 ,1, 黄涛,2, 卢国昌3, 岳涛4, 卢振西3, 黄锡霞5, 卫新璞6, 冯书堂7, 陈军8, 乌兰·卡格德尔8, 茹先古丽·阿不力孜8, 努尔胡马尔·木合塔尔8
,1, 黄涛,2, 卢国昌3, 岳涛4, 卢振西3, 黄锡霞5, 卫新璞6, 冯书堂7, 陈军8, 乌兰·卡格德尔8, 茹先古丽·阿不力孜8, 努尔胡马尔·木合塔尔8The evaluation of genomic homozygosity for Xinjiang inbred population by SNP panels
Rui Shi1, Yi Zhang1, Yachun Wang,1, Tao Huang,2, Guochang Lu3, Tao Yue4, Zhenxi Lu3, Xixia Huang5, Xinpu Wei6, Shutang Feng7, Jun Chen8, Wulan Kagedeer8, Ruxianguli Abulizi8, Nuerhumaer Muhetaer8通讯作者: 王雅春,博士,教授,研究方向:分子数量遗传学。E-mail:wangyachun@cau.edu.cn黄涛,博士,副教授,研究方向:动物遗传育种。E-mail:taohuagn100@sina.com. 卢国昌,新疆近交牛培育人。
编委: 赵要风
收稿日期:2020-03-15修回日期:2020-04-9网络出版日期:2020-05-20
| 基金资助: |
Editorial board:
Received:2020-03-15Revised:2020-04-9Online:2020-05-20
| Fund supported: |
作者简介 About authors
师睿,在读硕士研究生,专业方向:动物遗传育种与繁殖。E-mail:srandeffy@163.com。

摘要
新疆近交牛是经45年近亲繁育形成的近交群体,但由于繁育记录缺失,其原始亲本品种未知。为了明确新疆近交牛的遗传背景,并探索利用基因组信息评价牛群近交水平的可行性,本研究利用该群体及荷斯坦牛、新疆褐牛和哈萨克牛等16个国内外牛品种的SNP芯片数据,应用主成分分析和Admixture方法对塔城地区新疆近交牛的群体结构进行分析;通过进一步计算新疆近交牛、荷斯坦牛、新疆褐牛和哈萨克牛的群体遗传学参数以及基因组近交指标评估各群体近交程度;结合新疆近交牛的体型分类和基因组近交指标信息,探讨了个体近交程度与体型表现的关系;最后,基于对新疆近交牛和哈萨克牛高频长纯合片段区域的筛选,鉴定了新疆近交牛基因组特征区域。研究结果显示,新疆近交牛的遗传背景与哈萨克牛基本一致,近交牛基因组纯合程度明显高于其他群体,且基因纯合率越高的近交牛其体型越小,在一定程度上呈现了近交衰退对体型的影响。本研究还鉴定到与新疆近交牛基因组特征区域相关的6个基本生物学通路以及与重要经济性状相关的32个数量基因座(quantitative trait loci, QTL)。本研究结果为新疆近交牛这一特殊遗传资源的育种规划及未来该群体的开发利用提供了科学依据。
关键词:
Abstract
Xinjiang inbred cattle is a population which has been highly inbred for 45 years. However, the breed origin of this population cannot be traced back due to the lack of original records. To demonstrate the genetic background of Xinjiang inbred cattle, we analysed the worldwide genomic information of 16 cattle breeds using principal components analysis, and Admixture method. Furthermore, the shared SNP markers of Xinjiang inbred cattle, local Kazakh cattle, Holstein cattle, and Xinjiang Brown cattle were extracted to calculate population genetic parameters and genomic inbreeding indicators in order to evaluate the magnitude of inbreeding in each population. We also evaluated the relationship between inbreeding indicators and body size in the Xinjiang inbred population. Finally, the high frequency runs of homozygosity (ROH) regions for Xinjiang inbred cattle and local Kazakh population were selected for genes and QTL annotations. These results demonstrate that the ancestry proportions of inbreeding breed are similar to those of Kazakh cattle. The genomic homozygosity of Xinjiang inbred cattle is significantly higher than other populations; the inbreeding depression is observed in body size to a certain extent because body size decreased when corresponding homozygosity increased. Totally, six basic bio-pathways and 32 QTL regions that related to bovine economical traits were annotated. Our results provide the insights into breeding strategies, future protection, and utilization plan design for this special genetic material-Xinjiang inbred cattle.
Keywords:
PDF (1274KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
师睿, 张毅, 王雅春, 黄涛, 卢国昌, 岳涛, 卢振西, 黄锡霞, 卫新璞, 冯书堂, 陈军, 乌兰·卡格德尔, 茹先古丽·阿不力孜, 努尔胡马尔·木合塔尔. 利用SNP芯片信息评估新疆近交牛基因组纯合度. 遗传[J], 2020, 42(5): 493-505 doi:10.16288/j.yczz.20-071
Rui Shi.
