 ,1,2,3,4, 芦特1,2,3, 杜康1,2,3, 杨珺1,2,3, 康向阳,1,2,31
,1,2,3,4, 芦特1,2,3, 杜康1,2,3, 杨珺1,2,3, 康向阳,1,2,31 2
3
4
Variation of homologous recombination in Populus tomentosa with different genotypes
Geng Xining,1,2,3,4, Lu Te1,2,3, Du Kang1,2,3, Yang Jun1,2,3, Kang Xiangyang,1,2,31 2
3
4
通讯作者: 通讯作者: 康向阳,教授,博士生导师,研究方向:林木遗传育种。E-mail:kangxy@bjfu.edu.cn
第一联系人:
收稿日期:2020-07-6
| 基金资助: |
Received:2020-07-6
| Fund supported: |

摘要
同源重组是生物遗传变异的重要来源。受检测方法限制,高等植物同源重组发生及其产物——异源双链DNA(heteroduplex DNA, hDNA)鲜有报道。本研究采用构建抑制减数后分离群体检测同源重组产物hDNA的方法,以2个母本来源的基于抑制减数后分离获得的毛白杨(Populus tomentosa)杂种三倍体群体为研究材料,利用筛选出的110个简单序列重复(Simple sequence repeat, SSR)分子标记开展毛白杨不同基因型个体间9条染色体上hDNA发生及其遗传变异研究。结果表明,2个毛白杨雌株hDNA发生频率介于8.5%~87.2%之间,且hDNA发生频率与距着丝粒距离呈正相关关系,但同一染色体平均hDNA发生频率与染色体长度无相关关系;绝大多数的染色体检测出1~3次重组事件,少数检测出4次重组事件,极少数检测到5次重组事件;不同毛白杨基因型个体间同一染色体上hDNA发生频率总体相差不大,而在一些特定SSR位点间hDNA发生频率存在较大差异;与青杨杂种‘哲引3号’杨(P. pseudo-simonii×P. nigra ‘Zheyin3#’)相比,检测到的同源重组次数及hDNA发生频率和发生位置均存在较大差异。本研究首次对2个基因型毛白杨同源重组发生特征及其变异进行了研究,为揭示高等植物同源重组特点、种间和种内同源重组差异等提供了重要见解。
关键词:
Abstract
Keywords:
PDF (2208KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
耿喜宁, 芦特, 杜康, 杨珺, 康向阳. 不同基因型毛白杨同源重组变异研究. 遗传[J], 2021, 43(2): 182-193 doi:10.16288/j.yczz.20-205
Geng Xining.
在减数分裂期间,同源重组通过形成交叉互换(crossovers, COs)确保同源染色体的正确分离[1],同时,使同源染色体间DNA双链发生交换和基因转换,形成异源双链DNA(heteroduplex DNA, hDNA)而导致等位基因的重组,因此同源重组对于遗传多样性形成及物种进化等具有重要影响[2]。
作为一次减数分裂完成的产物——四分体是研究减数重组的理想型材料。酵母(Saccharomyces cerevisiae)在完成减数分裂后形成的四核子囊,保留了减数分裂后同源重组的完整信息,使得在酵母中进行同源重组研究[3,4]具有不可比拟的优势。拟南芥(Arabidopsis thaliana)qrt1突变体使花粉母细胞减数分裂发育而来的4个成熟花粉互不分离[5],再结合荧光标记技术,可实现植物同源重组的研究[6??~9]。借助于单细胞分离和单细胞基因组扩增技术,研究人员对分离后的玉米(Zea mays)、大麦(Hordeum vulgare)四分体进行了减数重组的研究[10,11]。Dong等[12]提出了一种通过阻止同源重组产物——hDNA减数后分离,进而采用共显性简单序列重复(simple sequence repeat, SSR)分子标记从DNA水平直接鉴定高等植物同源重组的新方法,首次对杨属(Populus)植物同源重组模式及其特点进行了研究,结果表明同源重组产物hDNA发生频率在青杨杂种‘哲引3号’杨(P. pseudo-simonii ×P. nigra ‘Zheyin3#’)中介于5.3%~76.6%之间。基于高通量测序构建的高密度遗传图谱的方法,研究人员发现同源重组发生在玉米不同基因型间具有一定差异[13]。而Dong等[12]研究仅涉及1个青杨杂种‘哲引3号’杨的同源重组特征,迄今未见有关杨属植物种间及种内不同基因型之间同源重组变异的研究报道。
本研究基于抑制减数后分离并结合共显性SSR分子标记实现同源重组产生的hDNA检测策略[12],以2个源于胚囊染色体加倍的毛白杨×银腺杨((P. tomentosa)×(P. alba ×P. glandulosa))全同胞杂种三倍体群体为材料,选择与Dong等[12]开展青杨杂种‘哲引3号’杨hDNA发生规律研究中同样的1~8号染色体以及可能为性染色体的杨树19号染色体(chromosome 19, Chr.19)[14,15]为对象,筛选与青杨杂种‘哲引3号杨’同源重组研究[12]相近数量的SSR标记对开展2个不同基因型毛白杨同源重组特征研究,探讨植物种内不同基因型之间同源重组变异特点,为揭示植物同源重组发生机制奠定基础。
1 材料与方法
1.1 材料
采用的实验材料为本实验室前期以毛白杨雌株3532(2n=2x=38)和3119(2n=2x=38)为母本,以银腺杨YX1为父本,通过高温处理诱导胚囊染色体加倍获得的2个三倍体群体[16]。随机选取2个三倍体群体的部分株系为研究材料,其中3532×YX1杂交组合包括45个三倍体子代,3119×YX1杂交组合包括47个三倍体子代。1.2 同源重组产物hDNA检测方法
杨属植物属蓼型胚囊植物,在胚囊发育期,功能大孢子需要经历3次有丝分裂,形成7-细胞8核的成熟配子体。施加高温处理诱导胚囊染色体加倍,即抑制减数后分离的结果(post-meiotic segregation restitution, PMR)[17,18],使获得的杨树三倍体细胞中的两套来源于母本的染色体保存了减数分裂期间发生的同源重组信息,即同源重组产物hDNA。根据Dong等[12]提出的hDNA检测策略,即在已经完成减数分裂的胚囊发育期,施加理化处理,使携带同源重组产物hDNA的姊妹染色单体不能进行正常的有丝分裂而共存于一个2n配子中,再授以单倍型异性配子,获得异源三倍体子代;筛选母本处于杂合状态且与父本有差异的多态性共显性SSR分子标记对三倍体进行遗传信息分析,根据2n配子是否保留母本的杂合遗传信息即可判断在该检测位点上是否有同源重组产物hDNA的产生。
1.3 1.3 DNA提取与SSR分子标记
利用植物基因组DNA提取试剂盒(天根生化科技(北京)有限公司),进行父母本和三倍体子代叶片基因组DNA的提取。TP-M13-SSR PCR技术[19]通过在上游引物引入一段通用的M13接头序列,可以使用荧光修饰的M13引物令扩增出的特异的PCR产物带上荧光,从而通过毛细管电泳进行检测。这种方法在较大程度上解决了分析通量较低、扩增产物检测流程繁琐、数据记录工作量过大等一系列问题。本研究根据该方法共需要3种引物,包括5′端接有M13序列(5′-TGTAAAACGACGGCCAGT-3′)的上游引物,普通下游引物以及标有荧光(ROX、FAM、TAMRA、HEX)的M13引物。PCR体系如下:ddH2O 7.2 μL,20 ng/μL模板DNA 2 μL,PCR Mix 10 μL (天根生化科技(北京)有限公司),10 μmol/L上游引物(5′端接有M13序列) 0.08 μL,10 μmol/L下游引物 0.32 mL,10 μmol/L M13荧光引物 0.4 μL。PCR扩增程序如下:94℃ 5 min;94℃ 30 s,待测引物的最适退火温度30 s,72℃ 30 s,25个循环;94℃ 30 s,53℃ 30 s,72℃ 30 s,8个循环;72℃ 8 min,4℃保存。父母本以及三倍体子代的PCR产物委托北京睿博兴科生物科技有限公司在ABI- 3730XL基因分析仪上进行测试,其结果应用GeneMarker 1.75软件[20]进行读取及分析。1.4 多态性引物筛选
本研究中的SSR引物有3种来源:(1)国际杨树基因组协会(InternationalPopulus Genome Consortium)公布的杨树SSR数据库(IPGC;1.5 重组事件的计数与统计
以位于一条染色体上的所有SSR检测位点处hDNA的产生与否为依据,对该条染色体上发生同源重组事件的次数进行统计分析。以相邻SSR位点间hDNA形成与否来确定同源重组事件是否发生,当在一条染色体上hDNA在相邻两SSR位点都产生时,判定在这2个检测位点区间发生了一次同源重组事件。此外,由于着丝粒附近发生hDNA的频率低,以着丝粒附近为区分,单独统计重组事件,即分布在着丝粒两侧的2个SSR位点均产生hDNA时,记为2次同源重组两次事件。1.6 统计与分析
根据异源三倍体的等位基因配置(abc、abd、aac、bbc、aad和bbd共6种等位基因配置),hDNA频率计算如下:其中,HF表示hDNA频率;N ab 表示在某一位点上传递母本杂合性基因型信息的三倍体数量;N aa 和N bb 表示在该位点上传递母本纯合基因型信息的三倍体数量。
为了描述一条染色体上在不同标记处的hDNA频率,利用Origin 2018 (OriginLab Cor., Northampton, MA, USA)绘制hDNA发生频率曲线、频率分布图和相关线性回归分析。
2 结果与分析
2.1 同源重组检测适宜引物筛选
通过对上述3种来源的SSR引物与毛果杨基因组P. trichocarpa v3.0(Phytozome v3.0;进一步对这1849对SSR引物进行筛选,获得母本毛白杨处于杂合状态且与父本银腺杨有差异的多态性SSR引物110对(每条引物的名称、所在染色体的具体位置及其在父母本中分别获得产物片段的大小等具体信息见附表1)。每条染色体上获得的多态性SSR引物为8~16对,其中Chr.07上获得的多态性引物最少,为8对;Chr.01和Chr.04上获得的多态性引物最多,为16对(表1)。这些标记数量与青杨杂种‘哲引3号’杨hDNA发生频率研究[12]中所用的数量相差不大,便于之后的杨树不同种间的同源重组对比研究。
Table 1
表1
表1多态性SSR引物与杨树基因组的BLAST分析
Table 1
