 ,中国农业科学院农业基因组研究所,“岭南现代农业”广东省实验室,深圳 518120
,中国农业科学院农业基因组研究所,“岭南现代农业”广东省实验室,深圳 518120Linking chromatin conformation to gene function
Qitong Huang, Qing Li, Yubo Zhang,Lingnan Guangdong Laboratory of Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518120, China通讯作者: 张玉波,博士,研究员,研究方向:动物三维基因组学。E-mail:ribon_001@163.com
编委: 吴强
收稿日期:2019-08-29修回日期:2019-10-29网络出版日期:2020-01-20
| 基金资助: |
Editorial board:
Received:2019-08-29Revised:2019-10-29Online:2020-01-20
| Fund supported: |
作者简介 About authors
黄其通,博士研究生,研究方向:动物三维基因组学。E-mail:miraclelive@qq.com。
李清,硕士,研究方向:动物三维基因组学。E-mail:liqing9102@163.com;黄其通和李清并列第一作者。。

摘要
在真核细胞中,DNA序列以染色质为载体,高度凝缩并存储于细胞核内,其复制、修复和转录表达等过程受到染色质构象的精准调控。越来越多的研究表明,特定的染色质构象可选择性激活或沉默基因,从而控制细胞自我维持或定向分化,决定细胞的组织特异性和细胞命运。因此,对染色质构象的深入研究已成为准确解析基因功能的一个关键切入点,也是当前基因组学研究所面临的一个巨大挑战。本文对染色质构象的研究历史、结构特征、动态调控机制进行了综述,并重点论述了不同维度构象特征对基因转录调控的影响,对该领域的研究难点进行了讨论,展望了其未来的发展方向,期望通过有效梳理染色质构象与基因调控之间的脉络关系,为未来该领域的研究提供参考。
关键词:
Abstract
DNA is highly compressed and packaged as chromatin within the nucleus in eukaryotes. DNA replication, DNA repair and transcription, and other biological processes are precisely regulated or determined by chromatin conformation. Activation or repression of different genes/transcription factors are tightly correlated to their chromatin conformation which contributes to cell self-maintenance, differentiation, specificity and identity. Therefore, the study of bridging chromatin conformation to gene function has became crucial for decoding genetic information and precision biology which is also a great challenge for current genomics research. Here, we review the multiple aspects of chromatin conformation, including its history, characteristics, dynamics and the impact on different dimension-chromatin architecture to gene function. Additionally, we discuss the limitation and challenges of current status of linking chromatin conformation to gene function study. We believe this is informative and foundational in combing the context between chromatin conformation and gene function as reference.
Keywords:
PDF (647KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黄其通, 李清, 张玉波. 染色质构象与基因功能. 遗传[J], 2020, 42(1): 1-17 doi:10.16288/j.yczz.19-257
Qitong Huang.
DNA是生物体的主要遗传物质。在真核细胞中,DNA序列与组蛋白结合形成核小体,并逐渐凝缩为染色质存储于细胞核内[1]。作为细胞核内DNA的载体,染色质的空间构象决定了基因组凝缩的空间特征,从而决定基因组生物学功能的分子基础——调控元件对特定转录因子的招募,及远端调控元件与靶基因的互作[2,3]。与蛋白质结构相似,按不同的空间尺寸,染色质构象可分为3个不同维度(图1):一维结构为核小体的物理定位,反映了不同基因组部位的可接近性;二维结构为核小体串进一步折叠或凝缩形成的染色质构象,如30 nm染色质纤维[4],重点研究邻近核小体之间的相互作用;三维结构为染色质的三维空间构象,是基因组范围内的、广泛的远距离互作结构,包括可能存在的跨越几kb到几Mb距离不等的染色质疆域(chromosome territory, CT)[5]、染色质区室(chromatin compartment A/B)[6],拓扑相关结构域(topologically associating domain, TAD)[7,8],染色质环(chromatin loop)[9],以及反式互作等。近年来,由于高通量测序技术的迅猛发展和多学科的交叉融合,探究染色质构象的新方法层出不穷[10,11],对染色质构象尤其是一维和三维构象的研究不断走向深入,为探索染色质构象背后的生物学意义打下了重要的基础。
图1
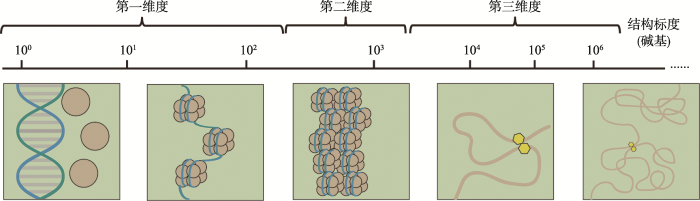 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1不同维度的染色质构象
Fig. 1Overview of chromatin structure at different scales
现有研究表明,染色质构象具有高度的异质性,不同的细胞状态或表型与特定的染色质构象之间存在着较强的关联性[12]。例如,胚胎干细胞的分化往往伴随着染色体内部互作的逐渐增强[13];癌症细胞中常见compartment A/B转置、TAD边界改变等现象[14,15]。染色质构象开始被科学家们认为是决定遗传信息传递的空间密码[16,17,18]。生物体通过染色质构象的改变,调控特定基因的复制、修复、转录表达等过程,进而决定细胞功能及其命运[19]。因此,准确捕捉染色质构象,并对其生物学意义进行深入解读,将成为人类进一步认识生命复杂行为的重要基础。
本文将以染色质构象与基因功能为主线,从一维到三维、多角度综述染色质构象的结构和功能特征,并对其可能的动态调控机制进行概述,以期为后续染色质构象与基因功能研究提供参考。
1 染色质的一维构象
核小体是染色质的基本组成单元,最早于1974年由Komberg[1]通过MNase酶切和电镜实验发现。在电镜视野下,核小体与核小体相连,呈现出经典的“串珠”结构(beads-on-a-string)[20,21]。但直到1997年,人们才成功获得核小体的高分辨(2.8 ?)晶体结构[22]。结构分析显示,核小体是由147 bp长的DNA缠绕组蛋白八聚体(组蛋白H2A、H2B、H3和H4各2个分子形成一个组蛋白八聚体) 1.65圈后形成,DNA通过14个小沟与组蛋白八聚体作用形成紧密的结构。核小体的发现与结构的确定是染色质构象研究中的一个里程碑事件,同时也成功开启了从染色质结构到功能的遗传信息解读序幕。相关研究表明,核小体在基因组上的分布有一定的偏好性:核小体在调控区域,如启动子、终止子和转录因子结合位点(TFBS)区域分布稀疏,在编码区分布密集[23,24,25]。而Saragosti等[26]关于SV40微小染色体的无核小体区和DNase超敏感区定位实验显示,核小体的缺失区域与活跃基因调控区——DNase超敏感区高度吻合。这些结果表明,核小体的定位影响基因表达,具有重要的生物学意义。同时,大量研究也表明了核小体的定位与DNA复制、修复、转录和可变剪接等过程密切相关[27,28,29]。例如,通过光镊实验观察单个RNA II型聚合酶(RNA polymerase-Ⅱ,RNA Pol Ⅱ)在转录延伸阶段的行为,Hodges等[27]发现编码区的核小体会使RNA Pol Ⅱ多次停顿,导致转录速率下降。而酵母启动子区域核小体的分析结果显示,启动子区域核小体的数量与启动子下游基因转录速率呈负相关关系[25]。
纵观目前研究进展,核小体定位影响基因表达的主要作用模式可概括为两类:一是识别标记,二是接触阻碍。通过研究核小体定位与转录起始位点(transcription start site, TSS)的关系,科学家们发现,启动子区域存在稳定开放的核小体缺乏区域(nucleosome free region, NFR),RNA Pol Ⅱ停靠在NFR的下游+1核小体处,起到调控基因转录表达的作用[30,31]。对于缺乏核心启动子元件的基因,转录复合物依然可正确识别转录起始位点,但在不改变核心启动子序列的情况下,替换NFR,却会导致TSS位置发生偏移[32,33]。这暗示着核小体定位很可能是转录机制准确识别TSS的一个重要标记。此外,也有相关证据表明,核小体定位与可变剪接过程中外显子的识别有关。如外显子上的核小体丰度明显高于内含子、假外显子区域,且丰度越高,剪接信号越弱[34,35]。但总体而言,目前对于核小体识别标记功能的具体机制尚不明确,有待进一步研究。核小体的阻碍效应主要与核小体的自身元件组蛋白密切相关。重构染色质的体外转录实验显示,当裸露的DNA与核心组蛋白(H2A、H2B和H4)形成核小体,并达到每200 bp DNA一个核小体的密度时,75%的转录将被抑制。而核小体与组蛋白H1的交联会加大这一抑制程度[36]。通过进一步研究,科学家们推测,核小体组蛋白的静电位阻很可能是这种阻碍效应的重要原因。核小体组蛋白带正电荷,而DNA带负电荷,根据同性相斥,组蛋白的静电位阻作用排斥其他蛋白质分子与核小体DNA结合,从而阻断RNA聚合酶、转录因子、DNA复制与修复酶等与DNA的接触机会,抑制了遗传信息的表达。而NFR以及两核小体间的linker DNA由于不存在组蛋白的静电位阻效应,转录因子等可顺利与顺式调控元件结合,从而启动基因转录[37]。
2 染色质的二维构象
通过形成核小体,DNA的长度压缩了6~7倍,但这还远远达不到存储于直径仅为10 μm的细胞核中的程度。这意味着,染色质拥有比核小体更高级的构象类型。1979年,Finch等[4] 最先通过电子显微镜观察到了直径为30 nm左右的染色质纤维,电镜结果提示该结构由11 nm核小体串珠结构组成。30 nm染色质纤维的发现正式使染色质构象研究推进到关注核小体间相互作用关系的二维层面。针对30 nm染色质纤维的结构模型,科学家们做了大量的工作,并提出了不同的猜想,如Solenoid模型[4,38]、Helical Ribbon模型[39]、two-start的Zigzag模型[40]等。2014年,通过冷冻电镜单颗粒三维重构技术,30 nm染色质纤维的高清晰三维结构被成功获取[41]。电镜结果显示,30 nm染色质纤维以4个核小体为结构单元;各单元之间通过相互扭曲折叠,从而形成一个左手双螺旋高级结构。其中,“四核小体”结构单元这一结果在染色质单分子力学研究过程中得到再次印证[42]。研究人员发现,“四核小体”结构是染色质折叠/去折叠过程中存在的一种稳定的中间状态,进而推测该结构单元可能是除核小体以外的,一个具有重要调控功能的结构与功能单元,可通过调控染色质纤维结构的紧密程度参与基因转录调控过程[42]。但事实上,以上所有关于30 nm染色质纤维的结果都是基于体外实验发现的,截止至今,科学家尚未真正在细胞内发现该结构的存在,因 此,关于染色质30 nm纤维的体内研究还有待进一步深入。目前,关于染色质二维构象与遗传信息传递功能方面的研究相对稀少,更多的是一些推测和猜想。例如通过Hi-CO方法,Ohno等[43]首次发现了核小体在折叠过程中存在两种基本的结构单元:α-四面体(α-tetrahedron)和β-菱形(β-rhombus)。其中,核小体相对距离更远的β-菱形单元更倾向于分布在代表启动子的H2A.Z组蛋白附近,并与转录激活标记如H3K18ac和H3K4ac的富集呈正相关关系。而结合观测到的随核小体相对距离增加,基因表达显著上调这一结果,Ohno等[43]推测基因的激活与染色质的二维构象β-菱形单元相关。
3 染色质的三维构象
染色质三维构象的研究起始时间较早,早在1885年Rabl[44]就观察到细胞核内存在不同的染色体区域,而后通过荧光染色技术、显微技术等手段,很多实验都证实了细胞核中存在不同的三维结构[45]。但由于缺乏微观的证据,染色质三维构象并没有获得太多的关注。随着测序技术的发展,“人类基因组计划”(human genome project, HGP)[46]和“人类基因组百科全书计划”(encyclopedia of DNA elements, ENCODE)[47]的完成,科学家们逐渐意识到,基因组在空间结构上并不是一维线性排开的,早前的线性分子模型已不足以揭示这些新发现的离散的调控元件以及结构变异与基因功能的联系。至此,三维基因组学研究热潮被正式掀起。染色质构象捕获技术(chromosome conformation capture, 3C)是Dekker等[48]于2002年开发的测定特定的点对点之间染色质交互作用的新技术。该技术第一次将认识DNA一维序列高度提升到三维水平,也成为了后续三维基因组测序技术开发的基础。目前,基于3C技术衍生出的构象捕获技术包括4C[49,50]、5C[51]、Hi-C[6]、ChIA- PET[52]、原位In situ Hi-C[9]、高效酶切DNase Hi-C[53]、杂交探针Capture-Hi-C[54]、单细胞Hi-C[55]、DLO Hi-C[56]等,为研究基因间的远距离互作,诠释转录因子与染色质相互作用的关系,以及细胞核内互作染色质的空间构象提供了重要的基础。此外,随着电镜技术的发展,超高分辨率电子显微镜技术也为染色质三维构象的研究提供了新的视角。基于上述技术和分析结果,科学家们提出了真核生物染色质三维层级假说模型。在不同的空间尺度上,这些层级结构依次为染色质疆域(chromosome territory, CT)[5]、染色质区室(chromatin compartment A/B)[6],拓扑相关结构域(topologically associating domain, TAD)[7,8]和染色质环(chromatin loop)[9](图2)。
图2
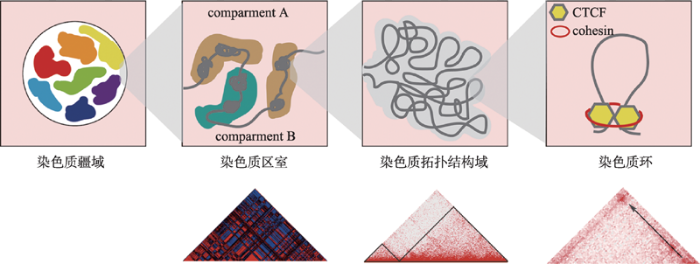 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2染色质的三维构象
Compartment A/B及TAD热图数据来源于小鼠胚胎干细胞eHi-C (专利号CN201610995880.X)数据,chromatin loop热图数据来源于人类淋巴母细胞(GM12878) in situ Hi-C数据[9]。
Fig. 2Three-dimensional conformation of chromatin
3.1 染色质疆域
在细胞核内,每条染色体的分布相对独立,它们各自占据着一块特定的、不重叠的区域,彼此间少有接触,而这个起着屏障作用的区域即是CT。大量研究显示,CT的径向定位具有偏好性,且在进化上高度保守。一般而言,小的、富含基因的染色体位于细胞核中心附近,而大的、缺乏基因的染色体位于细胞核边缘附近。Koehler等[57]通过对牛胚胎的研究发现,CT径向定位偏好性的首次出现与胚胎基因组的激活有关,并在囊胚中完全建立。此外,也有相关研究表明,CT的定位与细胞复制时间和转录活性相关。早期复制位点和活性基因倾向于在细胞核内部定位,而晚期复制位点和抑制基因则倾向于在核边缘定位[58,59]。这种现象与普遍认可的核纤层抑制基因表达的观点相符。但也存在例外,如夜行哺乳动物的视网膜杆状细胞中的CT定位与大多数白天活动的真核生物的相反,表现为异染色质定位于核中心,常染色质位于核外围[60]。3.2 染色质区室
2009年, Lieberman-Aiden[6]首次运用Hi-C技术揭示了人淋巴母细胞的核内三维构象,并提出了染色质空间构象的另一重要特征,即染色质由compartment A和compartment B交替分布构成。Compartment A结构具有基因密集、转录活跃、可接近性强(DNase Ⅰ高度敏感)等特征,而compartment B则恰恰相反,该区域折叠程度高,基因分布少,且多富集具有抑制活性的组蛋白如H3K27me3等[6]。通过关联分析,科学家们发现,compartment A和compartment B分别与细胞学上的常染色质和异染色质相对应。而除了在基因表达特征层面,compartment B在细胞核内的分布也与异染色质十分相像,与核纤层结合结构域(nuclear lamina-associated domains, LADs)有较大的重叠,主要定位于核纤层附近[61]。这些证据在一定程度上说明了compartment A/B实 质上是对常、异染色质具有不同亚细胞核定位的再发现。Compartment A/B具有一定的细胞特异性,伴随着细胞状态的改变,可发生compartment A/B的互换。例如,在人胚胎干细胞分化过程中,加州大学圣地亚哥分校的Bing Ren团队[62]发现至少有36%的基因组发生了compartment A/B的转换。而结合RNA-seq数据,可清晰的观察到compartment B到A的转变,使基因更倾向于高表达,而compartment A到B的转变,基因表达则倾向于被抑制。该研究表明compartment A/B参与基因转录表达的调控过程,对基因的特异性表达有重要的影响作用。此外,相关研究人员还发现,compartment A/B与DNA的复制时序相关。Compartment A到compartment B的转变和早期到晚期的复制时序变化相对应,且在发生时间上一致,而compartment B至compartment A的改变则早于晚期到早期复制时间的变化和转录激活[63]。进一步将compartment A/B细分成类亚区室(subcompartment),Rao等[9]的研究结果显示,活跃的A1、A2 subcompartment DNA复制发生时间更早,在细胞周期S期更早期的阶段,而抑制状态的B1、B2和B3的复制时间则与S期的中后期对应。
3.3 染色质拓扑结构域
TAD是基因组上长度从几百kb到几Mb不等的DNA片段,其内部含有一个或多个基因和调控元件,可视为一种高度自我互作的基因组单元。相关研究表明,TAD的边界富集了绝缘子结合蛋白CTCF、持家基因、tRNA、短散点元件(SINE)、H3K4me3、H3K36me3和黏连蛋白复合物(cohesin complex)等,科学家们推测这些因子与TAD的形成相关[7]。TAD在不同物种、不同细胞类型以及不同进化阶段中具有高度的保守性[64,65]。如50%~70%的TAD同时存在于小鼠和人类胚胎干细胞中[62],而随着分化的进行,TAD的位置几乎不会发生变化[66]。因此,TAD被认为是染色质折叠的基本结构单元。TAD与基因转录表达间的关系一直是科研界的研究热点。不同的研究表明,TAD与多种生物学现象间有着紧密的关联。如TAD边界与复制结构域高度吻合[67];在胚胎干细胞分化过程中,TAD经历了从无到有的变化过程[68];在小鼠细胞中,雌性X染色体的沉默伴随着TAD的重构[69,70];一些疾病的发生以及机体应激反应过程中伴随的TAD边界的移动或消失[71,72,73,74]。这些研究都在一定程度上暗示了,TAD可能是基因组发挥功能的一类基本单元,它们内部的协同调控及边界的屏障作用很可能是其发挥生物学功能的主要作用模式。
TAD内部的协同调控主要是指TAD将转录调控元件限定在同一个区域内,有效构建自主的基因调控域,独立调控该区域的基因表达(图3)。近年来,不少实验都为该观点提供了证据支持。如在不同细胞和组织中,同一TAD内部的基因表达均呈现出了趋同性,拥有更高的相关性[8,62,75]。而将报告基因随机插入到小鼠基因组中,Symmons等[76]发现报告基因的表达与同一TAD范围内基因的表达特征相仿。但也有科学家认为,这种作用其实是很微弱的,与可同时激活所有基因的操纵子不同,TAD的协同调控作用可能只影响单个TAD中的一个基因子集,在功能上具有协同效应的基因往往更倾向于位于同一个TAD内[8,77]。其中,协同影响调控元件活性的数量性状位点(quantitative trait loci, QTL)倾向于分布在同一个TAD中[78,79];相关基因簇被划入单个TAD中[80]即为有力佐证。当前,TAD内部协同调控作用模式为细胞中基因共表达现象做出了较好的解释。
