 ,中国医学科学院基础医学研究所病理生理学系,医学分子生物学国家重点实验室,北京 100730
,中国医学科学院基础医学研究所病理生理学系,医学分子生物学国家重点实验室,北京 100730Molecular mechanism of the 3D genome structure and function regulation during cell terminal differentiation
Ke Yang, Zheng Xue, Xiang Lv,State Key Laboratory of Medical Molecular Biology, Department of Pathophysiology, Institute of Basic Medical Sciences, Peking Union Medical College & Chinese Academy of Medical Sciences, Beijing 100730, China通讯作者: 吕湘,博士,教授,研究方向:造血调控与三维基因组。E-mail:lvxiang@pumc.edu.cn
编委: 方向东
收稿日期:2019-09-5修回日期:2019-12-13网络出版日期:2020-01-20
| 基金资助: |
Editorial board:
Received:2019-09-5Revised:2019-12-13Online:2020-01-20
| Fund supported: |
作者简介 About authors
杨科,博士研究生,研究方向:造血调控与三维基因组。E-mail:yangk_92@163.com。

摘要
真核细胞中的染色质DNA高度折叠形成复杂的三维结构,其空间组织方式对精准调控基因的表达和细胞发挥正常功能都起着重要的作用。细胞终末分化成熟过程中形态及基因表达谱常发生显著改变,同时伴随着明显的基因组三维结构变化。本文在简单介绍三维基因组多层次组织结构(染色质领域、A/B区室、拓扑相关结构域和成环构象等)基础上,重点综述了细胞终末分化过程中三维基因组结构变化与功能调控方面的研究进展,并探讨了当前三维基因组研究在解析细胞分化成熟过程时存在的问题和前景。
关键词:
Abstract
The eukaryotic chromatin is folded into highly complex three-dimensional (3D) structures, which plays an important role in the precise regulation of gene expression and normal physiological function. During differentiation and terminal maturation, cells usually undergo dramatic morphology and gene expression changes, accompanied by significant changes in the 3D structure of the genome. In this review, we provide a comprehensive view of the spatial hierarchical organization of the genome, including chromosome territories, A/B compartment, topologically associating domains (TADs) and looping, focusing on recent progresses in the dynamic 3D genomic structural changes and functional regulation during cell differentiation and terminal maturation. In the end, we summarize the unsolved issues as well as prospects of the 3D genome research in cell differentiation and maturation.
Keywords:
PDF (540KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
杨科, 薛征, 吕湘. 细胞终末分化过程中三维基因组结构与功能调控的分子机制. 遗传[J], 2020, 42(1): 32-44 doi:10.16288/j.yczz.19-270
Ke Yang.
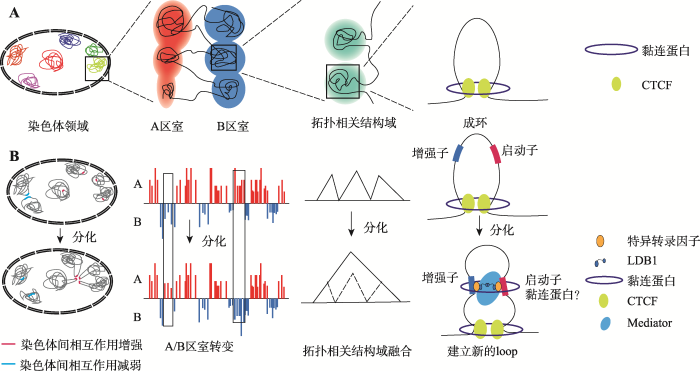
自遗传密码被破解以来,尤其是多个种属基因组测序完成后,基因组的一维序列信息得到了广泛研究和阐释。然而基因组在细胞核内并非以简单的一维线性排布,而是以复杂的三维结构存在,基因组的三维结构对基因的表达调控和细胞正常功能的发挥有着重要影响[1]。染色体构象捕获技术(chromosome conformation capture, 3C)[2]及由其衍生而来的“C”系列技术[3,4,5,6,7,8]的发展,使研究者们得以在DNA分子水平研究染色质之间的相互作用。3C技术主要是通过对交联固定后的核内基因组DNA进行限制性酶切连接,使一维序列上远离而空间上邻近的酶切片段有机会连接形成嵌合片段,再通过PCR检测定量连接产物,以校正后的定量结果来反映染色质片段间相互作用的强度。该技术建立后迅速从检测两个特定基因座位点间相互作用的3C方法发展到检测单个位点与整个基因组相互作用的(circular chromosome conformation capture/chromosome conformation capture-on-chip) 4C[3,4]、检测多对基因座位点间相互作用的(chromosome conformation capture carbon copy) 5C[6]、以及适用于全基因组染色质相互作用的(high-throughput chromosome conformation capture) Hi-C[5]等。Hi-C技术在酶切片段末端引入生物素标记后再进行连接反应,末端消化去除未连接端的生物素,实现对新形成嵌合片段的特异富集,测序获得全基因组范围的染色质相互作用信息。对Hi-C数据的深入解析使人们可以从染色体领域(chromosome territory, CT)、染色体间相互作用、A/B区室(compartment)、拓扑相关结构域(topologically associated domain, TAD)以及成环构象(looping)等不同层次揭示染色质的三维结构情况(图1A)。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1三维基因组的多层次组织结构及其在细胞分化前后的变化
A:三维基因组的多层次组织结构;B:细胞分化过程中的三维基因组结构变化。
Fig. 1The multi-level organization of three-dimensional genome and its variation during cell differentiation
细胞分化和发育过程中,随着细胞功能的改变,染色质三维结构也会发生相应变化。有关早期胚胎发育和细胞定向分化的三维基因组研究近期已有Zhang等[9]综述文章给予了详细介绍。细胞终末分化
成熟是干祖细胞发生特化、转变成为执行特定功能细胞的生理变化过程,该过程对应着基因表达谱的显著转变,并常伴随细胞核形态和大小的变化,进一步提示染色质三维结构可能发生了剧烈的转变。本文概述了染色质三维结构不同层次上的研究进展,重点讨论了细胞终末分化成熟过程中染色质三维结构功能的动态调节。
1 三维基因组的多层次组织结构
1.1 染色体领域
真核生物主要以染色体的形式承载遗传信息,染色体结构的存在为基因组以一定规律聚集或离散分布于细胞核中提供了基础和限制。在间期细胞核内,每条染色体占据一个相对独立但稳定的细胞核亚区域,称为染色体领域[10]。可以利用荧光原位杂交技术(fluorescent in situ hybridization, FISH),通过源于特定染色体全序列的探针来获得染色体领域的直观图像[11]。Hi-C分析也发现同一染色体内即使相距很远的位点间相互作用频率也明显高于染色体间的相互作用,提示染色体内的DNA呈聚集分布,从分子水平上支持了CT模型[5]。研究表明,CT在细胞核内所处的位置并非随机,长染色体倾向与长染色体相互接近,而短染色体之间也更为靠近[12]。在同一CT中富含基因或活跃转录的DNA片段更高频率定位于CT的外围, 甚至突出到CT之外[13,14],这样更有利于接近位于CT之间(interchromatin compartment, IC)的转录机器,并和邻近染色体上转录活性类似的基因共转录[15]。在发育过程中,基因的激活常伴随其在CT中的定位由内部转移到外围[16]。CT具有组织和细胞类型特异性[17],细胞分裂间期核内的各个CT位置相对稳定,但最近有研究发现有丝分裂前后以及两个子细胞之间CT的位置会发生一定程度的变化[18],这可能与细胞之间的个体差异有关。1.2 染色质核内定位与A/B区室
基因密度和转录活性不仅与其在CT中的定位有关,还与染色质在细胞核内的定位密切相关。基因密度低及不活跃转录的染色质倾向于定位在核周,其中与核纤层相互作用的区域称为核纤层蛋白相关结构域(lamin associated domains, LADs),富含H3K27me3和H3K9me2等非活性组蛋白修饰[19]。基因密度高或活跃表达的染色质则倾向定位于细胞核内部[20]。基因失去与核纤层的相互作用转而定位于核内部后,可以重新激活其表达,反之亦然[21]。在红系细胞中,LDB1复合物介导β-珠蛋白与其增强子基因座控制区(locus control region, LCR)间的相互作用,促进β-珠蛋白基因表达并定位于细胞核内部,敲低LDB1可抑制β-珠蛋白基因表达,其基因也从细胞核中部重新定位于核周[22]。此外,细胞核内邻近的活跃转录基因可以共定位并共享转录机器,形成转录工厂结构[23]。动态表达的基因在转录激活或抑制时移入或远离转录工厂,相比从头招募和组装转录复合物,将基因移动到预先装配好的转录工厂可能是更经济及常见的基因转录激活方式[23]。一个细胞核中通常存在多个不同类型的转录工厂[24],例如红系α-珠蛋白基因簇内的珠蛋白基因与其上游持家基因在空间上靠近并共同转录,是最早被鉴定的转录工厂之一[25]。类似的结构还有由不同染色体上的多个rRNA基因共定位,在核仁处形成的RNA聚合酶Ⅰ转录工厂[26]。转录工厂结构使细胞更有效的组织和调节自身基因的转录,协调细胞功能。Hi-C分析提供了另一种核内染色质分区定位的方法。其结果提示具有转录活性的片段之间有着更高的相互作用频率,包括松散的染色质、具有活性组蛋白修饰、高基因密度和高表达水平的DNA片段等。同样基因组的非活性区段更倾向于和非活性区段相互作用[5]。通过主成分分析(principal component analysis, PCA)将这种Hi-C揭示的相互作用倾向性量化,可将基因组区分为相对分隔的A/B 两个区室。其中A区室基因密度较高,更富集DNaseⅠ高敏位点和H3K4me3等表观遗传修饰标志,与活性常染色质相对应,而B区室多处于基因荒漠区或为异染色质。染色质间相互作用更多发生在同一个区室内的位点之间。最近有研究通过分子模拟提出A/B区室是经由染色质片段相分离过程形成的:当一段染色质结合在核纤层或其它核内小体上时,其他与之相同状态的染色质也会随之结合进而引发相分离,形成A/B区室[27]。
1.3 拓扑相关结构域
提高Hi-C分析的测序深度和分辨率可以发现每条染色体均由多个结构单元组成,称为拓扑相关结构域(TAD)[28,29]。早期在果蝇胚胎中的Hi-C分析即发现TAD结构,表现为多个和染色质表观修饰标志高度相关的结构域,其内部染色质相互作用明显高于相邻结构域之间的互作等特点[30]。哺乳动物90%的基因组被约2000个TADs覆盖,其长度范围从数百kb到数Mb不等[28]。同一TAD内的基因往往处于相似的活性或者非活性状态。基因组活性与非活性区段的转变频繁发生在TAD边界附近,提示TAD间的边界序列具有隔离功能[31]。在不同细胞类型中,TAD活性状态常作为一个整体发生转变,呈协同表达模式。支持TAD作为相对独立的调控单元协调邻近基因的表达。分裂期细胞中未能检测到TAD结构,推测TAD主要在细胞分裂间期起作用,参与协调转录等过程[32]。TAD位置及其边界序列在不同细胞类型间以及人和小鼠间均高度保守,进一步支持TAD边界及其隔离功能在哺乳动物细胞中具有重要意义。边界的存在使染色质片段间的相互作用限制在TAD内,从而将增强子等远距离调节元件功能局限于特定的靶基因[33]。TAD边界序列富含CTCF结合位点、转录起始位点和SINE元件[34]等序列。已知的TAD边界结合蛋白主要包括CTCF和黏连蛋白(cohesin)[35],CTCF可以起转录隔离的作用[36],阻断启动子和增强子的相互作用或表观遗传修饰标志向外延伸。黏连蛋白复合物形成环状结构,和CTCF共同参与TAD的形成。扰动CTCF/cohesin或其结合位点可增加邻近TAD间的相互作用,同时TAD内相互作用强度相对减弱,但并不完全破坏TAD边界,表明CTCF和cohesin在TAD的形成和维持中起一定作用[35,37~39],但不足以创建或消除TAD结构。
目前,TAD的鉴定主要建立在细胞群体Hi-C分析基础之上。单细胞Hi-C分析提示,TAD在单个细胞中可能不是固定的结构,而是群体细胞的染色质互作倾向性在总体上的反映[18,40]。最近,Bintu等[41]和Szabo等[42]基于高分辨成像的研究显示,即便在单细胞水平,类似TAD的结构也是存在的,这种差异也许与经典的分析流程对过于分散的单细胞Hi-C数据不太适用有关。
1.4 成环构象
细胞核内一维距离较远的DNA元件可以在空间上相互靠近,元件间的DNA片段环出,形成成环构象。Hi-C分析发现同一TAD中常存在多个染色质环,参与基因表达调节,其中尤以增强子和启动子间的成环构象在基因表达调控中的作用最为明确。β-珠蛋白基因簇是研究时空特异基因表达调控的理想模型[43]。其结构基因上游数十kb外的LCR作为超级增强子可显著增强簇内各类β-珠蛋白基因的表达。早前研究对远距离的LCR如何增强β-珠蛋白基因表达提出了成环模型(looping model),蛋白质联接介导模型(linking model)等多种假说[44]。3C技术从分子水平上证实了LCR和β-珠蛋白启动子在空间上靠近,支持增强子作用的成环模型[45]。启动子和增强子的相互作用常具有组织和发育阶段特异性,而非简单的一对一方式[46,47,48,49]。以LCR为例,在胚胎、胎儿和成年期分别与ε-、γ-和β-珠蛋白基因启动子空间靠近并激活其表达。反之,一个启动子也可与多个增强子互作。这种复杂的成环构象模式为基因选择性表达和精细调节提供了基础。TAD内部常有多个CTCF结合位点,可招募CTCF/cohesin等分子介导其相互作用,为基因组 成环构象的形成和维持提供基础[50]。敲除CTCF或cohesin抑制这些位点间成环构象的形成[51]。在CTCF参与的成环模型中,环状的cohesin复合物结合在染色体上,形成染色质小环,cohesin顺着小环两端向外延伸移动,挤压导致染色质环增大,直至遭遇两个汇聚的(即方向朝环内的)CTCF结合位点,并和CTCF共同稳定该环状结构[52]。转录事件本身及cohesin释放因子wapl共同参与cohesin在CTCF位点的募集[53]。在此基础上,转录因子招募互作蛋白或RNA聚合酶类形成复杂的转录复合物,介导增强子和启动子的相互作用及增强子转录调控功能,并通常将增强子的作用限制在CTCF/cohesin介导的成环构象及TAD边界以内。
核内染色质组织方式与基因转录活性密切相关,但其因果关系值得探讨。用DRB处理小鼠胎肝细胞抑制转录延伸对远距离基因共享转录工厂以及β-珠蛋白基因簇增强子与启动子的长距离相互作用均没有影响[54,55]。对转录起始的抑制因处理方法不同而效果有所差异,通过热激反应抑制转录起始显著影响转录工厂的形成并部分抑制β-珠蛋白启动子与LCR核心增强子HS2/3间的相互作用[55],而α-鹅膏蕈碱处理对β-珠蛋白启动子与LCR的相互作用没有影响[54]。这种差异可能和α-鹅膏蕈碱与热激具体抑制转录起始的方式不同有关。但两种转录起始抑制在总体上均对β-珠蛋白启动子与LCR相互作用形成的活性染色质结构域影响不大,表明染色质之间的相互作用并不是仅仅是基因转录的伴随现象。之后Deng等[56]在敲除GATA1的G1E细胞系中表达可识别GATA1基序的锌指结构与LDB1融合蛋白,人为把LCR和β-珠蛋白基因启动子联系在一起,促进了RNA聚合酶Pol II在β-珠蛋白基因启动子的募集,支持染色质成环构象可促进基因活跃转录。最近Ray等[57]的工作也发现热激不影响细胞的A/B分区和TAD结构,此时激活的基因多数在非热激状态已经建立增强子与启动子间相互作用,仅少部分与增强子启动子间的相互作用增强有关。综上提示,染色质三维结构是独立于转录事件的基因组特征,但又与基因转录密切相关并相互影响。
2 细胞终末分化过程中三维基因组结构变化
