 ,1
,1Transcriptome heterogeneity of porcine ear fibroblast and its potential influence on embryo development in nuclear transplantation
Jun Zhou1, Chengcheng Zhao1, Xiao Wu1, Junsong Shi2, Rong Zhou2, Zhenfang Wu1, Zicong Li,1通讯作者: 李紫聪,教授,博士生导师,研究方向:动物遗传育种与繁殖。E-mail:lizicongcong@163.com
责任编辑: 赵要风
收稿日期:2020-06-22修回日期:2020-08-11网络出版日期:2020-09-20
| 基金资助: |
Received:2020-06-22Revised:2020-08-11Online:2020-09-20
| Fund supported: |
作者简介 About authors
周俊,在读硕士研究生,专业方向:动物遗传育种与繁殖。E-mail:

摘要
同一来源的供体细胞之间存在异质性。许多研究已经表明体细胞核移植(somatic cell nuclear transfer, SCNT)效率与供体细胞有关。然而,鲜有在单细胞水平分析供体细胞异质性对核移植效率的潜在影响。本研究利用单细胞转录组测序技术对同一来源且随机挑选的52个猪耳组织成纤维细胞进行测序分析。结果表明有48个单细胞的基因表达模式相似,4个单细胞(编号为D11_1、D12_1、DW61_2和DW99_2)的基因表达模式与其他单细胞存在较大的差异,并且不存在基因表达模式完全相同的两个单细胞。以基因表达模式相似的48个单细胞作为对照,进一步分析了单细胞D11_1、D12_1、DW61_2和DW99_2的差异基因表达模式:首先利用R语言筛选4个单细胞的差异表达基因,并对前50差异表达基因进行汇总;然后对差异表达基因进行GO富集分析和KEGG通路分析。富集分析发现差异表达基因的主要分子功能包括能量代谢、蛋白质代谢和细胞对刺激的反应等;主要通路包括KEGG中富集的与细胞周期、细胞代谢、DNA复制相关的通路。根据以上研究结果并结合SCNT研究进展讨论了4个单细胞的差异基因表达模式对核移植胚胎发育效率的潜在影响。本研究揭示了猪耳组织成纤维细胞的转录组异质性,并提供了分析精英供体细胞的一种有效方法,为提高克隆效率带来新的思路。
关键词:
Abstract
There is heterogeneity among donor cells of the same source. Many studies have shown that donor cell affects the efficiency of somatic cell nuclear transfer (SCNT). However, the potential influence of donor cell heterogeneity on the efficiency of nuclear transplantation were rarely analyzed at the single-cell level. In this study, single-cell transcriptome sequencing was performed on 52 porcine ear fibroblasts randomly selected from the same source to compare their gene expression patterns. The results showed that 48 cells had similar gene expression patterns, whereas 4 cells (D11_1, D12_1, DW61_2, DW99_2) had significantly different gene expression patterns from those of other cells. There were no two cells with identical gene expression patterns. The gene expression patterns of D11_1, D12_1, DW61_2 and DW99_2 were analyzed, using the 48 cells with similar gene expression patterns as controls. Firstly, we used the R language statistics to select the differentially expressed genes in the 4 single cells, and identified the top 50 most significant differentially expressed genes. Then GO enrichment analysis and KEGG pathway analysis were performed on the differentially expressed genes. Enrichment analysis revealed that the main molecular functions of the differentially expressed genes included energy metabolism, protein metabolism and cell response to stimulation. The main pathways from KEGG enrichment were related to cell cycle, cell metabolism, and DNA replication. Finally, based on the above results and in consideration with the SCNT research progress, we discussed the potential effects of differential gene expression patterns of the 4 single cells on the embryonic development efficiency of nuclear transplantation. This study revealed transcriptional heterogeneity of porcine ear tissue fibroblasts and provided an effective method to analyze elite donor cells, thereby providing new ideas on improving the cloning efficiency of SCNT.
Keywords:
PDF (1479KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
周俊, 赵成成, 吴霄, 石俊松, 周荣, 吴珍芳, 李紫聪. 猪耳成纤维细胞转录组异质性及对核移植胚胎发育的潜在影响. 遗传[J], 2020, 42(9): 898-915 doi:10.16288/j.yczz.20-190
Jun Zhou.
体细胞核移植技术(somatic cell nuclear transfer, SCNT)又称为体细胞克隆技术。1997年,英国胚胎学家Wilmut利用SCNT技术成功克隆出第一只哺乳动物—克隆羊“多莉”[1],随后,通过该技术先后诞生了牛(Bos taurus)[2]、猪(Sus scrofa)[3]、猴(Macaca mulatta)[4]等20余种哺乳动物。SCNT技术在保护濒危物种[5]、优秀种繁扩繁[6]以及再生医学[7]等领域表现出巨大的应用价值,是生命科学研究的重要成果。但是,SCNT技术同样面临很多问题:以猪为例,体外培养条件下,囊胚率仅为20%左右,在不同品种或者卵母细胞质量差的情况下可能更低。即便胚胎成功附植,相较于体外受精(in vitro fertilization, IVF)胚胎,SCNT胚胎发育能力差、在受孕母猪体内发育异常的现象也十分普遍[8]。除此之外,大多数实验条件下,克隆猪的出生率只有1%左右,远低于人工授精(artificial insemination, AI)猪的出生效率(80%)[9],严重限制了SCNT技术的应用和普及。SCNT技术基础是将供体细胞移入去核的成熟卵母细胞中,最终发育成和供体细胞基因型相同的后代。因此供体细胞是影响体细胞核移植效率的关键因素。已有研究表明供体细胞会对SCNT效率产生影响:供体细胞的细胞周期[10]、传代数[11]以及性别[12]的不同都会导致克隆胚胎发育效率出现差异。因此,选择合适的供体细胞对提高SCNT胚胎发育效率十分重要。
供体细胞核在成熟的去核卵母细胞中异常的表观遗传重编程被认为是阻碍SCNT发育的主要原因[13,14]。而更容易被正确重编程的核供体细胞很可能是具有更大发育潜力的精英供体细胞。Zhai等[15]分别以猪骨髓间充质干细胞和猪胎儿成纤维细胞作为供体细胞进行核移植实验,发现发育效率较高的骨髓间充质干细胞含有更多的有利于重编程的表观遗传标记和较少的抑制重编程的表观遗传标记,暗示容易被正确重编程的供体细胞在SCNT过程中会有更好的发育潜能。同时,Yamanaka等[16]在鼠源诱导多能干细胞研究中就已经提出精英供体细胞的概念:同一来源的细胞中会存在一些更容易被正确重编程、发育潜力更大的精英供体细胞;同样,在克隆小鼠(Mus musculus)的研究中也发现,与其他品系的小鼠相比,129小鼠的基因组状态更不稳定,更容易被激活或抑制。用野生基因型和129小鼠基因型杂交得到的重组细胞做供体,可显著提高克隆动物的出生率[17],说明精英供体细胞具有独特的基因表达模式,只是其分子特征尚未探索清楚。另有研究显示,从供体细胞遗传而来的一些转录记忆可以导致克隆胚胎的发育缺陷[18],表明供体细胞在一定程度上决定了重构胚胎的发育命运。供体细胞的异质性是指供体细胞在基因组或表型水平上具有的不同特征。同时,因为细胞之间异质性的存在,不同的供体细胞发育潜力是有差异的。即便同一来源的供体细胞也存在更有利于胚胎发育的精英供体细胞。但是在实际研究中,同一来源的供体细胞之间表型特征差异不明显,分子层面的特征信息丢失严重,缺乏对常见供体细胞异质性的深入研究。
近年来,随着测序技术的发展,特别是低输入测序技术为体细胞核移植的研究提供了更多可能。而单细胞测序技术的诞生及发展[19,20,21]为人们进一步探究供体细胞之间的分子事件提供了便利。应用单细胞测序技术,可以以更精准的分辨率揭示供体细胞的异质性。基于此,本研究对来源相同的52个猪耳组织成纤维细胞进行单细胞转录组测序分析,发现了48个基因表达模式相似的“普通”细胞,4个基因表达模式互不相似且都与“普通”细胞的基因表达模式存在显著差异的“另类”细胞,揭示了猪耳组织成纤维细胞的转录组异质性,并结合已有研究结果讨论了供体细胞转录组异质性对核移植效率的潜在影响,为后续通过深入研究寻找精英供体细胞来提高猪体细胞核移植效率提供基础。
1 材料与方法
1.1 材料
杜洛克公猪(373日龄)耳组织样品由广东温氏食品集团华农温氏股份有限公司提供;总RNA提取试剂盒SMART-SeqTM v4 UltraTM Low Input RNA Kit for Sequencing购于北京诺禾致源生物信息科技有限公司;胎牛血清(fetal bovine serum, FBS)、氨基酸葡萄糖培养基(dulbecco's modified eagle medium, DMEM)、0.25%胰蛋白酶和乙二胺四乙酸均购自美国Gibco公司。单细胞转录组测序由北京诺禾致源生物信息科技有限公司在illumina平台完成。图片处理使用Photoshop 7.0/ACDSee 9.0软件;spliced reads比对使用HISAT软件[22];聚类分析、基因差异表达分析、GO富集分析和主成分分析(principal component analysis, PCA)均采用R语言,其中聚类分析使用软件包pheatmap,基因差异表达分析使用软件包DEGSeq (1.12.0)[23],富集分析采用软件包GOseq[24];KEGG富集使用软件KOBAS (2.0)。1.2 供体细胞单细胞培养与分离
将成年优良杜洛克种公猪的耳组织样品剪碎,用PBS洗涤两次之后,在100 mm的培养皿上用手术刀和剪子切碎,先用DMEM重悬,再用胰蛋白酶和乙二胺四乙酸消化1~2 h。将胰蛋白酶消化的细胞洗涤一次后,以300 g的离心率离心10 min,并将其接种于100 mm的细胞培养皿中,放入15% FBS和10 mg/L的青霉素-链霉素溶液的DMEM。在39℃、5%CO2饱和湿度的恒温培养箱中培养6~8 d。然后移除未附着的组织块,再将附着的细胞培养直至汇合,期间每隔3~7 d更换DMEM。最后利用显微操作法,在体视显微镜下吸取单个细胞,放于盛有裂解液的去DNase-RNase的离心管中,用于单细胞转录组测序。1.3 单细胞转录组数据获得
1.3.1 总RNA提取每个细胞样品保存在6 μL SMART-SeqTM v4 kit裂解液(北京诺禾致源生物信息科技有限公司)中。细胞样品经过体积测量后,使用SMART-SeqTM v4 UltraTM Low Input RNA Kit for Sequencing试剂盒(美国Clontech公司)进行细胞裂解,提取总RNA,并保存在RNase-Free水中。
1.3.2 单细胞cDNA文库构建及测序
对提取的总RNA直接进行First-stand cDNA的合成,然后对First-stand cDNA进行全长LD-PCR的扩增,利用AMPure XP beads纯化扩增后的双链cDNA (double-standed DNA, ds cDNA),使用Qubit进行ds cDNA定量检测;使用Covaris系统对ds cDNA进行超声打断,打断后的双链短片段进行末端修复、加A尾并连接测序接头,然后用AMPure XP beads纯化并选择片段大小在200 bp左右的文库;最后进行PCR富集得到最终的cDNA文库。使用Qubit2.0对文库进行初步定量,稀释文库至1 ng/μL,使用Agilent 2100对文库的插入片段长度进行检测。插入片段符合预期后,使用qPCR方法对文库的有效浓度进行准确定量,以保证文库质量。库检合格后,将不同文库按照有效浓度及目标下机数据量的需求合并后进行HiSeq测序。本研究最后扩增成功且完成单细胞测序的样品为52个,细胞样品编号见表1。