| 项目 | Chr.01 | Chr.02 | Chr.03 | Chr.04 | Chr.05 | Chr.06 | Chr.07 | Chr.08 | Chr.19 |
|---|---|---|---|---|---|---|---|---|---|
| 染色体长度(Mb) | 50.5 | 25.3 | 21.8 | 24.3 | 25.9 | 27.9 | 15.6 | 19.5 | 15.9 |
| 标记数量 | 16 | 12 | 9 | 16 | 13 | 14 | 8 | 9 | 13 |
新窗口打开|下载CSV
对筛选出的多态性SSR引物进一步分析,可以将这些引物分为两种类型:第一种为父母本无共同等位基因,以引物GCPM_3971-1 (图1A)为例,在母本3119和3532中,等位基因的配置是杂合的,具有193 bp和211 bp 2个位点(分别以“a”、“b”表示),而父本YX1的等位基因配置也是杂合的,有202 bp和218 bp 2个位点(分别以“c”、“d”表示)。PMR型三倍体子代T1、T2中遗传了母本的杂合位点,其遗传配置分别为“abc”和“abd”;PMR型三倍体子代T3、T4、T5、T6中遗传了母本的纯合位点,且在子代中表现出剂量效应(母本的峰高,即扩增量,约为父本的2倍),其遗传配置分别为“aac”、“bbc”、“aad”和“bbd”。第二种为母本检测到多态性的杂合基因型,但母本、父本有一共同等位基因,以引物GCPM_124 (图1B)为例,在母本3119和3532中,等位基因的配置是杂合的,具有207 bp和210 bp 2个位点(分别以“a”、“b”表示),而父本YX1的等位基因配置也是杂合的,具有201 bp和210 bp 2个位点(以“c”、“b”表示)。PMR型三倍体子代T1、T2中遗传了母本的杂合位点,其遗传配置分别为“abc”和“abb”;PMR型三倍体子代T3、T4、T5、T6中遗传了母本的纯合位点,且在子代中表现出剂量效应(母本的峰高约为父本的2倍或子代只有一个条带),其遗传配置分别为“aab”、“bbb”、“aac”和“bbc”。而ab型2n雌配子(三倍体子代T1和T2)检测到了母本毛白杨雌株3119和3532的所有杂合性信息,则表明毛白杨雌株3119和3532大孢子母细胞减数分裂时在该SSR位点处通过同源重组形成了hDNA。
图1
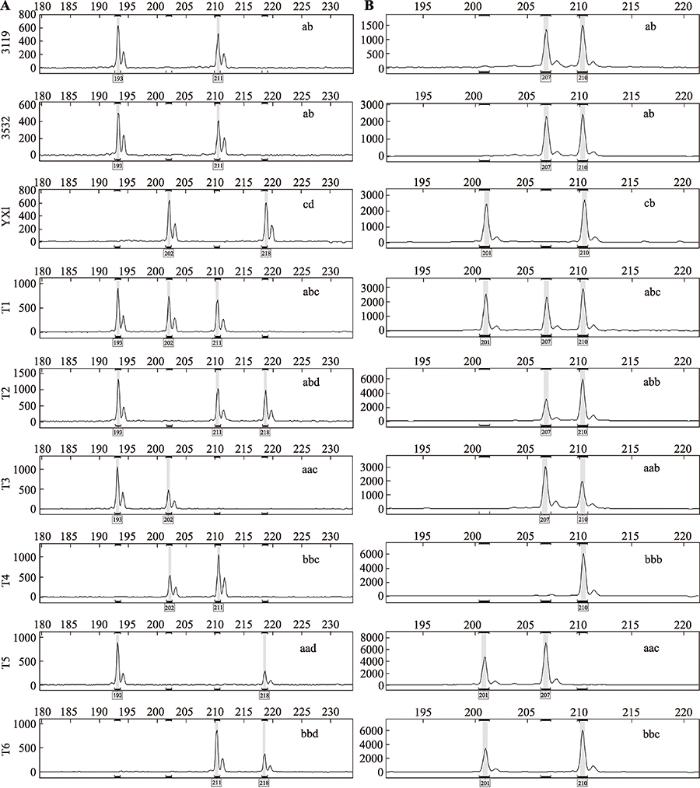 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1鉴定毛白杨hDNA的SSR位点毛细管电泳图
A:引物GCPM_3971-1在亲本和6个三倍体子代中的扩增结果;B:引物GCPM_124在亲本和6个三倍体子代中的扩增结果。
Fig. 1Capillary electrophoresis for the SSR markers used to identify hDNA in Populus tomentosa
2.2 不同基因型毛白杨同源重组特点
进一步利用筛选出的SSR标记,在来源于PMR型2n配子的三倍体群体中,利用公式(1)计算在Chr.01~08和Chr.19不同位置上发生同源重组而产生的hDNA频率(图2)。图2
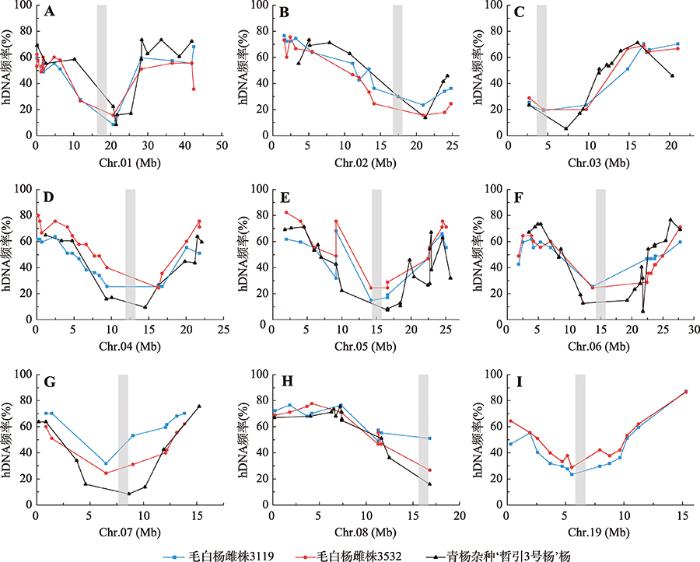 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2在9条染色体不同位置上检测到的hDNA频率(%)
A~I分别表示Chr.01~08和Chr.19上的hDNA发生频率(%)。灰色区域为假定着丝粒区,前8条染色体的着丝粒位置由毛果杨不同组织的甲基化程度推测得到 [ 22] ,Chr.19的着丝粒位置在5.3~6.7 Mb [ 23] 。
Fig. 2The hDNA frequency (%) detected at different locations on the nine chromosomes
2.2.1不同基因型毛白杨hDNA发生频率
在检测的9条染色体上,不同基因型毛白杨hDNA发生频率的结果如图2所示,不同SSR位点处发生的hDNA频率不同。其中,在毛白杨雌株3119中,GCPM_3408 (处于Chr.01上20.668 Mb)与GCPM_2453-1 SSR标记位点间的hDNA频率最小,在47株PMR三倍体中检测到只有4株源自ab型2n雌配子,即8.5%的大孢子母细胞在GCPM_3408与GCPM_2453-1 SSR标记位点间发生了重组事件并产生hDNA。而在MB70130(处于Chr.19上15.319 Mb)与MB125779 SSR标记位点间却有41株PMR型异源三倍体检测到源自ab型2n雌配子,即87.2%的大孢子母细胞在这个位点发生了重组事件并产生hDNA。这2个SSR位点处产生的hDNA频率相差10.0倍以上。在毛白杨雌株3532中,GCPM_3408 (处于Chr.01上20.668 Mb)与GCPM_2453-1 SSR标记位点间、GCPM_2768 (处于Chr.02上20.942 Mb)与PTSSR2445 SSR标记位点间的hDNA发生频率最小,在45株PMR三倍体中检测到只有7株源自ab型2n雌配子(图2),即15.6%的大孢子母细胞在这2个位点发生了重组事件并产生hDNA。而在MB70130 (处于Chr.19上15.319 Mb)与MB125779引物位点间却有39株PMR型异源三倍体被检测到源自ab型2n雌配子,即86.7%的大孢子母细胞在该位点发生了重组事件并产生hDNA。这2个SSR位点处产生的hDNA频率相差5.6倍。
在大多数SSR位点间,在2个基因型毛白杨雌株hDNA发生频率相差不大。但是在一些特定SSR位点间,2个基因型毛白杨雌株hDNA频率发生频率差异较大,如在Chr.01的PTSSR1686 (42.469 Mb)与GCPM_1263-1的SSR标记位点间,雌株3119产生的hDNA频率为68.1%,而雌株3532为35.6%。
在Chr.01~08上,毛白杨2个基因型雌株hDNA最大发生频率分别为76.6%、82.2%,最小发生频率分别为8.5%、15.6%,与青杨杂种‘哲引3号’杨最大、最小hDNA发生频率(76.6%、5.3%)相比[12],毛白杨表现出了更高的hDNA发生频率。
2.2.2hDNA发生频率与标记距着丝粒距离相关分析
由图2可知,在不同染色体上,2个基因型雌株的hDNA发生频率表现出了不同的发生模式。虽然在相同的SSR位点上,hDNA发生频率在2个基因型雌株上具有差异,但2个基因型雌株hDNA发生频率在同一条染色体上具有相近的趋势,即在近着丝粒区域,两种基因型雌株的hDNA发生频率均呈现出最低,随着位点与着丝粒距离增加,hDNA的发生频率也增大。对2个基因型雌株的SSR位点距着丝粒距离与hDNA发生频率进行线性回归分析(图3),结果表明在两种基因型中的9条染色体上,位点距着丝粒距离与hDNA发生频率之间存在显著的正相关关系,即hDNA发生频率随位点与着丝粒距离增大而增加。
图3
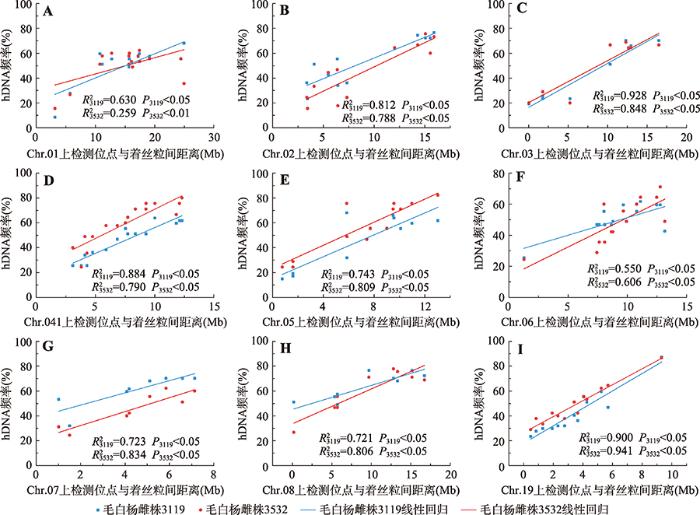 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3hDNA频率(%)及检测位点距着丝粒间距离的相关分析
A~I分别表示Chr.01~08和Chr.19上的hDNA发生频率(%)与检测位点距着丝粒间距离的线性回归。
Fig. 3Correlation analysis between the frequency of hDNA (%) and locus-centromere distance
2.2.3hDNA发生频率与染色体长度相关分析
对9条染色体上平均hDNA发生频率与染色体长度之间进行相关分析,结果发现,在这9条染色体上,两种基因型雌株均不存在显著正相关(图4),这说明染色体长度与平均hDNA发生频率无关。图4
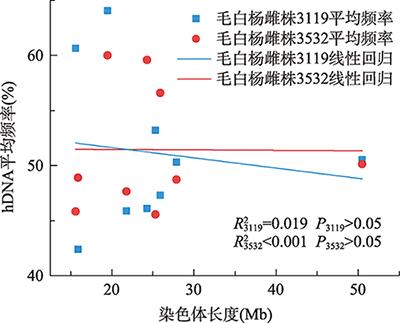 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4染色体长度与hDNA平均频率(%)间关系
Fig. 4Correlation analysis between the average frequency of hDNA (%) and physical length of the chromosome
2.2.32.3 重组次数统计模型示意图
根据本研究1.5描述的统计方法,以Chr.04为例,5株PMR型三倍体上检测位点的hDNA发生情况如图5所示,A~E分别表示Chr.04上发生了0~4次。图5
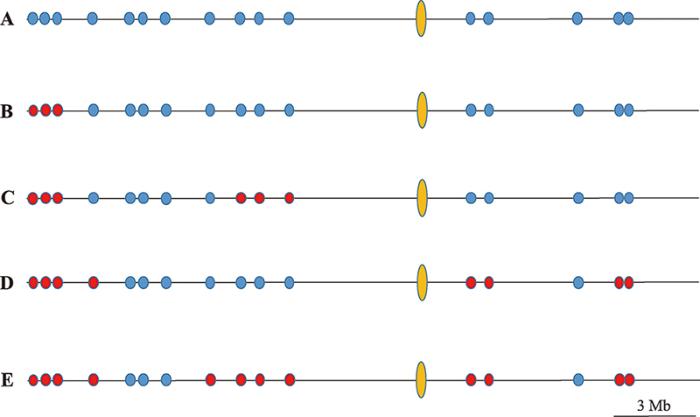 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5染色体长度与hDNA平均频率(%)间关系
A~E分别表示Chr.04上发生了0~4次重组事件。蓝色点表示该位点未产生hDNA,红色表示产生hDNA,黄色椭圆为着丝粒。
Fig. 5Model for statistics of recombination events
依据检测位点上的hDNA发生与否得出9条染色体上的重组事件发生次数,在毛白杨雌株3119中, Chr.03、06、08发生0~3次重组事件,Chr.01、02、04、05、07、19上发生0~4次重组事件(图6)。在毛白杨雌株3532中,Chr.02、03、08上发生0~3次重组事件,Chr.04、05、06、07和19上发生0~4次重组事件,只有Chr.01发生0~5次(图6)。
图6
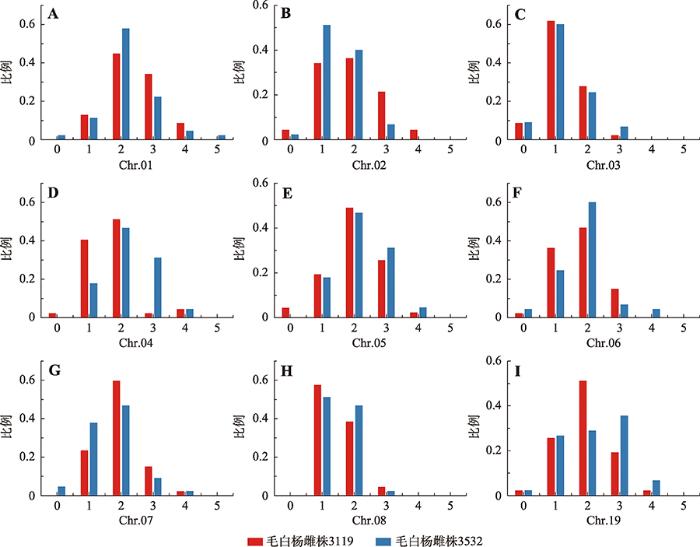 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6Chr.01~08和Chr.19上2个基因型毛白杨个体重组事件的比例
A~I分别表示Chr.01~08和Chr.19上的重组事件比例。
Fig. 6Ratio of recombination events on Chr.1~8 and Chr.19 in two genotypic individuals in P. tomentosa
另一方面,对毛白杨雌株3119为母本的47株PMR型杂种三倍体在检测的9条染色体(共计423条染色体)进行统计分析,结果表明1~3次重组事件在检测的9条染色体上发生比例最高,共计94.8% (计401条染色体);而检测到的最大重组事件次数是4次,发生了11次,占比2.6% (计11条染色体),分别发生在Chr.01、Chr.02、Chr.04、Chr.05、Chr.07和Chr.19上;未检测到重组事件的染色体占比2.6% (计11条染色体) (图6)。