内部互作频率高,组间互作频率低是TAD的一个基本特征,也是TAD屏障作用最直接的体现(图3)。由于大多数调控元件与启动子的空间交互受到TAD结构域的限制[81,82],科学家们推测TAD边界的保持对基因正确的时空表达具有重要的意义。目前,已有研究表明,TAD边界的破坏、移位都会造成基因表达的紊乱。例如敲除小鼠胚胎干细胞X染色体上的TAD边界,可明显观测到两个TAD间的互作增加,邻近TAD基因表达上调[8]。而Epha4基因所在TAD边界的敲除、倒位,可使Epha4基因的增强子分别错误地激活Pax3、Ihh和Wnt6基因,发生异位表达,进而导致短指、多指畸形和F综合症的发生[71]。此外,一些报道指出,TAD边界在降低转录噪音上有一定的贡献。许多启动子和增强子具有双向转录特性,从而产生转录噪音,而在TAD边界处,这种异向转录可被有效阻止[83]。这些结果都暗示着TAD边界是阻止基因组内活动扩散的物理屏障,通过阻断不必要的互作,使基因发挥正常的作用。但在最近的一篇报道中,一个有趣的现象却打破了这种认识。通过人为敲除TAD边界和CTCF位点诱发TAD融合,Alexandra等[84]发现TAD边界的消失并没能引起基因显著的表达变化。这说明TAD“屏障”的建立可能并不仅仅依赖于边界这一单一因素,TAD结构调节基因表达的强度和准确度有待进一步探究。
3.4 染色质环
根据染色质纤维的聚合特性,由于随机碰撞,两个相距较远的基因组位点会以非常低的频率相互接触[6,85]。然而,某些位点的偶然接触频率却明显高于预期。科学家们将这种连接高频互作位点的染色质环状结构命名为loop。Loop由CTCF和cohesin等因子介导形成[86,87],通常发生在同一个TAD或sub-TAD中[82]。根据两端连接元件的不同,loop可细分为启动子-增强子loop、启动子-启动子loop、增强子-增强子loop等,这不同的loop交互作用,共同形成一个复杂的调控网络[81,88,89]。Loop的形成与基因表达调控有着紧密的关联。其中,β-珠蛋白基因簇启动子与远端基因座控制区(locus control regions, LCR)的互作即为一个典型的案例。当β-珠蛋白基因簇与LCR形成染色质环时,基因表达被显著增强,反之,基因表达则处于一个极低水平[90,91]。而根据最新报道,美国丹娜-法伯癌症研究所Rani E. George课题组发现在治疗抵抗的癌细胞中,CTCF同源物BORIS调节的loop的变化可导致超级增强子(包含多个增强子,具有高转录因子密度,并能显著促进基因表达的基因组区域)的形成,从而驱动一类proneural转录因子的异位表达,最终产生抗ALK抑制的表型[92,93]。通过人为干预loop的形成与消失,越来越多的研究结果证明loop是基因转录发生变化的一个重要原因,而不是一个结果。如在转录因子GATA1的缺失的情况下,Deng等[91]通过将Lbd1加在一个锌指蛋白上,牵引该分子到达β珠蛋白启动子上的靶位点处,诱发形成一个位于增强子和启动子之间的染色质环,可使原本处于抑制状态的β-珠蛋白基因的表达获得明显的提高。而利用CRISPR/Cas9技术缺失前列腺癌风险区域,破坏该区域和HOXA13互作形成的loop结构后,原本被抑制的HOXA13表达显著上调,并成功激活HOXA13诱导的致癌基因GATA2的表达[94]。从这些结果可推测,loop是基因激活与沉默依赖的介导因素,在DNA编码信息传递到RNA上的这个基础过程中起着不可或缺的关键作用。
在细胞分化过程中,loop具有两种作用模式,分别是指导模型(instructive model)和许可模型(permissive model)[95] (图3)。指导模型认为,随着分化的推进,区域内转录因子的不同,导致细胞新形成具有组织特异性的loop,进而调控特定基因的表达。如α-珠蛋白基因和β-珠蛋白基因的增强子-启动子loop只存在于红细胞中,其互作频率在红细胞成熟过程中逐渐增强[96,97]。SatB1增强子与远端元件的互作只在SatB1高表达胸腺细胞中出现[98]。而许可模型则是指loop结构以沉默状态存在于祖细胞中,不同分化时期的转录因子选择性地将其激活,从而发挥特定的基因转录调控作用。如Shh与增强子ZRS的loop结构同时被观测到出现在胚胎干细胞和下肢细胞中,但只有在下肢细胞中,Shh才会表现为上调表达[7]。这两种模型在一定程度上解析了基因组织特异性表达的作用机理,也进一步暗示了loop是基因表达调控的必要不充分条件,具有重要的研究价值。
图3
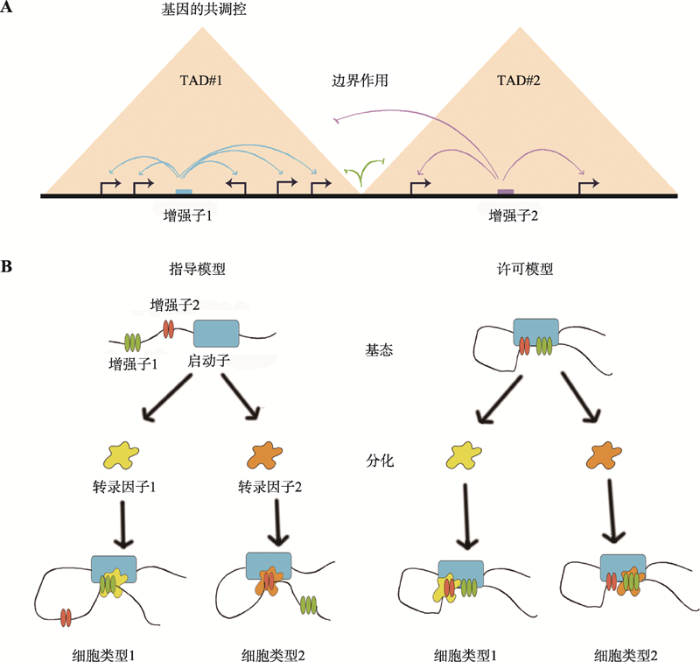 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3TAD与loop的作用模式
A:TAD作用模式;B:loop作用模式。
Fig. 3The function models of TAD and loop
4 染色质构象的动态调控
不同的细胞表型特征,背后往往对应着特定的染色质构象;不同维度、不同状态的染色质构象对DNA复制、转录及修复等过程起着重要的调控、甚至是决定作用。但实际上,DNA的高度压缩、折叠对这些过程中各种蛋白、调控因子“读取”和“访问”遗传信息起到的却是阻碍作用。细胞如何打破这种阻碍,如何实现染色质构象的动态变化成为了科学家研究的一个热点。基于目前研究报道,染色质构象的动态调控模式主要有以下4种:DNA甲基化、ATP依赖的染色质重塑复合物、组蛋白修饰或组蛋白变体以及相分离(图4)。图4
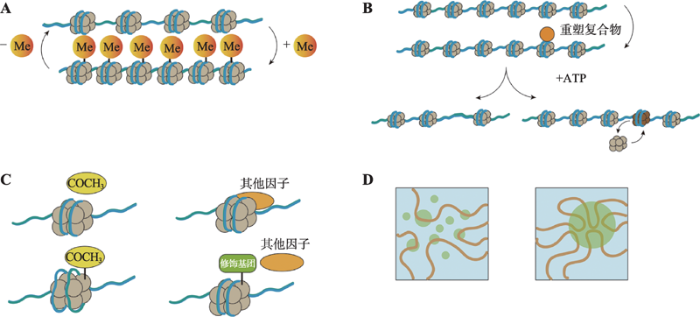 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4染色质构象动态调控机制
A:DNA甲基化;B:ATP依赖的染色质重塑复合物;C:组蛋白修饰;D:相分离。
Fig. 4Dynamic regulation of chromatin conformation
4.1 DNA甲基化
DNA甲基化是指在DNA甲基转移酶的作用 下,将甲基基团转移到腺嘌呤或胞嘧啶碱基上的一种修饰方式,主要发生在富含双核苷酸CpG岛的区域[99,100]。CpG岛的高甲基化会导致基因的表达下降或沉默,而染色质构象的变化是其中的一个重要原因。研究显示,基因上非启动子区的DNA甲基化能减少甲基化区域的H3K4me2/me3、H3K9Ac和H3K14Ac的修饰形式,从而使染色质结构紧密化,减少在甲基化区域的RNA Pol II的结合,最终影响转录速率[101]。而单碱基稀疏保守低甲基化CpG (scUMC)增高可减弱染色质环因子结合DNA的强度,从而减少DNA环绑定结两端的互作,导致相应DNA环调控的基因表达下降[102]。一般而言,DNA甲基化会引起染色质的高度压缩,形成非活跃状态的异染色质,从而导致基因沉默,去甲基化则会使染色质结构松散,进而激活基因的转录表达[103]。Compartment A的甲基化水平显著低于Compartment B即是该观点的一个印证[13]。但也有研究结果表明,DNA甲基化所导致的染色质构象变化并非一定带来基因表达下调,Flavahan等[104]对携带异柠檬酸脱氢酶基因(IDH)突变的脑瘤进行研究时发现,TAD边界CTCF位点的甲基化,可使CTCF结合减少,进而打破TAD边界,使原本被TAD边界分隔的两个调控元件发生强烈互作,激活相关基因的表达。4.2 ATP依赖的染色质重塑复合物
ATP依赖的染色质重塑复合物即是可利用ATP水解的能量来驱动核小体结构改变,从而改善转录因子等在染色质DNA局部的可接近性的蛋白因子。根据复合物中酶活中心蛋白亚基ATPase结构域相邻的其他结构域的组成,染色质重塑复合物被分为ISWI (imitation switch),SWI/SNF (mating type switching/sucrose non-fermenting),CHD (chromodomain helicase DNA-binding)和INO80 (inositol requiring 80)4个亚家族[105]。介导核小体“滑动”和“置换”是ATP依赖的染色质重塑复合物发挥染色质重塑功能的主要方式。研究表明,ATP依赖的染色质重塑复合物具有类DNA移位酶作用,即在DNA双链未解开的情况下可以使核小体沿着DNA滑动[106]。目前,最为详尽的一个移动模型为陈柱成和李雪明课题组[107]在研究不同核苷酸状态下Snf2-核小体复合物的冷冻电镜结构时提出的两步走“DNA波”模型,即第一步,ATP水解,Snf2张开,把DNA从入口端拉进,并在Snf2与核小体的结合点(SHL2)处储存1 bp DNA形变(“DNA波”);第二步,ATP结合,Snf2关闭,使得DNA形变向出口端传递,就像水波沿湖面传递一样,最终实现DNA对组蛋白的相对移动。而该模型在关于ISWI驱动核小体滑移机制的研究中也得到了证实[108]。介导核小体“置换”是指重塑复合物通过核小体中组蛋白变异体与经典组蛋白之间的替换,从而改变染色质构象。其中最典型的例子就是在酵母中Swr1催化H2AZ-H2B异源二聚体与核小体中经典H2A-H2B二聚体之间的替换[109],但其具体机制还有待进一步研究。
4.3 组蛋白修饰或组蛋白变体对染色质结构的影响
组蛋白的N端拖尾伸出核小体外,可被共价修饰,进而改变染色质构象,导致转录的激活或抑制。目前,该类修饰主要包括乙酰化、甲基化、磷酸化、泛素化等[110]。组蛋白修饰影响染色质构象的方式主要有两种:一是影响核心组蛋白的电荷平衡,如组蛋白乙酰化后可中和其部分的正电荷,松弛组蛋白对DNA的结合,使染色质重构,以便转录因子与DNA结合,进而激活转录[111,112,113];二是影响组蛋白与其他蛋白相互结合的功能,如核心组蛋白H4的Lys16乙酰化阻止其与SIR3的相互作用,因而抑制异染色质的形成[114];而组蛋白H3 N端尾部的Lys9甲基化可吸引异染色质蛋白1(heterochromatin protein 1, HP1),使邻近组蛋白H3的Lys9被甲基化,从而导致异染色质区域的扩展[115]。组蛋白变体是一类与经典的组蛋白序列高度相似,但功能却不同的变异体。目前,除H4外,H1、H2A、H2B和H3均有其相对应的组蛋白变体,它们通过与经典组蛋白发生置换,在染色质构象的动态调控中发挥着重要的作用[116]。例如,在胚胎干细胞中,H3.3 参与异染色质的形成[117]。同时也有研究表明,H2A.Z相比于H2A更能促进染色质高级结构的形成,并和异染色质蛋白HP1α协同作用维护组成型异染色质结构[118,119]。但对于组蛋白变体在转录调控过程中所发挥的功能及其机理至今还没有明确的解析。
4.4 相分离
相分离(phase separation)描述的是一种细胞里不同成分间相互碰撞、融合形成液滴,从而使一些成分被包裹在液滴内,一些成分被阻隔在液滴外的现象,它普遍存在细胞中,并与细胞结构的形成密切相关[120]。最新研究发现,相分离在染色质构象形成过程中具有突出贡献[121,122,123]。Strom等[121]在对早期果蝇胚胎和小鼠细胞进行研究时发现,相分离介导异染色质的形成,并提出了新异染色质形成模型。Strom等[121]认为HP1α蛋白发生液相分离,形成容纳染色体而排斥RNA聚合酶等分子的液相稳定区室,是异染色质形成的第一步。而该模型已获得体外实验、果蝇模型和人类细胞中的证据支持。为进一步研究相分离与染色质构象之间的关系,Shin等[124]开发了CasDrop体系,该体系通过人为调控相分离的发生,可直接观测相分离对染色质构象的影响。通过CasDrop,Shin等[124]发现相变易发生在染色质结构较为疏松、应变能较低的区域;染色质致密区域的较小的相变液滴则最终倾向于溶解;液滴对染色质的排斥会导致染色质结构的重塑。此外,Shin等[124]提出了染色质过滤模型(chromatin filter model),即结合在特定基因位点的液滴通过融合能够将距离较远的基因拉近,且排斥不含结合位点的背景基因区域,为研究染色质构象与基因转录表达关系提供了新的研究思路。但值得注意的是,相分离影响染色质构象方面的研究仍处于初期阶段,要真正揭示其中的关联必需更多的实验支持。5 挑战与展望
随着近年来不同染色质构象捕获技术的兴起,染色质构象研究领域出现了前所未有的发展,为进一步解读生物遗传密码开辟了新的途径,但同时也面临着大量的挑战。目前,在实验上,基于高通量测序的三维基因组数据仍存在信噪比低、分辨率不佳、数据质量参差不齐等问题。在数据分析上,基于bin矩阵的算法占据主导地位,该方法通过互作信号的中点坐标,将限制性片段分配到相应区间,并通过bin-bin交互观察计算相应结构,其计算结果深受bin本身大小的影响,容易产生偏差;此外,由于相关分析算法各有倚重,缺乏统一的度量和评判标准,不同软件分析结果的可重复性让人担忧,如对于同一数据同一分析目的,HICUP、FitHiC和Homer获取loop的数目和假阳性率相差甚远[125];如此种种,不仅使不同的数据之间缺乏可比性,造成大量数据资源的浪费,更让由此建立起来的假说和理论不断的受到考验和挑战;如Eileen Finn等[126]在利用模式动物果蝇研究高度重排的突变染色体时发现,大多数TAD变化对基因的表达几乎没有影响。这种TAD变化与基因表达之间的“脱钩”现象彻底颠覆了人们对TAD调控基因表达的认识。染色质构象研究是一个多学科交叉的研究领域。化学、物理、生物和计算机等多学科的参与为染色质构象的研究提供了不同的研究思路和角度,但与此同时也催生了一些新的问题,包括如何实现这些不同层面数据间的关联分析,去伪存真。目前,不同技术间的染色质构象分析结果差异较大,例如关于TAD的有无,高通量测序技术与超高分辨率ChromEMT电镜技术给出了截然相反的结果[127]。而尽管MERFISH超高分辨成像技术在单细胞层面观测到了类TAD结构域,其高度的异质性却与传统概念上的TAD的保守特征相悖,完全打破了人们对传统TAD的定义[128]。这到底是电镜技术缺陷使然,还是说TAD仅仅是个统计模型,并不存在于细胞核中,至今没有一个合理的解释。
染色质构象具有异质性,且时刻处于变化过程中,在单细胞水平捕捉到这种结构上的动态变化是染色质构象精确研究的基础也是目前面临的一大难题。截止目前,尚无一技术可实现高通量的、在未破碎的细胞即活细胞中直接观测到染色质构象变化。而标榜在单细胞水平捕捉染色质构象的单细胞Hi-C也因分辨率低下问题,无法捕捉到除compartment A/B以外的染色质高级构象[55]。这也就意味着,人们常说的染色质构象的动态变化实为群体细胞的“死”动态变化,并不能很好的揭示染色质构象的本质特征。如何在单细胞水平上实现高分辨率、高效、实时地监测染色质构象变化成为了一个亟待解决的问题。
揭示染色质构象与基因功能关系是染色质构象研究的最终目的,也是深入解读遗传密码关键的一步。染色质构象与基因功能的阐明要求多组学数据的联合分析,但目前同一样品多组学数据的同步分析实现起来还存在一定的难度。对于电镜下瞬时的构象特征,如何捕捉到与此相对应的转录组、蛋白组、代谢组、表观组的特征也成为了一个难题。由于细胞的特异性、取样时无法避免的时间误差,分别来源于不同细胞的多组学数据往往会导致多组学特征关联分析出现偏差。此外,针对多组学分析的成熟的算法亟待开发。目前,多组学数据的分析往往需要调用多个软件,设置十几个参数,过程纷繁复杂,各实验室流程各异,对后续的深入研究带来了极大的不便。
染色质构象变化是最基础的生物学现象,已成为联系不同生物学过程的重要桥梁和纽带。染色质构象研究的推进有赖于复制、转录、翻译等一系列生物学理论的完善,实验技术的改进,更需要多组学、多学科、多手段的有机整合。例如染色质构象捕获与基于免疫沉淀和tagmentation的文库制备相结合,可有效提高信噪比,在极少样本条件下,获取大量的构象信息[129];不同荧光分子和成像技术的引入,在提高分辨率的同时可在基因组尺度上实现其他分子如新生RNA行为的同步观测,为多组学数据的无缝整合提供了可能[130];大数据分析方法,如卷积神经网络[131]、自举式深度学习模型[132]等的应用可在一定程度上弥补分辨率不足的问题;而眺望未来,人工智能和深度学习算法的应用不仅能为特定的场景和特定的问题提供预测性模型和答案,更有可能一秒实现追本溯源,告诉我们生物学上到底发生了什么才会产生我们所看到的数据。我们有充分的理由相信,各领域的优势互补、不囿传统的大胆创新将成为推动“4D核体计划”[133]的强大动力,实现从空间(三维)和时间(第四维度)角度研究细胞核结构形成原理,进一步探究其对基因表达、细胞功能,以及对发育和疾病发生、发展的影响,揭示遗传信息最深层的奥秘。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1126/science.184.4139.868URLPMID:4825889 [本文引用: 2]
DOI:10.1016/j.gde.2012.11.008URL [本文引用: 1]

Transcription factors (TF) bind DNA sequence motifs, but the presence of a consensus DNA element is not sufficient to direct TF binding to chromatin. Recent genomic data have revealed that accessibility, as measured by DNase sensitivity and the presence of active histone marks, is necessary for TF binding. DNA sequence provides the initial specification of the accessibility of DNA elements within chromatin that permits TF binding. In yeast, it is known that poly(dA-dT) tracts directly encode low-nucleosome occupancy at promoters. Recent evidence suggests that CpG islands in mammals are inherently refractory to higher-order chromatin structure and remain accessible, despite favoring nucleosome formation in vitro. Taken together, these studies support a model for how accessibility originates and then propagates throughout regulatory cascades and development.
DOI:10.1038/nature11232URL [本文引用: 1]
DNase I hypersensitive sites (DHSs) are markers of regulatory DNA and have underpinned the discovery of all classes of cis-regulatory elements including enhancers, promoters, insulators, silencers and locus control regions. Here we present the first extensive map of human DHSs identified through genome-wide profiling in 125 diverse cell and tissue types. We identify similar to 2.9 million DHSs that encompass virtually all known experimentally validated cis-regulatory sequences and expose a vast trove of novel elements, most with highly cell-selective regulation. Annotating these elements using ENCODE data reveals novel relationships between chromatin accessibility, transcription, DNA methylation and regulatory factor occupancy patterns. We connect similar to 580,000 distal DHSs with their target promoters, revealing systematic pairing of different classes of distal DHSs and specific promoter types. Patterning of chromatin accessibility at many regulatory regions is organized with dozens to hundreds of co-activated elements, and the transcellular DNase I sensitivity pattern at a given region can predict cell-type-specific functional behaviours. The DHS landscape shows signatures of recent functional evolutionary constraint. However, the DHS compartment in pluripotent and immortalized cells exhibits higher mutation rates than that in highly differentiated cells, exposing an unexpected link between chromatin accessibility, proliferative potential and patterns of human variation.