干祖细胞经由终末分化过程最终成为执行特定生理功能的成熟细胞,其细胞类型多样,对应复杂独特的细胞功能,并经常伴随细胞及细胞核形态大小上的不同变化,提示各种不同类型的终末分化过程中可能具有不同特色的三维基因组组织方式。对其三维基因组结构的详细解析将有助于了解终末分化阶段发挥重要功能基因(简称终末功能基因)的表达维持和相关疾病发生的分子机制。另外,细胞进入终末分化后许多终末功能基因的表达即已开启,细分的终末成熟各阶段细胞在三维基因组结构上是否仍存在显著变化,还是在原构架基础上微调值得关注。近年来,研究人员对T-细胞[58,59]、B-细胞[60,61]、巨噬细胞[62]、(骨骼/平滑)肌细胞[63]、神经细胞[64,65]、表皮角质细胞[66]和胰岛β-细胞[67]等多种细胞终末分化成熟过程中的染色质三维结构进行研究,在不同层次上展示了基因组三维结构伴随终末分化中重要基因表达呈现的各自特异的变化模式(图1B)。2.1 细胞终末分化过程中的A/B区室翻转以及染色体间相互作用改变
基因组的A/B分区与染色质活性和抑制状态相对应,具有明显的组织特异性。细胞分化成熟过程往往伴随部分染色质区段的A/B分区转变。T细胞分化成熟过程经历造血干祖细胞(hematopoietic stem and progenitor cell, HSPC)、多潜能祖细胞(multipotent progenitor, MPP)、共同淋系祖细胞(common lymphoid progenitor, CLP)、早期T祖细胞(early T precursor, ETP)、CD4/CD8双阴T细胞2 (CD4 and CD8 double-negative 2, DN2)、DN3、DN4和CD4/CD8双阳T细胞(CD4 and CD8 double-positive, DP)等多个阶段。这个过程的基因组A/B分区持续发生转变,其中57.6%由B区室转变为A区室,37.4%由A区室转变为B区室,仅4.9%的区域在A/B分区上的存在反复。A/B区室的转变主要集中在DN2-DN3和DN4-DP阶段,提示这两个时间段可能是T细胞终末分化的关键。这与前期研究提出的DN2-DN3转变与T细胞命运决定相关,而DN4-DP转变是T细胞β选择关键步骤的观点相一致[68]。作为典型代表,T细胞分化和存活的关键转录因子BCL11b基因由造血干细胞中的B区室向CD4/CD8双阳性细胞中活性的A区室转变,同时基因定位由核周转向核内部。相反造血干细胞自我更新的关键因子Meis1基因则由活跃的A区室转变为抑制状态的B区室,基因定位向核周转移。该过程中基因的沉默和开启均早于其A/B分区的翻转,提示区室转变并非基因表达变化的上游因素[59]。除染色质A/B分区翻转外,同一区室内染色质相互作用强度和染色体间相互作用也伴随终末分化过程发生改变。小鼠皮质神经元终末分化时A/B区室均表现为大小渐增,其中A区室内部相互作用变弱、而B区室内部相互作用加强[65],与此前报道的神经细胞分化过程中异染色质聚集一致[69]。幼稚T细胞成熟分化为T辅Ⅰ、T辅Ⅱ细胞的过程中,11号染色体上调节元件和10号染色体γ干扰素基因启动子间的相互作用在分化后被染色体内相互作用所代替。在T辅Ⅰ细胞中,γ干扰素与其上游调控元件互作,产生更多的γ干扰素;在T辅Ⅱ细胞中,11号染色体上的调控元件与邻近的白介素基因相互作用并激活其表达[70,71]。
2.2 细胞终末分化过程中的TAD变化
研究表明TAD结构在不同细胞类型和物种间相对保守,但细胞分化过程中TAD内的局部染色质相互作用强度可发生变化。T细胞分化成熟过程中,TAD内部相互作用的强弱变化也主要发生在DN2- DN3和DN4-DP两个阶段,其变化与DNaseⅠ高敏位点信号及基因表达谱改变相吻合,但在时间上先后有别。沉默基因的表达下调先于相应TAD内染色质互作信号的减弱;相反激活基因TAD内染色质互作变强早于其基因表达上调[59],支持染色质互作只是基因转录的前提条件,转录激活还需要更多因素的参与。这种时序关系在转录抑制剂处理或热激引起转录改变时观察到的现象基本一致[54,55,57]。虽然TAD结构相对稳定保守,在一些细胞分化过程中也观察到相邻TAD融合和TAD分裂的情况。幼稚B细胞(na?ve B cells)向GC B细胞分化过程中TAD数目(>1500个)和平均大小(1.35 Mb)均无明显变化,但观察到其中171个TAD结构域与邻近TAD间融合形成更大的新型3D基因“城市”(city)结构。对三号染色体上一段6 Mb长的区域具体分析显示,该区段在幼稚B细胞中分为4个TAD结构域,其中多个基因在细胞分化为GC B细胞时跨越TAD边界建成新的染色质间相互作用,从而整段染色质融合形成一个大型TAD结构。跨界参与染色质相互作用的基因多在GC B细胞中特异表达上调并在其细胞特化过程中发挥重要作用。总体而言,相邻TAD的融合促进了基因的协同表达[60]。小鼠皮质神经元终末分化时也观察到TAD数目减少和平均大小增加,但同时一类不含CTCF的TAD边界明显增多。这类TAD边界附近富集终末阶段活跃表达基因的启动子,支持基因转录参与TAD边界形成,然而仅转录激活并不足以导致新TAD的出现[65]。
2.3 细胞终末分化过程的染色质成环情况
与TAD结构域的相对稳定保守相反的是,TAD内部染色质成环构象具有高度的细胞类型特异性,与不同细胞独特的基因表达谱形成密切相关。细胞终末分化过程中成环构象的研究有助于更深刻的理解细胞分化成熟的调控网络,鉴定新的调控元件和调节模式,以及发现终末分化过程中的关键基因。全基因组水平的成环构象分析对Hi-C数据量要求较高,ChIA-PET[7], capture Hi-C[8,72]和原位Hi-C[73]等方法的出现和优化以及二代测序效能的提高使在全基因组范围研究染色质成环构象成为可能。在PMA诱导的THP-1体外巨噬细胞分化模型中,通过差异loop分析发现多个分化后新形成的染色质环(获得型loop),其出现与基因表达上调显著相关。另外,对大部分未随分化改变的染色质环而言,启动子所关联的远端锚定位点上H3K27ac信号增强(激活型loop)也与基因表达上调显著相关。基因本体论(Gene Ontology, GO)分析显示获得型loop和激活型loop所对应的靶基因均富集在巨噬细胞功能相关通路上,包含IL-1β、MAFB和TLR1等参与巨噬细胞发育和功能的重要基因[62]。在人表皮角质细胞终末分化模型中观察到类似现象,基因表达上调与两种不同的增强子启动子成环构象有关:其中一类是在终末分化过程中新形成的染色质环,即获得型loop,同时伴随H3K27ac修饰水平增加;另一类环则在祖细胞阶段已经形成,并在终末分化过程中稳定存在(稳定型loop),与巨噬细胞分化有所不同的是,表皮角质细胞中这类染色质环的增强子呈组成型H3K27ac修饰[66]。由此提出成环构象参与分化过程中基因表达调节的两种模型:在远端调控元件和基因启动子间建立新的染色质相互作用,或者通过已有成环构象的重塑对靶基因进行调控。值得注意的是,前述巨噬细胞分化时有2070个基因上调,而位于获得型loop或激活型loop的启动子只有527个,且其中只有156个在分化过程中显著上调,提示成环构象只是参与基因转录调控的诸多要素之一[62]。
巨噬细胞分化成熟过程中,存在由多个增强子与同一启动子共同形成的、含有多个染色质环的3D基因组“社区”或“中心”(hubs)。类似的活性染色质中心在红系终末分化的关键基因α-珠蛋白和β-珠蛋白基因簇上也观察到过[74]。此外,在B细胞分化为GC B细胞的过程中还观察到启动子间成环构象以及启动子与自身基因3°-端的相互作用显著增多,伴随活跃染色质标志的富集和基因表达增加,其中富含GC B细胞特异表达基因[60]。3D基因组社区的组织方式有利于增加细胞核内局部增强子及其募集转录因子的浓度,共同调节分化过程中关键基因的激活。而一些关键调控元件又可同时调控与分化过程相关的多个基因,扮演染色质互作网络的核心元件。目前关于分化细胞中复杂成环构象形成的证据基本都来自对细胞群体的Hi-C分析,组成复杂成环构象的各种染色质相互作用是否同时存在于一个细胞当中尚有待通过新的实验或计算方法验证。
3 结构性因子与组织特异转录因子协同调控细胞分化成熟的染色质成环变换
CTCF是目前最广为人知的染色质成环介导因子。哺乳动物基因组上已发现的CTCF结合位点数多达5~6万。早期研究中发现的各种CTCF功能,包括转录激活、抑制、隔离和增强子阻断等,在很大程度上可以归结为CTCF作为一种结构性因子介导成环构象的形成。CTCF的具体功能取决于特定的辅因子种类和其所介导靠近的DNA序列性质[75]。一方面CTCF可与cohesin和TFIIIC等因子一起参与TAD边界的组建,发挥异染色质隔离和增强子阻断等功能;另一方面,它又可与cohesin、mediator和TAF3等因子共同介导启动子与增强子的相互作用,调控诸如神经细胞分化发育的基因启动子选择和胚胎干细胞到内胚层的分化等生物学过程[76]。细胞分化前后CTCF靶序列DNA甲基化水平,包括其经TET酶催化的各种氧化产物、CTCF自身Sumo-化修饰和多聚ADP化修饰、以及辅因子状态等均参与调节CTCF介导的成环构象形成[76,77]。近年研究显示非编码RNA在CTCF介导的染色质相互作用中也发挥重要作用[78]。最新研究对位于CTCF第11号锌指C-端的一段长38个氨基酸的RNA结合结构域进行敲除,发现胚胎干细胞中约一半的染色质环受到了明显影响[79]。同期文章还研究了CTCF第1及第10号锌指的RNA结合能力,其敲除亦可严重影响CTCF在染色质上的结合、成环构象形成和相应基因的表达[80]。CTCF还可以结合到基因5'-UTR序列和内含子上,调节RNA聚合酶Ⅱ的暂停和RNA加工,参与小鼠红系分化过程中myb基因的成环构象和转录延伸控制,并调节人淋巴细胞分化时CD45基因第5外显子的选择性剪接[81,82]。YY1是另一个在多种组织中普遍存在并介导增强子和启动子染色质相互作用的因子。YY1可以结合在活性的增强子和启动子上,通过二聚化介导增强子和启动子间的成环相互作用,YY1结合位点或者YY1自身的敲除均会破坏增强子和启动子之间的成环构象并降低基因表达[83]。参与染色质构象组织的结构性因子还包括核基质结合蛋白SATB1、SATB2和果蝇的BEAF-32、ZW5因子等,在红系珠蛋白基因簇、hsp70基因成环构象和介导gypsy逆转座子拷贝间互作中发挥作用。近年还发现锌指蛋白ZNF143可作为CTCF的重要辅因子广泛参与基因组成环构象的调节[84,85]。
组织特异转录因子在细胞分化特异的染色质高级构象形成当中发挥重要的作用。在单核细胞向巨噬细胞分化模型中,观察到巨噬细胞中Tn5转座酶敏感位点、新增和激活型成环构象锚定位点均富集AP-1家族转录因子基序,其富集程度甚至高于通常情况下最多见的CTCF基序。AP-1在远距离元件上的结合与染色质环另一端基因的表达激活正相关。提示AP-1及其辅因子介导了巨噬细胞特异成环构象形成并调节巨噬细胞分化[62]。细胞常利用多种因子分别参与不同类型染色质环的组织,协同调控细胞分化过程的基因表达。前述人表皮角质细胞终末分化模型中,发现仅稳定型loop上有cohesin的结合,组织特异的EHF因子也参与稳定型loop的形成并激活其基因表达;终末分化过程中新形成的染色质环上则缺乏cohesin结合,但高度富集C/EBP和KLF家族蛋白的结合基序,转录因子KLF4及其互作蛋白ZNF750在这类获得型loop的组建和基因表达激活中发挥关键作用[66]。
越来越多的研究[62,64,67]发现LIM结构域结合蛋白1 (LIM domain binding 1, LDB1)在细胞分化成熟过程的染色质高级构象动态组织中发挥关键作用。LDB1与组织特异的转录因子共同介导不同类型细胞中特异染色质成环构象形成,促进分化特异基因表达:胰岛β-细胞中LDB1与PDX1、FOXOA2、NKX6.1和NKX2.2等转录因子共同介导了多种终末功能基因启动子与增强子的相互作用,促进人与小鼠胰岛β-细胞终末分化状态的维持。LDB1的敲除严重影响成熟β-细胞的胰岛素分泌和血糖稳态[67]。在红系终末分化过程中,LMO2/TAL1/E2A和核心因子GATA1分别结合LCR和β-珠蛋白启动子,并各自募集LDB1因子,通过LDB1自身二聚化促使LCR和珠蛋白启动子成环,增加β-珠蛋白基因表达[56,86]。在PAX3诱导胚胎干细胞向肌细胞分化的模型中,以关键转录因子PAX3结合位点为中心建立染色质成环构象。质谱分析发现PAX3可与LDB1和cohesin复合物核心成分SMC1相互作用,LDB1可独立于SMC1募集到部分PAX3结合的增强子元件并介导成环构象形成。敲低LDB1表达可导致PAX3结合位点H3K4me1表观修饰的降低和成环构象破坏,进而抑制PAX3驱动的基因表达和肌细胞分化过程[63]。LDB1介导的成环构象在小鼠嗅觉受体的选择型表达中也发挥重要作用。小鼠基因组中共有1000多个嗅觉受体基因,分布在18条不同染色体上[87]。每个成熟的嗅感觉神经元只表达一个嗅觉受体基因[88]。在水平基底细胞(horizontal basal cells)分化最终产生嗅感觉神经元的过程中,染色质高级构象分析显示,随机的某个嗅觉受体基因与63个处于不同染色体上的增强子相互作用,反式激活该嗅觉受体基因的表达。LHX2蛋白结合在这些称为“希腊小岛”(Greek islands)的嗅觉受体基因增强子上,通过其LIM结构域和辅因子LDB1结合,稳定“希腊小岛”间的反式染色质相互作用并促进关联嗅觉受体基因的表达[64]。另外,LDB1还与多种神经细胞以及心肌、乳腺和垂体的分化发育有关。LDB1介导的成环构象及组织特异转录因子是否及如何参与调控在这些组织细胞分化成熟的过程尚有待进一步探究。
4 结语与展望
目前大部分细胞分化的染色质高级构象研究是细胞群体上平均信号水平的反映,考虑到细胞的异质性,尤其细胞分化作为一个动态过程异质性更为明显,在单细胞水平研究细胞分化的三维基因组结构变化是未来的发展方向之一[8,18,40,89]。Hi-C策略需要通过酶切连接的方法检测位点之间的相互作用,每个酶切片段只有两个末端可与空间邻近的片段连接,从而限制了单细胞Hi-C中复杂成环构象,如转录工厂或者活性染色质中心等结构的鉴定。开发和优化新的单细胞染色质构象检测技术和分析方法,利用诸如串联体连接分析法(concatemer ligation assay, COLA)[90]、不依赖邻位连接的SPRITE[91]和阮一骏教授团队新发展的ChIA-Drop技术[92]等,在单细胞全基因组水平实现对多个成环构象共定位情况的同时检测;结合高分辨显微成像和细胞原位捕获等技术研究介导染色质构象形成的复合物组成等均有助于极大地促进人们对细胞分化过程中三维基因组组织及其调控功能的认识。细胞分化成熟过程伴随终末功能基因表达的显著增加以及干性基因和其他组织细胞特异基因的表达抑制等转录组显著变化。细胞基因组三维组织方式的改变为其转录组变化提供了结构基础。从基因的染色质定位、染色体间的相互作用、A/B区室、TAD结构和成环构象等不同层次详细考察细胞终末分化过程的染色质高级构象动态变化不但能促进人们从理论上深入了解三维基因组多层次结构的建立过程和机制,加深对染色质高级构象结构和功能的认识;也有助于人们对细胞分化成熟过程的理解,发现分化成熟过程中的关键时期、新的关键基因或者调控模式,为体外培养细胞的高效终末分化和相关疾病的诊疗提供基础。
致谢
衷心感谢军事医学科学院生物工程研究所赵志虎研究员对本文提供的宝贵建议。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1360/N972014-00163URL [本文引用: 1]
DOI:10.1360/N972014-00163URL [本文引用: 1]
DOI:10.1126/science.1067799URLPMID:11847345 [本文引用: 1]

We describe an approach to detect the frequency of interaction between any two genomic loci. Generation of a matrix of interaction frequencies between sites on the same or different chromosomes reveals their relative spatial disposition and provides information about the physical properties of the chromatin fiber. This methodology can be applied to the spatial organization of entire genomes in organisms from bacteria to human. Using the yeast Saccharomyces cerevisiae, we could confirm known qualitative features of chromosome organization within the nucleus and dynamic changes in that organization during meiosis. We also analyzed yeast chromosome III at the G1 stage of the cell cycle. We found that chromatin is highly flexible throughout. Furthermore, functionally distinct AT- and GC-rich domains were found to exhibit different conformations, and a population-average 3D model of chromosome III could be determined. Chromosome III emerges as a contorted ring.