对毛白杨雌株3532为母本的45株PMR型杂种三倍体在检测的9条染色体(共计405条染色体)进行统计分析,结果表明1~3次重组事件在检测的9条染色体上发生比例最高,占比总计94.1% (计381条染色体)上;而4次重组事件发生在总计3.0%的染色体(计12条染色体);而检测到的最大重组事件次数是5次,占比0.2%,仅在Chr.01上发生1次;未检测到重组事件的染色体占比2.7%(计11条染色体) (图6)。在两种基因型雌株中,未检测到重组事件可能与SSR引物的覆盖密度不足有关。
2.2.33 讨论
在毛白杨Chr.01~08和Chr.19的所有检测SSR位点中,两种基因型个体雌配子形成过程中同源重组导致hDNA发生频率不同,分别介于8.5%~87.2%、15.6%~86.7%之间。hDNA发生频率与SSR位点所处的染色体位置有关。此外,从2个群体的92株三倍体所代表的雌配子中9条染色体上的同源重组事件分析上看,大多数的染色体至少经历了1~3次重组事件,少部分染色体经历了4~5次重组事件。2.2.33.1 杨树同源重组频率变异
从hDNA发生频率来看,Dong等[12]利用SSR标记检测PMR型青杨杂种‘哲引3号’杨的hDNA发生频率介于5.3%~76.6%之间。利用与上述研究相当数量的SSR标记,本研究发现毛白杨的hDNA发生频率在8.5%~87.2%之间,略高于青杨杂种‘哲引3号’的hDNA发生频率,这可能与物种间的差异有关。毛白杨的2个基因型个体在GCPM_3408与GCPM_ 2453-1 SSR标记位点间(Chr.01)上均表现出最低hDNA发生频率,分别为8.5%、15.6%。且hDNA最高发生频率的SSR位点在2个基因型个体中相同。另一方面,从hDNA发生的染色体水平上看,在毛白杨2个基因型个体中,9条染色体上hDNA平均发生频率基本一致,分别为51.2%、51.4%,其中最高分别为64.1%、60.0%;最低分别为42.4%、45.6%。这些可能与种内的重组发生的保守性有关。在毛白杨2个基因型个体中,hDNA发生频率与SSR所在染色体位置有关,即检测位点距着丝粒距离越近,hDNA发生频率越低,与‘哲引3号’杨以及其他植物的相关研究结果一致[12,24,25]。这应该是近着丝粒区域富集转座子和短序列重复,其甲基化程度较高,使得重组在近着丝粒区受到抑制的缘故[26]。杨树种内不同基因型个体之间也存在一定的变异,2个基因型个体在Chr.04上的平均hDNA发生频率为46.1%、59.6%,在Chr.07上分别为60.6%、45.8%;甚至在特定检测位点上hDNA发生频率存在较大差异,如在Chr.01的PTSSR1686与GCPM_ 1263-1 SSR标记位点间,毛白杨雌株3119产生的hDNA发生频率为68.1%,而毛白杨雌株3532为35.6%。造成杨树种间以及同种内不同基因型个体间在染色体相同区域hDNA发生频率差异的重要原因与染色体结(knob)、易染色质区、高度重复序列等一些抑制同源重组发生的染色体结构分布区分布等有关[13,27]。
有研究表明,联会复合体长度与染色体交换频率有关[28??~31]。在高等植物中,联会复合体长度与基因组大小[32]、染色体物理长度[10,33]存在正相关关系。本研究对染色体物理长度与hDNA频率进行了相关分析,但结果表明其相关性并未达到显著水平(图4),与水稻(Oryza sativa)中的相关研究结果类似[24],与青杨杂种‘哲引3号’杨的研究结果相一致[12]。此外,以往的研究结果表明只有染色体的长度较大时,联会复合体的长度与染色体物理长度才可能存在相关,而较短的染色体并未表现出类似规律[34,35]。而在杨树染色体中,只有Chr.01长度约为50.5 Mb,相对较大,其他18条染色体长度介于12.9~27.9 Mb之间。因此,本研究中大多数染色体较短,也可能是导致hDNA频率与染色体长度不相关的主要原因。
2.2.33.2 杨树同源重组事件
在减数分裂过程中,同源染色体间发生的联会对减数分裂I的正确分离具有重要作用,同源重组事件在该过程中至少发生一次[36?~38]。其中在拟南芥[39]中,Chr.04上COs发生次数均不超过3次。水稻中,12条染色体的COs平均在1.9~3.8次[24]。玉米的单细胞测序结果表明在Chr.01~10上发生了0~4次COs[10,40],同样,对大麦的四分体单细胞及双单倍体群体测序的结果表明在Mb分辨率下重组事件发生了0~4次[11]。董春波[41]利用SSR标记在青杨杂种三倍体群体Chr.01~08上检测到最高发生6次重组事件,但大部分染色体仅检测到1~3次的同源重组事件,占89.89%。本研究发现绝大多数的染色体至少经历了1~3次重组事件,与上述研究结果相似。少数的染色体至少经历了4~5次同源重组事件,其中5次同源重组事件只发生在毛白杨雌株3532中的Chr.01上,毛白杨雌株3119最多发生了4次重组事件,这可能与重组事件的发生在种内具有差异有关[42]。毛白杨同源重组事件发生次数略低于青杨杂种‘哲引3号’,检测到的同源重组事件发生次数最多的所在染色体与青杨杂种‘哲引3号’(Chr.06)的并不相同。这种结果可能与影响同源重组的基因组修饰在不同物种中存在差异有关[2]。本研究结果表明毛白杨减数分裂中具有较高的DNA重组频率发生,给子代带来更多的遗传变异,从而有利于物种加快进化以适应多变的环境;另一方面,不同基因型个体间重组频率存在着差异,这种差异对育种中亲本选配具有重要意义[43]。然而,影响毛白杨不同基因型间同源重组频率差异的遗传机制仍需进一步深入研究和解析。
附录:附加材料详见文章电子版www.chinagene.cn。
Supplemental Table 1附表1
附表1
Supplemental Table 1
| 标记名称 | 染色 体 | 位置 (Mb) | 重复 单元 | 引物序列(5?→3?) | Tm (℃) | GC含 量(%) | 3532中的 等位基因(bp) | 3119中的 等位基因(bp) | YX1中的 等位基因(bp) |
|---|---|---|---|---|---|---|---|---|---|
| LG_Ⅰ-18 | 1 | 0.047 | atta | F: CACGCTAAGATTGTCTCACA | 53.35 | 45.00 | 320/332 | 320/332 | 320/328 |
| R: TCACTGCATCAAGAAGATTG | 51.30 | 40.00 | |||||||
| LG_Ⅰ-22 | 1 | 0.059 | agcag | F: AGATAAGCATGGCTTGTTGT | 51.30 | 40.00 | 309/314 | 309/314 | 309 |
| R: CGTTACCATTCATTCCAAAT | 49.25 | 35.00 | |||||||
| GCPM_2935-2 | 1 | 0.275 | ttc | F: TCAGCTTCAGCTTCTTCTTC | 53.35 | 45.00 | 192/204 | 192/204 | 198 |
| R: AATTTGAAAAGGAAAAACCC | 47.20 | 30.00 | |||||||
| LG_Ⅰ-1223 | 1 | 1.241 | gca | F: ATATCACTCGCCAAGTCAAC | 53.35 | 45.00 | 190/193 | 190/193 | 196/199 |
| R: ATCACGCACAAGTTAGGTTT | 51.30 | 40.00 | |||||||
| LG_Ⅰ-1114 | 1 | 1.424 | ta | F: AAGTGGATCAGGTGATGAAG | 53.35 | 45.00 | 156/166 | 156/166 | 165 |
| R: CAATGTCTTTCGGTTCTTTC | 51.30 | 40.00 | |||||||
| LG_Ⅰ-1065 | 1 | 1.753 | agc | F: AATGGAGGTCCCTAGCTTAC | 55.40 | 50.00 | 383/386 | 383/386 | 386 |
| R: TGATATTGCTGGGCTATCTT | 51.30 | 40.00 | |||||||
| LG_Ⅰ-1027 | 1 | 1.834 | ta | F: TGAGCTTAGGAACGAAGAAG | 53.35 | 45.00 | 188/198 | 188/198 | 184/186 |
| R: CATCAATATGACTCCTCGGT | 53.35 | 45.00 | |||||||
| LG_Ⅰ-1125 | 1 | 1.835 | tca | F: GATGCTTACCCTTTGCTCTA | 53.35 | 45.00 | 400/409 | 400/409 | 400 |
| R: CATAATGGGCAAAGAATACC | 51.30 | 40.00 | |||||||
| GCPM_3979-1 | 1 | 4.749 | ga | F: TATCAATTTCCGTGGATTTC | 49.25 | 35.00 | 192/196 | 192/196 | 193 |
| R: GAAACTGAGAGGACTTGCTG | 55.40 | 50.00 | |||||||
| GCPM_124 | 1 | 6.328 | cac | F: TTTGAGCACTTCAACTACCA | 51.30 | 40.00 | 207/210 | 207/210 | 201/210 |
| R: TGTCTTCCCTTAGTCACCAC | 55.40 | 50.00 | |||||||
| GCPM_1960-1 | 1 | 11.783 | gaaa | F: TATCGAATCACAATGGCATA | 49.25 | 35.00 | 220/224 | 220/224 | 220 |
| R: TGCTCAGGTCATCTTTTCTT | 51.30 | 40.00 | |||||||
| GCPM_3408 | 1 | 20.668 | gt | F: ATGGAAGTTTGAGTCCACTG | 53.35 | 45.00 | 202/212 | 202/212 | 200/202 |
| R: CATGCATTACTTCAACCCTT | 51.30 | 40.00 | |||||||
