The applications of research progress of common wheat in teaching genetics
Na Zhao1, Bao Qi2, Qianli Dong3, Xiaoli Wang1通讯作者: 王晓丽,博士,教授,研究方向:细胞生物学。E-mail:1392313442@qq.com
责任编辑: 陈德富
收稿日期:2020-04-23修回日期:2020-08-8网络出版日期:2020-09-20
| 基金资助: |
Received:2020-04-23Revised:2020-08-8Online:2020-09-20
| Fund supported: |
作者简介 About authors
赵娜,博士,讲师,研究方向:多倍体植物基因组进化。E-mail:

摘要
普通小麦(T.aestivum L.)又称异源六倍体小麦,其基因组是由来自3个不同二倍体祖先且亲缘关系较近的基因组(A、B和D)构成。普通小麦的进化历程一直是遗传学教学中阐述物种形成和染色体数目变异机制的经典案例。近年来,伴随着科学技术的快速发展和应用,普通小麦的相关研究在细胞学水平、分子水平、基因组水平均取得了重大突破和进展。本文对普通小麦最新研究成果进行了梳理和总结,将相关前沿科学内容与遗传学各章节的理论教学相结合,并应用于遗传学的理论教学中。这不仅是对经典遗传学教材内容的补充和发展,同时也能够让学生认识到遗传学是一门不断发展的自然科学,在提高学生学习兴趣的同时,实现对遗传学基本内容和前沿科学动态的系统学习。
关键词:
Abstract
Common wheat (T. aestivum L.) is also known as allohexaploid wheat. Its genome is composed of A/B/D sub-genomes from three closely related diploid ancestors. The evolutionary history of common wheat is used as a classic example to illustrate the mechanism of species formation and chromosome number variation in the current genetics class. In recent years, with the rapid development and application of research technologies, there have been many breakthroughs in the study of common wheat, at the cytological, molecular and genomic level. Here, we summarize the latest research achievements on common wheat, and discuss our practice in combining them with the genetics teaching. Our approach is not only a supplement to the current genetics textbooks, but also enables students to realize that genetics is a constantly evolving natural science. We aim to enhance students’ interests in learning, as well as their systematic learning abilities on genetics and related scientific research frontiers.
Keywords:
PDF (572KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
赵娜, 亓宝, 董芊里, 王晓丽. 普通小麦相关研究进展在遗传学理论教学中的应用. 遗传[J], 2020, 42(9): 916-925 doi:10.16288/j.yczz.20-113
Na Zhao.
遗传学是现代生命科学领域中迅速发展的学科之一,也是高等院校生命学科相关专业必修的重要基础课程。自1900年诞生至今,遗传学经历了多个重要的发展阶段,特别是1953年DNA双螺旋结构的破解使得遗传学研究迅速进入分子水平,各种新概念和新技术被提出和应用。近年来基因组测序技术的快速发展和应用,促使遗传学研究进入了基因组层面。为了使遗传学教学内容与时俱进,本文对普通小麦的相关研究进展进行了梳理和总结,分别在基因组、染色体和基因等不同层面上解析普通小麦的遗传规律,并将其在遗传学不同章节理论教学中的应用进行了阐述(表1)。
Table 1
表1
表1普通小麦相关研究案例在遗传学理论教学中的应用框架
Table 1
| 教学案例 | 应用章节 | 教学目标 | 参考文献 |
|---|---|---|---|
| 普通小麦A、B、D亚基因组的分化时间以及杂交、加倍时间的重新界定 | 物种形成与进化 | (1)掌握多倍体的形成方式 (2)掌握普通小麦的形成与进化历程 | [1~4] |
| 荧光原位杂交技术在普通小麦基因组分型及易位系鉴定中的应用 | 染色体数目变异 染色体结构变异 | (1)掌握核型分析技术在普通小麦细胞学研究和遗传育种研究中的应用 (2)了解染色体结构变异和数目变异类型及细胞学特征 | [5~16] |
| 矮杆突变、落粒性突变在小麦育种中的应用 | 基因突变 | (1)了解矮杆突变和落粒性突变的分子遗传机制 (2)掌握基因突变的防护机制 | [17~22] |
| 甲基化修饰导致普通小麦LHS1-B基因、麦谷蛋白基因和麦醇溶蛋白基因的表达沉默 | 表观遗传学 | (1)掌握什么是表观遗传学 (2)了解DNA甲基化的作用机制 | [23,24] |
| 普通小麦全基因组测序及其在小麦性状解析中的应用进展 | 基因组学 | (1)掌握基因组测序的基本方法和策略 (2)了解普通小麦基因组的特征 | [25~40] |
新窗口打开|下载CSV
1 在物种形成与进化章节中的应用
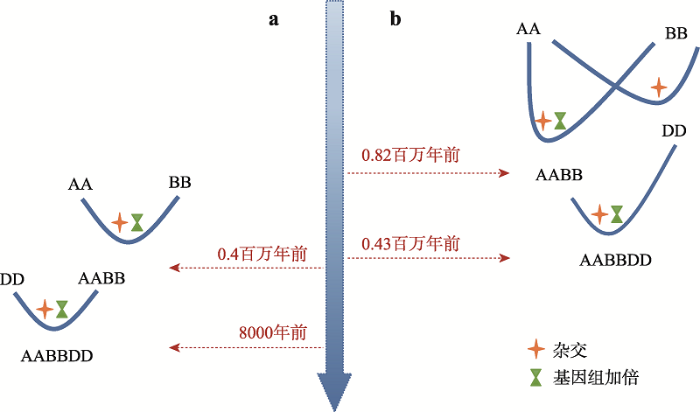
在本章节的导课中,每当提到多倍体物种的形成,同学们的回答通常是利用秋水仙素抑制细胞的有丝分裂,促使细胞内染色体加倍形成多倍体;或是通过杂交的方法培育同源三倍体无籽西瓜。同学们似乎忽视了在自然环境中多倍体物种的形成方式。事实上,多倍体植物在自然界的存在极具普遍性。在被子植物中,约有80%的物种在进化过程中经历过一次或多次基因组加倍过程,基因组多倍化的发生是自然界中大部分被子植物形成及进化过程中所经历的重要途经[41]。其中,重要粮食作物普通小麦(T.aestivum L.)的形成与进化恰好是展现自然环境中多倍体物种形成的典型案例。普通小麦是一个异源六倍体植物,基因组构成为AABBDD。遗传学教材介绍了普通小麦在形成过程中经历了两次异源多倍化事件:第一次发生在距今大约40万前[1],由二倍体的乌拉尔图小麦(T.urartu) (AA)和二倍体的拟斯卑尔托山羊草(A.speltoides) (BB)杂交加倍形成了异源四倍体圆锥小麦(T.turgidum) (AABB)[2];另一次发生在距今约8000~10,000年前,驯化后的异源四倍体小麦与二倍体粗山羊草(A.tauschii) (DD)再次发生杂交加倍事件形成异源六倍体小麦,即普通小麦(AABBDD)[3] (图1a)。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1普通小麦形成与进化历程新旧观点对比
a:旧观点;b:新观点。
Fig. 1New views on the formation and evolution of common wheat