Table 1
表1
表1扩增成功并测序的单细胞样品
Table 1
| 序号 | 样品编号 | 序号 | 样品编号 | 序号 | 样品编号 |
|---|---|---|---|---|---|
| 1 | D1_1 | 19 | D31_1 | 37 | DW22_2 |
| 2 | D1_3 | 20 | D32_3 | 38 | DW24_1 |
| 3 | D8_2 | 21 | D33_1 | 39 | DW31_1 |
| 4 | D9_2 | 22 | D36_3 | 40 | DW36_1 |
| 5 | D11_1 | 23 | D37_3 | 41 | DW36_2 |
| 6 | D12_1 | 24 | D40_2 | 42 | DW41_2 |
| 7 | D12_2 | 25 | D40_3 | 43 | DW45_1 |
| 8 | D13_1 | 26 | D43_3 | 44 | DW45_2 |
| 9 | D18_3 | 27 | D44_1 | 45 | DW58_2 |
| 10 | D20_1 | 28 | D45_3 | 46 | DW61_1 |
| 11 | D21_1 | 29 | D48_1 | 47 | DW61_2 |
| 12 | D22_1 | 30 | D52_3 | 48 | DW69_1 |
| 13 | D23_3 | 31 | D63_1 | 49 | DW69_2 |
| 14 | D25_1 | 32 | D63_2 | 50 | DW73-1 |
| 15 | D26_1 | 33 | D64_1 | 51 | DW99_1 |
| 16 | D27_1 | 34 | D66_1 | 52 | DW99_2 |
| 17 | D28_1 | 35 | DW16_1 | ||
| 18 | D28_2 | 36 | DW22_1 |
新窗口打开|下载CSV
1.4 单细胞转录组数据分析
1.4.1 测序数据质量控制fastq格式的原始数据先通过内部perl脚本进行处理。在此步骤中,删除包含适配器的reads、包含ploy-N的低质量的原始数据来获得干净的数据(clean reads),同时对Q20、Q30和GC内容进行计算,所有的下游分析都是基于高质量的清洁数据。1.4.2 差异基因表达分析参考基因组和基因模型注释文件直接从基因组网(http://www.ensembl.org/)下载。选取HISAT软件将过滤后的测序序列进行基因组定位分析。HISAT能够有效的比对到RNA-Seq测序数据中的spliced reads,是目前比对率最高且最准确的比对软件。先使用Hisat2 v2.0.4作为映射工具,它可以基于基因模型注释文件生成一个拼接连接的数据库,因此比其他非拼接映射工具具有更好的映射结果。然后使用软件HTSeq v0.9.1计算映射到每个基因的读取数字。然后根据基因的长度计算出每个基因的FPKM,并读取到该基因的计数。在进行差异基因表达分析之前,通过edgeR程序包[25]对每一个序列库标准化。利用R语言中的DEGSeq (1.20.0)软件包进行微分表达式分析并绘制4个“另类”细胞差异基因火山图、聚类软件包pheatmap绘制4个“另类”细胞差异基因聚类图。P值用Benjamini 和 Hochberg法进行调整,修正的P值为0.005和log2(fold change)为1,为显著差异表达的阈值。1.4.3 GO富集分析和KEGG通路分析通过R语言中的GOseq软件包对筛选得到的差异基因进行GO富集,展示差异基因在Gene Ontology (2 结果与分析
2.1 单细胞转录组测序揭示猪耳组织成纤维细胞间的异质性
按照PCA的前3个主要影响因素两两组合,对成功扩增且最终完成测序的52个单细胞进行主成分分析(图1),其中Dim1、Dim2、Dim3分别代表影响单细胞样本在图中相对位置的3个主要因素,相对位置较近的细胞表示基因表达模式相似。结果表明,D11_1、D12_1、DW61_2和DW99_2在图1中的位置与其他单细胞相比较“离群”,说明这4个细胞的基因表达模式与其他单细胞存在较大差异。同时将52个耳组织成纤维细胞的基因表达总体情况绘制成热图(图2),结果表明,在同一来源的猪耳成纤维细胞中,同一基因在不同细胞间的表达情况并不相同,并不存在基因表达模式完全一致的两个细胞,不同供体细胞之间存在转录组异质性。图1
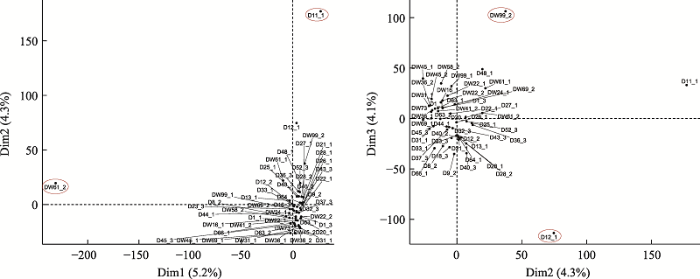 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图152个单细胞转录组主成分分析
每个点代表一个单细胞样品;横、纵坐标轴的刻度是相对距离,无实际意义;Dim1、Dim2、Dim3后的百分比代表横、纵的差异可以解释全面分析结果的百分比。红色圈出部分分别为单细胞D11_1、DW61_2、D12_1和DW99_2。
Fig. 1Principal component analysis of 52 single cell transcriptomes
图2
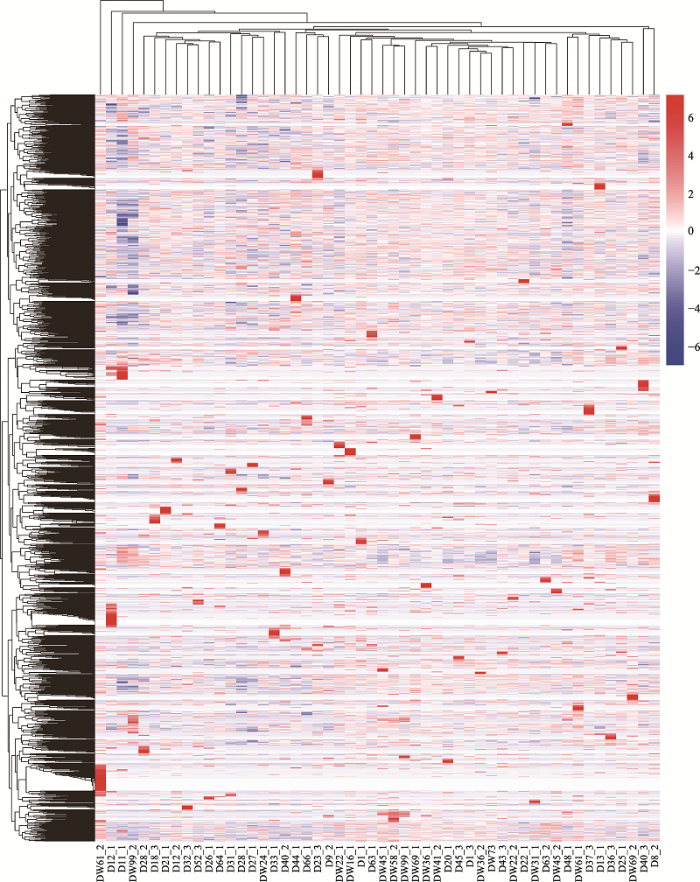 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图252个单细胞基因表达量热图
红色代表基因高表达,蓝色代表基因低表达。
Fig. 2Heat map of 52 single cell transcriptomes
2.2 D11_1、D12_1、DW61_2和DW99_2基因表达模式分析
将基因表达模式差异不明显的“普通”细胞作为对照组,使用R语言中的软件包DEGSeq (1.12.0)对D11_1、D12_1、DW61_2和DW99_2进行差异基因表达分析。D11_1与对照组比较,有1860个差异表达显著的基因,其中上调表达的基因有458个,下调表达的基因有1402个;D12_1与对照组比较, 有376个差异表达显著的基因,其中上调表达的基因有109个,下调表达的基因有267个;DW61_2与对照组比较,有330个差异表达显著的基因,其中上调表达的基因有277个,下调表达的基因有53个;DW99_2与对照组比较,有2225个差异表达显著的基因,其中上调表达的基因有316个,下调表达的基因有1909个。使用R语言中软件包DEGSeq (1.12.0)将4个“另类”细胞的差异基因的整体分布情况可视化(图3),使用R语言中的聚类软件包pheatmap将4个“另类”细胞的差异基因整体表达情况可视化(图4)。同时,将用软件包DEGSeq (1.12.0)筛选出的D11_1、D12_1、DW61_1和DW99_2前50个差异最显著的基因进行汇总:D11_1前50个差异最显著的基因功能主要集中于细胞增殖分化过程中一些有机物质的合成与能量代谢过程(表2);D12_1前50个差异最显著的基因功能主要集中于蛋白质及葡萄糖转运等过程(表3);DW61_2前50个差异最显著的基因功能比较多样,但有个别基因涉及转移酶活性(表4);DW99_2前50个差异最显著的基因功能主要集中于蛋白质编码、核酸修复与能量代谢方面(表5)。图3
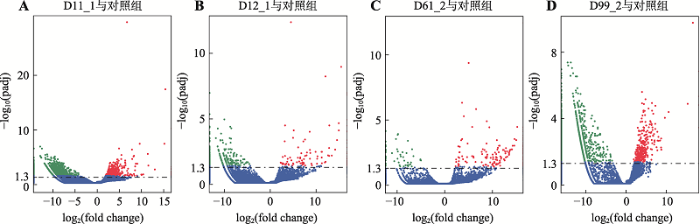 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3D11_1、D12_1、DW61_2和DW99_2差异基因火山图
A~D分别为D11_1、D12_1、DW61_2和DW99_2的差异基因火山图;有显著性差异表达的基因用红色点(上调)和绿色点(下调)表示,无显著性差异表达的基因用蓝色点表示;横坐标代表基因在不同样本中表达倍数变化;纵坐标代表基因表达量变化差异的统计学显著性;筛选标准padj < 0.05。
Fig. 3D11_1, D12_1, DW61_2 andDW99_2 differential genes expression volcano map
图4
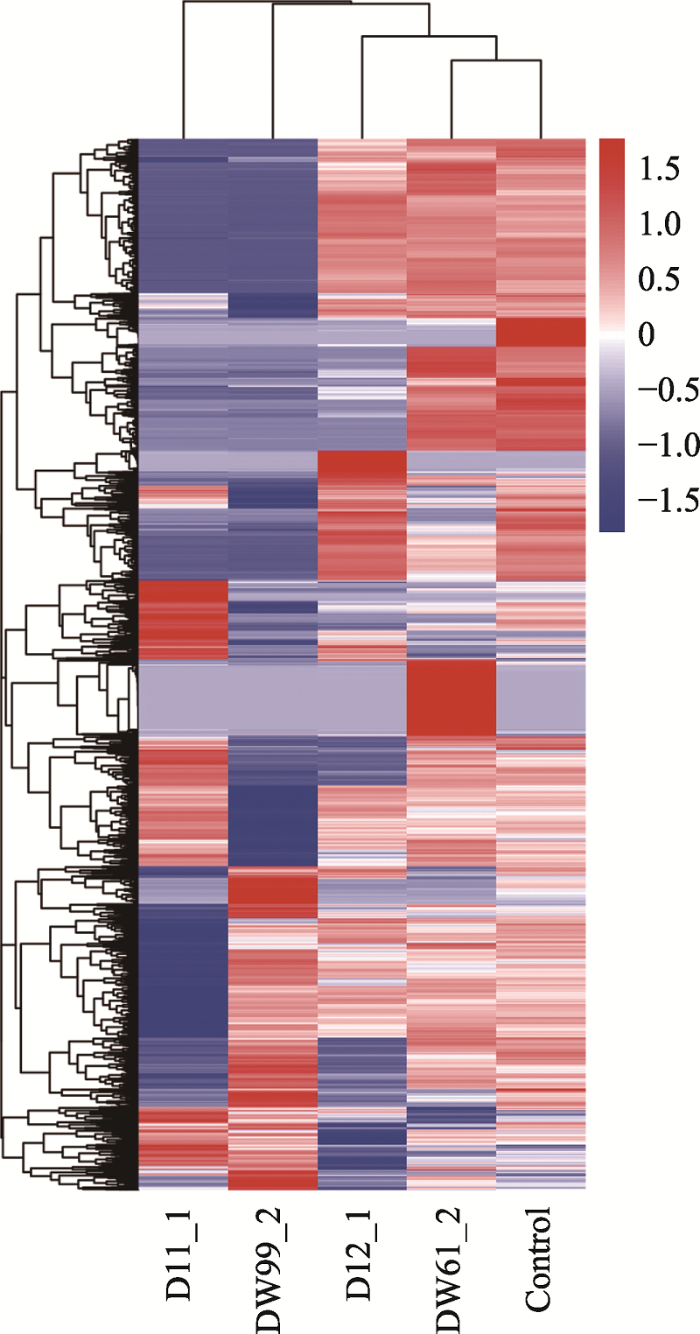 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4D11_1、D12_1、DW61_2和DW99_2差异基因聚类热图
整体FPKM层次聚类图,将log10(FPKM+1)值进行归一化转换(scale number)并进行聚类,红色表示高表达基因,蓝色表示低表达基因。颜色从红到蓝,表示log10(FPKM+1)从大到小。
Fig. 4D11_1, D12_1, DW61_2 and DW99_2 differential gene clustering heat map
Table 2
表2
表2D11_1前50个差异最显著的基因信息
Table 2
| 基因 | 基因全称 | 表达情况 |
|---|---|---|
| TMEM198 | Transmembrane protein 198 | ↑ |
| ALDOB | Aldolase, fructose-bisphosphate B | ↑ |
| UMOD | Uromodulin | ↑ |
| ASS1 | Argininosuccinate synthase 1 | ↑ |
| SLC5A12 | Solute carrier family 5 member 12 | ↑ |
| SLC34A1 | Sus scrofa solute carrier family 34 member 1 (SLC34A1), mRNA | ↑ |
| AGR2 | Anterior gradient protein 2 homolog precursor | ↑ |
| U6 | U6 spliceosomal RNA | ↑ |
| BHMT | Betaine-homocysteine S-methyltransferase 1 | ↑ |
| DDC | Dopa decarboxylase | ↑ |
| DAO | D-amino-acid oxidase | ↑ |
| SLC13A3 | Solute carrier family 13 member 3 | ↑ |
| CDH16 | Cadherin 16 | ↑ |
| CYP2D25 | Vitamin D(3) 25-hydroxylase | ↑ |
| PPARGC1B | PPARG coactivator 1 beta | ↑ |
| FBP1 | Fructose-1,6-bisphosphatase 1 | ↑ |
| G6PC | Glucose-6-phosphatase | ↑ |
| CLDN2 | Claudin-2 | ↑ |
| DMGDH | Dimethylglycine dehydrogenase | ↑ |
| FMO1 | Dimethylaniline monooxygenase [N-oxide-forming] 1 | ↑ |
| UPP2 | Uridine phosphorylase 2 | ↑ |
| CYP4A24 | Sus scrofa cytochrome P450,family 4,subfamily A,polypeptide 21 (CYP4A21), mRNA | ↑ |
| HNF4A | Hepatocyte nuclear factor 4-alpha | ↑ |
| ADSL | Adenylosuccinate lyase | ↓ |
| IGFBP6 | Insulin-like growth factor-binding protein 6 precursor | ↓ |
| ORMDL2 | ORMDL sphingolipid biosynthesis regulator 2 | ↓ |
| MRPS35 | Mitochondrial ribosomal protein S35 | ↓ |
| TM7SF3 | Transmembrane 7 superfamily member 3 | ↓ |
| DERA | Deoxyribose-phosphate aldolase | ↓ |
| LTBR | Tumor necrosis factor receptor superfamily member 3 precursor | ↓ |
| TULP3 | Tubby like protein 3 | ↓ |
| PPHLN1 | Periphilin 1 | ↓ |
| PUS7L | Pseudouridylate synthase 7 like | ↓ |
| SLC38A1 | Solute carrier family 38 member 1 | ↓ |
| NEDD1 | Neural precursor cell expressed, developmentally down-regulated 1 | ↓ |
| SELENOO | Sus scrofa selenoprotein O (SELENOO), mRNA | ↓ |
| SLC35B3 | Solute carrier family 35 member B3 | ↓ |
| FAM8A1 | Family with sequence similarity 8 member A1 | ↓ |
| MBOAT1 | Membrane bound O-acyltransferase domain containing 1 | ↓ |
| novel gene | Lysosomal thioesterase PPT2 precursor | ↓ |
| MAN2A2 | Mannosidase alpha class 2A member 2 | ↓ |
| HMG20A | High mobility group 20A | ↓ |
| CSPG4 | Chondroitin sulfate proteoglycan 4 | ↓ |
