 ,上海交通大学系统生物医学研究院比较生物医学研究中心,系统生物医学教育部重点实验室,上海 200240
,上海交通大学系统生物医学研究院比较生物医学研究中心,系统生物医学教育部重点实验室,上海 200240Probing 3D genome by CRISPR/Cas9
Peifeng Liu, Qiang Wu,Center for Comparative Biomedicine, Key Laboratory of Systems Biomedicine (Ministry of Education), Institute of Systems Biomedicine, Shanghai Jiao Tong University, Shanghai 200240, China通讯作者: 吴强,博士,教授,研究方向:分子遗传学。E-mail:qwu123@gmail.com
编委: 张飞雄
收稿日期:2019-08-22修回日期:2019-11-18网络出版日期:2020-01-20
| 基金资助: |
Editorial board:
Received:2019-08-22Revised:2019-11-18Online:2020-01-20
| Fund supported: |
作者简介 About authors
刘沛峰,博士研究生,专业方向:生物学。E-mail:lpfmail@foxmail.com。

摘要
CRISPR/Cas9系统在基因编辑方面具有巨大优势,能够低成本、可编程、方便快捷地用于动物、植物以及微生物的基因组靶向编辑和功能改造。三维基因组学是近年来兴起的一门研究染色质高级结构动态调控及基因组生物学功能的交叉学科。在三维基因组研究中,通常采用对DNA片段进行基因编辑以模拟基因组结构性变异,标记特定DNA片段,进而研究调控元件对于基因调控、细胞分化、组织发生、器官形成、个体发育的影响,最终阐明三维基因组的组装调控机制和生物学功能。因此,CRISPR及其衍生技术为研究三维基因组提供了极好的遗传学工具。本文主要综述了CRISPR片段编辑及其衍生技术在三维基因组调控与功能研究中的应用,以期为后续研究工作提供理论参考以及新的研究思路。
关键词:
Abstract
CRISPR/Cas9 system has significant advantages in gene editing strategy, offering cost-effective and efficient means to modify and edit the genomes of animals, plants, and microorganisms. Three-dimensional (3D) genome is an emerging and interdisciplinary field catapulted by combined technological breakthroughs of chromosome conformation capture with next-generation sequencing and live imaging with super-resolution microscopy. An important aspect of 3D genomics is to model structural variations and label specific genomic fragments to investigate the effects of manipulation of genomic elements on gene expression regulation, cell development and differentiation, and spatial location of chromosomal regions. Therefore, CRISPR/Cas9 system and its derivative technologies of DNA-fragment editing are excellent toolboxes for investigating dynamics and functions of the higher-order chromatin organization and three-dimensional genome structure. In this review, we describe the opportunities and challenges of CRISPR as well as its derivative technologies in 3D genome research, thereby providing some critical references and future research directions in the field.
Keywords:
PDF (819KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘沛峰, 吴强. CRISPR/Cas9基因编辑在三维基因组研究中的应用. 遗传[J], 2020, 42(1): 18-31 doi:10.16288/j.yczz.19-246
Peifeng Liu.
成簇规律间隔短回文重复序列及其相关蛋白(clustered regularly interspaced short palindromic repeats/CRISPR-associated protein, CRISPR/Cas)是古菌和细菌中一种用于抵抗噬菌体、病毒DNA等外来遗传因子的原核生物获得性免疫系统。该系统可以将外源入侵DNA的小片段整合到宿主基因组的CRISPR重复序列中靠近前导序列(leader sequence)的位置,形成CRISPR重复-间隔基因座。这种为外源DNA侵染所做的遗传记录使宿主产生遗传记忆并在下次面对同样的入侵者时可以进行防御[1]。当相同的外源DNA再次入侵时,CRISPR重复-间隔基因座会转录并加工为crRNA (CRISPR RNA),并引导DNA核酸内切酶Cas家族蛋白对入侵的核苷酸序列进行靶向切割。Cas9蛋白是Cas蛋白家族中的一员,Cas9蛋白参与组成的CRISPR/Cas9系统是CRISPR/Cas系统的子系统之一。自然界的CRISPR/ Cas9系统经过原始创新性改造可以在一个单链引导RNA (single guide RNA, sgRNA)介导下编辑基因组特定位点(图1)[2,3,4]。因其灵活、高效、方便、操作性强等特点,该基因组编辑系统很快被广泛应用于生物医药及人类健康等领域。近年来,随着进一步的研究发展,更多CRISPR/Cas9系统的应用方法被开发出来,在不同的研究领域发挥重要作用,三维基因组即是受益于CRISPR/Cas9系统的研究领域之一。
三维基因组是近年来兴起的研究生物体遗传物质空间结构及其动态调控和信息传递规律的学科,主要研究染色体内部的空间盘曲折叠或染色体间的立体相互关系对各种生命过程的影响及其调控机制。在真核细胞间期,不同染色体被规则排列在特定的“领地”内[5,6],而在一条染色体内部,不同染色质区域的开放状态、盘曲折叠的方式也不尽相同[7,8],形成不同的拓扑结构域[9,10]。以往人们只注重研究染色体上DNA的线性序列及其与蛋白质的结合和调控机制,因为线性序列承载了遗传信息的传递功能。但后来科学研究发现,基因组DNA一维线性序列中包含有三维基因组的架构信息,并且细胞核内复杂的三维基因组结构可以控制调控元件与靶基因之间的特异性相互作用,从而影响基因的时空表达,从立体层面调控着生命过程[11]。此外,许多疾病的发生发展已经被证明与三维基因组密切相关[12,13]。随着3C (chromosome conformation capture)、4C (circular chromosome conformation capture)、Hi-C (high- resolution chromosome conformation capture)和ChIA- PET (chromatin interaction analysis by paired-end tag sequencing)等各种构象捕获技术[14,15,16,17,18]以及高分辨率显微成像技术的发展,人们对染色体空间组织原则和三维架构规律的认识越来越深入。CRISPR/Cas9系统及其衍生技术更是极大地促进了三维基因组的研究。本文主要综述了CRISPR/Cas9基因编辑技术在三维基因组研究中的应用。
1 CRISPR/Cas9基因编辑机制
1.1 基因组单位点编辑
20世纪80年代开发的基因打靶技术实现了基因组中任何位点的编辑或改造,结合干细胞技术能够产生基因组靶向修饰的小鼠,有力推动了小鼠反向遗传学的发展。基因打靶技术通过直接同源重组的方法将外源DNA序列导入目标基因组,实现目标位点DNA序列的突变,删除或插入[19,20,21](图1A)。随后开发的锌指核酸酶(zinc finger nuclease, ZFN)和类转录激活因子效应物核酸酶(transcription activator- like effector nuclease, TALEN)分别将可以特异性识别DNA序列的锌指蛋白和类转录激活因子效应物与核酸内切酶Fok Ⅰ结合,造成基因组特定位点的双链断裂(double-strand break, DSB),接着利用细胞内的DNA修复系统,完成对基因组特定位点的改造。这两种技术的出现进一步促进了基因组的靶向切割和编辑领域的发展[22,23,24,25,26](图1,B和C)。2012年兴起的CRISPR/Cas9技术使得人们可以非常容易地靶向编辑几乎任何物种的基因组[2~4,27]。具体地讲,Cas9核酸内切酶在单个sgRNA的引导下能够切割基因组符合PAM (protospacer adjacent motif)规则的任意位点,引起DNA双链断裂损伤。在细胞内DNA损伤修复系统的作用下,单位点切割(单切)断裂处虽然被修复,但却可能会被引入突变,最终达到编辑基因组的效果(图1D)。图1
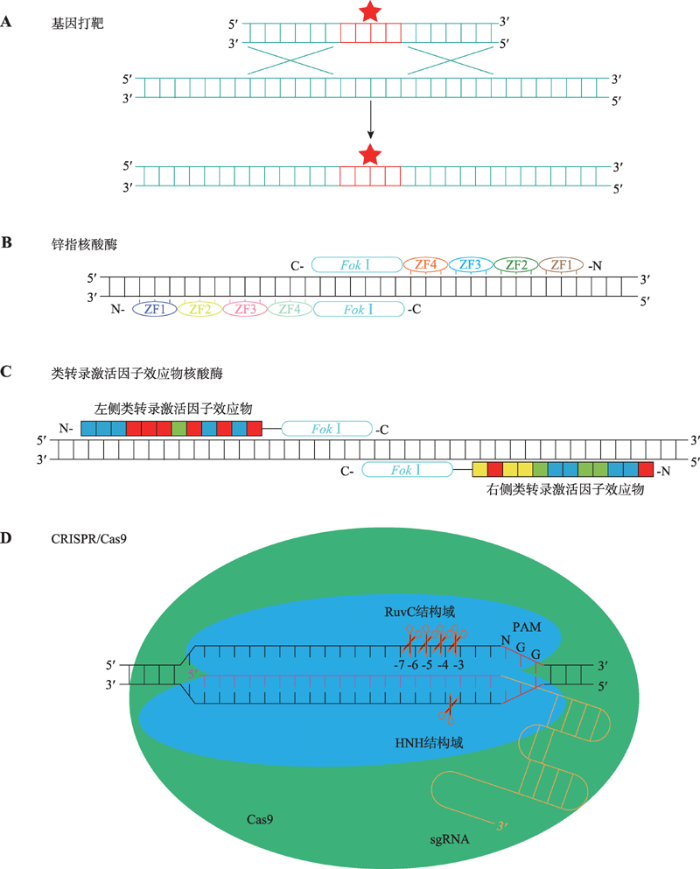 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1基因组编辑技术
A:基因打靶技术原理。基因打靶技术利用同源重组的方法将目的片段导入基因组。B:锌指核酸酶基因组编辑技术原理。将特异性识别DNA序列的锌指结构串联,与Fok Ⅰ核酸内切酶融合,实现基因组靶向切割。再利用DNA修复系统实现基因组编辑。C:类转录激活因子效应物核酸酶基因组编辑技术原理。将特异性识别DNA序列的类转录激活因子效应物串联,与Fok Ⅰ核酸内切酶融合,实现基因组靶向切割,再利用DNA修复系统实现基因组编辑。D:CRISPR/Cas9基因组编辑技术原理。Cas9蛋白可以被sgRNA引导至基因组特定位点,Cas9不具有核酸外切酶活性,仅仅具有核酸内切酶活性。Cas9的HNH和RuvC结构域分别催化互补链和非互补链的核酸内切反应造成DNA的双链断裂。HNH结构域造成的断裂位点位于PAM序列上游第3与第4个核苷酸之间,而RuvC结构域在PAM序列上游可造成多达4种不同的DNA断裂。
Fig. 1Schematic and summary of genome editing technologies
细胞的DNA修复系统主要包括同源重组(homologous recombination, HR)、非同源末端连接(nonhomologous end joining, NHEJ)和微同源末端连接(microhomology-mediated end joining, MMEJ)等。在没有模板的情况下,细胞主要选择NHEJ和MMEJ修复通路,产生随机突变进行基因组编辑。在提供模板的情况下,细胞利用HR修复通路产生遵照模板的精准编辑[27,28]。
1.2 CRISPR/Cas9双位点DNA片段编辑
单个sgRNA介导的单位点编辑很难造成基因组结构性变异(structural variation, SV),而一对sgRNA介导的双位点切割(CRISPR/Cas9双切系统)能够引起各种各样的染色质重排(chromosomal rearrangement),模拟基因组结构性变异。在一对sgRNA的引导下,Cas9核酸内切酶能够同时产生两个切口形成4个DNA双链断裂末端(Ⅰ~Ⅳ) (图2A)。如果断裂末端Ⅰ和断裂末端Ⅳ连接,则会造成两接口间的DNA片段删除(图2A)。由于DNA双链的Watson链和Crick链反向互补且都具有方向性,中间的DNA片段反转后仍然能够连接到基因组原位,也就是说断裂末端Ⅰ和Ⅲ以及断裂末端Ⅱ和Ⅳ相互连接,最终会形成DNA片段反转(图2A)。如果断裂末端Ⅲ和断裂末端Ⅱ连接,则会造成断裂末端Ⅰ和断裂末端Ⅳ中间的DNA片段重复或成环(图2A)。所以,CRISPR/Cas9双切系统能够形成DNA片段删除、反转、成环或重复,即所谓的DNA片段编辑[28,29,30]。由于三维基因组中存在大量的染色质重排,包括DNA片段删除、反转、重复、易位等,而这些染色质重排现象都可以利用DNA大片段编辑技术进行模拟,所以三维基因组结构和功能的研究为CRISPR/Cas9系统的DNA片段编辑技术提供了广阔的应用空间。同时,一对sgRNA编辑基因组的方法在应用于二倍体或多倍体生物中的高度同源序列时也表现出了足够的效率,这进一步扩展了CRISPR/ Cas9双切系统的应用前景[31]。图2
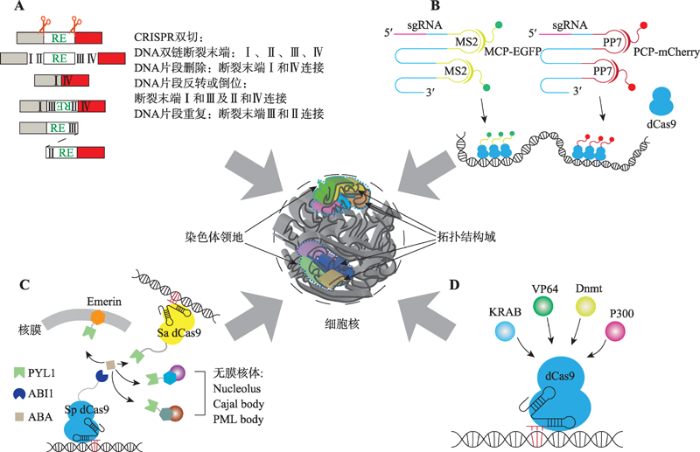 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2CRISPR在三维基因组研究中的技术方法
A:CRISPR介导的DNA片段编辑。通过一对sgRNA介导Cas9在基因组中进行靶向双位点切割,可以造成基因组片段删除、反转、重复或成环等现象,为三维基因组研究提供模型。B:CRISPR介导的基因组位点可视化。通过在sgRNA上加入MS2与PP7等茎环结构,招募融合了荧光蛋白的MCP、PCP等蛋白,将荧光信号特异性地标记在特定的基因组位点。C:CRISPR介导的基因组位点重定位。利用植物激素ABA介导的PYL1与ABI1的相互作用,将PYL1和ABI1分别结合在不同系统的dCas9或其他核体特有的蛋白上,可以将特定的基因组位点定位到新的目标位点。D:以CRISPR为平台的靶向效应系统。将激活或抑制转录以及表观遗传修饰相关蛋白结合在dCas9上,可以特异性地使基因组特定位点的转录水平以及表观遗传学修饰改变。
Fig. 2Application of CRISPR in 3D genomics
1.3 精准且可预测的基因编辑
通常只有同源重组通路介导的DNA修复被认为是精准的,所以CRISPR/Cas9基因编辑只有在提供模板的情况下通过HR通路实现精准编辑。但近年来的研究发现,经典的或传统的非同源末端连接修复通路(classic or canonic nonhomologous end joining, cNHEJ)也是细胞的精准修复通路,而选择性非同源末端连接修复通路(alternative nonhomologous end joining, aNHEJ或alt-NHEJ)和微同源末端连 接修复通路才是易错的(error-prone)[32]。所以,提高cNHEJ修复通路效率或抑制aNHEJ和MMEJ修复通路的效率,例如敲低CtIP或FANCD2等在aNHEJ修复通路中发挥作用的蛋白,可以在没有外源模板的情况下实现精准基因编辑[33]。与HR类似,MMEJ在修复过程中也利用了同源模板,不过其利用的同源序列较短,一般为5~25 bp左右。并且MMEJ使用的同源模板不是来自于外源导入而是位于DSB附近[34]。通过该通路,在不提供模板的情况下,精确的基因组编辑(如反转和删除)也可以被高质量地完成。Cas9切割DNA的功能是由HNH结构域以及RuvC结构域共同实现的。Cas9切割DNA时,HNH结构域切割sgRNA互补链,而RuvC结构域切割sgRNA非互补链。两个结构域制造的切点分别位于两条链上的PAM序列上游第3与第4个核苷酸之间,产生平头的双链DNA断裂末端。最近的研究发现,Cas9切割基因组时,RuvC结构域也有一定概率在PAM序列上游第4和第5个核苷酸间造成DNA断裂,从而在切割产物的5°端位置产生一个碱基突出的粘性末端。这些突出的粘性末端在细胞内源DNA聚合酶作用下能够被补平,并在DNA连接酶的作用下被精准连接。这就为CRISPR/Cas9基因编辑后可预测的单碱基插入提供了一条可能途径[32,33,35,36]。进一步利用DNA片段编辑方法研究发现,SpCas9核酸内切酶的RuvC结构域在PAM位点上游第5与第6以及第6与第7个核苷酸间均有一定几率造成DNA断裂,使切割产物产生两个或3个碱基突出的粘性末端,从而实现两个和3个碱基插入的可预测CRISPR/Cas9基因组编辑[37](图1D)。
因为单位点Cas9切割后产生的粘性末端很容易被完全修复并被再次切割,而一对sgRNA介导的双位点切割后产生的粘性末端在染色体重排后几乎不可能配对,所以在CRISPR/Cas9双切系统中,Cas9切割后产生的突出末端经过补平和连接后更容易被观察到[33,37]。也就是说,两个和3个碱基插入的效率在双切的情况下远远高于单切的情况[37],这就解释了为什么单切时几乎看不到一个以上碱基的可预测插入[35,36]。
