 ,1,2, 孙玉洁,1,2
,1,2, 孙玉洁,1,2The NOXA promoter could function as an active enhancer to regulate the expression of BCL2 in the apoptosis response
Zhongyong Qin1,2, Xiao Shi1,2, Pingping Cao1,2, Ying Chu1,2, Wei Guan4, Nan Yang1,3, He Cheng,1,2, Yujie Sun,1,2通讯作者: 孙玉洁,博士,教授,研究方向:增强子生物学功能、肿瘤耐药形成机制。E-mail:yujiesun@njmu.edu.cn程禾,博士,讲师,研究方向:乳腺癌耐药形成机制。E-mail:chenghe@njmu.edu.cn
编委: 方向东
收稿日期:2020-07-7修回日期:2020-10-4网络出版日期:2020-11-20
| 基金资助: |
Received:2020-07-7Revised:2020-10-4Online:2020-11-20
| Fund supported: |
作者简介 About authors
秦中勇,硕士,专业方向:基因远程调控。E-mail:

摘要
真核生物基因的转录受到近端启动子和远端增强子的共同调控,部分基因的启动子可兼具有增强子的活性。NOXA与BCL2分别是BCL2蛋白家族促凋亡和抗凋亡成员。本课题组前期研究发现,NOXA基因启动子与BCL2基因启动子在染色质三维空间结构上存在相互作用,且NOXA基因启动子区兼有启动子和增强子特征性的组蛋白修饰标记。为进一步探究NOXA启动子是否具有增强子活性、能否在细胞凋亡过程中作为增强子调控BCL2基因表达,本研究利用染色质构象捕获(chromosome conformation capture, 3C)、实时荧光定量PCR (quantitative real-time PCR, qRT-PCR)和荧光素酶报告基因等检测技术在喜树碱诱导的MCF-7细胞凋亡模型中证实,NOXA启动子兼具增强子活性,并可通过形成染色质环结构远程调控BCL2基因表达。NOXA启动子的调控属性与凋亡信号强弱密切相关,在较弱凋亡信号刺激下(1 μmol/L喜树碱处理),NOXA启动子主要发挥增强子功能;随着凋亡刺激信号的加强(10 μmol/L喜树碱处理),NOXA启动子活性增强,主要调控其基因自身的表达,促进细胞凋亡。染色质免疫共沉淀(chromatin immunoprecipitation, ChIP)证实NOXA启动子区启动子活性和增强子活性的动态变化与其组蛋白修饰标志一致。本研究为进一步探讨BCL2家族成员对细胞凋亡刺激做出协同反应的机制提供了新的线索。
关键词:
Abstract
The transcription of eukaryotic genes is regulated by both proximal promoters and distal enhancers. Some promoters also have enhancer activity. NOXA and BCL2 are pro-apoptotic and anti-apoptotic members of the BCL2 family of protein, respectively. Our previous study has found that the NOXA gene promoter and the BCL2 gene promoter interact at the level of three-dimensional chromatin structure. Moreover, the NOXA gene promoter region displays histone modifications characteristic of both promoters and enhancers. This study aimed to explore whether and when the NOXA promoter could act as an active enhancer to regulate BCL2 expression. Based on the apoptosis model of MCF-7 cells induced by camptothecin, we used chromosome conformation capture (3C), quantitative real-time PCR (qRT-PCR) and the luciferase reporter gene technology to demonstrate that the NOXA promoter could function as an active enhancer and physically interact with the BCL2 promoter through chromatin looping. The regulatory properties of the NOXA promoter were closely related to the strength of the apoptosis stimulation. Under weak apoptotic stimulation (1 μmol/L camptothecin treatment), the NOXA promoter mainly functioned as an enhancer; with the enhancement of apoptotic stimulation (10 μmol/L camptothecin treatment), the NOXA promoter activity increased and mainly regulated the expression of the gene itself to promote apoptosis. Chromatin immunoprecipitation (ChIP) confirmed that the dynamic changes of the promoter activity and enhancer activity in the NOXA promoter region are consistent with its histone modification marks. This study provides new clues for further exploring the mechanism underlying cooperative response of BCL2 family member to apoptosis stimuli.
Keywords:
PDF (1302KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
秦中勇, 石晓, 曹平平, 褚鹰, 管蔚, 杨楠, 程禾, 孙玉洁. 细胞凋亡反应中NOXA基因启动子发挥增强子功能调节BCL2基因表达. 遗传[J], 2020, 42(11): 1110-1121 doi:10.16288/j.yczz.20-213
Zhongyong Qin.
真核生物中基因转录是非常复杂的过程,反式作用因子需要通过识别各种类型的DNA调控序列来实现基因的转录调控,启动子和增强子是最具代表性也是研究最广泛的两类顺式转录调控元件[1,2,3,4]。启动子通常定位于基因的5?末端,包含TATA盒、下游核心启动子等元件组分,可以与RNA聚合酶II (RNAP II)结合,并确定转录起始位置和方向[5,6]。增强子大多具有组织或细胞特异性,可以在远端通过形成染色质环的方式与靶基因启动子充分接近,协助靶启动子募集转录因子和辅转录因子,从而加速靶基因的转录,以确保基因在时间空间上正确表达[7,8,9]。
区别增强子和启动子的主要依据它们相对于基因5?末端的位置以及特异性组蛋白修饰的富集[1,10,11]。从所在位置来看,增强子具有激活远端启动子的特性,而与其靶基因的位置和方向无关,启动子则只能启动近端基因转录。启动子和增强子可以通过区域内特异性的组蛋白修饰来进行预测和分析,活性增强子通常富含组蛋白H3 Lys4的一甲基化(H3K4me1)和组蛋白H3 Lys27乙酰化(H3K27ac)修饰[12,13,14,15],启动子则通常表现为组蛋白H3 Lys4的三甲基化(H3K4me3)修饰。因此,H3K27ac的存在伴随着高水平的H3K4me1和低水平的H3K4me3被视作活性增强子的标志。具有增强子功能的启动子兼有启动子和增强子特征性的组蛋白修饰,并与其他启动子形成远距离的相互作用[4,16]。虽然通过高通量的方法,越来越多的增强子样启动子被鉴定了出来,然而对其生物学功能的探究甚少[16,17,18],研究增强子样启动子在特定生理病理条件下发挥的功能和调控方式可以帮助人们全面认识转录调控网络。
