构型创造了条件。
2.2 A/B染色质区室
A/B染色质区室概念,最早是由Lieberman- Aiden等[11]于2009年发表Hi-C技术时提出。其在研究中,发现同一条染色体内的所有染色质区段的互作热图总体呈现“格子(plaid)”式排布。在此观察结果的基础上,作者对同一染色体内所有染色质区段之间的Hi-C互作值,以染色质区段为单位计算其与其他区段之间互作的相关性,并利用主成分分析(principal component analysis,以下简称PCA)将具有相似互作模式的染色质区段(与同一染色体内的其他染色质区段的互作)划归成“正”和“负”两个特征向量值组(eigenvalues),各组内的染色质区段分别对应A和B两个染色质区室。相同类型区室内的染色质区段呈现更相似的染色质互作模式。除以上依据Hi-C数据定义A/B染色质区室之外,Wang等[36,37]使用单细胞水平的FISH技术,也在细胞内成功验证了经由Hi-C划分出的区室A和区室B在细胞核中存在空间上的分离。如图5所示,水稻日本晴Chr.2互作热图上可以明显观察到特征向量的正负交替,即A/B区室的交替分布。图5
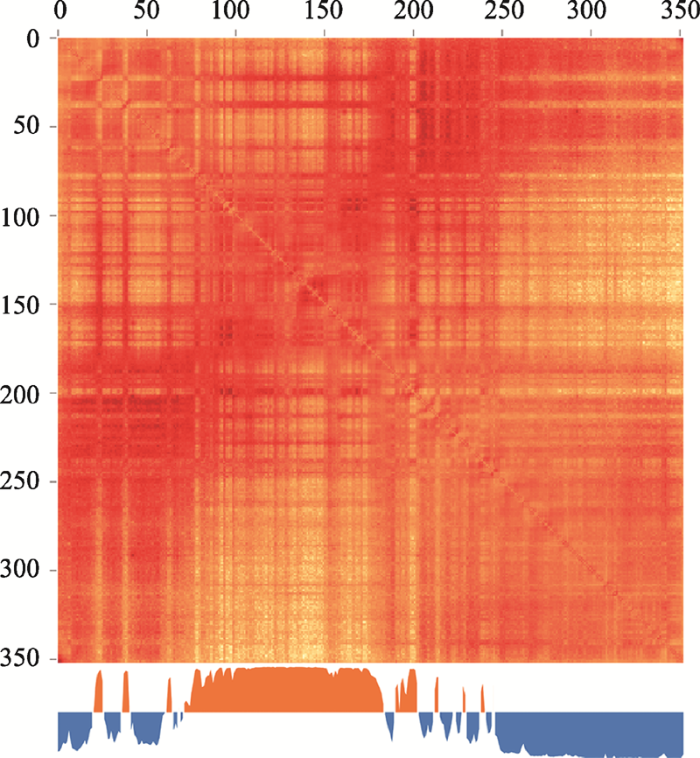 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5水稻2号染色体(Chr.2)中A/B染色质区室的划分及其对应的互作热图
Fig. 5Compartment A/B characterized in the Hi-C interaction heat maps of rice Chr.2 (Oryza sativa ssp. japonica)
近期的研究发现,植物物种的A/B染色质区室总体呈现与动物模型相似的染色质互作、基因组组成和表达以及表观遗传修饰的特征。首先,在染色质互作方面,植物物种中的同一条染色体内部,染色质区室B的不同染色质区段之间的互作用强度显著高于区室A的染色质区段[18,27,28,36]。但不同染色体间的染色质区室A的染色质区段之间的相互作用强度显著高于区室B的不同染色质区段之间的互 作。以上互作状态表明,染色质区室A的染色质的折叠状态更加松散,并且区室A的染色质区段应主要排布在相应染色体CT的外围区域[27,36];其次,在基因组组成和表达方面,染色质区室A内的基因组片段更加显著富集基因,而染色质区室B内的基因组片段反而显著富集转座子(transposable elements, TEs)。相应的染色质区室A内更高密度的基因拥有更高的转录表达水平;最后,在表观遗传修饰模式和水平方面,激活性组蛋白修饰标记(如H3K4me2、H3K4me3和H3K9ac等)在染色质区室A中显著富集,而抑制性组蛋白修饰(如H3K9me2)和甲基化的胞嘧啶(CG、CHG和CHH三种序列环境)在区室B中显著富集。综上所述,在常染色质和异染色质区段划分较为明确的植物物种中(如水稻、西红柿、高粱、棉花等)[15,23,28],由于常染色质和异染色质往往分别富集在染色质区室A和B中,同一染色体内的染色质区室A和B之间频繁交替出现的情况较少(non-interlaced; interlace);相反的,在常染色质和异染色质区段没有明确划分的植物物种中(如拟南芥),同一染色体内的染色质区室A和B之间存在频繁的交替(interlaced)[29]。
在其他以动物为模型的染色质构型研究中,不同细胞类型[38]、同一细胞类型的不同发育阶段(如受精卵的不同发育阶段)[39]以及不同处理条件下,同一染色质区段所在的染色质区室类型会发生A和B之间的转换[40]。对目前已经发表的植物染色质构型的研究结果进行总结分析后发现:(1)水稻混合细胞的Hi-C分析发现在冷胁迫处理后染色质区室A和B总体呈现较为稳定的状态[36];(2)水稻单细胞scHi-C分析发现卵细胞、精子和叶肉细胞的同一染色质区段所在的染色质区室类型有差别[29];(3)相较于常见的二倍体拟南芥细胞核染色质,同源染色体组加倍后形成的拟南芥四倍体中,出现了染色质区室A和B之间的转换以及伴随发生的组蛋白H3K4me3和H3K27me3修饰水平的变化[21];(4)对芸薹属(Brassica)内代表性二倍体物种芜菁(B. rapa)和甘蓝(B. oleracea)的染色质Hi-C构型数据分析发现,二者共线性区段内多数(61.62%)的染色质区室呈现保守状态(维持相同的染色质区室类型),但仍有约38.38%的区段发生了染色质区室类型的转换[22];(5)棉属(Gossypium)内自然进化过程中的异源多倍化(allopolyploidy)介导的不同亲本基因组融合和加倍,引发了其对应亲本亚基因组在多倍体陆地棉和海岛棉(G. hirsutum和G. barbadense)中的染色质区段发生染色质区室A和B之间的转换[23]。
2.3 拓扑偶联结构域TADs
上述介绍的CTs和compartment A/B层级结构,均是在较大的分辨率上(>500 kb的染色质区段)定义和观察到的染色质构型。基于足够分辨率的Hi-C染色质互作数据,Dixon等[41,42]率先在人类和小鼠中定义并解析了更精细层级的一种染色质构型单元—TADs:同一TAD内的不同顺式染色质区段呈现更强的区域内互作,而这些区段与其他相邻染色质区段(不在同一TAD内)的互作较弱。据此,TADs是染色质上自身相互作用很强,而和临近的染色质区域相互作用受到抑制的结构单元;在Hi-C互作图谱热图(对称互作热图对角线下的一半热图)上,每个TAD对应相应染色质区段的互作呈现出颜色较深的三角形(图6A)。近期将超分辨率显微镜应用于动物单细胞染色质构型的研究也发现了类似TAD的染色质结构单元(TAD-like structural unit),该结构也呈现出与TAD类似的,结构单元内相互作用显著更强的染色质互作模式[43,44]。图6
 新窗口打开|下载原图ZIP|生成PPT
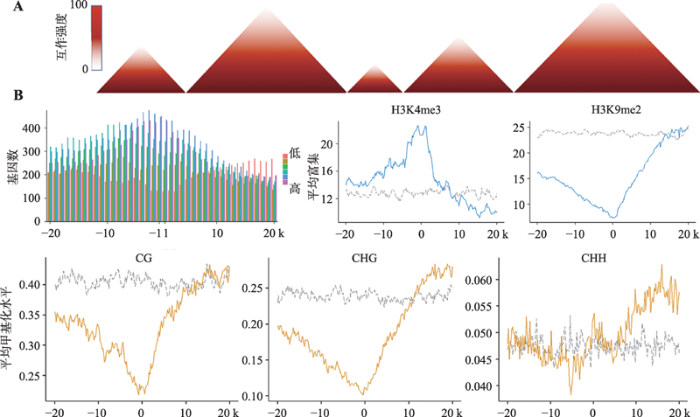
新窗口打开|下载原图ZIP|生成PPT图6TAD结构及水稻TAD boundaries附近的互作和表观遗传修饰特征
A:TAD结构模式图;B:水稻TAD boundaries附近不同表达水平基因的分布以及各种组蛋白的修饰。
Fig. 6Interaction patterns and epigenetic features of TADs and TAD boundaries in rice
目前,染色质TADs结构在动物细胞核内的形成机制[43,44,45,46,47,48]、组成和整体分布[42]、自身结构稳定性、维持基因组结构稳定性[49]、调控相应基因表达[42,50~53]等方面已经得到广泛研究和报道。对植物TADs在以上各个方面的特征总结发现:(1)哺乳动物染色质TADs主要是由CTCF(CTCCC-binding factor)和cohesin等绝缘蛋共同锚定于TAD边界处(TAD boundary),而TADs内部的cohesin来回滑动介导“loop extrusion”最终导致TADs的形成[54]。与果蝇(Drosophila)基因组相似,植物物种基因组虽然无编码CTCF的基因,但已发表的研究工作证实许多植物物种的染色质均可以形成TADs结构[15~17,19,20,22,24,25,34]。在目前已经鉴定的能够形成染色质TADs结构的植物物种中,包括水稻、玉米、番茄、高粱、棉花和甘蓝等,物种内能够参与形成TADs的保守性因子(类似CTCF的蛋白因子)还没有被鉴定出来。近期,Liu等[36]通过分析水稻TADs边界富集的DNA序列motif,获得了可能识别该DNA motif的蛋白因子—TCP转录因子(图6B),后者是否是形成TADs的候选保守因子,仍需后续的分子功能验证;(2)如上文提及,TADs的结构主要包含TAD边界和TAD内部(TAD-interior regions)两个区段。TADs内的两个结构区段所包含的DNA组成和其他表观遗传修饰的特征明显不同(植物和动物TADs一致):相较于TAD内部,TADs边界内的基因密度更高,染色质开放程度较高,转录表达水平较高;TAD边界和TAD内部往往分别富集激活(如H3K4me3等激活性组蛋白修饰等)和抑制性的表观遗传修饰(如H3K27me3等抑制性组蛋白修饰、5mC胞嘧啶甲基化等)[27,28,36]。此外,动物(以果蝇为例)和植物染色质TADs也呈现相似的在染色体上的分布状态,即激活性组蛋白修饰富集的转录激活TADs (active TADs)往往分布于常染色质区段;富集H3K9me2组蛋白修饰、HP1以及Su(var)3-9等蛋白的TADs (heterochromatin TADs)往往分布于异染色质区段;而富集H3K27me3组蛋白修饰和PcG (Polycomb group)蛋白的TADs (Polycomb- repressed TADs)除在异染色质区段内能检测到之外,也常在常染色区段出现[51];(3)在自身结构稳定性方面,动物TADs染色质结构在不同的细胞类型甚至于不同的物种之间都是相对保守的[41],但在生长发育过程中,同一细胞类型的TADs结构中的结构单元可能会出现合并(TADs merge)或者是分离(TADs split)的现象[39]。近期,植物中第一篇利用scHi-C技术探讨水稻单卵细胞、精子细胞、合子以及叶肉细胞染色质构型的研究,为分析同一植物物种的不同细胞类型和同一类型细胞的不同发育阶段、不同植物物种间的TADs染色质结构差异或动态变化,提供了实验技术及数据分析方面的强大支撑[29];(4)跨物种间的TADs位置的比较,能够反映出TADs维持基因组结构稳定的重要性。已有研究发现相对于人(Homo sapiens)的基因组组成,长臂猿(Nomascus leucogenys)的物种形成和进化过程中会出现染色体的融合,而通过比较两个物种的Hi-C数据所鉴定出的TADs,结果发现TADs边界往往出现在长臂猿物种内的融合位点处。该结果暗示了长臂猿染色体结构的变异可能是由TADs边界区的染色质较为脆弱而造成或是由自然选择将携带有TAD内部染色体断裂或融合的个体筛选掉而造成[49]。目前,在植物中TADs是否及如何参与染色质结构变异的研究仅局限在多倍体棉花物种(陆地棉和海岛棉)以及芸薹属内二倍体芜菁和甘蓝的物种形成过程中。其中,Wang等[23]通过分析和比较多倍体陆地棉和海岛棉物种与现存的二倍体亲本的Hi-C染色质构型特征,发现多倍体化后的基因组其开放染色质区会偏倚性的出现TADs的重排或结构变化;Xie等[22]分析并比较了芜菁和甘蓝Hi-C数据能够检测到的TADs的组成,发现两个物种分化后,物种间的共线性区段内多数的TADs均呈现保守状态。除此之外,在其他植物物种分化过程中是否及如何发生TADs的组成变化及后者与基因组结构稳定性之间的关系却鲜有报道。这也将成为植物染色质构型研究领域内今后发展的一个重点研究方向;(5)在调控基因表达方面,从TADs染色质结构特征考虑,无论是在动物或者植物细胞核中,将细胞核内的染色质通过TADs结构的形式进行打包,可以摆脱线性距离较远的影响,从而拉近结构内两位点之间的空间距离,还可以隔绝结构单元内调控元件与外部靶点的相互作用,从而可以特异性识别结构内的靶点[41,42,55~58]。