DOI:10.1038/ng1891URLPMID:17033624 [本文引用: 2]
Accumulating evidence converges on the possibility that chromosomes interact with each other to regulate transcription in trans. To systematically explore the epigenetic dimension of such interactions, we devised a strategy termed circular chromosome conformation capture (4C). This approach involves a circularization step that enables high-throughput screening of physical interactions between chromosomes without a preconceived idea of the interacting partners. Here we identify 114 unique sequences from all autosomes, several of which interact primarily with the maternally inherited H19 imprinting control region. Imprinted domains were strongly overrepresented in the library of 4C sequences, further highlighting the epigenetic nature of these interactions. Moreover, we found that the direct interaction between differentially methylated regions was linked to epigenetic regulation of transcription in trans. Finally, the patterns of interactions specific to the maternal H19 imprinting control region underwent reprogramming during in vitro maturation of embryonic stem cells. These observations shed new light on development, cancer epigenetics and the evolution of imprinting.
DOI:10.1038/ng1896URLPMID:17033623 [本文引用: 2]
The spatial organization of DNA in the cell nucleus is an emerging key contributor to genomic function. We developed 4C technology (chromosome conformation capture (3C)-on-chip), which allows for an unbiased genome-wide search for DNA loci that contact a given locus in the nuclear space. We demonstrate here that active and inactive genes are engaged in many long-range intrachromosomal interactions and can also form interchromosomal contacts. The active beta-globin locus in fetal liver preferentially contacts transcribed, but not necessarily tissue-specific, loci elsewhere on chromosome 7, whereas the inactive locus in fetal brain contacts different transcriptionally silent loci. A housekeeping gene in a gene-dense region on chromosome 8 forms long-range contacts predominantly with other active gene clusters, both in cis and in trans, and many of these intra- and interchromosomal interactions are conserved between the tissues analyzed. Our data demonstrate that chromosomes fold into areas of active chromatin and areas of inactive chromatin and establish 4C technology as a powerful tool to study nuclear architecture.
DOI:10.1126/science.1181369URLPMID:19815776 [本文引用: 4]
We describe Hi-C, a method that probes the three-dimensional architecture of whole genomes by coupling proximity-based ligation with massively parallel sequencing. We constructed spatial proximity maps of the human genome with Hi-C at a resolution of 1 megabase. These maps confirm the presence of chromosome territories and the spatial proximity of small, gene-rich chromosomes. We identified an additional level of genome organization that is characterized by the spatial segregation of open and closed chromatin to form two genome-wide compartments. At the megabase scale, the chromatin conformation is consistent with a fractal globule, a knot-free, polymer conformation that enables maximally dense packing while preserving the ability to easily fold and unfold any genomic locus. The fractal globule is distinct from the more commonly used globular equilibrium model. Our results demonstrate the power of Hi-C to map the dynamic conformations of whole genomes.
DOI:10.1101/gr.5571506URLPMID:16954542 [本文引用: 2]
Physical interactions between genetic elements located throughout the genome play important roles in gene regulation and can be identified with the Chromosome Conformation Capture (3C) methodology. 3C converts physical chromatin interactions into specific ligation products, which are quantified individually by PCR. Here we present a high-throughput 3C approach, 3C-Carbon Copy (5C), that employs microarrays or quantitative DNA sequencing using 454-technology as detection methods. We applied 5C to analyze a 400-kb region containing the human beta-globin locus and a 100-kb conserved gene desert region. We validated 5C by detection of several previously identified looping interactions in the beta-globin locus. We also identified a new looping interaction in K562 cells between the beta-globin Locus Control Region and the gamma-beta-globin intergenic region. Interestingly, this region has been implicated in the control of developmental globin gene switching. 5C should be widely applicable for large-scale mapping of cis- and trans- interaction networks of genomic elements and for the study of higher-order chromosome structure.
DOI:10.1038/nature08497URLPMID:19890323 [本文引用: 2]
Genomes are organized into high-level three-dimensional structures, and DNA elements separated by long genomic distances can in principle interact functionally. Many transcription factors bind to regulatory DNA elements distant from gene promoters. Although distal binding sites have been shown to regulate transcription by long-range chromatin interactions at a few loci, chromatin interactions and their impact on transcription regulation have not been investigated in a genome-wide manner. Here we describe the development of a new strategy, chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) for the de novo detection of global chromatin interactions, with which we have comprehensively mapped the chromatin interaction network bound by oestrogen receptor alpha (ER-alpha) in the human genome. We found that most high-confidence remote ER-alpha-binding sites are anchored at gene promoters through long-range chromatin interactions, suggesting that ER-alpha functions by extensive chromatin looping to bring genes together for coordinated transcriptional regulation. We propose that chromatin interactions constitute a primary mechanism for regulating transcription in mammalian genomes.
DOI:10.1038/ng.2871URLPMID:24413732 [本文引用: 3]
Gene expression during development and differentiation is regulated in a cell- and stage-specific manner by complex networks of intergenic and intragenic cis-regulatory elements whose numbers and representation in the genome far exceed those of structural genes. Using chromosome conformation capture, it is now possible to analyze in detail the interaction between enhancers, silencers, boundary elements and promoters at individual loci, but these techniques are not readily scalable. Here we present a high-throughput approach (Capture-C) to analyze cis interactions, interrogating hundreds of specific interactions at high resolution in a single experiment. We show how this approach will facilitate detailed, genome-wide analysis to elucidate the general principles by which cis-acting sequences control gene expression. In addition, we show how Capture-C will expedite identification of the target genes and functional effects of SNPs that are associated with complex diseases, which most frequently lie in intergenic cis-acting regulatory elements.
DOI:10.1038/s41580-019-0132-4URLPMID:31197269 [本文引用: 1]
In eukaryotes, the genome does not exist as a linear molecule but instead is hierarchically packaged inside the nucleus. This complex genome organization includes multiscale structural units of chromosome territories, compartments, topologically associating domains, which are often demarcated by architectural proteins such as CTCF and cohesin, and chromatin loops. The 3D organization of chromatin modulates biological processes such as transcription, DNA replication, cell division and meiosis, which are crucial for cell differentiation and animal development. In this Review, we discuss recent progress in our understanding of the general principles of chromatin folding, its regulation and its functions in mammalian development. Specifically, we discuss the dynamics of 3D chromatin and genome organization during gametogenesis, embryonic development, lineage commitment and stem cell differentiation, and focus on the functions of chromatin architecture in transcription regulation. Finally, we discuss the role of 3D genome alterations in the aetiology of developmental disorders and human diseases.
DOI:10.1101/cshperspect.a003889URLPMID:20300217 [本文引用: 1]
Chromosome territories (CTs) constitute a major feature of nuclear architecture. In a brief statement, the possible contribution of nuclear architecture studies to the field of epigenomics is considered, followed by a historical account of the CT concept and the final compelling experimental evidence of a territorial organization of chromosomes in all eukaryotes studied to date. Present knowledge of nonrandom CT arrangements, of the internal CT architecture, and of structural interactions with other CTs is provided as well as the dynamics of CT arrangements during cell cycle and postmitotic terminal differentiation. The article concludes with a discussion of open questions and new experimental strategies to answer them.
DOI:10.1007/s004180050169URLPMID:9387921 [本文引用: 1]
Spectral karyotyping (SKY) is a new fluorescence in situ hybridisation (FISH) technique that refers to the molecular cytogenetic analysis of metaphase preparations by means of spectral microscopy. For SKY of human metaphase chromosomes, 24 chromosome-specific painting probes are used in just one FISH experiment. The probes are labelled by degenerate oligonucleotide-primed PCR using three fluorochromes and two haptens. Each probe is differentially labelled with one, two, three or four fluorescent dyes, resulting in a unique spectral signature for every chromosome. After in situ hybridisation and immunodetection, a spectral image is acquired using a conventional fluorescence light microscope equipped with a custom-designed triple-bandpass filter and the SpectraCube, which is able to retrieve spectral information for every pixel in a digital CCD image. The 24-colour display and chromosome classification are based on the unique emission spectra of the chromosomes. Together with chromosome banding information from an inverted DAPI or a G-banded metaphase, a comprehensive overview of chromosomal aberrations is presented.
DOI:10.1016/j.cell.2012.02.002URL [本文引用: 1]
The extent to which the three-dimensional organization of the genome contributes to chromosomal translocations is an important question in cancer genomics. We generated a high-resolution Hi-C spatial organization map of the G1-arrested mouse pro-B cell genome and used high-throughput genome-wide translocation sequencing to map translocations from target DNA double-strand breaks (DSBs) within it. RAG endonuclease-cleaved antigen-receptor loci are dominant translocation partners for target DSBs regardless of genomic position, reflecting high-frequency DSBs at these loci and their colocalization in a fraction of cells. To directly assess spatial proximity contributions, we normalized genomic DSBs via ionizing radiation. Under these conditions, translocations were highly enriched in cis along single chromosomes containing target DSBs and within other chromosomes and subchromosomal domains in a manner directly related to pre-existing spatial proximity. By combining two high-throughput genomic methods in a genetically tractable system, we provide a new lens for viewing cancer genomes.
DOI:10.1007/s10577-011-9245-0URL [本文引用: 1]
The ability to visualize specific DNA sequences, on chromosomes and in nuclei, by fluorescence in situ hybridization (FISH) is fundamental to many aspects of genetics, genomics and cell biology. Probe selection is currently limited by the availability of DNA clones or the appropriate pool of DNA sequences for PCR amplification. Here, we show that liquid-phase probe pools from sequence capture technology can be adapted to generate fluorescently labelled pools of oligonucleotides that are very effective as repeat-free FISH probes in mammalian cells. As well as detection of small (15 kb) and larger (100 kb) specific loci in both cultured cells and tissue sections, we show that complex oligonucleotide pools can be used as probes to visualize features of nuclear organization. Using this approach, we dramatically reveal the disposition of exons around the outside of a chromosome territory core and away from the nuclear periphery.
DOI:10.1016/j.ceb.2007.04.016URL [本文引用: 1]
The establishment and maintenance of differential patterns of gene expression lie at the heart of development. How the precision of developmental gene regulation is achieved, despite the highly repetitive and complex nature of the mammalian genome, remains an important question. It is becoming increasingly clear that genetic regulation must be considered not only in the context of short- and long-range regulatory sequences and local chromatin structure, but also at the level of position within the nucleus. Recent studies have addressed the extent to which the location of a gene relative to its interphase chromosome territory affects its regulation or its capacity to be expressed. Two model systems have emphasized the role of this level of nuclear organization during development. Hox gene clusters have provided important insights into the dynamic repositioning of a locus relative to its chromosome territory during spatial and temporal patterning of gene expression. The inactive X chromosome has also become a useful paradigm for studying the differential chromatin status and chromosomal organization of the two X's within the same nucleus. Recent work suggests that chromosome territory reorganisation can be an important step in the gene silencing process.
DOI:10.1038/ng.496URLPMID:20010836 [本文引用: 1]
The discovery of interchromosomal interactions in higher eukaryotes points to a functional interplay between genome architecture and gene expression, challenging the view of transcription as a one-dimensional process. However, the extent of interchromosomal interactions and the underlying mechanisms are unknown. Here we present the first genome-wide analysis of transcriptional interactions using the mouse globin genes in erythroid tissues. Our results show that the active globin genes associate with hundreds of other transcribed genes, revealing extensive and preferential intra- and interchromosomal transcription interactomes. We show that the transcription factor Klf1 mediates preferential co-associations of Klf1-regulated genes at a limited number of specialized transcription factories. Our results establish a new gene expression paradigm, implying that active co-regulated genes and their regulatory factors cooperate to create specialized nuclear hot spots optimized for efficient and coordinated transcriptional control.