NOXA和BCL2基因定位于人18号染色体上,所编码的蛋白分别为BCL2蛋白家族重要的促凋亡和抗凋亡成员[19,20,21,22,23]。BCL2蛋白家族抗凋亡成员包括BCL-2、BCL-W、A1等,促凋亡成员又分为凋亡效应物(Bax、Bak)和凋亡激活物(Bim、Bid、Puma、NOXA等),各个成员转录水平在正常生理状态下的比例维持稳定,精密调节细胞凋亡。一旦细胞受到凋亡信号的刺激,如药物暴露、营养条件匮乏、氧化应激等刺激因素会打破两种基因的转录平衡[19,24,25],抗凋亡成员如BCL2的基因水平会被下调,而凋亡激活成员NOXA等会过度表达激活凋亡效应物的寡聚化,寡聚化的凋亡效应物会转移至线粒体外膜进行穿孔,释放细胞色素C,激活Caspase途径的细胞凋亡。
本课题组前期在Jurkat细胞中发现NOXA与BCL2基因均可被增强子mbr通过形成染色质环的方式所调控,而NOXA启动子与BCL2启动子存在空间上的相互作用,敲除mbr后两启动子的相互作用依然存在[26,27,28,29,30]。无独有偶,在乳腺癌细胞系MCF-7和正常乳腺上皮细胞MCF-10A中本课题组也检测到了NOXA和BCL2启动子的相互作用,同时在数据库查询发现NOXA启动子区兼具启动子和增强子特征性的组蛋白修饰。这一有趣的现象激发了对NOXA启动子的转录元件属性和BCL2家族基因调控新机制的思考。因此,本研究旨在探究NOXA启动子是否具有增强子活性、能否作为增强子调控BCL2基因表达及什么情况下发挥增强子功能。这对认识基因调控元件属性切换的条件和意义将有新的启发。
1 材料与方法
1.1 细胞培养
人乳腺癌细胞系MCF-7、正常乳腺上皮细胞MCF-10A购买自美国ATCC细胞库。MCF-10A所用培养液为DMEM-F12培养基,并添加终浓度为5%的马血清和4种生长因子(浓度为0.02 μg/mL EGF生长因子、10 μg/mL胰岛素、0.5 μg/mL氢化可地松、0.1 μg/mL霍乱毒素)以及两种抗生素:100 U/mL青霉素、100 μg/mL链霉素。MCF-7所用培养液为MEM培养基,并添加终浓度为10%胎牛血清、100 U/mL青霉素、100 μg/mL链霉素以及0.2 U/mL胰岛素。上述两种细胞在CO2浓度为5%、温度为37℃的恒温培养箱中进行培养。
1.2 载体构建
利用GenBank数据库查询NOXA、BCL2基因转录起始位点上游1000 bp至下游500 bp的序列为基因的启动子区,利用Prime 5.0软件设计合适的PCR引物,并选择合适的酶切位点。将PCR片段插入到pGL3-basic质粒荧光素酶报告基因上游检测启动子活性,插入pGL3-promoter质粒荧光素酶报告基因下游检测增强子活性,所用PCR片段与相应质粒使用同种限制酶切割,利用清洁回收试剂盒(广州美基生物)纯化后进行连接,转化导入大肠杆菌DH5α中,随后挑取单菌落扩增,鉴定阳性克隆。1.3 细胞凋亡模型
取处于对数生长期的MCF-7细胞和MCF-10A细胞,接种于6孔板和24孔板中,培养24 h后用不同浓度(0.0、0.1、0.5、1.0、5.0、10.0、15.0 μmol/L) 的喜树碱(camptothecin, CPT)处理10 h后,收取细胞,分别检测NOXA、BCL2基因的RNA水平以及各个喜树碱浓度下的细胞凋亡水平;同时将空载的对照组质粒、重构的pGL3-basic、pGL3-promoter质粒分别与内参海肾荧光素酶荧光素酶质粒pRL-SV40按质量比为1000∶1的比例,利用脂质体Lipfectamine 3000 (Thermo Fisher,美国)共转染进细胞,4~6 h后用上述浓度梯度的喜树碱处理相同时间(10 h),检测各浓度喜树碱下的NOXA启动子的启动子活性和增强子活性。综合分析细胞凋亡程度、NOXA和BCL2的RNA变化水平以及NOXA启动子不同方面的转录元件活性。1.4 RNA提取和 qRT-PCR
采用Trizol法提取细胞总RNA,利用试剂盒(南京诺唯赞生物科技股份有限公司)进行逆转录,各个样品取500 ng总RNA进行逆转录后,按1∶10稀释比例稀释各组cDNA,以稀释后的cDNA为模板,配制SYBR Green I Master Mix体系(Roche,瑞士),使用Light Cycler 480 II实时荧光定量PCR仪(Roche,瑞士)检测NOXA、BCL2以及内参基因β-Actin的表达变化,所用引物如下,RT-NOXA- F:5?-GCAGAGCTGGAAGTCGAGTGT-3?;RT-NOXA- R:5?-CTCTTTTGAAGGAGTCCCCTCAT-3?;RT-BCL2- F:5?-TCGCCCTGTGGATGACTGAG-3?;RT-BCL2- R:5?-CAGAGTCTTCAGAGACAGCCAGGA-3?;RT- β-Actin-F:5?-TCATGAAGTGTGACGTGGACAT-3?;RT-β-Actin-R:5?-CTCAGGAGGAGCAATGATCTTG- 3?。PCR扩增程序为:95℃ 10 min,95℃ 10 s,60℃ 30 s,72℃ 20 s;35个循环,采用2-ΔΔCt法分析定量PCR数据。1.5 细胞凋亡检测
使用凋亡检测试剂盒(南京诺唯赞生物科技股份有限公司)检测细胞凋亡。每组收集1×105个细胞,洗涤并重悬,加入Annexin V-FITC和PI Staining Solution避光、室温孵育染色;随后加入Binding Buffer,轻轻混匀,染色后样品在1 h内用流式细胞仪检测细胞凋亡情况(BD,美国)。流式细胞仪激发波长为488 nm;FITC的绿色荧光在FL1通道检测;PI的红色荧光在FL3 通道检测,每个样本采集10000个细胞。用Cell Quest软件进行数据分析,FL1为横坐标,FL3为纵坐标,根据FITC和PI荧光值确定两荧光参数阴阳界限,划定十字门。其中细胞可分为三个亚群:活细胞为双阴性(Annexin V-FITC-/PI-);早期凋亡细胞为Annexin V-FITC单阳性(Annexin V-FITC+/PI-);晚期凋亡细胞为Annexin V-FITC和PI双阳性(Annexin V-FITC+/PI+)。1.6 染色质免疫共沉淀(chromatin immunoprecipitation, ChIP)
使用ChIP试剂盒(CST,美国)进行实验,收集1×107个细胞,将细胞在室温下用含1 %甲醛的培养基交联固定10 min,接着用甘氨酸终止交联,裂解细胞后用超声破碎仪(SONICS,美国)将基因组DNA剪切成200~1000 bp的短片段,离心后取上清用ChIP Buffer稀释分别用H3K4me1和H3K4me3抗体(Millipore,美国)在4℃孵育过夜。次日,加入磁珠4℃孵育2 h后进行洗涤,之后在65℃水浴中解交联、吸附柱纯化DNA。最后取ChIP产物进行qRT-PCR分析,检测NOXA启动子区H3K4me1和H3K4me3药物处理前后的变化情况,所用引物为ChIP-F:5?- GTTGTTCTGAGGACGGAATG-3?;ChIP-R:5?-GCATAATGTTTGGCGAGGC-3?。 qRT-PCR扩增程序为:95℃ 10 min,95℃ 10 s,60℃ 30 s,72℃ 20 s;35个循环。以Input的Ct值作为校正值,采用2-ΔΔCt法进行相对定量分析。1.7 染色质构象捕获(chromosome conformation capture, 3C)
收集1×106个细胞,用2 mL含2%甲醛的培养基,在室温下交联固定细胞5 min,用浓度为0.125 mol/L的甘氨酸终止交联。随后用1.25 mL 3C裂解液(10 mmol/L Tris-HCl、pH=8.0、10 mmol/L NaCl、0.2% NP-40)在4℃条件下翻转孵育60 min。之后进行分步酶切:每管样品加入135.2 μL ddH2O、20 μL NEB buffer、4.4 μL 10% SDS在37℃摇床中孵育1 h;加入32.4 μL浓度10%的Triton在37℃摇床中孵育1 h;再加入8 μL Hind Ⅲ 限制性内切酶(NEB,美国)在37℃摇床中孵育过夜(22 h左右);然后每管加入38 μL 10% SDS (终浓度1.6%) 65℃水浴20 min使酶失活;随后取40 μL上一步酶切产物加入70 μL NEB Buffer、140 μL Triton、444 μL ddH2O进行在16℃水浴中连接过夜。最后连接产物用酚氯仿抽提纯化,取200 ng DNA为模板,用以下4条引物引物A:5?-GTCTAAGAGTAGCTCCTGTA-3?;引物B:5?-CTAAATCTAAACAGAGAACCCA-3?;引物C:5?-GTTTGAGAATGATGAATGATTTG-3?;引物D:5?-TAACATTCCTTCTGTGCTGCTA-3?的不同组合来扩增3C产物,Loading control扩增的序列不含所选酶切位点,以检测基因组DNA的完整性,所用引物为:Loading control-F:5?-GAAATCGTGCGTGACATTAA-3?;Loading control-R:5?-AAGGAAGGCTGGAAGAGTG-3?,上述PCR采用Hot Star (Qiagen,德国)体系扩增。1.8 双荧光素酶活性检测