近交群体的形成是由于群体内具有血缘关系的个体进行了交配,致使其后代基因纯合性增加。但随近交程度的增加,隐性致病基因的纯合概率也会上升,因此近交往往伴随着后代各种性状表型衰退,即近交衰退[1]。当群体连续20代以上进行亲子交配或全同胞交配后,近交系数可达99.8%以上,群体内基因达到高度纯合并且稳定时,该群体可称为近交系。我国已成功育成小鼠(Mus musculus)、鸡(Gallus gallus)和猪(Sus scrofa)等近交系动物,但由于牛(Bos taurus)的世代间隔长、繁殖效率低、育种成本高等原因,近交系牛罕有报道。
1974年,新疆塔城地区某家庭牧场以1头母牛和其子开始了近交牛培育,随后一直采用母子、父女及兄妹交配等高度近交的繁育模式,通过45年的封闭近亲繁育,形成了现有100头规模的近交群体。该牛群个体间在体型、生产性能等方面有很高的相似性,虽然与塔城地区哈萨克牛相比,生长较慢且体型较小,但是在恶劣的生存环境下能保持较正常的繁殖性能和生活力。马力鹏等[2]使用微卫星技术对该群体展开了鉴定研究,发现该群体具有较高纯合度,为遗传纯度较高的近交群体;经过当地科技局鉴定,2018年该群体被命名为新疆近交牛。但由于最初的繁育记录缺失,无法追溯和确认该群体原始亲本的品种。
近年来,高密度单核苷酸多态性(single nucleotide polymorphism, SNP)芯片被广泛应用于群体遗传学研究。基因组SNP能准确解析群体的遗传构成,以确定未知群体的品种来源[3]。如Gao等[4]利用SNP信息解析了中国不同品种牛只的血统组成。利用标记间的连锁不平衡(linkage disequilibrium, LD)程度,高密度SNP可以估计有效群体大小(effective population size, Ne)等群体遗传学参数[5];利用标记纯合度能够准确地反映个体真实基因组纯合度和分子近交水平,且较传统的系谱估计更加准确[6,7]。另外,基因组的长纯合片段(runs of homozygosity, ROH)也在多个物种的研究中[8,9,10]被用于定位功能基因和数量性状基因座(quantitative trait loci, QTL)。Kim等[8]对北美荷斯坦牛的研究发现,不同群体的ROH分布存在一定差异,能够反映群体的基因组特征。
本研究以国内外有代表性的牛品种为参考,对新疆近交牛进行遗传背景分析,并进一步使用多种群体遗传学指标,比较新疆近交牛、哈萨克牛(塔城)、新疆褐牛和荷斯坦牛之间的近交程度;通过筛选群体高频ROH位点,以寻找新疆近交牛基因组特征区域,旨在从多个角度对新疆近交牛进行遗传学分析,并为其繁育及未来群体的保护和利用提供科学依据。
1 材料与方法
1.1 试验动物
近交牛样本来自新疆塔城市恰夏镇,散养且与其他牛群无遗传交流。本研究选择牛群中健康且大于12月龄的牛69头,测量体高、体斜长、胸围和管围等表型指标,采集静脉血。为了便于群体间比较,本研究还采集了20头哈萨克牛(塔城)及50头新疆褐牛的样本,来源分别为新疆塔城市恰夏镇随机农户和新疆塔城种牛场。
1.2 基因组SNP检测及数据质控
采用GeneSeek Genomic Profiler (GGP) Bovine 50K芯片检测该新疆近交牛及哈萨克牛(塔城)样本。采用GeneSeek GGP Bovine 150K芯片测定新疆褐牛样本,50头随机选取的荷斯坦牛150K芯片数据来自中国奶牛基因组评估平台数据库(数据未发表)。此外,本研究通过整合已发表的数据,形成不同的群体组合共4个数据集用于新疆近交牛的基因组分析,具体信息见表1。
Table 1
表1
表1本研究使用的各样本数据
Table 1
| 群体/品种 | 缩写 | 类别 | 样本来源 | 数据集1 (310头) | 数据集2 (170头) | 数据集3 (69头) | 数据集4 (70头) | 数据来源 |
|---|---|---|---|---|---|---|---|---|
| 新疆近交牛 | XI | 中国北方牛 | 新疆塔城 | 20 | 50 | 69(40)* | 50 | 本研究 |
| 哈萨克牛(塔城) | TL | 中国北方牛 | 新疆塔城 | 20 | 20 | - | 20 | 本研究 |
| 新疆褐牛 | XB | 中国北方牛 | 新疆塔城 | 20 | 50 | - | - | 本研究 |
| 荷斯坦牛 | HO | 普通牛 | 北京大兴 | 20 | 50 | - | - | 本研究 |
| 瑞士褐牛 | BS | 普通牛 | 瑞士 | 20 | [4] | |||
| 西门塔尔牛 | SI | 普通牛 | 德国 | 20 | - | - | - | [4] |
| 加尔梅克牛 | JA | 普通牛 | 俄罗斯 | 20 | - | - | - | [11] |
| 延边牛 | YB | 中国北方牛 | 吉林延边 | 20 | - | - | - | [4] |
| 蒙古牛 | MG | 中国北方牛 | 内蒙古锡林郭勒 | 20 | - | - | - | [4] |