| SRP54 | Signal recognition particle 54 | ↓ |
| FOS | Proto-oncogene c-Fos | ↓ |
| SPTLC2 | Serine palmitoyltransferase long chain base subunit 2 | ↓ |
| ATXN3 | Ataxin-3 | ↓ |
新窗口打开|下载CSV
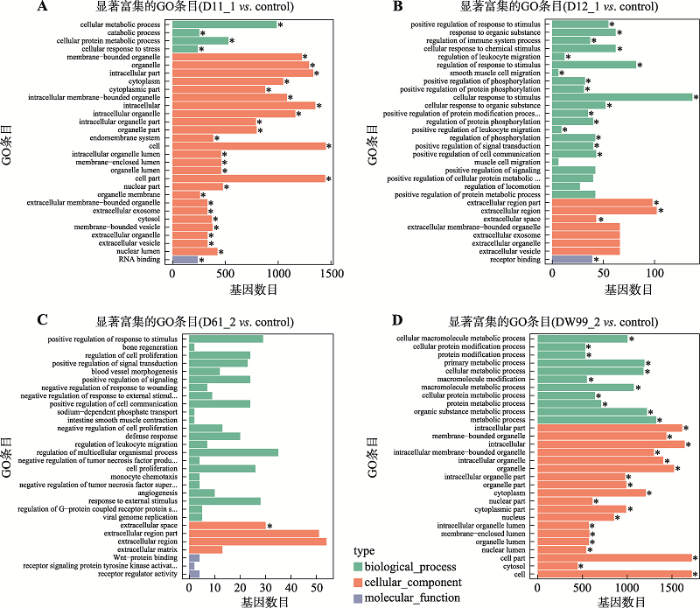
2.3 D11_1、D12_1、DW61_2和DW99_2差异基因GO富集分析和KEGG通路分析
对D11_1、D12_1、DW61_2和DW99_2测序得到的差异基因进行GO富集分析,这些差异基因在生物过程、细胞组分和分子功能方面的分布情况见图5。D11_1上调的差异表达基因主要集中于与细胞代谢有关的线粒体等细胞器和细胞器膜,下调的差异表达基因主要涉及与细胞代谢有关的蛋白代谢和物质运输生物过程。D12_1上调的差异表达基因无显著富集,而下调的差异表达基因主要富集于细胞对刺激的反应与蛋白代谢的细胞过程与应对刺激信号传导的膜的变化。DW61_2上调和下调的差异表达基因均无显著富集。DW99_2上调的差异表达基因主要富集于细胞有丝分裂有关的DNA复制、染色体分离、核酸代谢等生物过程和与之相关的一些胞内有机物质的变化,而下调的差异表达基因则主要涉及蛋白代谢和蛋白修饰等一些高分子修饰过程。结果显示D11_1和DW99_2的差异基因功能富集趋势更为明显,基因表达模式也更为“另类”。Table 3
表3
表3D12_1前50个差异最显著的基因概况
Table 3
| 基因 | 基因全称 | 表达情况 |
|---|---|---|
| PARVG | Gamma-parvin | ↑ |
| POU3F1 | POU class 3 homeobox 1 | ↑ |
| ASS1 | Argininosuccinate synthase 1 | ↑ |
| UMOD | Uromodulin | ↑ |
| PNMA2 | Paraneoplastic Ma antigen 2 | ↑ |
| ADGRG7 | Adhesion G protein-coupled receptor G7 | ↑ |
| KRT28 | Keratin 28 | ↑ |
| GSDMB | Gasdermin B | ↑ |
| U6 | U6 spliceosomal RNA | ↑ |
| RNF223 | Ring finger protein 223 | ↑ |
| TBX10 | T-box 10 | ↑ |
| TMPRSS2 | Transmembrane protease, serine 12 | ↑ |
| HTR1E | 5-hydroxytryptamine receptor 1E | ↑ |
| HIC2 | HIC ZBTB transcriptional repressor 2 | ↑ |
| SLC34A1 | Sus scrofa solute carrier family 34 member 1 (SLC34A1), mRNA. | ↑ |
| ALDOB | Aldolase, fructose-bisphosphate B | ↑ |
| CSN1S1 | Sus scrofa casein alpha s1 (CSN1S1), mRNA. | ↑ |
| SLC2A12 | Solute carrier family 2 member 12 | ↑ |
| CD53 | CD53 molecule | ↑ |
| NAGA | Alpha-N-acetylgalactosaminidase precursor | ↓ |
| ADSL | Adenylosuccinate lyase | ↓ |
| C12orf4 | Homolog isoform 2 | ↓ |
| SLC35B3 | Solute carrier family 35 member B3 | ↓ |
| LEMD2 | LEM domain containing 2 | ↓ |
| GOLGA5 | Golgin A5 | ↓ |
| GSTA4 | Glutathione S-transferase A4 | ↓ |
| FAM98C | Family with sequence similarity 98 member C | ↓ |
| LDLRAP1 | Low density lipoprotein receptor adaptor protein 1 | ↓ |
| PLK3 | Polo like kinase 3 | ↓ |
| SMOC2 | SPARC related modular calcium binding 2 | ↓ |
| SPG21 | Sus scrofa spastic paraplegia 21 (autosomal recessive, Mast syndrome) (SPG21), mRNA | ↓ |
| PCLAF | Sus scrofa PCNA-associated factor (LOC100514810), mRNA | ↓ |
| SERPIN2 | Serpin family B member 2 | ↓ |
| AEN | Apoptosis enhancing nuclease | ↓ |
| GCNT1 | Glucosaminyl (N-acetyl) transferase 1, core 2 | ↓ |
| PPP6C | Serine/threonine-protein phosphatase 6 catalytic subunit | ↓ |
| PCSK6 | Proprotein convertase subtilisin/kexin type 6 | ↓ |
| BOP1 | Block of proliferation 1 | ↓ |
| FAM49B | Protein FAM49B | ↓ |
| PLAT | Tissue-type plasminogen activator precursor | ↓ |
| SMOX | Spermine oxidase | ↓ |
| ASPN | Asporin precursor | ↓ |
| IL1R1 | Interleukin 1 receptor type 1 | ↓ |
新窗口打开|下载CSV
Table 4
表4
表4DW61_2前50个差异最显著的基因概况
Table 4
| 基因 | 基因全称 | 表达情况 |
|---|---|---|
| NPPB | Natriuretic peptides B Brain natriuretic peptide 32 Brain natriuretic peptide 26 | ↑ |
| GRIK2 | Glutamate ionotropic receptor kainate type subunit 2 | ↑ |
| PAX1 | Paired box 1 | ↑ |
| DOK5 | Docking protein 5 | ↑ |
| ANKR2 | Ankyrin repeat domain 2 | ↑ |
| SLC114 | Solute carrier family 16 member 14 | ↑ |
| GPR37 | G protein-coupled receptor 37 | ↑ |
| TRPV2 | Transient receptor potential cation channel subfamily V member 2 | ↑ |
| RHCE | Sus scrofa Rh blood group CcEe antigens (RHCE), mRNA. | ↑ |
| MFNG | MFNG O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase | ↑ |
| UBAPL | Ubiquitin associated protein 1 like | ↑ |
| ASPG | Asparaginase | ↑ |
| CRYBA1 | Crystallin beta A1 | ↑ |
| RECQL | ATP-dependent DNA helicase Q1 | ↓ |
| RIMKLB | Ribosomal modification protein rimK like family member B | ↓ |
| C1R | Complement C1r | ↓ |
| WASHC4 | WASH complex subunit 4 | ↓ |
| GNPTAB | N-acetylglucosamine-1-phosphate transferase alpha and beta subunits | ↓ |
| SELENOO | Sus scrofa selenoprotein O (SELENOO), mRNA. | ↓ |
| MAN2A2 | Mannosidase alpha class 2A member 2 | ↓ |
| STRA6 | Stimulated by retinoic acid 6 | ↓ |
| ISLR | Immunoglobulin superfamily containing leucine rich repeat | ↓ |
| HECTD1 | HECT domain E3 ubiquitin protein ligase 1 | ↓ |
| C14orf119 | Chromosome 14 open reading frame 119 | ↓ |
| NFAT5 | Nuclear factor of activated T-cells 5 | ↓ |
| E2F4 | E2F transcription factor 4 | ↓ |
| INPP5B | Inositol polyphosphate-5-phosphatase B | ↓ |
| SMOC2 | SPARC related modular calcium binding 2 | ↓ |
| MTHFD1L | Methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1 like | ↓ |
| LATS1 | Large tumor suppressor kinase 1 | ↓ |
| ME2 | Malic enzyme 2 | ↓ |
| TRIP4 | Thyroid hormone receptor interactor 4 | ↓ |
| LEO1 | LEO1 homolog, Paf1/RNA polymerase II complex component | ↓ |
| VPS39 | VPS39, HOPS complex subunit | ↓ |
| DPP8 | Dipeptidyl peptidase 8 | ↓ |
| HACD3 | 3-hydroxyacyl-CoA dehydratase 3 | ↓ |
| PRPF39 | Pre-mRNA processing factor 39 | ↓ |
新窗口打开|下载CSV
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5D11_1、D12_1、DW61_2和DW99_2差异表达基因的GO富集分析图
A~D分别为D11_1、D12_1、DW61_2和DW99_2与对照组差异表达基因的GO富集柱状图。纵坐标为富集的GO term,横坐标为该term中差异基因个数。不同颜色用来区分生物过程、细胞组分和分子功能,带“*”为显著富集的GO term,对富集最显著的30个GO term在图中展示,如果不足30条,则全部展示。
Fig. 5GO enrichment histogram of D11_1, D12_1, DW61_2 and DW99_2 differentially expressed genes
利用KEGG注释系统对D11_1、D12_1、DW61_2和DW99_2进行差异基因代谢通路富集分析。结果显示(图6),D11_1上调的基因主要作用于细胞代谢,而下调的基因主要作用于细胞凋亡;D12_1与DW61_2 KEGG均无显著通路富集;DW99_2 KEGG通路中上调的差异基因最显著的富集于调节DNA复制、细胞周期通路。
Table 5
表5
表5DW99_2前50个差异最显著的基因概况
Table 5
| 基因 | 基因全称 | 表达情况 |
|---|---|---|
| GBX2 | Gastrulation brain homeobox 2 | ↑ |
| PCDH12 | Protocadherin 12 | ↑ |
| ARHGEF9 | Cdc42 guanine nucleotide exchange factor 9 | ↑ |
| TRAM1L1 | Translocation associated membrane protein 1-like 1 | ↑ |
| U6 | U6 spliceosomal RNA | ↑ |
| DMTN | Dematin actin binding protein | ↑ |
| CEP72 | Centrosomal protein 72 | ↑ |
| YBX2 | Y-box binding protein 2 | ↑ |
| ZNF768 | Zinc finger protein 768 | ↑ |
| NOTCH4 | Neurogenic locus notch homolog protein 4 precursor | ↑ |
| GARNL3 | GTPase activating Rap/RanGAP domain like 3 | ↑ |
| MTBP | MDM2 binding protein | ↑ |
| UHRF1 | Ubiquitin like with PHD and ring finger domains 1 | ↑ |
| PACSIN2 | Protein kinase C and casein kinase substrate in neurons 2 | ↑ |