本实验采用Dual Luciferase Reporter Assay Kit (南京诺唯赞生物科技股份有限公司)进行,收取24孔板中的细胞,每孔加入100 μL 1×Cell Lysis Buffer,室温条件下裂解5~ 10 min后,吹打并吸取全部细胞裂解产物至1.5 mL EP管中,常温下12,000 g离心5 min,取上清用于后续检测。向100 μL Luciferase Substrate中加入20 μL上清,检测萤火虫荧光素酶读值,记为RLU1,随后加入100 μL经Stop & Reaction Buffer稀释的1× Renilla Substrate,该步操作可以终止萤火虫荧光素酶的反应并起始海肾荧光素酶的反应,将海肾荧光素酶的检测读值记为RLU2,计算每个样品RLU1/ RLU2的比值,并进行统计分析。1.9 统计学分析
实验数据以3次实验的平均值和标准误(mean± SEM)表示。组间比较用双尾 t 检验并计算P值, *表示P<0.05,差异有统计学意义;**表示P<0.01,差异显著。2 结果与分析
2.1 NOXA基因的启动子具有增强子活性
通过查询UCSC数据库[31]NOXA启动子区和BCL2启动子区组蛋白ChIP-seq数据,发现NOXA基因启动子区兼有增强子和启动子特征性的组蛋白修饰的标记H3K4me1、H3K4me3以及H3K27ac (图1A),提示其可能为增强子样启动子。利用双荧光素酶报告基因系统,分别构建不同类型的报告基因质粒,转染进正常乳腺上皮细胞MCF-10A和乳腺癌细胞MCF-7中检测这两个启动子的转录元件活性[32,33]。实验结果显示,NOXA启动子和BCL2启动子均能起始荧光素酶报告基因的转录(图1B);而NOXA启动子兼具增强子活性,BCL2启动子则未检测到增强子活性;为了进一步验证NOXA启动子作为增强子时是否具有方向性,将NOXA启动子序列翻转后插入pGL3-promoter质粒报告基因的下游,结果并未检测到荧光强度上调(图1C)。结果表明,与经典增强子有所不同,NOXA启动子发挥增强子功能时有方向效应,序列翻转后增强子活性消失。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1分析和鉴定NOXA基因启动子的增强子活性
A:UCSC数据库数据显示的NOXA基因启动子区(上图)和BCL2基因启动子区(下图)的组蛋白H3K4me1、H3K4me3、H3K27ac修饰特征。B:启动子活性验证。将NOXA启动子和BCL2启动子分别插入至pGL3-basic荧光素酶基因上游后转染进MCF-10A、MCF-7细胞中(对照为pGL3-basic空载质粒),验证二者的启动子活性;C:增强子活性验证。将NOXA启动子和BCL2启动子分别插入至pGL3-promoter荧光素酶基因下游后转染MCF-10A和MCF-7细胞(对照为pGL3-promoter空载质粒),检测二者的增强子活性,结果显示NOXA启动子兼具有增强子活性。D:翻转NOXA启动子后增强子活性验证。将NOXA启动子翻转后插入至pGL3-promoter荧光素酶基因下游后转染MCF-10A和MCF-7细胞(对照为pGL3-promoter空载质粒),检测NOXA启动子翻转后的增强子活性。*:P<0.05;**:P<0.01。
Fig. 1Determination of enhancer activity of the NOXA promoter
2.2 NOXA启动子与BCL2启动子存在相互作用
本课题组前期在Jurkat细胞中发现NOXA启动子与BCL2基因启动子存在空间上的相互作用,为了验证在乳腺癌MCF-7细胞中的情况,本研究开展了3C实验[34,35,36]。实验原理及引物设计如图2A所示,即细胞在甲醛交联状态下,利用限制性内切酶Hind Ⅲ进行切割后,再利用T4 DNA连接酶进行连接,如果两启动子序列在空间上足够接近,则可被分别设计在序列两端的单向引物的组合而扩增出PCR产物。实验结果显示,利用位于NOXA启动子的上游的引物A和位于BCL2启动子下游的引物C组合可成功扩增出217 bp的PCR的产物(图2B)。对此PCR产物的DNA测序结果证实产物DNA序列一部分来自NOXA基因启动子,另一部分来自BCL2基因启动子(图2C)。该实验结果提示NOXA启动子与BCL2启动子可以形成染色质环,从而在空间上靠近。结合荧光素酶报告基因的实验结果,提示NOXA启动子可能作为增强子调控BCL2的表达。图2
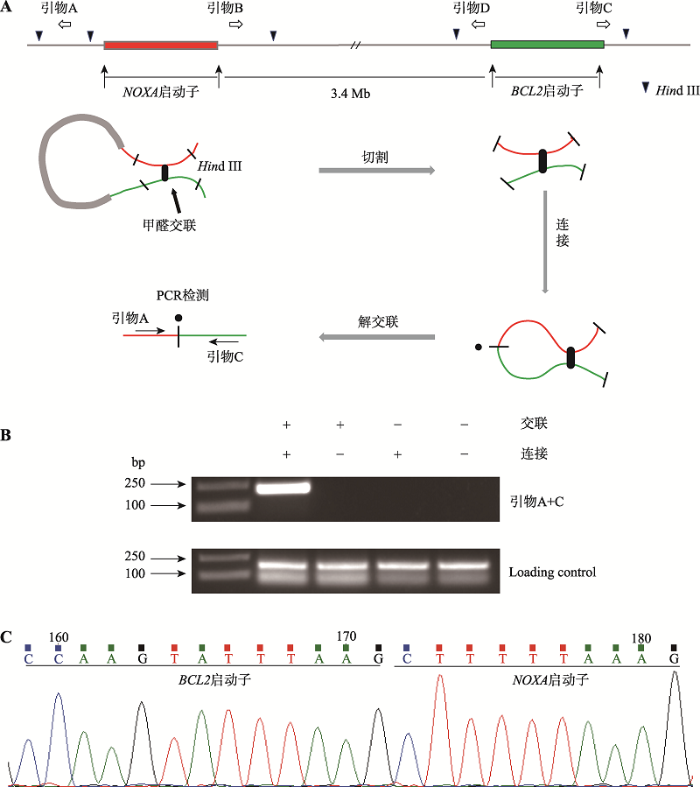 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图23C证明NOXA启动子与BCL2启动子存在空间互作
A:NOXA基因与BCL2基因的相对位置示意图。染色质构象捕获实验的原理以及所用的酶切位点和引物位置。黑色倒三角表示Hind Ⅲ限制性内切酶识别位点、白色空心箭头表示引物设计的位置及其扩增方向、红色标注的是NOXA启动子区、绿色标注的序列为BCL2启动子区。B:MCF-7细胞中的3C结果。上方DNA琼脂糖凝胶电泳图从左至右泳道依次为DNA Marker、交联连接、交联不连接、不交联连接、不交联不连接的3C产物PCR结果,交联且连接处的条带由引物A和引物C组合扩增产生。C:实验组PCR产物TA克隆后进行Sanger测序结果。
Fig. 2Identification of a spatial interaction between the NOXA promoter and BCL2 promoter by 3C
2.3 NOXA启动子作为增强子激活BCL2启动子
为验证NOXA启动子对BCL2启动子的调控作用,构建了不同类型的荧光素酶报告基因质粒,将BCL2基因启动子插入至报告基因上游,发挥启动子的作用;将NOXA启动子插入至报告基因的下游,验证其是否可以作为增强子促进上调BCL2启动子的转录活性(图3A)。结果显示在MCF-7细胞中,NOXA启动子位于荧光素酶报告基因下游时,未检测到报告基因的表达(图3B,②),排除了NOXA启动子反向激活报告基因的情况[37,38,39]。当BCL2启动子位于报告基因上游,NOXA启动子位于荧光素酶报告基因下游时(图3B,④),报告基因的活性比上游仅存在BCL2启动子时(图3B,③)上调了2.5倍左右,也即说明NOXA启动子可以作为增强子促进BCL2启动子转录,结合3C的结果可以说明NOXA启动子可作为增强子调控BCL2基因。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3荧光素酶报告基因实验证实NOXA启动子对BCL2启动子的增强子效应
A:不同类型的报告基因质粒模式图。分别显示各调控序列与报告基因的相对位置以及两个调控序列间的相对位置。当位于报告基因下游的NOXA调控序列具有增强子活性时,可增强上游BCL2基因启动子的活性。B:荧光素酶报告基因活性检测。分别将上述报告基因质粒转染至MCF-7细胞中,检测其荧光素酶报告基因的活性所得结果,以确定NOXA基因启动子序列对BCL2基因启动子的增强子效应。**:P<0.01。
Fig. 3Luciferase reporter gene experiments confirm the enhancer effect of the NOXA promoter on the BCL2 promoter
2.4 喜树碱诱导NOXA基因的启动子向增强子转变
喜树碱是重要的抗肿瘤药物[40,41],通过靶向抑制DNA拓扑异构酶I[42],引起细胞内DNA不可逆损伤,诱导细胞凋亡反应[40]。为建立紫杉醇诱导的细胞凋亡模型,分别用低浓度(1 μmol/L)和高浓度(10μmol/L)喜树碱处理MCF-7细胞10 h后用流式细胞术检测细胞凋亡情况。结果显示用1 μmol/L和10 μmol/L喜树碱处理时MCF-7细胞凋亡比例分别为6.6%和11.5% (图4A)。同步检测NOXA启动子的转录元件活性,发现NOXA启动子的增强子活性在1 μmol/L喜树碱刺激时处于最高水平,此时启动子活性无明显变化;10 μmol/L喜树碱处理细胞时NOXA启动子活性最强而增强子活性弱(图4,B和C)。同时依据ENCODE数据库[43]MCF-7细胞中特征性的组蛋白修饰峰图设计ChIP引物(图4D),检测NOXA基因启动子区的增强子和启动子特征性组蛋白修饰标记H3K4me1和H3K4me3的变化(图4,E和F),用H3K4me1/H3K4me3的比值来表征基因组上NOXA启动子区的转录元件属性,结果显示1 μmol/L喜树碱处理时H3K4me1/ H3K4me3的比值最高,此时NOXA启动子主要作为增强子发挥作用,而10 μmol/L喜树碱处理时H3K4me1/H3K4me3的比值最低,NOXA启动子发挥启动子本身的作用(图4G)。这与上述报告基因中得到启动子和增强子活性的变化结果是一致的。我们推断用1 μmol/L和10 μmol/L喜树碱处理细胞时,NOXA启动子发挥的转录元件的属性不同。在平行实验中,本研究利用qRT-PCR分析了NOXA和BCL2基因的表达情况。结果显示在不同药物浓度的处理下,BCL2的浓度均有下降,但在1 μmol/L喜树碱处理时,即NOXA启动子的增强子活性最强的时候,BCL2下降幅度最小,而用10 μmol/L喜树碱处理时,即NOXA启动子活性最强时,BCL2下降幅度较大,而此时NOXA mRNA处于最高水平。这些结果提示NOXA启动子在低浓度药物下可作为增强子调控BCL2,高浓度喜树碱刺激下NOXA主要作为启动子起始自身基因的表达(图4,H和I),进一步证实了NOXA启动子作为增强子对BCL2的调控作用。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4喜树碱诱导NOXA基因启动子向增强子转变