近几年,人们在小麦属基因组方面的研究有了新突破,为普通小麦的起源及进化历程提出了新观点[4]。通过普通小麦与近缘二倍体小麦物种基因组间的序列比对,研究人员重新阐述了小麦属A、B和D三个亚基因组之间的进化关系,提出二倍体小麦的A基因组和B基因组在距今约6.5个百万年前发生分化,在约5.5个百万年前发生第一次杂交并形成二倍体小麦的D基因组[4](图1b)。同时对普通小麦的形成与进化时间给与重新定义:乌拉尔图小麦(AA)与拟斯卑尔托山羊草A. (BB)在距今约0.82百万年内发生杂交加倍形成野生二粒小麦(AABB)。经过驯化的栽培四倍体小麦(AABB)与粗山羊草(DD)在距今0.43百万年内经历杂交加倍形成普通学小麦(AABBDD)[4]。上述研究结论重新界定了普通小麦A、B、D亚基因组的系统发生史和分化时间。通过图1的展示,使学生们清晰的理解普通小麦形成与进化历程。同时,突出强调“基因组加倍”可以克服远缘杂交导致的不孕性和不育性,使杂交后代F1能够正常的进行减数分裂,形成可育后代。人类已将“杂交”和“基因组加倍”技术应用到了粮食作物、蔬菜、水果的育种改良中,为人们创造了极其丰富的生活物资。基于上述讲解,向学生布置相关设计类作业题目,在强化知识点理解的同时,引导学生用所学的遗传学理论知识解决生活中的实际问题。同时,拓展讲解当前基因组学、生物信息学分析方法在物种的形成与进化研究中的应用,更重要的是鼓励学生敢于用新技术去解释旧问题,并富于敢于质疑和探索的学习精神。普通小麦形成与进化历程的重新界定也是对现有遗传学教材内容的补充和发展。
2 在染色体数目和结构变异章节中的应用
染色体核型分析技术,是细胞遗传学研究的基本方法,是当前高校遗传学实验教学中学生必须掌握的实验技能之一,在动植物育种、现代医学检测等方面应用十分广泛。常用的核型分析技术包括各类显带技术(G带、Q带、C带等)、荧光原位杂交(fluorescence in situ hybridization,FISH)技术等。同学们可以在显微镜下观察到诸如:染色体的长度、粗细;着丝粒(主缢痕)的位置;随体及次缢痕的有无、数目、位置;以及由于温度和药品处理所产生的染色体分带等相关信息[42],用于对细胞内的所有染色体的形态特征进行观察分析。本文中,我们将FISH在普通小麦染色体分型、易位系鉴定等方面的应用进行整理,作为“染色体数目变异”和“染色体结构变异”两个章节理论教学的拓展内容。2.1 小麦染色体分型
在针对植物多倍体的研究中,基因组原位杂交(genome in situ hybridization,GISH)技术和FISH技术成为了探索多倍体祖先基因组来源、确定种间进化关系以及分析基因组间杂交渐渗的重要手段[5]。近年来,刘宝教授课题组利用人工合成异源多倍体小麦,对早期核型稳定性对异源四倍体和六倍体小麦物染色体进化与物种形成的关系进行研究。通过FISH与GISH相结合的方法,不仅可以对普通小麦的3个亚基因组A、B和D中的21条染色体进行精确区分[6],也可对染色体水平发生的数目和结构变异进行精准识别[7]。通过对大量不同基因组构成的人工合成异源四倍体小麦单株(基因型为SlSlAA、SbSbDD、AADD,因基因组为BB的二倍体亲本现已灭绝,采用与B基因组最相近的S基因组二倍体亲本代替)进行精准染色体核型的分子细胞遗传学鉴定。在染色体水平上,观察到在异源四倍体小麦SlSlAA中仅发生了部分重复DNA序列和同源基因的拷贝数的变化,而其他两个基因型无论是在染色体数目还是染色体结构上均发生了很大的变化。上述研究结果为理解自然坏境下野生异源四倍体小麦仅有AABB的基因组构成模式提供细胞学证据[7]。
2.2 小麦易位系鉴定
1BL·1RS易位系是小麦品种改良中的经典案例。通过远缘杂交,黑麦的1RS染色体短臂被导入小麦基因组中,形成1BL·1RS易位系。由于1RS染色体上携带有抗条锈病(Yr9)、叶锈病(Lr26)、白粉病(Pm8)等抗性基因,以及提高产量和增强环境适应性的基因[8,9],因此1BL·1RS易位系在小麦抗病、抗逆和产量等方面均体现了独特的优势,在世界范围内得到广泛的应用[10,11]。21世纪初,在我国育成的小麦品种中,约有40%含有1BL·1RS易位系[12]。Schlegel[13]对全世界2470个小麦品种和试验品系调查显示,所有小麦材料中都带有外来基因,其中15%商业品种中携带黑麦1RS染色体。那么,针对国内外众多小麦品种,能够快速准确的对基因组中是否含有1RS易位染色体片段进行鉴定,对小麦品种的进一步改良具有重要意义。FISH技术可以快速证实小穗小麦种质10-A是小麦-黑麦1BL·1RS易位系[14],也可以在小麦品系与黑麦杂交的后代中快速筛选出含有1BL·1RS的易位系用于育种研究[15]。FISH技术亦可在染色体起源进化研究中提供了最直接的证据,在关于小麦4A/5A染色体易位的研究中,分布在二倍体小麦(T.urartu) 4A染色体上的Acc-2探针在5A染色体上也存在杂交信号,证实了4A与5A染色体间的易位,同时在一些近缘二倍体和多倍体小麦物种中杂交信号的存在,表明它们起源于同一个A基因组的祖先种[16]。上述案例表明,FISH为研究染色体数目和结构变异研究提供了高效且可靠的分析平台,大大提升了细胞学观察的分辨率和应用范围,使得人们对普通小麦乃至其他物种进行相对精准的细胞学研究成为可能,在研究物种进化、作物遗传育种等多方面领域提供可靠的技术支持。在当前大部分高校关于“染色体结构变异”这个章节的学习中,学生仅能通过减数分裂过程中同源染色体联会时是否会形成各种形态的“异构体”来判定染色体水平上的变异,例如:缺失、重复、倒位杂合体都会形成“圈”型染色体构态,易位杂合体可能会出现“十字形”、“8字形”等染色体异构体[42],但对于发生在染色体的具体位置和所参与的基因却无法准确判断。FISH技术能够解决上述问题,但是由于该技术耗材费用昂贵、技术细节精细、试验周期长,目前在高校本科开设相应的实验课程微乎其微。在我校的课程设置中,将在线实验课程观看与部分可行性实验操作相结合,有效地开展FISH技术等有难度的实验课程,使学生广泛了解高新技术在生物学领域的重要应用,掌握相关技术原理和操作要领,更重要的是培养学生对自然科学研究的兴趣。
3 在基因突变章节中的应用
基因突变是DNA分子中发生碱基对的替换、增添和缺失而引起的基因结构的改变。自然界中,基因突变普遍发生,是导致生物变异、构成生物多样性的重要原因,更是推动生物进化的动力之一。普通小麦从野生二倍体经历驯化选择至今,成为全世界范围重要的粮食作物,不仅经历了染色体数目和结构的变异,也发生了很多关键的基因突变事件。本文通过对小麦驯化史上两个重要的基因突变的研究进展进行梳理,帮助学生了解植物的天然突变在人类农业生产中的应用,引导学生思考如何开发和利用大量潜在的、有价值的基因资源,造福人类未来。3.1 株高
株高是影响小麦产量的关键因素之一,半矮秆基因Rht的发现和利用大幅度提升了世界小麦产量。首次将该基因引入小麦栽培种的美国科学家Norman Ernest Borlaug终其一生推动了举世闻名的“绿色革命”,为解决世界粮食问题、克服全球饥荒做出了巨大贡献。1999年,Peng等[17]首次克隆了小麦的半矮秆基因Rht-B1/Rht-D1,研究发现DELLA蛋白N端个别氨基酸的改变降低了与生长激素赤霉素受体蛋白GID的结合能力,从而降低了细胞核对赤霉素的敏感度,导致植株的矮化。自绿色革命以来,矮秆基因的研究和利用被越来越多的遗传学家和育种专家所重视,人们相继鉴定出主效半矮秆基因20余个[18],但真正用于育种和生产的并不多,育种学家仅对Rht-B1/Rht-D1进行了较强的选择和应用。在我国主产区的小麦品种中,约有24.3%携带Rht-B1,46.9%携带Rht-D1[19]。矮化育种在小麦生产应用中的作用是显而易见的,但也暴露出一定的弊端,Rht-B1/ Rht-D1虽然带来了抗倒伏的矮化表型,但同时也降低了小麦对土壤中氮素的利用效率。为了维持抗倒伏高产小麦对氮素的需求,在实际农业生产中,氮肥的施用量大幅增加,直接导致环境污染。因此,协同改良半矮化小麦的高产与氮肥高效利用性状才是解决问题的关键[20]。近期,中国科学院遗传与发育生物学研究所傅向东课题组在水稻的相关研究中有了重要突破:生长调节因子GRF4 (growth regulating factor 4)较高水平的表达不仅可以提高半矮杆水稻品种的氮素利用效率,同时也维持了半矮杆性状赋予的抗倒伏和高产的特性[21]。该研究为矮杆小麦的育种改良指明了目标和方向。