| GCPM_2453-1 | 1 | 28.263 | ggt | F: ACACCAAGAGCTGTAGCATT | 53.35 | 45.00 | 216/222 | 216/222 | 216/219 |
| R: ACAACATGGCCTAACTCATC | 53.35 | 45.00 | |||||||
| GCPM_1719 | 1 | 36.636 | aaag | F: AAGTGCTCATAACATCACCC | 53.35 | 45.00 | 216/228 | 216/228 | 227/234 |
| R: CTTTCCTCATTCCTGTTCTG | 53.35 | 45.00 | |||||||
| GCPM_1263-1 | 1 | 41.934 | ct | F: TGCATTAAGACATCACTTGC | 51.30 | 40.00 | 235/243 | 235/243 | 230/232 |
| R: TTCGCTTCTGTATTTCCTGT | 51.30 | 40.00 | |||||||
| PTSSR1686 | 1 | 42.469 | ttc | F: TCAGGATCGGAGTCGGAGTT | 57.45 | 55.00 | 165/168 | 165/168 | 165 |
| R: TGAGCAGAAACGACACCACT | 55.40 | 50.00 | |||||||
| GCPM_734 | 2 | 1.595 | cttt | F: GGCAATTTAGGTACAACAGC | 53.35 | 45.00 | 177/181 | 177/181 | 181 |
| R: ACAAGCGAATGCTAATTGAT | 49.25 | 35.00 | |||||||
| GCPM_2085-1 | 2 | 1.957 | ctt | F: GCAGGAACTAACAAGGTGAG | 55.40 | 50.00 | 154/157 | 154/157 | 154 |
| R: TCGGTAGCAATTGTCTGAAT | 51.30 | 40.00 | |||||||
| GCPM_3588-1 | 2 | 2.443 | aaca | F: TCGACCAACTATGTTTGACA | 51.30 | 40.00 | 184/184 | 184/184 | 191/195 |
| R: AGTTGTAACTTTGGCCTGAA | 51.30 | 40.00 |
新窗口打开|下载CSV
Supplemental Table 1(Continued)
附表1(续)
附表1(续)
Supplemental Table 1(Continued)
| 标记名称 | 染色 体 | 位置 (Mb) | 重复 单元 | 引物序列(5?→3?) | Tm (℃) | GC含 量(%) | 3532中的 等位基因(bp) | 3119中的 等位基因(bp) | YX1中的 等位基因(bp) |
|---|---|---|---|---|---|---|---|---|---|
| 标记名称 | 染色 体 | 位置 (Mb) | 重复 单元 | 引物序列(5?→3?) | Tm (℃) | GC含 量(%) | 3532中的 等位基因(bp) | 3119中的 等位基因(bp) | YX1中的 等位基因(bp) |
| GCPM_1857-1 | 2 | 3.205 | tat | F: TGCCAGGGTAAGAAATAAAA | 49.25 | 35.00 | 196/212 | 196/212 | 196/200 |
| R: GAACTGAACTGATCTTGTTGC | 53.66 | 42.86 | |||||||
| ORPM_222 | 2 | 5.481 | ag | F: TGCGAACATTTTTCTTGTGG | 51.30 | 40.00 | 201/205 | 201/205 | 205 |
| R: CGCAATAGAGCCTTTGGATG | 55.40 | 50.00 | |||||||
| GCPM_2637 | 2 | 11.126 | tta | F: GACACCGTTTCTTTTCTGAG | 53.35 | 45.00 | 213/216 | 213/216 | 217 |
| R: ACCAGATCTTCATCTTCCAA | 51.30 | 40.00 | |||||||
| PMGC_2709 | 2 | 12.013 | ga | F: ATTGTAATTATTGAACACATGCC | 50.64 | 30.43 | 234/236 | 234/236 | 200/214 |
| R: GTGCAGTTCAGAGTATTGTTG | 53.66 | 42.86 | |||||||
| ORPM_161 | 2 | 13.406 | ta | F: CGGGCTAGCTCAATTAGGAG | 57.45 | 55.00 | 205/219 | 205/219 | 205/207 |
| R: ATCACCGCGAGATTCTTGTC | 55.40 | 50.00 | |||||||
| ORPM_459 | 2 | 14.121 | att | F: TTGTGGCTGTGACCAGAAAG | 55.40 | 50.00 | 227/230 | 227/230 | 233/246 |
| R: CGAGCCTCAGAAAATTGCTT | 53.35 | 45.00 | |||||||
| GCPM_2768 | 2 | 20.942 | ga | F: TTATTTGGATCCTGAAATGG | 49.25 | 35.00 | 196/206 | 186/206 | 168/200 |
| R: GATGGTTCGGTATGTGAGTT | 53.35 | 45.00 | |||||||
| PTSSR2445 | 2 | 23.932 | ag | F: CTTTGATCCCACCAACCCCA | 57.45 | 55.00 | 230/244 | 230/244 | 240/242 |
| R: TAGGCAGCTCTTTGACTGCA | 55.40 | 50.00 | |||||||
| PTSSR774 | 2 | 24.833 | tga | F: TAAGGGCAAACATGGGAGGG | 57.45 | 55.00 | 239/242 | 239/242 | 236/239 |
| R: TAGGCAGAAGTAGCAGCAGC | 57.45 | 55.00 | |||||||
| GCPM_2840-1 | 3 | 2.708 | ta | F: GAATCCAAAACAAAACATCC | 49.25 | 35.00 | 227/231 | 227/231 | 225/231 |
| R: AGGCATCAAAACTAATTGGA | 49.25 | 35.00 | |||||||
| GCPM_134-1 | 3 | 2.708 | ta | F: AGGCATCAAAACTAATTGGA | 49.25 | 35.00 | 227/230 | 227/230 | 225/231 |
| R: GAATCCAAAACAAAACATCC | 49.25 | 35.00 | |||||||
| GCPM_2628-1 | 3 | 4.469 | agc | F: TAGGCCATGTTTTTGCTTAT | 49.25 | 35.00 | 211/214 | 211/214 | 211 |
| R: GTGCCTCATTTATTGGTAGG | 53.35 | 45.00 | |||||||
| ORPM_230 | 3 | 9.739 | ga | F: CTTCCCACCCCCAACATAAC | 57.45 | 55.00 | 213/219 | 213/219 | 211/215 |
| R: TGTCATTCCACAGAGAGTCTGG | 57.67 | 50.00 | |||||||
| GCPM_1184-1 | 3 | 14.831 | ct | F: TCTTGGCGAGAGAAGTAGAG | 55.40 | 50.00 | 156/158 | 156/158 | 154/164 |
| R: GGATTTGGTGAAAATTGAAG | 49.25 | 35.00 | |||||||
| GCPM_768-1 | 3 | 16.826 | cac | F: ATTCGCTTCTTCTTCCTCTT | 51.30 | 40.00 | 208/211 | 208/211 | 211 |
| R: TATGAATTCCTCGTTGAACC | 51.30 | 40.00 | |||||||
| GCPM_2434-2 | 3 | 17.172 | tct | F: AGAGAGAGAGGTATGAGGGC | 57.45 | 55.00 | 221/224 | 221/224 | 221 |
| R: GTTCGGTAAAGGTGATGGA | 53.01 | 47.37 | |||||||
| GCPM_1468-2 | 3 | 17.480 | gac | F: CAAGTCATGGCTTCTCAAAC | 53.35 | 45.00 | 192/195 | 192/195 | 195/198 |
| R: CTATTGATCCTTGAAGACGC | 53.35 | 45.00 | |||||||
| GCPM_3465 | 3 | 20.996 | gt | F: TCAATGATTGGTCTTGTTGA | 49.25 | 35.00 | 128/134 | 128/134 | 134/146 |
| R: CAGAATTCAGAATAGAACCCA | 51.71 | 38.10 | |||||||
| GCPM_2625-1 | 4 | 0.188 | gt | F: GATTTCTATTGTGGCAAAGG | 51.30 | 40.00 | 187/193 | 187/193 | 187 |
| R: TGTATTCCTCCACTCCACTC | 55.40 | 50.00 |
新窗口打开|下载CSV
Supplemental Table 1(Continued)
附表1(续)
附表1(续)
Supplemental Table 1(Continued)
| 标记名称 | 染色 体 | 位置 (Mb) | 重复 单元 | 引物序列(5?→3?) | Tm (℃) | GC含 量(%) | 3532中的 等位基因(bp) | 3119中的 等位基因(bp) | YX1中的 等位基因(bp) |