A:喜树碱处理后各组凋亡情况。分别以低浓度(1μmol/L)和高浓度(10μmol/L)喜树碱处理MCF-7细胞10 h,收获细胞后运用流式细胞术测定细胞凋亡。B和C:利用报告基因系统分别检测不同浓度喜树碱处理MCF-7细胞后10 h后,NOXA基因启动子和增强子活性变化。D:ENCODE数据库中的MCF-7细胞H3K4me1和H3K4me3修饰的ChIP-seq数据。红框圈出的部分为H3K4me1和H3K4me3组蛋白修饰重叠区域,在该部分设计合适的ChIP引物。E和F:利用ChIP验证不同浓度喜树碱处理MCF-7细胞后NOXA启动子区组蛋白修饰H3K4me1和H3K4me3的变化。G: ChIP数据H3K4me1/H3K4me3的比值变化。H:喜树碱处理MCF-7细胞BCL2基因的mRNA水平变化。I:喜树碱处理MCF-7细胞NOXA基因的mRNA水平变化。*:P<0.05;**:P<0.01。
Fig. 4Camptothecin induced enhancer activity from the NOXA gene promoter
3 讨论
本研究以NOXA和BCL2基因启动子间的相互作用为切入点,鉴定了一个新的具有增强子功能的启动子—NOXA启动子。NOXA启动子可通过形成染色质环结构远程调控BCL2基因表达。NOXA启动子的调控属性与凋亡信号强弱密切相关,在较弱凋亡信号刺激下(1 μmol/L喜树碱处理),NOXA启动子主要发挥增强子功能,随着凋亡刺激信号的加强(10 μmol/L喜树碱处理),NOXA启动子活性增强,主要调控其基因自身的表达,促进细胞凋亡。在喜树碱诱导的细胞凋亡模型中,细胞中BCL2的mRNA水平是呈药物浓度依赖性的下降,即药物浓度越高BCL2的转录水平下降越多,结合NOXA启动子表现出的增强子活性变化情况,我们推测这种现象的产生是由于细胞在应对不同程度的凋亡刺激时产生的不同反应。当轻微凋亡刺激时,促凋亡基因NOXA启动子活性较低,NOXA表达水平不高,而与此同时NOXA启动子表现较高增强子水平,使本该急剧下降的抗凋亡基因BCL2表达只有轻微下降;当强烈的凋亡刺激作用于细胞时,细胞凋亡不可挽回,此时NOXA启动子活性升高,其增强子活性降低,导致NOXA表达水平上升,BCL2表达水平进一步下降,细胞走向凋亡[19,21,44,45]。NOXA启动子作为BCL2的增强子在这里以一种“刹车机制”来发挥增强子作用,因其不是激活BCL2的表达而是拮抗BCL2的转录下调。本研究揭示了NOXA启动子作为增强子样启动子参与了细胞凋亡进程,在发挥启动子或增强子功能时受不同强弱凋亡信号的调节,对认识基因转录调控元件的转换和激活的调控机制及在生物学进程中的意义有新的启发。有意思的是NOXA基因启动子作为增强子时所调控的靶基因BCL2是同家族中与其蛋白功能相互拮抗的成员,这有助于人们更好地了解BCL2家族各成员之间的联系,为进一步探讨BCL2家族成员对细胞凋亡刺激做出协同反应的机制提供了新的线索。
细胞特异性染色质相互作用可以为细胞特异性的基因转录提供构象上的支持[33]。本课题组在多种细胞(MCF-7、MCF-10A、Jurkat细胞)均检测到了NOXA启动子与BCL2启动子在空间上的相互作用,并且在上述细胞中同时检测到了NOXA基因启动子兼具增强子活性,证明该种调控方式并不存在正常或肿瘤细胞特异性,暗示两者通过转录元件的调控是一种通用型转录调控模式。
增强子样启动子通过与靶基因启动子在空间上的相互作用来调节基因的表达[46,47],两者空间上的接近很可能是通过引入关键的转录因子,如ZNF143或YY1[48],据已有报道这两个因子参与了染色质环的形成,并在增强子样启动子上有着高丰度的结合[16,17]。本研究虽然初步探索了启动子向增强子转变的条件,但并未找到驱动这种转变的机制,具体的转变机制是未来需要进一步探讨的。值得注意的是,有文献报道在前列腺癌中长非编码RNA PCAT19的截短异构体的启动子同时也是一个增强子[17],作为增强子时调控全长PCAT19异构体的表达,而该启动子向增强子转换的因素是一个SNP,该SNP的不同风险位点介导了不同的转录因子的结合,从而决定了SNP所在区域是启动子还是增强子,这说明启动子向增强子的转变很可能是由于一种或多种关键转录因子所介导的,这是目前极少数涉及同一序列在启动子与增强子之间转换机制的报道。已有文献报道增强子样启动子结合的转录因子数量比普通启动子大,其种类也有所不同,这引发人们联想,是否在不同生理状态或刺激下增强子样启动子如NOXA启动子可选择性结合不同的转录因子从而实现其在启动子和增强子间的转换?本课题组将细胞凋亡过程中关键转录因子SATB1在MCF-7细胞中过量表达,发现NOXA启动子活性无明显变化,而NOXA启动子的增强子活性却大幅度增加(结果未列出),说明SATB1参与了NOXA启动子向增强子转化的过程,但是否为转化的驱动因素其具体机制如何则需要进一步的证明。
本研究工作为进一步探讨BCL2蛋白家族成员对细胞凋亡刺激做出协同反应的机制提供了新的线索。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/j.cell.2015.08.008URLPMID:26317464 [本文引用: 2]

With the explosion of genome-wide studies of regulated transcription, it has become clear that traditional definitions of enhancers and promoters need to be revisited. These control elements can now be characterized in terms of their local and regional architecture, their regulatory components, including histone modifications and associated binding factors, and their functional contribution to transcription. This Review discusses unifying themes between promoters and enhancers in transcriptional regulatory mechanisms.
DOI:10.1016/0092-8674(83)90410-5URLPMID:6305503 [本文引用: 1]
DOI:10.1101/gad.924301URLPMID:11581157 [本文引用: 1]
To investigate the basis for enhancer-promoter specificity, we compared the ability of enhancers to activate transcription in vivo from core promoters containing either downstream promoter element (DPE) or TATA box motifs. To eliminate position effects, we generated and analyzed pairs of sister Drosophila lines that contain a DPE- or TATA-dependent reporter gene at precisely the same genomic position relative to each enhancer. These studies revealed transcriptional enhancers that are specific for promoters that contain either DPE or TATA box elements. Thus, the core promoter not only mediates the initiation of transcription, but also functions as a regulatory element.