目前,在植物物种中TADs染色质结构作为一种重要且常见的染色质结构单元已经被广为接受,但是经典的模式植物拟南芥却是其中一个特例——其Hi-C染色质互作热图中没有经典的TAD互作模式[15~17,34]。有关拟南芥不存在经典TADs的理论猜测,主要来自于对其基因组组成的分析:模式植物拟南芥中的基因组相较于其他植物较小,其基因组内部含有较少的转座子(TEs)故而其染色体上的基因密度较高。据此,Liu等[5]推断,基因密度较低的植物物种,由于其基因在基因组上被转座子间隔开,因此需要形成局部的TADs染色质结构,才能够实现空间分离的基因的共表达调控。已经发现植物物种中的高转录密度和TADs结构的形成密切相关[5,36],而拟南芥的染色体臂上的转录密度则比较均匀,顺势调控区域往往位于启动子的附近,因此拟南芥中基因表达调控对TADs染色质结构的依赖性较弱。据此,以上有关基因组大小影响基因密度,进而影响染色质构型的推断得到了一定程度的支持。需要强调的是,以上提出的观点仅是基于已有研究的理论猜测,目前尚无定论。
2.4 染色质环
细胞核内比TADs更为精细的下一个层级的染色质构型特征是染色质环,这是一个更加局部的染色质结构单元。染色质环是一种非常常见的染色质内部的相互作用,该结构可以通过减小调控元件和靶基因之间的空间距离从而促进二者之间的相互作用[10,59]。在动物中的很多相关研究表明,主要发生在TADs结构内部的增强子和启动子之间的染色质环,在精确的转录调控过程中非常重要。动物中发现的一个增强子调控多个基因以及一个基因受到多个增强子的调控现象在植物物种鲜有报道[60]。植物中不同的染色质环在不同的物种、同一物种的不同生长发育过程阶段,同样扮演着非常重要的角色[57,61~64]。通过分析单基因分辨率的拟南芥Hi-C数据,在常染色体臂上总共检测到了20 000多个染色质环[35],其中已知的少数染色质环在转录调控过程中发挥着重要作用[65,66,67]。而基于水稻高分辨率的Hi-C数据,研究人员共检测到了51 319个染色质环[27]。在这些染色质环结构中,转录起始位点(transcription start sites, TSSs)倾向与下游区域形成染色质环,而TTSs倾向与其上游形成环状结构[35]。
这种现象类似于在酵母中报道的“gene globule”模型[68],并被后续定义为“基因自成环现象”。在拟南芥和水稻中的研究发现,自成环的基因往往要比未成环的基因更加活跃,且增强子-启动子形成的环状结构往往会激活其临近的基因[27]。
通过对5种植物基因组Hi-C的研究,科研人员发现玉米和高粱等具有较大基因组的物种拥有TADs结构外部的、长距离的染色质环状互作结构,其区别于动物TADs内较小的染色质环[28]。研究还发现,参与长距离染色质内部互作的区域往往和激活性的组蛋白修饰相关,并且富含基因。此外,对棉花的相关研究中,Wang等[23,24]发现,启动子区、潜在的增强子以及激活性表观遗传修饰的区域两两之间存在相互作用。他们将之分为启动子-启动子、增强子-增强子以及增强子-启动子等3种互作类型,并发现大部分参与的基因也都受到多个长距离相互作用的调节。最后,Lun等[15]利用高分辨率的ChIA-PET技术,捕获到了H3K4me3、RNAPII以及H3K9me2 3种靶蛋白的结合位点以及其介导的结合位点之间的相互作用,并将H3K4me3和RNAPII介导的染色质环分为PPI (promoter-promoter interaction) loop和BP (basal promoter) loop。该研究发现,PPI loop上的基因相较于BP loop上的基因表达量更高,且PPI loop上的基因在水稻基因组的进化过程中拥有更高的保守性;此外,H3K4me3和RNAPII共同介导的loop 基因相较于仅被K3K4me3或RNAPII介导的loop基因表达量更高;另外,H3K4me3以及RNAPII介导的染色质间的环状相互作用形成了复杂的空间转录单元,并且环状结构的形成促使了两端基因的共表达;最后,H3K9me2介导的染色质环(39~341 kb)相较于H3K4me3 (~18 kb)和RNAPII (11~27 kb)介导的染色质环更大。
2.5 IHIs/KEEs
利用Hi-C数据能够检测到植物细胞核特有的染色体内或染色体间的特殊染色质构型特征。在野生型拟南芥中,研究人员能够检测到10个非对角线的染色体(质)内或染色体(质)之间的强烈“网状”相互作用区段[18,19]。人们将这些强烈互作区域形成的结构称为KNOT,而将组成这种结构的区域命名为IHIs/KEEs[18]。对拟南芥中有关IHIs/KEEs的研究进行总结发现:(1)拟南芥的IHIs/KEEs是位于常染色体臂中的异染色质区域,富含TEs相关的重复序列、smRNAs以及H3K9me2、H3K27me1等异染色质标记[18,19];(2) Grob等[18]通过FISH技术证明KNOT结构在拟南芥细胞核中是真实存在的,并通过BLAT发现拟南芥中的IHIs/KEEs含有195 bp和70 bp的两个保守区域,前者对应于ATLANTYS3转座子(LTR/Gypsy家族),后者对应VANDAL6转座子(DNA MutR家族);(3)在拟南芥中IHIs/KEEs与端粒区存在着强烈的相互作用,而IHIs/KEEs与近着丝粒区虽然拥有着相似的序列特征和表观遗传修饰,但二者之间却并不存在显著的互作[18,19];(4)在拟南芥的表观遗传突变体中,IHIs/KEEs位点间的相互作用会发生动态变化[19]。在AtMORC6突变体中,IHIs/KEEs位点之间的相互作用增强。在met1和ddm1突变体中,IHIs/KEEs位点增多,但彼此之间的相互作用相对于野生型拟南芥来说出现了减弱的现象,在suvh4 suvh5 suvh6的“三突”拟南芥中,IHIs/KEEs位点同样出现了增多,但彼此之间的相互作用并没有发生显著的改变。以上结果表明,表观遗传修饰可能在IHIs/KEEs位点之间的相互作用中发挥着重要作用;(5) Grob等[18]猜测并证明了IHIs/KEEs位点是转座子转座插入的首选位点,该结果也暗示了IHIs/ KEEs在保护基因组的完整性中发挥着关键作用; (6)拟南芥中的端粒以及IHIs/KEEs并不会像其他异染色质区域会处在靠近细胞核膜的位置,而是会处在细胞核的内部[69]。除以上在拟南芥中鉴定到的IHIs/KEEs位点,Dong等[27]通过Hi-C技术在水稻中也鉴定到了81个IHIs/KEEs位点,并发现最显著的IHIs/KEEs位点处在水稻的Chr.1和Chr.12。通过探究水稻中的IHIs/ KEEs位点的特征发现,水稻中的IHIs/KEEs与拟南芥相似,也同样富集TEs、sRNAs、H3K9me2以及DNA甲基化(尤其是CHH序列环境)。但与拟南芥中鉴定到的两个保守序列不同,水稻中的IHIs/KEEs位点只含有一个163 bp的保守序列。此外,水稻中的IHIs/KEEs富含多种转座子类型,包括ATLANTYS2、RIRE3、SZ-64B和TRUNCATOR2等,其中ATLANTYS2也存在于拟南芥的IHIs/KEEs位点中。
最近在芸薹属植物芜菁和甘蓝中,Xie等[22]利用Hi-C技术分别鉴定到了31个和7个IHIs/KEEs位点。其中,芜菁中的10条染色体都含有IHIs/KEEs位点分布。与拟南芥和水稻中鉴定到的IHIs/KEEs位点不同,芜菁中的IHIs/KEEs位点在一些染色体上是成簇存在的。因此,从基因组进化的角度看,芜菁中的IHIs/KEEs位点经历了扩张,而与之相反的是,在甘蓝中的IHIs/KEEs位点则出现了收缩减少的现象。
3 结语与展望
虽然目前三维基因组学技术的应用使植物染色质构型的研究取得了突飞猛进的进步,但仍存在一些瓶颈问题:(1)二倍体植物细胞核内的染色体是以同源染色体对的形式出现(即染色体核型组成为2n)。但目前所有已发表的利用Hi-C及ChIA-PET技术探讨染色体(质)构型的研究,均未能区分同源染色体对内的各单条染色体的染色质空间构型。例如,水稻细胞核内实际共含有24条染色体,目前得到的染色体互作结果是将两条同源染色体的互作混合的平均状态,并非细胞内完全真实的染色体互作情况;(2)在进行scHi-C建库时,由于DNA含量相对较少,无法获得更高分辨率的Hi-C互作热图。因此,在分析染色体(质)构型特征时,无法进行拓扑偶联结构域和染色质环水平的特征分析;(3)目前植物中拓扑偶联结构域部分的识别与鉴定主要依靠Dixon等[50]和Crane等[70]为动物拓扑偶联结构域开发的算法,或采用Liu等[36]在鉴定水稻拓扑偶联结构域时所使用的算法。业内仍亟需对以上算法在植物中应用的有效性、差异性和可适用范围进行评估,进而总结或开发出一个更适用于植物细胞核染色质拓扑偶联结构域鉴定的算法。综上所述,染色质三维结构的特征及功能研究已经成为目前基因组学和细胞生物学研究的重要研究领域。随着鉴定和分析植物染色质构型技术的不断突破和进步(如scHi-C的出现),以动物细胞为模型的相关研究结论的普遍性以及植物细胞染色质构型的特征性,将在同一个水平上得到更为准确的验证,进一步促发人们对植物染色质构型的深入研究。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1104/pp.111.187161URLPMID:22095045 [本文引用: 4]
DOI:10.1046/j.1365-313x.2001.01194.xURLPMID:11851915 [本文引用: 2]

Chromosome painting, that is visualisation of chromosome segments or whole chromosomes based on fluorescence in situ hybridization (FISH) with chromosome-specific DNA probes is widely used for chromosome studies in mammals, birds, reptiles and insects. Attempts to establish chromosome painting in euploid plants have failed so far. Here, we report on chromosome painting in Arabidopsis thaliana (n = 5, 125 Mb C(-1)). Pools of contiguous 113-139 BAC clones spanning 2.6 and 13.3 Mb of the short and the long arm of chromosome 4 (17.5 Mb) were used to paint this entire chromosome during mitotic and meiotic divisions as well as in interphase nuclei. The possibility of identifying any particular chromosome region on pachytene chromosomes and within interphase nuclei using selected BACs is demonstrated by differential labelling. This approach allows us, for the first time, to paint an entire autosome of an euploid plant to study chromosome rearrangements, homologue association, interphase chromosome territories, as well as to identify homeologous chromosomes of related species.