2 CRISPR研究三维基因组的技术方法
CRISPR/Cas系统在三维基因组中的应用主要依靠的是其序列特异性结合的特性。利用该特性,改造或未被改造的多种Cas9蛋白可以被招募到基因组特定位点,进行切割或实现其他功能。2.1 CRISPR介导的DNA片段编辑
通过一对sgRNA介导的CRISPR/Cas9双位点切割,研究人员可以制作基因组调控元件插入、删除、反转的细胞或动植物模型,并利用这些模型研究三维基因组如何调控基因表达等生命过程,以及疾病发生发展的规律[29](图2A)。例如,染色质架构蛋白CTCF (CCCTC-binding factor)的结合位点反转、插入或敲除等都表明了CTCF对于细胞三维基因组架构维持和动态调控的重要性[11,38,39];CRISPR/Cas9双切系统介导的DNA大片段删除还能够筛选出基因组潜在的调控元件[40]。最近,利用DNA片段编辑技术发现了控制哺乳动物基因组复制事件时空特性的重要顺式元件[41]。删除与反转基因组部分片段会导致DNA复制时间的变化,说明这些片段包含着调控DNA复制的元件。单独删除与组合删除这些元件对DNA复制的影响不同,说明了这些元件在发挥作用时的相互依赖性。Hi-C结果也显示,这些元件的删除对于基因组区室划分以及拓扑结构域的强度都有影响。除此之外,多个靶向同一染色体不同位置的sgRNA介导的CRISPR/Cas9系统可以在老鼠或人类细胞中敲除一整条染色体,甚至将单倍体酵母的所有染色体融合在一起,创造出可以生存的只有一条染色体的酵母[42,43]。2.2 CRISPR介导的基因组位点可视化
生命信息传递以及基因组功能调节与染色质的空间组织架构和动态变化关系密切,而基因组位点在三维空间的直接可视化能够帮助理解基因组的空间组织架构以及其与基因表达调控等生命活动的关系。以往人们利用荧光原位杂交技术(fluorescence in situ hybridization, FISH)观察基因组位点在细胞核中的位置。而如今,利用CRISPR/Cas系统结合基因组的序列特异性,失活的dCas9 (nuclease-deactivated Cas9, D10A和H841A)荧光蛋白嵌合体可以被sgRNA招募到基因组的特定位点,从而使活细胞的特定染色体片段的时空动态可视化。Chen等[44]利用该方法观察了端粒在活细胞中的动态变化,发现端粒在受损后,在细胞核内的移动显著增加。同时他们还观察了编码基因MUC4在有丝分裂中的细胞核内的位置变化[44]。dCas9/sgRNA复合体的稳定性以及dCas9蛋白结合DNA的高亲和性使得该方法可以被方便高效广泛地应用于细胞基因组特定位点标记。在体外孵育组装的dCas9/sgRNA复合物也可以与细胞内的DNA杂交,并且这样在体外组装的系统可以在基因组不同的位点标记不同的颜色,从而同时检测多个位点的位置,而这是研究染色质相互作用的关键[45]。同时使用多种细菌的同源dCas9/sgRNA组合也可以构建多色CRISPR/dCas9可视化系统[46,47]。除此之外,另一种策略也可以解决多色标记问题:在sgRNA的支架上加入两个拷贝的MS2或PP7 RNA茎环结构(图2B)。MS2可以被MCP (MS2 coat protein)蛋白特异性识别,而PP7可以被PCP (PP7 coat protein)蛋白特异性识别。向细胞内转入MCP-EGFP和PCP- mCherry融合蛋白的表达载体后,不同基因组靶点就可以被EGFP和mCherry同时标记[48,49]。在此基础上,CRISPRainbow等更加高效的技术进一步促进了基因组可视化的发展[50]。此外,dCas9的N端与C端的核定位信号(nuclear localization sequences, NLS)插入,sgRNA支架上的U-A翻转、发夹结构延长等一些技术上的难题被攻克也使得可视化更加高效稳定[44,51]。染色体上重复的DNA元件可以被单独一种特异性的sgRNA结合CRISPR/dCas9系统稳定地显示,而非重复的DNA元件也可以被一系列串联覆盖目的片段的sgRNA协同CRISPR/dCas9系统标记出来。在sgRNA支架上加入更多的MS2等茎环结构可以增强单个位点的荧光强度,减少标记非重复区域时所需的sgRNA的数量[52]。提高sgRNA的表达水平并利用诸如强力霉素(doxycycline)介导的启动子等方法降低dCas9蛋白的表达可以降低信噪比。
2.3 CRISPR介导的基因组位点重定位
基因组位点在核内的位置与基因表达是否有联系?这是三维基因组学关注的关键问题之一。程序性的控制基因组空间架构对于研究细胞核内结构如何影响基因调控和细胞功能十分重要。参考LacI- LacO方法[53,54],依赖于CRISPR/dCas9系统的CRISPR- GO (CRISPR-genome organization)方法被建立起来。该方法利用植物激素脱落酸(abscisic acid, ABA)介导的PYL1 (pyrabactin resistance (PYR)-like)与ABI1 (ABA insensitive 1)的相互作用,将ABI1融合至dCas9蛋白上,而将PYL1融合至核膜蛋白Emerin上或其他核体如卡哈尔体(Cajal body)的标志物Coilin上。这样通过dCas9/sgRNA的靶向作用,就可以将基因组特定片段重定位至细胞核外周或与无膜核体共定位。该方法对于基因组重复和非重复区域都十分有效,并且在除去ABA后,重定位的效果是可逆的[55]。类似的,Morgan等[56]利用金黄色葡萄球菌(Staphylococcus aureus, Sa)和酿脓性链球菌(Streptococcus pyogenes, Sp)的dCas9,结合ABA信号通路相关组件,开发出了介导两个基因组位点共定位的工具CLOuD9 (chromatin loop reorganization using CRISPR-dCas9)。该工具可以可逆地介导染色质环的形成,改变特定区域基因的表达(图2C)。
2.4 以CRISPR为平台的靶向效应系统
CRISPR/Cas系统为蛋白靶向DNA片段提供了一个特异性良好的稳定平台。将dCas9与具有不同调节功能的效应因子结合,利用sgRNA可以将dCas9靶向传递到特定的基因组位点的特性,就可以实现特定位点的功能调控。研究人员在这一方面已经做了大量的尝试[57,58,59,60,61,62],其中以CRISPR interference (CRISPRi)最为典型[63]。在大肠杆菌和人类细胞中,CRISPRi通过sgRNA将dCas9靶向特定的蛋白编码区域,利用dCas9结合DNA的空间位阻,通过干扰RNA聚合酶介导的转录延伸抑制相关基因的表达。这种方法可逆且高效,可以同时针对多个基因进行干扰。在此基础上,在dCas9后加入转录抑制或激活因子(如KRAB或VP64),可以实现特定基因转录的抑制或激活[64]。除此之外,将激活或抑制效应因子与植物激素诱导系统和CRISPR/Cas系统相结合,可以实现可诱导可逆转的基因表达激活或抑制。两套植物激素诱导系统同时联用使得“AND”或“OR”这样的逻辑调控成为可能[65]。但是,dCas9融合蛋白在活体内的转导效率很低。另外,在体转导经常使用病毒载体,而编码dCas9/ gRNA和转录激活因子的序列长度超过了大部分常见病毒载体可以承受的范围,因此将靶向基因激活系统用于在体研究是一个难点。Liao等[62]开发了一个能够装载在一个病毒载体内、dgRNA (dead sgRNA, 识别序列为14或15 bp,可以抑制Cas9的切割活性)与MPH (MS2:P65:HSF1)转录激活复合物融合、具有高靶基因激活水平的系统,将其注射入表达Cas9的小鼠体内,可以有效地调节基因表达,改变基因位点的表观遗传学状态,从而改善小鼠模型的疾病表型。并且该系统因其使用的Cas9并未失去核酸酶活性,故可以同时介导不同基因的转录激活和基因敲除[62]。该系统不仅进一步推动了靶向基因激活系统的发展,同时也证明了CRISPR在疾病治疗应用中的前景。将dCas9与胞嘧啶脱氨酶融合在一起,可以作为靶向的单碱基编辑器[61,66,67]。这一技术可以用于修正某些疾病中的单碱基突变,同样也可以用于验证转录因子结合基序(motif)中的关键碱基,阐明转录因子与DNA的结合方式。
通过dCas9与具有甲基化、去甲基化、乙酰化和去乙酰化等功能的蛋白质的融合,CRISPR/Cas系统亦可用于特定位点表观遗传的修饰[60,68~70]。这种方法不会改变全基因组的表观遗传修饰,仅仅改变基因组特定位点和组蛋白的表观遗传状态,靶向操纵常染色质与异染色质状态的转变。因此,这样的方法可以操控特定位点的表观遗传状态,提供以往难以得到的证据与结论,在三维基因组研究中尤为重要。最后,通过靶向调控特定基因的表达,该技术亦具有癌症治疗的潜能。例如,将dCas9的C端融合KRAB (the Krüppel-associated box transcriptional repression domain)等表观遗传抑制因子,靶向抑制肝癌中表达上调且促进肿瘤发展的颗粒蛋白(Granulin, GRN)基因的表达,可以抑制肿瘤细胞增殖,减少肿瘤球的形成[70]。此外,使用dCas9靶向激活肿瘤细胞中肿瘤相关抗原(tumor-associated antigens, TAAs)的表达,可以诱导机体对肿瘤细胞产生更强的免疫识别,从而利用免疫系统清除肿瘤细胞[71](图2D)。
3 CRISPR研究三维基因组架构的机制
3.1 研究三维基因组的调控元件
人类基因组约2%的编码区包含2万多个基因,但这2万多个基因的时空表达却受到存在于98%的人类基因组非编码区中多达几百万个调控元件的精细控制。常见的基因组调控元件有启动子(promoter)、增强子(enhancer)、绝缘子(insulator)、基因座控制区(LCR: locus control region)等,而增强子、绝缘子、基因座控制区等远程调控元件通常通过与启动子之间的特异性空间相互作用调控基因表达,所以它们在基因表达调控以及三维基因组架构调节中发挥着重要作用。如何研究它们的功能及其具体机制是一个重要的难题,而CRISPR/Cas9系统则为解决这个难题提供了机遇。因为一个sgRNA介导的Cas9单位点切割,能够在编码区造成小的插入或删除,引起蛋白移码突变,从而破坏基因功能,所以利用一个sgRNA的CRISPR/Cas9单位点切割系统在研究基因组编码区的功能中发挥了重要作用。由于非编码区不存在移码突变,Cas9单切后通常不能完全破坏非编码区的功能,单位点切割的CRISPR/Cas9系统在非编码区研究中难以发挥作用。而利用两个sgRNA的CRISPR/Cas9双位点切割系统(CRISPR/Cas9双切系统)很容易把单个调控元件完全敲除,从而容易研究成千上万的三维基因组调控元件。另外CRISPR/ Cas9双切系统除了造成DNA片段敲除,还可以造成DNA片段的反转、重复、易位等染色体结构性变异,因此DNA片段编辑对于研究DNA元件在三维基因组调控与功能中作用机制非常重要。例如,秀丽线虫(Caenorhabditis elegans)中X染色体的计量补偿效应是性染色体特殊的空间构象造成的现象。此现象是由于计量补偿复合物(dosage compensation complex, DCC)被招募到rex (recruitment elements on X)位点引起的。使用CRISPR/Cas9技术敲除rex位点能够引起这种特殊的空间构象消失,同时DCC对X染色体基因表达的抑制作用也随之消失[72]。此外,将CRISPRi系统与单细胞RNA-seq联用,通过对假定的调控元件进行组合干扰,能够筛选出基因的调控元件[73]。
3.2 CTCF架构机制
利用CRISPR/Cas9双切系统对染色质架构蛋白CTCF结合的DNA片段进行删除或反转,结合染色质构象捕获技术,证明了CTCF对于三维基因组结构的重要性以及其DNA结合序列的方向性[11,74~76]。首先,一对方向收敛或者说正向-反向(convergent or forward-reverse)的CTCF的结合位点(CTCF-binding site, CBS)能够更容易地介导CTCF与cohesin一起控制染色质环形成。其次,一对方向发散或者说反向-正向(divergent or reverse-forward)的CBSs通常位于拓扑结构域(topologically associating domains, TADs)的边界。再次,串联排列方向一致的CBSs能够平衡启动子的选择[77]。CTCF/cohesin介导的染色质环保证了基因组的增强子与合适的启动子相互作用,隔绝错误的DNA调控元件之间的联系,维持正常的基因表达,而CTCF结合位点的删除或反转的转变会改变染色质环的结构从而影响基因表达、发育分化甚至导致畸形或癌症等疾病的发生[11,78,79]。3.3 增强子的方向性
哺乳动物基因组内许多增强子元件的附近都含有CBS,而CBS的方向可以决定CTCF结合DNA的方向,从而影响CTCF介导的染色质环的形成。这使得一些含有CBS的增强子方向特异性地与合适的启动子相互作用,如原钙粘蛋白(protocadherin, Pcdh)α基因簇的增强子HS5-1[80,81](图3A)。原钙粘蛋白基因座包含α、β、γ 3个基因簇,并且基因启动子与增强子区域含有大量的CBSs (图3A)。在原钙粘蛋白基因座中,除了αc2、β1、γc4和γc5的启动子外,每个启动子区都含有正向的CBS,而增强子通常含有反向的CBS。正向-反向的CBS可以介导CTCF与cohesin一起形成染色质环,控制启动子与增强子间的相互作用。原钙粘蛋白α基因簇的表达主要由其下游的两个增强子HS7以及HS5-1调控。原钙粘蛋白βγ基因簇位点的表达主要由其下游的超级增强子调控[82,83]。
图3
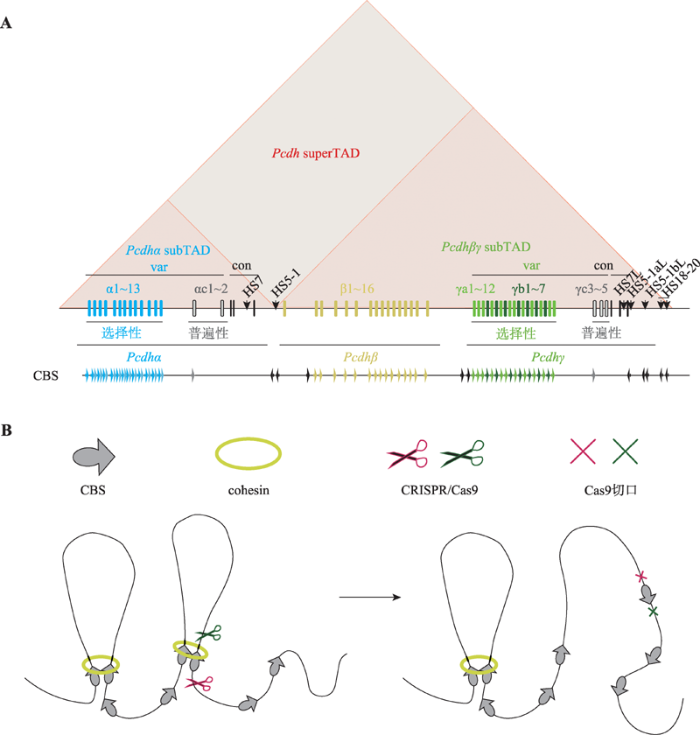 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3人类原钙粘蛋白基因簇结构以及CRISPR介导的基因组编辑
A:人类原钙粘蛋白基因簇。人类原钙粘蛋白基因座包含α、β、γ 3个基因簇,并且在基因启动子与增强子区域包含大量的CTCF位点(CTCF-binding site, CBS)。通过方向性结合这些CBSs,CTCF可以介导染色质环形成,调控原钙粘蛋白基因表达。B:CRISPR系统介导的基因组元件反转改变染色质拓扑结构。基因组折叠的染色质环挤出模型(chromatin loop extrusion):沿染色质滑动的cohesin可以将染色质挤出形成染色质环,而cohesin的滑动会被一对方向收敛或者说正向-反向(convergent or forward-reverse)的CBS上的CTCF阻滞,这使得染色质环挤出的进程终止,两个CBS在三维空间上被拉近。利用CRISPR/Cas9系统反转CBS方向能够改变染色质环化方向,证明了CTCF位点的方向性在三维基因组折叠中的关键作用。
Fig. 3Human protocadherin (Pcdh) gene clusters and CRISPR-mediated genome editing
原钙粘蛋白α基因簇的增强子HS5-1含有两个反向的CBSs,通常情况下,这两个反向的CBSs会介导HS5-1与上游的含有正向CBS的原钙粘蛋白α启动子形成染色质环,从而使得HS5-1激活原钙粘蛋白α而不是下游的原钙粘蛋白βγ基因簇表达。如果使用CRISPR/Cas9双切系统在小鼠与细胞内对HS5-1进行反转,可以观察到HS5-1与下游的βγ基因簇相互作用显著增加,同时,与上游的α基因簇启动子的相互作用减少(图3B)。原钙粘蛋白α的转录也相应地发生了显著下降[11,84]。这些现象都说明HS5-1在染色体原位环境中是以方向依赖性的方式调控原钙粘蛋白α基因簇的表达。
3.4 绝缘子作用机制
哺乳动物基因组中的绝缘子元件可以隔绝增强子与不合适的启动子间的相互作用,也可以分割常染色质与异染色质以及不同的TAD区域。那么其潜在的机制是怎样的呢?CRISPR/Cas9双切系统介导的DNA片段插入、删除与反转技术证明了CBS可以不依赖方向地实现绝缘子的功能[77]。在原钙粘蛋白α基因簇的启动子与增强子HS5-1之间插入单个或多个,正向或反向,成对的方向收敛或方向发散的CBSs后,使用定量高分辨率4C技术(quantitative high-resolution chromosome-conformation capture, QHR-4C)测定这些CBSs与基因组位点间的相互作用强弱。QHR-4C数据表明,这些CBSs都发挥了绝缘子的功能。这些插入的CBSs替代了与自己同向的CBSs的功能,和远处方向与自己相对的CBSs形成染色质环,从而破坏了原有的启动子-增强子相互作用,承担绝缘子的角色[77]。
删除原钙粘蛋白基因簇内原有的CBS会导致启动子或增强子跨过该CBS与更远处的DNA元件相互作用。这些结果说明原钙粘蛋白基因簇内原有的CBSs也作为绝缘子而存在着。
综合上面的现象可以发现,CBS其实是通过与其他方向相对的CBS形成染色质环从而隔离出一个个区室,防止不适当的DNA元件间相互作用,实现其绝缘子功能。
4 结语与展望