| EP300 | E1A binding protein p300 | ↓ |
| ADSL | Adenylosuccinate lyase | ↓ |
| PWP1 | PWP1 homolog, endonuclein | ↓ |
| IGFBP6 | Insulin-like growth factor-binding protein 6 precursor | ↓ |
| MMP19 | Matrix metallopeptidase 19 | ↓ |
| ESYT1 | Extended synaptotagmin 1 | ↓ |
| SMARCC2 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin subfamily c member 2 | ↓ |
| PTGES3 | Prostaglandin E synthase 3 | ↓ |
| MON2 | MON2 homolog, regulator of endosome-to-Golgi trafficking | ↓ |
| XPOT | Exportin for tRNA | ↓ |
| TMEM19 | Transmembrane protein 19 | ↓ |
| TBC1D15 | TBC1 domain family member 15 | ↓ |
| DNM1L | Dynamin 1 like | ↓ |
| FAR2 | Fatty acyl-CoA reductase 2 | ↓ |
| ARNTL2 | Aryl hydrocarbon receptor nuclear translocator like 2 | ↓ |
| TM7SF3 | Transmembrane 7 superfamily member 3 | ↓ |
| FGFR1OP2 | FGFR1 oncogene partner 2 | ↓ |
| AEBP2 | AE binding protein 2 | ↓ |
| LRP6 | LDL receptor related protein 6 | ↓ |
| C1R | Complement C1r | ↓ |
| NOP2 | Sus scrofa NOP2 nucleolar protein (NOP2), mRNA | ↓ |
新窗口打开|下载CSV
图6
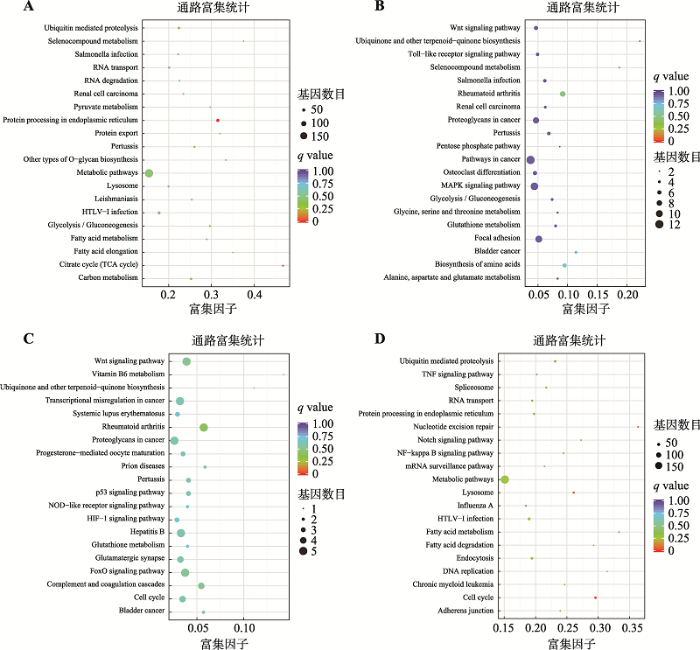 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6D11_1、D12_1、DW61_2和DW99_2差异表达基因的KEGG富集散点图
A~D分别代表D11_1、D12_1、DW61_2和DW99_2与对照组的差异表达基因KEGG富集散点图。纵轴表示pathway名称,横轴表示Rich factor,点的大小表示此pathway中差异表达基因个数多少,而点的颜色对应于不同的q value范围。
Fig. 6KEGG enrichment scatter plot of D11_1, D12_1, DW61_2 and DW99_2
3 讨论
单细胞转录组测序技术能够揭示单个细胞的基因表达动态,反映细胞间的异质性。在猪耳成纤维细胞异质性研究过程中,将单个细胞从耳组织块中分离出来是单细胞转录组测序的第一个关键步骤。为准确获得成纤维细胞,排除其他类型细胞对细胞异质性结果的干扰,本研究最终采用显微操作法进行单细胞分离。该方法适用于样本数量少的单细胞样品制备,能够在显微镜下观察成纤维细胞的形态,精准控制单个细胞的吸入与排出[26],确保52个单细胞样品均为成纤维细胞。在生物界异质性通常被解释为3个层面:(1)不同物种或生物体之间具有异质性;(2)同一生物体的不同器官或组织具有异质性;(3)同一器官或组织中的不同细胞具有异质性。事实上,早在1957年Novick等[27]就提出了细胞异质性的概念,最初是根据形态和功能上的差异将细胞群分化成不同的细胞亚群。单细胞测序技术的出现为细胞异质性的研究提供了全新的见解:每一个细胞都是独特的个体,被独一无二的DNA、RNA以及蛋白质所编码[28]。在医学领域,已有研究利用单细胞测序技术构建人类器官、组织的基因表达图谱[29]。这为SCNT研究提供一个可能:应用单细胞测序技术描述供体细胞核和卵母细胞质相互作用的动态过程,以更高的分辨率解析阻碍胚胎发育的分子原因。在体细胞核移植研究中,由于供体细胞异质性的存在,不同的供体核和卵母细胞质相互作用表现出不同应答模式,包括不同的染色质重构和表观遗传修饰重编程[15]。同样,在诱导多能干细胞的研究中,来自不同组织的供体细胞在重编程过程中表现出不同的敏感性[30,31],由不同供体细胞获得的多能干细胞拥有不同的转录模式[32]。先前大多数研究讨论了不同类型的供体细胞对克隆胚胎发育效率的影响[33,34],而尚不清楚来源相同的供体细胞异质性对核移植效率的影响。在实际研究中,往往把来源相同的供体细胞看成同质的,导致细胞内关键分子事件大量丢失。
本研究将精度对准细胞水平,用单细胞转录组数据反映不同供体细胞的基因表达模式:52个成功测序的猪耳成纤维细胞转录组热图反映同一组织供体细胞间的基因表达模式差异;通过对上述52个细胞进行主成分分析,发现 4个基因表达模式明显不同的“另类”细胞,证实了供体细胞间异质性的存在。同时,推测“另类”细胞的基因表达模式和精英供体细胞存在联系。D11_1与“普通”细胞相比,其上调基因GO富集于与细胞代谢相关的细胞器、细胞膜,尤其是线粒体,主要功能为促进细胞代谢。其下调基因在KEGG分析中主要集中在细胞凋亡通路。在正常受精胚胎中,精子来源的mtDNA在受精不久后被全部破坏,其mtRNA全部来源于卵母细胞[35]。但是在SCNT胚胎中却存在供体来源的mtDNA[36]。因此,这些供体来源的线粒体DNA可能是影响克隆效率的关键线索。同时,D11_1差异基因表达结果也暗示了通过研究线粒体相关基因寻找精英供体细胞的可能。在D11_1表达上调的差异基因中,ASS1基因[37,38]可以调节精氨酸合成,而L-精氨酸又是细胞信号传导、代谢功能分子(NO、多胺和肌酸)的主要合成前体[39],对胚胎发育十分重要。UMOD基因的表达能加快蛋白质代谢和高尔基体转运,增加胚胎发育能力[40,41]。在D11_1表达下调的差异基因中,ORM基因家族蛋白负向调节鞘脂代谢[42],其下调表达会促进细胞生长分化;LTBR基因的表达可促进细胞凋亡[43,44],其下调表达起到抑制细胞凋亡的作用;PPHLN1是一种介导表观遗传抑制修饰的多蛋白复合物[45],可以推动细胞重编程障碍H3K9me3[46]的表观抑制修饰,其下调表达有利于克隆胚胎正确重编程。因此,D11_1的基因表达模式可作为精英供体细胞的潜在参考。
DW61_2和D12_1与“普通”细胞比较,GO富集与KEGG富集均无显著趋势,其差异表达基因的功能多为抑制蛋白活性和糖代谢、阻遏细胞生长。如D12_1中LEMD2基因[47]的显著下调会抑制结合蛋白mRNA的转运,进而抑制细胞分裂;GOLGA5基因[48]的下调会影响高尔基体内部囊泡介导物质运输,抑制高尔基体和微管间的相互作用,抑制细胞有丝分裂;在DW61_2中表达下调的基因中,PYROXD1基因[49]编码二硫化物还原酶,其下调会抑制细胞生长,降低细胞活力;SMOC2基因[50,51]在胚胎发生和伤口愈合过程中高度表达。该基因产物是一种基质细胞蛋白,能促进基质组装,并能刺激内皮细胞的增殖和迁移,以及血管生成活性,其表达下调不利于胚胎发生和后续克隆动物的生长发育。因此,DW61_2、DW12_1的基因表达模式呈现抑制细胞活力,促进细胞凋亡的趋势,同时根据已有研究证据无法将这两个“另类”细胞与精英供体细胞相关联,其基因表达模式并不具备代表性。
DW99_2与对照组相比,GO富集上调的差异表达基因主要和DNA复制、染色体分离、核酸代谢等生物过程相关,KEGG分析结果显示显著富集于DNA复制、细胞周期更迭等通路,基因组处于活跃状态,明显区别于其他供体细胞。在显著表达上调基因中,GBX2基因和胚胎干细胞转录调控网络相关,其转录因子P52951可作为胚胎多能性因子[52,53],而多能性因子是影响克隆胚胎发育潜力的重要因素。ARHGEF9基因是一种蛋白质编码基因,其功能主要是调控外胚层分化,在细胞重编程中发挥关键作用[54,55]。PACSIN12基因的相关途径中有网格蛋白介导的内吞作用,值得注意的是,在小鼠克隆胚2细胞期停滞胚胎单细胞转录组测序研究中,也报道了与内吞作用相关的基因激活不足的现象[56],说明与内吞途径相关基因可能影响SCNT胚胎的发育效率。在体细胞重编程过程中,大量基因能否被成功激活直接影响克隆胚胎的发育命运。同时,已有研究表明供体细胞基因表达模式影响克隆效率[57]。因此,在供体细胞基因表达、核重编程、胚胎关键基因激活之间一定存在微妙的联系。DW99_2活跃基因组状态,可能有利细胞重编程,但是需要在不同情况下进行区分:先前关于SCNT胚胎重编程异常原因的报道主要集中于抑制性的表观遗传修饰[58,59],表现为协调胚胎发育所需基因失败,部分基因表达受到抑制,基因组不活跃。但是在克隆牛的研究中,供体细胞基因组的异常激活状态会阻碍克隆胚胎发育[18]。在克隆小鼠中,供体细胞异常激活的基因却会在胚胎发育中被成功重编程[60],说明供体细胞的基因组状态不完全决定SCNT胚胎的发育命运,不同物种之间是存在差异的,需要进一步研究猪供体细胞基因组状态与克隆胚胎发育命运的联系。
目前,关于供体细胞异质性对核移植胚胎发育效率的影响还有很多问题亟待解决。就研究广泛性而言,类似研究鲜有报道,同时,供体细胞异质性受物种、遗传变异、环境差异和物理刺激等多因素影响,需要更全面的研究来阐述造成这种差异的分子机制以及对核移植胚胎发育的影响。就研究深度而言,一方面需要有参考价值的精英供体细胞、克隆胚胎的遗传信息,另一方面,需要建立一种体系或技术,辅助研究人员根据供体细胞的分子特征筛选潜在精英供体细胞,并进行核移植实验来验证。在未来,可利用猪克隆胚胎活检技术(在2细胞、4细胞期对每个胚胎抽取一个卵裂球用于单细胞测序,每个胚胎剩余的部分继续培养观察其能否发育至囊胚,从而把抽取的卵裂球分成发育正常和异常两组样本)结合单细胞测序技术确定发育正常及异常的克隆胚胎的基因表达模式,通过分析本研究所获得的4个“另类”供体细胞与发育正常的克隆胚胎的基因表达模式的关联,从4个“另类”细胞中筛选出潜在的精英供体细胞,根据该精英供体细胞的基因表达特征设计可识别该精英供体细胞的探针或抗体,然后通过流式细胞仪分选等技术在供体细胞中筛选和富集精英供体细胞进行核移植,验证利用该精英供体细胞制备的克隆胚胎的发育是否高于普通供体细胞。最近,Li等[56]利用单细胞测序技术,通过比较核移植2细胞期正常发育胚胎、停滞发育胚胎,4细胞期正常发育胚胎、停滞发育胚胎的转录组数据,找出影响小鼠克隆胚胎重编程的潜在通路和关键基因,为在细胞水平研究克隆胚胎异常发育的原因提供了参考。相信随着单细胞测序技术的不断成熟,该技术在克隆研究中的应用有望取得进一步突破:一方面,单细胞多组学联合分析的发展有助于建立“基因组-转录组-代谢组-表型”的系统研究网路;另一方面,更多种类、更多数量的供体细胞命运将通过单细胞测序技术被追踪,形成“供体细胞-2细胞-4细胞-8细胞-囊胚-移植后胚胎”的完整研究体系,描绘供体细胞核和卵母细胞质相互作用的动态过程,综合分析影响克隆胚胎核移植效率的分子机制,进而提高克隆效率。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/385810a0URLPMID:9039911 [本文引用: 1]

Fertilization of mammalian eggs is followed by successive cell divisions and progressive differentiation, first into the early embryo and subsequently into all of the cell types that make up the adult animal. Transfer of a single nucleus at a specific stage of development, to an enucleated unfertilized egg, provided an opportunity to investigate whether cellular differentiation to that stage involved irreversible genetic modification. The first offspring to develop from a differentiated cell were born after nuclear transfer from an embryo-derived cell line that had been induced to become quiescent. Using the same procedure, we now report the birth of live lambs from three new cell populations established from adult mammary gland, fetus and embryo. The fact that a lamb was derived from an adult cell confirms that differentiation of that cell did not involve the irreversible modification of genetic material required for development to term. The birth of lambs from differentiated fetal and adult cells also reinforces previous speculation that by inducing donor cells to become quiescent it will be possible to obtain normal development from a wide variety of differentiated cells.