3.2 落粒性
落粒性是谷类作物另一个非常重要的性状,也是驯化研究的典型性状之一。野生小麦成熟后,穗轴会变脆,完整的穗会断裂成小穗,在风力作用下四处播散,有利于下一代的繁殖生长,但从人类农耕生产角度来讲,却是一个缺点。在大约1万年前,小麦起源地的居民就开始对穗轴不易断裂的突变个体进行选择,培育出易于收割的、具有落粒抗性的小麦品种。2017年,伴随着小麦全基因组测序结果的逐渐完善,小麦落粒性的分子机制得到了进一步解析。以色列特拉维夫大学Distelfeld研究团队[22]对普通小麦的异源四倍体祖先-野生二粒小麦进行基因组测序、组装。将其与驯化品种进行基因比对分析,发现了导致穗轴易断裂的两个关键基因TtBtr1-A和TtBtr1-B,二者突变导致穗轴断裂功能的丧失,使得破碎的麦穗转变为不易脱落的麦穗,达到了利于农民收割的目的,直接提高了小麦的收益。植株矮化和抗落粒性状是小麦驯化过程中人类对其自发突变进行选择和利用的两个经典案例。应用于教学之中,有助于学生深刻理解基因突变在物种驯化及品种形成过程中所扮演的重要角色和意义。
4 在表观遗传学章节中的应用
表观遗传学(epigenetics)是指在DNA序列不发生改变的情况下,因DNA甲基化、蛋白质的共价修饰、染色质重塑、非编码RNA调控等的修饰作用导致生物的性状产生可遗传的变异。该学科兴起于20世纪末,是遗传学发展最快的重要分支,部分经典遗传学教材中尚未列入此章节内容。因此,在教学中,我们以专题系列讲座的形式向学生介绍表观遗传学的前沿与发展。普通小麦复杂的基因组构成为表观遗传学研究提供了很多独特的研究案例。在本文中,我们主要针对普通小麦中DNA甲基化的相关研究进行总结,提出两个适用于本科理论教学的表观遗传学案例。4.1 DNA甲基化修饰对小麦部分同源等位基因功能的影响
普通小麦A、B、D基因组的4号染色体上存在3对部分同源等位基因(leafy hull sterile 1: LHS1-A、LHS1-B和LHS1-D),它们行使着相同的基因功能,其中位于A基因组上的LHS1-A因为一个较大DNA片段的插入导致其基因结构发生变异而丧失功能,位于B基因组的LHS1-B虽与LHS1-D序列相似,但因受到高密度的胞嘧啶甲基化修饰导致其在转录水平发生沉默而丧失基因功能,仅有LHS1-D行使正常的基因功能[23],维持普通小麦的正常生存和繁衍。通过此案例,学生们对表观遗传学一个重要的研究内容—“DNA甲基化”有了初步认知,了解到DNA甲基化是真核基因组中普遍存在的、能够遗传的化学修饰方式之一,往往通过对基因启动子区的修饰来抑制转录,参与基因表达调控过程。这个案例也从另一个角度体现了多倍体植物在应对遗传变异和表观遗传修饰等原因造成的部分同源等位基因功能丧失时的“防护”功能[42]。相对于二倍体或单倍体,普通小麦以更多的基因组构成为应对变异带来的损伤起到了一定的缓冲作用。4.2 DNA甲基化修饰的调控与小麦新品种选育
乳糜泻疾病(celiac disease),是一种对小麦等麦类作物籽粒中的麦谷蛋白和麦醇溶蛋白产生不良反应的肠道疾病。在西方国家,约有1%的人口患有此病。对于患者来说,安全的饮食策略就是杜绝食物中含有麦类蛋白成分,这给患者的生活带来诸多不便。科研人员从多角度针对这一难题展开研究,策略之一就是通过调控DNA甲基化的修饰程度实现低(或无)麦类蛋白的小麦品种的培育,为乳糜泻疾病的患者带来安全和便利。研究发现,麦谷蛋白基因和醇溶蛋白基因在小麦的胚乳中特异性高表达,源于5-甲基胞嘧啶DNA糖基化酶DEMETER(DME,去甲基化酶)的表达使两类基因处于较低水平的甲基化修饰状态[24]。鉴于此,研究人员通过RNAi的方法对发育过程中小麦胚乳的DME基因进行特异性的表达沉默,间接导致麦谷蛋白基因和麦醇溶蛋白基因处于高度甲基化状态[23],降低小麦种子中麦谷蛋白和麦醇溶蛋白的表达和积累,为育成乳糜泻患者可食用的小麦品种奠定了研究基础。上述研究属于用表观遗传理论指导作物遗传育种的一个典型案例。5 在基因组学章节中的应用
2000年,人类基因组测序的完成将遗传学带入到了基因组时代。而今,基因组学已经成为遗传学的重要组成部分,具备基因组学知识并掌握相关分析技术已成为目前从事生命科学领域研究型人才的基本要求。那么,如何加强基因组学课程的建设与完善,培养出符合时代需求的科技人才成为当前高等院校迫切要解决的问题。在遗传学课程中,基因组学作为一个章节的内容,课时有限,但信息量大,难点多。如何在有限的时间内,使学生迅速燃起对基因组学的热情,并理解基因组学的主要研究内容、熟悉基本分析方法,为将来更深入的学习基因组学奠定好基础,是遗传学教学过程中需要充分设计和思考的问题。在教学中,我们以普通小麦基因组测序的过程为案例之一,开启学生对基因组学的认知大门。5.1 普通小麦基因组测序
与遗传学研究中经典模式植物拟南芥(Arabidopsis thaliana)不同,普通小麦因其具有庞大的基因组,测序工作曾被认为是“不可能完成的任务”。普通小麦的3个亚基因组中共有21条染色体,总基因组量约为17 Gb,约为玉米(Zea mays)的7倍、水稻(Oryza sativa)的37倍、拟南芥的148倍。那么全世界科学家是如何攻克难关在小麦基因组测序上取得突破和进展的呢?在历时13年的时间里,由国际小麦基因组测序联盟(International Wheat Genome Sequencing Consortium, IWGSC, http://www.wheatgenome.org/)领导,全球20多个国家70多个研究机构约200名科学家共同参与并完成了这一世界性难题。普通小麦基因组测序之难,主要是由于它的六倍体基因组构成。在普通小麦的基因组中,每个功能基因都在A、B、D三个基因组上存在相同或相似功能的部分同源等位基因,它们在序列上具有很高的相似性。如何准确分辨某段DNA序列的基因组归属问题,成为序列拼装的难题之一。另外,普通小麦基因组含有大量的重复非编码DNA (repetitive non-coding DNA),约占总基因组序列的85%~90%[25],在3个亚基因组中,这些重复序列在A、B、D三个部分同源染色体上的排列顺序也都有所不同,使基因组组装工作变得更加复杂。
基于上述难题,在小麦基因组测序之初,捷克科学家Jaroslav Dolezel教授利用流式细胞仪分离技术,将普通小麦(中国春)的21条染色体进行了分离,以单条染色体(臂)为测序单位,排除了3个亚基因组间部分同源染色体相似性带来的困扰,分别构建BAC文库,后续物理图谱构建和基于BAC by BAC测序工作则由IWGSC的成员国共同分担完成[26]。随着高通量测序技术的出现和推广,在很大程度上加快了小麦基因组测序的速度。2012年,英国利物浦大学Neil Hall教授领导的研究小组利用全基因组鸟枪法测序技术,对中国春进行了5倍覆盖率的全基因组测序,组装基因组5.42 Gb,预测9.4~9.6万个基因,定位约2/3的基因,开创了小麦基因组测序的新局面[27]。2017年,Clavijo等[28]利用mate-pair文库和优化的组装算法,进一步提高了小麦基因组的组装质量和完整性,组装出近78%的中国春小麦基因组。同年底,美国霍普金斯大学Steven L.Salzberg研究小组利用二代、三代测序技术,组装出大约15 Gb的物理图谱,约占小麦全基因组的90%[29],国际小麦基因组测序联盟也在同年公布了中国春的参考基因组“IWGSC Ref Seq v1.0”,成为目前为止最完整的普通小麦参考图谱(https://wheat-urgi.versailles. inra.fr/Seq-Repository/Assemblies)。我国科学家在麦类作物基因组研究方面也做出了很多突出贡献,其中包括A和D基因组的精细图谱绘制,中国春AABBDD精细图谱的部分绘制工作[30,31,32,33]。直至2018年8月,普通小麦及其亲缘种的精细基因组序列图谱均已绘制完成[34],这为小麦的功能基因组学、比较基因组学和进化基因组学研究奠定了重要基础,更为小麦基因组育种提供了重要依据。