|---|---|---|---|---|---|---|---|---|---|
| 标记名称 | 染色 体 | 位置 (Mb) | 重复 单元 | 引物序列(5?→3?) | Tm (℃) | GC含 量(%) | 3532中的 等位基因(bp) | 3119中的 等位基因(bp) | YX1中的 等位基因(bp) |
| GCPM_1116-1 | 4 | 0.401 | tca | F: GGGGCTAATATCAGTTTCCT | 53.35 | 45.00 | 195/201 | 195/201 | 195 |
| R: TGAGAAAACCCTGGAAAATA | 49.25 | 35.00 | |||||||
| ORPM_394 | 4 | 0.667 | tgc | F: AAAAAGCCCCACAATTATTCA | 49.76 | 33.33 | 222/225 | 222/225 | 222/231 |
| R: GCAAGTTGCAATTGATGTCC | 53.35 | 45.00 | |||||||
| GCPM_3437 | 4 | 2.481 | ag | F: AAGATCTGGTGTTGTTTTGG | 51.30 | 40.00 | 234/236 | 234/236 | 236/242 |
| R: TTCTTAACAGAAGCCAGGAG | 53.35 | 45.00 | |||||||
| GCPM_3971-1 | 4 | 4.122 | taaa | F: CAGCCAGCCTCTTTAAGATA | 53.35 | 45.00 | 193/211 | 193/211 | 202/220 |
| R: ACAGCAATTCTGTACCTCGT | 53.35 | 45.00 | |||||||
| GCPM_1297-1 | 4 | 4.758 | tct | F: GGGATGAATCAGGAGATGTA | 53.35 | 45.00 | 135/141 | 135/141 | 140 |
| R: GAAGAAACCTGTGGGTGATA | 53.35 | 45.00 | |||||||
| ORPM_421 | 4 | 5.650 | ta | F: AAATGATGTTGCGATTTCCA | 49.25 | 35.00 | 213/221 | 213/221 | 219 |
| R: TCCCATCTCAACTACTCCAACA | 55.81 | 45.45 | |||||||
| PTSSR1738 | 4 | 6.603 | ctcttc | F: CTTCCTCGTTTGGCCCCTAA | 57.45 | 55.00 | 277/295 | 277/295 | 289/301 |
| R: ACCAGCTAGTTCGGGCTTTC | 57.45 | 55.00 | |||||||
| ORPM_201 | 4 | 7.736 | tc | F: GACTCCACCCAGTTCTGCTC | 59.50 | 60.00 | 214/226 | 214/226 | 224/226 |
| R: AACTTCCCATCGAATGATCG | 53.35 | 45.00 | |||||||
| PTSSR1873 | 4 | 8.381 | GA | F: GCGTGAGATGAGGGAGAGTG | 59.50 | 60.00 | 184/198 | 184/198 | 186/202 |
| R: CGTCAGGCGACAATTCCAAC | 57.45 | 55.00 | |||||||
| PTSSR1932 | 4 | 9.402 | TAC | F: CGGCAGGTCTGATGTCTTCA | 57.45 | 55.00 | 120/123 | 120/123 | 117/123 |
| R: CCATGGAGTCTAAAGGTGGCA | 57.57 | 52.38 | |||||||
| GCPM_3819-1 | 4 | 16.315 | gaa | F: ATCATCAAGTTCACGAAAGC | 51.30 | 40.00 | 240/243 | 240/243 | 237 |
| R: ACTGACCTACCACCATTTTG | 53.35 | 45.00 | |||||||
| GCPM_295-1 | 4 | 16.784 | aag | F: TAAGAGGATTACCAGGCAGA | 53.35 | 45.00 | 136/145 | 136/145 | 142/148 |
| R: CCCACTAACAATAAGCTTGG | 53.35 | 45.00 | |||||||
| PTSSR1623 | 4 | 20.046 | tga | F: CATCTCCCACCACCACAACA | 57.45 | 55.00 | 238/248 | 238/248 | 238 |
| R: CTCTGCGTAAACAACTGCGG | 57.45 | 55.00 | |||||||
| PTSSR2267 | 4 | 21.767 | tca | F: TGGAAGGCAAATCGTCGTCA | 55.40 | 50.00 | 182/197 | 182/197 | 190 |
| R: AAGTGGAGCAGACAGACACT | 55.40 | 50.00 | |||||||
| GCPM_3234-1 | 4 | 21.772 | agc | F: TCTCAATGTCGACACCATTA | 51.30 | 40.00 | 195/198 | 195/198 | 195 |
| R: TGCAAGAACTGCTATCACTG | 53.35 | 45.00 | |||||||
| GCPM_1255 | 5 | 1.974 | ag | F: GAACCTTAAAACCAGAACCC | 53.35 | 45.00 | 191/193 | 191/193 | 189/193 |
| R: GAGCCACAGAAATACTGCTC | 55.40 | 50.00 | |||||||
| GCPM_379-1 | 5 | 4.025 | tat | F: GGCCATTCATTAGATGTAGC | 53.35 | 45.00 | 153/156 | 153/156 | 153 |
| R: AAATCAAGATCCAAAGCAAA | 47.20 | 30.00 | |||||||
| GCPM_1063 | 5 | 5.996 | ca | F: AGTTAATTGCGCATGTTCTT | 49.25 | 35.00 | 168/188 | 168/188 | 170/176 |
| R: AAACAAACTCCAGCAAACAT | 49.25 | 35.00 | |||||||
| GCPM_3536-2 | 5 | 9.139 | ttg | F: AGATTCTTTTTCGGCTTCTT | 49.25 | 35.00 | 137/146 | 137/146 | 141/144 |
| R: AGAAGATGCTGGAGTTCAGA | 53.35 | 45.00 | |||||||
| GCPM_1838 | 5 | 9.156 | ga | F: GTTCAGCGAAAGCTAAAGAG | 53.35 | 45.00 | 142/146 | 142/146 | 138/168 |
| R: CACAGAATTACAGCTGATGC | 53.35 | 45.00 |
新窗口打开|下载CSV
Supplemental Table 1(Continued)
附表1(续)
附表1(续)
Supplemental Table 1(Continued)
| 标记名称 | 染色 体 | 位置 (Mb) | 重复 单元 | 引物序列(5?→3?) | Tm (℃) | GC含 量(%) | 3532中的 等位基因(bp) | 3119中的 等位基因(bp) | YX1中的 等位基因(bp) |
|---|---|---|---|---|---|---|---|---|---|
| 标记名称 | 染色 体 | 位置 (Mb) | 重复 单元 | 引物序列(5?→3?) | Tm (℃) | GC含 量(%) | 3532中的 等位基因(bp) | 3119中的 等位基因(bp) | YX1中的 等位基因(bp) |
| GCPM_2013-1 | 5 | 14.238 | At | F: TTTAGGGTCGTAGTGGAAAA | 51.30 | 40.00 | 238/240 | 238/240 | 250/254 |
| R: AAACCTGGAGGAGAATTAGAA | 51.71 | 38.10 | |||||||
| ORPM_136 | 5 | 16.608 | ct | F: TTTAAGCCTCCGAAAACCAA | 51.30 | 40.00 | 228/240 | 228/240 | 232 |
| R: CTGCAAGGCGAGGTTATTCT | 55.40 | 50.00 | |||||||
| ORPM_442 | 5 | 16.608 | ct | F: TGGTTTTCGGAGGCTTAAAA | 51.30 | 40.00 | 244/254 | 244/254 | 236 |
| R: CCTGCAGCGAGTATTAATTGG | 55.61 | 47.62 | |||||||
| GCPM_3151 | 5 | 22.434 | ag | F: ACCATCATTAACCCCACATA | 51.30 | 40.00 | 144/156 | 144/156 | 135/155 |
| R: AAAGAAACCAGACCACACAC | 53.35 | 45.00 | |||||||
| GCPM_2858-1 | 5 | 22.702 | tg | F: TGCAAGTCTTTTTAGGAACC | 51.30 | 40.00 | 254/256 | 254/256 | 254/260 |
| R: TTCAAAATGCATCAAAGTGT | 47.20 | 30.00 | |||||||
| GCPM_540-1 | 5 | 24.498 | taa | F: GATGGGGAGGTTATTTTCTT | 51.30 | 40.00 | 154/160 | 154/160 | 154 |