三维基因组研究对于人们深入理解发育、分化、基因表达调控等生命过程至关重要,而CRISPR/ Cas9系统则是研究三维基因组的重要工具。与其他技术相比,CRISPR/Cas9系统在精确性、可塑性、性价比和适用性等方面都有巨大的优势。但是如何利用这些优势并开发新的衍生技术则是未来努力的方向。随着CRISPR/Cas9介导的基因组编辑越来越精准和可预测以及一对sgRNA介导DNA片段编辑技术的广泛使用[33],研究者可以花费更少的精力得到想要的突变类型,这对于快速发展的三维基因组研究具有巨大的推动作用。而开发编辑效果更为准确且脱靶效应更少的Cas9突变体以及更准确的CRISPR编辑方法,将会有助于CRISPR/Cas9系统更多更好地应用于三维基因组研究[85,86]。。
利用CRISPR/Cas9系统对染色质架构蛋白进行高通量的筛选,可以找到在基因组复杂网络中在三维架构方面发挥重要功能的因子。使用CRISPR文库干扰或敲除一系列潜在的染色质架构蛋白后,在经典的基因座如原钙粘蛋白[80,81]和β-珠蛋白(β-globin)[11]等位点及其超级增强子区域中,利用染色质构象捕获等技术可以筛选出对染色质环或TADs等三维基因组重要组件的形成具有重要意义的蛋白。
对于原钙粘蛋白α基因簇内增强子HS5-1的反转降低了HS5-1与上游原钙粘蛋白α各启动子的相互作用,并且增加了HS5-1与下游原钙粘蛋白βγ基因簇的相互作用(图3B)。这样的改变虽然显著降低了原钙粘蛋白α的转录水平,却没有增强原钙粘蛋白βγ的转录水平[11]。增强子和启动子之间通过远程相互作用形成的染色质环调控基因转录水平是一个研究热点和难点。
虽然有很多研究证明染色质的拓扑结构性质与TAD对于基因表达十分重要[38,78,79],但是最近的研究发现,将果蝇的染色体高度重排后,果蝇的基因表达受到的影响并不大[87]。虽然重排后的基因组内出现了反转、删除和重复等许多结构性变异,拓扑结构性质发生巨大的变化,染色质环和TAD等三维基因组高级结构被破坏,但是表达受到影响的基因只是一小部分。TAD的变化并不能很好的预测基因表达的变化[87]。这样的发现挑战了三维基因组学的以往认知。这一发现是否说明TADs对于全基因组基因表达的影响没有以前预测的那么重要?是否是果蝇染色体的特殊性导致了这样的结果?染色质环等三维基因组结构是否只在小范围的尺度影响着基因表达?这些猜测都有待进一步的研究验证。
总之,CRISPR/Cas9系统在靶向三维基因组的精确性、易用性等方面都具有巨大的优势。相信随着CRISPR/Cas9基因编辑及其衍生技术的进一步发展,这些新兴遗传学技术在三维基因组研究中会发挥越来越重要的作用。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1126/science.1138140URLPMID:17379808 [本文引用: 1]

Clustered regularly interspaced short palindromic repeats (CRISPR) are a distinctive feature of the genomes of most Bacteria and Archaea and are thought to be involved in resistance to bacteriophages. We found that, after viral challenge, bacteria integrated new spacers derived from phage genomic sequences. Removal or addition of particular spacers modified the phage-resistance phenotype of the cell. Thus, CRISPR, together with associated cas genes, provided resistance against phages, and resistance specificity is determined by spacer-phage sequence similarity.
DOI:10.1126/science.1225829URLPMID:22745249 [本文引用: 2]
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems provide bacteria and archaea with adaptive immunity against viruses and plasmids by using CRISPR RNAs (crRNAs) to guide the silencing of invading nucleic acids. We show here that in a subset of these systems, the mature crRNA that is base-paired to trans-activating crRNA (tracrRNA) forms a two-RNA structure that directs the CRISPR-associated protein Cas9 to introduce double-stranded (ds) breaks in target DNA. At sites complementary to the crRNA-guide sequence, the Cas9 HNH nuclease domain cleaves the complementary strand, whereas the Cas9 RuvC-like domain cleaves the noncomplementary strand. The dual-tracrRNA:crRNA, when engineered as a single RNA chimera, also directs sequence-specific Cas9 dsDNA cleavage. Our study reveals a family of endonucleases that use dual-RNAs for site-specific DNA cleavage and highlights the potential to exploit the system for RNA-programmable genome editing.
DOI:10.1073/pnas.1208507109URLPMID:22949671 [本文引用: 1]
Clustered, regularly interspaced, short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems provide adaptive immunity against viruses and plasmids in bacteria and archaea. The silencing of invading nucleic acids is executed by ribonucleoprotein complexes preloaded with small, interfering CRISPR RNAs (crRNAs) that act as guides for targeting and degradation of foreign nucleic acid. Here, we demonstrate that the Cas9-crRNA complex of the Streptococcus thermophilus CRISPR3/Cas system introduces in vitro a double-strand break at a specific site in DNA containing a sequence complementary to crRNA. DNA cleavage is executed by Cas9, which uses two distinct active sites, RuvC and HNH, to generate site-specific nicks on opposite DNA strands. Results demonstrate that the Cas9-crRNA complex functions as an RNA-guided endonuclease with RNA-directed target sequence recognition and protein-mediated DNA cleavage. These findings pave the way for engineering of universal programmable RNA-guided DNA endonucleases.
DOI:10.1038/nprot.2013.143URL [本文引用: 2]
Targeted nucleases are powerful tools for mediating genome alteration with high precision. The RNA-guided Cas9 nuclease from the microbial clustered regularly interspaced short palindromic repeats (CRISPR) adaptive immune system can be used to facilitate efficient genome engineering in eukaryotic cells by simply specifying a 20-nt targeting sequence within its guide RNA. Here we describe a set of tools for Cas9-mediated genome editing via nonhomologous end joining (NHEJ) or homology-directed repair (HDR) in mammalian cells, as well as generation of modified cell lines for downstream functional studies. To minimize off-target cleavage, we further describe a double-nicking strategy using the Cas9 nickase mutant with paired guide RNAs. This protocol provides experimentally derived guidelines for the selection of target sites, evaluation of cleavage efficiency and analysis of off-target activity. Beginning with target design, gene modifications can be achieved within as little as 1-2 weeks, and modified clonal cell lines can be derived within 2-3 weeks.
DOI:10.1101/cshperspect.a003889URLPMID:20300217 [本文引用: 1]
Chromosome territories (CTs) constitute a major feature of nuclear architecture. In a brief statement, the possible contribution of nuclear architecture studies to the field of epigenomics is considered, followed by a historical account of the CT concept and the final compelling experimental evidence of a territorial organization of chromosomes in all eukaryotes studied to date. Present knowledge of nonrandom CT arrangements, of the internal CT architecture, and of structural interactions with other CTs is provided as well as the dynamics of CT arrangements during cell cycle and postmitotic terminal differentiation. The article concludes with a discussion of open questions and new experimental strategies to answer them.
DOI:10.1126/science.aat5641URLPMID:30166492 [本文引用: 1]
Three-dimensional genome structures play a key role in gene regulation and cell functions. Characterization of genome structures necessitates single-cell measurements. This has been achieved for haploid cells but has remained a challenge for diploid cells. We developed a single-cell chromatin conformation capture method, termed Dip-C, that combines a transposon-based whole-genome amplification method to detect many chromatin contacts, called META (multiplex end-tagging amplification), and an algorithm to impute the two chromosome haplotypes linked by each contact. We reconstructed the genome structures of single diploid human cells from a lymphoblastoid cell line and from primary blood cells with high spatial resolution, locating specific single-nucleotide and copy number variations in the nucleus. The two alleles of imprinted loci and the two X chromosomes were structurally different. Cells of different types displayed statistically distinct genome structures. Such structural cell typing is crucial for understanding cell functions.
DOI:10.1038/nature12753URL [本文引用: 1]
How a complex animal can arise from a fertilized egg is one of the oldest and most fascinating questions of biology, the answer to which is encoded in the genome. Body shape and organ development, and their integration into a functional organism all depend on the precise expression of genes in space and time. The orchestration of transcription relies mostly on surrounding control sequences such as enhancers, millions of which form complex regulatory landscapes in the non-coding genome. Recent research shows that high-order chromosome structures make an important contribution to enhancer functionality by triggering their physical interactions with target genes.