5.2 基因组信息在小麦性状解析中的应用
伴随着测序成本的逐年降低,基于基因组中单核苷酸多态性(single nucleotide polymorphism, SNP)为分子遗传标记的全基因组关联分析技术(genome- wide association study, GWAS)在挖掘小麦重要农艺性状基因方面获得一系列重要成果。科研人员对4302份优质面包小麦材料进行连续5年的产量试验,选择1092份材料开展连续2年不同环境下的田间产量数据调查,通过全基因组关联分析,检测到16个与小麦产量相关联的SNP标记,集中分布在染色体3B和6B上[35]。在针对192份普通小麦(包括87份栽培品种,80份地方品种和25份人工合成异源六倍体小麦) 4个地区两年间的田间性状调查,检测到分布于2A、2B、2D和6A染色体,与穗长相关联的4个SNP位点;3个与穗粒数相关联,位于2A、2B和7B的SNP位点,以及1个分布于7B染色体上的与穗数相关联的SNP位点[36]。与小麦种子品质[37]和抗锈病[38]等相关的部分SNP标记也被开发并定位于染色体上,详见表2。由此可见,基于小麦重测序开发的大量SNP分子标记在重要农艺性状基因的遗传定位和高效系统地克隆小麦的重要功能基因,解析小麦高产、抗逆、优质等重要性状的分子机制发挥了举足轻重的作用。加速了栽培小麦遗传改良和分子育种的进程,为促进小麦产量与品质的提升奠定了重要的理论基础。Table 2
表2
表2全基因组关联分析在小麦育种研究中的应用
Table 2
| 小麦性状 | SNP位点数 | 染色体定位 | 材料应用 | 参考文献 |
|---|---|---|---|---|
| 产量(grain yield) | 16 | 3B 6B | 1092株普通小麦 | [35] |
| 穗长(spike length) | 4 | 2A 2B 2D 6A | 192株普通小麦 | [36] |
| 穗粒数(kernels per spike) | 3 | 2A 2B 7B | ||
| 穗数(spikelet number) | 1 | 7B | ||
| 淀粉含量(starch content) | 1 | 5B | 1325株冬小麦 | [37] |
| 含水量(moisture) | 3 | 6A 1B | ||
| 条锈病(stripe rust) | 16 | 1A 1B 1D 2A 2B 5A 6A | 483株春小麦 | [38] |
| 叶锈病(leaf rust) | 18 | 1A 1B 2B 3A 3B 3D 5B 7A | ||
| 秆锈病(stem rust) | 27 | 1A 1B 1D 2A 2B 3A 3B 3D 5A 5B 6B 7A | ||
| 腥黑穗病(karnal bunt) | 15 | 2D 3B 4D 7B | 179株优良栽培小麦 | [39] |
| 镰刀菌茎基腐病(fusarium crown rot) | 5 | 5DL | 358株中国优质小麦 | [40] |
新窗口打开|下载CSV
通过上述关于普通小麦基因组测序过程及应用案例的讲解,使学生掌握了基因组测序的基本方法、策略和步骤,了解当前测序技术的发展及各种测序技术的特点,以及基因组信息在小麦基础理论研究和育种改良研究中的应用。上述案例在遗传学课堂上的应用发挥了抛砖引玉的作用,为学生未来学习计算机语言、基因组学、生物信息学等专业课程奠定基础;同时鼓励学生努力钻研,用掌握的基因组学相关知识和技术解决生活、生产中的实际问题。
6 结语
通过对普通小麦相关研究进展及应用进行梳理和总结,设计成教学案例贯穿于遗传学不同章节的内容之中(表1),使遗传学教学内容丰富化、前沿化、系统化,不仅有助于帮助学生建立系统的知识体系,开拓学生的学习视野,提高思考问题的深度,更有利于打破以往学生死啃书本的学习方法。这种尝试用同一种生物解析多种遗传现象的教学方法,充分体现了当前遗传学研究的前沿性和完整性,使遗传学教学具有连贯性和趣味性,为提高本科遗传学教学质量、培养创新型人才提供重要的支持。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1073/pnas.072223799URLPMID:12060759 [本文引用: 2]

The classic wheat evolutionary history is one of adaptive radiation of the diploid Triticum/Aegilops species (A, S, D), genome convergence and divergence of the tetraploid (Triticum turgidum AABB, and Triticum timopheevii AAGG) and hexaploid (Triticum aestivum, AABBDD) species. We analyzed Acc-1 (plastid acetyl-CoA carboxylase) and Pgk-1 (plastid 3-phosphoglycerate kinase) genes to determine phylogenetic relationships among Triticum and Aegilops species of the wheat lineage and to establish the timeline of wheat evolution based on gene sequence comparisons. Triticum urartu was confirmed as the A genome donor of tetraploid and hexaploid wheat. The A genome of polyploid wheat diverged from T. urartu less than half a million years ago (MYA), indicating a relatively recent origin of polyploid wheat. The D genome sequences of T. aestivum and Aegilops tauschii are identical, confirming that T. aestivum arose from hybridization of T. turgidum and Ae. tauschii only 8,000 years ago. The diploid Triticum and Aegilops progenitors of the A, B, D, G, and S genomes all radiated 2.5-4.5 MYA. Our data suggest that the Acc-1 and Pgk-1 loci have different histories in different lineages, indicating genome mosaicity and significant intraspecific differentiation. Some loci of the S genome of Aegilops speltoides and the G genome of T. timophevii are closely related, suggesting the same origin of some parts of their genomes. None of the Aegilops genomes analyzed is a close relative of the B genome, so the diploid progenitor of the B genome remains unknown.