URLPMID:9596577 [本文引用: 1]
URLPMID:10947985 [本文引用: 1]
DOI:10.1016/j.cell.2018.01.020URLPMID:29395327 [本文引用: 1]
Generation of genetically uniform non-human primates may help to establish animal models for primate biology and biomedical research. In this study, we have successfully cloned cynomolgus monkeys (Macaca fascicularis) by somatic cell nuclear transfer (SCNT). We found that injection of H3K9me3 demethylase Kdm4d mRNA and treatment with histone deacetylase inhibitor trichostatin A at one-cell stage following SCNT greatly improved blastocyst development and pregnancy rate of transplanted SCNT embryos in surrogate monkeys. For SCNT using fetal monkey fibroblasts, 6 pregnancies were confirmed in 21 surrogates and yielded 2 healthy babies. For SCNT using adult monkey cumulus cells, 22 pregnancies were confirmed in 42 surrogates and yielded 2 babies that were short-lived. In both cases, genetic analyses confirmed that the nuclear DNA and mitochondria DNA of the monkey offspring originated from the nucleus donor cell and the oocyte donor monkey, respectively. Thus, cloning macaque monkeys by SCNT is feasible using fetal fibroblasts.
DOI:10.1016/j.stem.2007.10.009URL [本文引用: 1]
Accessibility of human oocytes for research poses a serious ethical challenge to society. This fact categorically holds true when pursuing some of the most promising areas of research, such as somatic cell nuclear transfer and embryonic stem cell studies. One approach to overcoming this limitation is to use an oocyte from one species and a somatic cell from another. Recently, several attempts to capture the promises of this approach have met with varying success, ranging from establishing human embryonic stem cells to obtaining live offspring in animals. This review focuses on the challenges and opportunities presented by the formidable task of overcoming biological differences among species.
[本文引用: 1]
DOI:10.1016/j.cell.2013.05.006URL [本文引用: 1]
Reprogramming somatic cells into pluripotent embryonic stem cells (ESCs) by somatic cell nuclear transfer (SCNT) has been envisioned as an approach for generating patient-matched nuclear transfer (NT)-ESCs for studies of disease mechanisms and for developing specific therapies. Past attempts to produce human NT-ESCs have failed secondary to early embryonic arrest of SCNT embryos. Here, we identified premature exit from meiosis in human oocytes and suboptimal activation as key factors that are responsible for these outcomes. Optimized SCNT approaches designed to circumvent these limitations allowed derivation of human NT-ESCs. When applied to premium quality human oocytes, NT-ESC lines were derived from as few as two oocytes. NT-ESCs displayed normal diploid karyotypes and inherited their nuclear genome exclusively from parental somatic cells. Gene expression and differentiation profiles in human NT-ESCs were similar to embryo-derived ESCs, suggesting efficient reprogramming of somatic cells to a pluripotent state.
URLPMID:31347235 [本文引用: 1]
DOI:10.1071/RD13329URLPMID:25482653 [本文引用: 1]
During the last 17 years, considerable advancements have been achieved in the production of pigs, transgenic and non-transgenic, by methods of somatic cell nuclear transfer, in vitro fertilisation, intracytoplasmic sperm injection, microinjection and sperm-mediated gene transfer by artificial insemination. Therefore, a review of the overall efficiency for the developmental competence of embryos produced by these in vitro methods would be useful in order to obtain a more thorough overview of this growing area with respect to its development and present status. In this review a meta-analysis was used to analyse data collected from all published articles with a focus on zygotes and embryos for transfer, pregnancy, full-term development and piglets born. It was generally concluded that an increasing level of in vitro manipulation of porcine embryos decreased the overall efficiency for production of piglets. The techniques of nuclear transfer have been developed markedly through the increasing number of studies performed, and the results have become more stable. Prolonged in vitro culture period did not lead to any negative effect on nuclear transfer embryos after their transfer and it resulted in a similar or even higher litter size. More complete information is needed in future scientific articles about these in vitro manipulation techniques to establish a more solid basis for the evaluation of their status and to reveal and further investigate any eventual problems.
URLPMID:15226007 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/rda.13286URLPMID:30051521 [本文引用: 1]
Low efficiency of somatic cell nuclear transfer (SCNT) embryos is largely attributable to imperfect reprogramming of the donor nucleus. The differences in epigenetic reprogramming between female and male buffalo cloned embryos remain unclear. We explored the effects of donor cell sex differences on the development of SCNT embryos. We and then compared the expression of DNA methylation (5-methylcytosine-5mC and 5-hydroxymethylcytosine-5hmC) and the expression level of relevant genes, and histone methylation (H3K9me2 and H3K9me3) level in SCNT-female symbol and SCNT-male symbol preimplantation embryos with in vitro fertilization (IVF) counterparts. In the study, we showed that developmental potential of SCNT-female symbol embryos was greater than that of SCNT-male symbol embryos (p < 0.05). 5mC was mainly expressed in SCNT-female symbol embryos, whereas 5hmC was majorly expressed in SCNT-male symbol embryos (p < 0.05). The levels of DNA methylation (5mC and 5hmC), Dnmt3b, TET1 and TET3 in the SCNT-male symbol embryos were higher than those of SCNT-female symbol embryos (p < 0.05). In addition, there were no significant differences in the expression of H3K9me2 at eight-stage of the IVF, SCNT-female symbol and SCNT-male symbolembryos (p < 0.05). However, H3K9me3 was upregulated in SCNT-male symbol embryos at the eight-cell stage (p < 0.05). Thus, KDM4B ectopic expression decreased the level of H3K9me3 and significantly improved the developmental rate of two-cell, eight-cell and blastocysts of SCNT-male symbol embryos (p < 0.05). Overall, the lower levels of DNA methylation (5mC and 5hmC) and H3K9me3 may introduce the greater developmental potential in buffalo SCNT-female symbol embryos than that of SCNT-male symbol embryos.
DOI:10.1126/science.1063206URLPMID:11498580 [本文引用: 1]
Cloning of mammals by nuclear transfer (NT) results in gestational or neonatal failure with at most a few percent of manipulated embryos resulting in live births. Many of those that survive to term succumb to a variety of abnormalities that are likely due to inappropriate epigenetic reprogramming. Cloned embryos derived from donors, such as embryonic stem cells, that may require little or no reprogramming of early developmental genes develop substantially better beyond implantation than NT clones derived from somatic cells. Although recent experiments have demonstrated normal reprogramming of telomere length and X chromosome inactivation, epigenetic information established during gametogenesis, such as gametic imprints, cannot be restored after nuclear transfer. Survival of cloned animals to birth and beyond, despite substantial transcriptional dysregulation, is consistent with mammalian development being rather tolerant to epigenetic abnormalities, with lethality resulting only beyond a threshold of faulty gene reprogramming encompassing multiple loci.
[本文引用: 1]
[本文引用: 1]
DOI:10.1002/mrd.22935URLPMID:29205617 [本文引用: 2]
The type and pattern of epigenetic modification in donor cells can significantly affect the developmental competency of somatic cell nuclear transfer (SCNT) embryos. Here, we investigated the developmental capacity, gene expression, and epigenetic modifications of SCNT embryos derived from porcine bone marrow-derived mesenchymal stem cells (BMSCs) and fetal fibroblasts (FFs) donor cells compared to embryos obtained from in vitro fertilization (IVF). Compared to FFs, the donor BMSCs had more active epigenetic markers (Histone H3 modifications: H3K9Ac, H3K4me3, and H3K4me2) and fewer repressive epigenetic markers (H3K9me3, H3K9me2, and DNA methyltransferase 1). Embryos derived from BMSC nuclear-transfer (BMSC-NT embryos) and IVF embryos had significantly higher cleavage and blastocyst rates (BMSC-NT: 71.3 +/- 3.4%, 29.1 +/- 2.3%; IVF: 69.2 +/- 2.2%, 30.2 +/- 3.3%; respectively) than FF-NT embryos (58.1 +/- 3.4%, 15.1 +/- 1.5%, respectively). Bisulfite sequencing revealed that DNA methylation at the promoter regions of NANOG and POU5F1 was lower in BMSC-NT embryos (30.0%, 9.8%, respectively) than those in FF-NT embryos (34.2%, 28.0%, respectively). We also found that BMSC-NT embryos had more H3K9Ac and less H3K9me3 and 5-methylcytosine than FF-NT embryos. In conclusion, our finding comparing BMSCs versus FFs as donors for nuclear transfer revealed that differences in the initial epigenetic state of donor cells have a remarkable effect on overall nuclear reprogramming of SCNT embryos, wherein donor cells possessing a more open chromatin state are more conducive to nuclear reprogramming.
DOI:10.1038/nature08180URLPMID:19571877 [本文引用: 1]
Induced pluripotent stem cells offer unprecedented potential for disease research, drug screening, toxicology and regenerative medicine. However, the process of reprogramming is inefficient and often incomplete. Here I consider reasons for bottlenecks in induced pluripotent stem cell generation, and propose a model in which most or all cells have the potential to become pluripotent.
DOI:10.1095/biolreprod.103.017731URLPMID:12801984 [本文引用: 1]
Although it is widely assumed that the cell type and genotype of the donor cell affect the efficiency of somatic cell cloning, little systematic analysis has been done to verify this assumption. The present study was undertaken to examine whether donor cell type, donor genotype, or a combination thereof increased the efficiency of mouse cloning. Initially we assessed the developmental ability of embryos that were cloned from cumulus or immature Sertoli cells with six different genotypes (i.e., 2 x 6 factorial). Significantly better cleavage rates were obtained with cumulus cells than with Sertoli cells (P < 0.005, two-way ANOVA), which probably was due to the superior cell-cycle synchrony of cumulus cells at G0/G1. After embryo transfer, there was a significant effect of cell type on the birth rate, with Sertoli cells giving the better result (P < 0.005). Furthermore, there was a significant interaction (P < 0.05) between the cell type and genotype, which indicates that cloning efficiency is determined by a combination of these two factors. The highest mean birth rate (10.8 +/- 2.1%) was obtained with (B6 x 129)F1 Sertoli cells. In the second series of experiments, we examined whether the developmental ability of clones with the wild-type genotype (JF1) was improved when combined with the 129 genotype. Normal pups were cloned from cumulus and immature Sertoli cells of the (129 x JF1)F1 and (JF1 x 129)F1 genotypes, whereas no pups were born from cells with the (B6 x JF1)F1 genotype. The present study clearly demonstrates that the efficiency of somatic cell cloning, and in particular fetal survival after embryo transfer, may be improved significantly by choosing the appropriate combinations of cell type and genotype.
DOI:10.1096/fj.201900578RRURLPMID:31914649 [本文引用: 2]
Studies on the effects of transcriptional memory on clone reprogramming in mammals are limited. In the present study, we observed higher levels of active histone H3 lysine 4 trimethylation (H3K4me3 and 5-hydroxymethylcytosine) and repressive (5-methylcytosine) epigenetic modifications in bovine early cloned embryos than in in vitro fertilized embryos. We hypothesized that aberrant epigenetic modification may result in transcriptional disorders in bovine somatic cell nuclear transfer (SCNT) embryos. RNA sequencing results confirmed that both abnormal transcriptional silencing and transcriptional activation are involved in bovine SCNT reprogramming. The cloned embryos exhibited excessive transcription in RNA processing- and translation-related genes as well as transcriptional defects in reproduction-related genes whose transcriptional profiles were similar to those in donor cells. These results demonstrated the existence of active and silent memory genes inherited from donor cells in early bovine SCNT embryos. Further, H3K4me3-specific demethylase 5B (KDM5B) mRNA was injected into the reconstructed embryos to reduce the increased H3K4me3 modification. KDM5B overexpression not only reduced the transcriptional level of active memory genes, but also promoted the expression of silent memory genes; in particular, it rescued the expression of multiple development-related genes. These results showed that transcriptional memory acts as a reprogramming barrier and KDM5B improves SCNT reprogramming via bidirectional regulation effects on transcriptional memory genes in bovines.