DOI:10.3724/SP.J.1005.2011.01291URL [本文引用: 2]
DOI:10.3724/SP.J.1005.2011.01291URL [本文引用: 2]
URLPMID:6296253 [本文引用: 1]
The SV40 early region promoter, previously localized to the DNA segment bounded by the HpaII and HindIII restriction sites (nucleotides 346 and 5171), was further defined by construction of an extensive set of deletions within this region and measurement of their effects on (a) viral DNA replication, (b) virus multiplication and the ability to complement early and late mutations, (c) transformation of rat cells, (d) large T antigen formation, and (e) the location of the 5' ends of early mRNAs. One set of mutations is represented by deletions that begin at the HpaII site and extend unidirectionally for varying lengths toward the BglI site at ori. A second set of mutants contains deletions that start at ori and extend unidirectionally for varying lengths towards the HpaII site. A third set of mutants, with deletions or duplications of various lengths and boundaries, lie between the HpaII and BglI sites. Our studies indicate the following. (a) Ori, the sequence needed for initiating SV40 DNA replication, extends from the sequences needed for initiating SV40 DNA replication, extends from the sequences needed to bind T antigen to the palindrome in site II to nucleotide 34, the late region edge of the AT block. Flanking sequences adjacent to the AT block facilitate DNA replication. (b) The SV40 early region promoter comprises two functionally distinct nucleotide sequence elements. One is flanked by nucleotides 5231 and 107, and contains the RNA initiation sites at nucleotides 5231-5237, a positioning element resembling the TATAAATA consensus sequence about 20-25 nucleotides upstream, and an RNA polymerase II recognition sequence contributed by short GC-rich sequences clustered between nucleotides 35 and 107; we refer to this as the RNA polymerase II interaction site. The second distinct sequence element is contained within each of two 72-bp segments located between nucleotides 107 and 250; the behavior of this element suggests that it may influence the accessibility of RNA polymerase II for the interaction site or the efficiency of RNA chain initiation. Large T antigen binding sites I, II, and III overlap with the putative RNA polymerase II interaction site; since large T antigen does not prevent elongation of RNA transcripts initiated upstream, T antigen probably represses early region expression by preventing RNA polymerase II binding to the promoter.
DOI:10.1146/annurev.biochem.72.121801.161520URLPMID:12651739 [本文引用: 1]
The events leading to transcription of eukaryotic protein-coding genes culminate in the positioning of RNA polymerase II at the correct initiation site. The core promoter, which can extend ~35 bp upstream and/or downstream of this site, plays a central role in regulating initiation. Specific DNA elements within the core promoter bind the factors that nucleate the assembly of a functional preinitiation complex and integrate stimulatory and repressive signals from factors bound at distal sites. Although core promoter structure was originally thought to be invariant, a remarkable degree of diversity has become apparent. This article reviews the structural and functional diversity of the RNA polymerase II core promoter.
DOI:10.1146/annurev.cb.04.110188.001015URLPMID:2848550 [本文引用: 1]
DOI:10.1534/genetics.104.026955URLPMID:15342512 [本文引用: 1]
Eukaryotic enhancers act over very long distances, yet still show remarkable specificity for their own promoter. To better understand mechanisms underlying this enhancer-promoter specificity, we used transvection to analyze enhancer choice between two promoters, one located in cis to the enhancer and the other in trans to the enhancer, at the yellow gene of Drosophila melanogaster. Previously, we demonstrated that enhancers at yellow prefer to act on the cis-linked promoter, but that mutation of core promoter elements in the cis-linked promoter releases enhancers to act in trans. Here, we address the mechanism by which these elements affect enhancer choice. We consider and explicitly test three models that are based on promoter competency, promoter pairing, and promoter identity. Through targeted gene replacement of the endogenous yellow gene, we show that competency of the cis-linked promoter is a key parameter in the cis-trans choice of an enhancer. In fact, complete replacement of the yellow promoter with both TATA-containing and TATA-less heterologous promoters maintains enhancer action in cis.
DOI:10.1038/ng.545URLPMID:20208536 [本文引用: 1]
Chromatin plays a central role in eukaryotic gene regulation. We performed genome-wide mapping of epigenetically marked nucleosomes to determine their position both near transcription start sites and at distal regulatory elements, including enhancers. In prostate cancer cells, where androgen receptor binds primarily to enhancers, we found that androgen treatment dismisses a central nucleosome present at androgen receptor binding sites that is flanked by a pair of marked nucleosomes. A new quantitative model built on the behavior of such nucleosome pairs correctly identified regions bound by the regulators of the immediate androgen response, including androgen receptor and FOXA1. More importantly, this model also correctly predicted previously unidentified binding sites for other transcription factors present after prolonged androgen stimulation, including OCT1 and NKX3-1. Therefore, quantitative modeling of enhancer structure provides a powerful predictive method to infer the identity of transcription factors involved in cellular responses to specific stimuli.
DOI:10.1101/gad.308619.117URLPMID:29378788 [本文引用: 1]
Gene expression is regulated by promoters, which initiate transcription, and enhancers, which control their temporal and spatial activity. However, the discovery that mammalian enhancers also initiate transcription questions the inherent differences between enhancers and promoters. Here, we investigate the transcriptional properties of enhancers during Drosophila embryogenesis using characterized developmental enhancers. We show that while the timing of enhancer transcription is generally correlated with enhancer activity, the levels and directionality of transcription are highly varied among active enhancers. To assess how this impacts function, we developed a dual transgenic assay to simultaneously measure enhancer and promoter activities from a single element in the same embryo. Extensive transgenic analysis revealed a relationship between the direction of endogenous transcription and the ability to function as an enhancer or promoter in vivo, although enhancer RNA (eRNA) production and activity are not always strictly coupled. Some enhancers (mainly bidirectional) can act as weak promoters, producing overlapping spatio-temporal expression. Conversely, bidirectional promoters often act as strong enhancers, while unidirectional promoters generally cannot. The balance between enhancer and promoter activity is generally reflected in the levels and directionality of eRNA transcription and is likely an inherent sequence property of the elements themselves.
DOI:10.1038/nature12787URL [本文引用: 1]
Enhancers control the correct temporal and cell-type-specific activation of gene expression in multicellular eukaryotes. Knowing their properties, regulatory activity and targets is crucial to understand the regulation of differentiation and homeostasis. Here we use the FANTOM5 panel of samples, covering the majority of human tissues and cell types, to produce an atlas of active, in vivo-transcribed enhancers. We show that enhancers share properties with CpG-poor messenger RNA promoters but produce bidirectional, exosome-sensitive, relatively short unspliced RNAs, the generation of which is strongly related to enhancer activity. The atlas is used to compare regulatory programs between different cells at unprecedented depth, to identify disease-associated regulatory single nucleotide polymorphisms, and to classify cell-type-specific and ubiquitous enhancers. We further explore the utility of enhancer redundancy, which explains gene expression strength rather than expression patterns. The online FANTOM5 enhancer atlas represents a unique resource for studies on cell-type-specific enhancers and gene regulation.
DOI:10.1038/nature07829URLPMID:19295514 [本文引用: 1]
The human body is composed of diverse cell types with distinct functions. Although it is known that lineage specification depends on cell-specific gene expression, which in turn is driven by promoters, enhancers, insulators and other cis-regulatory DNA sequences for each gene, the relative roles of these regulatory elements in this process are not clear. We have previously developed a chromatin-immunoprecipitation-based microarray method (ChIP-chip) to locate promoters, enhancers and insulators in the human genome. Here we use the same approach to identify these elements in multiple cell types and investigate their roles in cell-type-specific gene expression. We observed that the chromatin state at promoters and CTCF-binding at insulators is largely invariant across diverse cell types. In contrast, enhancers are marked with highly cell-type-specific histone modification patterns, strongly correlate to cell-type-specific gene expression programs on a global scale, and are functionally active in a cell-type-specific manner. Our results define over 55,000 potential transcriptional enhancers in the human genome, significantly expanding the current catalogue of human enhancers and highlighting the role of these elements in cell-type-specific gene expression.
DOI:10.1073/pnas.1016071107URLPMID:21106759 [本文引用: 1]
Developmental programs are controlled by transcription factors and chromatin regulators, which maintain specific gene expression programs through epigenetic modification of the genome. These regulatory events at enhancers contribute to the specific gene expression programs that determine cell state and the potential for differentiation into new cell types. Although enhancer elements are known to be associated with certain histone modifications and transcription factors, the relationship of these modifications to gene expression and developmental state has not been clearly defined. Here we interrogate the epigenetic landscape of enhancer elements in embryonic stem cells and several adult tissues in the mouse. We find that histone H3K27ac distinguishes active enhancers from inactive/poised enhancer elements containing H3K4me1 alone. This indicates that the amount of actively used enhancers is lower than previously anticipated. Furthermore, poised enhancer networks provide clues to unrealized developmental programs. Finally, we show that enhancers are reset during nuclear reprogramming.
DOI:10.1016/j.molcel.2016.03.033URLPMID:27153539 [本文引用: 1]
The role of cytosine methylation in the structure and function of enhancers is not well understood. In this study, we investigate the role of DNA methylation at enhancers by comparing the epigenomes of the HCT116 cell line and its highly demethylated derivative, DKO1. Unlike promoters, a portion of regular and super- or stretch enhancers show active H3K27ac marks co-existing with extensive DNA methylation, demonstrating the unexpected presence of bivalent chromatin in both cultured and uncultured cells. Furthermore, our findings also show that bivalent regions have fewer nucleosome-depleted regions and transcription factor-binding sites than monovalent regions. Reduction of DNA methylation genetically or pharmacologically leads to a decrease of the H3K27ac mark. Thus, DNA methylation plays an unexpected dual role at enhancer regions, being anti-correlated focally at transcription factor-binding sites but positively correlated globally with the active H3K27ac mark to ensure structural enhancer integrity.