[本文引用: 1]
[本文引用: 1]
DOI:10.1126/science.1250092URLPMID:25035499 [本文引用: 4]
The allohexaploid bread wheat genome consists of three closely related subgenomes (A, B, and D), but a clear understanding of their phylogenetic history has been lacking. We used genome assemblies of bread wheat and five diploid relatives to analyze genome-wide samples of gene trees, as well as to estimate evolutionary relatedness and divergence times. We show that the A and B genomes diverged from a common ancestor ~7 million years ago and that these genomes gave rise to the D genome through homoploid hybrid speciation 1 to 2 million years later. Our findings imply that the present-day bread wheat genome is a product of multiple rounds of hybrid speciation (homoploid and polyploid) and lay the foundation for a new framework for understanding the wheat genome as a multilevel phylogenetic mosaic.
DOI:10.3390/genes1020166URL [本文引用: 2]
DOI:10.1534/genetics.111.127688URL [本文引用: 1]
Allopolyploidy has played a prominent role in organismal evolution, particularly in angiosperms. Allohexaploidization is a critical step leading to the formation of common wheat as a new species, Triticum aestivum, as well as for bestowing its remarkable adaptability. A recent study documented that the initial stages of wheat allohexaploidization was associated with rampant genetic and epigenetic instabilities at genomic regions flanking a retrotransposon family named Veju. Although this finding is in line with the prevailing opinion of rapid genomic instability associated with nascent plant allopolyploidy, its relevance to speciation of T. aestivum remains unclear. Here, we show that genetic instability at genomic regions flanking the Veju, flanking a more abundant retroelement BARE-1, as well as at a large number of randomly sampled genomic loci, is all extremely rare or nonexistent in preselected individuals representing three sets of independently formed nascent allohexaploid wheat lines, which had a transgenerationally stable genomic constitution analogous to that of T. aestivum. In contrast, extensive and transgenerationally heritable repatterning of DNA methylation at all three kinds of genomic loci were reproducibly detected. Thus, our results suggest that rampant genetic instability associated with nascent allohexaploidization in wheat likely represents incidental and anomalous phenomena that are confined to by-product individuals inconsequential to the establishment of the newly formed plants toward speciation of T. aestivum; instead, extensive and heritable epigenetic remodeling coupled with preponderant genetic stability is generally associated with nascent wheat allohexaploidy, and therefore, more likely a contributory factor to the speciation event(s).
DOI:10.1073/pnas.1319598110URLPMID:24218593 [本文引用: 2]
Polyploidy or whole-genome duplication is recurrent in plant evolution, yet only a small fraction of whole-genome duplications has led to successful speciation. A major challenge in the establishment of nascent polyploids is sustained karyotype instability, which compromises fitness. The three putative diploid progenitors of bread wheat, with AA, SS (S approximately B), and DD genomes occurred sympatrically, and their cross-fertilization in different combinations may have resulted in fertile allotetraploids with various genomic constitutions. However, only SSAA or closely related genome combinations have led to the speciation of tetraploid wheats like Triticum turgidum and Triticum timopheevii. We analyzed early generations of four newly synthesized allotetraploid wheats with genome compositions S(sh)S(sh)A(m)A(m), S(l)S(l)AA, S(b)S(b)DD, and AADD by combined fluorescence and genomic in situ hybridization-based karyotyping. Results of karyotype analyses showed that although S(sh)S(sh)A(m)A(m) and S(l)S(l)AA are characterized by immediate and persistent karyotype stability, massive aneuploidy and extensive chromosome restructuring are associated with S(b)S(b)DD and AADD in which parental subgenomes showed markedly different propensities for chromosome gain/loss and rearrangements. Although compensating aneuploidy and reciprocal translocation between homeologs prevailed, reproductive fitness was substantially compromised due to chromosome instability. Strikingly, localized genomic changes in repetitive DNA and copy-number variations in gene homologs occurred in both chromosome stable lines, S(sh)S(sh)A(m)A(m) and S(l)S(l)AA. Our data demonstrated that immediate and persistent karyotype stability is intrinsic to newly formed allotetraploid wheat with genome combinations analogous to natural tetraploid wheats. This property, coupled with rapid gene copy-number variations, may have laid the foundation of tetraploid wheat establishment.
DOI:10.1006/jcrs.2000.0336URL [本文引用: 1]
[本文引用: 1]
DOI:10.1023/A:1018361819215URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.2135/cropsci2006.03.0175URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00412-012-0384-7URL [本文引用: 2]
Fluorescence in situ hybridization (FISH) is a useful tool for physical mapping of chromosomes and studying evolutionary chromosome rearrangements. Here we report a robust method for single-copy gene FISH for wheat. FISH probes were developed from cDNA of cytosolic acetyl-CoA carboxylase (ACCase) gene (Acc-2) and mapped on chromosomes of bread wheat, Triticum aestivum L. (2n?=?6x?=?42, AABBDD), and related diploid and tetraploid species. Another nine full-length (FL) cDNA FISH probes were mapped and used to identify chromosomes of wheat species. The Acc-2 probe was detected on the long arms of each of the homoeologous group 3 chromosomes (3A, 3B, and 3D), on 5DL and 4AL of bread wheat, and on homoeologous and nonhomoeologous chromosomes of other species. In the species tested, FISH detected more Acc-2 gene or pseudogene sites than previously found by PCR and Southern hybridization analyses and showed presence/absence polymorphism of Acc-2 sequences. FISH with the Acc-2 probe revealed the 4A–5A translocation, shared by several related diploid and polyploid species and inherited from an ancestral A-genome species, and the T. timopheevii-specific 4At–3At translocation.
DOI:10.1038/22307URLPMID:10421366 [本文引用: 2]
World wheat grain yields increased substantially in the 1960s and 1970s because farmers rapidly adopted the new varieties and cultivation methods of the so-called 'green revolution'. The new varieties are shorter, increase grain yield at the expense of straw biomass, and are more resistant to damage by wind and rain. These wheats are short because they respond abnormally to the plant growth hormone gibberellin. This reduced response to gibberellin is conferred by mutant dwarfing alleles at one of two Reduced height-1 (Rht-B1 and Rht-D1) loci. Here we show that Rht-B1/Rht-D1 and maize dwarf-8 (d8) are orthologues of the Arabidopsis Gibberellin Insensitive (GAI) gene. These genes encode proteins that resemble nuclear transcription factors and contain an SH2-like domain, indicating that phosphotyrosine may participate in gibberellin signalling. Six different orthologous dwarfing mutant alleles encode proteins that are altered in a conserved amino-terminal gibberellin signalling domain. Transgenic rice plants containing a mutant GAI allele give reduced responses to gibberellin and are dwarfed, indicating that mutant GAI orthologues could be used to increase yield in a wide range of crop species.
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
【Objective】 Understanding the distribution of dwarfing genes in Chinese wheat will be crucial for yield improvement.【Method】A total of 239 Chinese wheat cultivars and advanced lines from major wheat regions were detected by STS markers to understand the distribution of the dwarfing genes Rht-B1b (Rht1) and Rht-D1b (Rht2). 【Result】The PCR-based markers could be used to test the presence of Rht-B1b and Rht-D1b in wheat cultivars. The average frequency was 24.3% for Rht-B1b gene and 46.9% for Rht-D1b gene, respectively. Frequencies in Northern Winter Wheat Zone, Yellow & Huai River Facultative Winter Wheat Region, Middle & Low Yangtze Valley Winter Wheat Region, Southwestern Winter Wheat Region, Northeastern Spring Wheat Region, Northern Spring Wheat Region, Northwestern Spring Wheat Region and Xinjiang Winter-Spring Wheat Region were 25.8%, 28%, 42.3%, 8.3%, 0%, 9.1%, 25% and 62.5% for Rht-B1b gene, and 35.5%, 69%, 23.1%, 38.9%, 0%, 72.7%, 37.5% and 12.5% for Rht-1Db gene, respectively. 【Conclusion】Molecular markers and pedigree information confirmed that Rht-B1b is from St2422/464 and Norin 10, Rht-D1b is from Norin 10, Suwon 86, Huixianhong, and Youbaomai.