DOI:10.1073/pnas.73.6.1897URLPMID:1064861 [本文引用: 3]
Chromatin prepared by brief digestion of nuclei with micrococcal nuclease, and extracted in 0.2 mM EDTA, appears in the electron microscope as filaments of about 100 A diameter which coil loosely. In 0.2 mM Mg++ these "nucleofilaments" condense into a supercoil or solenoidal structure of pitch about 110 A corresponding to the diameter of a nucleofilament. It is proposed that the x-ray reflections at orders of 110 A observed in chromatin originate in the spacing between turns of the solenoid rather than that between nucleosomes along the nucleofilament. The solenoidal structure appears to need histone H1 for its stabilization. Under certain conditions, isolated nucleosomes can also aggregate into a similar structure. The solenoidal structure can be correlated with the "thread" of diameter about 300 A observed by other workers in nuclei.
[本文引用: 2]
DOI:10.1126/science.1181369URLPMID:19815776 [本文引用: 6]
We describe Hi-C, a method that probes the three-dimensional architecture of whole genomes by coupling proximity-based ligation with massively parallel sequencing. We constructed spatial proximity maps of the human genome with Hi-C at a resolution of 1 megabase. These maps confirm the presence of chromosome territories and the spatial proximity of small, gene-rich chromosomes. We identified an additional level of genome organization that is characterized by the spatial segregation of open and closed chromatin to form two genome-wide compartments. At the megabase scale, the chromatin conformation is consistent with a fractal globule, a knot-free, polymer conformation that enables maximally dense packing while preserving the ability to easily fold and unfold any genomic locus. The fractal globule is distinct from the more commonly used globular equilibrium model. Our results demonstrate the power of Hi-C to map the dynamic conformations of whole genomes.
DOI:10.1038/nature11082URL [本文引用: 4]
The spatial organization of the genome is intimately linked to its biological function, yet our understanding of higher order genomic structure is coarse, fragmented and incomplete. In the nucleus of eukaryotic cells, interphase chromosomes occupy distinct chromosome territories, and numerous models have been proposed for how chromosomes fold within chromosome territories(1). These models, however, provide only few mechanistic details about the relationship between higher order chromatin structure and genome function. Recent advances in genomic technologies have led to rapid advances in the study of three-dimensional genome organization. In particular, Hi-C has been introduced as a method for identifying higher order chromatin interactions genome wide(2). Here we investigate the three-dimensional organization of the human and mouse genomes in embryonic stem cells and terminally differentiated cell types at unprecedented resolution. We identify large, megabase-sized local chromatin interaction domains, which we term 'topological domains', as a pervasive structural feature of the genome organization. These domains correlate with regions of the genome that constrain the spread of heterochromatin. The domains are stable across different cell types and highly conserved across species, indicating that topological domains are an inherent property of mammalian genomes. Finally, we find that the boundaries of topological domains are enriched for the insulator binding protein CTCF, housekeeping genes, transfer RNAs and short interspersed element (SINE) retrotransposons, indicating that these factors may have a role in establishing the topological domain structure of the genome.
DOI:10.1038/nature11049URL [本文引用: 5]
In eukaryotes transcriptional regulation often involves multiple long-range elements and is influenced by the genomic environment(1). A prime example of this concerns the mouse X-inactivation centre (Xic), which orchestrates the initiation of X-chromosome inactivation (XCI) by controlling the expression of the non-protein-coding Xist transcript. The extent of Xic sequences required for the proper regulation of Xist remains unknown. Here we use chromosome conformation capture carbon-copy (5C)(2) and super-resolution microscopy to analyse the spatial organization of a 4.5-megabases (Mb) region including Xist. We discover a series of discrete 200-kilobase to 1Mb topologically associating domains (TADs), present both before and after cell differentiation and on the active and inactive X. TADs align with, but do not rely on, several domain-wide features of the epigenome, such as H3K27me3 or H3K9me2 blocks and lamina-associated domains. TADs also align with coordinately regulated gene clusters. Disruption of a TAD boundary causes ectopic chromosomal contacts and long-range transcriptional misregulation. The Xist/Tsix sense/antisense unit illustrates how TADs enable the spatial segregation of oppositely regulated chromosomal neighbourhoods, with the respective promoters of Xist and Tsix lying in adjacent TADs, each containing their known positive regulators. We identify a novel distal regulatory region of Tsix within its TAD, which produces a long intervening RNA, Linx. In addition to uncovering a new principle of cis-regulatory architecture of mammalian chromosomes, our study sets the stage for the full genetic dissection of the X-inactivation centre.
DOI:10.1016/j.cell.2014.11.021URL [本文引用: 5]
We use in situ Hi-C to probe the 3D architecture of genomes, constructing haploid and diploid maps of nine cell types. The densest, in human lymphoblastoid cells, contains 4.9 billion contacts, achieving 1 kb resolution. We find that genomes are partitioned into contact domains (median length, 185 kb), which are associated with distinct patterns of histone marks and segregate into six subcompartments. We identify similar to 10,000 loops. These loops frequently link promoters and enhancers, correlate with gene activation, and show conservation across cell types and species. Loop anchors typically occur at domain boundaries and bind CTCF. CTCF sites at loop anchors occur predominantly (>90%) in a convergent orientation, with the asymmetric motifs "facing'' one another. The inactive X chromosome splits into two massive domains and contains large loops anchored at CTCF-binding repeats.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.tig.2015.03.010URLPMID:25887733 [本文引用: 1]
A decade of rapid method development has begun to yield exciting insights into the 3D architecture of the metazoan genome and the roles it may play in regulating transcription. Here we review core methods and new tools in the modern genomicist's toolbox at three length scales, ranging from single base pairs to megabase-scale chromosomal domains, and discuss the emerging picture of the 3D genome that these tools have revealed. Blind spots remain, especially at intermediate length scales spanning a few nucleosomes, but thanks in part to new technologies that permit targeted alteration of chromatin states and time-resolved studies, the next decade holds great promise for hypothesis-driven research into the mechanisms that drive genome architecture and transcriptional regulation.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.cell.2017.06.029URLPMID:28709003 [本文引用: 2]
High-order chromatin structure plays important roles in gene expression regulation. Knowledge of the dynamics of 3D chromatin structures during mammalian embryo development remains limited. We report the 3D chromatin architecture of mouse gametes and early embryos using an optimized Hi-C method with low-cell samples. We find that mature oocytes at the metaphase II stage do not have topologically associated domains (TADs). In sperm, extra-long-range interactions (>4 Mb) and interchromosomal interactions occur frequently. The high-order structures of both the paternal and maternal genomes in zygotes and two-cell embryos are obscure but are gradually re-established through development. The establishment of the TAD structure requires DNA replication but not zygotic genome activation. Furthermore, unmethylated CpGs are enriched in A compartment, and methylation levels are decreased to a greater extent in A compartment than in B compartment in embryos. In summary, the global reprogramming of chromatin architecture occurs during early mammalian development.
DOI:10.1186/s13059-015-0768-0URLPMID:26415882 [本文引用: 1]
Higher-order chromatin structure is often perturbed in cancer and other pathological states. Although several genetic and epigenetic differences have been charted between normal and breast cancer tissues, changes in higher-order chromatin organization during tumorigenesis have not been fully explored. To probe the differences in higher-order chromatin structure between mammary epithelial and breast cancer cells, we performed Hi-C analysis on MCF-10A mammary epithelial and MCF-7 breast cancer cell lines.
DOI:10.1101/gr.201517.115URLPMID:27053337 [本文引用: 1]
A three-dimensional chromatin state underpins the structural and functional basis of the genome by bringing regulatory elements and genes into close spatial proximity to ensure proper, cell-type-specific gene expression profiles. Here, we performed Hi-C chromosome conformation capture sequencing to investigate how three-dimensional chromatin organization is disrupted in the context of copy-number variation, long-range epigenetic remodeling, and atypical gene expression programs in prostate cancer. We find that cancer cells retain the ability to segment their genomes into megabase-sized topologically associated domains (TADs); however, these domains are generally smaller due to establishment of additional domain boundaries. Interestingly, a large proportion of the new cancer-specific domain boundaries occur at regions that display copy-number variation. Notably, a common deletion on 17p13.1 in prostate cancer spanning the TP53 tumor suppressor locus results in bifurcation of a single TAD into two distinct smaller TADs. Change in domain structure is also accompanied by novel cancer-specific chromatin interactions within the TADs that are enriched at regulatory elements such as enhancers, promoters, and insulators, and associated with alterations in gene expression. We also show that differential chromatin interactions across regulatory regions occur within long-range epigenetically activated or silenced regions of concordant gene activation or repression in prostate cancer. Finally, we present a novel visualization tool that enables integrated exploration of Hi-C interaction data, the transcriptome, and epigenome. This study provides new insights into the relationship between long-range epigenetic and genomic dysregulation and changes in higher-order chromatin interactions in cancer.
DOI:10.1023/A:1024966424909URL [本文引用: 1]
An important aim in biology is to understand how gene expression is regulated in the context of chromatin. Much progress has been made towards cracking the 'histone code', which describes the composition and organization of chromatin at high resolution. At the lower resolution provided by microscopy, nuclear compartmentalization has been linked to the control of gene expression and silencing. I will review two new techniques able to reveal the three-dimensional organization of individual loci, providing a view of the folding of the chromatin fibre at an intermediate level of resolution. Carter and colleagues and Tolhuis and colleagues have used the new techniques to demonstrate direct physical contact between the locus control region (LCR) and expressed genes in the active murine β-globin locus. The techniques will allow us to assess the role of locus organization when transcription is directed by distant regulatory elements. The new techniques (and their foreseeable descendants) will permit investigation of many genomic activities involving physical contact between separate regions of any genome. As such, they provide us with a new level of resolution at which to investigate the functional significance of chromatin organization as patterns of gene expression are initiated and modulated during development.
DOI:10.1038/srep41061URLPMID:28117353 [本文引用: 1]
Most of what we know about gene transcription comes from the view of cells as molecular machines: focusing on the role of molecular modifications to the proteins carrying out transcriptional reactions at a loci-by-loci basis. This view ignores a critical reality: biological reactions do not happen in an empty space, but in a highly complex, interrelated, and dense nanoenvironment that profoundly influences chemical interactions. We explored the relationship between the physical nanoenvironment of chromatin and gene transcription in vitro. We analytically show that changes in the fractal dimension, D, of chromatin correspond to simultaneous increases in chromatin accessibility and compaction heterogeneity. Using these predictions, we demonstrate experimentally that nanoscopic changes to chromatin D within thirty minutes correlate with concomitant enhancement and suppression of transcription. Further, we show that the increased heterogeneity of physical structure of chromatin due to increase in fractal dimension correlates with increased heterogeneity of gene networks. These findings indicate that the higher order folding of chromatin topology may act as a molecular-pathway independent code regulating global patterns of gene expression. Since physical organization of chromatin is frequently altered in oncogenesis, this work provides evidence pairing molecular function to physical structure for processes frequently altered during tumorigenesis.
DOI:10.1016/j.gde.2012.11.006URL [本文引用: 1]
Cell type specific transcriptional regulation must be adhered to in order to maintain cell identity throughout the lifetime of an organism, yet it must be flexible enough to allow for responses to endogenous and exogenous stimuli. This regulation is mediated not only by molecular factors (e.g. cell type specific transcription factors, histone and DNA modifications), but also on the level of chromatin and genome organization. In this review we focus on recent findings that have contributed to our understanding of higher order chromatin structure and genome organization within the nucleus. We highlight new findings on the dynamic positioning of genes relative to each other, as well as to their chromosome territory and the nuclear lamina, and how the position of genes correlates with their transcriptional activity.
DOI:10.1360/052014-108URL [本文引用: 1]
Chromatin is the basic element of eukaryotic chromosomes that package the genome in the nucleus, and its function is closely related to its three-dimensional (3D) structure. For example, many biological functions such as DNA replication, DNA damage, DNA repair, gene transcription and regulation, spread of long non-coding RNAs, and embryonic development, are processed in and affected by 3D chromatin architecture in nuclear space. In recent years, chromosome conformation capture (3C) and its derivative technologies combined with high-throughput sequencing such as ChIA-PET and Hi-C, have generated extensive chromatin interaction data. With these data, researchers have proposed a number of ways to reconstruct the 3D structures of chromsomal folding, which help to systematically study the chromatin 3D structures at different resolutions and to provide better understanding of chromatin functions with structural basis. In this review, we summarize recent advances in the methods for chromatin 3D structure modeling, and discuss its implications in chromatin function.
DOI:10.1360/052014-108URL [本文引用: 1]
Chromatin is the basic element of eukaryotic chromosomes that package the genome in the nucleus, and its function is closely related to its three-dimensional (3D) structure. For example, many biological functions such as DNA replication, DNA damage, DNA repair, gene transcription and regulation, spread of long non-coding RNAs, and embryonic development, are processed in and affected by 3D chromatin architecture in nuclear space. In recent years, chromosome conformation capture (3C) and its derivative technologies combined with high-throughput sequencing such as ChIA-PET and Hi-C, have generated extensive chromatin interaction data. With these data, researchers have proposed a number of ways to reconstruct the 3D structures of chromsomal folding, which help to systematically study the chromatin 3D structures at different resolutions and to provide better understanding of chromatin functions with structural basis. In this review, we summarize recent advances in the methods for chromatin 3D structure modeling, and discuss its implications in chromatin function.
DOI:10.1016/0092-8674(75)90149-xURLPMID:1122558 [本文引用: 1]
Electron microscopic and biochemical studies demonstrate that the fundamental structure of chromatin depleted of lysine-rich histones is composed of a flexible chain of spherical particles (nucleosomes), about 125 A in diameter, connected by DNA filaments. Such a chromatin preparation can be separated by centrifugation into two fractions which differ in the spacing of the nucleosomes; In one fraction almost all of the DNA is condensed in nucleosomes, while the other fraction contains long stretches of free DNA connecting regions where the nucleosomes are closely packed. The isolated nucleosomes contain about 200 base pairs of DNA and the four histones F2alpha1, F2alpha2, and F2b, and F3 in an overall histone/DNA ratio of 0.97; In such a structure the DNA is compacted slightly more than five times from its extended length; The same basic structure can be visualized in chromatin spilling out of lysed nuclei. However, in this latter case the nucleosomes are very closely packed, suggesting that histone F1 is involved in the superpacking of DNA in chromosomes and nuclei. The chromatin fiber appears to be a self-assembling structure, since the nucleosomal arrangement can be reconstituted in vitro from DNA and the four histones F2alpha1, F2alpha2, F2b and F3 only, irrespective of their cellular origin.
DOI:10.1126/science.183.4122.330URLPMID:4128918 [本文引用: 1]
Linear arrays of spherical chromatin particles (nu bodies) about 70 angstroms in diameter have been observed in preparations of isolated eukaryotic nuclei swollen in water, centrifuged onto carbon films, and positively or negatively stained. These bodies have been found in isolated rat thymus, rat liver, and chicken erythrocyte nuclei. Favorable views also reveal connecting strands about 15 angstroms wide between adjacent particles.
DOI:10.1038/38444URLPMID:9305837 [本文引用: 1]
The X-ray crystal structure of the nucleosome core particle of chromatin shows in atomic detail how the histone protein octamer is assembled and how 146 base pairs of DNA are organized into a superhelix around it. Both histone/histone and histone/DNA interactions depend on the histone fold domains and additional, well ordered structure elements extending from this motif. Histone amino-terminal tails pass over and between the gyres of the DNA superhelix to contact neighbouring particles. The lack of uniformity between multiple histone/DNA-binding sites causes the DNA to deviate from ideal superhelix geometry.
DOI:10.1038/ng1400URLPMID:15247917 [本文引用: 1]
The identification of nuclease-hypersensitive sites in an active globin gene and in the 5' regions of fruit fly heat shock genes first suggested that chromatin changes accompany gene regulation in vivo. Here we present evidence that the basic repeating units of eukaryotic chromatin, nucleosomes, are depleted from active regulatory elements throughout the Saccharomyces cerevisiae genome in vivo. We found that during rapid mitotic growth, the level of nucleosome occupancy is inversely proportional to the transcriptional initiation rate at the promoter. We also observed a partial loss of histone H3 and H4 tetramers from the coding regions of the most heavily transcribed genes. Alterations in the global transcriptional program caused by heat shock or a change in carbon source resulted in an increased nucleosome occupancy at repressed promoters, and a decreased nucleosome occupancy at promoters that became active. Nuclease-hypersensitive sites occur in species from yeast to humans and result from chromatin perturbation. Given the conservation of sequence and function among components of both chromatin and the transcriptional machinery, nucleosome depletion at promoters may be a fundamental feature of eukaryotic transcriptional regulation.
DOI:10.1016/j.molcel.2005.05.003URLPMID:15949447 [本文引用: 1]
In yeast cells, preferential accessibility of the HIS3-PET56 promoter region is determined by a general property of the DNA sequence, not by defined sequence elements. In vivo, this region is largely devoid of nucleosomes, and accessibility is directly related to reduced histone density. The HIS3-PET56 and DED1 promoter regions associate poorly with histones in vitro, indicating that intrinsic nucleosome stability is a major determinant of preferential accessibility. Specific and genome-wide analyses indicate that low nucleosome density is a very common feature of yeast promoter regions that correlates poorly with transcriptional activation. Thus, the yeast genome is organized into structurally distinct promoter and nonpromoter regions whose DNA sequences inherently differ with respect to nucleosome formation. This organization ensures that transcription factors bind preferentially to appropriate sites in promoters, rather than to the excess of irrelevant sites in nonpromoter regions.
DOI:10.1016/j.jinorgbio.2019.110769URLPMID:31326773 [本文引用: 2]
We synthesized two ruthenium(II) complexes: trans,trans-[Ru(im)2(tfa)2] (1) and trans,trans?[Ru(in)2(tfa)2] (2) where im?=?1H?imidazole, in?=?1H?indazole and tfa?=?tolfenamic acid, a potential nonsteroidal anti-inflammatory drug (NSAID). The NSAID was opted as bioactive ligand to understand its synergistic therapeutic effect in structurally analogous Ru(II)-compounds with KP418 (imidazolium trans?[tetrachloridobis(1H?imidazole)ruthenate(III)]) and KP1019 (indazolium trans?[tetrachloridobis(1H?indazole)ruthenate(III)]). The complexes were studied using various analytical methods and structure was determined by X-ray crystallography. Both the complexes display discrete mononuclear Ru(II) center in {RuN4O2} distorted octahedral geometry. The reactivity of the complexes was tested with potentially important biomolecules involved in metabolism of cancer cells, viz. l?arginine, dl?methionine, glutathione and L(+)ascorbate. Such studies intended to provide deeper insights on intracellular speciation and kinetic substitution encountered by Ru-drugs to target alternative cell death pathways. The complexes demonstrate a preferential binding affinity with calf thymus DNA (Kb?~?104?M-1) and human serum albumin (KHSA?=?105?M-1). Both the complexes showed potent inhibition of wild type yeast cell growth in a dose-dependent manner. Yeast cells were used as a powerful model system to study the molecular mechanism of pathobiology which shares a high degree of conservation of both cellular and molecular processes with human cells for assessing toxicity potential of the complexes. Fluorescence imaging studies reveal the localization of both complexes to yeast mitochondria despite its rigid cell wall and induce mitochondrial damage and formation of reactive oxygen species (ROS). The Micrococcal nuclease assay revealed complexes do not alter global nucleosome occupancy and probably target specific regions of the genome.
DOI:10.1016/0092-8674(80)90235-4URLPMID:6248237 [本文引用: 1]
Electron microscopic examination of SV40 chromatin prepared 44 hr post-infection led to the visualization of a nucleosome-free region (gap) in 15-20% of the minichromosomes. Minichromosomes with and without a gap displayed a mean number of 24 nucleosomes. Measurements carried out on dark field micrographs yielded for the gap a mean length of 249 +/- 13 bp, with a maximum value of 385 bp. The gap was mapped following digestion with three single-cut restriction endonucleases: Bgl l, Bam HI and Eco RI. It was located in the region of the origin of replication in accordance with previous biochemical data. To assess the situ existence of a nucleosome-free region, nuclei from infected cells were digested with DNase I. A highly sensitive region was thus revealed and mapped by secondary digestion with Eco RI. It was located in the same region as the gap, between 0.67 and 0.74 on the physical map. The sensitive region could be detected throughout the late phase of the virus cycle. These findings strongly suggest that a nucleosome-free region exists in the cells. The gap is not likely to be involved in replication, since it is asymmetric with respect to the Bgl I cleavage site, from which replication proceeds symmetrically.