DOI:10.1038/s41588-017-0015-6URLPMID:29255264 [本文引用: 1]
Enhancers act to regulate cell-type-specific gene expression by facilitating the transcription of target genes. In mammalian cells, active or primed enhancers are commonly marked by monomethylation of histone H3 at lysine 4 (H3K4me1) in a cell-type-specific manner. Whether and how this histone modification regulates enhancer-dependent transcription programs in mammals is unclear. In this study, we conducted SILAC mass spectrometry experiments with mononucleosomes and identified multiple H3K4me1-associated proteins, including many involved in chromatin remodeling. We demonstrate that H3K4me1 augments association of the chromatin-remodeling complex BAF to enhancers in vivo and that, in vitro, H3K4me1-marked nucleosomes are more efficiently remodeled by the BAF complex. Crystal structures of the BAF component BAF45C indicate that monomethylation, but not trimethylation, is accommodated by BAF45C's H3K4-binding site. Our results suggest that H3K4me1 has an active role at enhancers by facilitating binding of the BAF complex and possibly other chromatin regulators.
DOI:10.1038/ng.3884URLPMID:28581502 [本文引用: 3]
Gene expression in mammals is precisely regulated by the combination of promoters and gene-distal regulatory regions, known as enhancers. Several studies have suggested that some promoters might have enhancer functions. However, the extent of this type of promoters and whether they actually function to regulate the expression of distal genes have remained elusive. Here, by exploiting a high-throughput enhancer reporter assay, we unravel a set of mammalian promoters displaying enhancer activity. These promoters have distinct genomic and epigenomic features and frequently interact with other gene promoters. Extensive CRISPR-Cas9 genomic manipulation demonstrated the involvement of these promoters in the cis regulation of expression of distal genes in their natural loci. Our results have important implications for the understanding of complex gene regulation in normal development and disease.
DOI:10.1016/j.cell.2018.06.014URLPMID:30033362 [本文引用: 3]
The prostate cancer (PCa) risk-associated SNP rs11672691 is positively associated with aggressive disease at diagnosis. We showed that rs11672691 maps to the promoter of a short isoform of long noncoding RNA PCAT19 (PCAT19-short), which is in the third intron of the long isoform (PCAT19-long). The risk variant is associated with decreased and increased levels of PCAT19-short and PCAT19-long, respectively. Mechanistically, the risk SNP region is bifunctional with both promoter and enhancer activity. The risk variants of rs11672691 and its LD SNP rs887391 decrease binding of transcription factors NKX3.1 and YY1 to the promoter of PCAT19-short, resulting in weaker promoter but stronger enhancer activity that subsequently activates PCAT19-long. PCAT19-long interacts with HNRNPAB to activate a subset of cell-cycle genes associated with PCa progression, thereby promoting PCa tumor growth and metastasis. Taken together, these findings reveal a risk SNP-mediated promoter-enhancer switching mechanism underlying both initiation and progression of aggressive PCa.
DOI:10.1016/j.tibs.2018.03.004URLPMID:29673772 [本文引用: 1]
Gene expression in higher eukaryotes is precisely regulated in time and space through the interplay between promoters and gene-distal regulatory regions, known as enhancers. The original definition of enhancers implies the ability to activate gene expression remotely, while promoters entail the capability to locally induce gene expression. Despite the conventional distinction between them, promoters and enhancers share many genomic and epigenomic features. One intriguing finding in the gene regulation field comes from the observation that many core promoter regions display enhancer activity. Recent high-throughput reporter assays along with clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-related approaches have indicated that this phenomenon is common and might have a strong impact on our global understanding of genome organisation and gene expression regulation.
DOI:10.1038/sj.cdd.4401987URLPMID:16763616 [本文引用: 3]
Individual BCL2 family members couple apoptosis regulation and cell cycle control in unique ways. Antiapoptotic BCL2 and BCL-x(L) are antiproliferative by facilitating G0. BAX is proapoptotic and accelerates S-phase progression. The dual functions in apoptosis and cell cycle are coordinately regulated by the multi-domain BCL2 family members (MCL-1) and suggest that survival is maintained at the expense of proliferation. The role of BH3-only molecules in cell cycle is more variable. BAD antagonizes both the cell cycle and antiapoptotic functions of BCL2 and BCL-x(L) through BH3 binding. BID has biochemically separable functions in apoptosis and S-phase checkpoint, determined by post-translational modification. p53-induced PUMA is known only to have apoptotic function. Inhibition of apoptosis is oncogenic, whereas promotion of cell cycle arrest is tumor suppressive. Paradoxically, selected BCL2 family members can be both oncogenic and tumor suppressive. Which of the dual functions predominates is lineage specific and context dependent.
DOI:10.4143/crt.2017.134URLPMID:28701032 [本文引用: 1]
Purpose: We investigated B-cell lymphoma 2 (BCL2) regulation across DNA, RNA, protein, and methylation status according to molecular subtype of breast cancer using The Cancer Genome Atlas (TCGA) database. Materials and Methods: We analyzed clinical and biological data on 1,096 breast cancers from the TCGA database. Biological data included reverse phase protein array (RPPA), mRNA sequencing (mRNA-seq), mRNA microarray, methylation, copy number alteration linear, copy number alteration nonlinear, and mutation data. Results: The luminal A and luminal B subtypes showed upregulated expression of RPPA and mRNAseq and hypomethylation compared to the human epidermal growth factor receptor 2 (HER2) and triple-negative subtypes (all p < 0.001). No mutations were found in any subjects. High mRNA-seq and high RPPA were strongly associated with positive estrogen receptor, positive progesterone receptor (all p < 0.001), and negative HER2 (p < 0.001 and p=0.002, respectively). Correlation analysis revealed a strong positive correlation between protein and mRNA levels and a strong negative correlation between methylation and protein and mRNA levels (all p < 0.001). The high BCL2 group showed superior overall survival compared to the low BCL2 group (p=0.006). Conclusion: The regulation of BCL2 was mainly associated with methylation across the molecular subtypes of breast cancer, and luminal A and luminal B subtypes showed upregulated expression of BCL2 protein, mRNA, and hypomethylation. Although copy number alteration may have played a minor role, mutation status was not related to BCL2 regulation. Upregulation of BCL2 was associated with superior prognosis than downregulation of BCL2.
DOI:10.1016/j.bbcan.2017.06.004URLPMID:28647470 [本文引用: 2]
A remarkable characteristic of majority of cancer cells is that, they fail to undergo apoptosis, which in turn confers them a survival advantage over normal cells. Targeted cancer therapy aims at disrupting the functions of proteins that play an important role during cancer progression. Antiapoptotic protein, BCL2, is one such protein that is highly upregulated in many cancers as compared to normal cells, making it an ideal target for cancer therapy. Although, several BCL2 targeting agents have been investigated over the past 30 years, very few have exhibited any clinical significance. This mini-review outlines a road map of existing BCL2 inhibitors and their relevance in treating cancer, and discusses potential strategies for future research with respect to BCL2 specific cancer therapy.
DOI:10.1007/s10517-014-2696-5URLPMID:25403402 [本文引用: 1]
Expression of Bcl2 family genes was studied during the early phase of long-term potentiation in the CA1 field of rat hippocampal slices. The level of Bax mRNA and protein increased, while the content of Bcl2 mRNA and protein decreased 30 min after tetanization of the Schaffer collaterals. Our results suggest that proteins of the Bcl2 family play a role in the mechanisms of synaptic plasticity.
DOI:10.1016/j.tibs.2013.12.006URLPMID:24503222 [本文引用: 1]
During apoptotic cell death, cellular stress signals converge at the mitochondria to induce mitochondrial outer-membrane permeabilization (MOMP) through B cell lymphoma-2 (BCL-2) family proteins and their effectors. BCL-2 proteins function through protein-protein interactions, the mechanisms and structural aspects of which are only now being uncovered. Recently, the elucidation of the dynamic features underlying their function has highlighted their structural plasticity and the consequent complex thermodynamic landscape governing their protein-protein interactions. These studies show that canonical interactions involve a conserved, hydrophobic groove, whereas non-canonical interactions function allosterically outside the groove. We review the latest structural advances in understanding the interactions and functions of mammalian BCL-2 family members, and discuss new opportunities to modulate these proteins in health and disease.