DOI:10.1016/j.molcel.2013.02.011URLPMID:23473598 [本文引用: 1]
Mammalian genomes encode genetic information in their linear sequence, but appropriate expression of their genes requires chromosomes to fold into complex three-dimensional structures. Transcriptional control involves the establishment of physical connections among genes and regulatory elements, both along and between chromosomes. Recent technological innovations in probing the folding of chromosomes are providing new insights into the spatial organization of genomes and its role in gene regulation. It is emerging that folding of large complex chromosomes involves a hierarchy of structures, from chromatin loops that connect genes and enhancers to larger chromosomal domains and nuclear compartments. The larger these structures are along this hierarchy, the more stable they are within cells, while becoming more stochastic between cells. Here, we review the experimental and theoretical data on this hierarchy of structures and propose a key role for the recently discovered topologically associating domains.
DOI:10.1016/j.molcel.2016.05.018URLPMID:27259200 [本文引用: 1]
How eukaryotic chromosomes fold inside the nucleus is an age-old question that remains unanswered today. Early biochemical and microscopic studies revealed the existence of chromatin domains and loops as a pervasive feature of interphase chromosomes, but the biological implications of such organizational features were obscure. Genome-wide analysis of pair-wise chromatin interactions using chromatin conformation capture (3C)-based techniques has shed new light on the organization of chromosomes in interphase nuclei. Particularly, the finding of cell-type invariant, evolutionarily conserved topologically associating domains (TADs) in a broad spectrum of cell types has provided a new molecular framework for the study of animal development and human diseases. Here, we review recent progress in characterization of such chromatin domains and delineation of mechanisms of their formation in animal cells.
DOI:10.1016/j.cell.2016.02.007URLPMID:26967279 [本文引用: 1]
Proper expression of genes requires communication with their regulatory elements that can be located elsewhere along the chromosome. The physics of chromatin fibers imposes a range of constraints on such communication. The molecular and biophysical mechanisms by which chromosomal communication is established, or prevented, have become a topic of intense study, and important roles for the spatial organization of chromosomes are being discovered. Here we present a view of the interphase 3D genome characterized by extensive physical compartmentalization and insulation on the one hand and facilitated long-range interactions on the other. We propose the existence of topological machines dedicated to set up and to exploit a 3D genome organization to both promote and censor communication along and between chromosomes.
DOI:10.1016/j.cell.2015.07.038URLPMID:26276636 [本文引用: 7]
CTCF and the associated cohesin complex play a central role in insulator function and higher-order chromatin organization of mammalian genomes. Recent studies identified a correlation between the orientation of CTCF-binding sites (CBSs) and chromatin loops. To test the functional significance of this observation, we combined CRISPR/Cas9-based genomic-DNA-fragment editing with chromosome-conformation-capture experiments to show that the location and relative orientations of CBSs determine the specificity of long-range chromatin looping in mammalian genomes, using protocadherin (Pcdh) and β-globin as model genes. Inversion of CBS elements within the Pcdh enhancer reconfigures the topology of chromatin loops between the distal enhancer and target promoters and alters gene-expression patterns. Thus, although enhancers can function in an orientation-independent manner in reporter assays, in the native chromosome context, the orientation of at least some enhancers carrying CBSs can determine both the architecture of topological chromatin domains and enhancer/promoter specificity. These findings reveal how 3D chromosome architecture can be encoded by linear genome sequences.
DOI:10.1038/nrm.2016.138URLPMID:27826147 [本文引用: 1]
Genetic variation associated with disease often appears in non-coding parts of the genome. Understanding the mechanisms by which this phenomenon leads to disease is necessary to translate results from genetic association studies to the clinic. Assigning function to this type of variation is notoriously difficult because the human genome harbours a complex regulatory landscape with a dizzying array of transcriptional regulatory sequences, such as enhancers that have unpredictable, promiscuous and context-dependent behaviour. In this Review, we discuss how technological advances have provided increasingly detailed information on genome folding; for example, genome folding forms loops that bring enhancers and target genes into close proximity. We also now know that enhancers function within topologically associated domains, which are structural and functional units of chromosomes. Studying disease-associated mutations and chromosomal rearrangements in the context of the 3D genome will enable the identification of dysregulated target genes and aid the progression from descriptive genetic association results to discovering molecular mechanisms underlying disease.
.
DOI:10.1083/jcb.201611001URLPMID:28855250 [本文引用: 1]
Mammalian genomes are folded into unique topological structures that undergo precise spatiotemporal restructuring during healthy development. Here, we highlight recent advances in our understanding of how the genome folds inside the 3D nucleus and how these folding patterns are miswired during the onset and progression of mammalian disease states. We discuss potential mechanisms underlying the link among genome misfolding, genome dysregulation, and aberrant cellular phenotypes. We also discuss cases in which the endogenous 3D genome configurations in healthy cells might be particularly susceptible to mutation or translocation. Together, these data support an emerging model in which genome folding and misfolding is critically linked to the onset and progression of a broad range of human diseases.
DOI:10.1126/science.1067799URLPMID:11847345 [本文引用: 1]
We describe an approach to detect the frequency of interaction between any two genomic loci. Generation of a matrix of interaction frequencies between sites on the same or different chromosomes reveals their relative spatial disposition and provides information about the physical properties of the chromatin fiber. This methodology can be applied to the spatial organization of entire genomes in organisms from bacteria to human. Using the yeast Saccharomyces cerevisiae, we could confirm known qualitative features of chromosome organization within the nucleus and dynamic changes in that organization during meiosis. We also analyzed yeast chromosome III at the G1 stage of the cell cycle. We found that chromatin is highly flexible throughout. Furthermore, functionally distinct AT- and GC-rich domains were found to exhibit different conformations, and a population-average 3D model of chromosome III could be determined. Chromosome III emerges as a contorted ring.
DOI:10.1038/ng1891URLPMID:17033624 [本文引用: 1]
Accumulating evidence converges on the possibility that chromosomes interact with each other to regulate transcription in trans. To systematically explore the epigenetic dimension of such interactions, we devised a strategy termed circular chromosome conformation capture (4C). This approach involves a circularization step that enables high-throughput screening of physical interactions between chromosomes without a preconceived idea of the interacting partners. Here we identify 114 unique sequences from all autosomes, several of which interact primarily with the maternally inherited H19 imprinting control region. Imprinted domains were strongly overrepresented in the library of 4C sequences, further highlighting the epigenetic nature of these interactions. Moreover, we found that the direct interaction between differentially methylated regions was linked to epigenetic regulation of transcription in trans. Finally, the patterns of interactions specific to the maternal H19 imprinting control region underwent reprogramming during in vitro maturation of embryonic stem cells. These observations shed new light on development, cancer epigenetics and the evolution of imprinting.
DOI:10.1038/ng1896URLPMID:17033623 [本文引用: 1]
The spatial organization of DNA in the cell nucleus is an emerging key contributor to genomic function. We developed 4C technology (chromosome conformation capture (3C)-on-chip), which allows for an unbiased genome-wide search for DNA loci that contact a given locus in the nuclear space. We demonstrate here that active and inactive genes are engaged in many long-range intrachromosomal interactions and can also form interchromosomal contacts. The active beta-globin locus in fetal liver preferentially contacts transcribed, but not necessarily tissue-specific, loci elsewhere on chromosome 7, whereas the inactive locus in fetal brain contacts different transcriptionally silent loci. A housekeeping gene in a gene-dense region on chromosome 8 forms long-range contacts predominantly with other active gene clusters, both in cis and in trans, and many of these intra- and interchromosomal interactions are conserved between the tissues analyzed. Our data demonstrate that chromosomes fold into areas of active chromatin and areas of inactive chromatin and establish 4C technology as a powerful tool to study nuclear architecture.
DOI:10.1126/science.1181369URLPMID:19815776 [本文引用: 1]
We describe Hi-C, a method that probes the three-dimensional architecture of whole genomes by coupling proximity-based ligation with massively parallel sequencing. We constructed spatial proximity maps of the human genome with Hi-C at a resolution of 1 megabase. These maps confirm the presence of chromosome territories and the spatial proximity of small, gene-rich chromosomes. We identified an additional level of genome organization that is characterized by the spatial segregation of open and closed chromatin to form two genome-wide compartments. At the megabase scale, the chromatin conformation is consistent with a fractal globule, a knot-free, polymer conformation that enables maximally dense packing while preserving the ability to easily fold and unfold any genomic locus. The fractal globule is distinct from the more commonly used globular equilibrium model. Our results demonstrate the power of Hi-C to map the dynamic conformations of whole genomes.
DOI:10.1038/nature08497URLPMID:19890323 [本文引用: 1]
Genomes are organized into high-level three-dimensional structures, and DNA elements separated by long genomic distances can in principle interact functionally. Many transcription factors bind to regulatory DNA elements distant from gene promoters. Although distal binding sites have been shown to regulate transcription by long-range chromatin interactions at a few loci, chromatin interactions and their impact on transcription regulation have not been investigated in a genome-wide manner. Here we describe the development of a new strategy, chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) for the de novo detection of global chromatin interactions, with which we have comprehensively mapped the chromatin interaction network bound by oestrogen receptor alpha (ER-alpha) in the human genome. We found that most high-confidence remote ER-alpha-binding sites are anchored at gene promoters through long-range chromatin interactions, suggesting that ER-alpha functions by extensive chromatin looping to bring genes together for coordinated transcriptional regulation. We propose that chromatin interactions constitute a primary mechanism for regulating transcription in mammalian genomes.
DOI:10.1038/292154a0URLPMID:7242681 [本文引用: 1]
DOI:10.1038/317230a0URLPMID:2995814 [本文引用: 1]
A 'rescuable' plasmid containing globin gene sequences allowing recombination with homologous chromosomal sequences has enabled us to produce, score and clone mammalian cells with the plasmid integrated into the human beta-globin locus. The planned modification was achieved in about one per thousand transformed cells whether or not the target gene was expressed.
DOI:10.1016/0092-8674(86)90463-0URLPMID:3002636 [本文引用: 1]
We corrected a defective gene residing in the chromosome of a mammalian cell by injecting into the nucleus copies of the same gene carrying a different mutation. We determined how the number, the arrangement, and the chromosomal position of the integrated gene, as well as the number of injected molecules influence the gene-targeting frequency. Recombination between the newly introduced DNA and its chromosomal homolog occurred at a frequency of 1 in 10(3) cells receiving DNA. Correction events were mediated by either double reciprocal recombination or gene conversion. This resulted in sequences in the genome being replaced by sequences of the introduced DNA or, in separate experiments, sequences in the incoming DNA being replaced by chromosomal sequences. Both point mutations and deletion mutations were corrected; however, the nature of the mutation carried by the respective sequence influenced whether the integrated or injected sequence was corrected.
DOI:10.1073/pnas.93.3.1156URLPMID:8577732 [本文引用: 1]
A long-term goal in the field of restriction-modification enzymes has been to generate restriction endonucleases with novel sequence specificities by mutating or engineering existing enzymes. This will avoid the increasingly arduous task of extensive screening of bacteria and other microorganisms for new enzymes. Here, we report the deliberate creation of novel site-specific endonucleases by linking two different zinc finger proteins to the cleavage domain of Fok I endonuclease. Both fusion proteins are active and under optimal conditions cleave DNA in a sequence-specific manner. Thus, the modular structure of Fok I endonuclease and the zinc finger motifs makes it possible to create "artificial" nucleases that will cut DNA near a predetermined site. This opens the way to generate many new enzymes with tailor-made sequence specificities desirable for various applications.
DOI:10.1128/MCB.21.1.289-297.2001URLPMID:11113203 [本文引用: 1]
Chimeric nucleases that are hybrids between a nonspecific DNA cleavage domain and a zinc finger DNA recognition domain were tested for their ability to find and cleave their target sites in living cells. Both engineered DNA substrates and the nucleases were injected into Xenopus laevis oocyte nuclei, in which DNA cleavage and subsequent homologous recombination were observed. Specific cleavage required two inverted copies of the zinc finger recognition site in close proximity, reflecting the need for dimerization of the cleavage domain. Cleaved DNA molecules were activated for homologous recombination; in optimum conditions, essentially 100% of the substrate recombined, even though the DNA was assembled into chromatin. The original nuclease has an 18-amino-acid linker between the zinc finger and cleavage domains, and this enzyme cleaved in oocytes at paired sites separated by spacers in the range of 6 to 18 bp, with a rather sharp optimum at 8 bp. By shortening the linker, we found that the range of effective site separations could be narrowed significantly. With no intentional linker between the binding and cleavage domains, only binding sites exactly 6 bp apart supported efficient cleavage in oocytes. We also showed that two chimeric enzymes with different binding specificities could collaborate to stimulate recombination when their individual sites were appropriately placed. Because the recognition specificity of zinc fingers can be altered experimentally, this approach holds great promise for inducing targeted recombination in a variety of organisms.
DOI:10.1126/science.1178817URLPMID:19933106 [本文引用: 1]
TAL effectors of plant pathogenic bacteria in the genus Xanthomonas bind host DNA and activate genes that contribute to disease or turn on defense. Target specificity depends on an effector-variable number of typically 34 amino acid repeats, but the mechanism of recognition is not understood. We show that a repeat-variable pair of residues specifies the nucleotides in the target site, one pair to one nucleotide, with no apparent context dependence. Our finding represents a previously unknown mechanism for protein-DNA recognition that explains TAL effector specificity, enables target site prediction, and opens prospects for use of TAL effectors in research and biotechnology.
DOI:10.1126/science.1178811URLPMID:19933107 [本文引用: 1]
The pathogenicity of many bacteria depends on the injection of effector proteins via type III secretion into eukaryotic cells in order to manipulate cellular processes. TAL (transcription activator-like) effectors from plant pathogenic Xanthomonas are important virulence factors that act as transcriptional activators in the plant cell nucleus, where they directly bind to DNA via a central domain of tandem repeats. Here, we show how target DNA specificity of TAL effectors is encoded. Two hypervariable amino acid residues in each repeat recognize one base pair in the target DNA. Recognition sequences of TAL effectors were predicted and experimentally confirmed. The modular protein architecture enabled the construction of artificial effectors with new specificities. Our study describes the functionality of a distinct type of DNA binding domain and allows the design of DNA binding domains for biotechnology.
DOI:10.1534/genetics.110.120717URLPMID:20660643 [本文引用: 1]
Engineered nucleases that cleave specific DNA sequences in vivo are valuable reagents for targeted mutagenesis. Here we report a new class of sequence-specific nucleases created by fusing transcription activator-like effectors (TALEs) to the catalytic domain of the FokI endonuclease. Both native and custom TALE-nuclease fusions direct DNA double-strand breaks to specific, targeted sites.