| R: CAATATTGAGGAAAATCAAAGG | 50.22 | 31.82 | |||||||
| GCPM_4008-1 | 5 | 24.561 | acc | F: AGAGAGAAGCTTGTGTCCAG | 55.40 | 50.00 | 161/170 | 161/170 | 170 |
| R: TGAGGAAGCAGAAGTAGAGC | 55.40 | 50.00 | |||||||
| ORPM_25 | 5 | 25.091 | ta | F: AAGAGTTGAAGGCTGGACGA | 55.40 | 50.00 | 236/240 | 236/240 | 236 |
| R: AGACATGCATGAAGCCATGA | 53.35 | 45.00 | |||||||
| GCPM_547-1 | 6 | 1.831 | at | F: CCTCTTGAAAAGAAGCAAAA | 49.25 | 35.00 | 217/231 | 217/231 | 213 |
| R: ATCAAAAATGCCGATTAAAA | 45.15 | 25.00 | |||||||
| GCPM_139 | 6 | 2.516 | gt | F: ATGACATGACATGATTGGAA | 49.25 | 35.00 | 220/228 | 220/228 | 200/226 |
| R: CTTCTGCTGGAAGAAGAAAA | 51.30 | 40.00 | |||||||
| GCPM_2126 | 6 | 3.888 | ag | F: CACGTAAACAGCTTCCAAGT | 53.35 | 45.00 | 178/194 | 178/194 | 160/176 |
| R: TAATGATTCCAGCTATGGGT | 51.30 | 40.00 | |||||||
| GCPM_1072-2 | 6 | 4.204 | ttta | F: AGGAAAACAAAGGAGAGGAG | 53.35 | 45.00 | 150/154 | 150/154 | 142/154 |
| R: ATGCTTAAAAGGGGATCTCT | 51.30 | 40.00 | |||||||
| GCPM_3539-2 | 6 | 5.331 | aat | F: TATTCGGTACAAGACTTGGG | 53.35 | 45.00 | 215/218 | 215/218 | 212/215 |
| R: TAATTGTAAAGCGGCTATCG | 51.30 | 40.00 | |||||||
| GCPM_2642-2 | 6 | 6.965 | cca | F: GCTTAGCTGGATGAGAAGAA | 53.35 | 45.00 | 239/241 | 239/241 | 239 |
| R: ATAGTTACAGGCCACCATTG | 53.35 | 45.00 | |||||||
| PTSSR580 | 6 | 13.716 | atg | F: AATGCATCCTCAGCTCCAGG | 57.45 | 55.00 | 281/284 | 281/284 | 281 |
| R: CGGCGGCTGCAAACATTAAT | 55.40 | 50.00 | |||||||
| GCPM_1017-1 | 6 | 22.438 | ataa | F: GTTTAATTCCCACGTCGTTA | 51.30 | 40.00 | 171/175 | 171/175 | 175/177 |
| R: CGAATGAAGAAAAACCATTC | 49.25 | 35.00 | |||||||
| GCPM_2705-1 | 6 | 22.633 | ac | F: TTTGCACAGGTAAGTTGATG | 51.30 | 40.00 | 146/150 | 146/150 | 148/152 |
| R: ATTGACCATAGCAGACAACC | 53.35 | 45.00 | |||||||
| GCPM_2034 | 6 | 23.087 | ag | F: ACAAACTGCTTTGTTTGGTT | 49.25 | 35.00 | 176/190 | 176/190 | 186/196 |
| R: CTCCATTCATAAAATCGAGC | 51.30 | 40.00 | |||||||
| GCPM_1632-1 | 6 | 23.663 | ct | F: TTTCTCTCTCTGAAACCCCT | 53.35 | 45.00 | 211/225 | 211/225 | 203/221 |
| R: AGCGACTCACTGAGCTTTAG | 55.40 | 50.00 | |||||||
| ORPM_365 | 6 | 23.788 | tg | F: GGGTTGGACCTGCTCAAATA | 55.40 | 50.00 | 226/228 | 226/228 | 226/238 |
| R: CAGCTGCTTTTTCATGGCTA | 53.35 | 45.00 |
新窗口打开|下载CSV
Supplemental Table 1(Continued)
附表1(续)
附表1(续)
Supplemental Table 1(Continued)
| 标记名称 | 染色 体 | 位置 (Mb) | 重复 单元 | 引物序列(5?→3?) | Tm (℃) | GC含 量(%) | 3532中的 等位基因(bp) | 3119中的 等位基因(bp) | YX1中的 等位基因(bp) |
|---|---|---|---|---|---|---|---|---|---|
| GCPM_2627-1 | 6 | 24.930 | cac | F: TAAGTCCCACTACACCCAAC | 55.40 | 50.00 | 234/254 | 234/254 | 240/255 |
| R: GAGTTCGAGAGAGGGAATCT | 55.40 | 50.00 | |||||||
| GCPM_2615-1 | 6 | 27.782 | cttt | F: ATGTCAACGTCACTGACAAA | 51.30 | 40.00 | 231/237 | 231/237 | 229/236 |
| R: ATTAGGCAATGCAGAACACT | 51.30 | 40.00 | |||||||
| GCPM_1054-1 | 7 | 0.863 | ct | F: AGGTCTGTGCAAGGAATAAA | 51.30 | 40.00 | 159/165 | 159/165 | 154/165 |
| R: GTCTGTAATCAAGCCAAAGC | 53.35 | 45.00 | |||||||
| GCPM_741-1 | 7 | 1.416 | at | F: CCGTTTGATTAAAAAGATGC | 49.25 | 35.00 | 212/214 | 212/214 | 214 |
| R: TTATTGAGCTGATGATCCCT | 51.30 | 40.00 | |||||||
| WPMS_17 | 7 | 6.495 | cac | F: ACATCCGCCAATGCTTCGGTGTTT | 59.57 | 50.00 | 137/146 | 137/146 | 137/140 |
| R: GTGACGGTGGTGGCGGATTTTCTT | 61.28 | 54.17 | |||||||
| GCPM_3738-1 | 7 | 9.014 | ac | F: TGAACAAGACACCAAAATGA | 49.25 | 35.00 | 133/139 | 133/139 | 133 |
| R: TCTCGACTTTACCATCTCGT | 53.35 | 45.00 | |||||||
| GCPM_2741-1 | 7 | 12.076 | at | F: CAAGCAGTATCTTCCACTGA | 53.35 | 45.00 | 142/146 | 142/148 | 134 |
| R: TAGCCAACCACTCTAAAGGA | 53.35 | 45.00 | |||||||
| GCPM_3476-1 | 7 | 12.189 | ggt | F: GGGAATGTAAGGATGTGTTG | 53.35 | 45.00 | 202/220 | 202/220 | 205/211 |
| R: AACCAGAAAACGACAGTCAC | 53.35 | 45.00 | |||||||
| GCPM_3332-1 | 7 | 13.116 | ag | F: TCCACTGCCTATGAACTTTT | 51.30 | 40.00 | 138/154 | 138/154 | 138 |
| R: CACCCAATAGCTTCCATATT | 51.30 | 40.00 | |||||||
| GCPM_3727-1 | 7 | 13.848 | ct | F: TTGGGGTTAGTGACTAGTGG | 55.40 | 50.00 | 152/158 | 152/158 | 152/154 |
| R: CAAGCTGTGTAAAGACACCA | 53.35 | 45.00 | |||||||
| LG_Ⅷ-50 | 8 | 0.255 | ga | F: AACAACAGATCGCTGAAACT | 51.30 | 40.00 | 265/281 | 265/281 | 265/271 |
| R: CCATGTACTTAGGCATGGAT | 53.35 | 45.00 | |||||||
| GCPM_2992 | 8 | 1.833 | TA | F: GATCATAACCCAGGAAACAA | 51.30 | 40.00 | 237/241 | 237/241 | 241 |
| R: CGTGAATTTTTGGGATTTTA | 47.20 | 30.00 | |||||||
| GCPM_1344-2 | 8 | 3.669 | tat | F: GAGACCTGAGAAACAGAGGA | 55.40 | 50.00 | 227/230 | 227/230 | 230 |
| R: CAAATGCAACGCAATAATAA | 47.20 | 30.00 | |||||||
| GCPM_33-1 | 8 | 4.192 | tta | F: TTTGGCCTTAACTCTCCATA | 51.30 | 40.00 | 171/174 | 171/174 | 167/179 |