DOI:10.1242/dev.02779URLPMID:17251268 [本文引用: 1]
The relocalisation of some genes to positions outside chromosome territories, and the visible decondensation or unfolding of interphase chromatin, are two striking facets of nuclear reorganisation linked to gene activation that have been assumed to be related to each other. Here, in a study of nuclear reorganisation around the Hoxd cluster, we suggest that this may not be the case. Despite its very different genomic environment from Hoxb, Hoxd also loops out from its chromosome territory, and unfolds, upon activation in differentiating embryonic stem (ES) cells and in the tailbud of the embryo. However, looping out and decondensation are not simply two different manifestations of the same underlying change in chromatin structure. We show that, in the limb bud of the embryonic day 9.5 embryo, where Hoxd is also activated, there is visible decondensation of chromatin but no detectable movement of the region out from the chromosome territory. During ES cell differentiation, decondensed alleles can also be found inside of chromosome territories, and loci that have looped out of the territories can appear to still be condensed. We conclude that evolutionarily conserved chromosome remodelling mechanisms, predating the duplication of mammalian Hox loci, underlie Hox regulation along the rostrocaudal embryonic axis. However, we suggest that separate modes of regulation can modify Hoxd chromatin in different ways in different developmental contexts.
DOI:10.1371/journal.pbio.0030157URLPMID:15839726 [本文引用: 1]
Studies of higher-order chromatin arrangements are an essential part of ongoing attempts to explore changes in epigenome structure and their functional implications during development and cell differentiation. However, the extent and cell-type-specificity of three-dimensional (3D) chromosome arrangements has remained controversial. In order to overcome technical limitations of previous studies, we have developed tools that allow the quantitative 3D positional mapping of all chromosomes simultaneously. We present unequivocal evidence for a probabilistic 3D order of prometaphase chromosomes, as well as of chromosome territories (CTs) in nuclei of quiescent (G0) and cycling (early S-phase) human diploid fibroblasts (46, XY). Radial distance measurements showed a probabilistic, highly nonrandom correlation with chromosome size: small chromosomes-independently of their gene density-were distributed significantly closer to the center of the nucleus or prometaphase rosette, while large chromosomes were located closer to the nuclear or rosette rim. This arrangement was independently confirmed in both human fibroblast and amniotic fluid cell nuclei. Notably, these cell types exhibit flat-ellipsoidal cell nuclei, in contrast to the spherical nuclei of lymphocytes and several other human cell types, for which we and others previously demonstrated gene-density-correlated radial 3D CT arrangements. Modeling of 3D CT arrangements suggests that cell-type-specific differences in radial CT arrangements are not solely due to geometrical constraints that result from nuclear shape differences. We also found gene-density-correlated arrangements of higher-order chromatin shared by all human cell types studied so far. Chromatin domains, which are gene-poor, form a layer beneath the nuclear envelope, while gene-dense chromatin is enriched in the nuclear interior. We discuss the possible functional implications of this finding.
DOI:10.1038/nature12593URL [本文引用: 3]
Large-scale chromosome structure and spatial nuclear arrangement have been linked to control of gene expression and DNA replication and repair. Genomic techniques based on chromosome conformation capture (3C) assess contacts for millions of loci simultaneously, but do so by averaging chromosome conformations from millions of nuclei. Here we introduce single-cell Hi-C, combined with genome-wide statistical analysis and structural modelling of single-copy X chromosomes, to show that individual chromosomes maintain domain organization at the megabase scale, but show variable cell-to-cell chromosome structures at larger scales. Despite this structural stochasticity, localization of active gene domains to boundaries of chromosome territories is a hallmark of chromosomal conformation. Single-cell Hi-C data bridge current gaps between genomics and microscopy studies of chromosomes, demonstrating how modular organization underlies dynamic chromosome structure, and how this structure is probabilistically linked with genome activity patterns.
DOI:10.1016/j.cell.2013.07.018URL [本文引用: 1]
Reporter genes integrated into the genome are a powerful tool to reveal effects of regulatory elements and local chromatin context on gene expression. However, so far such reporter assays have been of low throughput. Here, we describe a multiplexing approach for the parallel monitoring of transcriptional activity of thousands of randomly integrated reporters. More than 27,000 distinct reporter integrations in mouse embryonic stem cells, obtained with two differentpromoters, show similar to 1,000-fold variation in expression levels. Data analysis indicates that lamina-associated domains act as attenuators of transcription, likely by reducing access of transcription factors to binding sites. Furthermore, chromatin compaction is predictive of reporter activity. We also found evidence for crosstalk between neighboring genes and estimate that enhancers can influence gene expression on average over similar to 20 kb. The multiplexed reporter assay is highly flexible in design and can be modified to query a wide range of aspects of gene regulation.
DOI:10.1073/pnas.072618599URLPMID:11930003 [本文引用: 1]
We demonstrate that the nuclear topological arrangement of chromosome territories (CTs) has been conserved during primate evolution over a period of about 30 million years. Recent evidence shows that the positioning of chromatin in human lymphocyte nuclei is correlated with gene density. For example, human chromosome 19 territories, which contain mainly gene-dense and early replicating chromatin, are located toward the nuclear center, whereas chromosome 18 territories, which consist mainly of gene-poor and later replicating chromatin, is located close to the nuclear border. In this study, we subjected seven different primate species to comparative analysis of the radial distribution pattern of human chromosome 18- and 19-homologous chromatin by three-dimensional fluorescence in situ hybridization. Our data demonstrate that gene-density-correlated radial chromatin arrangements were conserved during higher-primate genome evolution, irrespective of the major karyotypic rearrangements that occurred in different phylogenetic lineages. The evolutionarily conserved positioning of homologous chromosomes or chromosome segments in related species supports evidence for a functionally relevant higher-order chromatin arrangement that is correlated with gene-density.
DOI:10.1016/j.molcel.2010.03.016URLPMID:20513434 [本文引用: 1]
The three-dimensional organization of chromosomes within the nucleus and its dynamics during differentiation are largely unknown. To visualize this process in molecular detail, we generated high-resolution maps of genome-nuclear lamina interactions during subsequent differentiation of mouse embryonic stem cells via lineage-committed neural precursor cells into terminally differentiated astrocytes. This reveals that a basal chromosome architecture present in embryonic stem cells is cumulatively altered at hundreds of sites during lineage commitment and subsequent terminal differentiation. This remodeling involves both individual transcription units and multigene regions and affects many genes that determine cellular identity. Often, genes that move away from the lamina are concomitantly activated; many others, however, remain inactive yet become unlocked for activation in a next differentiation step. These results suggest that lamina-genome interactions are widely involved in the control of gene expression programs during lineage commitment and terminal differentiation.
DOI:10.1182/blood-2010-03-272252URLPMID:20570862 [本文引用: 1]
Ldb1 and erythroid partners SCL, GATA-1, and LMO2 form a complex that is required to establish spatial proximity between the β-globin locus control region and gene and for transcription activation during erythroid differentiation. Here we show that Ldb1 controls gene expression at multiple levels. Ldb1 stabilizes its erythroid complex partners on β-globin chromatin, even though it is not one of the DNA-binding components. In addition, Ldb1 is necessary for enrichment of key transcriptional components in the locus, including P-TEFb, which phosphorylates Ser2 of the RNA polymerase C-terminal domain for efficient elongation. Furthermore, reduction of Ldb1 results in the inability of the locus to migrate away from the nuclear periphery, which is necessary to achieve robust transcription of β-globin in nuclear transcription factories. Ldb1 contributes these critical functions at both embryonic and adult stages of globin gene expression. These results implicate Ldb1 as a factor that facilitates nuclear relocation for transcription activation.
DOI:10.1038/ng1423URLPMID:15361872 [本文引用: 2]
The intranuclear position of many genes has been correlated with their activity state, suggesting that migration to functional subcompartments may influence gene expression. Indeed, nascent RNA production and RNA polymerase II seem to be localized into discrete foci or 'transcription factories'. Current estimates from cultured cells indicate that multiple genes could occupy the same factory, although this has not yet been observed. Here we show that, during transcription in vivo, distal genes colocalize to the same transcription factory at high frequencies. Active genes are dynamically organized into shared nuclear subcompartments, and movement into or out of these factories results in activation or abatement of transcription. Thus, rather than recruiting and assembling transcription complexes, active genes migrate to preassembled transcription sites.
DOI:10.1021/cr300513pURLPMID:23597155 [本文引用: 1]
DOI:10.1128/MCB.02454-05URLPMID:16782894 [本文引用: 1]
RNA polymerases can be shared by a particular group of genes in a transcription "factory" in nuclei, where transcription may be coordinated in concert with the distribution of coexpressed genes in higher-eukaryote genomes. Moreover, gene expression can be modulated by regulatory elements working over a long distance. Here, we compared the conformation of a 130-kb chromatin region containing the mouse alpha-globin cluster and their flanking housekeeping genes in 14.5-day-postcoitum fetal liver and brain cells. The analysis of chromatin conformation showed that the active alpha1 and alpha2 globin genes and upstream regulatory elements are in close spatial proximity, indicating that looping may function in the transcriptional regulation of the mouse alpha-globin cluster. In fetal liver cells, the active alpha1 and alpha2 genes, but not the inactive zeta gene, colocalize with neighboring housekeeping genes C16orf33, C16orf8, MPG, and C16orf35. This is in sharp contrast with the mouse alpha-globin genes in nonexpressing cells, which are separated from the congregated housekeeping genes. A comparison of RNA polymerase II (Pol II) occupancies showed that active alpha1 and alpha2 gene promoters have a much higher RNA Pol II enrichment in liver than in brain. The RNA Pol II occupancy at the zeta gene promoter, which is specifically repressed during development, is much lower than that at the alpha1 and alpha2 promoters. Thus, the mouse alpha-globin gene cluster may be regulated through moving in or out active globin gene promoters and regulatory elements of a preexisting transcription factory in the nucleus, which is maintained by the flanking clustered housekeeping genes, to activate or inactivate alpha-globin gene expression.
DOI:10.1042/BST0341133URLPMID:17073768 [本文引用: 1]
Many cellular functions take place in discrete compartments, but our textbooks make little reference to any compartments involved in transcription. We review the evidence that active RNA polymerases and associated factors cluster into 'factories' that carry out many (perhaps all) of the functions required to generate mature transcripts. Clustering ensures high local concentrations and efficient interaction. Then, a gene must associate with the appropriate factory before it can be transcribed. Recent results show that the density and diameter of nucleoplasmic factories remain roughly constant as cells differentiate, despite large changes in the numbers of active polymerases and nucleoplasmic volumes.
DOI:10.1073/pnas.1717730115URLPMID:29967174 [本文引用: 1]
Mammalian chromatin is spatially organized at many scales showing two prominent features in interphase: (i) alternating regions (1-10 Mb) of active and inactive chromatin that spatially segregate into different compartments, and (ii) domains (<1 Mb), that is, regions that preferentially interact internally [topologically associating domains (TADs)] and are central to gene regulation. There is growing evidence that TADs are formed by active extrusion of chromatin loops by cohesin, whereas compartmentalization is established according to local chromatin states. Here, we use polymer simulations to examine how loop extrusion and compartmental segregation work collectively and potentially interfere in shaping global chromosome organization. A model with differential attraction between euchromatin and heterochromatin leads to phase separation and reproduces compartmentalization as observed in Hi-C. Loop extrusion, essential for TAD formation, in turn, interferes with compartmentalization. Our integrated model faithfully reproduces Hi-C data from puzzling experimental observations where altering loop extrusion also led to changes in compartmentalization. Specifically, depletion of chromatin-associated cohesin reduced TADs and revealed finer compartments, while increased processivity of cohesin strengthened large TADs and reduced compartmentalization; and depletion of the TAD boundary protein CTCF weakened TADs while leaving compartments unaffected. We reveal that these experimental perturbations are special cases of a general polymer phenomenon of active mixing by loop extrusion. Our results suggest that chromatin organization on the megabase scale emerges from competition of nonequilibrium active loop extrusion and epigenetically defined compartment structure.
DOI:10.1038/nature11082URL [本文引用: 2]
The spatial organization of the genome is intimately linked to its biological function, yet our understanding of higher order genomic structure is coarse, fragmented and incomplete. In the nucleus of eukaryotic cells, interphase chromosomes occupy distinct chromosome territories, and numerous models have been proposed for how chromosomes fold within chromosome territories(1). These models, however, provide only few mechanistic details about the relationship between higher order chromatin structure and genome function. Recent advances in genomic technologies have led to rapid advances in the study of three-dimensional genome organization. In particular, Hi-C has been introduced as a method for identifying higher order chromatin interactions genome wide(2). Here we investigate the three-dimensional organization of the human and mouse genomes in embryonic stem cells and terminally differentiated cell types at unprecedented resolution. We identify large, megabase-sized local chromatin interaction domains, which we term 'topological domains', as a pervasive structural feature of the genome organization. These domains correlate with regions of the genome that constrain the spread of heterochromatin. The domains are stable across different cell types and highly conserved across species, indicating that topological domains are an inherent property of mammalian genomes. Finally, we find that the boundaries of topological domains are enriched for the insulator binding protein CTCF, housekeeping genes, transfer RNAs and short interspersed element (SINE) retrotransposons, indicating that these factors may have a role in establishing the topological domain structure of the genome.
DOI:10.1038/nature11049URL [本文引用: 1]
In eukaryotes transcriptional regulation often involves multiple long-range elements and is influenced by the genomic environment(1). A prime example of this concerns the mouse X-inactivation centre (Xic), which orchestrates the initiation of X-chromosome inactivation (XCI) by controlling the expression of the non-protein-coding Xist transcript. The extent of Xic sequences required for the proper regulation of Xist remains unknown. Here we use chromosome conformation capture carbon-copy (5C)(2) and super-resolution microscopy to analyse the spatial organization of a 4.5-megabases (Mb) region including Xist. We discover a series of discrete 200-kilobase to 1Mb topologically associating domains (TADs), present both before and after cell differentiation and on the active and inactive X. TADs align with, but do not rely on, several domain-wide features of the epigenome, such as H3K27me3 or H3K9me2 blocks and lamina-associated domains. TADs also align with coordinately regulated gene clusters. Disruption of a TAD boundary causes ectopic chromosomal contacts and long-range transcriptional misregulation. The Xist/Tsix sense/antisense unit illustrates how TADs enable the spatial segregation of oppositely regulated chromosomal neighbourhoods, with the respective promoters of Xist and Tsix lying in adjacent TADs, each containing their known positive regulators. We identify a novel distal regulatory region of Tsix within its TAD, which produces a long intervening RNA, Linx. In addition to uncovering a new principle of cis-regulatory architecture of mammalian chromosomes, our study sets the stage for the full genetic dissection of the X-inactivation centre.
DOI:10.1016/j.cell.2012.01.010URL [本文引用: 1]
Chromosomes are the physical realization of genetic information and thus form the basis for its readout and propagation. Here we present a high-resolution chromosomal contact map derived from a modified genome-wide chromosome conformation capture approach applied to Drosophila embryonic nuclei. The data show that the entire genome is linearly partitioned into well-demarcated physical domains that overlap extensively with active and repressive epigenetic marks. Chromosomal contacts are hierarchically organized between domains. Global modeling of contact density and clustering of domains show that inactive domains are condensed and confined to their chromosomal territories, whereas active domains reach out of the territory to form remote intra-and interchromosomal contacts. Moreover, we systematically identify specific long-range intrachromosomal contacts between Polycomb-repressed domains. Together, these observations allow for quantitative prediction of the Drosophila chromosomal contact map, laying the foundation for detailed studies of chromosome structure and function in a genetically tractable system.