DOI:10.3724/SP.J.1005.2013.01265URL [本文引用: 2]
Bacteria and archaea have evolved an adaptive immune system, known as typeⅡprokaryotic clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) system, which uses short RNA to direct the degradation of target sequences present in invading viral and plasmid DNAs. Recent advances in CRISPR/Cas system provide an improved method for genome editing, showing robust and specific RNA-guided endonuclease activity at targeted endogenous genomic loci. It is the latest technology to modify genome DNA specifically and effectively following zinc finger nucleases (ZFNs) and TALE nucleases (TALENs). Compared with ZFNs and TALENs, CRISPR/Cas is much simpler and easier to engineer. This review summarizes recent progress, and discusses the prospects of CRISPR/Cas system, with an emphasis on its structure, principle, applications and potential challenges.
DOI:10.3724/SP.J.1005.2013.01265URL [本文引用: 2]
Bacteria and archaea have evolved an adaptive immune system, known as typeⅡprokaryotic clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) system, which uses short RNA to direct the degradation of target sequences present in invading viral and plasmid DNAs. Recent advances in CRISPR/Cas system provide an improved method for genome editing, showing robust and specific RNA-guided endonuclease activity at targeted endogenous genomic loci. It is the latest technology to modify genome DNA specifically and effectively following zinc finger nucleases (ZFNs) and TALE nucleases (TALENs). Compared with ZFNs and TALENs, CRISPR/Cas is much simpler and easier to engineer. This review summarizes recent progress, and discusses the prospects of CRISPR/Cas system, with an emphasis on its structure, principle, applications and potential challenges.
.
DOI:10.1016/j.jgg.2016.03.006URLPMID:27210040 [本文引用: 2]
The genomes are organized into ordered and hierarchical topological structures in interphase nuclei. Within discrete territories of each chromosome, topologically associated domains (TADs) play important roles in various nuclear processes such as gene regulation. Inside TADs separated by relatively constitutive boundaries, distal elements regulate their gene targets through specific chromatin-looping contacts such as long-distance enhancer-promoter interactions. High-throughput sequencing studies have revealed millions of potential regulatory DNA elements, which are much more abundant than the mere ~20,000 genes they control. The recently emerged CRISPR-Cas9 genome editing technologies have enabled efficient and precise genetic and epigenetic manipulations of genomes. The multiplexed and high-throughput CRISPR capabilities facilitate the discovery and dissection of gene regulatory elements. Here, we describe the applications of CRISPR for genome, epigenome, and 3D genome editing, focusing on CRISPR DNA-fragment editing with Cas9 and a pair of sgRNAs to investigate topological folding of chromatin TADs and developmental gene regulation.
.
DOI:10.1093/jmcb/mjv016URLPMID:25757625 [本文引用: 2]
The human genome contains millions of DNA regulatory elements and a large number of gene clusters, most of which have not been tested experimentally. The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated nuclease 9 (Cas9) programed with a synthetic single-guide RNA (sgRNA) emerges as a method for genome editing in virtually any organisms. Here we report that targeted DNA fragment inversions and duplications could easily be achieved in human and mouse genomes by CRISPR with two sgRNAs. Specifically, we found that, in cultured human cells and mice, efficient precise inversions of DNA fragments ranging in size from a few tens of bp to hundreds of kb could be generated. In addition, DNA fragment duplications and deletions could also be generated by CRISPR through trans-allelic recombination between the Cas9-induced double-strand breaks (DSBs) on two homologous chromosomes (chromatids). Moreover, junctions of combinatorial inversions and duplications of the protocadherin (Pcdh) gene clusters induced by Cas9 with four sgRNAs could be detected. In mice, we obtained founders with alleles of precise inversions, duplications, and deletions of DNA fragments of variable sizes by CRISPR. Interestingly, we found that very efficient inversions were mediated by microhomology-mediated end joining (MMEJ) through short inverted repeats. We showed for the first time that DNA fragment inversions could be transmitted through germlines in mice. Finally, we applied this CRISPR method to a regulatory element of the Pcdhα cluster and found a new role in the regulation of members of the Pcdhγ cluster. This simple and efficient method should be useful in manipulating mammalian genomes to study millions of regulatory DNA elements as well as vast numbers of gene clusters.
DOI:10.16288/j.yczz.15-291URLPMID:26496751 [本文引用: 1]
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated nuclease 9 (Cas9) system from bacteria and archaea emerged recently as a new powerful technology of genome editing in virtually any organism. Due to its simplicity and cost effectiveness, a revolutionary change of genetics has occurred. Here, we summarize the recent development of DNA fragment editing methods by CRISPR/Cas9 and describe targeted DNA fragment deletions, inversions, duplications, insertions, and translocations. The efficient method of DNA fragment editing provides a powerful tool for studying gene function, regulatory elements, tissue development, and disease progression. Finally, we discuss the prospects of CRISPR/Cas9 system and the potential applications of other types of CRISPR system.
DOI:10.16288/j.yczz.15-291URLPMID:26496751 [本文引用: 1]
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated nuclease 9 (Cas9) system from bacteria and archaea emerged recently as a new powerful technology of genome editing in virtually any organism. Due to its simplicity and cost effectiveness, a revolutionary change of genetics has occurred. Here, we summarize the recent development of DNA fragment editing methods by CRISPR/Cas9 and describe targeted DNA fragment deletions, inversions, duplications, insertions, and translocations. The efficient method of DNA fragment editing provides a powerful tool for studying gene function, regulatory elements, tissue development, and disease progression. Finally, we discuss the prospects of CRISPR/Cas9 system and the potential applications of other types of CRISPR system.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s10943-020-00982-0URLPMID:31953788 [本文引用: 2]
An international survey was conducted of authors (N?=?288) in the religion-health (R-H) research field concerning the disclosure of their theistic orientation (T-O) (i.e., whether they believe in God[s], a Higher Power, or a universal spirit) in their journal articles. Most (74%) of the respondents said they never disclose their T-O in this context; e.g., because they feel the information is private (20%), irrelevant (36%), unimportant (56%), and/or likely to make them appear less credible (36%). Atheists were four times less likely than deists and gnostic theists were to disclose their T-O; authors who conducted experimental research and published more frequently were also less likely to disclose their T-O. When disclosure did occur, it was more likely to take place informally within the narrative of manuscripts. Most (66%) of the respondents did not view their T-O as a competing interest (CI). Agnostic theism and the absence of theistic belief were less likely to be experienced as CIs than gnostic theism, deism, and atheism were. The respondents predominantly disagreed both that T-O should be characterized as a CI (48%) and that authors in the R-H field should disclose their T-O as such (59%). Only 18% of the authors in this study who did perceive their T-O as a CI reported that they formally disclose that information to journals or publishers, while the majority (59%) of those authors said they never disclose the information in this context at all. The discussion focuses on reasons as to why authors might choose not to do so. Recommendations are offered for the R-H field.
DOI:10.1016/j.molcel.2018.06.021URLPMID:30033371 [本文引用: 4]
Chromosomal rearrangements including large DNA-fragment inversions, deletions, and duplications by Cas9 with paired sgRNAs are important to investigate genome structural variations and developmental gene regulation, but little is known about the underlying mechanisms. Here, we report that disrupting CtIP or FANCD2, which have roles in alternative non-homologous end joining, enhances precise DNA-fragment deletion. By analyzing the inserted nucleotides at the junctions of DNA-fragment editing of deletions, inversions, and duplications and characterizing the cleaved products, we find that Cas9 endonucleolytically cleaves the noncomplementary strand with a flexible scissile profile upstream of the -3 position of the PAM site in?vivo and in?vitro, generating double-strand break ends with 5' overhangs of 1-3 nucleotides. Moreover, we find that engineered Cas9 nucleases have distinct cleavage profiles. Finally, Cas9-mediated nucleotide insertions are nonrandom and are equal to the combined sequences upstream of both PAM sites with predicted frequencies. Thus, precise and predictable DNA-fragment editing could be achieved by perturbing DNA repair genes and using appropriate PAM configurations.
DOI:10.1016/j.tig.2008.08.007URLPMID:18809224 [本文引用: 1]
DNA double-strand breaks are normal consequences of cell division and differentiation and must be repaired faithfully to maintain genome stability. Two mechanistically distinct pathways are known to efficiently repair double-strand breaks: homologous recombination and Ku-dependent non-homologous end joining. Recently, a third, less characterized repair mechanism, named microhomology-mediated end joining (MMEJ), has received increasing attention. MMEJ repairs DNA breaks via the use of substantial microhomology and always results in deletions. Furthermore, it probably contributes to oncogenic chromosome rearrangements and genetic variation in humans. Here, we summarize the genetic attributes of MMEJ from several model systems and discuss the relationship between MMEJ and 'alternative end joining'. We propose a mechanistic model for MMEJ and highlight important questions for future research.
DOI:10.1038/nbt.4317URLPMID:30480667 [本文引用: 2]
The DNA mutation produced by cellular repair of a CRISPR-Cas9-generated double-strand break determines its phenotypic effect. It is known that the mutational outcomes are not random, but depend on DNA sequence at the targeted location. Here we systematically study the influence of flanking DNA sequence on repair outcome by measuring the edits generated by >40,000 guide RNAs (gRNAs) in synthetic constructs. We performed the experiments in a range of genetic backgrounds and using alternative CRISPR-Cas9 reagents. In total, we gathered data for >109 mutational outcomes. The majority of reproducible mutations are insertions of a single base, short deletions or longer microhomology-mediated deletions. Each gRNA has an individual cell-line-dependent bias toward particular outcomes. We uncover sequence determinants of the mutations produced and use these to derive a predictor of Cas9 editing outcomes. Improved understanding of sequence repair will allow better design of gene editing experiments.
DOI:10.1038/s41586-018-0686-xURLPMID:30405244 [本文引用: 2]
Following Cas9 cleavage, DNA repair without a donor template is generally considered stochastic, heterogeneous and impractical beyond gene disruption. Here, we show that template-free Cas9 editing is predictable and capable of precise repair to a predicted genotype, enabling correction of disease-associated mutations in humans. We constructed a library of 2,000 Cas9 guide RNAs paired with DNA target sites and trained inDelphi, a machine learning model that predicts genotypes and frequencies of 1- to 60-base-pair deletions and 1-base-pair insertions with high accuracy (r?=?0.87) in five human and mouse cell lines. inDelphi predicts that 5-11% of Cas9 guide RNAs targeting the human genome are 'precise-50', yielding a single genotype comprising greater than or equal to 50% of all major editing products. We experimentally confirmed precise-50 insertions and deletions in 195 human disease-relevant alleles, including correction in primary patient-derived fibroblasts of pathogenic alleles to wild-type genotype for Hermansky-Pudlak syndrome and Menkes disease. This study establishes an approach for precise, template-free genome editing.
DOI:10.1038/s41421-019-0120-zURLPMID:31636963 [本文引用: 3]
DOI:10.1016/j.cell.2014.09.030URL [本文引用: 2]
The pluripotent state of embryonic stem cells (ESCs) is produced by active transcription of genes that control cell identity and repression of genes encoding lineage-specifying developmental regulators. Here, we use ESC cohesin ChIA-PET data to identify the local chromosomal structures at both active and repressed genes across the genome. The results produce a map of enhancer-promoter interactions and reveal that super-enhancer-driven genes generally occur within chromosome structures that are formed by the looping of two interacting CTCF sites co-occupied by cohesin. These looped structures form insulated neighborhoods whose integrity is important for proper expression of local genes. We also find that repressed genes encoding lineage-specifying developmental regulators occur within insulated neighborhoods. These results provide insights into the relationship between transcriptional control of cell identity genes and control of local chromosome structure.
DOI:10.1038/s41588-019-0466-zURLPMID:31358994 [本文引用: 1]
The genome is organized in three-dimensional units called topologically associating domains (TADs), through a process dependent on the cooperative action of cohesin and the DNA-binding factor CTCF. Genomic rearrangements of TADs have been shown to cause gene misexpression and disease, but genome-wide depletion of CTCF has no drastic effects on transcription. Here, we investigate TAD function in vivo in mouse limb buds at the Sox9-Kcnj2 locus. We show that the removal of all major CTCF sites at the boundary and within the TAD resulted in a fusion of neighboring TADs, without major effects on gene expression. Gene misexpression and disease phenotypes, however, were achieved by redirecting regulatory activity through inversions and/or the repositioning of boundaries. Thus, TAD structures provide robustness and precision but are not essential for developmental gene regulation. Aberrant disease-related gene activation is not induced by a mere loss of insulation but requires CTCF-dependent redirection of enhancer-promoter contacts.
DOI:10.1038/s41588-018-0221-xURLPMID:30262816 [本文引用: 1]
The regulatory specificity of enhancers and their interaction with gene promoters is thought to be controlled by their sequence and the binding of transcription factors. By studying Pitx1, a regulator of hindlimb development, we show that dynamic changes in chromatin conformation can restrict the activity of enhancers. Inconsistent with its hindlimb-restricted expression, Pitx1 is controlled by an enhancer (Pen) that shows activity in forelimbs and hindlimbs. By Capture Hi-C and three-dimensional modeling of the locus, we demonstrate that forelimbs and hindlimbs have fundamentally different chromatin configurations, whereby Pen and Pitx1 interact in hindlimbs and are physically separated in forelimbs. Structural variants can convert the inactive into the active conformation, thereby inducing Pitx1 misexpression in forelimbs, causing partial arm-to-leg transformation in mice and humans. Thus, tissue-specific three-dimensional chromatin conformation can contribute to enhancer activity and specificity in vivo and its disturbance can result in gene misexpression and disease.
DOI:10.1016/j.cell.2018.11.036URLPMID:30595451 [本文引用: 1]
The temporal order of DNA replication (replication timing [RT]) is highly coupled with genome architecture, but cis-elements regulating either remain elusive. We created a series of CRISPR-mediated deletions and inversions of a pluripotency-associated topologically associating domain (TAD) in mouse ESCs. CTCF-associated domain boundaries were dispensable for RT. CTCF protein depletion weakened most TAD boundaries but had no effect on RT or A/B compartmentalization genome-wide. By contrast, deletion of three intra-TAD CTCF-independent 3D contact sites caused a domain-wide early-to-late RT shift, an A-to-B compartment switch, weakening of TAD architecture, and loss of transcription. The dispensability of TAD boundaries and the?necessity of these "early replication control elements" (ERCEs) was validated by deletions and inversions at additional domains. Our results demonstrate that discrete cis-regulatory elements orchestrate domain-wide RT, A/B compartmentalization, TAD architecture, and transcription, revealing fundamental principles linking genome structure and function.
DOI:10.1186/s13059-017-1354-4URLPMID:29178945 [本文引用: 1]
The CRISPR/Cas9 system has become an efficient gene editing method for generating cells carrying precise gene mutations, including the rearrangement and deletion of chromosomal segments. However, whether an entire chromosome could be eliminated by this technology is still unknown.