DOI:10.1371/journal.pbio.0030157URLPMID:15839726 [本文引用: 1]
Studies of higher-order chromatin arrangements are an essential part of ongoing attempts to explore changes in epigenome structure and their functional implications during development and cell differentiation. However, the extent and cell-type-specificity of three-dimensional (3D) chromosome arrangements has remained controversial. In order to overcome technical limitations of previous studies, we have developed tools that allow the quantitative 3D positional mapping of all chromosomes simultaneously. We present unequivocal evidence for a probabilistic 3D order of prometaphase chromosomes, as well as of chromosome territories (CTs) in nuclei of quiescent (G0) and cycling (early S-phase) human diploid fibroblasts (46, XY). Radial distance measurements showed a probabilistic, highly nonrandom correlation with chromosome size: small chromosomes-independently of their gene density-were distributed significantly closer to the center of the nucleus or prometaphase rosette, while large chromosomes were located closer to the nuclear or rosette rim. This arrangement was independently confirmed in both human fibroblast and amniotic fluid cell nuclei. Notably, these cell types exhibit flat-ellipsoidal cell nuclei, in contrast to the spherical nuclei of lymphocytes and several other human cell types, for which we and others previously demonstrated gene-density-correlated radial 3D CT arrangements. Modeling of 3D CT arrangements suggests that cell-type-specific differences in radial CT arrangements are not solely due to geometrical constraints that result from nuclear shape differences. We also found gene-density-correlated arrangements of higher-order chromatin shared by all human cell types studied so far. Chromatin domains, which are gene-poor, form a layer beneath the nuclear envelope, while gene-dense chromatin is enriched in the nuclear interior. We discuss the possible functional implications of this finding.
DOI:10.1038/nature23884URLPMID:28905911 [本文引用: 1]
The 4D Nucleome Network aims to develop and apply approaches to map the structure and dynamics of the human and mouse genomes in space and time with the goal of gaining deeper mechanistic insights into how the nucleus is organized and functions. The project will develop and benchmark experimental and computational approaches for measuring genome conformation and nuclear organization, and investigate how these contribute to gene regulation and other genome functions. Validated experimental technologies will be combined with biophysical approaches to generate quantitative models of spatial genome organization in different biological states, both in cell populations and in single cells.
DOI:10.1038/s41477-018-0199-5URLPMID:30061747 [本文引用: 4]
Information and function of a genome are not only decorated with epigenetic marks in the linear DNA sequence but also in their non-random spatial organization in the nucleus. Recent research has revealed that three-dimensional (3D) chromatin organization is highly correlated with the functionality of the genome, contributing to many cellular processes. Driven by the improvements in chromatin conformation capture methods and visualization techniques, the past decade has been an exciting period for the study of plants' 3D genome structures, and our knowledge in this area has been substantially advanced. This Review describes our current understanding of plant chromatin organization and positioning beyond the nucleosomal level, and discusses future directions.
DOI:10.1038/nrg.2016.112URLPMID:27739532 [本文引用: 1]
Understanding how chromatin is organized within the nucleus and how this 3D architecture influences gene regulation, cell fate decisions and evolution are major questions in cell biology. Despite spectacular progress in this field, we still know remarkably little about the mechanisms underlying chromatin structure and how it can be established, reset and maintained. In this Review, we discuss the insights into chromatin architecture that have been gained through recent technological developments in quantitative biology, genomics and cell and molecular biology approaches and explain how these new concepts have been used to address important biological questions in development and disease.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.tplants.2018.03.014URLPMID:29703667 [本文引用: 1]
After linear sequences of genomes and epigenomic landscape data, the 3D organization of chromatin in the nucleus is the next level to be explored. Different organisms present a general hierarchical organization, with chromosome territories at the top. Chromatin interaction maps, obtained by chromosome conformation capture (3C)-based methodologies, for eight plant species reveal commonalities, but also differences, among them and with animals. The smallest structures, found in high-resolution maps of the Arabidopsis genome, are single genes. Epigenetic marks (histone modification and DNA methylation), transcriptional activity, and chromatin interaction appear to be correlated, and whether structure is the cause or consequence of the function of interacting regions is being actively investigated.
DOI:10.1186/s13059-015-0738-6URLPMID:26294115 [本文引用: 3]
Methods that use high-throughput sequencing have begun to reveal features of the three-dimensional structure of genomes at a resolution that goes far beyond that of traditional microscopy. Integration of these methods with other molecular tools has advanced our knowledge of both global and local chromatin packing in plants, and has revealed how patterns of chromatin packing correlate with the genomic and epigenomic landscapes. This update reports recent progress made in this area in plants, and suggests new research directions.
DOI:10.1126/science.1181369URLPMID:19815776 [本文引用: 4]
We describe Hi-C, a method that probes the three-dimensional architecture of whole genomes by coupling proximity-based ligation with massively parallel sequencing. We constructed spatial proximity maps of the human genome with Hi-C at a resolution of 1 megabase. These maps confirm the presence of chromosome territories and the spatial proximity of small, gene-rich chromosomes. We identified an additional level of genome organization that is characterized by the spatial segregation of open and closed chromatin to form two genome-wide compartments. At the megabase scale, the chromatin conformation is consistent with a fractal globule, a knot-free, polymer conformation that enables maximally dense packing while preserving the ability to easily fold and unfold any genomic locus. The fractal globule is distinct from the more commonly used globular equilibrium model. Our results demonstrate the power of Hi-C to map the dynamic conformations of whole genomes.
DOI:10.1038/nature08497URLPMID:19890323 [本文引用: 2]
Genomes are organized into high-level three-dimensional structures, and DNA elements separated by long genomic distances can in principle interact functionally. Many transcription factors bind to regulatory DNA elements distant from gene promoters. Although distal binding sites have been shown to regulate transcription by long-range chromatin interactions at a few loci, chromatin interactions and their impact on transcription regulation have not been investigated in a genome-wide manner. Here we describe the development of a new strategy, chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) for the de novo detection of global chromatin interactions, with which we have comprehensively mapped the chromatin interaction network bound by oestrogen receptor alpha (ER-alpha) in the human genome. We found that most high-confidence remote ER-alpha-binding sites are anchored at gene promoters through long-range chromatin interactions, suggesting that ER-alpha functions by extensive chromatin looping to bring genes together for coordinated transcriptional regulation. We propose that chromatin interactions constitute a primary mechanism for regulating transcription in mammalian genomes.
DOI:10.1126/science.1067799URLPMID:11847345 [本文引用: 1]
We describe an approach to detect the frequency of interaction between any two genomic loci. Generation of a matrix of interaction frequencies between sites on the same or different chromosomes reveals their relative spatial disposition and provides information about the physical properties of the chromatin fiber. This methodology can be applied to the spatial organization of entire genomes in organisms from bacteria to human. Using the yeast Saccharomyces cerevisiae, we could confirm known qualitative features of chromosome organization within the nucleus and dynamic changes in that organization during meiosis. We also analyzed yeast chromosome III at the G1 stage of the cell cycle. We found that chromatin is highly flexible throughout. Furthermore, functionally distinct AT- and GC-rich domains were found to exhibit different conformations, and a population-average 3D model of chromosome III could be determined. Chromosome III emerges as a contorted ring.
DOI:10.16288/j.yczz.17-152URLPMID:28936982 [本文引用: 1]
Highest-throughput chromosome conformation capture (Hi-C) is one of the key assays for genome- wide chromatin interaction studies. It is a time-consuming process that involves many steps and many different kinds of reagents, consumables, and equipments. At present, the reproducibility is unsatisfactory. By optimizing the key steps of the Hi-C experiment, such as crosslinking, pretreatment of digestion, inactivation of restriction enzyme, and in situ ligation etc., we established a robust Hi-C procedure and prepared two biological replicates of Hi-C libraries from the GM12878 cells. After preliminary quality control by Sanger sequencing, the two replicates were high-throughput sequenced. The bioinformatics analysis of the raw sequencing data revealed the mapping-ability and pair-mate rate of the raw data were around 90% and 72%, respectively. Additionally, after removal of self-circular ligations and dangling-end products, more than 96% of the valid pairs were reached. Genome-wide interactome profiling shows clear topological associated domains (TADs), which is consistent with previous reports. Further correlation analysis showed that the two biological replicates strongly correlate with each other in terms of both bin coverage and all bin pairs. All these results indicated that the optimized Hi-C procedure is robust and stable, which will be very helpful for the wide applications of the Hi-C assay.
DOI:10.16288/j.yczz.17-152URLPMID:28936982 [本文引用: 1]
Highest-throughput chromosome conformation capture (Hi-C) is one of the key assays for genome- wide chromatin interaction studies. It is a time-consuming process that involves many steps and many different kinds of reagents, consumables, and equipments. At present, the reproducibility is unsatisfactory. By optimizing the key steps of the Hi-C experiment, such as crosslinking, pretreatment of digestion, inactivation of restriction enzyme, and in situ ligation etc., we established a robust Hi-C procedure and prepared two biological replicates of Hi-C libraries from the GM12878 cells. After preliminary quality control by Sanger sequencing, the two replicates were high-throughput sequenced. The bioinformatics analysis of the raw sequencing data revealed the mapping-ability and pair-mate rate of the raw data were around 90% and 72%, respectively. Additionally, after removal of self-circular ligations and dangling-end products, more than 96% of the valid pairs were reached. Genome-wide interactome profiling shows clear topological associated domains (TADs), which is consistent with previous reports. Further correlation analysis showed that the two biological replicates strongly correlate with each other in terms of both bin coverage and all bin pairs. All these results indicated that the optimized Hi-C procedure is robust and stable, which will be very helpful for the wide applications of the Hi-C assay.
DOI:10.1038/s41467-018-07882-8URLPMID:30602773 [本文引用: 5]
Wave-particle duality is an inherent peculiarity of the quantum world. The double-slit experiment has been frequently used for understanding different aspects of this fundamental concept. The occurrence of interference rests on the lack of which-way information and on the absence of decoherence mechanisms, which could scramble the wave fronts. Here, we report on the observation of two-center interference in the molecular-frame photoelectron momentum distribution upon ionization of the neon dimer by a strong laser field. Postselection of ions, which are measured in coincidence with electrons, allows choosing the symmetry of the residual ion, leading to observation of both, gerade and ungerade, types of interference.