| 哈萨克牛(伊犁) | KA | 中国北方牛 | 新疆伊犁 | 20 | - | - | - | [4] |
| 秦川牛 | QC | 中国中原牛 | 陕西宝鸡 | 20 | - | - | - | [4] |
| 晋南牛 | JN | 中国中原牛 | 山西运城 | 14 | - | - | - | [4] |
| 南阳牛 | NY | 中国中原牛 | 河南南阳 | 20 | - | - | - | [4] |
| 鲁西牛 | LX | 中国中原牛 | 山东梁山 | 16 | - | - | - | [4] |
| 温岭牛 | WL | 瘤牛 | 浙江温州 | 20 | - | - | - | [4] |
| 吉尔牛 | GI | 瘤牛 | 印度 | 20 | - | - | - | [12] |
新窗口打开|下载CSV
数据集1用于分析新疆近交牛的遗传背景,包括本研究所检测的3个群体、荷斯坦牛以及已发表的国内外代表性牛品种12个,共310头牛。为了使各品种样本数均衡,避免样本量差异对结果的影响,本研究从新疆近交牛、荷斯坦牛和新疆褐牛群体中各随机挑选20个样本进行Admixture分析。使用PLINK v1.90[13]进行质量控制,标准如下:最小等位基因频率≥0.05,个体基因型缺失率<10%,标记基因型缺失率<10%,哈迪-温伯格检验显著性P> 10E-05,然后保留16个牛群共享的位点。质量控制后保留常染色体上共9476个SNP。
数据集2用于新疆近交牛的基因组近交程度分析,包括新疆近交牛、哈萨克牛(塔城)、新疆褐牛和荷斯坦牛共170头。数据质量控制标准同上。另外,为保证各群体的SNP密度相同,本研究将荷斯坦牛和新疆褐牛的SNP位点进行了筛选,仅保留了与新疆近交牛和哈萨克牛(塔城)共享的位点,最终保留了常染色体上共33,590个SNP。
数据集3用于近交衰退分析,仅包含新疆近交牛,质量控制过程和保留的SNP数量与数据集2相同。分析中将该数据集划分为两个子集分别分析。
数据集4用于基因组特征区域分析,是在数据集2的基础上保留了新疆近交牛和哈萨克牛(塔城),经过相同的质量控制过程后,保留了常染色体上40,723个位点。
1.3 统计分析
1.3.1 新疆近交牛群体遗传学分析本研究使用主成分分析(principal component analysis, PCA)以及Admixture1.3软件[3]分析遗传背景结构。Admixture采用非监督型算法,设定祖先血统数量K=2~5。使用PLINK计算4个群体LD指标r2[14]。依据各标记间的距离(0~10 Mb),计算不同标记距离下r2的均值,以展示不同群体LD的衰减程度。使用SNeP软件[15]中的Sved方法[16],基于LD的信息对4个群体的Ne进行估计。
1.3.2 同态相同位点分析
使用PLINK软件计算两两个体之间的同态相同位点(identical by state, IBS)比例,最终生成N×N大小的IBS矩阵。个体间IBS比例反映了个体之间遗传关系远近以及群体的近交历史。
1.3.3 基因组近交分析
ROH指连续纯合的染色体片段,它是由于亲代的同源单倍型传递给后代所致,其长度和频率能够反映群体的交配历史。另外,较长的ROH片段可以指示较近的亲缘关系。因为较长ROH片段的存在说明其没有被多个世代染色体的随机重组分割成不同小段。相反,较短的ROH则可以反映较远的亲缘关系[17]。基于ROH的近交系数也被证明与系谱近交系数之间存在高度正相关(0.75)[18],因此可以用于衡量基因组的近交水平。近交可能导致ROH片段的最小长度增加[19]。
本研究利用PLINK软件扫描基因组中的ROH,扫描参数为:扫描窗口为30个SNP,定义的ROH长度大于0.5 Mb,连续SNP之间的距离小于1 Mb,扫描的ROH中最多允许出现一个杂合子。根据50K芯片数据的研究[9],将所得ROH片段依据其长度进行分组:0.5~5 Mb,5~10 Mb和10 Mb以上,用于比较不同群体的ROH分布差异。
采用2种方法计算个体基因组近交系数:(1) ROH覆盖率(ROH%),即个体ROH总长占染色体总长的比例;(2)纯合位点占比(HOM%),即个体的纯合SNP位点数占总位点数的比例。
1.3.4 近交衰退分析
为了评估新疆近交牛的近交衰退效应,本研究将新疆近交牛按体型分为大、中、小3组,统计分析各组的基因组近交系数差异。分组方法采用K-means聚类算法[20],基于4个体型表型指标对个体进行聚类划分。考虑到年龄对体型的影响,本研究将牛只按月龄分为:(1)≥12月龄的牛只共69头;(2)进一步筛选≥24月龄的牛只共40头。对两个数据集分别进行近交衰退分析。
1.3.5 新疆近交牛的基因组特征区域分析
本研究分析了新疆近交牛和哈萨克牛(塔城)的ROH在各染色体上的分布特征,以定位近交群体内ROH出现频率(ROHf)较高的位点,ROHf是统计群体水平下ROH覆盖特定位点的频率(N覆盖/N)。最终筛选了两群体全基因组水平ROHf前1%的SNPs,用于确定新疆近交牛的基因组特征区域。