DOI:10.1038/s41586-018-0382-xURLPMID:30069045 [本文引用: 1]
Eukaryotic genomes are generally organized in multiple chromosomes. Here we have created a functional single-chromosome yeast from a Saccharomyces cerevisiae haploid cell containing sixteen linear chromosomes, by successive end-to-end chromosome fusions and centromere deletions. The fusion of sixteen native linear chromosomes into a single chromosome results in marked changes to the global three-dimensional structure of the chromosome due to the loss of all centromere-associated inter-chromosomal interactions, most telomere-associated inter-chromosomal interactions and 67.4% of intra-chromosomal interactions. However, the single-chromosome and wild-type yeast cells have nearly identical transcriptome and similar phenome profiles. The giant single chromosome can support cell life, although this strain shows reduced growth across environments, competitiveness, gamete production and viability. This synthetic biology study demonstrates an approach to exploration of eukaryote evolution with respect to chromosome structure and function.
DOI:10.1016/j.cell.2013.12.001URL [本文引用: 3]
The spatiotemporal organization and dynamics of chromatin play critical roles in regulating genome function. However, visualizing specific, endogenous genomic loci remains challenging in living cells. Here, we demonstrate such an imaging technique by repurposing the bacterial CRISPR/Cas system. Using an EGFP-tagged endonuclease-deficient Cas9 protein and a structurally optimized small guide (sg) RNA, we show robust imaging of repetitive elements in telomeres and coding genes in living cells. Furthermore, an array of sgRNAs tiling along the target locus enables the visualization of nonrepetitive genomic sequences. Using this method, we have studied telomere dynamics during elongation or disruption, the subnuclear localization of the MUC4 loci, the cohesion of replicated MUC4 loci on sister chromatids, and their dynamic behaviors during mitosis. This CRISPR imaging tool has potential to significantly improve the capacity to study the conformation and dynamics of native chromosomes in living human cells.
DOI:10.1073/pnas.1515692112URLPMID:26324940 [本文引用: 1]
Direct visualization of genomic loci in the 3D nucleus is important for understanding the spatial organization of the genome and its association with gene expression. Various DNA FISH methods have been developed in the past decades, all involving denaturing dsDNA and hybridizing fluorescent nucleic acid probes. Here we report a novel approach that uses in vitro constituted nuclease-deficient clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated caspase 9 (Cas9) complexes as probes to label sequence-specific genomic loci fluorescently without global DNA denaturation (Cas9-mediated fluorescence in situ hybridization, CASFISH). Using fluorescently labeled nuclease-deficient Cas9 (dCas9) protein assembled with various single-guide RNA (sgRNA), we demonstrated rapid and robust labeling of repetitive DNA elements in pericentromere, centromere, G-rich telomere, and coding gene loci. Assembling dCas9 with an array of sgRNAs tiling arbitrary target loci, we were able to visualize nonrepetitive genomic sequences. The dCas9/sgRNA binary complex is stable and binds its target DNA with high affinity, allowing sequential or simultaneous probing of multiple targets. CASFISH assays using differently colored dCas9/sgRNA complexes allow multicolor labeling of target loci in cells. In addition, the CASFISH assay is remarkably rapid under optimal conditions and is applicable for detection in primary tissue sections. This rapid, robust, less disruptive, and cost-effective technology adds a valuable tool for basic research and genetic diagnosis.
DOI:10.1073/pnas.1420024112URLPMID:25713381 [本文引用: 1]
The intranuclear location of genomic loci and the dynamics of these loci are important parameters for understanding the spatial and temporal regulation of gene expression. Recently it has proven possible to visualize endogenous genomic loci in live cells by the use of transcription activator-like effectors (TALEs), as well as modified versions of the bacterial immunity clustered regularly interspersed short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) system. Here we report the design of multicolor versions of CRISPR using catalytically inactive Cas9 endonuclease (dCas9) from three bacterial orthologs. Each pair of dCas9-fluorescent proteins and cognate single-guide RNAs (sgRNAs) efficiently labeled several target loci in live human cells. Using pairs of differently colored dCas9-sgRNAs, it was possible to determine the intranuclear distance between loci on different chromosomes. In addition, the fluorescence spatial resolution between two loci on the same chromosome could be determined and related to the linear distance between them on the chromosome's physical map, thereby permitting assessment of the DNA compaction of such regions in a live cell.
DOI:10.1093/nar/gkv1533URLPMID:26740581 [本文引用: 1]
In order to elucidate the functional organization of the genome, it is vital to directly visualize the interactions between genomic elements in living cells. For this purpose, we engineered the Cas9 protein from Staphylococcus aureus (SaCas9) for the imaging of endogenous genomic loci, which showed a similar robustness and efficiency as previously reported for Streptococcus pyogenes Cas9 (SpCas9). Imaging readouts allowed us to characterize the DNA-binding activity of SaCas9 and to optimize its sgRNA scaffold. Combining SaCas9 and SpCas9, we demonstrated two-color CRISPR imaging with the capability to resolve genomic loci spaced by <300 kb. Combinatorial color-mixing further enabled us to code multiple genomic elements in the same cell. Our results highlight the potential of combining SpCas9 and SaCas9 for multiplexed CRISPR-Cas9 applications, such as imaging and genome engineering.
DOI:10.1038/ncomms11707URLPMID:27222091 [本文引用: 1]
Imaging systems that allow visualization of specific loci and nuclear structures are highly relevant for investigating how organizational changes within the nucleus play a role in regulating gene expression and other cellular processes. Here we present a live imaging system for targeted detection of genomic regions. Our approach involves generating chimaeric transcripts of viral RNAs (MS2 and PP7) and single-guide RNAs (sgRNAs), which when co-expressed with a cleavage-deficient Cas9 can recruit fluorescently tagged viral RNA-binding proteins (MCP and PCP) to specific genomic sites. This allows for rapid, stable, low-background visualization of target loci. We demonstrate the efficiency and flexibility of our method by simultaneously labelling major and minor satellite regions as well as two individual loci on mouse chromosome 12. This system provides a tool for dual-colour labelling, which is important for tracking the dynamics of chromatin interactions and for validating epigenetic processes identified in fixed cells.
DOI:10.1016/j.molcel.2018.02.007URLPMID:29526697 [本文引用: 1]
Imaging (fluorescence in situ hybridization [FISH]) and genome-wide chromosome conformation capture (Hi-C) are two major approaches to the study of higher-order genome organization in the nucleus. Intra-chromosomal and inter-chromosomal interactions (referred to as non-homologous chromosomal?contacts [NHCCs]) have been observed by several FISH-based studies, but locus-specific NHCCs have not been detected by Hi-C. Due to crosslinking, neither of these approaches assesses spatiotemporal properties. Toward resolving the discrepancies between imaging and Hi-C, we sought to understand the spatiotemporal properties of NHCCs in living cells by CRISPR/Cas9 live-cell imaging (CLING). In mammalian cells, we find that NHCCs are stable and occur as frequently as intra-chromosomal interactions, but NHCCs occur at farther spatial distance that could explain their lack of detection in Hi-C. By revealing the spatiotemporal properties in living cells, our study provides fundamental insights into the biology of NHCCs.
DOI:10.1038/nbt.3526URLPMID:27088723 [本文引用: 1]
A lack of techniques to image multiple genomic loci in living cells has limited our ability to investigate chromosome dynamics. Here we describe CRISPRainbow, a system for labeling DNA in living cells based on nuclease-dead (d) Cas9 combined with engineered single guide RNA (sgRNA) scaffolds that bind sets of fluorescent proteins. We demonstrate simultaneous imaging of up to six chromosomal loci in individual live cells and document large differences in the dynamic properties of different chromosomal loci.
DOI:10.1093/nar/gkw066URLPMID:26850639 [本文引用: 1]
Visualization of chromosomal dynamics is important for understanding many fundamental intra-nuclear processes. Efficient and reliable live-cell multicolor labeling of chromosomal loci can realize this goal. However, the current methods are constrained mainly by insufficient labeling throughput, efficiency, flexibility as well as photostability. Here we have developed a new approach to realize dual-color chromosomal loci imaging based on a modified single-guide RNA (sgRNA) of the CRISPR/Cas9 system. The modification of sgRNA was optimized by structure-guided engineering of the original sgRNA, consisting of RNA aptamer insertions that bind fluorescent protein-tagged effectors. By labeling and tracking telomeres, centromeres and genomic loci, we demonstrate that the new approach is easy to implement and enables robust dual-color imaging of genomic elements. Importantly, our data also indicate that the fast exchange rate of RNA aptamer binding effectors makes our sgRNA-based labeling method much more tolerant to photobleaching than the Cas9-based labeling method. This is crucial for continuous, long-term tracking of chromosomal dynamics. Lastly, as our method is complementary to other live-cell genomic labeling systems, it is therefore possible to combine them into a plentiful palette for the study of native chromatin organization and genome ultrastructure dynamics in living cells.
DOI:10.1038/ncomms14725URLPMID:28290446 [本文引用: 1]
Imaging chromatin dynamics is crucial to understand genome organization and its role in transcriptional regulation. Recently, the RNA-guidable feature of CRISPR-Cas9 has been utilized for imaging of chromatin within live cells. However, these methods are mostly applicable to highly repetitive regions, whereas imaging regions with low or no repeats remains as a challenge. To address this challenge, we design single-guide RNAs (sgRNAs) integrated with up to 16 MS2 binding motifs to enable robust fluorescent signal amplification. These engineered sgRNAs enable multicolour labelling of low-repeat-containing regions using a single sgRNA and of non-repetitive regions with as few as four unique sgRNAs. We achieve tracking of native chromatin loci throughout the cell cycle and determine differential positioning of transcriptionally active and inactive regions in the nucleus. These results demonstrate the feasibility of our approach to monitor the position and dynamics of both repetitive and non-repetitive genomic regions in live cells.
DOI:10.1371/journal.pgen.1000039URLPMID:18369458 [本文引用: 1]
The spatial organisation of the genome in the nucleus has a role in the regulation of gene expression. In vertebrates, chromosomal regions with low gene-density are located close to the nuclear periphery. Correlations have also been made between the transcriptional state of some genes and their location near the nuclear periphery. However, a crucial issue is whether this level of nuclear organisation directly affects gene function, rather than merely reflecting it. To directly investigate whether proximity to the nuclear periphery can influence gene expression in mammalian cells, here we relocate specific human chromosomes to the nuclear periphery by tethering them to a protein of the inner nuclear membrane. We show that this can reversibly suppress the expression of some endogenous human genes located near the tethering sites, and even genes further away. However, the expression of many other genes is not detectably reduced and we show that location at the nuclear periphery is not incompatible with active transcription. The dampening of gene expression around the nuclear periphery is dependent on the activity of histone deacetylases. Our data show that the radial position within the nucleus can influence the expression of some, but not all, genes. This is compatible with the suggestion that re-localisation of genes relative to the peripheral zone of the nucleus could be used by metazoans to modulate the expression of selected genes during development and differentiation.
.
DOI:10.1083/jcb.200706060URLPMID:18195101 [本文引用: 1]
The peripheral nuclear lamina, which is largely but not entirely associated with inactive chromatin, is considered to be an important determinant of nuclear structure and gene expression. We present here an inducible system to target a genetic locus to the nuclear lamina in living mammalian cells. Using three-dimensional time-lapse microscopy, we determined that targeting of the locus requires passage through mitosis. Once targeted, the locus remains anchored to the nuclear periphery in interphase as well as in daughter cells after passage through a subsequent mitosis. Upon transcriptional induction, components of the gene expression machinery are recruited to the targeted locus, and we visualized nascent transcripts at the nuclear periphery. The kinetics of transcriptional induction at the nuclear lamina is similar to that observed at an internal nuclear region. This new cell system provides a powerful approach to study the dynamics of gene function at the nuclear periphery in living cells.
DOI:10.1016/j.cell.2018.09.013URLPMID:30318144 [本文引用: 1]
Programmable control of spatial genome organization is a powerful approach for studying how nuclear structure affects gene regulation and cellular function. Here, we develop a versatile CRISPR-genome organization (CRISPR-GO) system that can efficiently control the spatial positioning of genomic loci relative to specific nuclear compartments, including the nuclear periphery, Cajal bodies, and promyelocytic leukemia (PML) bodies. CRISPR-GO is chemically inducible and reversible, enabling interrogation of real-time dynamics of chromatin interactions with nuclear compartments in living cells. Inducible repositioning of genomic loci to the nuclear periphery allows for dissection of mitosis-dependent and -independent relocalization events and also for interrogation of the relationship between gene position and gene expression. CRISPR-GO mediates rapid de novo formation of Cajal bodies at desired chromatin loci and causes significant repression of endogenous gene expression over long distances (30-600 kb). The CRISPR-GO system offers a programmable platform to investigate large-scale spatial genome organization and function.
DOI:10.1038/ncomms15993URLPMID:28703221 [本文引用: 1]
Chromatin looping is key to gene regulation, yet no broadly applicable methods to selectively modify chromatin loops have been described. We have engineered a method for chromatin loop reorganization using CRISPR-dCas9 (CLOuD9) to selectively and reversibly establish chromatin loops. We demonstrate the power of this technology to selectively modulate gene expression at targeted loci.
DOI:10.1038/cr.2013.122URL [本文引用: 1]
Technologies allowing for specific regulation of endogenous genes are valuable for the study of gene functions and have great potential in therapeutics. We created the CRISPR-on system, a two-component transcriptional activator consisting of a nuclease-dead Cas9 (dCas9) protein fused with a transcriptional activation domain and single guide RNAs (sgRNAs) with complementary sequence to gene promoters. We demonstrate that CRISPR-on can efficiently activate exogenous reporter genes in both human and mouse cells in a tunable manner. In addition, we show that robust reporter gene activation in vivo can be achieved by injecting the system components into mouse zygotes. Furthermore, we show that CRISPR-on can activate the endogenous IL1RN, SOX2, and OCT4 genes. The most efficient gene activation was achieved by clusters of 3-4 sgRNAs binding to the proximal promoters, suggesting their synergistic action in gene induction. Significantly, when sgRNAs targeting multiple genes were simultaneously introduced into cells, robust multiplexed endogenous gene activation was achieved. Genome-wide expression profiling demonstrated high specificity of the system.
DOI:10.1016/j.stem.2016.07.001URLPMID:27524438 [本文引用: 1]
Overexpression of exogenous fate-specifying transcription factors can directly reprogram differentiated somatic cells to target cell types. Here, we?show that similar reprogramming can also be achieved through the direct activation of endogenous genes using engineered CRISPR/Cas9-based transcriptional activators. We use this approach to induce activation of the endogenous Brn2, Ascl1, and Myt1l genes (BAM factors) to convert mouse embryonic fibroblasts to induced neuronal cells. This direct activation of endogenous genes rapidly remodeled the epigenetic state of the target loci and induced sustained endogenous gene expression during reprogramming. Thus, transcriptional activation and epigenetic remodeling of endogenous master transcription factors are sufficient for conversion between cell types. The rapid and sustained activation of endogenous genes in their native chromatin context by this approach may facilitate reprogramming with transient methods that avoid genomic integration and provides a new strategy for overcoming epigenetic barriers to cell fate specification.