DOI:10.1084/jem.20030613URLPMID:14699081 [本文引用: 1]
Hypoxia is a common cause of cell death and is implicated in many disease processes including stroke and chronic degenerative disorders. In response to hypoxia, cells express a variety of genes, which allow adaptation to altered metabolic demands, decreased oxygen demands, and the removal of irreversibly damaged cells. Using polymerase chain reaction-based suppression subtractive hybridization to find genes that are differentially expressed in hypoxia, we identified the BH3-only Bcl-2 family protein Noxa. Noxa is a candidate molecule mediating p53-induced apoptosis. We show that Noxa promoter responds directly to hypoxia via hypoxia-inducible factor (HIF)-1alpha. Suppression of Noxa expression by antisense oligonucleotides rescued cells from hypoxia-induced cell death and decreased infarction volumes in an animal model of ischemia. Further, we show that reactive oxygen species and resultant cytochrome c release participate in Noxa-mediated hypoxic cell death. Altogether, our results show that Noxa is induced by HIF-1alpha and mediates hypoxic cell death.
DOI:10.1016/j.molcel.2004.12.030URLPMID:15694340 [本文引用: 1]
Apoptosis is initiated when Bcl-2 and its prosurvival relatives are engaged by proapoptotic BH3-only proteins via interaction of its BH3 domain with a groove on the Bcl-2-like proteins. These interactions have been considered promiscuous, but our analysis of the affinity of eight BH3 peptides for five Bcl-2-like proteins has revealed that the interactions vary over 10,000-fold in affinity, and accordingly, only certain protein pairs associate inside cells. Bim and Puma potently engaged all the prosurvival proteins comparably. Bad, however, bound tightly to Bcl-2, Bcl-xL, and Bcl-w but only weakly to A1 and not to Mcl-1. Strikingly, Noxa bound only Mcl-1 and A1. In accord with their complementary binding, Bad and Noxa cooperated to induce potent killing. The results suggest that apoptosis relies on selective interactions between particular subsets of these proteins and that it should be feasible to discover BH3-mimetic drugs that inactivate specific prosurvival targets.
DOI:10.1371/journal.pone.0139170URLPMID:26422397 [本文引用: 1]
Aberrant expression of special AT-rich binding protein 1 (SATB1), a global genomic organizer, has been associated with various cancers, which raises the question of how higher-order chromatin structure contributes to carcinogenesis. Disruption of apoptosis is one of the hallmarks of cancer. We previously demonstrated that SATB1 mediated specific long-range chromosomal interactions between the mbr enhancer located within 3'-UTR of the BCL2 gene and the promoter to regulate BCL2 expression during early apoptosis. In the present study, we used chromosome conformation capture (3C) assays and molecular analyses to further investigate the function of the SATB1-mediated higher-order chromatin structure in co-regulation of the anti-apoptotic BCL2 gene and the pro-apoptotic NOXA gene located 3.4Mb downstream on Chromosome 18. We demonstrated that the mbr enhancer spatially juxtaposed the promoters of BCL2 and NOXA genes through SATB1-mediated chromatin-loop in Jurkat cells. Decreased SATB1 levels switched the mbr-BCL2 loop to mbr-NOXA loop, and thus changed expression of these two genes. The SATB1-mediated dynamic switch of the chromatin loop structures was essential for the cooperative expression of the BCL2 and NOXA genes in apoptosis. Notably, the role of SATB1 was specific, since inhibition of SATB1 degradation by caspase-6 inhibitor or caspase-6-resistant SATB1 mutant reversed expression of BCL-2 and NOXA in response to apoptotic stimulation. This study reveals the critical role of SATB1-organized higher-order chromatin structure in regulating the dynamic equilibrium of apoptosis-controlling genes with antagonistic functions and suggests that aberrant SATB1 expression might contribute to cancer development by disrupting the co-regulated genes in apoptosis pathways.
DOI:10.1016/j.gene.2006.05.002URLPMID:16777355 [本文引用: 1]
The bcl-2 major breakpoint region (mbr), located within the 3'-UTR of the bcl-2 gene, is the site of the most common chromosomal translocation, t(14;18) (q32;q21), which occurs in follicular lymphoma. The mbr forms a triplex DNA structure under physiological conditions and the transcription factor special AT-rich sequence-binding protein 1 (SATB1) binds immediately downstream of the mbr. These observations raise the possibility that the mbr may be involved in regulation of bcl-2 gene expression. We investigated the role of the bcl-2 mbr on reporter gene activity and the relevance of SATB1 to this function in a variety of cell lines. We found that the mbr up-regulated reporter gene expression. Deletion of the 37-bp AT-rich SATB1 binding site abolished the bcl-2 mbr regulation of reporter gene expression. Overexpression of SATB1 enhanced bcl-2 mbr up-regulation of the reporter gene activity. Our data strongly demonstrated that the bcl-2 mbr possessed regulatory function that was related to SATB1.
DOI:10.1038/sj.onc.1210069URLPMID:17057736 [本文引用: 1]
BCL2 expression is finely tuned by a variety of environmental and endogenous stimuli and regulated at both transcriptional and post-transcriptional levels. Our previous investigations demonstrated that the BCL2 major breakpoint region (mbr) in the 3'-UTR upregulates reporter gene expression, which implies that this region possessed intrinsic regulatory function. However, the effect of the mbr on BCL2 expression, and the underlying regulatory mechanisms, remain to be elucidated. To assess the direct effect of the mbr on the transcriptional activity of the BCL2 gene, we employed targeted homologous recombination to establish a mbr(+)/mbr(-) heterozygous Nalm-6 cell line and then compared the transcriptional activity and apoptotic effect on transcription between the wild type and targeted alleles. We found that deletion of the mbr significantly decreased the transcriptional activity of the corresponding allele in the mbr(+)/mbr(-) cell. The BCL2 allele deleted of the mbr had a slower response to apoptotic stimuli than did the wild type allele. The regulatory function of the mbr was mediated through SATB1. Overexpression of SATB1 increased BCL2 expression, while knockdown of SATB1 with RNAi decreased BCL2 expression. Our results clearly indicated that the mbr could positively regulate BCL2 gene expression and this regulatory function was closely related to SATB1.
DOI:10.1093/nar/gkr023URLPMID:21310710 [本文引用: 1]
The 279-bp major breakpoint region (mbr) within the 3'-untranslated region (3'-UTR) of the BCL2 gene is a binding site of special AT-rich sequence binding protein 1 (SATB1) that is well known to participate in the long-range regulation of gene transcription. Our previous studies have revealed that the mbr could regulate BCL2 transcription over a 200-kb distance and this regulatory function was closely related to SATB1. This study is to explore the underlying mechanism and its relevance to cellular apoptosis. With chromosome conformation capture (3C) and chromatin immunoprecipitation (ChIP) assays we demonstrated that the mbr could physically interact with BCL2 promoter through SATB1-mediated chromatin looping, which was required for epigenetic modifications of the promoter, CREB accessibility and high expression of the BCL2 gene. During early apoptosis, SATB1 was a key regulator of BCL2 expression. Inhibition of SATB1 cleavage by treatment of cells with a caspase-6 inhibitor or overexpression of mutant SATB1 that was resistant to caspase-6, inhibited disassembly of the SATB1-mediated chromatin loop and restored the BCL2 mRNA level in Jurkat cells. These data revealed a novel mechanism of BCL2 regulation and mechanistically link SATB1-mediated long-range interaction with the regulation of a gene controlling apoptosis pathway for the first time.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.ygeno.2008.02.003URL [本文引用: 1]
Abstract
The University of California Santa Cruz (UCSC) Genome Bioinformatics website consists of a suite of free, open-source, on-line tools that can be used to browse, analyze, and query genomic data. These tools are available to anyone who has an Internet browser and an interest in genomics. The website provides a quick and easy-to-use visual display of genomic data. It places annotation tracks beneath genome coordinate positions, allowing rapid visual correlation of different types of information. Many of the annotation tracks are submitted by scientists worldwide; the others are computed by the UCSC Genome Bioinformatics group from publicly available sequence data. It also allows users to upload and display their own experimental results or annotation sets by creating a custom track. The suite of tools, downloadable data files, and links to documentation and other information can be found at http://genome.ucsc.edu/.[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.cell.2011.12.014URL [本文引用: 2]
Higher-order chromosomal organization for transcription regulation is poorly understood in eukaryotes. Using genome-wide Chromatin Interaction Analysis with Paired-End-Tag sequencing (ChIA-PET), we mapped long-range chromatin interactions associated with RNA polymerase II in human cells and uncovered widespread promoter-centered intragenic, extragenic, and intergenic interactions. These interactions further aggregated into higher-order clusters, wherein proximal and distal genes were engaged through promoter-promoter interactions. Most genes with promoter-promoter interactions were active and transcribed cooperatively, and some interacting promoters could influence each other implying combinatorial complexity of transcriptional controls. Comparative analyses of different cell lines showed that cell-specific chromatin interactions could provide structural frameworks for cell-specific transcription, and suggested significant enrichment of enhancer-promoter interactions for cell-specific functions. Furthermore, genetically-identified disease-associated noncoding elements were found to be spatially engaged with corresponding genes through long-range interactions. Overall, our study provides insights into transcription regulation by three-dimensional chromatin interactions for both housekeeping and cell-specific genes in human cells.