| R: AGTTGCTCTCAAAGAAGGTT | 51.30 | 40.00 | |||||||
| GCPM_13-1 | 8 | 7.309 | taa | F: ATTGTTCTTGTTGAAGGACG | 51.30 | 40.00 | 207/213 | 207/213 | 204/210 |
| R: AGAGCAAACAAATTGATGGT | 49.25 | 35.00 | |||||||
| ORPM_126 | 8 | 11.293 | [ct]4cct | F: GCCGAAGTTGACGATAGCTC | 57.45 | 55.00 | 215/222 | 215/222 | 215 |
| R: GAGTTAAACCCACCCTGCAA | 55.40 | 50.00 | |||||||
| ORPM_455 | 8 | 11.293 | ga | F: GAGTTAAACCCACCCTGCAA | 55.40 | 50.00 | 215/225 | 215/225 | 215 |
| R: GCCGAAGTTGACGATAGCTC | 57.45 | 55.00 | |||||||
| GCPM_2958-1 | 8 | 11.639 | ct | F: AAAGAGGATTACTTAGGCGG | 53.35 | 45.00 | 201/215 | 201/215 | 217/219 |
| R: ACGAGAACATTCATCAATCC | 51.30 | 40.00 | |||||||
| GCPM_3474-1 | 8 | 16.814 | agc | F: GATCCGAAAACAACAACAAT | 49.25 | 35.00 | 123/135 | 123/135 | 123 |
| R: ACCCCTTTCTCTTCTCAATC | 53.35 | 45.00 |
新窗口打开|下载CSV
Supplemental Table 1(Continued)
附表1(续)
附表1(续)
Supplemental Table 1(Continued)
| 标记名称 | 染色 体 | 位置 (Mb) | 重复 单元 | 引物序列(5?→3?) | Tm (℃) | GC含 量(%) | 3532中的 等位基因(bp) | 3119中的 等位基因(bp) | YX1中的 等位基因(bp) |
|---|---|---|---|---|---|---|---|---|---|
| 标记名称 | 染色 体 | 位置 (Mb) | 重复 单元 | 引物序列(5?→3?) | Tm (℃) | GC含 量(%) | 3532中的 等位基因(bp) | 3119中的 等位基因(bp) | YX1中的 等位基因(bp) |
| MB50065 | 19 | 0.301 | A | F: CACCAAAATTAAAGGGCCAA | 204/218 | 204/218 | 201 | ||
| R: GCATGCTAGTGCCTTTTCCT | |||||||||
| MB41376 | 19 | 1.940 | at | F: GAGCAGGTGACTCGTGAACA | 251/255 | 251/255 | 251 | ||
| R: GGAGAGTGATAAAACCAACAATCC | |||||||||
| MB41583 | 19 | 2.580 | gcg | F: CAAGTTGGCTTGGATGGATT | 144/162 | 144/162 | 146/149 | ||
| R: AACCAGCCCAACAAATCAAG | |||||||||
| MB55349 | 19 | 3.680 | tc | F: GGACACGTGATGTCATGGAG | 264/268 | 264/268 | 258/260 | ||
| R: TCAAATGCAAAGCCCTTTTT | |||||||||
| MB54907 | 19 | 4.713 | ga | F: TAGCAAAAGCAGCAGCAGAG | 262/300 | 262/300 | 262/378 | ||
| R: CAAGTGCGTGAAAAACATGG | |||||||||
| MB54763 | 19 | 5.159 | tta | F: AGTTGAATTTGCGCTTCGAT | 128/134 | 128/134 | 128 | ||
| R: TAGGGTGTGGCCAACATACA | |||||||||
| MB84322 | 19 | 5.526 | tta | F: GCATGCTGTTTGCATCTAAAG | 234/248 | 234/248 | 234 | ||
| R: ATTAAACCTCGGGTCCTGGT | |||||||||
| MB120414 | 19 | 7.927 | tg | F: GTGGCGAAGCTTTATTCCTG | 297/311 | 297/311 | 307/311 | ||
| R: TAAAGCCCAAAGCAGTCACC | |||||||||
| LG_XIX-11 | 19 | 8.770 | atca | F: CAACATGAAATGAGCTGCTA | 398/404 | 398/404 | 404/408 | ||
| R: TCCACATGATGTCTGATTTG | |||||||||
| MB125350 | 19 | 9.649 | tc | F: AAAGAGTCAACGGGCAAAAA | 284/290 | 284/290 | 292/300 | ||
| R: AGTGGCAGCTGAATGGAAAT | |||||||||
| MB37553 | 19 | 10.266 | ta | F: TGACCAAGAGATTGCCACAG | 298/306 | 298/306 | 317/343 | ||
| R: GAAAGTTTCATGTGCCGGAT | |||||||||
| MB125779 | 19 | 11.237 | t | F: TTGAAGTATGATTAGCATCCTCG | 112/115 | 112/115 | 115/123 | ||
| R: TCCCTCTGGTTACCATTTGC | |||||||||
| MB70130 | 19 | 15.319 | a | F: TGCTCCAGAGGTTACTCCGT | 233/239 | 233/239 | 233 | ||
| R: TCCAATGATGGATCTGGTGA |
新窗口打开|下载CSV
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/j.tcb.2007.07.007URL [本文引用: 1]
[本文引用: 2]
DOI:10.1186/gb-2011-12-4-r36URLPMID:21481229 [本文引用: 1]

BACKGROUND: When mismatches in heteroduplex DNA formed during meiotic recombination are left unrepaired, post-meiotic segregation of the two mismatched alleles occurs during the ensuing round of mitosis. This gives rise to somatic mosaicism in multicellular organisms and leads to unexpected allelic combinations among progeny. Despite its implications for inheritance, post-meiotic segregation has been studied at only a few loci. RESULTS: By genotyping tens of thousands of genetic markers in yeast segregants and their clonal progeny, we analyzed post-meiotic segregation at a genome-wide scale. We show that post-meiotic segregation occurs in close to 10% of recombination events. Although the overall number of markers affected in a single meiosis is small, the rate of post-meiotic segregation is more than five orders of magnitude larger than the base substitution mutation rate. Post-meiotic segregation took place with equal relative frequency in crossovers and non-crossovers, and usually at the edges of gene conversion tracts. Furthermore, post-meiotic segregation tended to occur in markers that are isolated from other heterozygosities and preferentially at polymorphism types that are relatively uncommon in the yeast species. CONCLUSIONS: Overall, our survey reveals the genome-wide characteristics of post-meiotic segregation. The results show that post-meiotic segregation is widespread in meiotic recombination and could be a significant determinant of allelic inheritance and allele frequencies at the population level.