DOI:10.1126/science.1172926URLPMID:19644123 [本文引用: 2]
RNA polymerase II (Pol II) must overcome the barriers imposed by nucleosomes during transcription elongation. We have developed an optical tweezers assay to follow individual Pol II complexes as they transcribe nucleosomal DNA. Our results indicate that the nucleosome behaves as a fluctuating barrier that locally increases pause density, slows pause recovery, and reduces the apparent pause-free velocity of Pol II. The polymerase, rather than actively separating DNA from histones, functions instead as a ratchet that rectifies nucleosomal fluctuations. We also obtained direct evidence that transcription through a nucleosome involves transfer of the core histones behind the transcribing polymerase via a transient DNA loop. The interplay between polymerase dynamics and nucleosome fluctuations provides a physical basis for the regulation of eukaryotic transcription.
DOI:10.1038/nsmb.1658URLPMID:19684599 [本文引用: 1]
Chromatin structure influences transcription, but its role in subsequent RNA processing is unclear. Here we present analyses of high-throughput data that imply a relationship between nucleosome positioning and exon definition. First, we have found stable nucleosome occupancy within human and Caenorhabditis elegans exons that is stronger in exons with weak splice sites. Conversely, we have found that pseudoexons--intronic sequences that are not included in mRNAs but are flanked by strong splice sites--show nucleosome depletion. Second, the ratio between nucleosome occupancy within and upstream from the exons correlates with exon-inclusion levels. Third, nucleosomes are positioned central to exons rather than proximal to splice sites. These exonic nucleosomal patterns are also observed in non-expressed genes, suggesting that nucleosome marking of exons exists in the absence of transcription. Our analysis provides a framework that contributes to the understanding of splicing on the basis of chromatin architecture.
DOI:10.1038/nsmb.1659URLPMID:19684600 [本文引用: 1]
An increasing body of evidence indicates that transcription and splicing are coupled, and it is accepted that chromatin organization regulates transcription. Little is known about the cross-talk between chromatin structure and exon-intron architecture. By analysis of genome-wide nucleosome-positioning data sets from humans, flies and worms, we found that exons show increased nucleosome-occupancy levels with respect to introns, a finding that we link to differential GC content and nucleosome-disfavoring elements between exons and introns. Analysis of genome-wide chromatin immunoprecipitation data in humans and mice revealed four specific post-translational histone modifications enriched in exons. Our findings indicate that previously described enrichment of H3K36me3 modifications in exons reflects a more fundamental phenomenon, namely increased nucleosome occupancy along exons. Our results suggest an RNA polymerase II-mediated cross-talk between chromatin structure and exon-intron architecture, implying that exon selection may be modulated by chromatin structure.
DOI:10.1038/nature06929URLPMID:18408708 [本文引用: 1]
Comparative genomics of nucleosome positions provides a powerful means for understanding how the organization of chromatin and the transcription machinery co-evolve. Here we produce a high-resolution reference map of H2A.Z and bulk nucleosome locations across the genome of the fly Drosophila melanogaster and compare it to that from the yeast Saccharomyces cerevisiae. Like Saccharomyces, Drosophila nucleosomes are organized around active transcription start sites in a canonical -1, nucleosome-free region, +1 arrangement. However, Drosophila does not incorporate H2A.Z into the -1 nucleosome and does not bury its transcriptional start site in the +1 nucleosome. At thousands of genes, RNA polymerase II engages the +1 nucleosome and pauses. How the transcription initiation machinery contends with the +1 nucleosome seems to be fundamentally different across major eukaryotic lines.
DOI:10.1038/ng2117URLPMID:17873876 [本文引用: 1]
We present the first complete high-resolution map of nucleosome occupancy across the whole Saccharomyces cerevisiae genome, identifying over 70,000 positioned nucleosomes occupying 81% of the genome. On a genome-wide scale, the persistent nucleosome-depleted region identified previously in a subset of genes demarcates the transcription start site. Both nucleosome occupancy signatures and overall occupancy correlate with transcript abundance and transcription rate. In addition, functionally related genes can be clustered on the basis of the nucleosome occupancy patterns observed at their promoters. A quantitative model of nucleosome occupancy indicates that DNA structural features may account for much of the global nucleosome occupancy.
DOI:10.1093/nar/gkp116URLPMID:19264803 [本文引用: 1]
Nucleosome depletion at transcription start sites (TSS) has been documented genome-wide in multiple eukaryotic organisms. However, the mechanisms that mediate this nucleosome depletion and its functional impact on transcription remain largely unknown. We have studied these issues at human MHC class II (MHCII) genes. Activation-induced nucleosome free regions (NFR) encompassing the TSS were observed at all MHCII genes. Nucleosome depletion was exceptionally strong, attaining over 250-fold, at the promoter of the prototypical HLA-DRA gene. The NFR was induced primarily by the transcription factor complex that assembles on the conserved promoter-proximal enhancer situated upstream of the TSS. Functional analyses performed in the context of native chromatin demonstrated that displacing the NFR without altering the sequence of the core promoter induced a shift in the position of the TSS. The NFR thus appears to play a critical role in transcription initiation because it directs correct TSS positioning in vivo. Our results provide support for a novel mechanism in transcription initiation whereby the position of the TSS is controlled by nucleosome eviction rather than by promoter sequence.
DOI:10.1016/j.tig.2009.06.002URL [本文引用: 1]
The DNA of eukaryotic genomes is wrapped in nucleosomes, which strongly distort and occlude the DNA from access to most DNA-binding proteins. An understanding of the mechanisms that control nucleosome positioning along the DNA is thus essential to understanding the binding and action of proteins that carry out essential genetic functions. New genome-wide data on in vivo and in vitro nucleosome positioning greatly advance our understanding of several factors that can influence nucleosome positioning, including DNA sequence preferences, DNA methylation, histone variants and post-translational modifications, higher order chromatin structure, and the actions of transcription factors, chromatin remodelers and other DNA-binding proteins. We discuss how these factors function and ways in which they might be integrated into a unified framework that accounts for both the preservation of nucleosome positioning and the dynamic nucleosome repositioning that occur across biological conditions, cell types, developmental processes and disease.
DOI:10.1038/emboj.2010.71URLPMID:20407423 [本文引用: 1]
How are short exonic sequences recognized within the vast intronic oceans in which they reside? Despite decades of research, this remains one of the most fundamental, yet enigmatic, questions in the field of pre-mRNA splicing research. For many years, studies aiming to shed light on this process were focused at the RNA level, characterizing the manner by which splicing factors and auxiliary proteins interact with splicing signals, thereby enabling, facilitating and regulating splicing. However, we increasingly understand that splicing is not an isolated process; rather it occurs co-transcriptionally and is presumably also regulated by transcription-related processes. In fact, studies by our group and others over the past year suggest that DNA structure in terms of nucleosome positioning and specific histone modifications, which have a well established role in transcription, may also have a role in splicing. In this review we discuss evidence for the coupling between transcription and splicing, focusing on recent findings suggesting a link between chromatin structure and splicing, and highlighting challenges this emerging field is facing.
DOI:10.1016/j.molcel.2009.10.008URLPMID:19854133 [本文引用: 1]
Core RNA-processing reactions in eukaryotic cells occur cotranscriptionally in a chromatin context, but the relationship between chromatin structure and pre-mRNA processing is poorly understood. We observed strong nucleosome depletion around human polyadenylation sites (PAS) and nucleosome enrichment just downstream of PAS. In genes with multiple alternative PAS, higher downstream nucleosome affinity was associated with higher PAS usage, independently of known PAS motifs that function at the RNA level. Conversely, exons were associated with distinct peaks in nucleosome density. Exons flanked by long introns or weak splice sites exhibited stronger nucleosome enrichment, and incorporation of nucleosome density data improved splicing simulation accuracy. Certain histone modifications, including H3K36me3 and H3K27me2, were specifically enriched on exons, suggesting active marking of exon locations at the chromatin level. Together, these findings provide evidence for extensive functional connections between chromatin structure and RNA processing.
DOI:10.1126/science.1718039URLPMID:1718039 [本文引用: 1]
The relation between chromatin structure and transcriptional activity was examined by in vitro transcription analysis of chromatin reconstituted in the absence or presence of histone H1. To maintain well-defined template DNA, purified components were used in the reconstitution of chromatin. Reconstitution of nucleosomal cores to an average density of 1 nucleosome per 200 base pairs of DNA resulted in a mild reduction of basal RNA polymerase II transcription to 25 to 50 percent of that obtained with naked DNA templates. This nucleosome-mediated repression was due to nucleosomal cores located at the RNA start site and could not be counteracted by the sequence-specific transcription activators Sp1 and GAL4-VP16. When H1 was incorporated into the chromatin at 0.5 to 1.0 molecule per nucleosome (200 base pairs of DNA), RNA synthesis was reduced to 1 to 4 percent of that observed with chromatin containing only nucleosomal cores, and this H1-mediated repression could be counteracted by the addition of Sp1 or GAL4-VP16 (antirepression). With naked DNA templates, transcription was increased by a factor of 3 and 8 by Sp1 and GAL4-VP-16, respectively (true activation). With H1-repressed chromatin templates, however, the magnitude of transcriptional activation mediated by Sp1 and GAL4-VP16 was 90 and more than 200 times higher, respectively, because of the combined effects of true activation and antirepression. The data provide direct biochemical evidence that support and clarify previously proposed models in which there is depletion or reconfiguration of nucleosomal cores and histone H1 at the promoter regions of active genes.
DOI:10.1038/nrg2522URLPMID:19204718 [本文引用: 1]
Knowing the precise locations of nucleosomes in a genome is key to understanding how genes are regulated. Recent 'next generation' ChIP-chip and ChIP-Seq technologies have accelerated our understanding of the basic principles of chromatin organization. Here we discuss what high-resolution genome-wide maps of nucleosome positions have taught us about how nucleosome positioning demarcates promoter regions and transcriptional start sites, and how the composition and structure of promoter nucleosomes facilitate or inhibit transcription. A detailed picture is starting to emerge of how diverse factors, including underlying DNA sequences and chromatin remodelling complexes, influence nucleosome positioning.
DOI:10.1016/0092-8674(85)90025-xURLPMID:4075395 [本文引用: 1]
X-ray diffraction patterns have been obtained from partially oriented samples of 300A chromatin filaments. The chromatin was prepared by methods that preserve its structure, and conditions were found in which the 300A filaments spontaneously form ordered aggregates, so that it was not necessary to pull fibers. The diffraction patterns show a meridional band at 110A, and equatorial bands at 340, 57, 37, and 27A. These patterns, together with patterns calculated from the known 7A electron density map of the nucleosome core particle, imply side-to-side packing of nucleosomes in the direction of the 300A filament, and radial packing around it. These results are consistent with the "solenoid" model of Finch and Klug, and are inconsistent with many other proposed models.
.
DOI:10.1083/jcb.99.1.42URLPMID:6736132 [本文引用: 1]
Both intact and nuclease-isolated chromatin fibers have been examined at different degrees of salt-induced compaction, using a variety of preparation techniques. The results suggest that the initial folding step in nucleosome packing involves the formation of a zig-zag ribbon as has been proposed by others (Thoma F., T. Koller, and A. Klug, 1979, J. Cell Biol., 83:403-427; Worcel A., S. Strogartz, and D. Riley, 1981, Proc. Natl. Acad. Sci. USA, 78:1461-1465), and that subsequent compaction occurs by coiling of the ribbon to form a double helical structure. This type of folding generates a fiber in which the nucleosome-nucleosome contacts established in the zig-zag ribbon are maintained and in which the histone H1 molecules occupy equivalent sites. The diameter of the fiber is not dependent upon the nucleosome repeat length. Direct mass values for individual isolated fibers obtained from electron scattering measurements showed that the mass per length was dependent on ionic strength, and ranged from 6.0 X 10(4) daltons/nm at 10 mM NaCl to 27 X 10(4) daltons/nm at 150 mM salt. These values are equivalent to 2.5 nucleosomes/11 nm at 10 mM NaCl and to 11.6 nucleosomes/11 nm at 150 mM salt and are consistent with the range of packing ratios for the proposed helical ribbon.
DOI:10.1126/science.1103124URLPMID:15567867 [本文引用: 1]
Chromatin folding determines the accessibility of DNA constituting eukaryotic genomes and consequently is profoundly important in the mechanisms of nuclear processes such as gene regulation. Nucleosome arrays compact to form a 30-nanometer chromatin fiber of hitherto disputed structure. Two competing classes of models have been proposed in which nucleosomes are either arranged linearly in a one-start higher order helix or zigzag back and forth in a two-start helix. We analyzed compacted nucleosome arrays stabilized by introduction of disulfide cross-links and show that the chromatin fiber comprises two stacks of nucleosomes in accord with the two-start model.
DOI:10.1126/science.1251413URLPMID:24763583 [本文引用: 1]
The hierarchical packaging of eukaryotic chromatin plays a central role in transcriptional regulation and other DNA-related biological processes. Here, we report the 11-angstrom-resolution cryogenic electron microscopy (cryo-EM) structures of 30-nanometer chromatin fibers reconstituted in the presence of linker histone H1 and with different nucleosome repeat lengths. The structures show a histone H1-dependent left-handed twist of the repeating tetranucleosomal structural units, within which the four nucleosomes zigzag back and forth with a straight linker DNA. The asymmetric binding and the location of histone H1 in chromatin play a role in the formation of the 30-nanometer fiber. Our results provide mechanistic insights into how nucleosomes compact into higher-order chromatin fibers.
DOI:10.1016/j.molcel.2016.08.024URLPMID:27666592 [本文引用: 2]
In eukaryotes, the packaging of genomic DNA into chromatin plays a critical role in gene regulation. However, the dynamic organization of chromatin fibers and its regulatory mechanisms remain poorly understood. Using single-molecule force spectroscopy, we reveal that the tetranucleosomes-on-a-string appears as a stable secondary structure during hierarchical organization of chromatin fibers. The stability of the tetranucleosomal unit is attenuated by histone chaperone FACT (facilitates chromatin transcription) in?vitro. Consistent with in?vitro observations, our genome-wide analysis further shows that FACT facilitates gene transcription by destabilizing the tetranucleosomal unit of chromatin fibers in yeast. Additionally, we found that the linker histone H1 not only enhances the stability but also facilitates the folding and unfolding kinetics of the outer nucleosomal wrap. Our study demonstrates that the tetranucleosome is a regulatory structural unit of chromatin fibers beyond the nucleosome and provides crucial mechanistic insights into the structure and dynamics of chromatin fibers during gene transcription.
DOI:10.1016/j.cell.2018.12.014URLPMID:30661750 [本文引用: 2]
Elucidating the global and local rules that govern genome-wide, hierarchical chromatin architecture remains a critical challenge. Current high-throughput chromosome conformation capture (Hi-C) technologies have identified large-scale chromatin structural motifs, such as topologically associating domains and looping. However, structural rules at the smallest or nucleosome scale remain poorly understood. Here, we coupled nucleosome-resolved Hi-C technology with simulated annealing-molecular dynamics (SA-MD) simulation to reveal 3D spatial distributions of nucleosomes and their genome-wide orientation in chromatin. Our method, called Hi-CO, revealed distinct nucleosome folding motifs across the yeast genome. Our results uncovered two types of basic secondary structural motifs in nucleosome folding: α-tetrahedron and β-rhombus analogous to α helix and β sheet motifs in protein folding. Using mutants and cell-cycle-synchronized cells, we further uncovered motifs with specific nucleosome positioning and orientation coupled to epigenetic features at individual loci. By illuminating molecular-level structure-function relationships in eukaryotic chromatin, our findings establish organizational principles of nucleosome folding.
[本文引用: 1]
DOI:10.1101/cshperspect.a003889URLPMID:20300217 [本文引用: 1]
Chromosome territories (CTs) constitute a major feature of nuclear architecture. In a brief statement, the possible contribution of nuclear architecture studies to the field of epigenomics is considered, followed by a historical account of the CT concept and the final compelling experimental evidence of a territorial organization of chromosomes in all eukaryotes studied to date. Present knowledge of nonrandom CT arrangements, of the internal CT architecture, and of structural interactions with other CTs is provided as well as the dynamics of CT arrangements during cell cycle and postmitotic terminal differentiation. The article concludes with a discussion of open questions and new experimental strategies to answer them.
DOI:10.1038/nature03001URLPMID:15496913 [本文引用: 1]
The sequence of the human genome encodes the genetic instructions for human physiology, as well as rich information about human evolution. In 2001, the International Human Genome Sequencing Consortium reported a draft sequence of the euchromatic portion of the human genome. Since then, the international collaboration has worked to convert this draft into a genome sequence with high accuracy and nearly complete coverage. Here, we report the result of this finishing process. The current genome sequence (Build 35) contains 2.85 billion nucleotides interrupted by only 341 gaps. It covers approximately 99% of the euchromatic genome and is accurate to an error rate of approximately 1 event per 100,000 bases. Many of the remaining euchromatic gaps are associated with segmental duplications and will require focused work with new methods. The near-complete sequence, the first for a vertebrate, greatly improves the precision of biological analyses of the human genome including studies of gene number, birth and death. Notably, the human genome seems to encode only 20,000-25,000 protein-coding genes. The genome sequence reported here should serve as a firm foundation for biomedical research in the decades ahead.
DOI:10.1126/science.1105136URLPMID:15499007 [本文引用: 1]
The ENCyclopedia Of DNA Elements (ENCODE) Project aims to identify all functional elements in the human genome sequence. The pilot phase of the Project is focused on a specified 30 megabases (approximately 1%) of the human genome sequence and is organized as an international consortium of computational and laboratory-based scientists working to develop and apply high-throughput approaches for detecting all sequence elements that confer biological function. The results of this pilot phase will guide future efforts to analyze the entire human genome.
DOI:10.1126/science.1067799URLPMID:11847345 [本文引用: 1]
We describe an approach to detect the frequency of interaction between any two genomic loci. Generation of a matrix of interaction frequencies between sites on the same or different chromosomes reveals their relative spatial disposition and provides information about the physical properties of the chromatin fiber. This methodology can be applied to the spatial organization of entire genomes in organisms from bacteria to human. Using the yeast Saccharomyces cerevisiae, we could confirm known qualitative features of chromosome organization within the nucleus and dynamic changes in that organization during meiosis. We also analyzed yeast chromosome III at the G1 stage of the cell cycle. We found that chromatin is highly flexible throughout. Furthermore, functionally distinct AT- and GC-rich domains were found to exhibit different conformations, and a population-average 3D model of chromosome III could be determined. Chromosome III emerges as a contorted ring.
DOI:10.1038/ng1896URLPMID:17033623 [本文引用: 1]
The spatial organization of DNA in the cell nucleus is an emerging key contributor to genomic function. We developed 4C technology (chromosome conformation capture (3C)-on-chip), which allows for an unbiased genome-wide search for DNA loci that contact a given locus in the nuclear space. We demonstrate here that active and inactive genes are engaged in many long-range intrachromosomal interactions and can also form interchromosomal contacts. The active beta-globin locus in fetal liver preferentially contacts transcribed, but not necessarily tissue-specific, loci elsewhere on chromosome 7, whereas the inactive locus in fetal brain contacts different transcriptionally silent loci. A housekeeping gene in a gene-dense region on chromosome 8 forms long-range contacts predominantly with other active gene clusters, both in cis and in trans, and many of these intra- and interchromosomal interactions are conserved between the tissues analyzed. Our data demonstrate that chromosomes fold into areas of active chromatin and areas of inactive chromatin and establish 4C technology as a powerful tool to study nuclear architecture.
DOI:10.1038/ng1891URLPMID:17033624 [本文引用: 1]
Accumulating evidence converges on the possibility that chromosomes interact with each other to regulate transcription in trans. To systematically explore the epigenetic dimension of such interactions, we devised a strategy termed circular chromosome conformation capture (4C). This approach involves a circularization step that enables high-throughput screening of physical interactions between chromosomes without a preconceived idea of the interacting partners. Here we identify 114 unique sequences from all autosomes, several of which interact primarily with the maternally inherited H19 imprinting control region. Imprinted domains were strongly overrepresented in the library of 4C sequences, further highlighting the epigenetic nature of these interactions. Moreover, we found that the direct interaction between differentially methylated regions was linked to epigenetic regulation of transcription in trans. Finally, the patterns of interactions specific to the maternal H19 imprinting control region underwent reprogramming during in vitro maturation of embryonic stem cells. These observations shed new light on development, cancer epigenetics and the evolution of imprinting.