DOI:10.1128/MMBR.00006-15URLPMID:26223848 [本文引用: 1]
In humans, nearly two meters of genomic material must be folded to fit inside each micrometer-scale cell nucleus while remaining accessible for gene transcription, DNA replication, and DNA repair. This fact highlights the need for mechanisms governing genome organization during any activity and to maintain the physical organization of chromosomes at all times. Insight into the functions and three-dimensional structures of genomes comes mostly from the application of visual techniques such as fluorescence in situ hybridization (FISH) and molecular approaches including chromosome conformation capture (3C) technologies. Recent developments in both types of approaches now offer the possibility of exploring the folded state of an entire genome and maybe even the identification of how complex molecular machines govern its shape. In this review, we present key methodologies used to study genome organization and discuss what they reveal about chromosome conformation as it relates to transcription regulation across genomic scales in mammals.
DOI:10.1186/s13059-017-1281-4URLPMID:28830561 [本文引用: 2]
The merging of two diverged genomes can result in hybrid offspring that phenotypically differ greatly from both parents. In plants, interspecific hybridization plays important roles in evolution and speciation. In addition, many agricultural and horticultural species are derived from interspecific hybridization. However, the detailed mechanisms responsible for non-additive phenotypic novelty in hybrids remain elusive.
DOI:10.1016/j.molcel.2014.07.009URLPMID:25132176 [本文引用: 7]
Chromosomes are folded, spatially organized, and regulated by epigenetic marks. How chromosomal architecture is connected to the epigenome is not well understood. We show that chromosomal architecture of Arabidopsis is tightly linked to the epigenetic state. Furthermore, we show how physical constraints, such as nuclear size, correlate with the folding principles of chromatin. We also describe a nuclear structure, termed KNOT, in which genomic regions of all five Arabidopsis chromosomes interact. These KNOT ENGAGED ELEMENT (KEE) regions represent heterochromatic islands within euchromatin. Similar to PIWI-interacting RNA clusters, such as flamenco in Drosophila, KEEs represent preferred landing sites for transposable elements, which may be part of a transposon defense mechanism in the Arabidopsis nucleus.
DOI:10.1016/j.molcel.2014.07.008URLPMID:25132175 [本文引用: 5]
Chromosomes form 3D structures that are critical to the regulation of cellular and genetic processes. Here, we present a study of global chromatin interaction patterns in Arabidopsis thaliana. Our genome-wide approach confirmed interactions that were previously observed by other methods as well as uncovered long-range interactions such as those among small heterochromatic regions embedded in euchromatic arms. We also found that interactions are correlated with various epigenetic marks that are localized in active or silenced chromatin. Arabidopsis chromosomes do not contain large local interactive domains that resemble the topological domains described in animals but, instead, contain relatively small interactive regions scattered around the genome that contain H3K27me3 or H3K9me2. We generated interaction maps in mutants that are defective in specific epigenetic pathways and found altered interaction patterns that correlate with changes in the epigenome. These analyses provide further insights into molecular mechanisms of epigenetic regulation of the genome.
DOI:10.1101/gr.170332.113URLPMID:25367294 [本文引用: 1]
The spatial arrangement of interphase chromosomes in the nucleus is important for gene expression and genome function in animals and in plants. The recently developed Hi-C technology is an efficacious method to investigate genome packing. Here we present a detailed Hi-C map of the three-dimensional genome organization of the plant Arabidopsis thaliana. We find that local chromatin packing differs from the patterns seen in animals, with kilobasepair-sized segments that have much higher intrachromosome interaction rates than neighboring regions, representing a dominant local structural feature of genome conformation in A. thaliana. These regions, which appear as positive strips on two-dimensional representations of chromatin interaction, are enriched in epigenetic marks H3K27me3, H3.1, and H3.3. We also identify more than 400 insulator-like regions. Furthermore, although topologically associating domains (TADs), which are prominent in animals, are not an obvious feature of A. thaliana genome packing, we found more than 1000 regions that have properties of TAD boundaries, and a similar number of regions analogous to the interior of TADs. The insulator-like, TAD-boundary-like, and TAD-interior-like regions are each enriched for distinct epigenetic marks and are each correlated with different gene expression levels. We conclude that epigenetic modifications, gene density, and transcriptional activity combine to shape the local packing of the A. thaliana nuclear genome.
DOI:10.1093/nar/gkz511URLPMID:31184697 [本文引用: 1]
Autopolyploidy is widespread in higher plants and important for agricultural yield and quality. However, the effects of genome duplication on the chromatin organization and transcriptional regulation are largely unknown in plants. Using High-throughput Chromosome Conformation Capture (Hi-C), we showed that autotetraploid Arabidopsis presented more inter-chromosomal interactions and fewer short-range chromatin interactions compared with its diploid progenitor. In addition, genome duplication contributed to the switching of some loose and compact structure domains with altered H3K4me3 and H3K27me3 histone modification status. 539 genes were identified with altered transcriptions and chromatin interactions in autotetraploid Arabidopsis. Especially, we found that genome duplication changed chromatin looping and H3K27me3 histone modification in Flowering Locus C. We propose that genome doubling modulates the transcription genome-wide by changed chromatin interactions and at the specific locus by altered chromatin loops and histone modifications.
DOI:10.1038/s41477-019-0479-8URLPMID:31383969 [本文引用: 4]
The non-random three-dimensional (3D) organization of the genome in the nucleus is critical to gene regulation and genome function. Using high-throughput chromatin conformation capture, we generated chromatin interaction maps for Brassica rapa and Brassica oleracea at a high resolution and characterized the conservation and divergence of chromatin organization in these two species. Large-scale chromatin structures, including A/B compartments and topologically associating domains, are notably conserved between B. rapa and B. oleracea, yet their KNOT structures are highly divergent. We found that genes retained in less fractionated subgenomes exhibited stronger interaction strengths, and diploidization-resistant duplicates retained in pairs or triplets are more likely to be colocalized in both B. rapa and B. oleracea. These observations suggest that spatial constraint in duplicated genes is correlated to their biased retention in the diploidization process. In addition, we found strong similarities in the epigenetic modification and Gene Ontology terms of colocalized paralogues, which were largely conserved across B. rapa and B. oleracea, indicating functional constraints on their 3D positioning in the nucleus. This study presents an investigation of the spatial organization of genomes in Brassica and provides insights on the role of 3D organization in the genome evolution of this genus.
DOI:10.1038/s41477-017-0096-3URLPMID:29379149 [本文引用: 4]
The formation of polyploids significantly increases the complexity of transcriptional regulation, which is expected to be reflected in sophisticated higher-order chromatin structures. However, knowledge of three-dimensional (3D) genome structure and its dynamics during polyploidization remains poor. Here, we characterize 3D genome architectures for diploid and tetraploid cotton, and find the existence of A/B compartments and topologically associated domains (TADs). By comparing each subgenome in tetraploids with its extant diploid progenitor, we find that genome allopolyploidization has contributed to the switching of A/B compartments and the reorganization of TADs in both subgenomes. We also show that the formation of TAD boundaries during polyploidization preferentially occurs in open chromatin, coinciding with the deposition of active chromatin modification. Furthermore, analysis of inter-subgenomic chromatin interactions has revealed the spatial proximity of homoeologous genes, possibly associated with their coordinated expression. This study advances our understanding of chromatin organization in plants and sheds new light on the relationship between 3D genome evolution and transcriptional regulation.
DOI:10.1038/ng.3807URLPMID:28263319 [本文引用: 2]
Comparative population genomics offers an excellent opportunity for unraveling the genetic history of crop domestication. Upland cotton (Gossypium hirsutum) has long been an important economic crop, but a genome-wide and evolutionary understanding of the effects of human selection is lacking. Here, we describe a variation map for 352 wild and domesticated cotton accessions. We scanned 93 domestication sweeps occupying 74 Mb of the A subgenome and 104 Mb of the D subgenome, and identified 19 candidate loci for fiber-quality-related traits through a genome-wide association study. We provide evidence showing asymmetric subgenome domestication for directional selection of long fibers. Global analyses of DNase I-hypersensitive sites and 3D genome architecture, linking functional variants to gene transcription, demonstrate the effects of domestication on cis-regulatory divergence. This study provides new insights into the evolution of gene organization, regulation and adaptation in a major crop, and should serve as a rich resource for genome-based cotton improvement.
DOI:10.1038/nature22043URLPMID:28447635 [本文引用: 1]
Cereal grasses of the Triticeae tribe have been the major food source in temperate regions since the dawn of agriculture. Their large genomes are characterized by a high content of repetitive elements and large pericentromeric regions that are virtually devoid of meiotic recombination. Here we present a high-quality reference genome assembly for barley (Hordeum vulgare L.). We use chromosome conformation capture mapping to derive the linear order of sequences across the pericentromeric space and to investigate the spatial organization of chromatin in the nucleus at megabase resolution. The composition of genes and repetitive elements differs between distal and proximal regions. Gene family analyses reveal lineage-specific duplications of genes involved in the transport of nutrients to developing seeds and the mobilization of carbohydrates in grains. We demonstrate the importance of the barley reference sequence for breeding by inspecting the genomic partitioning of sequence variation in modern elite germplasm, highlighting regions vulnerable to genetic erosion.
DOI:10.1111/jipb.12907URLPMID:31944575
The balance between cellular carbon (C) and nitrogen (N) must be tightly coordinated to sustain optimal growth and development in plants. In chloroplasts, photosynthesis converts inorganic C to organic C, which is important for maintenance of C content in plant cells. However, little is known about the role of chloroplasts in C/N balance. Here, we identified a nuclear-encoded protein LOW PHOTOSYNTHETIC EFFICIENCY2 (LPE2) that it is required for photosynthesis and C/N balance in Arabidopsis. LPE2 is specifically localized in the chloroplast. Both of loss-of-function mutants, lpe2-1 and lpe2-2, showed lower photosynthetic activity, characterized by slower electron transport and lower PSII quantum yield than the wild type. Notably, LPE2 is predicted to encode the plastid ribosomal protein S21 (RPS21). Deficiency of LPE2 significantly perturbed the thylakoid membrane composition and plastid protein accumulation, although the transcription of plastid genes is not affected obviously. More interestingly, transcriptome analysis indicated that the loss of LPE2 altered the expression of C and N response related genes in nucleus, which is confirmed by quantitive RT-PCR. Moreover, deficiency of LPE2 suppressed the response of C/N balance in physiological level. Taken together, our findings suggest that LPE2 plays dual roles in photosynthesis and the response to C/N balance. This article is protected by copyright.
DOI:10.1111/tpj.13925URLPMID:29660196 [本文引用: 6]
The non-random spatial packing of chromosomes in the nucleus plays a critical role in orchestrating gene expression and genome function. Here, we present a Hi-C analysis of the chromatin interaction patterns in rice (Oryza sativa L.) at hierarchical architectural levels. We confirm that rice chromosomes occupy their own territories with certain preferential inter-chromosomal associations. Moderate compartment delimitation and extensive TADs (Topologically Associated Domains) were determined to be associated with heterogeneous genomic compositions and epigenetic marks in the rice genome. We found subtle features including chromatin loops, gene loops, and off-/near-diagonal intensive interaction regions. Gene chromatin loops associated with H3K27me3 could be positively involved in gene expression. In addition to insulated enhancing effects for neighbor gene expression, the identified rice gene loops could bi-directionally (+/-) affect the expression of looped genes themselves. Finally, web-interleaved off-diagonal IHIs/KEEs (Interactive Heterochromatic Islands or KNOT ENGAGED ELEMENTs) could trap transposable elements (TEs) via the enrichment of silencing epigenetic marks. In parallel, the near-diagonal FIREs (Frequently Interacting Regions) could positively affect the expression of involved genes. Our results suggest that the chromatin packing pattern in rice is generally similar to that in Arabidopsis thaliana but with clear differences at specific structural levels. We conclude that genomic composition, epigenetic modification, and transcriptional activity could act in combination to shape global and local chromatin packing in rice. Our results confirm recent observations in rice and A.?thaliana but also provide additional insights into the patterns and features of chromatin organization in higher plants.