基于UCSC数据库(http://hgdownload.soe.ucsc.edu/goldenPath/bosTau9/bigZips/genes/),在候选SNPs上下游100 kb内注释相关基因,使用DAVID (https://david.ncifcrf.gov/)进行GO(gene ontology)富集分析,并使用牛QTL数据库(https://www.animalgenome.org/cgi-bin/QTLdb/)定位QTLs。
2 结果与分析
2.1 新疆近交牛群体遗传学研究
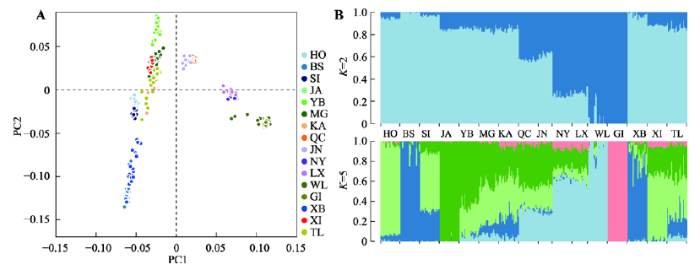
2.1.1 遗传背景分析本研究使用PLINK对16个牛群的基因组信息进行PCA分析,结果显示,第一主成分(PC1)和第二主成分(PC2)分别占总变异40.49%和12.22%,新疆近交牛(XI)与哈萨克牛(塔城,TL)、哈萨克牛(伊犁,KA)和蒙古牛(MG)聚在一起(图1A)。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1新疆近交牛与其他品种的遗传关系
A:主成分分析;B:Admixture分析结果(K=2或5)。图中每一列表示一个个体,不同颜色片段的长度表示该个体基因组中某个祖先所占的比例,K表示本研究假定的祖先群体个数。HO:荷斯坦牛;BS:瑞士褐牛;SI:西门塔尔牛;JA:加尔梅克牛;YB:延边牛;MG:蒙古牛;KA:哈萨克牛(伊犁);QC:秦川牛;JN:晋南牛;NY:南阳牛;LX:鲁西牛;WL:温岭牛;GI:吉尔牛;XB:新疆褐牛;XI:新疆近交牛;TL:哈萨克牛(塔城)。
Fig. 1Genetic relationships between Xinjiang inbreeding population and other breeds
本研究利用Admixture分析了16个牛群的系祖结构。分析显示(图1B),当K=2时,普通牛群体和瘤牛群体(温岭牛和吉尔牛)明显分开,新疆近交牛、哈萨克牛(塔城)和中国北方牛品种(蒙古牛、延边牛和哈萨克牛(伊犁))血统组成相似,普通牛血统占84%,瘤牛血统占16%;中原牛(秦川牛、晋南牛、南阳牛和鲁西牛)含有较高比例瘤牛血统(40%~77%);当K=5时,新疆近交牛与哈萨克牛(塔城)的血统构成已有些许不同;哈萨克牛(塔城)深蓝色的系祖占比达到20%,与中国北方牛群的遗传组成类似,尤其与哈萨克牛(伊犁)最接近。
2.1.2 连锁不平衡分析
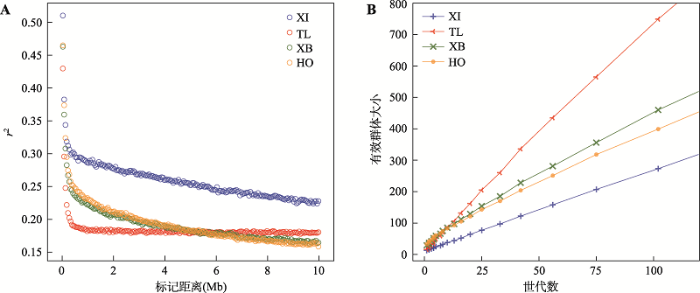
本研究利用PLINK检测了新疆近交牛、哈萨克牛(塔城)、新疆褐牛和荷斯坦牛4个群体的LD特征,结果表明,4个群体均呈现LD逐渐衰减模式(图2A),物理距离较近的SNP位点之间的r2较高,当标记间距离增加时,r2则迅速下降。但新疆近交牛的LD程度明显高于其他3个牛群,当标记之间物理距离为8 Mb时,其他牛群标记之间的关联系数<0.20,而近交牛则为0.25。相反,哈萨克牛(塔城)的LD衰减速度最快,在1 Mb之前标记之间r2<0.2,反映出该群体近交程度最低。
2.1.3 有效群体大小
基于群体LD信息,本研究使用SNeP软件估计了新疆近交牛、哈萨克牛(塔城)、新疆褐牛和荷斯坦牛的历史Ne。结果显示4个群体的历史Ne都随着世代数的增加而增加(图2B)。哈萨克牛(塔城)的Ne在10~102世代前都明显大于其他3个群体,从102世代前的749头减小至10世代前的86头。新疆褐牛和荷斯坦牛两个群体的Ne低于哈萨克牛(塔城),新疆近交牛的Ne则在各世代中都是最小的,体现出该群体内SNP标记间存在较强LD。在10个世代以内,新疆近交牛和哈萨克牛(塔城)的Ne皆小于新疆褐牛和荷斯坦牛,当前世代下4个群体的Ne分别为13、16、29和33头。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2各群体的连锁不平衡(LD)衰减以及历史有效群体大小(Ne)
A:4个牛群的连锁不平衡(LD)衰减;B:常染色体估计4个牛群不同世代的有效群体大小(Ne)。XI:新疆近交牛;TL:哈萨克牛(塔城);XB:新疆褐牛;HO:荷斯坦牛。
Fig. 2Decay map of linkage disequilibrium and estimated effective population size (Ne) for four populations
2.2 同态相同位点与个体间亲缘关系
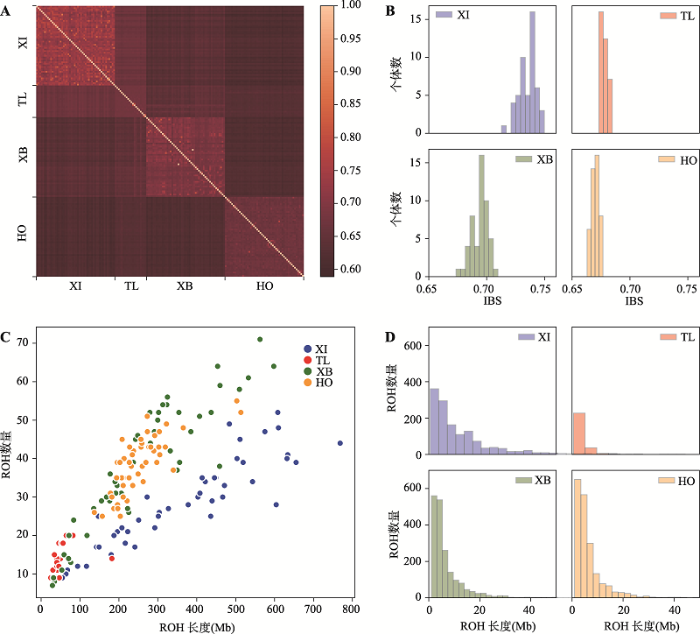
IBS可以直观反映个体间的基因组相似性和亲缘关系,本研究通过PLINK计算了新疆近交牛、哈萨克牛(塔城)、新疆褐牛和荷斯坦牛4个群体个体间的IBS矩阵。从热图可以看出,对角线上呈现3个亮度高且内部相似的模块,反映群体内较群体间具有更高的基因组相似性(图3A)。新疆近交牛个体之间基因组相似性最高(IBS=0.735±0.008),反映出该群体通过几个世代的近交,个体间亲缘关系非常近。新疆褐牛(IBS=0.694±0.007)内也具有较高的相似性,而荷斯坦牛(IBS=0.670±0.003)和哈萨克牛(塔城) (IBS=0.678±0.003)个体间相似性较低(图3B)。2.3 基因组近交程度