DOI:10.1101/gr.5571506URLPMID:16954542 [本文引用: 1]
Physical interactions between genetic elements located throughout the genome play important roles in gene regulation and can be identified with the Chromosome Conformation Capture (3C) methodology. 3C converts physical chromatin interactions into specific ligation products, which are quantified individually by PCR. Here we present a high-throughput 3C approach, 3C-Carbon Copy (5C), that employs microarrays or quantitative DNA sequencing using 454-technology as detection methods. We applied 5C to analyze a 400-kb region containing the human beta-globin locus and a 100-kb conserved gene desert region. We validated 5C by detection of several previously identified looping interactions in the beta-globin locus. We also identified a new looping interaction in K562 cells between the beta-globin Locus Control Region and the gamma-beta-globin intergenic region. Interestingly, this region has been implicated in the control of developmental globin gene switching. 5C should be widely applicable for large-scale mapping of cis- and trans- interaction networks of genomic elements and for the study of higher-order chromosome structure.
DOI:10.1038/nature08497URLPMID:19890323 [本文引用: 1]
Genomes are organized into high-level three-dimensional structures, and DNA elements separated by long genomic distances can in principle interact functionally. Many transcription factors bind to regulatory DNA elements distant from gene promoters. Although distal binding sites have been shown to regulate transcription by long-range chromatin interactions at a few loci, chromatin interactions and their impact on transcription regulation have not been investigated in a genome-wide manner. Here we describe the development of a new strategy, chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) for the de novo detection of global chromatin interactions, with which we have comprehensively mapped the chromatin interaction network bound by oestrogen receptor alpha (ER-alpha) in the human genome. We found that most high-confidence remote ER-alpha-binding sites are anchored at gene promoters through long-range chromatin interactions, suggesting that ER-alpha functions by extensive chromatin looping to bring genes together for coordinated transcriptional regulation. We propose that chromatin interactions constitute a primary mechanism for regulating transcription in mammalian genomes.
DOI:10.1038/nmeth.3205URLPMID:25437436 [本文引用: 1]
High-throughput methods based on chromosome conformation capture have greatly advanced our understanding of the three-dimensional (3D) organization of genomes but are limited in resolution by their reliance on restriction enzymes. Here we describe a method called DNase Hi-C for comprehensively mapping global chromatin contacts. DNase Hi-C uses DNase I for chromatin fragmentation, leading to greatly improved efficiency and resolution over that of Hi-C. Coupling this method with DNA-capture technology provides a high-throughput approach for targeted mapping of fine-scale chromatin architecture. We applied targeted DNase Hi-C to characterize the 3D organization of 998 large intergenic noncoding RNA (lincRNA) promoters in two human cell lines. Our results revealed that expression of lincRNAs is tightly controlled by complex mechanisms involving both super-enhancers and the Polycomb repressive complex. Our results provide the first glimpse of the cell type-specific 3D organization of lincRNA genes.
DOI:10.1038/ng.496URLPMID:20010836 [本文引用: 1]
The discovery of interchromosomal interactions in higher eukaryotes points to a functional interplay between genome architecture and gene expression, challenging the view of transcription as a one-dimensional process. However, the extent of interchromosomal interactions and the underlying mechanisms are unknown. Here we present the first genome-wide analysis of transcriptional interactions using the mouse globin genes in erythroid tissues. Our results show that the active globin genes associate with hundreds of other transcribed genes, revealing extensive and preferential intra- and interchromosomal transcription interactomes. We show that the transcription factor Klf1 mediates preferential co-associations of Klf1-regulated genes at a limited number of specialized transcription factories. Our results establish a new gene expression paradigm, implying that active co-regulated genes and their regulatory factors cooperate to create specialized nuclear hot spots optimized for efficient and coordinated transcriptional control.
DOI:10.1038/nature12593URL [本文引用: 2]
Large-scale chromosome structure and spatial nuclear arrangement have been linked to control of gene expression and DNA replication and repair. Genomic techniques based on chromosome conformation capture (3C) assess contacts for millions of loci simultaneously, but do so by averaging chromosome conformations from millions of nuclei. Here we introduce single-cell Hi-C, combined with genome-wide statistical analysis and structural modelling of single-copy X chromosomes, to show that individual chromosomes maintain domain organization at the megabase scale, but show variable cell-to-cell chromosome structures at larger scales. Despite this structural stochasticity, localization of active gene domains to boundaries of chromosome territories is a hallmark of chromosomal conformation. Single-cell Hi-C data bridge current gaps between genomics and microscopy studies of chromosomes, demonstrating how modular organization underlies dynamic chromosome structure, and how this structure is probabilistically linked with genome activity patterns.
DOI:10.1038/s41588-018-0111-2URLPMID:29700467 [本文引用: 1]
Chromosome conformation capture (3C) technologies can be used to investigate 3D genomic structures. However, high background noise, high costs, and a lack of straightforward noise evaluation in current methods impede the advancement of 3D genomic research. Here we developed a simple digestion-ligation-only Hi-C (DLO Hi-C) technology to explore the 3D landscape of the genome. This method requires only two rounds of digestion and ligation, without the need for biotin labeling and pulldown. Non-ligated DNA was efficiently removed in a cost-effective step by purifying specific linker-ligated DNA fragments. Notably, random ligation could be quickly evaluated in an early quality-control step before sequencing. Moreover, an in situ version of DLO Hi-C using a four-cutter restriction enzyme has been developed. We applied DLO Hi-C to delineate the genomic architecture of THP-1 and K562 cells and uncovered chromosomal translocations. This technology may facilitate investigation of genomic organization, gene regulation, and (meta)genome assembly.
DOI:10.1016/j.yexcr.2009.02.016URLPMID:19254712 [本文引用: 1]
Gene-dense chromosome territories (CTs) are typically located more interior, gene-poor CTs more peripheral in mammalian cell nuclei. Here, we show that this gene-density correlated CT positioning holds for the most gene-rich and gene-poor bovine chromosomes 19 and 20, respectively, in bovine fibroblast and lymphocyte nuclei. In order to determine the period at which this non-random CT order is established during development, we performed fluorescence in situ hybridization, on three-dimensionally preserved bovine preimplantation embryos generated by in vitro fertilization and investigated the distribution of BTA 19 and 20 CTs. Radial arrangements of CTs 19 and 20 were the same up to the 8-cell stage. At the 10- to 16-cell stage, however, a significant difference became apparent with CTs 19 localized more internally and CTs 20 more peripherally. Since major genome activation in bovine embryos occurs at the 8- to 16-cell stage, our findings demonstrate a temporal correlation between transcriptional activation and a major rearrangement of chromatin topography in blastomere nuclei.
DOI:10.1007/978-1-59745-406-3_15URLPMID:18951171 [本文引用: 1]
Fluorescence in situ hybridization (FISH) of specific DNA probes has become a widely used technique mostly for chromosome analysis and for studies of the chromosomal location of specific DNA segments in metaphase preparations as well as in interphase nuclei. FISH on 3D-preserved nuclei (3D-FISH) in combination with 3D-microscopy and image reconstruction is an efficient tool to analyze the spatial arrangement of targeted DNA sequences in the nucleus. Recent developments of a "new generation" of confocal microscopes that allow the distinct visualization of at least five different fluorochromes within one experiment opened the way for multicolor 3D-FISH experiments. Thus, numerous differently labeled nuclear targets can be delineated simultaneously and their spatial interrelationships can be analyzed on the level of individual nuclei.In this chapter, we provide protocols for the preparation of complex DNA-probe sets suitable for 3D-FISH with up to six different fluorochromes, for 3D-FISH on cultured mammalian cells (growing in suspension or adherently) as well as on tissue sections, and for 3D immuno-FISH.In comparison with FISH on metaphase chromosomes and conventional interphase cytogenetics, FISH on 3D-preserved nuclei requires special demands with regard to probe quality, fixation, and pretreatment steps of cells in order to achieve the two goals, namely the best possible preservation of the nuclear structure and at the same time an efficient probe accessibility.
DOI:10.1016/j.cell.2008.09.026URLPMID:18854147 [本文引用: 1]
There is no doubt that genomes are organized nonrandomly in the nucleus of higher eukaryotes. But what is the functional relevance of this nonrandomness? In this Essay, we explore the biological meaning of spatial gene positioning by examining the functional link between the activity of a gene and its radial position in the nucleus.
DOI:10.1016/j.cell.2009.01.052URLPMID:19379699 [本文引用: 1]
We show that the nuclear architecture of rod photoreceptor cells differs fundamentally in nocturnal and diurnal mammals. The rods of diurnal retinas possess the conventional architecture found in nearly all eukaryotic cells, with most heterochromatin situated at the nuclear periphery and euchromatin residing toward the nuclear interior. The rods of nocturnal retinas have a unique inverted pattern, where heterochromatin localizes in the nuclear center, whereas euchromatin, as well as nascent transcripts and splicing machinery, line the nuclear border. The inverted pattern forms by remodeling of the conventional one during terminal differentiation of rods. The inverted rod nuclei act as collecting lenses, and computer simulations indicate that columns of such nuclei channel light efficiently toward the light-sensing rod outer segments. Comparison of the two patterns suggests that the conventional architecture prevails in eukaryotic nuclei because it results in more flexible chromosome arrangements, facilitating positional regulation of nuclear functions.
DOI:10.1101/gr.099655.109URLPMID:20430782 [本文引用: 1]
To identify evolutionarily conserved features of replication timing and their relationship to epigenetic properties, we profiled replication timing genome-wide in four human embryonic stem cell (hESC) lines, hESC-derived neural precursor cells (NPCs), lymphoblastoid cells, and two human induced pluripotent stem cell lines (hiPSCs), and compared them with related mouse cell types. Results confirm the conservation of coordinately replicated megabase-sized "replication domains" punctuated by origin-suppressed regions. Differentiation-induced replication timing changes in both species occur in 400- to 800-kb units and are similarly coordinated with transcription changes. A surprising degree of cell-type-specific conservation in replication timing was observed across regions of conserved synteny, despite considerable species variation in the alignment of replication timing to isochore GC/LINE-1 content. Notably, hESC replication timing profiles were significantly more aligned to mouse epiblast-derived stem cells (mEpiSCs) than to mouse ESCs. Comparison with epigenetic marks revealed a signature of chromatin modifications at the boundaries of early replicating domains and a remarkably strong link between replication timing and spatial proximity of chromatin as measured by Hi-C analysis. Thus, early and late initiation of replication occurs in spatially separate nuclear compartments, but rarely within the intervening chromatin. Moreover, cell-type-specific conservation of the replication program implies conserved developmental changes in spatial organization of chromatin. Together, our results reveal evolutionarily conserved aspects of developmentally regulated replication programs in mammals, demonstrate the power of replication profiling to distinguish closely related cell types, and strongly support the hypothesis that replication timing domains are spatially compartmentalized structural and functional units of three-dimensional chromosomal architecture.
DOI:10.1038/nature14222URLPMID:25693564 [本文引用: 3]
Higher-order chromatin structure is emerging as an important regulator of gene expression. Although dynamic chromatin structures have been identified in the genome, the full scope of chromatin dynamics during mammalian development and lineage specification remains to be determined. By mapping genome-wide chromatin interactions in human embryonic stem (ES) cells and four human ES-cell-derived lineages, we uncover extensive chromatin reorganization during lineage specification. We observe that although self-associating chromatin domains are stable during differentiation, chromatin interactions both within and between domains change in a striking manner, altering 36% of active and inactive chromosomal compartments throughout the genome. By integrating chromatin interaction maps with haplotype-resolved epigenome and transcriptome data sets, we find widespread allelic bias in gene expression correlated with allele-biased chromatin states of linked promoters and distal enhancers. Our results therefore provide a global view of chromatin dynamics and a resource for studying long-range control of gene expression in distinct human cell lineages.
DOI:10.1038/s41588-019-0474-zURLPMID:31406346 [本文引用: 1]
In mammalian cells, chromosomes are partitioned into megabase-sized topologically associating domains (TADs). TADs can be in either A (active) or B (inactive) subnuclear compartments, which exhibit early and late replication timing (RT), respectively. Here, we show that A/B compartments change coordinately with RT changes genome wide during mouse embryonic stem cell (mESC) differentiation. While A to B compartment changes and early to late RT changes were temporally inseparable, B to A changes clearly preceded late to early RT changes and transcriptional activation. Compartments changed primarily by boundary shifting, altering the compartmentalization of TADs facing the A/B compartment interface, which was conserved during reprogramming and confirmed in individual cells by single-cell Repli-seq. Differentiating mESCs altered single-cell Repli-seq profiles gradually but uniformly, transiently resembling RT profiles of epiblast-derived stem cells (EpiSCs), suggesting that A/B compartments might also change gradually but uniformly toward a primed pluripotent state. These results provide insights into how megabase-scale chromosome organization changes in individual cells during differentiation.
DOI:10.1016/j.febslet.2015.08.044URLPMID:26348399 [本文引用: 1]
Recent studies have shown that chromosomes in a range of organisms are compartmentalized in different types of chromatin domains. In mammals, chromosomes form compartments that are composed of smaller Topologically Associating Domains (TADs). TADs are thought to represent functional domains of gene regulation but much is still unknown about the mechanisms of their formation and how they exert their regulatory effect on embedded genes. Further, similar domains have been detected in other organisms, including flies, worms, fungi and bacteria. Although in all these cases these domains appear similar as detected by 3C-based methods, their biology appears to be quite distinct with differences in the protein complexes involved in their formation and differences in their internal organization. Here we outline our current understanding of such domains in different organisms and their roles in gene regulation.
DOI:10.1016/j.molcel.2016.05.018URLPMID:27259200 [本文引用: 1]
How eukaryotic chromosomes fold inside the nucleus is an age-old question that remains unanswered today. Early biochemical and microscopic studies revealed the existence of chromatin domains and loops as a pervasive feature of interphase chromosomes, but the biological implications of such organizational features were obscure. Genome-wide analysis of pair-wise chromatin interactions using chromatin conformation capture (3C)-based techniques has shed new light on the organization of chromosomes in interphase nuclei. Particularly, the finding of cell-type invariant, evolutionarily conserved topologically associating domains (TADs) in a broad spectrum of cell types has provided a new molecular framework for the study of animal development and human diseases. Here, we review recent progress in characterization of such chromatin domains and delineation of mechanisms of their formation in animal cells.
DOI:10.1186/s13059-015-0642-0URLPMID:25886366 [本文引用: 1]
The three-dimensional organization of the genome is tightly connected to its biological function. The Hi-C approach was recently introduced as a method that can be used to identify higher-order chromatin interactions genome-wide. The aim of this study was to determine genome-wide chromatin interaction frequencies using the Hi-C approach in mouse sperm cells and embryonic fibroblasts.
DOI:10.1038/nature13986URL [本文引用: 1]
Eukaryotic chromosomes replicate in a temporal order known as the replication-timing program(1). In mammals, replication timing is cell-type-specific with at least half the genome switching replication timing during development, primarily in units of 400-800 kilobases (replication domains'), whose positions are preserved in different cell types, conserved between species, and appear to confine long-range effects of chromosome rearrangements(2-7). Early and late replication correlate, respectively, with open and closed three-dimensional chromatin compartments identified by high-resolution chromosome conformation capture (Hi-C), and, to a lesser extent, late replication correlates with lamina-associated domains (LADs)(4,5,8,9). Recent Hi-C mapping has unveiled substructure within chromatin compartments called topologically associating domains (TADs) that are largely conserved in their positions between cell types and are similar in size to replication domains(8,10). However, TADs can be further sub-stratified into smaller domains, challenging the significance of structures at any particular scale(11,12). Moreover, attempts to reconcile TADs and LADs to replication-timing data have not revealed a common, underlying domain structure(8,9,13). Here we localize boundaries of replication domains to the early-replicating border of replication-timing transitions and map their positions in 18 human and 13 mouse cell types. We demonstrate that, collectively, replication domain boundaries share a near one-to-one correlation with TAD boundaries, whereas within a cell type, adjacent TADs that replicate at similar times obscure replication domain boundaries, largely accounting for the previously reported lack of alignment. Moreover, cell-type-specific replication timing of TADs partitions the genome into two large-scale sub-nuclear compartments revealing that replication-timing transitions are indistinguishable from late-replicating regions in chromatin composition and lamina association and accounting for the reduced correlation of replication timing to LADs and heterochromatin. Our results reconcile cell-type-specific sub-nuclear compartmentalization and replication timing with developmentally stable structural domains and offer a unified model for large-scale chromosome structure and function.
DOI:10.1016/j.cell.2017.06.029URLPMID:28709003 [本文引用: 1]
High-order chromatin structure plays important roles in gene expression regulation. Knowledge of the dynamics of 3D chromatin structures during mammalian embryo development remains limited. We report the 3D chromatin architecture of mouse gametes and early embryos using an optimized Hi-C method with low-cell samples. We find that mature oocytes at the metaphase II stage do not have topologically associated domains (TADs). In sperm, extra-long-range interactions (>4 Mb) and interchromosomal interactions occur frequently. The high-order structures of both the paternal and maternal genomes in zygotes and two-cell embryos are obscure but are gradually re-established through development. The establishment of the TAD structure requires DNA replication but not zygotic genome activation. Furthermore, unmethylated CpGs are enriched in A compartment, and methylation levels are decreased to a greater extent in A compartment than in B compartment in embryos. In summary, the global reprogramming of chromatin architecture occurs during early mammalian development.
DOI:10.1016/j.cell.2018.05.007URLPMID:29887375 [本文引用: 1]
Mammalian chromosomes are partitioned into A/B compartments and topologically associated domains (TADs). The inactive X (Xi) chromosome, however, adopts a distinct conformation without evident compartments or TADs. Here, through exploration of an architectural protein, structural-maintenance-of-chromosomes hinge domain containing 1 (SMCHD1), we probe how the Xi is reconfigured during X?chromosome inactivation. A/B compartments are first?fused into "S1" and "S2" compartments, coinciding with Xist spreading into gene-rich domains. SMCHD1 then binds S1/S2 compartments and merges them to create a compartment-less architecture. Contrary to current views, TADs remain on?the?Xi but in an attenuated state. Ablating SMCHD1 results in a persistent S1/S2 organization and?strengthening of TADs. Furthermore, loss of SMCHD1 causes regional defects in Xist spreading and erosion of heterochromatic silencing. We present a stepwise model for Xi folding, where SMCHD1 attenuates a?hidden layer of Xi architecture to facilitate Xist spreading.
DOI:10.1038/nature18589URLPMID:27437574 [本文引用: 1]
X-chromosome inactivation (XCI) involves major reorganization of the X chromosome as it becomes silent and heterochromatic. During female mammalian development, XCI is triggered by upregulation of the non-coding Xist RNA from one of the two X chromosomes. Xist coats the chromosome in cis and induces silencing of almost all genes via its A-repeat region, although some genes (constitutive escapees) avoid silencing in most cell types, and others (facultative escapees) escape XCI only in specific contexts. A role for Xist in organizing the inactive X (Xi) chromosome has been proposed. Recent chromosome conformation capture approaches have revealed global loss of local structure on the Xi chromosome and formation of large mega-domains, separated by a region containing the DXZ4 macrosatellite. However, the molecular architecture of the Xi chromosome, in both the silent and expressed regions,remains unclear. Here we investigate the structure, chromatin accessibility and expression status of the mouse Xi chromosome in highly polymorphic clonal neural progenitors (NPCs) and embryonic stem cells. We demonstrate a crucial role for Xist and the DXZ4-containing boundary in shaping Xi chromosome structure using allele-specific genome-wide chromosome conformation capture (Hi-C) analysis, an assay for transposase-accessible chromatin with high throughput sequencing (ATAC-seq) and RNA sequencing. Deletion of the boundary disrupts mega-domain formation, and induction of Xist RNA initiates formation of the boundary and the loss of DNA accessibility. We also show that in NPCs, the Xi chromosome lacks active/inactive compartments and topologically associating domains (TADs), except around genes that escape XCI. Escapee gene clusters display TAD-like structures and retain DNA accessibility at promoter-proximal and CTCF-binding sites. Furthermore, altered patterns of facultative escape genes indifferent neural progenitor clones are associated with the presence of different TAD-like structures after XCI. These findings suggest a key role for transcription and CTCF in the formation of TADs in the context of the Xi chromosome in neural progenitors.