URL [本文引用: 1]
【Objective】 Understanding the distribution of dwarfing genes in Chinese wheat will be crucial for yield improvement.【Method】A total of 239 Chinese wheat cultivars and advanced lines from major wheat regions were detected by STS markers to understand the distribution of the dwarfing genes Rht-B1b (Rht1) and Rht-D1b (Rht2). 【Result】The PCR-based markers could be used to test the presence of Rht-B1b and Rht-D1b in wheat cultivars. The average frequency was 24.3% for Rht-B1b gene and 46.9% for Rht-D1b gene, respectively. Frequencies in Northern Winter Wheat Zone, Yellow & Huai River Facultative Winter Wheat Region, Middle & Low Yangtze Valley Winter Wheat Region, Southwestern Winter Wheat Region, Northeastern Spring Wheat Region, Northern Spring Wheat Region, Northwestern Spring Wheat Region and Xinjiang Winter-Spring Wheat Region were 25.8%, 28%, 42.3%, 8.3%, 0%, 9.1%, 25% and 62.5% for Rht-B1b gene, and 35.5%, 69%, 23.1%, 38.9%, 0%, 72.7%, 37.5% and 12.5% for Rht-1Db gene, respectively. 【Conclusion】Molecular markers and pedigree information confirmed that Rht-B1b is from St2422/464 and Norin 10, Rht-D1b is from Norin 10, Suwon 86, Huixianhong, and Youbaomai.
DOI:10.1360/TB-2019-0043URL [本文引用: 1]
DOI:10.1038/s41586-018-0415-5URLPMID:30111841 [本文引用: 1]
Enhancing global food security by increasing the productivity of green revolution varieties of cereals risks increasing the collateral environmental damage produced by inorganic nitrogen fertilizers. Improvements in the efficiency of nitrogen use of crops are therefore essential; however, they require an in-depth understanding of the co-regulatory mechanisms that integrate growth, nitrogen assimilation and carbon fixation. Here we show that the balanced opposing activities and physical interactions of the rice GROWTH-REGULATING FACTOR 4 (GRF4) transcription factor and the growth inhibitor DELLA confer homeostatic co-regulation of growth and the metabolism of carbon and nitrogen. GRF4 promotes and integrates nitrogen assimilation, carbon fixation and growth, whereas DELLA inhibits these processes. As a consequence, the accumulation of DELLA that is characteristic of green revolution varieties confers not only yield-enhancing dwarfism, but also reduces the efficiency of nitrogen use. However, the nitrogen-use efficiency of green revolution varieties and grain yield are increased by tipping the GRF4-DELLA balance towards increased GRF4 abundance. Modulation of plant growth and metabolic co-regulation thus enables novel breeding strategies for future sustainable food security and a new green revolution.
DOI:10.1126/science.aan0032URLPMID:28684525 [本文引用: 2]
Wheat (Triticum spp.) is one of the founder crops that likely drove the Neolithic transition to sedentary agrarian societies in the Fertile Crescent more than 10,000 years ago. Identifying genetic modifications underlying wheat's domestication requires knowledge about the genome of its allo-tetraploid progenitor, wild emmer (T. turgidum ssp. dicoccoides). We report a 10.1-gigabase assembly of the 14 chromosomes of wild tetraploid wheat, as well as analyses of gene content, genome architecture, and genetic diversity. With this fully assembled polyploid wheat genome, we identified the causal mutations in Brittle Rachis 1 (TtBtr1) genes controlling shattering, a key domestication trait. A study of genomic diversity among wild and domesticated accessions revealed genomic regions bearing the signature of selection under domestication. This reference assembly will serve as a resource for accelerating the genome-assisted improvement of modern wheat varieties.
DOI:10.1105/tpc.107.051813URLPMID:17586655 [本文引用: 3]
Bread wheat (Triticum aestivum) is a hexaploid species with A, B, and D ancestral genomes. Most bread wheat genes are present in the genome as triplicated homoeologous genes (homoeologs) derived from the ancestral species. Here, we report that both genetic and epigenetic alterations have occurred in the homoeologs of a wheat class E MADS box gene. Two class E genes are identified in wheat, wheat SEPALLATA (WSEP) and wheat LEAFY HULL STERILE1 (WLHS1), which are homologs of Os MADS45 and Os MADS1 in rice (Oryza sativa), respectively. The three wheat homoeologs of WSEP showed similar genomic structures and expression profiles. By contrast, the three homoeologs of WLHS1 showed genetic and epigenetic alterations. The A genome WLHS1 homoeolog (WLHS1-A) had a structural alteration that contained a large novel sequence in place of the K domain sequence. A yeast two-hybrid analysis and a transgenic experiment indicated that the WLHS1-A protein had no apparent function. The B and D genome homoeologs, WLHS1-B and WLHS1-D, respectively, had an intact MADS box gene structure, but WLHS1-B was predominantly silenced by cytosine methylation. Consequently, of the three WLHS1 homoeologs, only WLHS1-D functions in hexaploid wheat. This is a situation where three homoeologs are differentially regulated by genetic and epigenetic mechanisms.
DOI:10.1073/pnas.1217927109URLPMID:23184965 [本文引用: 2]
Wheat supplies about 20% of the total food calories consumed worldwide and is a national staple in many countries. Besides being a key source of plant proteins, it is also a major cause of many diet-induced health issues, especially celiac disease. The only effective treatment for this disease is a total gluten-free diet. The present report describes an effort to develop a natural dietary therapy for this disorder by transcriptional suppression of wheat DEMETER (DME) homeologs using RNA interference. DME encodes a 5-methylcytosine DNA glycosylase responsible for transcriptional derepression of gliadins and low-molecular-weight glutenins (LMWgs) by active demethylation of their promoters in the wheat endosperm. Previous research has demonstrated these proteins to be the major source of immunogenic epitopes. In this research, barley and wheat DME genes were cloned and localized on the syntenous chromosomes. Nucleotide diversity among DME homeologs was studied and used for their virtual transcript profiling. Functional conservation of DME enzyme was confirmed by comparing the motif and domain structure within and across the plant kingdom. Presence and absence of CpG islands in prolamin gene sequences was studied as a hallmark of hypo- and hypermethylation, respectively. Finally the epigenetic influence of DME silencing on accumulation of LMWgs and gliadins was studied using 20 transformants expressing hairpin RNA in their endosperm. These transformants showed up to 85.6% suppression in DME transcript abundance and up to 76.4% reduction in the amount of immunogenic prolamins, demonstrating the possibility of developing wheat varieties compatible for the celiac patients.
[本文引用: 2]
[本文引用: 2]
DOI:10.1038/nprot.2007.310URLPMID:17853881 [本文引用: 1]
Flow cytometry (FCM) using DNA-selective fluorochromes is now the prevailing method for the measurement of nuclear DNA content in plants. Ease of sample preparation and high sample throughput make it generally better suited than other methods such as Feulgen densitometry to estimate genome size, level of generative polyploidy, nuclear replication state and endopolyploidy (polysomaty). Here we present four protocols for sample preparation (suspensions of intact cell nuclei) and describe the analysis of nuclear DNA amounts using FCM. We consider the chemicals and equipment necessary, the measurement process, data analysis, and describe the most frequent problems encountered with plant material such as the interference of secondary metabolites. The purpose and requirement of internal and external standardization are discussed. The importance of using a correct terminology for DNA amounts and genome size is underlined, and its basic principles are explained.