利用PLINK在新疆近交牛、哈萨克牛(塔城)、新疆褐牛和荷斯坦牛的常染色体上进行了ROH扫描。经统计后,4个群体各自的ROH数量和ROH长度之间呈正比关系(图3C),新疆近交牛的斜率明显小于新疆褐牛和荷斯坦牛,尽管ROH数量较少,但每个个体ROH的总长度较大。哈萨克牛(塔城) ROH长度的分布集中于0.5~20 Mb,且ROH数量多于新疆褐牛和荷斯坦牛,而新疆近交牛ROH分布较为分散,且长ROH的数量明显要比其他牛群多(图3D)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3各群体中同态相同(IBS)比例以及长纯合片段(ROH)的分布情况
A:不同个体两两之间的同态相同(IBS)关系(浅色表示同态相同程度高,深色则表示程度低);B:不同牛群内个体与剩余群体的IBS平均值分布情况;C:各群体中长纯合片段(ROH)数量与总长度之间的关系,Pop:品种名称缩写;D:各群体中ROH数量和长度的分布情况。XI:新疆近交牛;TL:哈萨克牛(塔城);XB:新疆褐牛;HO:荷斯坦牛。
Fig. 3Distributions of identity by state (IBS) and runs of homozygosity (ROH) for four populations
对ROH进行分组后,本研究统计了4个群体不同长度的ROH在常染色体上的分布。结果可见,新疆近交牛(图4A) ROH数量虽然较少,但其长ROH片段在各染色体上的分布要比其他群体多,且ROH%也更高。对于新疆近交牛而言,1号染色体ROH数量最多,13号染色体数量最少;27号染色体的ROH%最高,7号染色体最低。但是,新疆近交牛28号染色体虽然ROH数量最少,但其覆盖率则达到了第2,仅次于27号染色体。哈萨克牛(塔城)的ROH数量及ROH%都处于较低水平,且29号染色体上未检测到ROH(图4B)。新疆褐牛的6号染色体ROH数量最多(图4C),而荷斯坦牛ROH数量随染色体编号增加呈现递减趋势(图4D)。除了哈萨克牛(塔城)外,其余3个群体ROH%都随染色体编号增加呈上升趋势。
图4
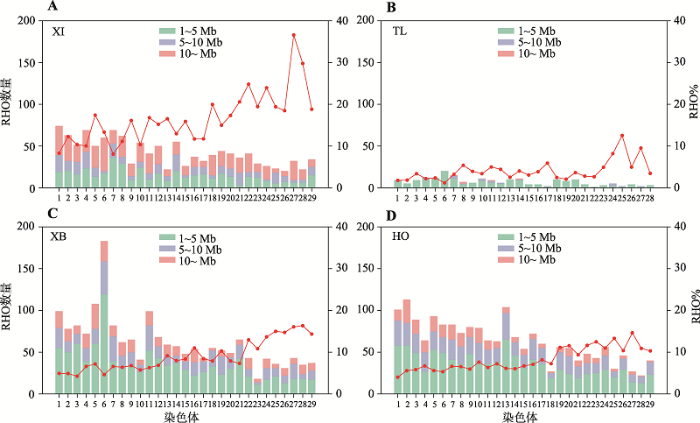 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4各群体长纯合片段(ROH)在常染色体上的分布
A:新疆近交牛不同长度的长纯合片段(ROH)在常染色体的分布情况;B:哈萨克牛(塔城)不同长度的长纯合片段(ROH)在常染色体的分布情况;C:新疆褐牛不同长度的长纯合片段(ROH)在常染色体的分布情况;D:荷斯坦牛不同长度的长纯合片段(ROH)在常染色体的分布情况。红线表示各染色体的ROH%。XI:新疆近交牛;TL:哈萨克牛(塔城);XB:新疆褐牛;HO:荷斯坦牛。
Fig. 4Chromosomal distributions for runs of homozygosity (ROH) in four populations
基于ROH与SNP纯合位点数量,本研究计算了4个群体HOM%和ROH%,从结果可以看出,新疆近交牛的两个指标都显著高于其余群体(表2)。各群体之间的HOM%皆有差异,新疆近交牛与哈萨克牛(塔城)之间差异达到0.104,荷斯坦牛的HOM%最小,为0.541。在ROH%指标中,新疆褐牛与荷斯坦牛之间不存在差异,且哈萨克牛(塔城)的ROH%最小,为0.021,与新疆近交牛相差0.115。
Table 2
表2
表2各群体基因组近交指标
Table 2
| 牛群 | HOM% | ROH% | ||
|---|---|---|---|---|
| 平均值 | 标准差 | 平均值 | 标准差 | |
| 新疆近交牛(XI) | 0.681a | 0.040 | 0.136a | 0.079 |
| 哈萨克牛(塔城,TL) | 0.577b | 0.006 | 0.021b | 0.013 |
| 新疆褐牛(XB) | 0.596c | 0.025 | 0.102c | 0.060 |
| 荷斯坦牛(HO) | 0.541d | 0.011 | 0.102c | 0.028 |
新窗口打开|下载CSV
2.4 近交对新疆近交牛体型性状的影响
本研究利用新疆近交牛的4个体型参数进行K-means聚类后,统计了不同月龄分组下体型与近交指标的关系(表3)。结果显示,在≥12月龄分组情况下,HOM%和ROH%随体型的减小皆呈现上升趋势;≥24月龄分组的结果仍是相同趋势(由于筛选≥24月龄个体,无小体型牛分组)。Table 3
表3
表3新疆近交牛不同体型分组各参数平均值及标准差
Table 3
| 体型分类 | 个体数 | 平均值±标准差 | |||||
|---|---|---|---|---|---|---|---|
| 体高(cm) | 体斜长(cm) | 胸围(cm) | 管围(cm) | HOM% | ROH% | ||