DOI:10.1126/science.1067799URLPMID:11847345 [本文引用: 1]
We describe an approach to detect the frequency of interaction between any two genomic loci. Generation of a matrix of interaction frequencies between sites on the same or different chromosomes reveals their relative spatial disposition and provides information about the physical properties of the chromatin fiber. This methodology can be applied to the spatial organization of entire genomes in organisms from bacteria to human. Using the yeast Saccharomyces cerevisiae, we could confirm known qualitative features of chromosome organization within the nucleus and dynamic changes in that organization during meiosis. We also analyzed yeast chromosome III at the G1 stage of the cell cycle. We found that chromatin is highly flexible throughout. Furthermore, functionally distinct AT- and GC-rich domains were found to exhibit different conformations, and a population-average 3D model of chromosome III could be determined. Chromosome III emerges as a contorted ring.
DOI:10.1101/pdb.top098210URLPMID:29438064 [本文引用: 1]
Biological reactions work well only within a narrow concentration range of hydrogen ions. Paradoxically, however, many of these reactions themselves generate or consume protons. Buffers are substances that undergo reversible protonation within a particular pH range and therefore maintain the concentration of hydrogen ions within acceptable limits. This introduction describes standard buffers used in molecular cloning, including Tris buffers, Good buffers, and phosphate buffers.
DOI:10.1007/s00412-016-0593-6URLPMID:27130552 [本文引用: 1]
It has been more than a decade since the first chromosome conformation capture (3C) assay was described. The assay was originally devised to measure the frequency with which two genomic loci interact within the three-dimensional (3D) nuclear space. Over time, this method has evolved both qualitatively and quantitatively, from detection of pairwise interaction of two unique loci to generating maps for the global chromatin interactome. Combined with the analysis of the epigenetic chromatin context, these advances led to the unmasking of general genome folding principles. The evolution of 3C-based methods has been supported first by the revolution in ChIP and then by sequencing-based approaches, methods that were primarily tools to study the unidimensional genome. The gradual improvement of 3C-based methods illustrates how the field adapted to the need to gradually address more subtle questions, beginning with enquiries of reductionist nature to reach more holistic perspectives, as the technology advanced, in a process that is greatly improving our knowledge on genome behavior and regulation. Here, we describe the evolution of 3C and other 3C-based methods for the analysis of chromatin interactions, along with a brief summary of their contribution in uncovering the significance of the three-dimensional world within the nucleus. We also discuss their inherent limitations and caveats in order to provide a critical view of the power and the limits of this technology.
DOI:10.1134/S0006297913040020URL [本文引用: 1]
DOI:10.1016/j.molcel.2015.04.006URLPMID:26028540 [本文引用: 1]
Anti-sense transcription originating upstream of mammalian protein-coding genes is a well-documented phenomenon, but remarkably little is known about the regulation or function of anti-sense promoters and the non-coding RNAs they generate. Here we define at nucleotide resolution the divergent transcription start sites (TSSs) near mouse mRNA genes. We find that coupled sense and anti-sense TSSs precisely define the boundaries of a nucleosome-depleted region (NDR) that is highly enriched in transcription factor (TF) motifs. Notably, as the distance between sense and anti-sense TSSs increases, so does the size of the NDR, the level of signal-dependent TF binding, and gene activation. We further discover a group of anti-sense TSSs in macrophages with an enhancer-like chromatin signature. Interestingly, this signature identifies divergent promoters that are activated during immune challenge. We propose that anti-sense promoters serve as platforms for TF binding and establishment of active chromatin to further regulate or enhance sense-strand mRNA expression.
DOI:10.1371/journal.pone.0043283URLPMID:22916237 [本文引用: 1]
Transient plasmid transfection is a common approach in studies in cultured mammalian cells. To examine behavior of transfected plasmids, we analyzed their transcriptional landscape by deep sequencing. We have found that the entire plasmid sequence is transcribed at different levels. Spurious transcription may have undesirable effects as some plasmids, when co-transfected, inhibited expression of luciferase reporters in a dose-dependent manner. In one case, we attributed this effect to a Kan/Neo resistance cassette, which generated a unique population of edited sense and antisense small RNAs. The unexpected complexity of expression from transiently transfected plasmids underscores the importance of appropriate experimental controls.
DOI:10.2174/092986706777585004URLPMID:16842195 [本文引用: 2]
The review provides a detailed discussion of recent advances in the medicinal chemistry of camptothecin, a potent antitumor agent that targets topoisomerase I. Thousands of CPT derivatives have been synthesized. Two of them, Topotecan and Irinotecan, are commercially approved for use in clinic as antitumor agents while more are still in clinic trials. This review summarizes the current status of the modern synthetic approaches to CPT, the mechanism of action of CPT, the structure-activity relationship(SAR), a number of novel CPT analogs and their biologic activity. There is a systematic evaluation of A-, B- and E-ring- modified camptothecins reported recently.
DOI:10.1080/14786410412331299005URLPMID:15938148 [本文引用: 1]
Topoisomerase I (Topo-I) is a major target for anticancer drug discovery and design. As a result, Topo-I inhibitors constitute an important class of the current anticancer drugs. To date, all of the Topo-I inhibitors that have been clinically evaluated are analogues of camptothecin (CPT), an extract of the Chinese tree Camptotheca acuminata. CPT has shown significant antitumor activity to lung, ovarian, breast, pancreas and stomach cancers. In this article the, phytochemical aspect, and various structural modifications are comprehensively reviewed as in rings A, B, C, D and E. Biological activity of camptothecin, other than anticancer, reported till the year 2003 has also been discussed.
DOI:10.1038/nrc1977URLPMID:16990856 [本文引用: 1]
Nuclear DNA topoisomerase I (TOP1) is an essential human enzyme. It is the only known target of the alkaloid camptothecin, from which the potent anticancer agents irinotecan and topotecan are derived. As camptothecins bind at the interface of the TOP1-DNA complex, they represent a paradigm for interfacial inhibitors that reversibly trap macromolecular complexes. Several camptothecin and non-camptothecin derivatives are being developed to further increase anti-tumour activity and reduce side effects. The mechanisms and molecular determinants of tumour response to TOP1 inhibitors are reviewed, and rational combinations of TOP1 inhibitors with other drugs are considered based on current knowledge of repair and checkpoint pathways that are associated with TOP1-mediated DNA damage.
[本文引用: 1]
[本文引用: 1]
DOI:10.1098/rsob.180002URLPMID:29769323 [本文引用: 1]
The ability of a cell to undergo mitochondrial apoptosis is governed by pro- and anti-apoptotic members of the BCL-2 protein family. The equilibrium of pro- versus anti-apoptotic BCL-2 proteins ensures appropriate regulation of programmed cell death during development and maintains organismal health. When unbalanced, the BCL-2 family can act as a barrier to apoptosis and facilitate tumour development and resistance to cancer therapy. Here we discuss the BCL-2 family, their deregulation in cancer and recent pharmaceutical developments to target specific members of this family as cancer therapy.
DOI:10.1016/j.canlet.2018.03.007URLPMID:29526801 [本文引用: 1]
Here we report that BCL2 blocks DNA double strand break (DSB) repair via nonhomologous end-joining (NHEJ), through sequestration of KU80 protein outside the nucleus. We find that this effect is associated with a repair switch to the error-prone PARP1-dependent end-joining (PARP1-EJ). We present in-vitro proof-of-concept for therapeutic targeting of this switch using PARP inhibitor to specifically enhance the radiosensitivity of BCL2-overexpressing cells. Given its erroneous behavior, PARP1-EJ might allow for the accumulation of genetic alterations and tumor progression. Consistently, we report an inverse correlation between BCL2 expression and biochemical recurrence-free survival of 10.259 prostate cancer (PCa) patients who underwent primary radical-prostatectomy for localized disease. Further, we evaluated retrospectively the impact of BCL2 expression on clinical outcome of 1.426 PCa patients, who had been given salvage radiotherapy at relapse after radical prostatectomy. In line with its role in blocking NHEJ, BCL2 over-expressers showed significantly better response to salvage radiotherapy compared to low-expressers. Collectively, our findings identify BCL2 status in PCa as a putative predictor of (i) radiotherapy response and (ii) response to treatment with PARP inhibitor olaparib as a radiosensitizing agent.
URLPMID:28417999 [本文引用: 1]
[本文引用: 1]
URLPMID:29224777 [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}