DOI:10.1038/emboj.2013.237URL [本文引用: 1]
To ensure proper gene regulation within constrained nuclear space, chromosomes facilitate access to transcribed regions, while compactly packaging all other information. Recent studies revealed that chromosomes are organized into megabase-scale domains that demarcate active and inactive genetic elements, suggesting that compartmentalization is important for genome function. Here, we show that very specific long-range interactions are anchored by cohesin/CTCF sites, but not cohesin-only or CTCF-only sites, to form a hierarchy of chromosomal loops. These loops demarcate topological domains and form intricate internal structures within them. Post-mitotic nuclei deficient for functional cohesin exhibit global architectural changes associated with loss of cohesin/CTCF contacts and relaxation of topological domains. Transcriptional analysis shows that this cohesin-dependent perturbation of domain organization leads to widespread gene deregulation of both cohesin-bound and non-bound genes. Our data thereby support a role for cohesin in the global organization of domain structure and suggest that domains function to stabilize the transcriptional programmes within them.
DOI:10.1126/science.1236083URL [本文引用: 1]
Mitotic chromosomes are among the most recognizable structures in the cell, yet for over a century their internal organization remains largely unsolved. We applied chromosome conformation capture methods, 5C and Hi-C, across the cell cycle and revealed two distinct three-dimensional folding states of the human genome. We show that the highly compartmentalized and cell type-specific organization described previously for nonsynchronous cells is restricted to interphase. In metaphase, we identified a homogenous folding state that is locus-independent, common to all chromosomes, and consistent among cell types, suggesting a general principle of metaphase chromosome organization. Using polymer simulations, we found that metaphase Hi-C data are inconsistent with classic hierarchical models and are instead best described by a linearly organized longitudinally compressed array of consecutive chromatin loops.
DOI:10.1016/j.ceb.2013.02.005URL [本文引用: 1]
It is now well accepted that cell-type specific gene regulation is under the purview of enhancers. Great strides have been made recently to characterize and identify enhancers both genetically and epigenetically for multiple cell types and species, but efforts have just begun to link enhancers to their target promoters. Mapping these interactions and understanding how the 3D landscape of the genome constrains such interactions is fundamental to our understanding of mammalian gene regulation. Here, we review recent progress in mapping long-range regulatory interactions in mammalian genomes, focusing on transcriptional enhancers and chromatin organization principles.
DOI:10.1016/j.molcel.2012.08.031URLPMID:23041285 [本文引用: 1]
The mechanisms responsible for the establishment of physical domains in metazoan chromosomes are poorly understood. Here we find that physical domains in Drosophila chromosomes are demarcated at regions of active transcription and high gene density that are enriched for transcription factors and specific combinations of insulator proteins. Physical domains contain different types of chromatin defined by the presence of specific proteins and epigenetic marks, with active chromatin preferentially located at the borders and silenced chromatin in the interior. Domain boundaries participate in long-range interactions that may contribute to the clustering of regions of active or silenced chromatin in the nucleus. Analysis of transgenes suggests that chromatin is more accessible and permissive to transcription at the borders than inside domains, independent of the presence of active or silencing histone modifications. These results suggest that the higher-order physical organization of chromatin may impose an additional level of regulation over classical epigenetic marks.
DOI:10.1073/pnas.1317788111URLPMID:24335803 [本文引用: 2]
Recent studies of genome-wide chromatin interactions have revealed that the human genome is partitioned into many self-associating topological domains. The boundary sequences between domains are enriched for binding sites of CTCC-binding factor (CTCF) and the cohesin complex, implicating these two factors in the establishment or maintenance of topological domains. To determine the role of cohesin and CTCF in higher-order chromatin architecture in human cells, we depleted the cohesin complex or CTCF and examined the consequences of loss of these factors on higher-order chromatin organization, as well as the transcriptome. We observed a general loss of local chromatin interactions upon disruption of cohesin, but the topological domains remain intact. However, we found that depletion of CTCF not only reduced intradomain interactions but also increased interdomain interactions. Furthermore, distinct groups of genes become misregulated upon depletion of cohesin and CTCF. Taken together, these observations suggest that CTCF and cohesin contribute differentially to chromatin organization and gene regulation.
DOI:10.1016/j.cell.2009.06.001URLPMID:19563753 [本文引用: 1]
CTCF is a highly conserved zinc finger protein implicated in diverse regulatory functions, including transcriptional activation/repression, insulation, imprinting, and X chromosome inactivation. Here we re-evaluate data supporting these roles in the context of mechanistic insights provided by recent genome-wide studies and highlight evidence for CTCF-mediated intra- and interchromosomal contacts at several developmentally regulated genomic loci. These analyses support a primary role for CTCF in the global organization of chromatin architecture and suggest that CTCF may be a heritable component of an epigenetic system regulating the interplay between DNA methylation, higher-order chromatin structure, and lineage-specific gene expression.
DOI:10.1016/j.cell.2017.09.026URLPMID:28985562 [本文引用: 1]
The human genome folds to create thousands of intervals, called &quot;contact domains,&quot; that exhibit enhanced contact frequency within themselves. &quot;Loop domains&quot; form because of tethering between two loci-almost always bound by CTCF and cohesin-lying on the same chromosome. &quot;Compartment domains&quot; form when genomic intervals with similar histone marks co-segregate. Here, we explore the effects of degrading cohesin. All loop domains are eliminated, but neither compartment domains nor histone marks are affected. Loss of loop domains does not lead to widespread ectopic gene activation but does affect a significant minority of active genes. In particular, cohesin loss causes superenhancers to co-localize, forming hundreds of links within and across chromosomes and affecting the regulation of nearby genes. We then restore cohesin and monitor the re-formation of each loop. Although re-formation rates vary greatly, many megabase-sized loops recovered in under an hour, consistent with a model where loop extrusion is rapid.
DOI:10.1016/j.cell.2017.05.004URLPMID:28525758
The molecular mechanisms underlying folding of mammalian chromosomes remain poorly understood. The transcription factor CTCF is a candidate regulator of chromosomal structure. Using the auxin-inducible degron system in mouse embryonic stem cells, we show that CTCF is absolutely and dose-dependently required for looping between CTCF target sites and insulation of topologically associating domains (TADs). Restoring CTCF reinstates proper architecture on altered chromosomes, indicating a powerful instructive function for CTCF in?chromatin folding. CTCF remains essential for TAD organization in non-dividing cells. Surprisingly, active and inactive genome compartments remain properly segregated upon CTCF depletion, revealing that compartmentalization of mammalian chromosomes emerges independently of proper insulation of TADs. Furthermore, our data support that CTCF mediates transcriptional insulator function through enhancer blocking but not as a direct barrier to heterochromatin spreading. Beyond defining the functions of CTCF in chromosome folding, these results provide new fundamental insights into the rules governing mammalian genome organization.
DOI:10.1038/nature24281URLPMID:29094699 [本文引用: 1]
Imaging and chromosome conformation capture studies have revealed several layers of chromosome organization, including segregation into megabase-sized active and inactive compartments, and partitioning into sub-megabase domains (TADs). It remains unclear, however, how these layers of organization form, interact with one another and influence genome function. Here we show that deletion of the cohesin-loading factor Nipbl in mouse liver leads to a marked reorganization of chromosomal folding. TADs and associated Hi-C peaks vanish globally, even in the absence of transcriptional changes. By contrast, compartmental segregation is preserved and even reinforced. Strikingly, the disappearance of TADs unmasks a finer compartment structure that accurately reflects the underlying epigenetic landscape. These observations demonstrate that the three-dimensional organization of the genome results from the interplay of two independent mechanisms: cohesin-independent segregation of the genome into fine-scale compartments, defined by chromatin state; and cohesin-dependent formation of TADs, possibly by loop extrusion, which helps to guide distant enhancers to their target genes.
DOI:10.1038/nature21429URLPMID:28289288 [本文引用: 2]
The folding of genomic DNA from the beads-on-a-string-like structure of nucleosomes into higher-order assemblies is crucially linked to nuclear processes. Here we calculate 3D structures of entire mammalian genomes using data from a new chromosome conformation capture procedure that allows us to first image and then process single cells. The technique enables genome folding to be examined at a scale of less than 100?kb, and chromosome structures to be validated. The structures of individual topological-associated domains and loops vary substantially from cell to cell. By contrast, A and B compartments, lamina-associated domains and active enhancers and promoters are organized in a consistent way on a genome-wide basis in every cell, suggesting that they could drive chromosome and genome folding. By studying genes regulated by pluripotency factor and nucleosome remodelling deacetylase (NuRD), we illustrate how the determination of single-cell genome structure provides a new approach for investigating biological processes.
DOI:10.1126/science.362.6413.494URLPMID:30361376 [本文引用: 1]
Activation of stretch-sensitive baroreceptor neurons exerts acute control over heart rate and blood pressure. Although this homeostatic baroreflex has been described for more than 80 years, the molecular identity of baroreceptor mechanosensitivity remains unknown. We discovered that mechanically activated ion channels PIEZO1 and PIEZO2 are together required for baroreception. Genetic ablation of both Piezo1 and Piezo2 in the nodose and petrosal sensory ganglia of mice abolished drug-induced baroreflex and aortic depressor nerve activity. Awake, behaving animals that lack Piezos had labile hypertension and increased blood pressure variability, consistent with phenotypes in baroreceptor-denervated animals and humans with baroreflex failure. Optogenetic activation of Piezo2-positive sensory afferents was sufficient to initiate baroreflex in mice. These findings suggest that PIEZO1 and PIEZO2 are the long-sought baroreceptor mechanosensors critical for acute blood pressure control.
DOI:10.1126/sciadv.aar5255URLPMID:29507889 [本文引用: 1]
Cueva de los Aviones (southeast Spain) is a site of the Neandertal-associated Middle Paleolithic of Europe. It has yielded ochred and perforated marine shells, red and yellow colorants, and shell containers that feature residues of complex pigmentatious mixtures. Similar finds from the Middle Stone Age of South Africa have been widely accepted as archaeological proxies for symbolic behavior. U-series dating of the flowstone capping the Cueva de los Aviones deposit shows that the symbolic finds made therein are 115,000 to 120,000 years old and predate the earliest known comparable evidence associated with modern humans by 20,000 to 40,000 years. Given our findings, it is possible that the roots of symbolic material culture may be found among the common ancestor of Neandertals and modern humans, more than half-a-million years ago.
DOI:10.1002/iub.129URLPMID:18767169 [本文引用: 1]
The mammalian beta-globin locus is a multigene locus containing several globin genes and a number of regulatory elements. During development, the expression of the genes changes in a process called &quot;switching.&quot; The most important regulatory element in the locus is the locus control region (LCR) upstream of the globin genes that is essential for high-level expression of these genes. The discovery of the LCR initially raised the question how this element could exert its effect on the downstream globin genes. The question was solved by the finding that the LCR and activate globin genes are in physical contact, forming a chromatin structure named the active chromatin hub (ACH). Here we discuss the significance of ACH formation, provide an overview of the proteins implicated in chromatin looping at the beta-globin locus, and evaluate the relationship between nuclear organization and beta-globin gene expression.
DOI:10.1101/gad.13.19.2465URLPMID:10521391 [本文引用: 1]
DOI:10.1016/j.molcel.2019.12.027URLPMID:31954095 [本文引用: 1]
A comprehensive catalog of cancer driver mutations is essential for understanding tumorigenesis and developing therapies. Exome-sequencing studies have mapped many protein-coding drivers, yet few non-coding drivers are known because genome-wide discovery is challenging. We developed a driver discovery method, ActiveDriverWGS, and analyzed 120,788 cis-regulatory modules (CRMs) across 1,844 whole tumor genomes from the ICGC-TCGA PCAWG project. We found 30 CRMs with enriched SNVs and indels (FDR?&lt; 0.05). These frequently mutated regulatory elements (FMREs) were ubiquitously active in human tissues, showed long-range chromatin interactions and mRNA abundance associations with target genes, and were enriched in motif-rewiring mutations and structural variants. Genomic deletion of one FMRE in human cells caused proliferative deficiencies and transcriptional deregulation of cancer genes CCNB1IP1, CDH1, and CDKN2B, validating observations in FMRE-mutated tumors. Pathway analysis revealed further sub-significant FMREs at cancer genes and processes, indicating an unexplored landscape of infrequent driver mutations in the non-coding genome.
DOI:10.1016/j.cell.2011.12.014URL [本文引用: 1]
Higher-order chromosomal organization for transcription regulation is poorly understood in eukaryotes. Using genome-wide Chromatin Interaction Analysis with Paired-End-Tag sequencing (ChIA-PET), we mapped long-range chromatin interactions associated with RNA polymerase II in human cells and uncovered widespread promoter-centered intragenic, extragenic, and intergenic interactions. These interactions further aggregated into higher-order clusters, wherein proximal and distal genes were engaged through promoter-promoter interactions. Most genes with promoter-promoter interactions were active and transcribed cooperatively, and some interacting promoters could influence each other implying combinatorial complexity of transcriptional controls. Comparative analyses of different cell lines showed that cell-specific chromatin interactions could provide structural frameworks for cell-specific transcription, and suggested significant enrichment of enhancer-promoter interactions for cell-specific functions. Furthermore, genetically-identified disease-associated noncoding elements were found to be spatially engaged with corresponding genes through long-range interactions. Overall, our study provides insights into transcription regulation by three-dimensional chromatin interactions for both housekeeping and cell-specific genes in human cells.
DOI:10.1038/nature11279URL [本文引用: 1]
The vast non-coding portion of the human genome is full of functional elements and disease-causing regulatory variants. The principles defining the relationships between these elements and distal target genes remain unknown. Promoters and distal elements can engage in looping interactions that have been implicated in gene regulation(1). Here we have applied chromosome conformation capture carbon copy (5C(2)) to interrogate comprehensively interactions between transcription start sites (TSSs) and distal elements in 1% of the human genome representing the ENCODE pilot project regions(3). 5C maps were generated for GM12878, K562 and HeLa-S3 cells and results were integrated with data from the ENCODE consortium(4). In each cell line we discovered >1,000 long-range interactions between promoters and distal sites that include elements resembling enhancers, promoters and CTCF-bound sites. We observed significant correlations between gene expression, promoter-enhancer interactions and the presence of enhancer RNAs. Long-range interactions show marked asymmetry with a bias for interactions with elements located similar to 120 kilobases upstream of the TSS. Long-range interactions are often not blocked by sites bound by CTCF and cohesin, indicating that many of these sites do not demarcate physically insulated gene domains. Furthermore, only similar to 7% of looping interactions are with the nearest gene, indicating that genomic proximity is not a simple predictor for long-range interactions. Finally, promoters and distal elements are engaged in multiple long-range interactions to form complex networks. Our results start to place genes and regulatory elements in three-dimensional context, revealing their functional relationships.