| ≥12月龄 | |||||||
| 小 | 10 | 86.30±9.99 | 96.60±10.72 | 111.70±11.97 | 12.40±1.41 | 0.612±0.053 | 0.188±0.143 |
| 中 | 28 | 102.56±4.15 | 122.41±8.21 | 141.24±7.09 | 14.59±1.08 | 0.611±0.032 | 0.186±0.088 |
| 大 | 31 | 111.96±4.66 | 140.56±8.12 | 155.84±9.30 | 16.00±0.98 | 0.593±0.032 | 0.130±0.084 |
| ≥24月龄 | |||||||
| 中 | 17 | 102.12±3.23 | 120.69±6.31 | 140.59±7.63 | 14.47±1.15 | 0.607±0.026 | 0.167±0.067 |
| 大 | 23 | 111.25±4.94 | 139.90±8.36 | 154.64±9.79 | 15.93±1.02 | 0.594±0.032 | 0.132±0.086 |
新窗口打开|下载CSV
2.5 新疆近交牛基因组特征区域
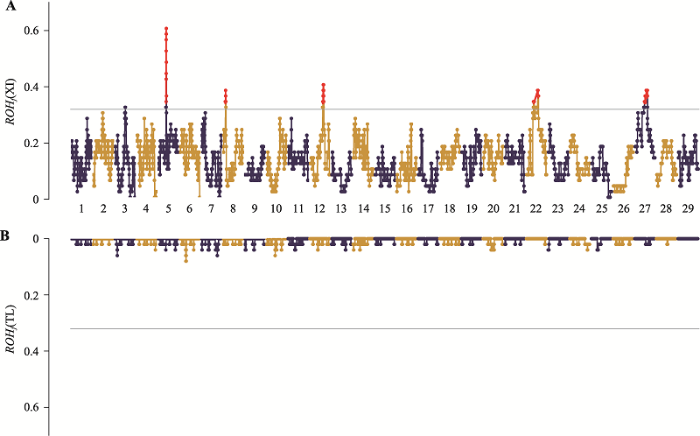
新疆近交牛的ROHf均值为0.131,最大值达到0.600,这意味着30个个体的ROH覆盖到位于5号染色体的高频位点(图5A)。而哈萨克牛(塔城)的均值仅有0.005,最大为0.080,在整个基因组范围内ROHf皆小于阈值(图5B)。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5两个群体ROHf在基因组上的分布图
A:新疆近交牛ROHf在各染色体上的分布;B:哈萨克牛(塔城) ROHf在各染色体上的分布。灰色横线表示ROHf >0.32作为位点选择阈值,即前1%的SNP位点。XI:新疆近交牛;TL:哈萨克牛(塔城)。
Fig. 5The genomic distributions of ROHf in two populations
本研究在新疆近交牛中检测到8个高频ROH区域,包含332个SNP位点并注释到了130个相关基因。设定GO富集分析阈值为0.05,找到的4个生物学过程通路主要与神经和免疫系统功能相关,两个分子生物功能通路与膜结构相关(表4)。
Table 4
表4
表4GO富集分析结果
Table 4
| GO条目 | 通路名称 | 基因数 | P值 |
|---|---|---|---|
| GO生物学过程 | |||
| GO:0051965 | 突触组装正调控 | 4 | 7.65E-04 |
| GO:0007409 | 轴突生成 | 4 | 2.05E-03 |
| GO:0006979 | 氧化应激反应 | 3 | 2.28E-02 |
| GO:0032727 | α干扰素生成正调控 | 2 | 3.03E-02 |
| GO分子生物功能 | |||
| GO:0030176 | 内质网膜组成 | 4 | 2.19E-03 |
| GO:0005923 | 细胞紧密连接 | 3 | 3.11E-02 |
新窗口打开|下载CSV
本研究在高频ROH区域还定位到32个与牛重要经济性状相关的QTLs(附表1)。其中11个与产奶性状相关,7个与体型性状相关,5个与繁殖性状相关,5个与肉质和胴体性状相关以及4个与长寿性状相关。所有QTLs均分布于5、12和22号染色体上,其中22号染色体上定位到了23个QTLs。这些QTLs分布的染色体与新疆近交牛ROHf的峰值位点所在染色体相一致(图5A)。
3 讨论
3.1 新疆近交牛群体遗传学研究及同态相同位点分析
本研究PCA分析结果表明,新疆近交牛、哈萨克牛(塔城)、哈萨克牛(伊犁)和蒙古牛的遗传背景相似,这与它们的地理位置分布相一致,都处于中国北部。综合PCA与Admixture结果,本研究推测新疆近交牛与哈萨克牛遗传关系最近,这与其培育人对牛群近交培育历史的记述内容相一致。不同群体间LD程度的差异是由有效群体数量以及群体的历史所造成的[21,22],通常本地品种的LD程度较小,因为其相对于基因交流具有普遍性的品种(例如荷斯坦牛)而言Ne的数量较大[23]。本研究中各群体随着标记距离增加,LD程度迅速下降,该趋势与已有报道一致[24,25,26]。新疆近交牛LD程度较高,说明该群体染色体上相邻位点间的关联性强,这反映了其培育起始于少量系祖,呈现群体奠基者效应(founder effect)。而哈萨克牛(塔城) LD程度较低,可能与当地牛选育程度低有关。另有研究显示野猪与家猪相比,在所有常染色体上的LD程度最小,且衰减速度最快[23]。本研究中,选育程度较低的哈萨克牛(塔城) LD特征与之相似。