DOI:10.1038/nature11650URL [本文引用: 1]
Bread wheat (Triticum aestivum) is a globally important crop, accounting for 20 per cent of the calories consumed by humans. Major efforts are underway worldwide to increase wheat production by extending genetic diversity and analysing key traits, and genomic resources can accelerate progress. But so far the very large size and polyploid complexity of the bread wheat genome have been substantial barriers to genome analysis. Here we report the sequencing of its large, 17-gigabase-pair, hexaploid genome using 454 pyrosequencing, and comparison of this with the sequences of diploid ancestral and progenitor genomes. We identified between 94,000 and 96,000 genes, and assigned two-thirds to the three component genomes (A, B and D) of hexaploid wheat. High-resolution synteny maps identified many small disruptions to conserved gene order. We show that the hexaploid genome is highly dynamic, with significant loss of gene family members on polyploidization and domestication, and an abundance of gene fragments. Several classes of genes involved in energy harvesting, metabolism and growth are among expanded gene families that could be associated with crop productivity. Our analyses, coupled with the identification of extensive genetic variation, provide a resource for accelerating gene discovery and improving this major crop.
DOI:10.1101/gr.217117.116URLPMID:28420692 [本文引用: 1]
Advances in genome sequencing and assembly technologies are generating many high-quality genome sequences, but assemblies of large, repeat-rich polyploid genomes, such as that of bread wheat, remain fragmented and incomplete. We have generated a new wheat whole-genome shotgun sequence assembly using a combination of optimized data types and an assembly algorithm designed to deal with large and complex genomes. The new assembly represents >78% of the genome with a scaffold N50 of 88.8 kb that has a high fidelity to the input data. Our new annotation combines strand-specific Illumina RNA-seq and Pacific Biosciences (PacBio) full-length cDNAs to identify 104,091 high-confidence protein-coding genes and 10,156 noncoding RNA genes. We confirmed three known and identified one novel genome rearrangements. Our approach enables the rapid and scalable assembly of wheat genomes, the identification of structural variants, and the definition of complete gene models, all powerful resources for trait analysis and breeding of this key global crop.
DOI:10.1093/gigascience/gix093URLPMID:29048480 [本文引用: 1]
Ginseng, which contains ginsenosides as bioactive compounds, has been regarded as an important traditional medicine for several millennia. However, the genetic background of ginseng remains poorly understood, partly because of the plant's large and complex genome composition. We report the entire genome sequence of Panax ginseng using next-generation sequencing. The 3.5-Gb nucleotide sequence contains more than 60% repeats and encodes 42 006 predicted genes. Twenty-two transcriptome datasets and mass spectrometry images of ginseng roots were adopted to precisely quantify the functional genes. Thirty-one genes were identified to be involved in the mevalonic acid pathway. Eight of these genes were annotated as 3-hydroxy-3-methylglutaryl-CoA reductases, which displayed diverse structures and expression characteristics. A total of 225 UDP-glycosyltransferases (UGTs) were identified, and these UGTs accounted for one of the largest gene families of ginseng. Tandem repeats contributed to the duplication and divergence of UGTs. Molecular modeling of UGTs in the 71st, 74th, and 94th families revealed a regiospecific conserved motif located at the N-terminus. Molecular docking predicted that this motif captures ginsenoside precursors. The ginseng genome represents a valuable resource for understanding and improving the breeding, cultivation, and synthesis biology of this key herb.
DOI:10.1038/nature11997URL [本文引用: 1]
Bread wheat (Triticum aestivum, AABBDD) is one of the most widely cultivated and consumed food crops in the world. However, the complex polyploid nature of its genome makes genetic and functional analyses extremely challenging. The A genome, as a basic genome of bread wheat and other polyploid wheats, for example, T. turgidum (AABB), T. timopheevii (AAGG) and T. zhukovskyi (AAGGA(m)A(m)), is central to wheat evolution, domestication and genetic improvement(1). The progenitor species of the A genome is the diploid wild einkorn wheat T. urartu(2), which resembles cultivated wheat more extensively than do Aegilops speltoides (the ancestor of the B genome(3)) and Ae. tauschii (the donor of the D genome(4)), especially in the morphology and development of spike and seed. Here we present the generation, assembly and analysis of a whole-genome shotgun draft sequence of the T. urartu genome. We identified protein-coding gene models, performed genome structure analyses and assessed its utility for analysing agronomically important genes and for developing molecular markers. Our T. urartu genome assembly provides a diploid reference for analysis of polyploid wheat genomes and is a valuable resource for the genetic improvement of wheat.
DOI:10.1038/s41586-018-0108-0URLPMID:29743678 [本文引用: 1]
Triticum urartu (diploid, AA) is the progenitor of the A subgenome of tetraploid (Triticum turgidum, AABB) and hexaploid (Triticum aestivum, AABBDD) wheat(1,2). Genomic studies of T. urartu have been useful for investigating the structure, function and evolution of polyploid wheat genomes. Here we report the generation of a high-quality genome sequence of T. urartu by combining bacterial artificial chromosome (BAC)-by-BAC sequencing, single molecule real-time whole-genome shotgun sequencing (3) , linked reads and optical mapping(4,5). We assembled seven chromosome-scale pseudomolecules and identified protein-coding genes, and we suggest a model for the evolution of T. urartu chromosomes. Comparative analyses with genomes of other grasses showed gene loss and amplification in the numbers of transposable elements in the T. urartu genome. Population genomics analysis of 147 T. urartu accessions from across the Fertile Crescent showed clustering of three groups, with differences in altitude and biostress, such as powdery mildew disease. The T. urartu genome assembly provides a valuable resource for studying genetic variation in wheat and related grasses, and promises to facilitate the discovery of genes that could be useful for wheat improvement.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nature24486URLPMID:29143815 [本文引用: 1]
Aegilops tauschii is the diploid progenitor of the D genome of hexaploid wheat (Triticum aestivum, genomes AABBDD) and an important genetic resource for wheat. The large size and highly repetitive nature of the Ae. tauschii genome has until now precluded the development of a reference-quality genome sequence. Here we use an array of advanced technologies, including ordered-clone genome sequencing, whole-genome shotgun sequencing, and BioNano optical genome mapping, to generate a reference-quality genome sequence for Ae. tauschii ssp. strangulata accession AL8/78, which is closely related to the wheat D genome. We show that compared to other sequenced plant genomes, including a much larger conifer genome, the Ae. tauschii genome contains unprecedented amounts of very similar repeated sequences. Our genome comparisons reveal that the Ae. tauschii genome has a greater number of dispersed duplicated genes than other sequenced genomes and its chromosomes have been structurally evolving an order of magnitude faster than those of other grass genomes. The decay of colinearity with other grass genomes correlates with recombination rates along chromosomes. We propose that the vast amounts of very similar repeated sequences cause frequent errors in recombination and lead to gene duplications and structural chromosome changes that drive fast genome evolution.
DOI:10.1126/science.361.6403.636URLPMID:30115791 [本文引用: 1]
DOI:10.3389/fpls.2020.00197URLPMID:32194596 [本文引用: 2]
Untangling the genetic architecture of grain yield (GY) and yield stability is an important determining factor to optimize genomics-assisted selection strategies in wheat. We conducted in-depth investigation on the above using a large set of advanced bread wheat lines (4,302), which were genotyped with genotyping-by-sequencing markers and phenotyped under contrasting (irrigated and stress) environments. Haplotypes-based genome-wide-association study (GWAS) identified 58 associations with GY and 15 with superiority index Pi (measure of stability). Sixteen associations with GY were
DOI:10.3389/fpls.2018.01584URLPMID:30429867 [本文引用: 2]
Rapid detection of allelic variation and identification of advantage haplotypes responsible for spike related traits play a crucial role in wheat yield improvement. The released genome sequence of hexaploid wheat (Chinese Spring) provides an extraordinary opportunity for rapid detection of natural variation and promotes breeding application. Here, selection signals detection and genome-wide association study (GWAS) were conducted for spike related traits. Based on the genotyping results by 90K SNP chip, 192 common wheat samples from southwest China were analyzed. One hundred and forty-six selective windows and one hundred and eighty-four significant SNPs (51 for spike length, 28 for kernels per spike, 39 for spikelet number, 30 for thousand kernel weight, and 36 for spike number per plant) were detected. Furthermore, tightly linkage and environmental stability window clusters and SNP clusters were also obtained. As a result, four SNP clusters associated with spike length were detected on chromosome 2A, 2B, 2D, and 6A. Two SNP clusters correlated to kernels per spike were detected on 2A and 2B. One pleiotropy SNP cluster correlated to spikelet number and kernels per spike was detected on 7B. According to the genome sequence, these SNP clusters and their overlapped/flanking QTLs which have been reported previously were integrated to a physical map. The candidate genes responsible for spike length, kernels per spike and spikelet number were predicted. Based on the genotypes of cultivars in south China, two advantage haplotypes associated with spike length and one advantage haplotype associated with kernels per spike/spikelet number were detected which have not been effectively transited into cultivars. According to these haplotypes, KASP markers were developed and diagnosed across landraces and cultivars which were selected from south and north China. Consequently, KASP assay, consistent with the GWAS results, provides reliable haplotypes for MAS in wheat yield improvement.