DOI:10.1016/j.molp.2017.11.005URLPMID:29175436 [本文引用: 4]
The spatial organization of the genome plays an important role in the regulation of gene expression. However, the core structural features of animal genomes, such as topologically associated domains (TADs) and chromatin loops, are not prominent in the extremely compact Arabidopsis genome. In this study, we examine?the chromatin architecture, as well as their DNA methylation, histone modifications, accessible chromatin, and gene expression, of maize, tomato, sorghum, foxtail millet, and rice with genome sizes ranging from 0.4?to 2.4 Gb. We found that these plant genomes can be divided into mammalian-like A/B compartments. At higher resolution, the chromosomes of these plants can be further partitioned to local A/B compartments that reflect their euchromatin, heterochromatin, and polycomb status. Chromatins in all these plants are organized into domains that are not conserved across species. They show similarity to the Drosophila compartment domains, and are clustered into active, polycomb, repressive, and intermediate types based on their transcriptional activities and epigenetic signatures, with domain border overlaps with the local A/B compartment junctions. In the large maize and tomato genomes, we observed extensive chromatin loops. However, unlike the mammalian chromatin loops that are enriched at the TAD border, plant chromatin loops are often formed between gene islands outside the repressive domains?and are closely associated with active compartments. Our study indicates that plants have complex and unique 3D chromatin architectures, which require?further study to elucidate their biological functions.
DOI:10.1038/s41477-019-0471-3URLPMID:31332313 [本文引用: 4]
Chromatin conformation capture (3C)1 and high-throughput 3C (Hi-C)2 assays allow the study of three-dimensional (3D) genome structures in cell populations or tissues, based on average proximities of folded DNA. However, differences between cells can be observed only by single-cell measurements that avoid ensemble averaging3-5. To study 3D chromatin organization and dynamics before and after fertilization in flowering plants, we analysed the 3D genomes of rice eggs, sperm cells, unicellular zygotes and shoot mesophyll cells. We show that chromatin architectures of rice eggs and sperm cells are comparable to those of mesophyll cells and are reorganized after fertilization. The rice single-cell 3D genomes display specific features of chromosome compartments and telomere/centromere configuration compared to those in mammalian single cells. Active and silent chromatin domains combine to form multiple foci in the nuclear space. Notably, the 3D genomes of the eggs and unicellular zygotes contain a compact silent centre (CSC) that is absent in sperm cells. CSC appears to be reorganized after fertilization, and may be involved in the regulation of zygotic genome activation (ZGA). Our results reveal specific 3D genome features of plant gametes and the unicellular zygote, and provide a spatial chromatin basis for ZGA and epigenetic regulation in plants.
DOI:10.1038/s41467-019-10602-5URLPMID:31201335
Chromatin loops connect regulatory elements to their target genes. They serve as bridges between transcriptional regulation and phenotypic variation in mammals. However, spatial organization of regulatory elements and its impact on gene expression in plants remain unclear. Here, we characterize epigenetic features of active promoter proximal regions and candidate distal regulatory elements to construct high-resolution chromatin interaction maps for maize via long-read chromatin interaction analysis by paired-end tag sequencing (ChIA-PET). The maps indicate that chromatin loops are formed between regulatory elements, and that gene pairs between promoter proximal regions tend to be co-expressed. The maps also demonstrated the topological basis of quantitative trait loci which influence gene expression and phenotype. Many promoter proximal regions are involved in chromatin loops with distal regulatory elements, which regulate important agronomic traits. Collectively, these maps provide a high-resolution view of 3D maize genome architecture, and its role in gene expression and phenotypic variation.
[本文引用: 1]
DOI:10.1007/bf00303033URLPMID:3219911 [本文引用: 1]
Specific chromosome domains in interphase nuclei of neurons and glia were studied by three-dimensional (3-D) reconstruction of serial optical sections from in situ hybridized human CNS tissue. Overall patterns of centromere organization, delineated with alphoid repeats, were comparable to those seen in mouse, and are clearly conserved in mammalian evolution. Cloned probes from other individual chromosome domains were used to define interphase organization more precisely. Homologous chromosomes were spatially separated in nuclei. In large neurons, probes specific for 9q12, or 1q12 showed that at least one homolog was always compartmentalized together with centromeres on the nucleolus, while the second signal either abutted the nucleolus or was on the nuclear membrane. A telomeric Yq12 sequence also localized together with perinucleolar centromeres in a completely non-Rabl orientation. In astrocytes, these three chromosome regions were on the membrane and not necessarily associated with nucleoli. Therefore there are different patterns of interphase chromosome organization in functionally distinct cell types. In contrast to the above domains, a 1p36.3 telomeric sequence embedded in a large Alu-rich and early replicating chromosome region, was always found in an interior euchromatic nuclear compartment in both neurons and glial cells. In double hybridizations with 1q12 and 1p36.3 probes, 1p arms were clearly separated in all cells, and arms projected radially into the interior nucleoplasm with non-Rabl orientations. There was no absolute or rigid position for each 1p arm with respect to each other or to the major dendrite, indicating that individual chromosome arms may be dynamically positioned even in highly differentiated cell types. We suggest that centromeric and other highly repeated non-transcribed sequence domains may act as general organizing centers for cell type specific interphase patterns that are conserved in mammalian evolution. Such centers would allow selected groups of chromosome arms to extend into (and contract from) an interior, presumably transcriptionally active, nuclear compartment.
URLPMID:11739653 [本文引用: 1]
The intranuclear arrangements of centromeres and telomeres during meiotic interphase and early prophase I of meiosis in Arabidopsis thaliana were analysed by fluorescent in situ hybridisation to spread pollen mother cells and embryo-sac mother cells. Meiocyte identification, staging and progression were established by spreading and sectioning techniques, including various staining procedures and bromodeoxyuridine labeling of replicating DNA. Centromere regions of Arabidopsis are unpaired, widely dispersed and peripherally located in nuclei during meiotic interphase, and they remain unpaired and unassociated throughout leptotene. Eventually they associate pairwise during zygotene, as part of the nucleus-wide synapsis of homologous chromosomes. Telomeres, by contrast, show a persistent association with the nucleolus throughout meiotic interphase. Variation in telomere signal number indicates that telomeres undergo pairing during this interval, preceding the onset of general chromosome synapsis. During leptotene the paired telomeres lose their association with the nucleolus and become widely dispersed. As the chromosomes synapse during zygotene, the telomeres reveal a loose clustering within one hemisphere, which may represent a degenerate or relic bouquet configuration. We propose that in Arabidopsis the classical leptotene/zygotene bouquet is absent and is replaced functionally by nucleolus-associated telomere clustering.
DOI:10.1073/pnas.212325299URLPMID:12384572 [本文引用: 3]
Heterochromatin in the model plant Arabidopsis thaliana is confined to small pericentromeric regions of all five chromosomes and to the nucleolus organizing regions. This clear differentiation makes it possible to study spatial arrangement and functional properties of individual chromatin domains in interphase nuclei. Here, we present the organization of Arabidopsis chromosomes in young parenchyma cells. Heterochromatin segments are organized as condensed chromocenters (CCs), which contain heavily methylated, mostly repetitive DNA sequences. In contrast, euchromatin contains less methylated DNA and emanates from CCs as loops spanning 0.2-2 Mbp. These loops are rich in acetylated histones, whereas CCs contain less acetylated histones. We identified individual CCs and loops by fluorescence in situ hybridization by using rDNA clones and 131 bacterial artificial chromosome DNA clones from chromosome 4. CC and loops together form a chromosome territory. Homologous CCs and territories were associated frequently. Moreover, a considerable number of nuclei displayed perfect alignment of homologous subregions, suggesting physical transinteractions between the homologs. The arrangement of interphase chromosomes in Arabidopsis provides a well defined system to investigate chromatin organization and its role in epigenetic processes.
DOI:10.1101/gr.204032.116URLPMID:27225844 [本文引用: 3]
The three-dimensional packing of the genome plays an important role in regulating gene expression. We have used Hi-C, a genome-wide chromatin conformation capture (3C) method, to analyze Arabidopsis thaliana chromosomes dissected into subkilobase segments, which is required for gene-level resolution in this species with a gene-dense genome. We found that the repressive H3K27me3 histone mark is overrepresented in the promoter regions of genes that are in conformational linkage over long distances. In line with the globally dispersed distribution of RNA polymerase II in A. thaliana nuclear space, actively transcribed genes do not show a strong tendency to associate with each other. In general, there are often contacts between 5' and 3' ends of genes, forming local chromatin loops. Such self-loop structures of genes are more likely to occur in more highly expressed genes, although they can also be found in silent genes. Silent genes with local chromatin loops are highly enriched for the histone variant H3.3 at their 5' and 3' ends but depleted of repressive marks such as heterochromatic histone modifications and DNA methylation in flanking regions. Our results suggest that, different from animals, a major theme of genome folding in A. thaliana is the formation of structural units that correspond to gene bodies.
DOI:10.1038/s41477-017-0005-9URLPMID:28848243 [本文引用: 8]
The non-random three-dimensional organization of genomes is critical for many cellular processes. Recently, analyses of genome-wide chromatin packing in the model dicot plant Arabidopsis thaliana have been reported 1-4 . At a kilobase scale, the A. thaliana chromatin interaction network is highly correlated with a range of genomic and epigenomic features 1-4 . Surprisingly, topologically associated domains (TADs), which appear to be a prevalent structural feature of genome packing in many animal species, are not prominent in the A. thaliana genome 1,2,4-6 . Using a genome-wide chromatin conformation capture approach, Hi-C (ref. 7 ), we report high-resolution chromatin packing patterns of another model plant, rice. We unveil new structural features of chromatin organization at both chromosomal and local levels compared to A. thaliana, with thousands of distinct TADs that cover about a quarter of the rice genome. The rice TAD boundaries are associated with euchromatic epigenetic marks and active gene expression, and enriched with a sequence motif that can be recognized by plant-specific TCP proteins. In addition, we report chromosome decondensation in rice seedlings undergoing cold stress, despite local chromatin packing patterns remaining largely unchanged. The substantial variation found already in a comparison of two plant species suggests that chromatin organization in plants might be more diverse than in multicellular animals.
DOI:10.1126/science.aaf8084URLPMID:27445307 [本文引用: 1]
The spatial organization of chromatin critically affects genome function. Recent chromosome-conformation-capture studies have revealed topologically associating domains (TADs) as a conserved feature of chromatin organization, but how TADs are spatially organized in individual chromosomes remains unknown. Here, we developed an imaging method for mapping the spatial positions of numerous genomic regions along individual chromosomes and traced the positions of TADs in human interphase autosomes and X chromosomes. We observed that chromosome folding deviates from the ideal fractal-globule model at large length scales and that TADs are largely organized into two compartments spatially arranged in a polarized manner in individual chromosomes. Active and inactive X chromosomes adopt different folding and compartmentalization configurations. These results suggest that the spatial organization of chromatin domains can change in response to regulation.