DOI:10.1016/j.cell.2013.11.039URL [本文引用: 1]
A key finding of the ENCODE project is that the enhancer landscape of mammalian cells undergoes marked alterations during ontogeny. However, the nature and extent of these changes are unclear. As part of the NIH Mouse Regulome Project, we here combined DNaseI hypersensitivity, ChIP-seq, and ChIA-PET technologies to map the promoter-enhancer interactomes of pluripotent ES cells and differentiated B lymphocytes. We confirm that enhancer usage varies widely across tissues. Unexpectedly, we find that this feature extends to broadly transcribed genes, including Myc and Pim1 cell-cycle regulators, which associate with an entirely different set of enhancers in ES and B cells. By means of high-resolution CpG methylomes, genome editing, and digital footprinting, we show that these enhancers recruit lineage-determining factors. Furthermore, we demonstrate that the turning on and off of enhancers during development correlates with promoter activity. We propose that organisms rely on a dynamic enhancer landscape to control basic cellular functions in a tissue-specific manner.
DOI:10.1038/nature12644URL [本文引用: 1]
A large number of cis-regulatory sequences have been annotated in the human genome(1,2), but defining their target genes remains a challenge(3). One strategy is to identify the long-range looping interactions at these elements with the use of chromosome conformation capture (3C)-based techniques(4). However, previous studies lack either the resolution or coverage to permit a whole-genome, unbiased view of chromatin interactions. Here we report a comprehensive chromatin interaction map generated in human fibroblasts using a genome-wide 3C analysis method (Hi-C)(5). We determined over one million long-range chromatin interactions at 5-10-kb resolution, and uncovered general principles of chromatin organization at different types of genomic features. We also characterized the dynamics of promoter-enhancer contacts after TNF-alpha signalling in these cells. Unexpectedly, we found that TNF-alpha-responsive enhancers are already in contact with their target promoters before signalling. Such pre-existing chromatin looping, which also exists in other cell types with different extracellular signalling, is a strong predictor of gene induction. Our observations suggest that the three-dimensional chromatin landscape, once established in a particular cell type, is relatively stable and could influence the selection or activation of target genes by a ubiquitous transcription activator in a cell-specific manner.
DOI:10.1016/j.cell.2013.07.034URL [本文引用: 1]
During cell division, transcription factors (TFs) are removed from chromatin twice, during DNA synthesis and during condensation of chromosomes. How TFs can efficiently find their sites following these stages has been unclear. Here, we have analyzed the binding pattern of expressed TFs in human colorectal cancer cells. We find that binding of TFs is highly clustered and that the clusters are enriched in binding motifs for several major TF classes. Strikingly, almost all clusters are formed around cohesin, and loss of cohesin decreases both DNA accessibility and binding of TFs to clusters. We show that cohesin remains bound in S phase, holding the nascent sister chromatids together at the TF cluster sites. Furthermore, cohesin remains bound to the cluster sites when TFs are evicted in early M phase. These results suggest that cohesin-binding functions as a cellular memory that promotes re-establishment of TF clusters after DNA replication and chromatin condensation.
.
DOI:10.1074/jbc.M110.207365URLPMID:21454523 [本文引用: 1]
The β-globin locus undergoes dynamic chromatin interaction changes in differentiating erythroid cells that are thought to be important for proper globin gene expression. However, the underlying mechanisms are unclear. The CCCTC-binding factor, CTCF, binds to the insulator elements at the 5' and 3' boundaries of the locus, but these sites were shown to be dispensable for globin gene activation. We found that, upon induction of differentiation, cohesin and the cohesin loading factor Nipped-B-like (Nipbl) bind to the locus control region (LCR) at the CTCF insulator and distal enhancer regions as well as at the specific target globin gene that undergoes activation upon differentiation. Nipbl-dependent cohesin binding is critical for long-range chromatin interactions, both between the CTCF insulator elements and between the LCR distal enhancer and the target gene. We show that the latter interaction is important for globin gene expression in vivo and in vitro. Furthermore, the results indicate that such cohesin-mediated chromatin interactions associated with gene regulation are sensitive to the partial reduction of Nipbl caused by heterozygous mutation. This provides the first direct evidence that Nipbl haploinsufficiency affects cohesin-mediated chromatin interactions and gene expression. Our results reveal that dynamic Nipbl/cohesin binding is critical for developmental chromatin organization and the gene activation function of the LCR in mammalian cells.
DOI:10.1073/pnas.1518552112URLPMID:26499245 [本文引用: 1]
We recently used in situ Hi-C to create kilobase-resolution 3D maps of mammalian genomes. Here, we combine these maps with new Hi-C, microscopy, and genome-editing experiments to study the physical structure of chromatin fibers, domains, and loops. We find that the observed contact domains are inconsistent with the equilibrium state for an ordinary condensed polymer. Combining Hi-C data and novel mathematical theorems, we show that contact domains are also not consistent with a fractal globule. Instead, we use physical simulations to study two models of genome folding. In one, intermonomer attraction during polymer condensation leads to formation of an anisotropic &quot;tension globule.&quot; In the other, CCCTC-binding factor (CTCF) and cohesin act together to extrude unknotted loops during interphase. Both models are consistent with the observed contact domains and with the observation that contact domains tend to form inside loops. However, the extrusion model explains a far wider array of observations, such as why loops tend not to overlap and why the CTCF-binding motifs at pairs of loop anchors lie in the convergent orientation. Finally, we perform 13 genome-editing experiments examining the effect of altering CTCF-binding sites on chromatin folding. The convergent rule correctly predicts the affected loops in every case. Moreover, the extrusion model accurately predicts in silico the 3D maps resulting from each experiment using only the location of CTCF-binding sites in the WT. Thus, we show that it is possible to disrupt, restore, and move loops and domains using targeted mutations as small as a single base pair.
DOI:10.1038/nature22063URLPMID:28424523 [本文引用: 1]
Mammalian genomes are spatially organized by CCCTC-binding factor (CTCF) and cohesin into chromatin loops and topologically associated domains, which have important roles in gene regulation and recombination. By binding to specific sequences, CTCF defines contact points for cohesin-mediated long-range chromosomal cis-interactions. Cohesin is also present at these sites, but has been proposed to be loaded onto DNA elsewhere and to extrude chromatin loops until it encounters CTCF bound to DNA. How cohesin is recruited to CTCF sites, according to this or other models, is unknown. Here we show that the distribution of cohesin in the mouse genome depends on transcription, CTCF and the cohesin release factor Wings apart-like (Wapl). In CTCF-depleted fibroblasts, cohesin cannot be properly recruited to CTCF sites but instead accumulates at transcription start sites of active genes, where the cohesin-loading complex is located. In the absence of both CTCF and Wapl, cohesin accumulates in up to 70 kilobase-long regions at 3'-ends of active genes, in particular if these converge on each other. Changing gene expression modulates the position of these 'cohesin islands'. These findings indicate that transcription can relocate mammalian cohesin over long distances on DNA, as previously reported for yeast cohesin, that this translocation contributes to positioning cohesin at CTCF sites, and that active genes can be freed from cohesin either by transcription-mediated translocation or by Wapl-mediated release.
DOI:10.1371/journal.pone.0001661URLPMID:18286208 [本文引用: 3]
A relationship exists between nuclear architecture and gene activity and it has been proposed that the activity of ongoing RNA polymerase II transcription determines genome organization in the mammalian cell nucleus. Recently developed 3C and 4C technology allowed us to test the importance of transcription for nuclear architecture. We demonstrate that upon transcription inhibition binding of RNA polymerase II to gene regulatory elements is severely reduced. However, contacts between regulatory DNA elements and genes in the beta-globin locus are unaffected and the locus still interacts with the same genomic regions elsewhere on the chromosome. This is a general phenomenon since the great majority of intra- and interchromosomal interactions with the ubiquitously expressed Rad23a gene are also not affected. Our data demonstrate that without transcription the organization and modification of nucleosomes at active loci and the local binding of specific trans-acting factors is unaltered. We propose that these parameters, more than transcription or RNA polymerase II binding, determine the maintenance of long-range DNA interactions.
DOI:10.1101/gad.454008URLPMID:18172162 [本文引用: 3]
Nascent transcription occurs at nuclear foci of concentrated, hyperphosphorylated RNA polymerase II (RNAPII). We investigate RNAPII localization, distal gene co-association, and Hbb locus conformation during inhibition of transcription. Our results show distal active genes remain associated with RNAPII foci and each other in the absence of elongation. When initiation is inhibited, active genes dissociate from RNAPII foci and each other, suggesting initiation is necessary to tether distal active genes to shared foci. In the absence of transcription RNAPII foci remain, indicating they are not simple accumulations of RNAPII on transcribed genes but exist as independent nuclear subcompartments.
DOI:10.1016/j.cell.2012.03.051URL [本文引用: 2]
Chromatin loops juxtapose distal enhancers with active promoters, but their molecular architecture and relationship with transcription remain unclear. In erythroid cells, the locus control region (LCR) and beta-globin promoter form a chromatin loop that requires transcription factor GATA1 and the associated molecule Ldb1. We employed artificial zinc fingers (ZF) to tether Ldb1 to the b-globin promoter in GATA1 null erythroblasts, in which the b-globin locus is relaxed and inactive. Remarkably, targeting Ldb1 or only its self-association domain to the b-globin promoter substantially activated b-globin transcription in the absence of GATA1. Promotertethered Ldb1 interacted with endogenous Ldb1 complexes at the LCR to form a chromatin loop, causing recruitment and phosphorylation of RNA polymerase II. ZF-Ldb1 proteins were inactive at alleles lacking the LCR, demonstrating that their activities depend on long-range interactions. Our findings establish Ldb1 as a critical effector of GATA1-mediated loop formation and indicate that chromatin looping causally underlies gene regulation.
DOI:10.1073/pnas.1901244116URLPMID:31506350 [本文引用: 2]
Heat shock (HS) initiates rapid, extensive, and evolutionarily conserved changes in transcription that are accompanied by chromatin decondensation and nucleosome loss at HS loci. Here we have employed in situ Hi-C to determine how heat stress affects long-range chromatin conformation in human and Drosophila cells. We found that compartments and topologically associating domains (TADs) remain unchanged by an acute HS. Knockdown of Heat Shock Factor 1 (HSF1), the master transcriptional regulator of the HS response, identified HSF1-dependent genes and revealed that up-regulation is often mediated by distal HSF1 bound enhancers. HSF1-dependent genes were usually found in the same TAD as the nearest HSF1 binding site. Although most interactions between HSF1 binding sites and target promoters were established in the nonheat shock (NHS) condition, a subset increased contact frequency following HS. Integrating information about HSF1 binding strength, RNA polymerase abundance at the HSF1 bound sites (putative enhancers), and contact frequency with a target promoter accurately predicted which up-regulated genes were direct targets of HSF1 during HS. Our results suggest that the chromatin conformation necessary for a robust HS response is preestablished in NHS cells of diverse metazoan species.
DOI:10.1016/j.cell.2017.09.001URLPMID:28938112 [本文引用: 1]
It is now established that Bcl11b specifies T?cell fate. Here, we show that in developing T?cells, the Bcl11b enhancer repositioned from the lamina to the nuclear interior. Our search for factors that relocalized the Bcl11b enhancer identified a non-coding RNA named ThymoD (thymocyte differentiation factor). ThymoD-deficient mice displayed a block at the onset of T?cell development and developed lymphoid malignancies. We found that ThymoD transcription promoted demethylation at CTCF bound sites and activated cohesin-dependent looping to reposition the Bcl11b enhancer from the lamina to the nuclear interior and?to juxtapose the Bcl11b enhancer and promoter?into a single-loop domain. These large-scale changes in nuclear architecture were associated with the deposition of activating epigenetic marks across the loop domain, plausibly facilitating phase separation. These data indicate how, during developmental progression and tumor suppression, non-coding transcription orchestrates chromatin folding and compartmentalization to direct with high precision enhancer-promoter communication.
DOI:10.1016/j.immuni.2018.01.013URLPMID:29466755 [本文引用: 3]
How chromatin reorganization coordinates differentiation and lineage commitment from hematopoietic stem and progenitor cells (HSPCs) to mature immune cells has not been well understood. Here, we carried out an integrative analysis of chromatin accessibility, topologically associating domains, AB compartments, and gene expression from HSPCs to CD4+CD8+ T?cells. We found that abrupt genome-wide changes at all three levels of chromatin organization occur during the transition from double-negative stage 2 (DN2) to DN3, accompanying the T lineage commitment. The transcription factor BCL11B, a critical regulator of T?cell commitment, is associated with increased chromatin interaction, and Bcl11b deletion compromised chromatin interaction at its target genes. We propose that these large-scale and concerted changes in chromatin organization present an energy barrier to prevent the cell from reversing its fate to earlier stages or redirecting to alternatives and thus lock the cell fate into the T lineages.
DOI:10.1016/j.immuni.2016.08.012URLPMID:27637145 [本文引用: 3]
During the humoral immune response, B cells undergo a dramatic change in phenotype to enable antibody affinity maturation in germinal centers (GCs). Using genome-wide chromosomal conformation capture (Hi-C), we found that GC B cells undergo massive reorganization of the genomic architecture that encodes the GC B cell transcriptome. Coordinate expression of genes that specify the GC B cell phenotype-most prominently BCL6-was achieved through a multilayered chromatin reorganization process involving (1) increased promoter connectivity, (2) formation of enhancer networks, (3) 5' to 3' gene looping, and (4) merging of gene neighborhoods that share active epigenetic marks. BCL6 was an anchor point for the formation of GC-specific gene and enhancer loops on chromosome 3. Deletion of a GC-specific, highly interactive locus control region upstream of Bcl6 abrogated GC formation in mice. Thus, large-scale and multi-tiered genomic three-dimensional reorganization is required for coordinate expression of phenotype-driving gene sets that determine the unique characteristics of GC B cells.
DOI:10.1016/j.molcel.2017.07.013URLPMID:28803781 [本文引用: 1]
50 years ago, Vincent Allfrey and colleagues discovered that lymphocyte activation triggers massive acetylation of chromatin. However, the molecular mechanisms driving epigenetic accessibility are still unknown. We here show that stimulated lymphocytes decondense chromatin by three differentially regulated steps. First, chromatin is repositioned away from the nuclear periphery in response to global acetylation. Second, histone nanodomain clusters decompact into mononucleosome fibers through a mechanism that requires Myc and continual energy input. Single-molecule imaging shows that this step lowers transcription factor residence time and non-specific collisions during sampling for DNA targets. Third, chromatin interactions shift from long range to predominantly short range, and CTCF-mediated loops and contact domains double in numbers. This architectural change facilitates cognate promoter-enhancer contacts and also requires Myc and continual ATP production. Our results thus define the nature and transcriptional impact of chromatin decondensation and reveal an unexpected role for Myc in the establishment of nuclear topology in mammalian cells.
DOI:10.1016/j.molcel.2017.08.006URLPMID:28890333 [本文引用: 5]
The three-dimensional arrangement of the human genome comprises a complex network of structural and regulatory chromatin loops important for coordinating changes in transcription during human development. To better understand the mechanisms underlying context-specific 3D chromatin structure and transcription during cellular differentiation, we generated comprehensive in?situ Hi-C maps of DNA loops in human monocytes and differentiated macrophages. We demonstrate that dynamic looping events are regulatory rather than structural in nature and uncover widespread coordination of dynamic enhancer activity at preformed and acquired DNA loops. Enhancer-bound loop formation and enhancer activation of preformed loops together form multi-loop activation hubs at key macrophage genes. Activation hubs connect 3.4 enhancers per promoter and exhibit a strong enrichment for activator protein 1 (AP-1)-binding events, suggesting that multi-loop activation hubs involving cell-type-specific transcription factors represent an important class of regulatory chromatin structures for the spatiotemporal control of transcription.