DOI:10.1038/nprot.2014.006URLPMID:24385147 [本文引用: 1]
Emerging methods for the accurate quantification of gene expression in individual cells hold promise for revealing the extent, function and origins of cell-to-cell variability. Different high-throughput methods for single-cell RNA-seq have been introduced that vary in coverage, sensitivity and multiplexing ability. We recently introduced Smart-seq for transcriptome analysis from single cells, and we subsequently optimized the method for improved sensitivity, accuracy and full-length coverage across transcripts. Here we present a detailed protocol for Smart-seq2 that allows the generation of full-length cDNA and sequencing libraries by using standard reagents. The entire protocol takes approximately 2 d from cell picking to having a final library ready for sequencing; sequencing will require an additional 1-3 d depending on the strategy and sequencer. The current limitations are the lack of strand specificity and the inability to detect nonpolyadenylated (polyA(-)) RNA.
URLPMID:31768052 [本文引用: 1]
DOI:10.1038/nmeth.1315URLPMID:19349980 [本文引用: 1]
Next-generation sequencing technology is a powerful tool for transcriptome analysis. However, under certain conditions, only a small amount of material is available, which requires more sensitive techniques that can preferably be used at the single-cell level. Here we describe a single-cell digital gene expression profiling assay. Using our mRNA-Seq assay with only a single mouse blastomere, we detected the expression of 75% (5,270) more genes than microarray techniques and identified 1,753 previously unknown splice junctions called by at least 5 reads. Moreover, 8-19% of the genes with multiple known transcript isoforms expressed at least two isoforms in the same blastomere or oocyte, which unambiguously demonstrated the complexity of the transcript variants at whole-genome scale in individual cells. Finally, for Dicer1(-/-) and Ago2(-/-) (Eif2c2(-/-)) oocytes, we found that 1,696 and 1,553 genes, respectively, were abnormally upregulated compared to wild-type controls, with 619 genes in common.
DOI:10.1038/nmeth.3317URLPMID:25751142 [本文引用: 1]
HISAT (hierarchical indexing for spliced alignment of transcripts) is a highly efficient system for aligning reads from RNA sequencing experiments. HISAT uses an indexing scheme based on the Burrows-Wheeler transform and the Ferragina-Manzini (FM) index, employing two types of indexes for alignment: a whole-genome FM index to anchor each alignment and numerous local FM indexes for very rapid extensions of these alignments. HISAT's hierarchical index for the human genome contains 48,000 local FM indexes, each representing a genomic region of approximately 64,000 bp. Tests on real and simulated data sets showed that HISAT is the fastest system currently available, with equal or better accuracy than any other method. Despite its large number of indexes, HISAT requires only 4.3 gigabytes of memory. HISAT supports genomes of any size, including those larger than 4 billion bases.
DOI:10.1093/bioinformatics/btp612URL [本文引用: 1]
DOI:10.1186/gb-2010-11-2-r14URLPMID:20132535 [本文引用: 1]
We present GOseq, an application for performing Gene Ontology (GO) analysis on RNA-seq data. GO analysis is widely used to reduce complexity and highlight biological processes in genome-wide expression studies, but standard methods give biased results on RNA-seq data due to over-detection of differential expression for long and highly expressed transcripts. Application of GOseq to a prostate cancer data set shows that GOseq dramatically changes the results, highlighting categories more consistent with the known biology.
DOI:10.1093/bioinformatics/btp616URLPMID:19910308 [本文引用: 1]
SUMMARY: It is expected that emerging digital gene expression (DGE) technologies will overtake microarray technologies in the near future for many functional genomics applications. One of the fundamental data analysis tasks, especially for gene expression studies, involves determining whether there is evidence that counts for a transcript or exon are significantly different across experimental conditions. edgeR is a Bioconductor software package for examining differential expression of replicated count data. An overdispersed Poisson model is used to account for both biological and technical variability. Empirical Bayes methods are used to moderate the degree of overdispersion across transcripts, improving the reliability of inference. The methodology can be used even with the most minimal levels of replication, provided at least one phenotype or experimental condition is replicated. The software may have other applications beyond sequencing data, such as proteome peptide count data. AVAILABILITY: The package is freely available under the LGPL licence from the Bioconductor web site (http://bioconductor.org).
URLPMID:21631906 [本文引用: 1]
URLPMID:16590055 [本文引用: 1]
DOI:10.1039/c6mb00388eURLPMID:27460751 [本文引用: 1]
A single cell creates surprising heterogeneity in a multicellular organism. While every organismal cell shares almost an identical genome, molecular interactions in cells alter the use of DNA sequences to modulate the gene of interest for specialization of cellular functions. Each cell gains a unique identity through molecular coding across the DNA, RNA, and protein conversions. On the other hand, loss of cellular identity leads to critical diseases such as cancer. Most cell identity dissection studies are based on bulk molecular assays that mask differences in individual cells. To probe cell-to-cell variability in a population, we discuss single cell approaches to decode the genetic, epigenetic, transcriptional, and translational mechanisms for cell identity formation. In combination with molecular instructions, the physical principles behind cell identity determination are examined. Deciphering and reprogramming cellular types impact biology and medicine.
URLPMID:31653841 [本文引用: 1]
DOI:10.1016/j.cell.2007.11.019URLPMID:18035408 [本文引用: 1]
Successful reprogramming of differentiated human somatic cells into a pluripotent state would allow creation of patient- and disease-specific stem cells. We previously reported generation of induced pluripotent stem (iPS) cells, capable of germline transmission, from mouse somatic cells by transduction of four defined transcription factors. Here, we demonstrate the generation of iPS cells from adult human dermal fibroblasts with the same four factors: Oct3/4, Sox2, Klf4, and c-Myc. Human iPS cells were similar to human embryonic stem (ES) cells in morphology, proliferation, surface antigens, gene expression, epigenetic status of pluripotent cell-specific genes, and telomerase activity. Furthermore, these cells could differentiate into cell types of the three germ layers in vitro and in teratomas. These findings demonstrate that iPS cells can be generated from adult human fibroblasts.
DOI:10.1073/pnas.0711983105URLPMID:18287077 [本文引用: 1]
The generation of patient-specific pluripotent stem cells has the potential to accelerate the implementation of stem cells for clinical treatment of degenerative diseases. Technologies including somatic cell nuclear transfer and cell fusion might generate such cells but are hindered by issues that might prevent them from being used clinically. Here, we describe methods to use dermal fibroblasts easily obtained from an individual human to generate human induced pluripotent stem (iPS) cells by ectopic expression of the defined transcription factors KLF4, OCT4, SOX2, and C-MYC. The resultant cell lines are morphologically indistinguishable from human embryonic stem cells (HESC) generated from the inner cell mass of a human preimplantation embryo. Consistent with these observations, human iPS cells share a nearly identical gene-expression profile with two established HESC lines. Importantly, DNA fingerprinting indicates that the human iPS cells were derived from the donor material and are not a result of contamination. Karyotypic analyses demonstrate that reprogramming of human cells by defined factors does not induce, or require, chromosomal abnormalities. Finally, we provide evidence that human iPS cells can be induced to differentiate along lineages representative of the three embryonic germ layers indicating the pluripotency of these cells. Our findings are an important step toward manipulating somatic human cells to generate an unlimited supply of patient-specific pluripotent stem cells. In the future, the use of defined factors to change cell fate may be the key to routine nuclear reprogramming of human somatic cells.
DOI:10.1038/nbt.1667URLPMID:20644536 [本文引用: 1]
Induced pluripotent stem cells (iPSCs) have been derived from various somatic cell populations through ectopic expression of defined factors. It remains unclear whether iPSCs generated from different cell types are molecularly and functionally similar. Here we show that iPSCs obtained from mouse fibroblasts, hematopoietic and myogenic cells exhibit distinct transcriptional and epigenetic patterns. Moreover, we demonstrate that cellular origin influences the in vitro differentiation potentials of iPSCs into embryoid bodies and different hematopoietic cell types. Notably, continuous passaging of iPSCs largely attenuates these differences. Our results suggest that early-passage iPSCs retain a transient epigenetic memory of their somatic cells of origin, which manifests as differential gene expression and altered differentiation capacity. These observations may influence ongoing attempts to use iPSCs for disease modeling and could also be exploited in potential therapeutic applications to enhance differentiation into desired cell lineages.
DOI:10.1095/biolreprod65.5.1558URLPMID:11673275 [本文引用: 1]
The type of donor cell most suitable for producing cloned animals is one of the topics under debate in the field of nuclear transfer. To provide useful information to answer this question, G2/M- and G0/G1-stage fetal fibroblasts were used as donor cells for nuclear transfer. In vitro-matured oocytes derived from abattoir ovaries were used as recipient cytoplasts. In both groups, nuclear envelope breakdown and premature chromosome condensation were completed within 1-2 h after donor cells were injected into the cytoplasm of oocytes. Microtubules were organized around condensed chromosomes and formed a spindle within 1-1.5 h after activation. Decondensation of chromosomes could be seen within 2-4 h after activation. Reformation of the new nuclear envelope occurred 4-6 h after activation and was followed by nuclear swelling and formation of a pronucleus-like structure (PN) 8-12 h after activation. Most (80.6%) of the reconstructed oocytes derived from G2/M cells extruded polar body-like structures (PB). However, a much lower frequency of PB (21.7%) was observed in the reconstructed oocytes derived from G0/G1 donors. A variety of PN and PB combinations were observed in reconstructed oocytes derived from G2/M-stage donors, including 1PN+0PB, 1PN+1PB, 1PN+2PB, 2PN+0PB, 2PN+1PB, 2PN+2PB, and 3PN+1PB. Chromosomes of most embryos (10/13) derived from G2/M stage were diploid. The percentage of cleavage and blastocysts and the average nuclear number of blastocysts in the G2/M and G0/G1 groups were not different. These results demonstrate that the G2/M stage can be morphologically remodeled by cytoplasm of MII oocytes in pigs. To maintain normal ploidy, the extra chromosomes derived from G2/M-stage cells could be expelled by oocytes as a second polar body. G2/M-stage fibroblast nuclei could direct reconstructed embryos to develop to the blastocyst stage.
DOI:10.1038/nbt0402-366URLPMID:11923842 [本文引用: 1]
We have developed a method to produce live somatic clones in the rabbit, one of the mammalian species considered up to now as difficult to clone. To do so, we have modified current cloning protocols proven successful in other species by taking into account both the rapid kinetics of the cell cycle of rabbit embryos and the narrow window of time for their implantation after transfer into foster recipients. Although our method still has a low level of efficiency, it has produced several clones now proven to be fertile. Our work indicates that cloning can probably be carried out successfully in any mammalian species by taking into account physiological features of their oocytes and embryos. Our results will contribute to extending the use of rabbit models for biomedical research.
DOI:10.1038/46466URLPMID:10586873 [本文引用: 1]
DOI:10.1126/science.289.5482.1188URLPMID:10947985 [本文引用: 1]
Pig cloning will have a marked impact on the optimization of meat production and xenotransplantation. To clone pigs from differentiated cells, we microinjected the nuclei of porcine (Sus scrofa) fetal fibroblasts into enucleated oocytes, and development was induced by electroactivation. The transfer of 110 cloned embryos to four surrogate mothers produced an apparently normal female piglet. The clonal provenance of the piglet was indicated by her coat color and confirmed by DNA microsatellite analysis.
DOI:10.1007/s12022-018-9516-9URLPMID:29453600 [本文引用: 1]
High-grade neuroendocrine carcinomas (HGNECs) of the urinary bladder encompass small cell (SCNEC) and large cell neuroendocrine carcinomas (LCNEC). Currently, recommended initial management is with systemic chemotherapy, followed by consolidative therapy with either radical cystectomy or radiotherapy in patients with localized disease. Nevertheless, survival in this setting remains poor. We therefore evaluated the potential to modify arginine metabolism as an alternative, targeted therapy approach in these carcinomas. In humans, arginine is a semi-essential amino acid and its synthesis enzyme argininosuccinate synthetase (ASS1) represents the rate-limiting step in arginine biosynthesis. Neoplasms that show low to absent ASS1 expression require extracellular arginine for cancer cell survival, and thus can be targeted using arginine-degrading enzymes such as pegylated arginine deiminase (ADI-PEG 20). An initial study by our group of 19 patients demonstrated that a high percentage of SCNEC lack ASS1 expression. Herein, we evaluated an expanded cohort of 74 radical cystectomy patients with HGNEC, including 63 SCNEC, 5 LCNEC, and 6 mixed morphology HGNEC patients. ASS1 expression was assessed through immunohistochemistry. Fifty-eight (of 74, 78%) patients with HGNEC showed absent ASS1 expression, including all patients with LCNEC and mixed morphology (11 of 11, 100%). Ten-year survival from disease-specific death was not statistically significant between ASS1-expressing and ASS1-deficient cases (p = 0.75). Our results show that HGNEC of the bladder may be candidates for arginine deprivation therapy using drugs such as ADI-PEG 20. Further studies are needed to validate these findings and to determine the therapeutic efficacy of such agents.