DOI:10.1016/j.cell.2015.04.004URLPMID:25959774 [本文引用: 2]
Mammalian genomes are organized into megabase-scale topologically associated domains (TADs). We demonstrate that disruption of TADs can rewire long-range regulatory architecture and result in pathogenic phenotypes. We show that distinct human limb malformations are caused by deletions, inversions, or duplications altering the structure of the TAD-spanning WNT6/IHH/EPHA4/PAX3 locus. Using CRISPR/Cas genome editing, we generated mice with corresponding rearrangements. Both in mouse limb tissue and patient-derived fibroblasts, disease-relevant structural changes cause ectopic interactions between promoters and non-coding DNA, and a cluster of limb enhancers normally associated with Epha4 is misplaced relative to TAD boundaries and drives ectopic limb expression of another gene in the locus. This rewiring occurred only if the variant disrupted a CTCF-associated boundary domain. Our results demonstrate the functional importance of TADs for orchestrating gene expression via genome architecture and indicate criteria for predicting the pathogenicity of human structural variants, particularly in non-coding regions of the human genome.
DOI:10.1093/bioinformatics/btu747URLPMID:25391400 [本文引用: 1]
Genome-wide proximity ligation assays, e.g. Hi-C and its variant TCC, have recently become important tools to study spatial genome organization. Removing biases from chromatin contact matrices generated by such techniques is a critical preprocessing step of subsequent analyses. The continuing decline of sequencing costs has led to an ever-improving resolution of the Hi-C data, resulting in very large matrices of chromatin contacts. Such large-size matrices, however, pose a great challenge on the memory usage and speed of its normalization. Therefore, there is an urgent need for fast and memory-efficient methods for normalization of Hi-C data. We developed Hi-Corrector, an easy-to-use, open source implementation of the Hi-C data normalization algorithm. Its salient features are (i) scalability-the software is capable of normalizing Hi-C data of any size in reasonable times; (ii) memory efficiency-the sequential version can run on any single computer with very limited memory, no matter how little; (iii) fast speed-the parallel version can run very fast on multiple computing nodes with limited local memory.
DOI:10.1016/j.tig.2016.01.003URLPMID:26862051 [本文引用: 1]
Spatial organization is an inherent property of the vertebrate genome to accommodate the roughly 2m of DNA in the nucleus of a cell. In this nonrandom organization, topologically associating domains (TADs) emerge as a fundamental structural unit that is thought to guide regulatory elements to their cognate promoters. In this review we summarize the most recent findings about TADs and the boundary regions separating them. We discuss how the disruption of these structures by genomic rearrangements can result in gene misexpression and disease.
DOI:10.1126/science.aad9024URLPMID:26940867 [本文引用: 1]
Oncogenes are activated through well-known chromosomal alterations such as gene fusion, translocation, and focal amplification. In light of recent evidence that the control of key genes depends on chromosome structures called insulated neighborhoods, we investigated whether proto-oncogenes occur within these structures and whether oncogene activation can occur via disruption of insulated neighborhood boundaries in cancer cells. We mapped insulated neighborhoods in T cell acute lymphoblastic leukemia (T-ALL) and found that tumor cell genomes contain recurrent microdeletions that eliminate the boundary sites of insulated neighborhoods containing prominent T-ALL proto-oncogenes. Perturbation of such boundaries in nonmalignant cells was sufficient to activate proto-oncogenes. Mutations affecting chromosome neighborhood boundaries were found in many types of cancer. Thus, oncogene activation can occur via genetic alterations that disrupt insulated neighborhoods in malignant cells.
DOI:10.1101/gr.212803.116URLPMID:28057745 [本文引用: 1]
Understanding how regulatory sequences interact in the context of chromosomal architecture is a central challenge in biology. Chromosome conformation capture revealed that mammalian chromosomes possess a rich hierarchy of structural layers, from multi-megabase compartments to sub-megabase topologically associating domains (TADs) and sub-TAD contact domains. TADs appear to act as regulatory microenvironments by constraining and segregating regulatory interactions across discrete chromosomal regions. However, it is unclear whether other (or all) folding layers share similar properties, or rather TADs constitute a privileged folding scale with maximal impact on the organization of regulatory interactions. Here, we present a novel algorithm named CaTCH that identifies hierarchical trees of chromosomal domains in Hi-C maps, stratified through their reciprocal physical insulation, which is a single and biologically relevant parameter. By applying CaTCH to published Hi-C data sets, we show that previously reported folding layers appear at different insulation levels. We demonstrate that although no structurally privileged folding level exists, TADs emerge as a functionally privileged scale defined by maximal boundary enrichment in CTCF and maximal cell-type conservation. By measuring transcriptional output in embryonic stem cells and neural precursor cells, we show that the likelihood that genes in a domain are coregulated during differentiation is also maximized at the scale of TADs. Finally, we observe that regulatory sequences occur at genomic locations corresponding to optimized mutual interactions at the same scale. Our analysis suggests that the architectural functionality of TADs arises from the interplay between their ability to partition interactions and the specific genomic position of regulatory sequences.
DOI:10.1101/gr.163519.113URL [本文引用: 1]
Long-range regulatory interactions play an important role in shaping gene-expression programs. However, the genomic features that organize these activities are still poorly characterized. We conducted a large operational analysis to chart the distribution of gene regulatory activities along the mouse genome, using hundreds of insertions of a regulatory sensor. We found that enhancers distribute their activities along broad regions and not in a gene-centric manner, defining large regulatory domains. Remarkably, these domains correlate strongly with the recently described TADs, which partition the genome into distinct self-interacting blocks. Different features, including specific repeats and CTCF-binding sites, correlate with the transition zones separating regulatory domains, and may help to further organize promiscuously distributed regulatory influences within large domains. These findings support a model of genomic organization where TADs confine regulatory activities to specific but large regulatory domains, contributing to the establishment of specific gene expression profiles.
DOI:10.1038/nature11243URL [本文引用: 1]
The laboratory mouse is the most widely used mammalian model organism in biomedical research. The 2.6 x 10(9) bases of the mouse genome possess a high degree of conservation with the human genome(1), so a thorough annotation of the mouse genome will be of significant value to understanding the function of the human genome. So far, most of the functional sequences in the mouse genome have yet to be found, and the cis-regulatory sequences in particular are still poorly annotated. Comparative genomics has been a powerful tool for the discovery of these sequences(2), but on its own it cannot resolve their temporal and spatial functions. Recently, ChIP-Seq has been developed to identify cis-regulatory elements in the genomes of several organisms including humans, Drosophila melanogaster and Caenorhabditis elegans(3-5). Here we apply the same experimental approach to a diverse set of 19 tissues and cell types in the mouse to produce a map of nearly 300,000 murine cis-regulatory sequences. The annotated sequences add up to 11% of the mouse;[GRAPHICS];genome, and include more than 70% of conserved non-coding sequences. We define tissue-specific enhancers and identify potential transcription factors regulating gene expression in each tissue or cell type. Finally, we show that much of the mouse genome is organized into domains of coordinately regulated enhancers and promoters. Our results provide a resource for the annotation of functional elements in the mammalian genome and for the study of mechanisms regulating tissue-specific gene expression.
DOI:10.1016/j.cell.2015.07.048URLPMID:26300125 [本文引用: 1]
Deciphering the impact of genetic variants on gene regulation is fundamental to understanding human disease. Although gene regulation often involves long-range interactions, it is unknown to what extent non-coding genetic variants influence distal molecular phenotypes. Here, we integrate chromatin profiling for three histone marks in lymphoblastoid cell lines (LCLs) from 75 sequenced individuals with LCL-specific Hi-C and ChIA-PET-based chromatin contact maps to uncover one of the largest collections of local and distal histone quantitative trait loci (hQTLs). Distal QTLs are enriched within topologically associated domains and exhibit largely concordant variation of?chromatin state coordinated by proximal and distal?non-coding genetic variants. Histone QTLs are enriched for common variants associated with autoimmune diseases and enable identification of putative target genes of disease-associated variants from genome-wide association studies. These analyses provide insights into how genetic variation can affect human disease phenotypes by coordinated changes in chromatin at interacting regulatory elements.
DOI:10.1016/j.cell.2015.08.001URLPMID:26300124 [本文引用: 1]
Chromatin state variation at gene regulatory elements is abundant across individuals, yet we understand little about the genetic basis of this variability. Here, we profiled several histone modifications, the transcription factor (TF) PU.1, RNA polymerase II, and gene expression in lymphoblastoid cell lines from 47 whole-genome sequenced individuals. We observed that distinct cis-regulatory elements exhibit coordinated chromatin variation across individuals in?the form of variable chromatin modules (VCMs) at?sub-Mb scale. VCMs were associated with thousands of genes and preferentially cluster within chromosomal contact domains. We mapped strong proximal and weak, yet more ubiquitous, distal-acting chromatin quantitative trait loci (cQTL) that frequently explain this variation. cQTLs were associated with molecular activity at clusters of cis-regulatory elements and mapped preferentially within TF-bound regions. We propose that local, sequence-independent chromatin variation emerges as a result of genetic perturbations in cooperative interactions between cis-regulatory elements that are located within the same genomic domain.
.
DOI:10.1002/jcp.25996URLPMID:28504305 [本文引用: 1]
Alterations in nuclear morphology are common in cancer progression. However, the degree to which gross morphological abnormalities translate into compromised higher-order chromatin organization is poorly understood. To explore the functional links between gene expression and chromatin structure in breast cancer, we performed RNA-seq gene expression analysis on the basal breast cancer progression model based on human MCF10A cells. Positional gene enrichment identified the major histone gene cluster at chromosome 6p22 as one of the most significantly upregulated (and not amplified) clusters of genes from the normal-like MCF10A to premalignant MCF10AT1 and metastatic MCF10CA1a cells. This cluster is subdivided into three sub-clusters of histone genes that are organized into hierarchical topologically associating domains (TADs). Interestingly, the sub-clusters of histone genes are located at TAD boundaries and interact more frequently with each other than the regions in-between them, suggesting that the histone sub-clusters form an active chromatin hub. The anchor sites of loops within this hub are occupied by CTCF, a known chromatin organizer. These histone genes are transcribed and processed at a specific sub-nuclear microenvironment termed the major histone locus body (HLB). While the overall chromatin structure of the major HLB is maintained across breast cancer progression, we detected alterations in its structure that may relate to gene expression. Importantly, breast tumor specimens also exhibit a coordinate pattern of upregulation across the major histone gene cluster. Our results provide a novel insight into the connection between the higher-order chromatin organization of the major HLB and its regulation during breast cancer progression.
DOI:10.1038/nature11279URL [本文引用: 2]
The vast non-coding portion of the human genome is full of functional elements and disease-causing regulatory variants. The principles defining the relationships between these elements and distal target genes remain unknown. Promoters and distal elements can engage in looping interactions that have been implicated in gene regulation(1). Here we have applied chromosome conformation capture carbon copy (5C(2)) to interrogate comprehensively interactions between transcription start sites (TSSs) and distal elements in 1% of the human genome representing the ENCODE pilot project regions(3). 5C maps were generated for GM12878, K562 and HeLa-S3 cells and results were integrated with data from the ENCODE consortium(4). In each cell line we discovered >1,000 long-range interactions between promoters and distal sites that include elements resembling enhancers, promoters and CTCF-bound sites. We observed significant correlations between gene expression, promoter-enhancer interactions and the presence of enhancer RNAs. Long-range interactions show marked asymmetry with a bias for interactions with elements located similar to 120 kilobases upstream of the TSS. Long-range interactions are often not blocked by sites bound by CTCF and cohesin, indicating that many of these sites do not demarcate physically insulated gene domains. Furthermore, only similar to 7% of looping interactions are with the nearest gene, indicating that genomic proximity is not a simple predictor for long-range interactions. Finally, promoters and distal elements are engaged in multiple long-range interactions to form complex networks. Our results start to place genes and regulatory elements in three-dimensional context, revealing their functional relationships.
DOI:10.1038/nature12644URL [本文引用: 2]
A large number of cis-regulatory sequences have been annotated in the human genome(1,2), but defining their target genes remains a challenge(3). One strategy is to identify the long-range looping interactions at these elements with the use of chromosome conformation capture (3C)-based techniques(4). However, previous studies lack either the resolution or coverage to permit a whole-genome, unbiased view of chromatin interactions. Here we report a comprehensive chromatin interaction map generated in human fibroblasts using a genome-wide 3C analysis method (Hi-C)(5). We determined over one million long-range chromatin interactions at 5-10-kb resolution, and uncovered general principles of chromatin organization at different types of genomic features. We also characterized the dynamics of promoter-enhancer contacts after TNF-alpha signalling in these cells. Unexpectedly, we found that TNF-alpha-responsive enhancers are already in contact with their target promoters before signalling. Such pre-existing chromatin looping, which also exists in other cell types with different extracellular signalling, is a strong predictor of gene induction. Our observations suggest that the three-dimensional chromatin landscape, once established in a particular cell type, is relatively stable and could influence the selection or activation of target genes by a ubiquitous transcription activator in a cell-specific manner.
DOI:10.1016/j.molcel.2015.09.018URLPMID:26593720 [本文引用: 1]
Upon recruitment to active enhancers and promoters, RNA polymerase II (Pol II) generates short non-coding transcripts of unclear function. The mechanisms that control the length and the amount of ncRNAs generated by cis-regulatory elements are largely unknown. Here, we show that the adaptor protein WDR82 and its associated complexes actively limit such non-coding transcription. WDR82 targets the SET1 H3K4 methyltransferases and the nuclear protein phosphatase 1 (PP1) complexes to the initiating Pol II. WDR82 and PP1 also interact with components of the transcriptional termination and RNA processing machineries. Depletion of WDR82, SET1, or the PP1 subunit required for its nuclear import caused distinct but overlapping transcription termination defects at highly expressed genes and active enhancers and promoters, thus enabling the increased synthesis of unusually long ncRNAs. These data indicate that transcription initiated from cis-regulatory elements is tightly coordinated with termination mechanisms that impose the synthesis of short RNAs.
DOI:10.1038/s41588-019-0466-zURLPMID:31358994 [本文引用: 1]
The genome is organized in three-dimensional units called topologically associating domains (TADs), through a process dependent on the cooperative action of cohesin and the DNA-binding factor CTCF. Genomic rearrangements of TADs have been shown to cause gene misexpression and disease, but genome-wide depletion of CTCF has no drastic effects on transcription. Here, we investigate TAD function in vivo in mouse limb buds at the Sox9-Kcnj2 locus. We show that the removal of all major CTCF sites at the boundary and within the TAD resulted in a fusion of neighboring TADs, without major effects on gene expression. Gene misexpression and disease phenotypes, however, were achieved by redirecting regulatory activity through inversions and/or the repositioning of boundaries. Thus, TAD structures provide robustness and precision but are not essential for developmental gene regulation. Aberrant disease-related gene activation is not induced by a mere loss of insulation but requires CTCF-dependent redirection of enhancer-promoter contacts.
DOI:10.1016/S0006-3495(99)77119-5URLPMID:10545385 [本文引用: 1]
The motion of subchromosomal foci and of whole chromosome territories in live human cell nuclei was investigated in four-dimensional space-time images. Visualization of subchromosomal foci was achieved by incorporating Cy3-dUTP into the nuclear DNA of two different cell types after microinjection. A subsequent segregation of the labeled cell nuclei led to the presence of only a few labeled chromosome territories on a background of nonlabeled chromatin (Zink et al.,1998. Hum. Genet. 102:241-251). This procedure yielded many distinct signals in a given cell nucleus. Motion analysis in four-dimensional space-time images was performed using single-particle tracking and a statistical approach to the detection of a possible directional motion of foci relative to the center of mass of a chromosome territory. The accuracy of the analysis was tested using simulated data sets that closely mirrored the experimental setup and using microparticles of known size. Application of the analysis tools to experimental data showed that mutual diffusion-like movements between foci located on different chromosomes were more pronounced than inside the territories. In the time range observed, movements of individual foci could best be described by a random diffusion process. The statistical test for joint directed motion of several foci inside chromosome territories revealed that foci occasionally switched from random to directional motion inside the territories.
DOI:10.1016/j.cell.2015.11.024URLPMID:26686651 [本文引用: 1]
Spatial genome organization and its effect on transcription remains a fundamental question. We applied an advanced chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) strategy to comprehensively map higher-order chromosome folding and specific chromatin interactions mediated by CCCTC-binding factor (CTCF) and RNA polymerase II (RNAPII) with haplotype specificity and nucleotide resolution in different human cell lineages. We find that CTCF/cohesin-mediated interaction anchors serve as structural foci for spatial organization of constitutive genes concordant with CTCF-motif orientation, whereas RNAPII interacts within these structures by selectively drawing cell-type-specific genes toward CTCF foci for coordinated transcription. Furthermore, we show that haplotype variants and allelic interactions have differential effects on chromosome configuration, influencing gene expression, and may provide mechanistic insights into functions associated with disease susceptibility. 3D genome simulation suggests a model of chromatin folding around chromosomal axes, where CTCF is involved in defining the interface between condensed and open compartments for structural regulation. Our 3D genome strategy thus provides unique insights in the topological mechanism of human variations and diseases.
DOI:10.1016/j.cell.2014.11.021URL [本文引用: 1]
We use in situ Hi-C to probe the 3D architecture of genomes, constructing haploid and diploid maps of nine cell types. The densest, in human lymphoblastoid cells, contains 4.9 billion contacts, achieving 1 kb resolution. We find that genomes are partitioned into contact domains (median length, 185 kb), which are associated with distinct patterns of histone marks and segregate into six subcompartments. We identify similar to 10,000 loops. These loops frequently link promoters and enhancers, correlate with gene activation, and show conservation across cell types and species. Loop anchors typically occur at domain boundaries and bind CTCF. CTCF sites at loop anchors occur predominantly (>90%) in a convergent orientation, with the asymmetric motifs "facing'' one another. The inactive X chromosome splits into two massive domains and contains large loops anchored at CTCF-binding repeats.
DOI:10.1038/nature12716URL [本文引用: 1]
In multicellular organisms, transcription regulation is one of the central mechanisms modelling lineage differentiation and cell-fate determination(1). Transcription requires dynamic chromatin configurations between promoters and their corresponding distal regulatory elements(2). It is believed that their communication occurs within large discrete foci of aggregated RNA polymerases termed transcription factories in three-dimensional nuclear space(3). However, the dynamic nature of chromatin connectivity has not been characterized at the genome-wide level. Here, through a chromatin interaction analysis with paired-end tagging approach(3-5) using an antibody that primarily recognizes the pre-initiation complexes of RNA polymerase II6, we explore the transcriptional interactomes of three mouse cells of progressive lineage commitment, including pluripotent embryonic stem cells(7), neural stem cells(8) and neurosphere stem/progenitor cells(9). Our global chromatin connectivity maps reveal approximately 40,000 long-range interactions, suggest precise enhancer-promoter associations and delineate cell-type-specific chromatin structures. Analysis of the complex regulatory repertoire shows that there are extensive colocalizations among promoters and distal-acting enhancers. Most of the enhancers associate with promoters located beyond their nearest active genes, indicating that the linear juxtaposition is not the only guiding principle driving enhancer target selection. Although promoter-enhancer interactions exhibit high cell-type specificity, promoters involved in interactions are found to be generally common and mostly active among different cells. Chromatin connectivity networks reveal that the pivotal genes of reprogramming functions are transcribed within physical proximity to each other in embryonic stem cells, linking chromatin architecture to coordinated gene expression. Our study sets the stage for the full-scale dissection of spatial and temporal genome structures and their roles in orchestrating development.
DOI:10.1016/j.cell.2011.12.014URLPMID:22265404 [本文引用: 1]
Higher-order chromosomal organization for transcription regulation is poorly understood in eukaryotes. Using genome-wide Chromatin Interaction Analysis with Paired-End-Tag sequencing (ChIA-PET), we mapped long-range chromatin interactions associated with RNA polymerase II in human cells and uncovered widespread promoter-centered intragenic, extragenic, and intergenic interactions. These interactions further aggregated into higher-order clusters, wherein proximal and distal genes were engaged through promoter-promoter interactions. Most genes with promoter-promoter interactions were active and transcribed cooperatively, and some interacting promoters could influence each other implying combinatorial complexity of transcriptional controls. Comparative analyses of different cell lines showed that cell-specific chromatin interactions could provide structural frameworks for cell-specific transcription, and suggested significant enrichment of enhancer-promoter interactions for cell-specific functions. Furthermore, genetically-identified disease-associated noncoding elements were found to be spatially engaged with corresponding genes through long-range interactions. Overall, our study provides insights into transcription regulation by three-dimensional chromatin interactions for both housekeeping and cell-specific genes in human cells.