DOI:10.1038/mt.2015.200URLPMID:26527377 [本文引用: 1]
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most prevalent myopathies, affecting males and females of all ages. Both forms of the disease are linked by epigenetic derepression of the D4Z4 macrosatellite repeat array at chromosome 4q35, leading to aberrant expression of D4Z4-encoded RNAs in skeletal muscle. Production of full-length DUX4 (DUX4-fl) mRNA from the derepressed D4Z4 array results in misexpression of DUX4-FL protein and its transcriptional targets, and apoptosis, ultimately leading to accumulated muscle pathology. Returning the chromatin at the FSHD locus to its nonpathogenic, epigenetically repressed state would simultaneously affect all D4Z4 RNAs, inhibiting downstream pathogenic pathways, and is thus an attractive therapeutic strategy. Advances in CRISPR/Cas9-based genome editing make it possible to target epigenetic modifiers to an endogenous disease locus, although reports to date have focused on more typical genomic regions. Here, we demonstrate that a CRISPR/dCas9 transcriptional inhibitor can be specifically targeted to the highly repetitive FSHD macrosatellite array and alter the chromatin to repress expression of DUX4-fl in primary FSHD myocytes. These results implicate the promoter and exon 1 of DUX4 as potential therapeutic targets and demonstrate the utility of CRISPR technology for correction of the epigenetic dysregulation in FSHD.
247.
[本文引用: 2]
DOI:10.1038/nature17946URLPMID:27096365 [本文引用: 2]
Current genome-editing technologies introduce double-stranded (ds) DNA breaks at a target locus as the first step to gene correction. Although most genetic diseases arise from point mutations, current approaches to point mutation correction are inefficient and typically induce an abundance of random insertions and deletions (indels) at the target locus resulting from the cellular response to dsDNA breaks. Here we report the development of 'base editing', a new approach to genome editing that enables the direct, irreversible conversion of one target DNA base into another in a programmable manner, without requiring dsDNA backbone cleavage or a donor template. We engineered fusions of CRISPR/Cas9 and a cytidine deaminase enzyme that retain the ability to be programmed with a guide RNA, do not induce dsDNA breaks, and mediate the direct conversion of cytidine to uridine, thereby effecting a C→T (or G→A) substitution. The resulting 'base editors' convert cytidines within a window of approximately five nucleotides, and can efficiently correct a variety of point mutations relevant to human disease. In four transformed human and murine cell lines, second- and third-generation base editors that fuse uracil glycosylase inhibitor, and that use a Cas9 nickase targeting the non-edited strand, manipulate the cellular DNA repair response to favour desired base-editing outcomes, resulting in permanent correction of ~15-75% of total cellular DNA with minimal (typically ≤1%) indel formation. Base editing expands the scope and efficiency of genome editing of point mutations.
1507.
[本文引用: 3]
DOI:10.1016/j.cell.2013.02.022URL [本文引用: 1]
Targeted gene regulation on a genome-wide scale is a powerful strategy for interrogating, perturbing, and engineering cellular systems. Here, we develop a method for controlling gene expression based on Cas9, an RNA-guided DNA endonuclease from a type II CRISPR system. We show that a catalytically dead Cas9 lacking endonuclease activity, when coexpressed with a guide RNA, generates a DNA recognition complex that can specifically interfere with transcriptional elongation, RNA polymerase binding, or transcription factor binding. This system, which we call CRISPR interference (CRISPRi), can efficiently repress expression of targeted genes in Escherichia coli, with no detectable off-target effects. CRISPRi can be used to repress multiple target genes simultaneously, and its effects are reversible. We also show evidence that the system can be adapted for gene repression in mammalian cells. This RNA-guided DNA recognition platform provides a simple approach for selectively perturbing gene expression on a genome-wide scale.
DOI:10.1016/j.cell.2013.06.044URLPMID:23849981 [本文引用: 1]
The genetic interrogation and reprogramming of cells requires methods for robust and precise targeting of genes for expression or repression. The CRISPR-associated catalytically inactive dCas9 protein offers a general platform for RNA-guided DNA targeting. Here, we show that fusion of dCas9 to effector domains with distinct regulatory functions enables stable and efficient transcriptional repression or activation in human and yeast cells, with the site of delivery determined solely by a coexpressed short guide (sg)RNA. Coupling of dCas9 to a transcriptional repressor domain can robustly silence expression of multiple endogenous genes. RNA-seq analysis indicates that CRISPR interference (CRISPRi)-mediated transcriptional repression is highly specific. Our results establish that the CRISPR system can be used as a modular and flexible DNA-binding platform for the recruitment of proteins to a target DNA sequence, revealing the potential of CRISPRi as a general tool for the precise regulation of gene expression in eukaryotic cells.
DOI:10.1038/nmeth.4042URLPMID:27776111 [本文引用: 1]
The ability to dynamically manipulate the transcriptome is important for studying how gene networks direct cellular functions and how network perturbations cause disease. Nuclease-dead CRISPR-dCas9 transcriptional regulators, while offering an approach for controlling individual gene expression, remain incapable of dynamically coordinating complex transcriptional events. Here, we describe a flexible dCas9-based platform for chemical-inducible complex gene regulation. From a screen of chemical- and light-inducible dimerization systems, we identified two potent chemical inducers that mediate efficient gene activation and repression in mammalian cells. We combined these inducers with orthogonal dCas9 regulators to independently control expression of different genes within the same cell. Using this platform, we further devised AND, OR, NAND, and NOR dCas9 logic operators and a diametric regulator that activates gene expression with one inducer and represses with another. This work provides a robust CRISPR-dCas9-based platform for enacting complex transcription programs that is suitable for large-scale transcriptome engineering.
DOI:10.1126/science.aav9973URLPMID:30819928 [本文引用: 1]
Genome editing holds promise for correcting pathogenic mutations. However, it is difficult to determine off-target effects of editing due to single-nucleotide polymorphism in individuals. Here we developed a method named GOTI (genome-wide off-target analysis by two-cell embryo injection) to detect off-target mutations by editing one blastomere of two-cell mouse embryos using either CRISPR-Cas9 or base editors. Comparison of the whole-genome sequences of progeny cells of edited and nonedited blastomeres at embryonic day 14.5 showed that off-target single-nucleotide variants (SNVs) were rare in embryos edited by CRISPR-Cas9 or adenine base editor, with a frequency close to the spontaneous mutation rate. By contrast, cytosine base editing induced SNVs at more than 20-fold higher frequencies, requiring a solution to address its fidelity.
DOI:10.1126/science.aaw7166URLPMID:30819931 [本文引用: 1]
Cytosine and adenine base editors (CBEs and ABEs) are promising new tools for achieving the precise genetic changes required for disease treatment and trait improvement. However, genome-wide and unbiased analyses of their off-target effects in vivo are still lacking. Our whole-genome sequencing analysis of rice plants treated with the third-generation base editor (BE3), high-fidelity BE3 (HF1-BE3), or ABE revealed that BE3 and HF1-BE3, but not ABE, induce substantial genome-wide off-target mutations, which are mostly the C→T type of single-nucleotide variants (SNVs) and appear to be enriched in genic regions. Notably, treatment of rice with BE3 or HF1-BE3 in the absence of single-guide RNA also results in the rise of genome-wide SNVs. Thus, the base-editing unit of BE3 or HF1-BE3 needs to be optimized in order to attain high fidelity.
DOI:10.1038/nbt.3199URLPMID:25849900 [本文引用: 1]
Technologies that enable targeted manipulation of epigenetic marks could be used to precisely control cell phenotype or interrogate the relationship between the epigenome and transcriptional control. Here we describe a programmable, CRISPR-Cas9-based acetyltransferase consisting of the nuclease-null dCas9 protein fused to the catalytic core of the human acetyltransferase p300. The fusion protein catalyzes acetylation of histone H3 lysine 27 at its target sites, leading to robust transcriptional activation of target genes from promoters and both proximal and distal enhancers. Gene activation by the targeted acetyltransferase was highly specific across the genome. In contrast to previous dCas9-based activators, the acetyltransferase activates genes from enhancer regions and with an individual guide RNA. We also show that the core p300 domain can be fused to other programmable DNA-binding proteins. These results support targeted acetylation as a causal mechanism of transactivation and provide a robust tool for manipulating gene regulation.
DOI:10.1038/nmeth.3325URLPMID:25775043
Understanding of mammalian enhancers is limited by the lack of a technology to rapidly and thoroughly test the cell type-specific function. Here, we use a nuclease-deficient Cas9 (dCas9)-histone demethylase fusion to functionally characterize previously described and new enhancer elements for their roles in the embryonic stem cell state. Further, we distinguish the mechanism of action of dCas9-LSD1 at enhancers from previous dCas9-effectors.
DOI:10.1016/j.ecoenv.2019.110160URLPMID:31951899 [本文引用: 2]
Although much has been determined about the molecular mechanisms of hexavalent chromium [Cr(VI)]-induced hepatotoxicity, more remains to be explored. In particular, explicit epigenetic alterations of microRNAs (miRNAs) which can negatively regulate mRNAs at post transcriptional level remain understudied. In the present study, cell apoptosis was determined using Annexin V/propidium iodide (PI) staining, while proliferative growth was analyzed by colony formation assay and proliferating cell nuclear antigen (PCNA) detection. miRNA microarray was performed to compare the global miRNAs expression patterns. miR-21-5p mimics (mi)/inhibitor (in), and PDCD4-siRNAs were transfected into L02 hepatocytes. Our results revealed that Cr(VI) induced apoptosis and inhibited proliferation in L02 hepatocytes via reactive oxygen species (ROS), the formation of which is closely related to mitochondrial damage, especially the inhibition of mitochondrial respiratory chain complex (MRCC). We also confirmed that ROS-mediated miR-21-5p inhibition participated in cell apoptosis and proliferative inhibition induced by Cr(VI). Furthermore, programmed cell death protein 4 (PDCD4), the up-regulation of which was related to ROS over-production, was predicted and verified as a target of miR-21-5p. Transcription factor PDCD4 silencing suppressed apoptosis and stimulated cell proliferation. In conclusion, from the perspective of epigenetics, the present study revealed that ROS-mediated miR-21-5p regulated the proliferation and apoptosis of Cr(VI)-exposed L02 hepatocytes via targeting PDCD4, which provided the new targets for molecular intervention and treatment of liver damage in Cr(VI)-exposed population.
DOI:10.1038/s41590-019-0500-4URLPMID:31611701 [本文引用: 1]
Immunotherapy has transformed cancer treatment. However, current immunotherapy modalities face various limitations. In the present study, we developed multiplexed activation of endogenous genes as an immunotherapy (MAEGI), a new form of immunotherapy that elicits antitumor immunity through multiplexed activation of endogenous genes in tumors. We leveraged CRISPR activation (CRISPRa) to directly augment the in situ expression of endogenous genes, and thereby the presentation of tumor antigens, leading to dramatic antitumor immune responses. Deploying this as a cell-based vaccination strategy showed efficacy in both prophylactic and therapeutic settings. Intratumoral adeno-associated virus delivery of CRISPRa libraries elicited strong antitumor immunity across multiple cancer types. Precision targeting of mutated gene sets eradicated a large fraction of established tumors at both local and distant sites. This treatment modality led to alterations in the tumor microenvironment, marked by enhanced T cell infiltration and antitumor immune signatures. Multiplexed endogenous gene activation is a versatile and highly scalable strategy to elicit potent immune responses against cancer, distinct from all existing cancer therapies.
DOI:10.1038/nature14450URLPMID:26030525 [本文引用: 1]
The three-dimensional organization of a genome plays a critical role in regulating gene expression, yet little is known about the machinery and mechanisms that determine higher-order chromosome structure. Here we perform genome-wide chromosome conformation capture analysis, fluorescent in situ hybridization (FISH), and RNA-seq to obtain comprehensive three-dimensional (3D) maps of the Caenorhabditis elegans genome and to dissect X chromosome dosage compensation, which balances gene expression between XX hermaphrodites and XO males. The dosage compensation complex (DCC), a condensin complex, binds to both hermaphrodite X chromosomes via sequence-specific recruitment elements on X (rex sites) to reduce chromosome-wide gene expression by half. Most DCC condensin subunits also act in other condensin complexes to control the compaction and resolution of all mitotic and meiotic chromosomes. By comparing chromosome structure in wild-type and DCC-defective embryos, we show that the DCC remodels hermaphrodite X chromosomes into a sex-specific spatial conformation distinct from autosomes. Dosage-compensated X chromosomes consist of self-interacting domains (~1 Mb) resembling mammalian topologically associating domains (TADs). TADs on X chromosomes have stronger boundaries and more regular spacing than on autosomes. Many TAD boundaries on X chromosomes coincide with the highest-affinity rex sites and become diminished or lost in DCC-defective mutants, thereby converting the topology of X to a conformation resembling autosomes. rex sites engage in DCC-dependent long-range interactions, with the most frequent interactions occurring between rex sites at DCC-dependent TAD boundaries. These results imply that the DCC reshapes the topology of X chromosomes by forming new TAD boundaries and reinforcing weak boundaries through interactions between its highest-affinity binding sites. As this model predicts, deletion of an endogenous rex site at a DCC-dependent TAD boundary using CRISPR/Cas9 greatly diminished the boundary. Thus, the DCC imposes a distinct higher-order structure onto X chromosomes while regulating gene expression chromosome-wide.
390.
[本文引用: 1]
DOI:10.1016/j.molcel.2015.09.023URLPMID:26527277 [本文引用: 1]
CCCTC-binding factor (CTCF) is an architectural protein involved in the three-dimensional (3D) organization of chromatin. In this study, we assayed the 3D genomic contact profiles of a large number of CTCF binding sites with high-resolution 4C-seq. As recently reported, our data also suggest that chromatin loops preferentially form between CTCF binding sites oriented in a convergent manner. To directly test this, we used CRISPR/Cas9 genome editing to delete core CTCF binding sites in three loci, including the CTCF site in the Sox2 super-enhancer. In all instances, CTCF and cohesin recruitment were lost, and chromatin loops with distal, convergent CTCF sites were disrupted or destabilized. Re-insertion of oppositely oriented CTCF recognition sequences restored CTCF and cohesin recruitment, but did not re-establish chromatin loops. We conclude that CTCF binding polarity plays a functional role in the formation of higher-order chromatin structure.
DOI:10.1073/pnas.1518552112URLPMID:26499245
We recently used in situ Hi-C to create kilobase-resolution 3D maps of mammalian genomes. Here, we combine these maps with new Hi-C, microscopy, and genome-editing experiments to study the physical structure of chromatin fibers, domains, and loops. We find that the observed contact domains are inconsistent with the equilibrium state for an ordinary condensed polymer. Combining Hi-C data and novel mathematical theorems, we show that contact domains are also not consistent with a fractal globule. Instead, we use physical simulations to study two models of genome folding. In one, intermonomer attraction during polymer condensation leads to formation of an anisotropic "tension globule." In the other, CCCTC-binding factor (CTCF) and cohesin act together to extrude unknotted loops during interphase. Both models are consistent with the observed contact domains and with the observation that contact domains tend to form inside loops. However, the extrusion model explains a far wider array of observations, such as why loops tend not to overlap and why the CTCF-binding motifs at pairs of loop anchors lie in the convergent orientation. Finally, we perform 13 genome-editing experiments examining the effect of altering CTCF-binding sites on chromatin folding. The convergent rule correctly predicts the affected loops in every case. Moreover, the extrusion model accurately predicts in silico the 3D maps resulting from each experiment using only the location of CTCF-binding sites in the WT. Thus, we show that it is possible to disrupt, restore, and move loops and domains using targeted mutations as small as a single base pair.