DOI:10.1038/s41467-019-10318-6URLPMID:31127120 [本文引用: 2]
Chromatin looping allows enhancer-bound regulatory factors to influence transcription. Large domains, referred to as topologically associated domains, participate in genome organization. However, the mechanisms underlining interactions within these?domains, which control gene expression, are not fully understood. Here we report that activation of embryonic myogenesis is associated with establishment of long-range chromatin interactions centered on Pax3-bound loci. Using mass spectrometry and genomic studies, we identify the ubiquitously expressed LIM-domain binding protein 1 (Ldb1) as the mediator of looping interactions at a subset of Pax3 binding sites. Ldb1 is recruited to Pax3-bound elements independently of CTCF-Cohesin, and is necessary for efficient deposition of H3K4me1 at these sites and chromatin looping. When Ldb1 is deleted in Pax3-expressing cells in vivo, specification of migratory myogenic progenitors is severely impaired. These results highlight Ldb1 requirement for Pax3 myogenic activity and demonstrate how transcription factors can promote formation of sub-topologically associated domain interactions involved in lineage specification.
DOI:10.1038/s41586-018-0845-0URLPMID:30626972 [本文引用: 3]
The genome is partitioned into topologically associated domains and genomic compartments with shared chromatin valence. This architecture is constrained by the DNA polymer, which precludes interactions between genes on different chromosomes. Here we report a marked divergence from this pattern of nuclear organization that occurs in mouse olfactory sensory neurons. Chromatin conformation capture using in situ Hi-C on fluorescence-activated cell-sorted olfactory sensory neurons and their progenitors shows that olfactory receptor gene clusters from 18 chromosomes make specific and robust interchromosomal contacts that increase with differentiation of the cells. These contacts are orchestrated by intergenic olfactory receptor enhancers, the 'Greek islands', which first contribute to the formation of olfactory receptor compartments and then form a multi-chromosomal super-enhancer that associates with the single active olfactory receptor gene. The Greek-island-bound transcription factor LHX2 and adaptor protein LDB1 regulate the assembly and maintenance of olfactory receptor compartments, Greek island hubs and olfactory receptor transcription, providing mechanistic insights into and functional support for the role of trans interactions in gene expression.
DOI:10.1016/j.cell.2017.09.043URLPMID:29053968 [本文引用: 3]
Chromosome conformation capture technologies have revealed important insights into genome folding. Yet, how spatial genome architecture is related to gene expression and cell fate remains unclear. We comprehensively mapped 3D chromatin organization during mouse neural differentiation in?vitro and in?vivo, generating the highest-resolution Hi-C maps available to date. We found that transcription is correlated with chromatin insulation and long-range interactions, but dCas9-mediated activation is insufficient for creating TAD boundaries de novo. Additionally, we discovered long-range contacts between gene bodies of exon-rich, active genes in all cell types. During neural differentiation, contacts between active TADs become less pronounced while inactive TADs interact more strongly. An extensive Polycomb network in stem cells is disrupted, while?dynamic interactions between neural transcription factors appear in?vivo. Finally, cell type-specific enhancer-promoter contacts are established concomitant to gene expression. This work shows that multiple factors influence the dynamics of chromatin interactions in development.
DOI:10.1038/ng.3935URLPMID:28805829 [本文引用: 3]
Chromosome conformation is an important feature of metazoan gene regulation; however, enhancer-promoter contact remodeling during cellular differentiation remains poorly understood. To address this, genome-wide promoter capture Hi-C (CHi-C) was performed during epidermal differentiation. Two classes of enhancer-promoter contacts associated with differentiation-induced genes were identified. The first class ('gained') increased in contact strength during differentiation in concert with enhancer acquisition of the H3K27ac activation mark. The second class ('stable') were pre-established in undifferentiated cells, with enhancers constitutively marked by H3K27ac. The stable class was associated with the canonical conformation regulator cohesin, whereas the gained class was not, implying distinct mechanisms of contact formation and regulation. Analysis of stable enhancers identified a new, essential role for a constitutively expressed, lineage-restricted ETS-family transcription factor, EHF, in epidermal differentiation. Furthermore, neither class of contacts was observed in pluripotent cells, suggesting that lineage-specific chromatin structure is established in tissue progenitor cells and is further remodeled in terminal differentiation.
.
DOI:10.1172/JCI88016URLPMID:27941246 [本文引用: 3]
The recognition of β cell dedifferentiation in type 2 diabetes raises the translational relevance of mechanisms that direct and maintain β cell identity. LIM domain-binding protein 1 (LDB1) nucleates multimeric transcriptional complexes and establishes promoter-enhancer looping, thereby directing fate assignment and maturation of progenitor populations. Many terminally differentiated endocrine cell types, however, remain enriched for LDB1, but its role is unknown. Here, we have demonstrated a requirement for LDB1 in maintaining the terminally differentiated status of pancreatic β cells. Inducible ablation of LDB1 in mature β cells impaired insulin secretion and glucose homeostasis. Transcriptomic analysis of LDB1-depleted β cells revealed the collapse of the terminally differentiated gene program, indicated by a loss of β cell identity genes and induction of the endocrine progenitor factor neurogenin 3 (NEUROG3). Lineage tracing confirmed that LDB1-depleted, insulin-negative β cells express NEUROG3 but do not adopt alternate endocrine cell fates. In primary mouse islets, LDB1 and its LIM homeodomain-binding partner islet 1 (ISL1) were coenriched at chromatin sites occupied by pancreatic and duodenal homeobox 1 (PDX1), NK6 homeobox 1 (NKX6.1), forkhead box A2 (FOXA2), and NK2 homeobox 2 (NKX2.2) - factors that co-occupy active enhancers in 3D chromatin domains in human islets. Indeed, LDB1 was enriched at active enhancers in human islets. Thus, LDB1 maintains the terminally differentiated state of β cells and is a component of active enhancers in both murine and human islets.
DOI:10.1038/ni.1887URLPMID:20644572 [本文引用: 1]
The development of T cells in the thymus involves several differentiation and proliferation events, during which hematopoietic precursors give rise to T cells ready to respond to antigen stimulation and undergo effector differentiation. This review addresses signaling and transcriptional checkpoints that control the intrathymic journey of T cell precursors. We focus on the divergence of alphabeta and gammadelta lineage cells and the elaboration of the alphabeta T cell repertoire, with special emphasis on the emergence of transcriptional programs that direct lineage decisions.
DOI:10.1016/j.cell.2013.01.009URL [本文引用: 1]
Eukaryotic cells have a layer of heterochromatin at the nuclear periphery. To investigate mechanisms regulating chromatin distribution, we analyzed heterochromatin organization in different tissues and species, including mice with mutations in the lamin B receptor (Lbr) and lamin A (Lmna) genes that encode nuclear envelope (NE) proteins. We identified LBR- and lamin-A/C-dependent mechanisms tethering heterochromatin to the NE. The two tethers are sequentially used during cellular differentiation and development: first the LBR -and then the lamin-A/C-dependent tether. The absence of both LBR and lamin A/C leads to loss of peripheral heterochromatin and an inverted architecture with heterochromatin localizing to the nuclear interior. Myoblast transcriptome analyses indicated that selective disruption of the LBR- or lamin-A-dependent heterochromatin tethers have opposite effects on muscle gene expression, either increasing or decreasing, respectively. These results show how changes in NE composition contribute to regulating heterochromatin positioning, gene expression, and cellular differentiation during development.
DOI:10.1038/ni1115URLPMID:15378057 [本文引用: 1]
The T helper type 2 (T(H)2) locus control region is important in the regulation of the genes encoding the cytokines interleukins 4, 5 and 13. Using the chromosome conformation capture technique, we found that in T cells, natural killer cells, B cells and fibroblasts, the promoters for the genes encoding T(H)2 cytokines are located in close spatial proximity, forming an initial chromatin core configuration. In CD4(+) T cells and natural killer cells, but not B cells and fibroblasts, the T(H)2 locus control region participates in this configuration. The transcription factors GATA3 and STAT6 are essential for the establishment and/or maintenance of these interactions. Intrachromosomal interactions in the T(H)2 cytokine locus may form the basis for the coordinated transcriptional regulation of cytokine-encoding genes by the T(H)2 locus control region.
DOI:10.1038/nature03574URLPMID:15880101 [本文引用: 1]
The T-helper-cell 1 and 2 (T(H)1 and T(H)2) pathways, defined by cytokines interferon-gamma (IFN-gamma) and interleukin-4 (IL-4), respectively, comprise two alternative CD4+ T-cell fates, with functional consequences for the host immune system. These cytokine genes are encoded on different chromosomes. The recently described T(H)2 locus control region (LCR) coordinately regulates the T(H)2 cytokine genes by participating in a complex between the LCR and promoters of the cytokine genes Il4, Il5 and Il13. Although they are spread over 120 kilobases, these elements are closely juxtaposed in the nucleus in a poised chromatin conformation. In addition to these intrachromosomal interactions, we now describe interchromosomal interactions between the promoter region of the IFN-gamma gene on chromosome 10 and the regulatory regions of the T(H)2 cytokine locus on chromosome 11. DNase I hypersensitive sites that comprise the T(H)2 LCR developmentally regulate these interchromosomal interactions. Furthermore, there seems to be a cell-type-specific dynamic interaction between interacting chromatin partners whereby interchromosomal interactions are apparently lost in favour of intrachromosomal ones upon gene activation. Thus, we provide an example of eukaryotic genes located on separate chromosomes associating physically in the nucleus via interactions that may have a function in coordinating gene expression.
DOI:10.1038/ng.3286URLPMID:25938943 [本文引用: 1]
Transcriptional control in large genomes often requires looping interactions between distal DNA elements, such as enhancers and target promoters. Current chromosome conformation capture techniques do not offer sufficiently high resolution to interrogate these regulatory interactions on a genomic scale. Here we use Capture Hi-C (CHi-C), an adapted genome conformation assay, to examine the long-range interactions of almost 22,000 promoters in 2 human blood cell types. We identify over 1.6 million shared and cell type-restricted interactions spanning hundreds of kilobases between promoters and distal loci. Transcriptionally active genes contact enhancer-like elements, whereas transcriptionally inactive genes interact with previously uncharacterized elements marked by repressive features that may act as long-range silencers. Finally, we show that interacting loci are enriched for disease-associated SNPs, suggesting how distal mutations may disrupt the regulation of relevant genes. This study provides new insights and accessible tools to dissect the regulatory interactions that underlie normal and aberrant gene regulation.
DOI:10.1016/j.cell.2014.11.021URL [本文引用: 1]
We use in situ Hi-C to probe the 3D architecture of genomes, constructing haploid and diploid maps of nine cell types. The densest, in human lymphoblastoid cells, contains 4.9 billion contacts, achieving 1 kb resolution. We find that genomes are partitioned into contact domains (median length, 185 kb), which are associated with distinct patterns of histone marks and segregate into six subcompartments. We identify similar to 10,000 loops. These loops frequently link promoters and enhancers, correlate with gene activation, and show conservation across cell types and species. Loop anchors typically occur at domain boundaries and bind CTCF. CTCF sites at loop anchors occur predominantly (>90%) in a convergent orientation, with the asymmetric motifs "facing'' one another. The inactive X chromosome splits into two massive domains and contains large loops anchored at CTCF-binding repeats.
DOI:10.1038/ng1244URLPMID:14517543 [本文引用: 1]
Efficient transcription of genes requires a high local concentration of the relevant trans-acting factors. Nuclear compartmentalization can provide an effective means to locally increase the concentration of rapidly moving trans-acting factors; this may be achieved by spatial clustering of chromatin-associated binding sites for such factors. Here we analyze the structure of an erythroid-specific spatial cluster of cis-regulatory elements and active beta-globin genes, the active chromatin hub (ACH; ref. 6), at different stages of development and in erythroid progenitors. We show, in mice and humans, that a core ACH is developmentally conserved and consists of the hypersensitive sites (HS1-HS6) of the locus control region (LCR), the upstream 5' HS-60/-62 and downstream 3' HS1. Globin genes switch their interaction with this cluster during development, correlating with the switch in their transcriptional activity. In mouse erythroid progenitors that are committed to but do not yet express beta-globin, only the interactions between 5' HS-60/-62, 3' HS1 and hypersensitive sites at the 5' side of the LCR are stably present. After induction of differentiation, these sites cluster with the rest of the LCR and the gene that is activated. We conclude that during erythroid differentiation, cis-regulatory DNA elements create a developmentally conserved nuclear compartment dedicated to RNA polymerase II transcription of beta-globin genes.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nrg3663URL [本文引用: 2]
The eukaryotic genome is organized in the three-dimensional nuclear space in a specific manner that is both a cause and a consequence of its function. This organization is partly established by a special class of architectural proteins, of which CCCTC-binding factor (CTCF) is the best characterized. Although CTCF has been assigned various roles that are often contradictory, new results now help to draw a unifying model to explain the many functions of this protein. CTCF creates boundaries between topologically associating domains in chromosomes and, within these domains, facilitates interactions between transcription regulatory sequences. Thus, CTCF links the architecture of the genome to its function.
DOI:10.1101/gad.277863.116URLPMID:27083996 [本文引用: 1]
The role of the zinc finger protein CTCF in organizing the genome within the nucleus is now well established. Widely separated sites on DNA, occupied by both CTCF and the cohesin complex, make physical contacts that create large loop domains. Additional contacts between loci within those domains, often also mediated by CTCF, tend to be favored over contacts between loci in different domains. A large number of studies during the past 2 years have addressed the questions: How are these loops generated? What are the effects of disrupting them? Are there rules governing large-scale genome organization? It now appears that the strongest and evolutionarily most conserved of these CTCF interactions have specific rules for the orientation of the paired CTCF sites, implying the existence of a nonequilibrium mechanism of generation. Recent experiments that invert, delete, or inactivate one of a mating CTCF pair result in major changes in patterns of organization and gene expression in the surrounding regions. What remain to be determined are the detailed molecular mechanisms for re-establishing loop domains and maintaining them after replication and mitosis. As recently published data show, some mechanisms may involve interactions with noncoding RNAs as well as protein cofactors. Many CTCF sites are also involved in other functions such as modulation of RNA splicing and specific regulation of gene expression, and the relationship between these activities and loop formation is another unanswered question that should keep investigators occupied for some time.
DOI:10.1016/j.cell.2013.05.028URL [本文引用: 1]
In mammals, dosage compensation between XX and XY individuals occurs through X chromosome inactivation (XCI). The noncoding Xist RNA is expressed and initiates XCI only when more than one X chromosome is present. Current models invoke a dependency on the X-to-autosome ratio (X:A), but molecular factors remain poorly defined. Here, we demonstrate that molecular titration between an X-encoded RNA and an autosomally encoded protein dictates Xist induction. In pre-XCI cells, CTCF protein represses Xist transcription. At the onset of XCI, Jpx RNA is upregulated, binds CTCF, and extricates CTCF from one Xist allele. We demonstrate that CTCF is an RNA-binding protein and is titrated away from the Xist promoter by Jpx RNA. Thus, Jpx activates Xist by evicting CTCF. The functional antagonism via molecular titration reveals a role for long noncoding RNA in epigenetic regulation.