DOI:10.1016/j.cell.2014.05.050URL [本文引用: 1]
Distal enhancers commonly contact target promoters via chromatin looping. In erythroid cells, the locus control region (LCR) contacts beta-type globin genes in a developmental stage-specific manner to stimulate transcription. Previously, we induced LCR-promoter looping by tethering the self-association domain (SA) of Ldb1 to the beta-globin promoter via artificial zinc fingers. Here, we show that targeting the SA to a developmentally silenced embryonic globin gene in adult murine erythroblasts triggers its transcriptional reactivation. This activity depends on the LCR, consistent with an LCR-promoter looping mechanism. Strikingly, targeting the SA to the fetal gamma-globin promoter in primary adult human erythroblasts increases gamma-globin promoter-LCR contacts, stimulating transcription to approximately 85% of total beta-globin synthesis, with a reciprocal reduction in adult beta-globin expression. Our findings demonstrate that forced chromatin looping can override a stringent developmental gene expression program and suggest a novel approach to control the balance of globin gene transcription for therapeutic applications.
DOI:10.1016/j.cell.2012.03.051URL [本文引用: 2]
Chromatin loops juxtapose distal enhancers with active promoters, but their molecular architecture and relationship with transcription remain unclear. In erythroid cells, the locus control region (LCR) and beta-globin promoter form a chromatin loop that requires transcription factor GATA1 and the associated molecule Ldb1. We employed artificial zinc fingers (ZF) to tether Ldb1 to the b-globin promoter in GATA1 null erythroblasts, in which the b-globin locus is relaxed and inactive. Remarkably, targeting Ldb1 or only its self-association domain to the b-globin promoter substantially activated b-globin transcription in the absence of GATA1. Promotertethered Ldb1 interacted with endogenous Ldb1 complexes at the LCR to form a chromatin loop, causing recruitment and phosphorylation of RNA polymerase II. ZF-Ldb1 proteins were inactive at alleles lacking the LCR, demonstrating that their activities depend on long-range interactions. Our findings establish Ldb1 as a critical effector of GATA1-mediated loop formation and indicate that chromatin looping causally underlies gene regulation.
DOI:10.1038/s41586-019-1472-0URLPMID:31391581 [本文引用: 1]
The CCCTC-binding factor (CTCF), which anchors DNA loops that organize the genome into structural domains, has a central role in gene control by facilitating or constraining interactions between genes and their regulatory elements1,2. In cancer cells, the disruption of CTCF binding at specific loci by somatic mutation3,4 or DNA hypermethylation5 results in the loss of loop anchors and consequent activation of oncogenes. By contrast, the germ-cell-specific paralogue of CTCF, BORIS (brother of the regulator of imprinted sites, also known as CTCFL)6, is overexpressed in several cancers7-9, but its contributions to the malignant phenotype remain unclear. Here we show that aberrant upregulation of BORIS promotes chromatin interactions in ALK-mutated, MYCN-amplified neuroblastoma10 cells that develop resistance to ALK inhibition. These cells are reprogrammed to a distinct phenotypic state during the acquisition of resistance, a process defined by the initial loss of MYCN expression followed by subsequent overexpression of BORIS and a concomitant switch in cellular dependence from MYCN to BORIS. The resultant BORIS-regulated alterations in chromatin looping lead to the formation of super-enhancers that drive the ectopic expression of a subset of proneural transcription factors that ultimately define the resistance phenotype. These results identify a previously unrecognized role of BORIS-to promote regulatory chromatin interactions that support specific cancer phenotypes.
DOI:10.1002/ame2.12032URLPMID:30891562 [本文引用: 1]
Enhancer is a positive regulator for spatiotemporal development in eukaryotes. As a cluster, super-enhancer is closely related to cell identity- and fate-determined processes. Both of them function tightly depending on their targeted transcription factors, cofactors, and genes through distal genomic interactions. They have been recognized as critical components and played positive roles in transcriptional regulatory network or factory. Recent advances of next-generation sequencing have dramatically expanded our ability and knowledge to interrogate the molecular mechanism of enhancer and super-enhancer for transcription. Here, we review the history, importance, advances and challenges on enhancer and super-enhancer field. This will benefit our understanding of their function mechanism for transcription underlying precise gene expression.
DOI:10.1016/j.celrep.2017.10.048URLPMID:29117547 [本文引用: 1]
Prostate cancer (PCa) is the leading cancer among men in the United States, with genetic factors contributing to ~42% of the susceptibility to PCa. We analyzed a PCa risk region located at 7p15.2 to gain insight into the mechanisms by which this noncoding region may affect gene regulation and contribute to PCa risk. We performed Hi-C analysis and demonstrated that this region has long-range interactions with the HOXA locus, located ~873 kb away. Using the CRISPR/Cas9 system, we deleted a 4-kb region encompassing several PCa risk-associated SNPs and performed RNA-seq to investigate transcriptomic changes in prostate cells lacking the regulatory element. Our results suggest that the risk element affects the expression of HOXA13 and HOTTIP, but not other genes in the HOXA locus, via a repressive loop. Forced expression of HOXA13 was performed to gain further insight into the mechanisms by which this risk element affects PCa risk.
DOI:10.1038/nature12753URL [本文引用: 1]
How a complex animal can arise from a fertilized egg is one of the oldest and most fascinating questions of biology, the answer to which is encoded in the genome. Body shape and organ development, and their integration into a functional organism all depend on the precise expression of genes in space and time. The orchestration of transcription relies mostly on surrounding control sequences such as enhancers, millions of which form complex regulatory landscapes in the non-coding genome. Recent research shows that high-order chromosome structures make an important contribution to enhancer functionality by triggering their physical interactions with target genes.
DOI:10.1038/ng1244URLPMID:14517543 [本文引用: 1]
Efficient transcription of genes requires a high local concentration of the relevant trans-acting factors. Nuclear compartmentalization can provide an effective means to locally increase the concentration of rapidly moving trans-acting factors; this may be achieved by spatial clustering of chromatin-associated binding sites for such factors. Here we analyze the structure of an erythroid-specific spatial cluster of cis-regulatory elements and active beta-globin genes, the active chromatin hub (ACH; ref. 6), at different stages of development and in erythroid progenitors. We show, in mice and humans, that a core ACH is developmentally conserved and consists of the hypersensitive sites (HS1-HS6) of the locus control region (LCR), the upstream 5' HS-60/-62 and downstream 3' HS1. Globin genes switch their interaction with this cluster during development, correlating with the switch in their transcriptional activity. In mouse erythroid progenitors that are committed to but do not yet express beta-globin, only the interactions between 5' HS-60/-62, 3' HS1 and hypersensitive sites at the 5' side of the LCR are stably present. After induction of differentiation, these sites cluster with the rest of the LCR and the gene that is activated. We conclude that during erythroid differentiation, cis-regulatory DNA elements create a developmentally conserved nuclear compartment dedicated to RNA polymerase II transcription of beta-globin genes.
DOI:10.1038/sj.emboj.7601654URLPMID:17380126 [本文引用: 1]
To understand how mammalian genes are regulated from their natural chromosomal environment, we have analysed the molecular events occurring throughout a 150 kb chromatin segment containing the alpha globin gene locus as it changes from a poised, silent state in erythroid progenitors, to the fully activated state in late, erythroid cells. Active transcription requires the late recruitment of general transcription factors, mediator and Pol II not only to the promoter but also to its remote regulatory elements. Natural mutants of the alpha cluster show that whereas recruitment of the pre-initiation complex to the upstream elements occurs independently, recruitment to the promoter is largely dependent on the regulatory elements. An improved, quantitative chromosome conformation capture analysis demonstrates that this recruitment is associated with a conformational change, in vivo, apposing the promoter with its remote regulators, consistent with a chromosome looping mechanism. These findings point to a general mechanism by which a gene can be held in a poised state until the appropriate stage for expression, coordinating the level and timing of gene expression during terminal differentiation.
DOI:10.1038/NMETH.2173URL [本文引用: 1]
Regulatory DNA elements can control the expression of distant genes via physical interactions. Here we present a cost-effective methodology and computational analysis pipeline for robust characterization of the physical organization around selected promoters and other functional elements using chromosome conformation capture combined with high-throughput sequencing (4C-seq). Our approach can be multiplexed and routinely integrated with other functional genomics assays to facilitate physical characterization of gene regulation.
DOI:10.1007/bf00286715URLPMID:2777259 [本文引用: 1]
The dinucleotide CpG is a "hotspot" for mutation in the human genome as a result of (1) the modification of the 5' cytosine by cellular DNA methyltransferases and (2) the consequent high frequency of spontaneous deamination of 5-methyl cytosine (5mC) to thymidine. DNA methylation thus contributes significantly, albeit indirectly, to the incidence of human genetic disease. We have attempted to estimate for the first time the in vivo rate of deamination of 5mC from the measured rate of 5mC deamination in vitro and the known error frequency of the cellular G/T mismatch-repair system. The accuracy and utility of this estimate (md) was then assessed by comparison with clinical data, and an improved estimate of md (1.66 X 10(-16) s-1) was derived. Comparison of the CpG mutation rates exhibited by globin gene and pseudogene sequences from human, chimpanzee and macaque provided further estimates of md, all of which were consistent with the first. Use of this value in a mathematical model then permitted the estimation of the length of time required to produce the level of "CpG suppression" currently found in the "bulk DNA" of vertebrate genomes. This time span, approximately 450 million years, corresponds closely to the estimated time since the emergence and adaptive radiation of the vertebrates and thus coincides with the probable advent of heavily methylated genomes. An accurate estimate of the 5mC deamination rate is important not only for clinical medicine but also for studies of gene evolution. Our data suggest both that patterns of vertebrate gene methylation may be comparatively stable over relatively long periods of evolutionary time, and that the rate of CpG deamination can, under certain limited conditions, serve as a "molecular clock".
DOI:10.3724/SP.J.1005.2014.0403URL [本文引用: 1]
DNA methylation plays important roles in cell differentiation, embryonic development, host adaptations to environmental factors, and pathogenesis through regulation of gene transcription and imprinting, X-inactivation, and defense of foreign genetic material invasion, is currently one of the hottest research fields on epigenetics. In the past few years, a number of important findings on DNA methylation have been achieved. These findings include discovery of TETs-catalyzed cytosine hydroxymethylation and its functions in the early embryonic development; the relationship between active and passive DNA demethylation; establishment and maintenance of DNA methylation patterns and their associations with histone modifications, chromatin configuration, polycomb group proteins and non-coding RNA bindings. DNA methylation has become a new potential biomarker and therapy target.
DOI:10.3724/SP.J.1005.2014.0403URL [本文引用: 1]
DNA methylation plays important roles in cell differentiation, embryonic development, host adaptations to environmental factors, and pathogenesis through regulation of gene transcription and imprinting, X-inactivation, and defense of foreign genetic material invasion, is currently one of the hottest research fields on epigenetics. In the past few years, a number of important findings on DNA methylation have been achieved. These findings include discovery of TETs-catalyzed cytosine hydroxymethylation and its functions in the early embryonic development; the relationship between active and passive DNA demethylation; establishment and maintenance of DNA methylation patterns and their associations with histone modifications, chromatin configuration, polycomb group proteins and non-coding RNA bindings. DNA methylation has become a new potential biomarker and therapy target.
DOI:10.1038/nsmb840URLPMID:15467727 [本文引用: 1]
Transcriptional silencing in mammals is often associated with promoter methylation. However, a considerable number of genomic methylated CpGs exist in transposable elements, which are frequently found in intronic regions. To determine whether intragenic methylation influences transcription efficiency, we used the Cre/loxP-based system, RMCE, to introduce a transgene, methylated exclusively in a region downstream of the promoter, into a specific genomic site. This methylation pattern was maintained in vivo, and yielded a clear decrease in transgene expression relative to an unmethylated control. Notably, RNA polymerase II (Pol II) was depleted exclusively in the methylated region, as was histone H3 di- and trimethylated on Lys4 and acetylated on Lys9 and Lys14. As the methylated region adopts a closed chromatin structure in vivo, we propose that dense intragenic DNA methylation in mammalian cells initiates formation of a chromatin structure that reduces the efficiency of Pol II elongation.
DOI:10.1186/s13059-017-1296-xURLPMID:28859663 [本文引用: 1]
Whole-genome bisulfite sequencing (WGBS) is the gold standard for studying landscape DNA methylation. Current computational methods for WGBS are mainly designed for gene regulatory regions with multiple under-methylated CpGs (UMCs), such as promoters and enhancers.
.
DOI:10.1038/jid.2011.364URLPMID:22327261 [本文引用: 1]
DNA methylation is one of several epigenetic mechanisms that can lead to stable and heritable changes in gene expression without changing the underlying DNA sequence. In this issue, Roberson et al. demonstrate that lesional skin in psoriasis is accompanied by changes in DNA methylation, which revert back to baseline with treatment, indicating that DNA methylation in psoriasis is dynamic.
DOI:10.1038/nature16490URLPMID:26700815 [本文引用: 1]
Gain-of-function IDH mutations are initiating events that define major clinical and prognostic classes of gliomas. Mutant IDH protein produces a new onco-metabolite, 2-hydroxyglutarate, which interferes with iron-dependent hydroxylases, including the TET family of 5'-methylcytosine hydroxylases. TET enzymes catalyse a key step in the removal of DNA methylation. IDH mutant gliomas thus manifest a CpG island methylator phenotype (G-CIMP), although the functional importance of this altered epigenetic state remains unclear. Here we show that human IDH mutant gliomas exhibit hypermethylation at cohesin and CCCTC-binding factor (CTCF)-binding sites, compromising binding of this methylation-sensitive insulator protein. Reduced CTCF binding is associated with loss of insulation between topological domains and aberrant gene activation. We specifically demonstrate that loss of CTCF at a domain boundary permits a constitutive enhancer to interact aberrantly with the receptor tyrosine kinase gene PDGFRA, a prominent glioma oncogene. Treatment of IDH mutant gliomaspheres with a demethylating agent partially restores insulator function and downregulates PDGFRA. Conversely, CRISPR-mediated disruption of the CTCF motif in IDH wild-type gliomaspheres upregulates PDGFRA and increases proliferation. Our study suggests that IDH mutations promote gliomagenesis by disrupting chromosomal topology and allowing aberrant regulatory interactions that induce oncogene expression.
.
DOI:10.1002/path.4585URLPMID:26174723 [本文引用: 1]
Over the past decade, chromatin remodelling emerged as one of the most important causes of both abnormal development and cancer. Although much has been written about one or another of the complexes, no recent concise summary of the chromatin remodelling families as a whole is available. In this short review, we introduce the family members, briefly summarize their role in developmental abnormalities and neoplasia, and outline the different ways in which these families remodel chromatin.
DOI:10.1016/j.mrfmmm.2006.08.015URLPMID:17306844 [本文引用: 1]
The inter-relationship between DNA repair and ATP dependent chromatin remodeling has begun to become very apparent with recent discoveries. ATP dependent remodeling complexes mobilize nucleosomes along DNA, promote the exchange of histones, or completely displace nucleosomes from DNA. These remodeling complexes are often categorized based on the domain organization of their catalytic subunit. The biochemical properties and structural information of several of these remodeling complexes are reviewed. The different models for how these complexes are able to mobilize nucleosomes and alter nucleosome structure are presented incorporating several recent findings. Finally the role of histone tails and their respective modifications in ATP-dependent remodeling are discussed.
DOI:10.1038/s41586-019-1029-2URLPMID:30867599 [本文引用: 1]
Chromatin remodellers include diverse enzymes with distinct biological functions, but nucleosome-sliding activity appears to be a common theme1,2. Among the remodelling enzymes, Snf2 serves as the prototype to study the action of this protein family. Snf2 and related enzymes share two conserved RecA-like lobes3, which by themselves are able to couple ATP hydrolysis to chromatin remodelling. The mechanism by which these enzymes couple ATP hydrolysis to translocate the nucleosome along the DNA remains unclear2,4-8. Here we report the structures of Saccharomyces cerevisiae Snf2 bound to the nucleosome in the presence of ADP and ADP-BeFx. Snf2 in the ADP-bound state adopts an open conformation similar to that in the apo state, and induces a one-base-pair DNA bulge at superhelix location 2 (SHL2), with the tracking strand showing greater distortion than the guide strand. The DNA distortion propagates to the proximal end, leading to staggered translocation of the two strands. The binding of ADP-BeFx triggers a closed conformation of the enzyme, resetting the nucleosome to a relaxed state. Snf2 shows altered interactions with the DNA in different nucleotide states, providing the structural basis for DNA translocation. Together, our findings suggest a fundamental mechanism for the DNA translocation that underlies chromatin remodelling.
DOI:10.1038/s41594-019-0199-9URLPMID:30872815 [本文引用: 1]
Chromatin remodelers are diverse enzymes, and different models have been proposed to explain how these proteins work. Here we report the 3.3??-resolution cryogenic electron microscopy (cryo-EM) structures of Saccharomyces cerevisiae ISWI (ISW1) in complex with the nucleosome in adenosine diphosphate (ADP)-bound and ADP-BeFx-bound states. The data show that after nucleosome binding, ISW1 is activated by substantial rearrangement of the catalytic domains, with the regulatory AutoN domain packing the first RecA-like core and the NegC domain being disordered. The high-resolution structure reveals local DNA distortion and translocation induced by ISW1 in the ADP-bound state, which is essentially identical to that induced by the Snf2 chromatin remodeler, suggesting a common mechanism of DNA translocation. The histone core remains largely unperturbed, and prevention of histone distortion by crosslinking did not inhibit the activity of yeast ISW1 or its human homolog. Together, our findings suggest a general mechanism of chromatin remodeling involving local DNA distortion without notable histone deformation.
DOI:10.1126/science.1090701URLPMID:14645854 [本文引用: 1]
The conserved histone variant H2AZ has an important role in the regulation of gene expression and the establishment of a buffer to the spread of silent heterochromatin. How histone variants such as H2AZ are incorporated into nucleosomes has been obscure. We have found that Swr1, a Swi2/Snf2-related adenosine triphosphatase, is the catalytic core of a multisubunit, histone-variant exchanger that efficiently replaces conventional histone H2A with histone H2AZ in nucleosome arrays. Swr1 is required for the deposition of histone H2AZ at specific chromosome locations in vivo, and Swr1 and H2AZ commonly regulate a subset of yeast genes. These findings define a previously unknown role for the adenosine triphosphate-dependent chromatin remodeling machinery.
DOI:10.1093/nar/gkl338URLPMID:16714444 [本文引用: 1]
Histone post-translational modifications occur, not only in the N-terminal tail domains, but also in the core domains. While modifications in the N-terminal tail function largely through the regulation of the binding of non-histone proteins to chromatin, based on their location in the nucleosome, core domain modifications may also function through distinct mechanisms involving structural alterations to the nucleosome. This article reviews the recent developments in regards to these novel histone modifications and discusses their important role in the regulation of chromatin structure.
DOI:10.1016/j.bpj.2016.11.015URLPMID:27931745 [本文引用: 1]
The N-terminal tail of histone H4 is an indispensable mediator for inter-nucleosome interaction, which is required for chromatin fiber condensation. H4K16 acetylation (H4K16Ac) activates gene transcription by influencing both chromatin structure and interplay with nonhistone proteins. To understand the influence of H4K16Ac on inter-nucleosome interaction, we performed a simulation study for the H4 tail in the context of two nucleosomes in neighboring unit cells in the crystal structure. The binding conformation of H4 tail with/without K16Ac was sampled by replica exchange with solute tempering, and the free energy landscape was explored by metadynamics. The results indicate two important features of H4K16: 1) it is the first button to anchor the H4 tail on the adjacent nucleosome; and 2) it is the only acetylation site interacting with the acidic patch. H4K16Ac disrupts the electrostatic interactions of K16, weakens H4 tail-acidic patch binding, and significantly increases H4 tail conformation diversity. Our study suggests that H4K16Ac directly reduces the inter-nucleosome interaction mediated by the H4 tail, which might further encourage the binding of nonhistone proteins on the acidic patch.