DOI:10.16288/j.yczz.19-072URLPMID:31257199 [本文引用: 1]
UDP-glucuronosyltransferases (UGTs) are an important family of phase 2 drug-metabolizing enzymes that catalyze the glucuronidation of numerous endogenous or exogenous small compounds. The aberrant expression of UGT isoforms causes many diseases, such as hyperbilirubinemia and affect drug efficacy or toxicity. Understanding mechanisms of UGT gene regulation will provide scientific foundations for disease prevention and personalized or precision medicine. Vertebrate UGT family genes can be divided into UGT1 and UGT2 subfamilies. Similar to the protocadherin, immunoglobulin, and T-cell receptor gene clusters and different from the UGT2 gene cluster, the UGT1 gene cluster is organized into variable and constant regions. The UGT1 variable region contains a tandem array of variable exons, each of which can be alternatively spliced to a single set of 4 downstream constant exons, generating at least nine UGT1 mRNAs that could be translated into different UGT1 glucuronyltransferase isoforms. Our previous work reveals that the relative orientations and locations of CTCF binding sites play a key role in the three-dimensional organization of the mammalian genomes in cell nuclei. Thus in order to study the transcriptional mechanisms of UGT1 gene cluster, the distributions and orientations of CTCF binding sites (CBSs) are analyzed and compared between human and mouse UGT1 gene clusters. We find that the CBSs in the UGT1 gene cluster are not conserved between human and mouse species. We show that CTCF and cohesin regulate the transcription of the UGT1 gene cluster by knocking down the CTCF or the cohesin subunit SMC3 in the human A549 cell line. By using CRISPR DNA-fragment editing, we deleted and inverted hCBS1. By RNA-seq experiments, we find that hCBS1 deletion results in a significant decrease of levels of the UGT1A6, UGT1A7, and UGT1A9 gene expression and that hCBS1 inversion results in a significant decrease of levels of the UGT1A7 gene expression. Our data suggest that the CTCF binding site hCBS1 plays an important regulatory role in the regulation of UGT1 gene expression, providing an experimental basis for further mechanistic studies of the 3D genome regulation of the UGT1 gene cluster.
DOI:10.16288/j.yczz.19-072URLPMID:31257199 [本文引用: 1]
UDP-glucuronosyltransferases (UGTs) are an important family of phase 2 drug-metabolizing enzymes that catalyze the glucuronidation of numerous endogenous or exogenous small compounds. The aberrant expression of UGT isoforms causes many diseases, such as hyperbilirubinemia and affect drug efficacy or toxicity. Understanding mechanisms of UGT gene regulation will provide scientific foundations for disease prevention and personalized or precision medicine. Vertebrate UGT family genes can be divided into UGT1 and UGT2 subfamilies. Similar to the protocadherin, immunoglobulin, and T-cell receptor gene clusters and different from the UGT2 gene cluster, the UGT1 gene cluster is organized into variable and constant regions. The UGT1 variable region contains a tandem array of variable exons, each of which can be alternatively spliced to a single set of 4 downstream constant exons, generating at least nine UGT1 mRNAs that could be translated into different UGT1 glucuronyltransferase isoforms. Our previous work reveals that the relative orientations and locations of CTCF binding sites play a key role in the three-dimensional organization of the mammalian genomes in cell nuclei. Thus in order to study the transcriptional mechanisms of UGT1 gene cluster, the distributions and orientations of CTCF binding sites (CBSs) are analyzed and compared between human and mouse UGT1 gene clusters. We find that the CBSs in the UGT1 gene cluster are not conserved between human and mouse species. We show that CTCF and cohesin regulate the transcription of the UGT1 gene cluster by knocking down the CTCF or the cohesin subunit SMC3 in the human A549 cell line. By using CRISPR DNA-fragment editing, we deleted and inverted hCBS1. By RNA-seq experiments, we find that hCBS1 deletion results in a significant decrease of levels of the UGT1A6, UGT1A7, and UGT1A9 gene expression and that hCBS1 inversion results in a significant decrease of levels of the UGT1A7 gene expression. Our data suggest that the CTCF binding site hCBS1 plays an important regulatory role in the regulation of UGT1 gene expression, providing an experimental basis for further mechanistic studies of the 3D genome regulation of the UGT1 gene cluster.
URLPMID:10 [本文引用: 3]
DOI:10.1016/j.cell.2015.04.004URLPMID:25959774 [本文引用: 2]
Mammalian genomes are organized into megabase-scale topologically associated domains (TADs). We demonstrate that disruption of TADs can rewire long-range regulatory architecture and result in pathogenic phenotypes. We show that distinct human limb malformations are caused by deletions, inversions, or duplications altering the structure of the TAD-spanning WNT6/IHH/EPHA4/PAX3 locus. Using CRISPR/Cas genome editing, we generated mice with corresponding rearrangements. Both in mouse limb tissue and patient-derived fibroblasts, disease-relevant structural changes cause ectopic interactions between promoters and non-coding DNA, and a cluster of limb enhancers normally associated with Epha4 is misplaced relative to TAD boundaries and drives ectopic limb expression of another gene in the locus. This rewiring occurred only if the variant disrupted a CTCF-associated boundary domain. Our results demonstrate the functional importance of TADs for orchestrating gene expression via genome architecture and indicate criteria for predicting the pathogenicity of human structural variants, particularly in non-coding regions of the human genome.
DOI:10.1126/science.aad9024URLPMID:26940867 [本文引用: 2]
Oncogenes are activated through well-known chromosomal alterations such as gene fusion, translocation, and focal amplification. In light of recent evidence that the control of key genes depends on chromosome structures called insulated neighborhoods, we investigated whether proto-oncogenes occur within these structures and whether oncogene activation can occur via disruption of insulated neighborhood boundaries in cancer cells. We mapped insulated neighborhoods in T cell acute lymphoblastic leukemia (T-ALL) and found that tumor cell genomes contain recurrent microdeletions that eliminate the boundary sites of insulated neighborhoods containing prominent T-ALL proto-oncogenes. Perturbation of such boundaries in nonmalignant cells was sufficient to activate proto-oncogenes. Mutations affecting chromosome neighborhood boundaries were found in many types of cancer. Thus, oncogene activation can occur via genetic alterations that disrupt insulated neighborhoods in malignant cells.
DOI:10.1073/pnas.1219280110URLPMID:23204437 [本文引用: 2]
The closely linked human protocadherin (Pcdh) α, β, and γ gene clusters encode 53 distinct protein isoforms, which are expressed in a combinatorial manner to generate enormous diversity on the surface of individual neurons. This diversity is a consequence of stochastic promoter choice and alternative pre-mRNA processing. Here, we show that Pcdhα promoter choice is achieved by DNA looping between two downstream transcriptional enhancers and individual promoters driving the expression of alternate Pcdhα isoforms. In addition, we show that this DNA looping requires specific binding of the CTCF/cohesin complex to two symmetrically aligned binding sites in both the transcriptionally active promoters and in one of the enhancers. These findings have important implications regarding enhancer/promoter interactions in the generation of complex Pcdh cell surface codes for the establishment of neuronal identity and self-avoidance in individual neurons.
[本文引用: 2]
[本文引用: 2]
DOI:10.1016/s0092-8674(00)80789-8URLPMID:10380929 [本文引用: 1]
We have identified 52 novel human cadherin-like genes organized into three closely linked clusters. Comparison of the genomic DNA sequences with those of representative cDNAs reveals a striking genomic organization similar to that of immunoglobulin and T cell receptor gene clusters. The N-terminal extracellular and transmembrane domains of each cadherin protein are encoded by a distinct and unusually large exon. These exons are organized in a tandem array. By contrast, the C-terminal cytoplasmic domain of each protein is identical and is encoded by three small exons located downstream from the cluster of N-terminal exons. This unusual organization has interesting implications regarding the molecular code required to establish complex networks of neuronal connections in the brain and the mechanisms of cell-specific cadherin-like gene expression.
DOI:10.13703/j.0255-2930.2017.10.012URLPMID:29354976 [本文引用: 1]
The acupoint effect of Sanyinjiao (SP 6) and Xuanzhong (GB 39) as well as the internal relation of Sanyinjiao (SP 6)-Xuanzhong (GB 39) were analyzed in this paper to explore the relationship between opposite acupoints and acupoint effect. It was found both Sanyinjiao (SP 6) and Xuanzhong (GB 39) had acupoint effects, and the two acupoints had close relationship in acupoint effects (specificity along meridian and specificity of acupoints), acupoint-meridian relationship (anatomical locations, meridians and organs, indications). It is indicated the opposite acupoint of Sanyinjiao (SP 6)-Xuanzhong (GB 39) had related aspects in acupoint effects, which is related with acupoint effect through specificity and relationship of acupoint-meridian.
DOI:10.13703/j.0255-2930.2017.10.012URLPMID:29354976 [本文引用: 1]
The acupoint effect of Sanyinjiao (SP 6) and Xuanzhong (GB 39) as well as the internal relation of Sanyinjiao (SP 6)-Xuanzhong (GB 39) were analyzed in this paper to explore the relationship between opposite acupoints and acupoint effect. It was found both Sanyinjiao (SP 6) and Xuanzhong (GB 39) had acupoint effects, and the two acupoints had close relationship in acupoint effects (specificity along meridian and specificity of acupoints), acupoint-meridian relationship (anatomical locations, meridians and organs, indications). It is indicated the opposite acupoint of Sanyinjiao (SP 6)-Xuanzhong (GB 39) had related aspects in acupoint effects, which is related with acupoint effect through specificity and relationship of acupoint-meridian.
DOI:10.1007/s13238-019-0623-2URLPMID:31037510 [本文引用: 1]
Cockayne syndrome (CS) is a rare autosomal recessive?inherited?disorder?characterized by a variety of clinical features, including?increased sensitivity?to?sunlight,?progressive neurological abnormalities, and the appearance?of?premature aging. However, the pathogenesis of CS remains unclear due to the limitations of current disease models. Here, we generate integration-free induced pluripotent stem cells (iPSCs) from fibroblasts from a CS patient bearing mutations in CSB/ERCC6 gene and further derive isogenic gene-corrected CS-iPSCs (GC-iPSCs) using the CRISPR/Cas9 system. CS-associated phenotypic defects are recapitulated in CS-iPSC-derived mesenchymal?stem?cells (MSCs) and neural stem cells (NSCs), both of which display increased susceptibility to DNA damage stress. Premature aging defects in CS-MSCs are rescued by the targeted correction of mutant ERCC6. We next map the transcriptomic landscapes in CS-iPSCs and GC-iPSCs and their somatic stem cell derivatives (MSCs and NSCs) in the absence or presence of ultraviolet (UV)?and replicative stresses, revealing that defects in DNA repair account for CS pathologies. Moreover, we generate autologous GC-MSCs free of pathogenic mutation under a cGMP (Current Good Manufacturing Practice)-compliant condition, which hold potential for use as improved biomaterials for future stem cell replacement therapy for CS. Collectively, our models demonstrate novel disease features and molecular mechanisms and lay a foundation for the development of novel therapeutic strategies to treat CS.
DOI:10.1038/s41587-018-0011-0URLPMID:30742127 [本文引用: 1]
Broad use of CRISPR-Cas12a (formerly Cpf1) nucleases1 has been hindered by the requirement for an extended TTTV protospacer adjacent motif (PAM)2. To address this limitation, we engineered an enhanced Acidaminococcus sp. Cas12a variant (enAsCas12a) that has a substantially expanded targeting range, enabling targeting of many previously inaccessible PAMs. On average, enAsCas12a exhibits a twofold higher genome editing activity on sites with canonical TTTV PAMs compared to wild-type AsCas12a, and we successfully grafted a subset of mutations from enAsCas12a onto other previously described AsCas12a variants3 to enhance their activities. enAsCas12a improves the efficiency of multiplex gene editing, endogenous gene activation and C-to-T base editing, and we engineered a high-fidelity version of enAsCas12a (enAsCas12a-HF1) to reduce off-target effects. Both enAsCas12a and enAsCas12a-HF1 function in HEK293T and primary human T cells when delivered as ribonucleoprotein (RNP) complexes. Collectively, enAsCas12a provides an optimized version of Cas12a that should enable wider application of Cas12a enzymes for gene and epigenetic editing.
DOI:10.1016/j.cell.2017.06.050URLPMID:28757251 [本文引用: 1]
Cas13a, a type VI-A CRISPR-Cas RNA-guided RNA ribonuclease, degrades invasive RNAs targeted by CRISPR RNA (crRNA) and has potential applications in RNA technology. To understand how Cas13a is?activated to cleave RNA, we have determined the?crystal structure of Leptotrichia buccalis (Lbu) Cas13a bound to crRNA and its target RNA, as well as the cryo-EM structure of the LbuCas13a-crRNA complex. The crRNA-target RNA duplex binds in a positively charged central channel of the nuclease (NUC) lobe, and Cas13a protein and crRNA undergo a significant conformational change upon target RNA binding. The guide-target RNA duplex formation triggers HEPN1 domain to move toward HEPN2 domain, activating the HEPN catalytic site of Cas13a protein, which subsequently cleaves both single-stranded target and collateral RNAs in a non-specific manner. These findings reveal how Cas13a of type VI CRISPR-Cas systems defend against RNA phages and set the stage for its development as a tool for RNA manipulation.
DOI:10.1038/s41588-019-0462-3URLPMID:31308546 [本文引用: 2]
Chromatin topology is intricately linked to gene expression, yet its functional requirement remains unclear. Here, we comprehensively assessed the interplay between genome topology and gene expression using highly rearranged chromosomes (balancers) spanning ~75% of the Drosophila genome. Using transheterozyte (balancer/wild-type) embryos, we measured allele-specific changes in topology and gene expression in cis, while minimizing trans effects. Through genome sequencing, we resolved eight large nested inversions, smaller inversions, duplications and thousands of deletions. These extensive rearrangements caused many changes to chromatin topology, disrupting long-range loops, topologically associating domains (TADs) and promoter interactions, yet these are not predictive of changes in expression. Gene expression is generally not altered around inversion breakpoints, indicating that mis-appropriate enhancer-promoter activation is a rare event. Similarly, shuffling or fusing TADs, changing intra-TAD connections and disrupting long-range inter-TAD loops does not alter expression for the majority of genes. Our results suggest that properties other than chromatin topology ensure productive enhancer-promoter interactions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}