DOI:10.1186/s13059-015-0741-yURLPMID:26316348 [本文引用: 1]
Analysis of Hi-C data has shown that the genome can be divided into two compartments called A/B compartments. These compartments are cell-type specific and are associated with open and closed chromatin. We show that A/B compartments can reliably be estimated using epigenetic data from several different platforms: the Illumina 450 k DNA methylation microarray, DNase hypersensitivity sequencing, single-cell ATAC sequencing and single-cell whole-genome bisulfite sequencing. We do this by exploiting that the structure of long-range correlations differs between open and closed compartments. This work makes A/B compartment assignment readily available in a wide variety of cell types, including many human cancers.
DOI:10.1016/j.cell.2017.06.029URLPMID:28709003 [本文引用: 2]
High-order chromatin structure plays important roles in gene expression regulation. Knowledge of the dynamics of 3D chromatin structures during mammalian embryo development remains limited. We report the 3D chromatin architecture of mouse gametes and early embryos using an optimized Hi-C method with low-cell samples. We find that mature oocytes at the metaphase II stage do not have topologically associated domains (TADs). In sperm, extra-long-range interactions (>4 Mb) and interchromosomal interactions occur frequently. The high-order structures of both the paternal and maternal genomes in zygotes and two-cell embryos are obscure but are gradually re-established through development. The establishment of the TAD structure requires DNA replication but not zygotic genome activation. Furthermore, unmethylated CpGs are enriched in A compartment, and methylation levels are decreased to a greater extent in A compartment than in B compartment in embryos. In summary, the global reprogramming of chromatin architecture occurs during early mammalian development.
DOI:10.1038/nature24281URLPMID:29094699 [本文引用: 1]
Imaging and chromosome conformation capture studies have revealed several layers of chromosome organization, including segregation into megabase-sized active and inactive compartments, and partitioning into sub-megabase domains (TADs). It remains unclear, however, how these layers of organization form, interact with one another and influence genome function. Here we show that deletion of the cohesin-loading factor Nipbl in mouse liver leads to a marked reorganization of chromosomal folding. TADs and associated Hi-C peaks vanish globally, even in the absence of transcriptional changes. By contrast, compartmental segregation is preserved and even reinforced. Strikingly, the disappearance of TADs unmasks a finer compartment structure that accurately reflects the underlying epigenetic landscape. These observations demonstrate that the three-dimensional organization of the genome results from the interplay of two independent mechanisms: cohesin-independent segregation of the genome into fine-scale compartments, defined by chromatin state; and cohesin-dependent formation of TADs, possibly by loop extrusion, which helps to guide distant enhancers to their target genes.
DOI:10.1038/nature11082URL [本文引用: 3]
The spatial organization of the genome is intimately linked to its biological function, yet our understanding of higher order genomic structure is coarse, fragmented and incomplete. In the nucleus of eukaryotic cells, interphase chromosomes occupy distinct chromosome territories, and numerous models have been proposed for how chromosomes fold within chromosome territories(1). These models, however, provide only few mechanistic details about the relationship between higher order chromatin structure and genome function. Recent advances in genomic technologies have led to rapid advances in the study of three-dimensional genome organization. In particular, Hi-C has been introduced as a method for identifying higher order chromatin interactions genome wide(2). Here we investigate the three-dimensional organization of the human and mouse genomes in embryonic stem cells and terminally differentiated cell types at unprecedented resolution. We identify large, megabase-sized local chromatin interaction domains, which we term 'topological domains', as a pervasive structural feature of the genome organization. These domains correlate with regions of the genome that constrain the spread of heterochromatin. The domains are stable across different cell types and highly conserved across species, indicating that topological domains are an inherent property of mammalian genomes. Finally, we find that the boundaries of topological domains are enriched for the insulator binding protein CTCF, housekeeping genes, transfer RNAs and short interspersed element (SINE) retrotransposons, indicating that these factors may have a role in establishing the topological domain structure of the genome.
DOI:10.1038/nature11049URL [本文引用: 4]
In eukaryotes transcriptional regulation often involves multiple long-range elements and is influenced by the genomic environment(1). A prime example of this concerns the mouse X-inactivation centre (Xic), which orchestrates the initiation of X-chromosome inactivation (XCI) by controlling the expression of the non-protein-coding Xist transcript. The extent of Xic sequences required for the proper regulation of Xist remains unknown. Here we use chromosome conformation capture carbon-copy (5C)(2) and super-resolution microscopy to analyse the spatial organization of a 4.5-megabases (Mb) region including Xist. We discover a series of discrete 200-kilobase to 1Mb topologically associating domains (TADs), present both before and after cell differentiation and on the active and inactive X. TADs align with, but do not rely on, several domain-wide features of the epigenome, such as H3K27me3 or H3K9me2 blocks and lamina-associated domains. TADs also align with coordinately regulated gene clusters. Disruption of a TAD boundary causes ectopic chromosomal contacts and long-range transcriptional misregulation. The Xist/Tsix sense/antisense unit illustrates how TADs enable the spatial segregation of oppositely regulated chromosomal neighbourhoods, with the respective promoters of Xist and Tsix lying in adjacent TADs, each containing their known positive regulators. We identify a novel distal regulatory region of Tsix within its TAD, which produces a long intervening RNA, Linx. In addition to uncovering a new principle of cis-regulatory architecture of mammalian chromosomes, our study sets the stage for the full genetic dissection of the X-inactivation centre.
DOI:10.1126/sciadv.aar5255URLPMID:29507889 [本文引用: 2]
Cueva de los Aviones (southeast Spain) is a site of the Neandertal-associated Middle Paleolithic of Europe. It has yielded ochred and perforated marine shells, red and yellow colorants, and shell containers that feature residues of complex pigmentatious mixtures. Similar finds from the Middle Stone Age of South Africa have been widely accepted as archaeological proxies for symbolic behavior. U-series dating of the flowstone capping the Cueva de los Aviones deposit shows that the symbolic finds made therein are 115,000 to 120,000 years old and predate the earliest known comparable evidence associated with modern humans by 20,000 to 40,000 years. Given our findings, it is possible that the roots of symbolic material culture may be found among the common ancestor of Neandertals and modern humans, more than half-a-million years ago.
DOI:10.1126/science.362.6421.1442URLPMID:30573631 [本文引用: 2]
The endosomal sorting complexes required for transport (ESCRTs) catalyze reverse-topology scission from the inner face of membrane necks in HIV budding, multivesicular endosome biogenesis, cytokinesis, and other pathways. We encapsulated ESCRT-III subunits Snf7, Vps24, and Vps2 and the AAA+ ATPase (adenosine triphosphatase) Vps4 in giant vesicles from which membrane nanotubes reflecting the correct topology of scission could be pulled. Upon ATP release by photo-uncaging, this system generated forces within the nanotubes that led to membrane scission in a manner dependent upon Vps4 catalytic activity and Vps4 coupling to the ESCRT-III proteins. Imaging of scission revealed Snf7 and Vps4 puncta within nanotubes whose presence followed ATP release, correlated with force generation and nanotube constriction, and preceded scission. These observations directly verify long-standing predictions that ATP-hydrolyzing assemblies of ESCRT-III and Vps4 sever membranes.
DOI:10.1093/nar/gkx738URLPMID:28977568 [本文引用: 1]
Topologically associated domains (TADs) are 3D genomic structures with high internal interactions that play important roles in genome compaction and gene regulation. Their genomic locations and their association with CCCTC-binding factor (CTCF)-binding sites and transcription start sites (TSSs) were recently reported. However, the relationship between TADs and other genomic elements has not been systematically evaluated. This was addressed in the present study, with a focus on the enrichment of these genomic elements and their ability to predict the TAD boundary region. We found that consensus CTCF-binding sites were strongly associated with TAD boundaries as well as with the transcription factors (TFs) Zinc finger protein (ZNF)143 and Yin Yang (YY)1. TAD boundary-associated genomic elements include DNase I-hypersensitive sites, H3K36 trimethylation, TSSs, RNA polymerase II, and TFs such as Specificity protein 1, ZNF274 and SIX homeobox 5. Computational modeling with these genomic elements suggests that they have distinct roles in TAD boundary formation. We propose a structural model of TAD boundaries based on these findings that provides a basis for studying the mechanism of chromatin structure formation and gene regulation.
DOI:10.1146/annurev-cellbio-100616-060531URLPMID:28783961 [本文引用: 1]
Animal development depends on not only the linear genome sequence that embeds millions of cis-regulatory elements, but also the three-dimensional (3D) chromatin architecture that orchestrates the interplay between cis-regulatory elements and their target genes. Compared to our knowledge of the cis-regulatory sequences, the understanding of the 3D genome organization in human and other eukaryotes is still limited. Recent advances in technologies to map the 3D genome architecture have greatly accelerated the pace of discovery. Here, we review emerging concepts of chromatin organization in mammalian cells, discuss the dynamics of chromatin conformation during development, and highlight important roles for chromatin organization in cancer and other human diseases.
DOI:10.1016/j.cell.2012.01.010URL [本文引用: 1]
Chromosomes are the physical realization of genetic information and thus form the basis for its readout and propagation. Here we present a high-resolution chromosomal contact map derived from a modified genome-wide chromosome conformation capture approach applied to Drosophila embryonic nuclei. The data show that the entire genome is linearly partitioned into well-demarcated physical domains that overlap extensively with active and repressive epigenetic marks. Chromosomal contacts are hierarchically organized between domains. Global modeling of contact density and clustering of domains show that inactive domains are condensed and confined to their chromosomal territories, whereas active domains reach out of the territory to form remote intra-and interchromosomal contacts. Moreover, we systematically identify specific long-range intrachromosomal contacts between Polycomb-repressed domains. Together, these observations allow for quantitative prediction of the Drosophila chromosomal contact map, laying the foundation for detailed studies of chromosome structure and function in a genetically tractable system.
DOI:10.15252/embj.201798004URLPMID:29217591 [本文引用: 1]
Mammalian genomes are spatially organized into compartments, topologically associating domains (TADs), and loops to facilitate gene regulation and other chromosomal functions. How compartments, TADs, and loops are generated is unknown. It has been proposed that cohesin forms TADs and loops by extruding chromatin loops until it encounters CTCF, but direct evidence for this hypothesis is missing. Here, we show that cohesin suppresses compartments but is required for TADs and loops, that CTCF defines their boundaries, and that the cohesin unloading factor WAPL and its PDS5 binding partners control the length of loops. In the absence of WAPL and PDS5 proteins, cohesin forms extended loops, presumably by passing CTCF sites, accumulates in axial chromosomal positions (vermicelli), and condenses chromosomes. Unexpectedly, PDS5 proteins are also required for boundary function. These results show that cohesin has an essential genome-wide function in mediating long-range chromatin interactions and support the hypothesis that cohesin creates these by loop extrusion, until it is delayed by CTCF in a manner dependent on PDS5 proteins, or until it is released from DNA by WAPL.