DOI:10.1016/j.ejcb.2009.11.006URLPMID:20004495 [本文引用: 1]
Protein function is considerably altered by posttranslational modification. In recent years, cycles of acetylation/deacetylation emerged as fundamental regulators adjusting biological activity of many proteins. Particularly, protein deacetylation by Sirtuins, a family of atypical histone deacetylases (HDACs), was demonstrated to regulate fundamental cell biological processes including gene expression, genome stability, mitosis, nutrient metabolism, aging, mitochondrial function and cell motility. Given this wealth of biological functions, perhaps not unexpectedly then, pharmacological compounds targeting Sirtuin activity are now prime therapeutic agents for alleviating severity of major diseases encompassing diabetes, cancer, cardiovascular and neurodegenerative disorders in many organs. In this review, we will focus on the brain and its physiological and pathological processes governed by Sirtuin-mediated deacetylation. Besides discussing Sirtuin function in neurodegenerative diseases, emphasis will be given on the mounting evidence deciphering key developmental brain functions for Sirtuins in neuronal motility, neuroprotection and oligodendrocyte differentiation. In this respect, we will particularly highlight functions of the unconventional family member SIRT2 in post-mitotic neurons and glial cells.
DOI:10.1073/pnas.0500136102URLPMID:15795371 [本文引用: 1]
The histone code hypothesis holds that covalent posttranslational modifications of histone tails are interpreted by the cell to yield a rich combinatorial transcriptional output. This hypothesis has been the subject of active debate in the literature. Here, we investigated the combinatorial complexity of the acetylation code at the four lysine residues of the histone H4 tail in budding yeast. We constructed yeast strains carrying all 15 possible combinations of mutations among lysines 5, 8, 12, and 16 to arginine in the histone H4 tail, mimicking positively charged, unacetylated lysine states, and characterized the resulting genome-wide changes in gene expression by using DNA microarrays. Only the lysine 16 mutation had specific transcriptional consequences independent of the mutational state of the other lysines (affecting approximately 100 genes). In contrast, for lysines 5, 8, and 12, expression changes were due to nonspecific, cumulative effects seen as increased transcription correlating with an increase in the total number of mutations (affecting approximately 1,200 genes). Thus, acetylation of histone H4 is interpreted by two mechanisms: a specific mechanism for lysine 16 and a nonspecific, cumulative mechanism for lysines 5, 8, and 12.
.
DOI:10.1074/jbc.M110532200URLPMID:11714726 [本文引用: 1]
Heterochromatin at yeast telomeres and silent mating (HM) loci represses adjacent genes and is formed by the binding and spreading of silencing information regulators (SIR proteins) along histones. This involves the interaction between the C terminus of SIR3 and the N terminus of histone H4. Since H4 is hypoacetylated in heterochromatin we wished to determine whether acetylation is involved in regulating the contacts between SIR3 and H4. Binding of H4 peptide (residues 1-34) acetylated at lysines Lys-5, Lys-8, Lys-12, and Lys-16 to an immobilized SIR3 protein fragment (residues 510-970) was investigated using surface plasmon resonance. We find that acetylation of H4 lysines reduces binding (K(a)) of H4 to SIR3 in a cumulative manner so that the fully acetylated peptide binding is decreased approximately 50-fold relative to unacetylated peptide. Thus, by affecting SIR3-H4 binding, acetylation may regulate the formation of heterochromatin. These data help explain the hypoacetylated state of histone H4 in heterochromatin of eukaryotes.
DOI:10.1038/nature722URLPMID:11882902 [本文引用: 1]
Specific modifications to histones are essential epigenetic markers---heritable changes in gene expression that do not affect the DNA sequence. Methylation of lysine 9 in histone H3 is recognized by heterochromatin protein 1 (HP1), which directs the binding of other proteins to control chromatin structure and gene expression. Here we show that HP1 uses an induced-fit mechanism for recognition of this modification, as revealed by the structure of its chromodomain bound to a histone H3 peptide dimethylated at Nzeta of lysine 9. The binding pocket for the N-methyl groups is provided by three aromatic side chains, Tyr21, Trp42 and Phe45, which reside in two regions that become ordered on binding of the peptide. The side chain of Lys9 is almost fully extended and surrounded by residues that are conserved in many other chromodomains. The QTAR peptide sequence preceding Lys9 makes most of the additional interactions with the chromodomain, with HP1 residues Val23, Leu40, Trp42, Leu58 and Cys60 appearing to be a major determinant of specificity by binding the key buried Ala7. These findings predict which other chromodomains will bind methylated proteins and suggest a motif that they recognize.
DOI:10.1007/s13311-013-0229-yURL [本文引用: 1]
This review highlights recent discoveries that have shaped the emerging viewpoints in the field of epigenetic influences in the central nervous system (CNS), focusing on the following questions: i) How is the CNS shaped during development when precursor cells transition into morphologically and molecularly distinct cell types, and is this event driven by epigenetic alterations?; ii) How do epigenetic pathways control CNS function?; iii) What happens to "epigenetic memory" during aging processes, and do these alterations cause CNS dysfunction?; iv) Can one restore normal CNS function by manipulating the epigenome using pharmacologic agents, and will this ameliorate aging-related neurodegeneration? These and other still unanswered questions remain critical to understanding the impact of multifaceted epigenetic machinery on the age-related dysfunction of CNS.
DOI:10.1016/j.cell.2013.08.061URL [本文引用: 1]
Polycomb repressive complex 2 (PRC2) regulates gene expression during lineage specification through trimethylation of lysine 27 on histone H3 (H3K27me3). In Drosophila, polycomb binding sites are dynamic chromatin regions enriched with the histone variant H3.3. Here, we show that, in mouse embryonic stem cells (ESCs), H3.3 is required for proper establishment of H3K27me3 at the promoters of developmentally regulated genes. Upon H3.3 depletion, these promoters show reduced nucleosome turnover measured by deposition of de novo synthesized histones and reduced PRC2 occupancy. Further, we show H3.3-dependent interaction of PRC2 with the histone chaperone, Hira, and that Hira localization to chromatin requires H3.3. Our data demonstrate the importance of H3.3 in maintaining a chromatin landscape in ESCs that is important for proper gene regulation during differentiation. Moreover, our findings support the emerging notion that H3.3 has multiple functions in distinct genomic locations that are not always correlated with an "active'' chromatin state.
DOI:10.1038/nsb767URLPMID:11850638 [本文引用: 1]
Explaining the determinants involved in regulating the equilibrium between different chromatin structural states is fundamental to understanding differential gene expression. Histone variant H2A.Z is essential to chromatin architecture in higher eukaryotes but its role has not yet been explained. We show here that H2A.Z facilitates the intramolecular folding of nucleosomal arrays while simultaneously inhibiting the formation of highly condensed structures that result from intermolecular association. This makes a case for H2A.Z playing a fundamental role in creating unique chromatin domains poised for transcriptional activation. These results provide new insights into understanding how chromatin fiber dynamics can be altered by core histone variants to potentially regulate genomic function.
DOI:10.1016/j.molcel.2004.10.023URLPMID:15546624 [本文引用: 1]
Controlling the degree of higher order chromatin folding is a key element in partitioning the metazoan genome into functionally distinct chromosomal domains. However, the mechanism of this fundamental process is poorly understood. Our recent studies suggested that the essential histone variant H2A.Z and the silencing protein HP1alpha may function together to establish a specialized conformation at constitutive heterochromatic domains. We demonstrate here that HP1alpha is a unique chromatin binding protein. It prefers to bind to condensed higher order chromatin structures and alters the chromatin-folding pathway in a novel way to locally compact individual chromatin fibers without crosslinking them. Strikingly, both of these features are enhanced by an altered nucleosomal surface created by H2A.Z (the acidic patch). This shows that the surface of the nucleosome can regulate the formation of distinct higher order chromatin structures mediated by an architectural chromatin binding protein.
.
URLPMID:31444270 [本文引用: 1]
Liquid-liquid phase separation (LLPS) facilitates the formation of condensed biological assemblies with well-delineated physical boundaries, but without lipid membrane barriers. LLPS is increasingly recognized as a common mechanism for cells to organize and maintain different cellular compartments in addition to classical membrane-delimited organelles. Membraneless condensates have many distinct features that are not present in membrane-delimited organelles and that are likely indispensable for the viability and function of living cells. Malformation of membraneless condensates is increasingly linked to human diseases. In this review, we summarize commonly used methods to investigate various forms of LLPS occurring both in 3D aqueous solution and on 2D membrane bilayers, such as LLPS condensates arising from intrinsically disordered proteins or structured modular protein domains. We then discuss, in the context of comparisons with membrane-delimited organelles, the potential functional implications of membraneless condensate formation in cells. We close by highlighting some challenges in the field devoted to studying LLPS-mediated membraneless condensate formation.
DOI:10.1038/nature22989URLPMID:28636597 [本文引用: 3]
Constitutive heterochromatin is an important component of eukaryotic genomes that has essential roles in nuclear architecture, DNA repair and genome stability, and silencing of transposon and gene expression. Heterochromatin is highly enriched for repetitive sequences, and is defined epigenetically by methylation of histone H3 at lysine 9 and recruitment of its binding partner heterochromatin protein 1 (HP1). A prevalent view of heterochromatic silencing is that these and associated factors lead to chromatin compaction, resulting in steric exclusion of regulatory proteins such as RNA polymerase from the underlying DNA. However, compaction alone does not account for the formation of distinct, multi-chromosomal, membrane-less heterochromatin domains within the nucleus, fast diffusion of proteins inside the domain, and other dynamic features of heterochromatin. Here we present data that support an alternative hypothesis: that the formation of heterochromatin domains is mediated by phase separation, a phenomenon that gives rise to diverse non-membrane-bound nuclear, cytoplasmic and extracellular compartments. We show that Drosophila HP1a protein undergoes liquid-liquid demixing in vitro, and nucleates into foci that display liquid properties during the first stages of heterochromatin domain formation in early Drosophila embryos. Furthermore, in both Drosophila and mammalian cells, heterochromatin domains exhibit dynamics that are characteristic of liquid phase-separation, including sensitivity to the disruption of weak hydrophobic interactions, and reduced diffusion, increased coordinated movement and inert probe exclusion at the domain boundary. We conclude that heterochromatic domains form via phase separation, and mature into a structure that includes liquid and stable compartments. We propose that emergent biophysical properties associated with phase-separated systems are critical to understanding the unusual behaviours of heterochromatin, and how chromatin domains in general regulate essential nuclear functions.
DOI:10.1038/nature22822URLPMID:28636604 [本文引用: 1]
Gene silencing by heterochromatin is proposed to occur in part as a result of the ability of heterochromatin protein 1 (HP1) proteins to spread across large regions of the genome, compact the underlying chromatin and recruit diverse ligands. Here we identify a new property of the human HP1α protein: the ability to form phase-separated droplets. While unmodified HP1α is soluble, either phosphorylation of its N-terminal extension or DNA binding promotes the formation of phase-separated droplets. Phosphorylation-driven phase separation can be promoted or reversed by specific HP1α ligands. Known components of heterochromatin such as nucleosomes and DNA preferentially partition into the HP1α droplets, but molecules such as the transcription factor TFIIB show no preference. Using a single-molecule DNA curtain assay, we find that both unmodified and phosphorylated HP1α induce rapid compaction of DNA strands into puncta, although with different characteristics. We show by direct protein delivery into mammalian cells that an HP1α mutant incapable of phase separation in vitro forms smaller and fewer nuclear puncta than phosphorylated HP1α. These findings suggest that heterochromatin-mediated gene silencing may occur in part through sequestration of compacted chromatin in phase-separated HP1 droplets, which are dissolved or formed by specific ligands on the basis of nuclear context.
DOI:10.1021/acs.biochem.8b00401URLPMID:29644850 [本文引用: 1]
In eukaryotic cells, structures called heterochromatin play critical roles in nuclear processes ranging from gene repression to chromosome segregation. Biochemical and in vivo studies over the past several decades have implied that the diverse functions of heterochromatin rely on the ability of these structures to spread across large regions of the genome, to compact the underlying DNA, and to recruit different types of activities. Recent observations have suggested that heterochromatin may possess liquid droplet-like properties. Here, we discuss how these observations provide a new perspective on the mechanisms for the assembly, regulation, and functions of heterochromatin.
DOI:10.1016/j.cell.2018.10.057URLPMID:30500535 [本文引用: 3]
Phase transitions involving biomolecular liquids are a fundamental mechanism underlying intracellular organization. In the cell nucleus, liquid-liquid phase separation of intrinsically disordered proteins (IDPs) is implicated in assembly of the nucleolus, as well as transcriptional clusters, and other nuclear bodies. However, it remains unclear whether and how physical forces associated with nucleation, growth, and wetting of liquid condensates can directly restructure chromatin. Here, we use CasDrop, a novel CRISPR-Cas9-based optogenetic technology, to show that various IDPs phase separate into liquid condensates that mechanically exclude chromatin as they grow and preferentially form in low-density, largely euchromatic regions. A minimal physical model explains how this stiffness sensitivity arises from lower mechanical energy associated with deforming softer genomic regions. Targeted genomic loci can nonetheless be mechanically pulled together through surface tension-driven coalescence. Nuclear condensates may thus function as mechano-active chromatin filters, physically pulling in targeted genomic loci while pushing out non-targeted regions of the neighboring genome. VIDEO ABSTRACT.
DOI:10.1038/nmeth.4325URLPMID:28604721 [本文引用: 1]
Hi-C is a genome-wide sequencing technique used to investigate 3D chromatin conformation inside the nucleus. Computational methods are required to analyze Hi-C data and identify chromatin interactions and topologically associating domains (TADs) from genome-wide contact probability maps. We quantitatively compared the performance of 13 algorithms in their analyses of Hi-C data from six landmark studies and simulations. This comparison revealed differences in the performance of methods for chromatin interaction identification, but more comparable results for TAD detection between algorithms.
DOI:10.1038/s41588-019-0462-3URLPMID:31308546 [本文引用: 1]
Chromatin topology is intricately linked to gene expression, yet its functional requirement remains unclear. Here, we comprehensively assessed the interplay between genome topology and gene expression using highly rearranged chromosomes (balancers) spanning ~75% of the Drosophila genome. Using transheterozyte (balancer/wild-type) embryos, we measured allele-specific changes in topology and gene expression in cis, while minimizing trans effects. Through genome sequencing, we resolved eight large nested inversions, smaller inversions, duplications and thousands of deletions. These extensive rearrangements caused many changes to chromatin topology, disrupting long-range loops, topologically associating domains (TADs) and promoter interactions, yet these are not predictive of changes in expression. Gene expression is generally not altered around inversion breakpoints, indicating that mis-appropriate enhancer-promoter activation is a rare event. Similarly, shuffling or fusing TADs, changing intra-TAD connections and disrupting long-range inter-TAD loops does not alter expression for the majority of genes. Our results suggest that properties other than chromatin topology ensure productive enhancer-promoter interactions.
DOI:10.1126/science.357.6349.422URLPMID:28751612 [本文引用: 1]
Ribosome stalling leads to recruitment of the ribosome quality control complex (RQC), which targets the partially synthesized polypeptide for proteasomal degradation through the action of the ubiquitin ligase Ltn1p. A second core RQC component, Rqc2p, modifies the nascent polypeptide by adding a carboxyl-terminal alanine and threonine (CAT) tail through a noncanonical elongation reaction. Here we examined the role of CAT-tailing in nascent-chain degradation in budding yeast. We found that Ltn1p efficiently accessed only nascent-chain lysines immediately proximal to the ribosome exit tunnel. For substrates without Ltn1p-accessible lysines, CAT-tailing enabled degradation by exposing lysines sequestered in the ribosome exit tunnel. Thus, CAT-tails do not serve as a degron, but rather provide a fail-safe mechanism that expands the range of RQC-degradable substrates.
DOI:10.1126/science.aau1783URLPMID:30361340 [本文引用: 1]
The spatial organization of chromatin is pivotal for regulating genome functions. We report an imaging method for tracing chromatin organization with kilobase- and nanometer-scale resolution, unveiling chromatin conformation across topologically associating domains (TADs) in thousands of individual cells. Our imaging data revealed TAD-like structures with globular conformation and sharp domain boundaries in single cells. The boundaries varied from cell to cell, occurring with nonzero probabilities at all genomic positions but preferentially at CCCTC-binding factor (CTCF)- and cohesin-binding sites. Notably, cohesin depletion, which abolished TADs at the population-average level, did not diminish TAD-like structures in single cells but eliminated preferential domain boundary positions. Moreover, we observed widespread, cooperative, multiway chromatin interactions, which remained after cohesin depletion. These results provide critical insight into the mechanisms underlying chromatin domain and hub formation.
DOI:10.1038/nmeth.3999URLPMID:27643841 [本文引用: 1]
Genome conformation is central to gene control but challenging to interrogate. Here we present HiChIP, a protein-centric chromatin conformation method. HiChIP improves the yield of conformation-informative reads by over 10-fold and lowers the input requirement over 100-fold relative to that of ChIA-PET. HiChIP of cohesin reveals multiscale genome architecture with greater signal-to-background ratios than those of in situ Hi-C.
DOI:10.1038/s41586-019-1035-4URLPMID:30886393 [本文引用: 1]
The establishment of cell types during development requires precise interactions between genes and distal regulatory sequences. We have a limited understanding of how these interactions look in three dimensions, vary across cell types in complex tissue, and relate to transcription. Here we describe optical reconstruction of chromatin architecture (ORCA), a method that can trace the DNA path in single cells with nanoscale accuracy and genomic resolution reaching two kilobases. We used ORCA to study a Hox gene cluster in cryosectioned Drosophila embryos and labelled around 30 RNA species in parallel. We identified cell-type-specific physical borders between active and Polycomb-repressed DNA, and unexpected Polycomb-independent borders. Deletion of Polycomb-independent borders led to ectopic enhancer-promoter contacts, aberrant gene expression, and developmental defects. Together, these results illustrate an approach for high-resolution, single-cell DNA domain analysis in vivo, identify domain structures that change with cell identity, and show that border elements contribute to the formation of physical domains in Drosophila.
DOI:10.1038/s41467-018-03113-2URLPMID:29467363 [本文引用: 1]
Although Hi-C technology is one of the most popular tools for studying 3D genome organization, due to sequencing cost, the resolution of most Hi-C datasets are coarse and cannot be used to link distal regulatory elements to their target genes. Here we develop HiCPlus, a computational approach based on deep convolutional neural network, to infer high-resolution Hi-C interaction matrices from low-resolution Hi-C data. We demonstrate that HiCPlus can impute interaction matrices highly similar to the original ones, while only using 1/16 of the original sequencing reads. We show that?the models learned from one cell type can be applied to make predictions in other cell or tissue types. Our work not only provides a computational framework to enhance Hi-C data resolution but also reveals features underlying the formation of 3D chromatin interactions.
DOI:10.1093/nar/gkz167URLPMID:30869141 [本文引用: 1]
Interactions between regulatory elements are of crucial importance for the understanding of transcriptional regulation and the interpretation of disease mechanisms. Hi-C technique has been developed for genome-wide detection of chromatin contacts. However, unless extremely deep sequencing is performed on a very large number of input cells, which is technically limited and expensive, current Hi-C experiments do not have high enough resolution to resolve contacts between regulatory elements. Here, we develop DeepTACT, a bootstrapping deep learning model, to integrate genome sequences and chromatin accessibility data for the prediction of chromatin contacts between regulatory elements. DeepTACT can infer not only promoter-enhancer interactions, but also promoter-promoter interactions. In tests based on promoter capture Hi-C data, DeepTACT shows better performance over existing methods. DeepTACT analysis also identifies a class of hub promoters, which are correlated with transcriptional activation across cell lines, enriched in housekeeping genes, functionally related to fundamental biological processes, and capable of reflecting cell similarity. Finally, the utility of chromatin contacts in the study of human diseases is illustrated by the association of IFNA2?to coronary artery disease via an integrative analysis of GWAS data and interactions predicted by DeepTACT.
DOI:10.1038/nature23884URLPMID:28905911 [本文引用: 1]
The 4D Nucleome Network aims to develop and apply approaches to map the structure and dynamics of the human and mouse genomes in space and time with the goal of gaining deeper mechanistic insights into how the nucleus is organized and functions. The project will develop and benchmark experimental and computational approaches for measuring genome conformation and nuclear organization, and investigate how these contribute to gene regulation and other genome functions. Validated experimental technologies will be combined with biophysical approaches to generate quantitative models of spatial genome organization in different biological states, both in cell populations and in single cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}