DOI:10.1016/j.molcel.2019.07.039URLPMID:31522987 [本文引用: 1]
Mammalian genomes are folded into topologically associating domains (TADs), consisting of chromatin loops anchored by CTCF and cohesin. Some loops are cell-type specific. Here we asked whether CTCF loops are established by a universal or locus-specific mechanism. Investigating the molecular determinants of CTCF clustering, we found that CTCF self-association in?vitro is RNase sensitive and that an internal RNA-binding region (RBRi) mediates CTCF clustering and RNA interaction in?vivo. Strikingly, deleting the RBRi impairs about half of all chromatin loops in mESCs and causes deregulation of gene expression. Disrupted loop formation correlates with diminished clustering and chromatin binding of RBRi mutant CTCF, which in turn results in a failure to halt cohesin-mediated extrusion. Thus, CTCF loops fall into at least two classes: RBRi-independent and RBRi-dependent loops. We speculate that evidence for RBRi-dependent loops may provide a molecular mechanism for establishing cell-specific CTCF loops, potentially regulated by RNA(s) or other RBRi-interacting partners.
DOI:10.1016/j.molcel.2019.08.015URLPMID:31522988 [本文引用: 1]
The function of the CCCTC-binding factor (CTCF) in the organization of the genome has become an important area of investigation, but the mechanisms by which CTCF dynamically contributes to genome organization are not clear. We previously discovered that CTCF binds to large numbers of endogenous RNAs, promoting its self-association. In this regard, we now report two independent features that disrupt CTCF association with chromatin: inhibition of transcription and disruption of CTCF-RNA interactions through mutations of 2 of its 11 zinc fingers that are not required for CTCF binding to its cognate DNA site: zinc finger 1 (ZF1) or zinc finger 10 (ZF10). These mutations alter gene expression profiles as CTCF mutants lose their ability to form chromatin loops and thus the ability to insulate chromatin domains and to mediate CTCF long-range genomic interactions. Our results point to the importance of CTCF-mediated RNA interactions as a structural component of genome organization.
DOI:10.1038/emboj.2011.450URL [本文引用: 1]
The key haematopoietic regulator Myb is essential for coordinating proliferation and differentiation. ChIP-Sequencing and Chromosome Conformation Capture (3C)-Sequencing were used to characterize the structural and protein-binding dynamics of the Myb locus during erythroid differentiation. In proliferating cells expressing Myb, enhancers within the Myb-Hbs1l intergenic region were shown to form an active chromatin hub (ACH) containing the Myb promoter and first intron. This first intron was found to harbour the transition site from transcription initiation to elongation, which takes place around a conserved CTCF site. Upon erythroid differentiation, Myb expression is downregulated and the ACH destabilized. We propose a model for Myb activation by distal enhancers dynamically bound by KLF1 and the GATA1/TAL1/LDB1 complex, which primarily function as a transcription elongation element through chromatin looping. The EMBO Journal (2012) 31, 986-999. doi: 10.1038/emboj.2011.450; Published online 13 December 2011
DOI:10.1038/nature10442URLPMID:21964334 [本文引用: 1]
Alternative splicing of pre-messenger RNA is a key feature of transcriptome expansion in eukaryotic cells, yet its regulation is poorly understood. Spliceosome assembly occurs co-transcriptionally, raising the possibility that DNA structure may directly influence alternative splicing. Supporting such an association, recent reports have identified distinct histone methylation patterns, elevated nucleosome occupancy and enriched DNA methylation at exons relative to introns. Moreover, the rate of transcription elongation has been linked to alternative splicing. Here we provide the first evidence that a DNA-binding protein, CCCTC-binding factor (CTCF), can promote inclusion of weak upstream exons by mediating local RNA polymerase II pausing both in a mammalian model system for alternative splicing, CD45, and genome-wide. We further show that CTCF binding to CD45 exon 5 is inhibited by DNA methylation, leading to reciprocal effects on exon 5 inclusion. These findings provide a mechanistic basis for developmental regulation of splicing outcome through heritable epigenetic marks.
DOI:10.1016/j.cell.2017.11.008URLPMID:29224777 [本文引用: 1]
There is considerable evidence that chromosome structure plays important roles in gene control, but we have limited understanding of the proteins that contribute to structural interactions between gene promoters and their enhancer elements. Large DNA loops that encompass genes and their regulatory elements depend on CTCF-CTCF interactions, but most enhancer-promoter interactions do not employ this structural protein. Here, we show that the ubiquitously expressed transcription factor Yin Yang 1 (YY1) contributes to enhancer-promoter structural interactions in a manner analogous to DNA interactions mediated by CTCF. YY1 binds to active enhancers and promoter-proximal elements and forms dimers that facilitate the interaction of these DNA elements. Deletion of YY1 binding sites or depletion of YY1 protein disrupts enhancer-promoter looping and gene expression. We propose that YY1-mediated enhancer-promoter interactions are a general feature of mammalian gene control.
DOI:10.1101/gr.176586.114URL [本文引用: 1]
Increasing evidence suggests that interactions between regulatory genomic elements play an important role in regulating gene expression. We generated a genome-wide interaction map of regulatory elements in human cells (ENCODE tier I cells, K562, GM12878) using Chromatin Interaction Analysis by Paired-End Tag sequencing (ChIA-PET) experiments targeting six broadly distributed factors. Bound regions covered 80% of DNase I hypersensitive sites including 99.7% of TSS and 98% of enhancers. Correlating this map with ChIP-seq and RNA-seq data sets revealed cohesin, CTCF, and ZNF143 as key components of three-dimensional chromatin structure and revealed how the distal chromatin state affects gene transcription. Comparison of interactions between cell types revealed that enhancer promoter interactions were highly cell-type-specific. Construction and comparison of distal and proximal regulatory networks revealed stark differences in structure and biological function. Proximal binding events are enriched at genes with housekeeping functions, while distal binding events interact with genes involved in dynamic biological processes including response to stimulus. This study reveals new mechanistic and functional insights into regulatory region organization in the nucleus.
DOI:10.7868/S0026898416030034URLPMID:27414788 [本文引用: 1]
ZNF143 is a ubiquitously expressed transcription factor conserved in vertebrates and might regulate the expression of numerous genes. But its function in mediating chromatin interactions remains elusive. By integrated analysis of public datasets, we provided evidence that a majority of ZNF143 binding sites (BSs) were involved in CTCF-mediated chromatin interaction networks (CTCF-CINs) by overlapping with cohesin-BSs and CTCF-BSs. We further showed that only a very few CTCF-CINs were associated with ZNF143 alone, whereas those associated with ZNF143 and cohesin simultaneously were highly overlapped with constitutive, conserved CTCF-BSs and enriched at boundaries of chromatin topologically associating domains. These observations implicate that as an important partner of CTCF, ZNF143 helps it establish the conserved chromatin structure by cooperating with cohesin.
DOI:10.1016/j.cell.2014.05.050URL [本文引用: 1]
Distal enhancers commonly contact target promoters via chromatin looping. In erythroid cells, the locus control region (LCR) contacts beta-type globin genes in a developmental stage-specific manner to stimulate transcription. Previously, we induced LCR-promoter looping by tethering the self-association domain (SA) of Ldb1 to the beta-globin promoter via artificial zinc fingers. Here, we show that targeting the SA to a developmentally silenced embryonic globin gene in adult murine erythroblasts triggers its transcriptional reactivation. This activity depends on the LCR, consistent with an LCR-promoter looping mechanism. Strikingly, targeting the SA to the fetal gamma-globin promoter in primary adult human erythroblasts increases gamma-globin promoter-LCR contacts, stimulating transcription to approximately 85% of total beta-globin synthesis, with a reciprocal reduction in adult beta-globin expression. Our findings demonstrate that forced chromatin looping can override a stringent developmental gene expression program and suggest a novel approach to control the balance of globin gene transcription for therapeutic applications.
DOI:10.1016/j.cell.2011.03.040URL [本文引用: 1]
Constitutive heterochromatin is traditionally viewed as the static form of heterochromatin that silences pericentromeric and telomeric repeats in a cell cycle-and differentiation-independent manner. Here, we show that, in the mouse olfactory epithelium, olfactory receptor (OR) genes are marked in a highly dynamic fashion with the molecular hallmarks of constitutive heterochromatin, H3K9me3 and H4K20me3. The cell type and developmentally dependent deposition of these marks along the OR clusters are, most likely, reversed during the process of OR choice to allow for monogenic and monoallelic OR expression. In contrast to the current view of OR choice, our data suggest that OR silencing takes place before OR expression, indicating that it is not the product of an OR-elicited feedback signal. Our findings suggest that chromatin-mediated silencing lays a molecular foundation upon which singular and stochastic selection for gene expression can be applied.
DOI:10.1146/annurev-cellbio-100814-125308URLPMID:26359778 [本文引用: 1]
The sense of smell collects vital information about the environment by detecting a multitude of chemical odorants. Breadth and sensitivity are provided by a huge number of chemosensory receptor proteins, including more than 1,400 olfactory receptors (ORs). Organizing the sensory information generated by these receptors so that it can be processed and evaluated by the central nervous system is a major challenge. This challenge is overcome by monogenic and monoallelic expression of OR genes. The single OR expressed by each olfactory sensory neuron determines the neuron's odor sensitivity and the axonal connections it will make to downstream neurons in the olfactory bulb. The expression of a single OR per neuron is accomplished by coupling a slow chromatin-mediated activation process to a fast negative-feedback signal that prevents activation of additional ORs. Singular OR activation is likely orchestrated by a network of interchromosomal enhancer interactions and large-scale changes in nuclear architecture.
DOI:10.1038/nature21711URLPMID:28355183 [本文引用: 1]
Chromatin is reprogrammed after fertilization to produce a totipotent zygote with the potential to generate a new organism. The maternal genome inherited from the oocyte and the paternal genome provided by sperm coexist as separate haploid nuclei in the zygote. How these two epigenetically distinct genomes are spatially organized is poorly understood. Existing chromosome conformation capture-based methods are not applicable to oocytes and zygotes owing to a paucity of material. To study three-dimensional chromatin organization in rare cell types, we developed a single-nucleus Hi-C (high-resolution chromosome conformation capture) protocol that provides greater than tenfold more contacts per cell than the previous method. Here we show that chromatin architecture is uniquely reorganized during the oocyte-to-zygote transition in mice and is distinct in paternal and maternal nuclei within single-cell zygotes. Features of genomic organization including compartments, topologically associating domains (TADs) and loops are present in individual oocytes when averaged over the genome, but the presence of each feature at a locus varies between cells. At the sub-megabase level, we observed stochastic clusters of contacts that can occur across TAD boundaries but average into TADs. Notably, we found that TADs and loops, but not compartments, are present in zygotic maternal chromatin, suggesting that these are generated by different mechanisms. Our results demonstrate that the global chromatin organization of zygote nuclei is fundamentally different from that of other interphase cells. An understanding of this zygotic chromatin 'ground state' could potentially provide insights into reprogramming cells to a state of totipotency.
DOI:10.1073/pnas.1609643113URLPMID:27432957 [本文引用: 1]
During interphase, the inactive X chromosome (Xi) is largely transcriptionally silent and adopts an unusual 3D configuration known as the &quot;Barr body.&quot; Despite the importance of X chromosome inactivation, little is known about this 3D conformation. We recently showed that in humans the Xi chromosome exhibits three structural features, two of which are not shared by other chromosomes. First, like the chromosomes of many species, Xi forms compartments. Second, Xi is partitioned into two huge intervals, called &quot;superdomains,&quot; such that pairs of loci in the same superdomain tend to colocalize. The boundary between the superdomains lies near DXZ4, a macrosatellite repeat whose Xi allele extensively binds the protein CCCTC-binding factor. Third, Xi exhibits extremely large loops, up to 77 megabases long, called &quot;superloops.&quot; DXZ4 lies at the anchor of several superloops. Here, we combine 3D mapping, microscopy, and genome editing to study the structure of Xi, focusing on the role of DXZ4 We show that superloops and superdomains are conserved across eutherian mammals. By analyzing ligation events involving three or more loci, we demonstrate that DXZ4 and other superloop anchors tend to colocate simultaneously. Finally, we show that deleting DXZ4 on Xi leads to the disappearance of superdomains and superloops, changes in compartmentalization patterns, and changes in the distribution of chromatin marks. Thus, DXZ4 is essential for proper Xi packaging.
DOI:10.1016/j.cell.2018.05.024URLPMID:29887377 [本文引用: 1]
Eukaryotic genomes are packaged into a 3-dimensional structure in the nucleus. Current methods for studying genome-wide structure are based on proximity ligation. However, this approach can fail to detect known structures, such as interactions with nuclear bodies, because these DNA regions can be too far apart to directly ligate. Accordingly, our overall understanding of genome organization remains incomplete. Here, we develop split-pool recognition of interactions by tag extension (SPRITE), a method that enables genome-wide detection of higher-order interactions within the nucleus. Using SPRITE, we recapitulate known structures identified by proximity ligation and identify additional interactions occurring across larger distances, including two hubs of inter-chromosomal interactions that are arranged around the nucleolus and nuclear speckles. We show that a substantial fraction of the genome exhibits preferential organization relative to these nuclear bodies. Our results generate a global model whereby nuclear bodies act as inter-chromosomal hubs that shape the overall packaging of DNA in the nucleus.
DOI:10.1038/s41586-019-0949-1URLPMID:30778195 [本文引用: 1]
The genomes of multicellular organisms are extensively folded into 3D chromosome territories within the nucleus1. Advanced 3D genome-mapping methods that combine proximity ligation and high-throughput sequencing (such as chromosome conformation capture, Hi-C)2, and chromatin immunoprecipitation techniques (such as chromatin interaction analysis by paired-end tag sequencing, ChIA-PET)3, have revealed topologically associating domains4 with frequent chromatin contacts, and have identified chromatin loops mediated by specific protein factors for insulation and regulation of transcription5-7. However, these methods rely on pairwise proximity ligation and reflect population-level views, and thus cannot reveal the detailed nature of chromatin interactions. Although single-cell Hi-C8 potentially overcomes this issue, this method may be limited by the sparsity of data that is inherent to current single-cell assays. Recent advances in microfluidics have opened opportunities for droplet-based genomic analysis9 but this approach has not yet been adapted for chromatin interaction analysis. Here we describe a strategy for multiplex chromatin-interaction analysis via droplet-based and barcode-linked sequencing, which we name ChIA-Drop. We demonstrate the robustness of ChIA-Drop in capturing complex chromatin interactions with single-molecule precision, which has not been possible using methods based on population-level pairwise contacts. By applying ChIA-Drop to Drosophila cells, we show that chromatin topological structures predominantly consist of multiplex chromatin interactions with high heterogeneity; ChIA-Drop also reveals promoter-centred multivalent interactions, which provide topological insights into transcription.

{kind=link}
{kind=link}