URLPMID:29422017 [本文引用: 1]
DOI:10.1038/s41598-018-35077-0URLPMID:30425285 [本文引用: 1]
Abnormalities in gene expression that negatively affect embryonic development are frequently observed in cloned embryos generated by somatic cell nuclear transfer (SCNT). In the present study, we successfully produced a cell-penetrating peptide (CPP)-conjugated with coactivator-associated arginine methyltransferase 1 (CARM1) protein from mammalian cells and confirmed introduction into donor somatic cells and cloned 8-cell embryos within 3 hours after addition to culture medium. In addition, H3R17 dimethylation and embryonic development up to the blastocyst stage were increased in the group treated with exogenous CPP-CARM1 protein compared with the untreated group (control). Interestingly, the number of total cells and trophectoderm in blastocysts as well as implantation rate were significantly increased in the CPP-CARM1 protein-treated group. However, the cell number of inner cell mass (ICM) was not changed compared with the control group; similarly, expression of pluripotency-related genes Oct4 and Nanog (ICM markers) was not significantly different between groups. On the other hand, expression of the implantation-related gene Cdx2 (trophectoderm marker) was transiently increased after treatment with CPP-CARM1 protein. On the basis of these results, we conclude that supplementation with exogenous CPP-CARM1 protein improves embryonic development of cloned embryos through regulation of histone methylation and gene expression. In addition, our results suggest that CPP-CARM1 protein may be a useful tool for strengthening implantation of mammalian embryos.
URLPMID:28418009 [本文引用: 1]
DOI:10.1371/journal.pone.0178321URLPMID:28609449 [本文引用: 1]
Chronic kidney disease (CKD) has a prevalence of approximately 10% in adult populations. CKD can progress to end-stage renal disease (ESRD) and this is usually fatal unless some form of renal replacement therapy (chronic dialysis or renal transplantation) is provided. There is an inherited predisposition to CKD with several genetic risk markers now identified. The UMOD gene has been associated with CKD of varying aetiologies. An AmpliSeq next generation sequencing panel was developed to facilitate comprehensive sequencing of the UMOD gene, covering exonic and regulatory regions. SNPs and CpG sites in the genomic region encompassing UMOD were evaluated for association with CKD in two studies; the UK Wellcome Trust Case-Control 3 Renal Transplant Dysfunction Study (n = 1088) and UK-ROI GENIE GWAS (n = 1726). A technological comparison of two Ion Torrent machines revealed 100% allele call concordance between S5 XL and PGM machines. One SNP (rs183962941), located in a non-coding region of UMOD, was nominally associated with ESRD (p = 0.008). No association was identified between UMOD variants and estimated glomerular filtration rate. Analysis of methylation data for over 480,000 CpG sites revealed differential methylation patterns within UMOD, the most significant of these was cg03140788 p = 3.7 x 10-10.
DOI:10.1016/j.jbior.2018.08.002URLPMID:30193828 [本文引用: 1]
Sphingolipids comprise a diverse family of lipids that perform multiple functions in both structure of cellular membranes and intra- and inter-cellular signaling. The diversity of this family is generated by an array of enzymes that produce individual classes and molecular species of family members and enzymes which catabolize those lipids for recycling pathways. However, all of these lipids begin their lives with a single step, the condensation of an amino acid, almost always serine, and a fatty acyl-CoA, almost always the 16-carbon, saturated fatty acid, palmitate. The enzyme complex that accomplishes this condensation is serine palmitoyltransferase (SPT), a membrane-bound component of the endoplasmic reticulum. This places SPT in the unique position of regulating the production of the entire sphingolipid pool. Understanding how SPT activity is regulated is currently a central focus in the field of sphingolipid biology. In this review we examine the regulation of SPT activity by a set of small, membrane-bound proteins of the endoplasmic reticulum, the Orms (in yeast) and ORMDLs (in vertebrates). We discuss what is known about how these proteins act as homeostatic regulators by monitoring cellular levels of sphingolipid, but also how the Orms/ORMDLs regulate SPT in response to other stimuli. Finally, we discuss the intriguing connection between one of the mammalian ORMDL isoforms, ORMDL3, and the pervasive pulmonary disease, asthma, in humans.
DOI:10.1007/978-1-4419-6612-4_39URLPMID:21153342 [本文引用: 1]
DOI:10.1016/j.intimp.2017.05.031URLPMID:28575727 [本文引用: 1]
Lymphotoxin-beta receptor (LTbetaR) signaling is involved in hepatitis B virus (HBV) infection, hepatitis and liver carcinogenesis. However, the potential association between LTBR polymorphisms and HBV infection remains unclear. This study investigated the associations between LTBR polymorphisms and chronic HBV infection and HBV-related hepatocellular carcinoma (HCC). The study included 409 patients with chronic HBV infection, 73 HBV infection resolvers, and 197 healthy controls. Two polymorphisms rs12354 and rs3759333 were selected and genotyped by polymerase chain reaction-ligase detection reaction method. The frequencies of rs12354 genotype GT and allele T in HBV infection resolvers were significantly higher than those in patients with chronic HBV infection and healthy controls (genotype GT: 38.4% vs. 22.2% and 38.4% vs. 20.8%, P=0.004 and P=0.004, respectively; allele T: 20.5% vs. 13.1% and 20.5% vs. 12.9%, P=0.017 and P=0.028, respectively). The frequencies of rs3759333 genotypes and alleles between HBV patients, HBV infection resolvers and healthy controls had no statistical difference. The genotype and allele frequencies of rs12354 and rs3759333 had no statistical differences between chronic hepatitis B and HBV-related HCC patients. The serum LTbetaR levels and the overall survival rate between HBV-related HCC patients carrying different rs12354 and rs3759333 genotypes had no statistical differences. These results suggest that the LTBR rs12354 polymorphism might be associated with the spontaneous resolution of HBV infection. Additional studies with large sample size are needed to confirm and extend these findings.
DOI:10.1038/ncomms7087URLPMID:25608663 [本文引用: 1]
Intrahepatic cholangiocarcinoma (iCCA) is a fatal bile duct cancer with dismal prognosis and limited therapeutic options. By performing RNA- and exome-sequencing analyses, we report a novel fusion event, FGFR2-PPHLN1 (16%), and damaging mutations in the ARAF oncogene (11%). Here we demonstrate that the chromosomal translocation t(10;12)(q26;q12) leading to FGFR2-PPHLN1 fusion possesses transforming and oncogenic activity, which is successfully inhibited by a selective FGFR2 inhibitor in vitro. Among the ARAF mutations, N217I and G322S lead to activation of the pathway and N217I shows oncogenic potential in vitro. Screening of a cohort of 107 iCCA patients reveals that FGFR2 fusions represent the most recurrent targetable alteration (45%, 17/107), while they are rarely present in other primary liver tumours (0/100 of hepatocellular carcinoma (HCC); 1/21 of mixed iCCA-HCC). Taken together, around 70% of iCCA patients harbour at least one actionable molecular alteration (FGFR2 fusions, IDH1/2, ARAF, KRAS, BRAF and FGF19) that is amenable for therapeutic targeting.
DOI:10.1016/j.stem.2015.10.001URLPMID:26526725 [本文引用: 1]
The extremely low efficiency of human embryonic stem cell (hESC) derivation using somatic cell nuclear transfer (SCNT) limits its potential application. Blastocyst formation from human SCNT embryos occurs at a low rate and with only some oocyte donors. We previously showed in mice that reduction of histone H3 lysine 9 trimethylation (H3K9me3) through ectopic expression of the H3K9me3 demethylase Kdm4d greatly improves SCNT embryo development. Here we show that overexpression of a related H3K9me3 demethylase KDM4A improves human SCNT, and that, as in mice, H3K9me3 in the human somatic cell genome is an SCNT reprogramming barrier. Overexpression of KDM4A significantly improves the blastocyst formation rate in human SCNT embryos by facilitating transcriptional reprogramming, allowing efficient derivation of SCNT-derived ESCs using adult Age-related Macular Degeneration (AMD) patient somatic nuclei donors. This conserved mechanistic insight has potential applications for improving SCNT in a variety of contexts, including regenerative medicine.
DOI:10.1002/mgg3.181URLPMID:26788539 [本文引用: 1]
BACKGROUND: Juvenile-onset cataracts are known among the Hutterites of North America. Despite being identified over 30 years ago, this autosomal recessive condition has not been mapped, and the disease gene is unknown. METHODS: We performed whole exome sequencing of three Hutterite-type cataract trios and follow-up genotyping and mapping in four extended kindreds. RESULTS: Trio exomes enabled genome-wide autozygosity mapping, which localized the disease gene to a 9.5-Mb region on chromosome 6p. This region contained two candidate variants, LEMD2 c.T38G and MUC21 c.665delC. Extended pedigrees recruited for variant genotyping revealed multiple additional relatives with juvenile-onset cataract, as well as six deceased relatives with both cataracts and sudden cardiac death. The candidate variants were genotyped in 84 family members, including 17 with cataracts; only the variant in LEMD2 cosegregated with cataracts (LOD = 9.62). SNP-based fine mapping within the 9.5 Mb linked region supported this finding by refining the cataract locus to a 0.5- to 2.9-Mb subregion (6p21.32-p21.31) containing LEMD2 but not MUC21. LEMD2 is expressed in mouse and human lenses and encodes a LEM domain-containing protein; the c.T38G missense mutation is predicted to mutate a highly conserved residue within this domain (p.Leu13Arg). CONCLUSION: We performed a genetic and genomic study of Hutterite-type cataract and found evidence for an association of this phenotype with sudden cardiac death. Using combined genetic and genomic approaches, we mapped cataracts to a small portion of chromosome 6 and propose that they result from a homozygous missense mutation in LEMD2.
.
[本文引用: 1]
DOI:10.1016/j.ajhg.2016.09.005URLPMID:27745833 [本文引用: 1]
This study establishes PYROXD1 variants as a cause of early-onset myopathy and uses biospecimens and cell lines, yeast, and zebrafish models to elucidate the fundamental role of PYROXD1 in skeletal muscle. Exome sequencing identified recessive variants in PYROXD1 in nine probands from five families. Affected individuals presented in infancy or childhood with slowly progressive proximal and distal weakness, facial weakness, nasal speech, swallowing difficulties, and normal to moderately elevated creatine kinase. Distinctive histopathology showed abundant internalized nuclei, myofibrillar disorganization, desmin-positive inclusions, and thickened Z-bands. PYROXD1 is a nuclear-cytoplasmic pyridine nucleotide-disulphide reductase (PNDR). PNDRs are flavoproteins (FAD-binding) and catalyze pyridine-nucleotide-dependent (NAD/NADH) reduction of thiol residues in other proteins. Complementation experiments in yeast lacking glutathione reductase glr1 show that human PYROXD1 has reductase activity that is strongly impaired by the disease-associated missense mutations. Immunolocalization studies in human muscle and zebrafish myofibers demonstrate that PYROXD1 localizes to the nucleus and to striated sarcomeric compartments. Zebrafish with ryroxD1 knock-down recapitulate features of PYROXD1 myopathy with sarcomeric disorganization, myofibrillar aggregates, and marked swimming defect. We characterize variants in the oxidoreductase PYROXD1 as a cause of early-onset myopathy with distinctive histopathology and introduce altered redox regulation as a primary cause of congenital muscle disease.
DOI:10.2147/OPTH.S126459URLPMID:28356709 [本文引用: 1]
Primary glaucomas are among the most common eye diseases that may potentially result in bilateral blindness. Both genetics and environmental factors are reported to be involved in the etiology of primary glaucomas. Secreted protein acidic and rich in cysteine (SPARC)-related modular calcium binding protein 2 (SMOC2) is a matricellular glycoprotein encoded by the SMOC2 gene and known to regulate the expression of extracellular matrix (ECM) proteins and matrix metalloproteinases (MMPs), which play an important role in the pathogenesis of primary glaucomas. The frequencies of alleles and genotypes of SMOC2 variants were examined in 406 Saudi subjects, including primary open angle glaucoma (POAG, n=140) and primary angle closure glaucoma (PACG, n=64) patients and 202 matched healthy controls using the polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) technique. Genotyping of SMOC2 polymorphism (rs13208776) revealed a significantly higher frequency of the heterozygous genotype GA (P<0.01) and a lower frequency of wild type GG genotype (P=0.05) in glaucoma patients compared to the controls. Upon stratification of the patients on the basis of types of glaucoma, PACG patients had a significantly higher frequency of GA genotype as compared to the controls (P<0.01), whereas there was no significant difference between the POAG patient and control groups in frequencies of SMOC2 alleles and genotypes. Further, there was no significant difference in frequency distribution of alleles and genotypes between male and female patients. This study indicates that the GA genotype of SMOC2 (G>A) polymorphism is significantly associated with PACG and may be a risk factor. However, further large-scale studies in the Saudi population as well as in other ethnic populations are needed to confirm this association.
DOI:10.7150/jca.20775URLPMID:29151969 [本文引用: 1]
Secreted modular calcium binding protein-2 (SMOC2), a recently identified matricellular protein that belongs to the SPARC protein family, has been reported to be downregulated in various cancers. The purpose of this study was to investigate the clinical significance and biological function of SMOC2 in human hepatocellular carcinoma. Real-time quantitative PCR and western blotting analyses revealed that SMOC2 mRNA and protein levels were significantly downregulated in human HCC tissues compared to the matched adjacent normal tissues. Clinicopathological analysis indicated that SMOC2 expression was significantly associated with tumor size, number of tumors, tumor-node-metastasis (TNM) stage and distant metastasis. Kaplan-Meier survival analysis showed that high tumor SMOC2 expression was associated with improved overall survival and disease-free survival in patients with HCC. Functional analyses (cell proliferation and colony formation assays, cell migration and invasion assays, cell cycle and apoptosis assays) demonstrated that stable overexpression of SMOC2 using a lentiviral vector significantly inhibited cell proliferation, colony formation, migration and invasion, and induced G0/G1 phase arrest in HCC cells in vitro. In addition, experiments with a mouse model revealed the suppressed effect of SMOC2 on HCC tumorigenicity and metastases in vivo. These results suggest that SMOC2 functions as a tumor suppressor during the development of HCC and may represent an effective prognostic factor and novel therapeutic target for HCC.