DOI:10.1038/s41598-020-60203-2URLPMID:32099054 [本文引用: 2]
Genome-wide association study (GWAS) and genomic prediction (GP) are extensively employed to accelerate genetic gain and identify QTL in plant breeding. In this study, 1,317 spring barley and 1,325 winter wheat breeding lines from a commercial breeding program were genotyped with the Illumina 9 K barley or 15 K wheat SNP-chip, and phenotyped in multiple years and locations. For GWAS, in spring barley, a QTL on chr. 4H associated with powdery mildew and ramularia resistance were found. There were several SNPs on chr. 4H showing genome-wide significance with yield traits. In winter wheat, GWAS identified two SNPs on chr. 6A, and one SNP on chr. 1B, significantly associated with quality trait moisture, as well as one SNP located on chr. 5B associated with starch content in the seeds. The significant SNPs identified by multiple trait GWAS were generally the same as those found in single trait GWAS. GWAS including genotype-location information in the model identified significant SNPs in each tested location, which were not found previously when including all locations in the GWAS. For GP, in spring barley, GP using the Bayesian Power Lasso model had higher accuracy than ridge regression BLUP in powdery mildew and yield traits, whereas the prediction accuracies were similar using Bayesian Power Lasso model and rrBLUP for yield traits in winter wheat.
DOI:10.3389/fpls.2020.00748URLPMID:32582265 [本文引用: 2]
Among several important wheat foliar diseases, Stripe rust (YR), Leaf rust (LR), and Stem rust (SR) have always been an issue of concern to the farmers and wheat breeders. Evolution of virulent pathotypes of these rusts has posed frequent threats to an epidemic. Pyramiding rust-resistant genes are the most economical and environment-friendly approach in postponing this inevitable threat. To achieve durable long term resistance against the three rusts, an attempt in this study was made searching for novel sources of resistant alleles in a panel of 483 spring wheat genotypes. This is a unique and comprehensive study where evaluation of a diverse panel comprising wheat germplasm from various categories and adapted to different wheat agro-climatic zones was challenged with 18 pathotypes of the three rusts with simultaneous screening in field conditions. The panel was genotyped using 35K SNP array and evaluated for each rust at two locations for two consecutive crop seasons. High heritability estimates of disease response were observed between environments for each rust type. A significant effect of population structure in the panel was visible in the disease response. Using a compressed mixed linear model approach, 25 genomic regions were found associated with resistance for at least two rusts. Out of these, seven were associated with all the three rusts on chromosome groups 1 and 6 along with 2B. For resistance against YR, LR, and SR, there were 16, 18, and 27 QTL (quantitative trait loci) identified respectively, associated at least in two out of four environments. Several of these regions got annotated with resistance associated genes viz. NB-LRR, E3-ubiquitin protein ligase, ABC transporter protein, etc. Alien introgressed (on 1B and 3D) and pleiotropic (on 7D) resistance genes were captured in seedling and adult plant disease responses, respectively. The present study demonstrates the use of genome-wide association for identification of a large number of favorable alleles for leaf, stripe, and stem rust resistance for broadening the genetic base. Quick conversion of these QTL into user-friendly markers will accelerate the deployment of these resistance loci in wheat breeding programs.
DOI:10.1038/s41598-020-62711-7URLPMID:32265455 [本文引用: 1]
This study was initiated to identify genomic regions conferring resistance to Karnal Bunt (KB) disease in wheat through a genome-wide association study (GWAS) on a set of 179 pre-breeding lines (PBLs). A GWAS of 6,382 high-quality DArTseq SNPs revealed 15 significant SNPs (P-value <10(-3)) on chromosomes 2D, 3B, 4D and 7B that were associated with KB resistance in individual years. In particular, two SNPs (chromosome 4D) had the maximum R(2) values: SNP 1114200 | F | 0-63:T > C at 1.571 cM and R(2) of 12.49% and SNP 1103052 | F | 0-61:C > A at 1.574 cM and R(2) of 9.02%. These two SNPs displayed strong linkage disequilibrium (LD). An in silico analysis of SNPs on chromosome 4D identified two candidate gene hits, TraesCS4D02G352200 (TaNox8; an NADPH oxidase) and TraesCS4D02G350300 (a rhomboid-like protein belonging to family S54), with SNPs 1103052 | F | 0-61:C > A and 1101835 | F | 0-5:C > A, respectively, both of which function in biotic stress tolerance. The epistatic interaction analysis revealed significant interactions among 4D and 7B loci. A pedigree analysis of confirmed resistant PBLs revealed that Aegilops species is one of the parents and contributed the D genome in these resistant PBLs. These identified lines can be crossed with any elite cultivar across the globe to incorporate novel KB resistance identified on 4B.
DOI:10.1007/s00122-020-03577-1URLPMID:32172298 [本文引用: 2]
KEY MESSAGE: Genome-wide association study (GWAS) on 358 Chinese wheat germplasms and validation in a biparental population identified a novel significant genomic region on 5DL for FCR resistance. Fusarium crown rot (FCR) is a chronic and severe disease in many dryland wheat-producing areas worldwide. In the last few years, the incidence and severity of FCR progressively increased in China, and the disease has currently become a new threat to local wheat crops. Here, we report a genome-wide association study (GWAS) on a set of 358 Chinese germplasms with the wheat 55 K SNP array. A total of 104 SNPs on chromosomes 1BS, 1DS, 2AL, 5AL, 5DS, 5DL, 6BS and 7BL were significantly associated with seedling resistance to FCR in the association panel. Of these SNPs, a novel 13.78 Mb region targeted by five SNPs on chromosome arm 5DL was continually detected in all three trials. The effects of this region on FCR resistance was confirmed in biparental population. qRT-PCR showed that within this 5DL region, several genes encoding TIR-NBS-LRR proteins and proteins related to mycotoxins deoxynivalenol (DON) detoxification increased rapidly in the disease-resistant variety 04 Zhong 36 than the susceptible variety Xinmai 26 after inoculation. Our study provides new insights into gene discovery and creation of new cultivars with desirable alleles for improving FCR resistance in wheat.
DOI:10.1126/science.264.5157.421URLPMID:17836906 [本文引用: 1]
Three published estimates of the frequency of polyploidy in angiosperms (30 to 35 percent, 47 percent, and 70 to 80 percent) were tested by estimating the genome size of extinct woody angiosperms with the use of fossil guard cell size as a proxy for cellular DNA content. The inferred chromosome numbers of these extinct species suggest that seven to nine is the primitive haploid chromosome number of angiosperms and that most angiosperms (approximately 70 percent) have polyploidy in their history.
[本文引用: 3]
[本文引用: 3]

{kind=link}
{kind=link}