本研究估计的历史Ne趋势与荷斯坦牛和绵羊的结果相一致,随着历史世代数增加,Ne逐渐增大[27,28]。基因交流具有普遍性的群体中Ne较低,可能是由于人工选择使得群体中优势基因型比例上升,人为导致了高强度LD,因此Ne下降[27]。估计10世代以内Ne时,标记间的物理距离约为5.33 Mb,此距离之后新疆褐牛和荷斯坦牛LD程度低于新疆近交牛和塔城哈萨克牛,导致Ne估计值较大。本研究Ne的估计依赖于LD,由于估计近世代时标记间LD程度低(除新疆近交牛外r2 < 0.06),Ne估计的准确性可能会受到影响[27]。此外,样本群体大小也会显著影响Ne的估计值[29],本研究仅使用了50头荷斯坦牛,与其他同品种研究相比偏小[30,31],导致结果偏低。尽管如此,各群体Ne的增加趋势及大小也与图2A各群体LD特征相一致,该结果可以用来进行不同群体之间的比较。
Di Gaetano等[32]对意大利人种的遗传结构分析发现,IBS群体均值的分布情况能够分辨不同的亚群体遗传背景。本研究通过比较群体间的IBS,从结果可以看出,荷斯坦牛与其他3个群体关系最远,而新疆近交牛、哈萨克牛(塔城)与新疆褐牛都具有一定亲缘关系,这也与各群体的历史及当前塔城地区新疆褐牛为主推品种的现状相一致。
3.2 新疆近交牛基因组近交程度以及对体型的影响
本研究结果表明,群体ROH数量与ROH长度之间存在正比关系,这与多篇研究报道的结果相一致[9,33]。新疆近交牛的长ROH数量明显多于其他群体,该结果也印证了图3C中新疆近交牛的斜率较小。哈萨克牛(塔城)样本数量较少(仅20头)可能导致本研究未能在其29号染色体上检测到ROH。长ROH片段预示着近期的近交[17],本研究结果也证明了这一点。也有研究表明ROH数量与染色体长度之间呈正比[34],本研究中新疆褐牛(图4C)和荷斯坦牛(图4D)的趋势较为明显,哈萨克牛(塔城)由于样本数量较少而未呈现明显趋势。HOM%和ROH%的结果说明了新疆近交牛的近交程度最高。杨湛澄等[9]利用Illumina 54K芯片检测了中国荷斯坦牛的ROH%和HOM%,其中ROH%为0.064,略小于本研究中荷斯坦群体的结果。这可能是因为本研究ROH检测的参数较为宽松,包含了长度<1 Mb的ROH,因此ROH%偏高。此外,Purfield等[18]研究表明,芯片密度也会影响ROH的检测结果,因此本研究在各群体提取出相同密度的标记进行比较。杨湛澄等[9]研究表明,荷斯坦公牛HOM%的范围为0.625~0.750,高于本研究母牛的结果(0.541)。究其原因可能是本研究荷斯坦牛所使用的标记是从150K芯片中提取的部分标记,导致一些纯合位点被忽略,致使HOM%较低。
为避免月龄对体型大小评估的影响,本研究分别对≥12月龄和≥24月龄的个体进行分组。HOM%和ROH%在不同体型分组间的差异都未达到显著性水平(P>0.05),这可能与体型数据样本量少有关。但本研究结果表明:个体位点纯合率越高,其体型就越小。这说明近交对体型性状有影响,在一定程度上造成了牛只生长方面的近交衰退。
3.3 新疆近交牛基因组特征区域检测
研究表明,经过多个世代的近交,新疆近交牛的SNP位点纯合率上升[1],也导致ROH检出概率提高,致使ROHf明显高于哈萨克牛(塔城)。ROHf在基因组中的分布差异较大,该分布特征与其他多个物种的研究相一致[8,9,10]。Kim等[8]利用50K芯片数据对北美荷斯坦牛的ROH进行了检测,发现未经选择的荷斯坦牛群体各SNP的ROHf皆小于0.09,且未检出高频区域。人工选择在一定程度上导致了近交,从而使位点纯合率上升,而未经选择的群体中,ROH在染色体上的覆盖率较低。本研究也未能在哈萨克牛(塔城)中检测出高频ROH区域,这说明其近交程度较新疆近交牛低,新疆近交牛多个世代的近交繁育导致了高频ROH区域的出现。新疆近交牛基因组特征区域鉴定的基因主要参与了基本的生物学过程和分子功能,且定位的QTLs与重要经济性状相关联,说明多个世代的近交可能对这些区域的遗传特征产生了影响。本研究也发现新疆近交牛的体型大小与位点纯合率存在一定趋势,有可能纯合位点区域覆盖了与生长和体型性状相关的基因或QTLs,积累了有害突变,致使性状表型出现衰退。
本研究从多个方面证明了新疆近交牛在基因组上达到了很高的近交纯度,该群体是我国难得的反刍动物近交资源。目前尚未对新疆近交牛的重要经济性状进行系统记录,若能积累准确且充足的表型数据,该群体在研究大型反刍动物近交衰退的表型表现及其遗传调控机制方面将具有重大意义。
附录
附表1详见文章电子版www.chinagene.cn。Supplemental Table 1
附表1
附表1 32个QTLs的详细信息
Supplemental Table 1
| 染色体 | 位置(Mb) | QTL类型 | ID号* |
|---|---|---|---|
| 5 | 20.22~48.75 | 肉质和胴体性状相关 | 22866 |
| 5 | 47.54~90.49 | 肉质和胴体性状相关 | 22864 |
| 5 | 48.08~48.99 | 肉质和胴体性状相关 | 24694 |
| 5 | 48.08~48.99 | 繁殖性状相关 | 24565 |
| 5 | 48.08~48.99 | 长寿性状相关 | 24524 |