DOI:10.1101/gr.233874.117URLPMID:29914971 [本文引用: 2]
The relationship between evolutionary genome remodeling and the three-dimensional structure of the genome remain largely unexplored. Here, we use the heavily rearranged gibbon genome to examine how evolutionary chromosomal rearrangements impact genome-wide chromatin interactions, topologically associating domains (TADs), and their epigenetic landscape. We use high-resolution maps of gibbon-human breaks of synteny (BOS), apply Hi-C in gibbon, measure an array of epigenetic features, and perform cross-species comparisons. We find that gibbon rearrangements occur at TAD boundaries, independent of the parameters used to identify TADs. This overlap is supported by a remarkable genetic and epigenetic similarity between BOS and TAD boundaries, namely presence of CpG islands and SINE elements, and enrichment in CTCF and H3K4me3 binding. Cross-species comparisons reveal that regions orthologous to BOS also correspond with boundaries of large (400-600 kb) TADs in human and other mammalian species. The colocalization of rearrangement breakpoints and TAD boundaries may be due to higher chromatin fragility at these locations and/or increased selective pressure against rearrangements that disrupt TAD integrity. We also examine the small portion of BOS that did not overlap with TAD boundaries and gave rise to novel TADs in the gibbon genome. We postulate that these new TADs generally lack deleterious consequences. Last, we show that limited epigenetic homogenization occurs across breakpoints, irrespective of their time of occurrence in the gibbon lineage. Overall, our findings demonstrate remarkable conservation of chromatin interactions and epigenetic landscape in gibbons, in spite of extensive genomic shuffling.
DOI:10.1016/j.molcel.2016.05.018URLPMID:27259200 [本文引用: 2]
How eukaryotic chromosomes fold inside the nucleus is an age-old question that remains unanswered today. Early biochemical and microscopic studies revealed the existence of chromatin domains and loops as a pervasive feature of interphase chromosomes, but the biological implications of such organizational features were obscure. Genome-wide analysis of pair-wise chromatin interactions using chromatin conformation capture (3C)-based techniques has shed new light on the organization of chromosomes in interphase nuclei. Particularly, the finding of cell-type invariant, evolutionarily conserved topologically associating domains (TADs) in a broad spectrum of cell types has provided a new molecular framework for the study of animal development and human diseases. Here, we review recent progress in characterization of such chromatin domains and delineation of mechanisms of their formation in animal cells.
DOI:10.1038/s41467-017-02525-wURLPMID:29335486 [本文引用: 1]
Despite an abundance of new studies about topologically associating domains (TADs), the role of genetic information in TAD formation is still not fully understood. Here we use our software, HiCExplorer (hicexplorer.readthedocs.io) to annotate >2800 high-resolution (570?bp) TAD boundaries?in Drosophila?melanogaster. We identify eight DNA motifs enriched at boundaries, including a motif bound by the M1BP protein, and two new boundary motifs. In contrast to mammals, the CTCF motif is only enriched on a small fraction of boundaries flanking inactive chromatin while most active boundaries contain the motifs bound by the M1BP or Beaf-32 proteins. We demonstrate that boundaries can be accurately predicted using only the motif sequences at open chromatin sites. We propose that DNA sequence guides the genome architecture by allocation of boundary proteins in the genome. Finally, we present an interactive online database to access and explore the spatial organization of fly, mouse and human genomes, available at http://chorogenome.ie-freiburg.mpg.de .
DOI:10.1101/gr.212803.116URLPMID:28057745
Understanding how regulatory sequences interact in the context of chromosomal architecture is a central challenge in biology. Chromosome conformation capture revealed that mammalian chromosomes possess a rich hierarchy of structural layers, from multi-megabase compartments to sub-megabase topologically associating domains (TADs) and sub-TAD contact domains. TADs appear to act as regulatory microenvironments by constraining and segregating regulatory interactions across discrete chromosomal regions. However, it is unclear whether other (or all) folding layers share similar properties, or rather TADs constitute a privileged folding scale with maximal impact on the organization of regulatory interactions. Here, we present a novel algorithm named CaTCH that identifies hierarchical trees of chromosomal domains in Hi-C maps, stratified through their reciprocal physical insulation, which is a single and biologically relevant parameter. By applying CaTCH to published Hi-C data sets, we show that previously reported folding layers appear at different insulation levels. We demonstrate that although no structurally privileged folding level exists, TADs emerge as a functionally privileged scale defined by maximal boundary enrichment in CTCF and maximal cell-type conservation. By measuring transcriptional output in embryonic stem cells and neural precursor cells, we show that the likelihood that genes in a domain are coregulated during differentiation is also maximized at the scale of TADs. Finally, we observe that regulatory sequences occur at genomic locations corresponding to optimized mutual interactions at the same scale. Our analysis suggests that the architectural functionality of TADs arises from the interplay between their ability to partition interactions and the specific genomic position of regulatory sequences.
DOI:10.1016/j.cell.2015.04.004URLPMID:25959774 [本文引用: 1]
Mammalian genomes are organized into megabase-scale topologically associated domains (TADs). We demonstrate that disruption of TADs can rewire long-range regulatory architecture and result in pathogenic phenotypes. We show that distinct human limb malformations are caused by deletions, inversions, or duplications altering the structure of the TAD-spanning WNT6/IHH/EPHA4/PAX3 locus. Using CRISPR/Cas genome editing, we generated mice with corresponding rearrangements. Both in mouse limb tissue and patient-derived fibroblasts, disease-relevant structural changes cause ectopic interactions between promoters and non-coding DNA, and a cluster of limb enhancers normally associated with Epha4 is misplaced relative to TAD boundaries and drives ectopic limb expression of another gene in the locus. This rewiring occurred only if the variant disrupted a CTCF-associated boundary domain. Our results demonstrate the functional importance of TADs for orchestrating gene expression via genome architecture and indicate criteria for predicting the pathogenicity of human structural variants, particularly in non-coding regions of the human genome.
DOI:10.1016/j.celrep.2016.04.085URLPMID:27210764 [本文引用: 1]
Topologically associating domains (TADs) are fundamental structural and functional building blocks of human interphase chromosomes, yet the mechanisms of TAD formation remain unclear. Here, we propose that loop extrusion underlies TAD formation. In this process, cis-acting loop-extruding factors, likely cohesins, form progressively larger loops but stall at TAD boundaries due to interactions with boundary proteins, including CTCF. Using polymer simulations, we show that this model produces TADs and finer-scale features of Hi-C data. Each TAD emerges from multiple loops dynamically formed through extrusion, contrary to typical illustrations of single static loops. Loop extrusion both explains diverse experimental observations-including the preferential orientation of CTCF motifs, enrichments of architectural proteins at TAD boundaries, and boundary deletion experiments-and makes specific predictions for the depletion of CTCF versus cohesin. Finally, loop extrusion has potentially far-ranging consequences for processes such as enhancer-promoter interactions, orientation-specific chromosomal looping, and compaction of mitotic chromosomes.
DOI:10.1016/j.cell.2014.02.009URL [本文引用: 1]
Comparative genome analyses reveal that organismal complexity scales not with gene number but with gene regulation. Recent efforts indicate that the human genome likely contains hundreds of thousands of enhancers, with a typical gene embedded in a milieu of tens of enhancers. Proliferation of cis-regulatory DNAs is accompanied by increased complexity and functional diversification of transcriptional machineries recognizing distal enhancers and core promoters and by the highorder spatial organization of genetic elements. We review progress in unraveling one of the outstanding mysteries of modern biology: the dynamic communication of remote enhancers with target promoters in the specification of cellular identity.
DOI:10.1016/j.molcel.2013.02.011URLPMID:23473598
Mammalian genomes encode genetic information in their linear sequence, but appropriate expression of their genes requires chromosomes to fold into complex three-dimensional structures. Transcriptional control involves the establishment of physical connections among genes and regulatory elements, both along and between chromosomes. Recent technological innovations in probing the folding of chromosomes are providing new insights into the spatial organization of genomes and its role in gene regulation. It is emerging that folding of large complex chromosomes involves a hierarchy of structures, from chromatin loops that connect genes and enhancers to larger chromosomal domains and nuclear compartments. The larger these structures are along this hierarchy, the more stable they are within cells, while becoming more stochastic between cells. Here, we review the experimental and theoretical data on this hierarchy of structures and propose a key role for the recently discovered topologically associating domains.
DOI:10.1038/nrg3454URLPMID:23657480 [本文引用: 1]
How DNA is organized in three dimensions inside the cell nucleus and how this affects the ways in which cells access, read and interpret genetic information are among the longest standing questions in cell biology. Using newly developed molecular, genomic and computational approaches based on the chromosome conformation capture technology (such as 3C, 4C, 5C and Hi-C), the spatial organization of genomes is being explored at unprecedented resolution. Interpreting the increasingly large chromatin interaction data sets is now posing novel challenges. Here we describe several types of statistical and computational approaches that have recently been developed to analyse chromatin interaction data.
DOI:10.3390/genes7100071URLPMID:27669308 [本文引用: 1]
Proximity ligation assays such as circularized chromosome conformation capture and high-throughput chromosome capture assays have shed light on the structural organization of the interphase genome. Functional topologically associating domains (TADs) that constitute the building blocks of genomic organization are disrupted and reconstructed during the cell cycle. Epigenetic memory, as well as the sequence of chromosomes, regulate TAD reconstitution. Sub-TAD domains that are invariant across cell types have been identified, and contacts between these domains, rather than looping, are speculated to drive chromatin folding. Replication domains are established simultaneously with TADs during the cell cycle and the two correlate well in terms of characteristic features, such as lamin association and histone modifications. CCCTC-binding factor (CTCF) and cohesin cooperate across different cell types to regulate genes and genome organization. CTCF elements that demarcate TAD boundaries are commonly disrupted in cancer and promote oncogene activation. Chromatin looping facilitates interactions between distant promoters and enhancers, and the resulting enhanceosome complex promotes gene expression. Deciphering the chromatin tangle requires comprehensive integrative analyses of DNA- and protein-dependent factors that regulate genomic organization.
DOI:10.1016/j.cell.2014.11.021URL [本文引用: 1]
We use in situ Hi-C to probe the 3D architecture of genomes, constructing haploid and diploid maps of nine cell types. The densest, in human lymphoblastoid cells, contains 4.9 billion contacts, achieving 1 kb resolution. We find that genomes are partitioned into contact domains (median length, 185 kb), which are associated with distinct patterns of histone marks and segregate into six subcompartments. We identify similar to 10,000 loops. These loops frequently link promoters and enhancers, correlate with gene activation, and show conservation across cell types and species. Loop anchors typically occur at domain boundaries and bind CTCF. CTCF sites at loop anchors occur predominantly (>90%) in a convergent orientation, with the asymmetric motifs "facing'' one another. The inactive X chromosome splits into two massive domains and contains large loops anchored at CTCF-binding repeats.
DOI:10.1016/j.tplants.2016.07.013URLPMID:27593567 [本文引用: 1]
Higher eukaryotes typically contain many different cell types, displaying different cellular functions that are influenced by biotic and abiotic cues. The different functions are characterized by specific gene expression patterns mediated by regulatory sequences such as transcriptional enhancers. Recent genome-wide approaches have identified thousands of enhancers in animals, reviving interest in enhancers in gene regulation. Although the regulatory roles of plant enhancers are as crucial as those in animals, genome-wide approaches have only very recently been applied to plants. Here we review characteristics of enhancers at the DNA and chromatin level in plants and other species, their similarities and differences, and techniques widely used for genome-wide discovery of enhancers in animal systems that can be implemented in plants.
.