DOI:10.3389/fnana.2017.00050URLPMID:28785208 [本文引用: 1]
The cerebellum (Cb) is an exquisite structure that controls elaborate motor behaviors and is essential for sensory-motor learning. During development, the Cb is derived from rhombomere 1 (r1). Within this embryonic compartment, precursors in r1 are patterned by signaling cues originating from the isthmus organizer (IsO) and subsequently undergo complex morphogenic movements to establish their final position in the mature Cb. The transcription factor Gbx2 is expressed in the developing Cb and is intimately involved in organizing and patterning the Cb. Nevertheless, how precursors expressing Gbx2 at specific embryonic time points contribute to distinct cell types in the adult Cb is unresolved. In this study, we used Genetic Inducible Fate Mapping (GIFM) to mark Gbx2-expressing precursors with fine temporal resolution and to subsequently track this lineage through embryogenesis. We then determined the terminal neuronal fate of the Gbx2 lineage in the adult Cb. Our analysis demonstrates that the Gbx2 lineage contributes to the Cb with marking over the course of five stages: Embryonic day 7.5 (E7.5) through E11.5. The Gbx2 lineage gives rise to Purkinje cells, granule neurons, and deep cerebellar neurons across these marking stages. Notably, the contribution of the Gbx2 lineage shifts as development proceeds with each marking stage producing a distinct profile of mature neurons in the adult Cb. These findings demonstrate the relationship between the temporal expression of Gbx2 and the terminal cell fate of neurons in the Cb. Based on these results, Gbx2 is critical to Cb development, not only for its well-defined role in positioning and maintaining the IsO, but also for guiding the development of Cb precursors and determining the identity of Cb neurons.
DOI:10.1016/j.ydbio.2015.08.010URLPMID:26297811 [本文引用: 1]
The thalamus and habenula, two important nodes of the forebrain circuitry, are derived from a single developmental compartment, called prosomere 2, in the diencephalon. Habenular and thalamic neurons display distinct molecular identity, neurochemistry, and connectivity. Furthermore, their progenitors exhibit distinctive neurogenic patterns with a marked delay in the onset of neurogenesis in the thalamus. However, the progenitors in prosomere 2 express many common developmental regulators and the mechanism underlying the specification and differentiation of these two populations of neurons remains unknown. Gbx2, coding for a homeodomain transcription factor, is initially expressed in thalamic neuronal precursors that have just exited the cell cycle, and its expression is maintained in many mature thalamic neurons in adults. Deletion of Gbx2 severely disrupts histogenesis of the thalamus and abolishes thalamocortical projections in mice. Here, by using genome-wide transcriptional profiling, we show that Gbx2 promotes thalamic but inhibits habenular molecular characters. Remarkably, although Gbx2 is expressed in postmitotic neuronal precursors, deletion of Gbx2 changes gene expression and cell proliferation in dividing progenitors in the developing thalamus. These defects are partially rescued by the mosaic presence of wild-type cells, demonstrating a cell non-autonomous role of Gbx2 in regulating the development of thalamic progenitors. Our results suggest that Gbx2 is essential for the acquisition of the thalamic neuronal identity by repressing habenular identity through a feedback signaling from postmitotic neurons to progenitors.
DOI:10.1212/NXG.0000000000000148URLPMID:28589176 [本文引用: 1]
OBJECTIVE: We aimed to generate a review and description of the phenotypic and genotypic spectra of ARHGEF9 mutations. METHODS: Patients with mutations or chromosomal disruptions affecting ARHGEF9 were identified through our clinics and review of the literature. Detailed medical history and examination findings were obtained via a standardized questionnaire, or if this was not possible by reviewing the published phenotypic features. RESULTS: A total of 18 patients (including 5 females) were identified. Six had de novo, 5 had maternally inherited mutations, and 7 had chromosomal disruptions. All females had strongly skewed X-inactivation in favor of the abnormal X-chromosome. Symptoms presented in early childhood with delayed motor development alone or in combination with seizures. Intellectual disability was severe in most and moderate in patients with milder mutations. Males with severe intellectual disability had severe, often intractable, epilepsy and exhibited a particular facial dysmorphism. Patients with mutations in exon 9 affecting the protein's PH domain did not develop epilepsy. CONCLUSIONS: ARHGEF9 encodes a crucial neuronal synaptic protein; loss of function of which results in severe intellectual disability, epilepsy, and a particular facial dysmorphism. Loss of only the protein's PH domain function is associated with the absence of epilepsy.
DOI:10.1007/s00415-017-8539-3URLPMID:28620718 [本文引用: 1]
Mutations or structural genomic alterations of the X-chromosomal gene ARHGEF9 have been described in male and female patients with intellectual disability. Hyperekplexia and epilepsy were observed to a variable degree, but incompletely described. Here, we expand the phenotypic spectrum of ARHGEF9 by describing a large Ethiopian-Jewish family with epilepsy and intellectual disability. The four affected male siblings, their unaffected parents and two unaffected female siblings were recruited and phenotyped. Parametric linkage analysis was performed using SNP microarrays. Variants from exome sequencing in two affected individuals were confirmed by Sanger sequencing. All affected male siblings had febrile seizures from age 2-3 years and intellectual disability. Three developed afebrile seizures between age 7-17 years. Three showed focal seizure semiology. None had hyperekplexia. A novel ARHGEF9 variant (c.967G>A, p.G323R, NM_015185.2) was hemizygous in all affected male siblings and heterozygous in the mother. This family reveals that the phenotypic spectrum of ARHGEF9 is broader than commonly assumed and includes febrile seizures and focal epilepsy with intellectual disability in the absence of hyperekplexia or other clinically distinguishing features. Our findings suggest that pathogenic variants in ARHGEF9 may be more common than previously assumed in patients with intellectual disability and mild epilepsy.
DOI:10.1016/j.omtn.2019.12.035URLPMID:32045876 [本文引用: 2]
Terminally differentiated somatic cells can be reprogrammed into a totipotent state through somatic cell nuclear transfer (SCNT). The incomplete reprogramming is the major reason for developmental arrest of SCNT embryos at early stages. In our studies, we found that pathways for autophagy, endocytosis, and apoptosis were incompletely activated in nuclear transfer (NT) 2-cell arrest embryos, whereas extensively inhibited pathways for stem cell pluripotency maintenance, DNA repair, cell cycle, and autophagy may result in NT 4-cell embryos arrest. As for NT normal embryos, a significant shift in expression of developmental transcription factors (TFs) Id1, Pou6f1, Cited1, and Zscan4c was observed. Compared with pluripotent gene Ascl2 being activated only in NT 2-cell, Nanog, Dppa2, and Sall4 had major expression waves in normal development of both NT 2-cell and 4-cell embryos. Additionally, Kdm4b/4d and Kdm5b had been confirmed as key markers in NT 2-cell and 4-cell embryos, respectively. Histone acetylases Kat8, Elp6, and Eid1 were co-activated in NT 2-cell and 4-cell embryos to facilitate normal development. Gadd45a as a key driver functions with Tet1 and Tet2 to improve the efficiency of NT reprogramming. Taken together, our findings provided an important theoretical basis for elucidating the potential molecular mechanisms and identified reprogramming driver factor to improve the efficiency of SCNT reprogramming.
DOI:10.1095/biolreprod.103.017731URLPMID:12801984 [本文引用: 1]
Although it is widely assumed that the cell type and genotype of the donor cell affect the efficiency of somatic cell cloning, little systematic analysis has been done to verify this assumption. The present study was undertaken to examine whether donor cell type, donor genotype, or a combination thereof increased the efficiency of mouse cloning. Initially we assessed the developmental ability of embryos that were cloned from cumulus or immature Sertoli cells with six different genotypes (i.e., 2 x 6 factorial). Significantly better cleavage rates were obtained with cumulus cells than with Sertoli cells (P < 0.005, two-way ANOVA), which probably was due to the superior cell-cycle synchrony of cumulus cells at G0/G1. After embryo transfer, there was a significant effect of cell type on the birth rate, with Sertoli cells giving the better result (P < 0.005). Furthermore, there was a significant interaction (P < 0.05) between the cell type and genotype, which indicates that cloning efficiency is determined by a combination of these two factors. The highest mean birth rate (10.8 +/- 2.1%) was obtained with (B6 x 129)F1 Sertoli cells. In the second series of experiments, we examined whether the developmental ability of clones with the wild-type genotype (JF1) was improved when combined with the 129 genotype. Normal pups were cloned from cumulus and immature Sertoli cells of the (129 x JF1)F1 and (JF1 x 129)F1 genotypes, whereas no pups were born from cells with the (B6 x JF1)F1 genotype. The present study clearly demonstrates that the efficiency of somatic cell cloning, and in particular fetal survival after embryo transfer, may be improved significantly by choosing the appropriate combinations of cell type and genotype.
DOI:10.1530/REP-15-0338URLPMID:26515777 [本文引用: 1]
Aberrant epigenetic reprogramming is the main obstacle to the development of somatic cell nuclear transfer (SCNT) embryos and the generation of induced pluripotent stem (iPS) cells, which results in the low reprogramming efficiencies of SCNT and iPS. Histone H3 lysine 27 trimethylation (H3K27me3), as a repressive epigenetic mark, plays important roles in mammalian development and iPS induction. However, the reprogramming of H3K27me3 in pig remains elusive. In this study, we showed that H3K27me3 levels in porcine early cloned embryos were higher than that in IVF embryos. Then GSK126 and GSK-J4, two small molecule inhibitors of H3K27me3 methylase (EZH2) and demethylases (UTX/JMJD3), were used to regulate the H3K27me3 level. The results showed that H3K27me3 level was reduced in cloned embryos after treatment of PEF with 0.75 muM GSK126 for 48 h, incubation of one-cell reconstructed oocytes with 0.1 muM GSK126 and injection of antibody for EZH2 into oocyte. Meanwhile, the development of the cloned embryos was significantly improved after these treatments. On the contrary, GSK-J4 treatment increased the H3K27me3 level in cloned embryos and decreased the cloned embryonic development. Furthermore, iPS efficiency was both increased after reducing the H3K27me3 level in donor cells and in early reprogramming phase. In summary, our results suggest that H3K27me3 acts as an epigenetic barrier in SCNT and iPS reprogramming, and reduction of H3K27me3 level in donor cells and in early reprogramming phase can enhance both porcine SCNT and iPS efficiency.
DOI:10.1016/j.cell.2014.09.055URL [本文引用: 1]
Mammalian oocytes can reprogram somatic cells into a totipotent state enabling animal cloning through somatic cell nuclear transfer (SCNT). However, the majority of SCNT embryos fail to develop to term due to undefined reprogramming defects. Here, we identify histone H3 lysine 9 trimethylation (H3K9me3) of donor cell genome as a major barrier for efficient reprogramming by SCNT. Comparative transcriptome analysis identified reprogramming resistant regions (RRRs) that are expressed normally at 2-cell mouse embryos generated by in vitro fertilization (IVF) but not SCNT. RRRs are enriched for H3K9me3 in donor somatic cells and its removal by ectopically expressed H3K9me3 demethylase Kdm4d not only reactivates the majority of RRRs, but also greatly improves SCNT efficiency. Furthermore, use of donor somatic nuclei depleted of H3K9 methyltransferases markedly improves SCNT efficiency. Our study thus identifies H3K9me3 as a critical epigenetic barrier in SCNT-mediated reprogramming and provides a promising approach for improving mammalian cloning efficiency.
DOI:10.1186/s12864-018-5091-1URLPMID:30305014 [本文引用: 1]
BACKGROUND: Nuclear reprogramming reinstates totipotency or pluripotency in somatic cells by changing their gene transcription profile. This technology is widely used in medicine, animal husbandry and other industries. However, certain deficiencies severely restrict the applications of this technology. RESULTS: Using single-embryo RNA-seq, our study provides complete transcriptome blueprints of embryos generated by cumulus cell (CC) donor nuclear transfer (NT), embryos generated by mouse embryonic fibroblast (MEF) donor NT and in vivo embryos at each stage (zygote, 2-cell, 4-cell, 8-cell, morula, and blastocyst). According to the results from further analyses, NT embryos exhibit RNA processing and translation initiation defects during the zygotic genome activation (ZGA) period, and protein kinase activity and protein phosphorylation are defective during blastocyst formation. Two thousand three constant genes are not able to be reprogrammed in CCs and MEFs. Among these constant genes, 136 genes are continuously mis-transcribed throughout all developmental stages. These 136 differential genes may be reprogramming barrier genes (RBGs) and more studies are needed to identify. CONCLUSIONS: These embryonic transcriptome blueprints provide new data for further mechanistic studies of somatic nuclear reprogramming. These findings may improve the efficiency of somatic cell nuclear transfer.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}