| 5 | 48.08~48.99 | 肉质和胴体性状相关 | 24695 |
| 5 | 48.16~48.75 | 肉质和胴体性状相关 | 22878 |
| 12 | 52.26~60.51 | 产奶性状相关 | 56344 |
| 12 | 52.26~60.51 | 产奶性状相关 | 56385 |
| 22 | 31.48~34.13 | 产奶性状相关 | 126214 |
| 22 | 31.48~34.13 | 体型性状相关 | 126019 |
| 22 | 31.48~34.13 | 繁殖性状相关 | 126008 |
| 22 | 31.48~34.13 | 繁殖性状相关 | 126007 |
| 22 | 31.48~34.13 | 繁殖性状相关 | 126006 |
| 22 | 31.48~34.13 | 繁殖性状相关 | 126005 |
| 22 | 31.48~34.13 | 长寿性状相关 | 125994 |
| 22 | 31.48~34.13 | 长寿性状相关 | 125993 |
| 22 | 31.48~34.13 | 体型性状相关 | 125379 |
| 22 | 31.48~34.13 | 产奶性状相关 | 126225 |
| 22 | 31.48~34.13 | 体型性状相关 | 125955 |
| 22 | 31.48~34.13 | 体型性状相关 | 125925 |
| 22 | 31.48~34.13 | 体型性状相关 | 125877 |
| 22 | 31.48~34.13 | 体型性状相关 | 125876 |
| 22 | 31.48~34.13 | 产奶性状相关 | 126357 |
| 22 | 31.48~34.13 | 产奶性状相关 | 126236 |
| 22 | 31.48~34.13 | 长寿性状相关 | 125992 |
| 22 | 33.98~33.98 | 产奶性状相关 | 51582 |
| 22 | 33.98~33.98 | 产奶性状相关 | 51581 |
| 22 | 33.98~33.98 | 体型性状相关 | 51583 |
| 22 | 33.98~33.98 | 产奶性状相关 | 51580 |
| 22 | 8.96~41.99 | 产奶性状相关 | 56580 |
| 22 | 8.96~41.99 | 产奶性状相关 | 56581 |
新窗口打开|下载CSV
致谢
衷心感谢新疆石河子大学谢苏和公红斌同学,新疆农业大学胥磊、徐庆磊、杨明路和邱文卿同学,以及中国农业大学张海亮和胡丽蓉同学在本研究样品采集过程中给予的大力帮助;感谢新疆近交牛的培育人卢国昌以及老先生的爱人李秀英、儿子卢振宇(邮箱: zhangll1000@163.com)、儿媳朱晓静、孙子芦金龙等一家人对近交牛培育的支持。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
DOI:10.3168/jds.S0022-0302(05)72861-7URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 4]
[本文引用: 6]
[本文引用: 6]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
DOI:10.1038/hdy.1974.89URL [本文引用: 1]
[本文引用: 1]
DOI:10.1016/0040-5809(71)90011-6URL [本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1101/gr.6023607URL [本文引用: 1]
DOI:10.1038/nrg777URL [本文引用: 1]
DOI:10.1038/s41598-019-49830-6URL [本文引用: 2]
DOI:10.1038/ng1001-229URL [本文引用: 1]
[本文引用: 1]
DOI:10.1534/genetics.107.084277URL [本文引用: 1]
DOI:10.3724/SP.J.1005.2012.00050URL [本文引用: 3]
DOI:10.3724/SP.J.1005.2012.00050URL [本文引用: 3]
DOI:10.1016/j.livsci.2014.10.015URL [本文引用: 1]
DOI:10.1016/0040-5809(75)90028-3URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1371/journal.pone.0043759URL [本文引用: 1]
DOI:10.1017/S1751731115002943URL [本文引用: 1]
DOI:10.3168/jds.2018-14805URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}