DOI:10.1093/jxb/erw168URLPMID:27129951 [本文引用: 1]
The three-dimensional organization of the eukaryotic nucleus and its chromosomal conformation have emerged as important features in the complex network of mechanisms behind gene activity and genome connectivity dynamics, which can be evidenced in the regionalized chromosomal spatial distribution and the clustering of diverse genomic regions with similar expression patterns. The development of chromatin conformation capture (3C) techniques has permitted the elucidation of commonalities between the eukaryotic phyla, as well as important differences among them. The growing number of studies in the field performed in plants has shed light on the structural and regulatory features of these organisms. For instance, it has been proposed that plant chromatin can be arranged into different conformations such as Rabl, Rosette-like, and Bouquet, and that both short- and long-range chromatin interactions occur in Arabidopsis. In this review, we compile the current knowledge about chromosome architecture characteristics in plants, as well as the molecular events and elements (including long non-coding RNAs, histone and DNA modifications, chromatin remodeling complexes, and transcription factors) shaping the genome three-dimensional conformation. Furthermore, we discuss the developmental outputs of genome topology-mediated gene expression regulation. It is becoming increasingly clear that new tools and techniques with higher resolution need to be developed and implemented in Arabidopsis and other model plants in order to better understand chromosome architecture dynamics, from an integrative perspective with other fields of plant biology such as development, stress biology, and finally agriculture.
DOI:10.1038/nsmb.2474URL
Although genomes are defined by their sequence, the linear arrangement of nucleotides is only their most basic feature. A fundamental property of genomes is their topological organization in three-dimensional space in the intact cell nucleus. The application of imaging methods and genome-wide biochemical approaches, combined with functional data, is revealing the precise nature of genome topology and its regulatory functions in gene expression and genome maintenance. The emerging picture is one of extensive self-enforcing feedback between activity and spatial organization of the genome, suggestive of a self-organizing and self-perpetuating system that uses epigenetic dynamics to regulate genome function in response to regulatory cues and to propagate cell-fate memory.
DOI:10.1101/gad.281964.116URLPMID:27340173
The relevance of three-dimensional (3D) genome organization for transcriptional regulation and thereby for cellular fate at large is now widely accepted. Our understanding of the fascinating architecture underlying this function is based on microscopy studies as well as the chromosome conformation capture (3C) methods, which entered the stage at the beginning of the millennium. The first decade of 3C methods rendered unprecedented insights into genome topology. Here, we provide an update of developments and discoveries made over the more recent years. As we discuss, established and newly developed experimental and computational methods enabled identification of novel, functionally important chromosome structures. Regulatory and architectural chromatin loops throughout the genome are being cataloged and compared between cell types, revealing tissue invariant and developmentally dynamic loops. Architectural proteins shaping the genome were disclosed, and their mode of action is being uncovered. We explain how more detailed insights into the 3D genome increase our understanding of transcriptional regulation in development and misregulation in disease. Finally, to help researchers in choosing the approach best tailored for their specific research question, we explain the differences and commonalities between the various 3C-derived methods.
DOI:10.1073/pnas.0906067107URLPMID:20133699 [本文引用: 1]
Recent genomewide association studies have found multiple genetic variants on chromosome 8q24 that are significantly associated with an increased susceptibility to prostate, colorectal, and breast cancer. These risk loci are located in a "gene desert," a few hundred kilobases telomeric to the Myc gene. To date, the biological mechanism(s) underlying these associations remain unclear. It has been speculated that these 8q24 genetic variant(s) might affect Myc expression by altering its regulation or amplification status. Here, we show that multiple enhancer elements are present within this region and that they can regulate transcription of Myc. We also demonstrate that one such enhancer element physically interacts with the Myc promoter via transcription factor Tcf-4 binding and acts in an allele specific manner to regulate Myc expression.
DOI:10.1002/wdev.265URLPMID:28251841 [本文引用: 1]
The evolution of gene regulation is considered one of the main drivers causing the astonishing morphological diversity in the animal kingdom. Gene regulation in animals heavily depends upon cis-regulatory elements, discrete pieces of DNA that interact with target promoters to regulate gene expression. In the last years, Chromosome Conformation Capture experiments (4C-seq, 5C, and HiC) in several organisms have shown that the genomes of many bilaterian animals are organized in the 3D chromatin space in compartments called topologically associated domains (TADs). The appearance of the architectural protein CTCF in the bilaterian ancestor likely facilitated the origin of this chromatin 3D organization. TADs play a critical role favoring the contact of cis-regulatory elements with their proper target promoters (that often lay within the same TAD) and preventing undesired regulatory interactions with promoters located in neighboring TADs. We propose that TAD may have had a major influence in the history of the evolution of gene regulation. They have contributed to the increment of regulatory complexity in bilaterians by allowing newly evolved cis-regulatory elements to find target promoters in a range of hundreds of kilobases. In addition, they have conditioned the mechanisms of evolution of gene regulation. These mechanisms include the appearance, removal, or relocation of TAD borders. Such architectural changes have been able to wire or unwire promoters with different sets of cis-regulatory elements in a single mutational event. We discuss the contribution of these architectural changes to the generation of critical genomic 3D structures required for new regulatory mechanisms associated to morphological novelties. WIREs Dev Biol 2017, 6:e265. doi: 10.1002/wdev.265 For further resources related to this article, please visit the WIREs website.
DOI:10.1038/emboj.2012.324URL [本文引用: 1]
Gene activation in eukaryotes frequently involves interactions between chromosomal regions. We have investigated whether higher-order chromatin structures are involved in the regulation of the Arabidopsis floral repressor gene FLC, a target of several chromatin regulatory pathways. Here, we identify a gene loop involving the physical interaction of the 5' and 3' flanking regions of the FLC locus using chromosome conformation capture. The FLC loop is unaffected by mutations disrupting conserved chromatin regulatory pathways leading to very different expression states. However, the loop is disrupted during vernalization, the cold-induced, Polycomb-dependent epigenetic silencing of FLC. Loop disruption parallels timing of the cold-induced FLC transcriptional shut-down and upregulation of FLC antisense transcripts, but does not need a cold-induced PHD protein required for the epigenetic silencing. We suggest that gene loop disruption is an early step in the switch from an expressed to a Polycomb-silenced state. The EMBO Journal (2013) 32, 140-148. doi:10.1038/emboj.2012.324; Published online 7 December 2012
DOI:10.1016/j.molcel.2014.06.011URLPMID:25018019 [本文引用: 1]
The eukaryotic epigenome is shaped by the genome topology in three-dimensional space. Dynamic reversible variations in this epigenome structure directly influence the transcriptional responses to developmental cues. Here, we show that the Arabidopsis long intergenic noncoding RNA (lincRNA) APOLO is transcribed by RNA polymerases II and V in response to auxin, a phytohormone controlling numerous facets of plant development. This dual APOLO transcription regulates the formation of a chromatin loop encompassing the promoter of its neighboring gene PID, a key regulator of polar auxin transport. Altering APOLO expression affects chromatin loop formation, whereas RNA-dependent DNA methylation, active DNA demethylation, and Polycomb complexes control loop dynamics. This dynamic chromatin topology determines PID expression patterns. Hence, the dual transcription of a lincRNA influences local chromatin topology and directs dynamic auxin-controlled developmental outputs on neighboring genes. This mechanism likely underscores the adaptive success of plants in diverse environments and may be widespread in eukaryotes.
DOI:10.1016/j.cell.2015.05.048URLPMID:26119342 [本文引用: 1]
We describe a Hi-C-based method, Micro-C, in which micrococcal nuclease is used instead of restriction enzymes to fragment chromatin, enabling nucleosome resolution chromosome folding maps. Analysis of Micro-C maps for budding yeast reveals abundant self-associating domains similar to those reported in other species, but not previously observed in yeast. These structures, far shorter than topologically associating domains in mammals, typically encompass one to five genes in yeast. Strong boundaries between self-associating domains occur at promoters of highly transcribed genes and regions of rapid histone turnover that are typically bound by the RSC chromatin-remodeling complex. Investigation of chromosome folding in mutants confirms roles for RSC, "gene looping" factor Ssu72, Mediator, H3K56 acetyltransferase Rtt109, and the N-terminal tail of H4 in folding of the yeast genome. This approach provides detailed structural maps of a eukaryotic genome, and our findings provide insights into the machinery underlying chromosome compaction.
DOI:10.1101/gr.215186.116URLPMID:28385710 [本文引用: 1]
The nuclear space is not a homogeneous biochemical environment. Many studies have demonstrated that the transcriptional activity of a gene is linked to its positioning within the nuclear space. Following the discovery of lamin-associated domains (LADs), which are transcriptionally repressed chromatin regions, the nonrandom positioning of chromatin at the nuclear periphery and its biological relevance have been studied extensively in animals. However, it remains unknown whether comparable chromatin organizations exist in plants. Here, using a strategy using restriction enzyme-mediated chromatin immunoprecipitation, we present genome-wide identification of nonrandom domain organization of chromatin at the peripheral zone of Arabidopsis thaliana nuclei. We show that in various tissues, 10%-20% of the regions on the chromosome arms are anchored at the nuclear periphery, and these regions largely overlap between different tissues. Unlike LADs in animals, the identified domains in plants are not gene-poor or A/T-rich. These domains are enriched with silenced protein-coding genes, transposable element genes, and heterochromatic marks, which collectively define a repressed environment. In addition, these domains strongly correlate with our genome-wide chromatin interaction data set (Hi-C) by largely explaining the patterns of chromatin compartments, revealed on Hi-C maps. Moreover, our results reveal a spatial compartment of different DNA methylation pathways that regulate silencing of transposable elements, where the CHH methylation of transposable elements located at the nuclear periphery and in the interior are preferentially mediated by CMT2 and DRM methyltransferases, respectively. Taken together, the results demonstrate functional partitioning of the Arabidopsis genome in the nuclear space.
DOI:10.1038/nature14450URLPMID:26030525 [本文引用: 1]
The three-dimensional organization of a genome plays a critical role in regulating gene expression, yet little is known about the machinery and mechanisms that determine higher-order chromosome structure. Here we perform genome-wide chromosome conformation capture analysis, fluorescent in situ hybridization (FISH), and RNA-seq to obtain comprehensive three-dimensional (3D) maps of the Caenorhabditis elegans genome and to dissect X chromosome dosage compensation, which balances gene expression between XX hermaphrodites and XO males. The dosage compensation complex (DCC), a condensin complex, binds to both hermaphrodite X chromosomes via sequence-specific recruitment elements on X (rex sites) to reduce chromosome-wide gene expression by half. Most DCC condensin subunits also act in other condensin complexes to control the compaction and resolution of all mitotic and meiotic chromosomes. By comparing chromosome structure in wild-type and DCC-defective embryos, we show that the DCC remodels hermaphrodite X chromosomes into a sex-specific spatial conformation distinct from autosomes. Dosage-compensated X chromosomes consist of self-interacting domains (~1 Mb) resembling mammalian topologically associating domains (TADs). TADs on X chromosomes have stronger boundaries and more regular spacing than on autosomes. Many TAD boundaries on X chromosomes coincide with the highest-affinity rex sites and become diminished or lost in DCC-defective mutants, thereby converting the topology of X to a conformation resembling autosomes. rex sites engage in DCC-dependent long-range interactions, with the most frequent interactions occurring between rex sites at DCC-dependent TAD boundaries. These results imply that the DCC reshapes the topology of X chromosomes by forming new TAD boundaries and reinforcing weak boundaries through interactions between its highest-affinity binding sites. As this model predicts, deletion of an endogenous rex site at a DCC-dependent TAD boundary using CRISPR/Cas9 greatly diminished the boundary. Thus, the DCC imposes a distinct higher-order structure onto X chromosomes while regulating gene expression chromosome-wide.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}