DOI:10.1371/journal.pgen.1002305URLPMID:21980306 [本文引用: 1]
Meiotic DNA double-strand breaks (DSBs) initiate crossover (CO) recombination, which is necessary for accurate chromosome segregation, but DSBs may also repair as non-crossovers (NCOs). Multiple recombination pathways with specific intermediates are expected to lead to COs and NCOs. We revisited the mechanisms of meiotic DSB repair and the regulation of CO formation, by conducting a genome-wide analysis of strand-transfer intermediates associated with recombination events. We performed this analysis in a SK1 x S288C Saccharomyces cerevisiae hybrid lacking the mismatch repair (MMR) protein Msh2, to allow efficient detection of heteroduplex DNAs (hDNAs). First, we observed that the anti-recombinogenic activity of MMR is responsible for a 20% drop in CO number, suggesting that in MMR-proficient cells some DSBs are repaired using the sister chromatid as a template when polymorphisms are present. Second, we observed that a large fraction of NCOs were associated with trans-hDNA tracts constrained to a single chromatid. This unexpected finding is compatible with dissolution of double Holliday junctions (dHJs) during repair, and it suggests the existence of a novel control point for CO formation at the level of the dHJ intermediate, in addition to the previously described control point before the dHJ formation step. Finally, we observed that COs are associated with complex hDNA patterns, confirming that the canonical double-strand break repair model is not sufficient to explain the formation of most COs. We propose that multiple factors contribute to the complexity of recombination intermediates. These factors include repair of nicks and double-stranded gaps, template switches between non-sister and sister chromatids, and HJ branch migration. Finally, the good correlation between the strand transfer properties observed in the absence of and in the presence of Msh2 suggests that the intermediates detected in the absence of Msh2 reflect normal intermediates.
URLPMID:8197459 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nprot.2007.491URLPMID:18193020 [本文引用: 1]
In most organisms, one crossover (CO) event inhibits the chances of another nearby event. The term used to describe this phenomenon is 'CO interference'. Here, we describe a protocol for quickly generating large data sets that are amenable to CO interference analysis in the flowering plant, Arabidopsis thaliana. We employ a visual assay that utilizes transgenic marker constructs encoding pollen-expressed fluorescent proteins of three colors in the quartet mutant background. In this genetic background, male meiotic products--the pollen grains--remain physically attached thereby facilitating tetrad analysis. We have developed a library of mapped marker insertions that, when crossed together, create adjacent intervals that can be rapidly and simultaneously screened for COs. This assay system is capable of detecting and differentiating single COs as well as two-, three- and four-strand double COs. We also describe how to analyze the data that are produced by this method. To generate and score a double interval in a wild-type and mutant background using this protocol will take 22-27 weeks.
DOI:10.1038/nprot.2013.131URLPMID:24113785 [本文引用: 1]
During meiosis, reciprocal exchange between homologous chromosomes occurs as a result of crossovers (COs). CO frequency varies within genomes and is subject to genetic, epigenetic and environmental control. As robust measurement of COs is limited by their low numbers, typically 1-2 per chromosome, we adapted flow cytometry for use with Arabidopsis transgenic fluorescent protein-tagged lines (FTLs) that express eCFP, dsRed or eYFP fluorescent proteins in pollen. Segregation of genetically linked transgenes encoding fluorescent proteins of distinct colors can be used to detect COs. The fluorescence of up to 80,000 pollen grains per individual plant can be measured in 10-15 min using this protocol. A key element of CO control is interference, which inhibits closely spaced COs. We describe a three-color assay for the measurement of CO frequency in adjacent intervals and calculation of CO interference. We show that this protocol can be used to detect changes in CO frequency and interference in the fancm zip4 double mutant. By enabling high-throughput measurement of CO frequency and interference, these methods will facilitate genetic dissection of meiotic recombination control.
URLPMID:25800954 [本文引用: 3]
DOI:10.3389/fpls.2017.01620URLPMID:29018459 [本文引用: 2]
Meiotic recombination is a fundamental mechanism to generate novel allelic combinations which can be harnessed by breeders to achieve crop improvement. The recombination landscape of many crop species, including the major crop barley, is characterized by a dearth of recombination in 65% of the genome. In addition, segregation distortion caused by selection on genetically linked loci is a frequent and undesirable phenomenon in double haploid populations which hampers genetic mapping and breeding. Here, we present an approach to directly investigate recombination at the DNA sequence level by combining flow-sorting of haploid pollen nuclei of barley with single-cell genome sequencing. We confirm the skewed distribution of recombination events toward distal chromosomal regions at megabase resolution and show that segregation distortion is almost absent if directly measured in pollen. Furthermore, we show a bimodal distribution of inter-crossover distances, which supports the existence of two classes of crossovers which are sensitive or less sensitive to physical interference. We conclude that single pollen nuclei sequencing is an approach capable of revealing recombination patterns in the absence of segregation distortion.
[本文引用: 11]
[本文引用: 2]
DOI:10.1101/gr.7076308URLPMID:18256239 [本文引用: 1]
The genus Populus consists of dioecious woody species with largely unknown genetic mechanisms for gender determination. We have discovered genetic and genomic features in the peritelomeric region of chromosome XIX that suggest this region of the Populus genome is in the process of developing characteristics of a sex chromosome. We have identified a gender-associated locus that consistently maps to this region. Furthermore, comparison of genetic maps across multiple Populus families reveals consistently distorted segregation within this region. We have intensively characterized this region using an F(1) interspecific cross involving the female genotype that was used for genome sequencing. This region shows suppressed recombination and high divergence between the alternate haplotypes, as revealed by dense map-based genome assembly using microsatellite markers. The suppressed recombination, distorted segregation, and haplotype divergence were observed only for the maternal parent in this cross. Furthermore, the progeny of this cross showed a strongly male-biased sex ratio, in agreement with Haldane's rule that postulates that the heterogametic sex is more likely to be absent, rare, or sterile in interspecific crosses. Together, these results support the role of chromosome XIX in sex determination and suggest that sex determination in Populus occurs through a ZW system in which the female is the heterogametic gender.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[学位论文].
[本文引用: 1]
[本文引用: 1]
[学位论文].
[本文引用: 1]
DOI:10.1038/72708URLPMID:10657137 [本文引用: 1]
A poor man's approach to genotyping for research and high-throughput diagnostics.
[本文引用: 1]
DOI:10.1111/j.1469-8137.2008.02663.xURLPMID:19121044 [本文引用: 1]
In this study, 148 428 simple sequence repeat (SSR) primer pairs were designed from the unambiguously mapped sequence scaffolds of the Nisqually-1 genome. The physical position of the priming sites were identified along each of the 19 Populus chromosomes, and it was specified whether the priming sequences belong to intronic, intergenic, exonic or UTR regions. A subset of 150 SSR loci were amplified and a high amplification success rate (72%) was obtained in P. tremuloides, which belongs to a divergent subgenus of Populus relative to Nisqually-1. PCR reactions showed that the amplification success rate of exonic primer pairs was much higher than that of the intronic/intergenic primer pairs. Applying ANOVA and regression analyses to the flanking sequences of microsatellites, the repeat lengths, the GC contents of the repeats, the repeat motif numbers, the repeat motif length and the base composition of the repeat motif, it was determined that only the base composition of the repeat motif and the repeat motif length significantly affect the microsatellite variability in P. tremuloides samples. The SSR primer resource developed in this study provides a database for selecting highly transferable SSR markers with known physical position in the Populus genome and provides a comprehensive genetic tool to extend the genome sequence of Nisqually-1 to genetic studies in different Populus species.
DOI:10.1186/1471-2164-13-27URLPMID:22251412 [本文引用: 1]
BACKGROUND: DNA cytosine methylation is an epigenetic modification that has been implicated in many biological processes. However, large-scale epigenomic studies have been applied to very few plant species, and variability in methylation among specialized tissues and its relationship to gene expression is poorly understood. RESULTS: We surveyed DNA methylation from seven distinct tissue types (vegetative bud, male inflorescence [catkin], female catkin, leaf, root, xylem, phloem) in the reference tree species black cottonwood (Populus trichocarpa). Using 5-methyl-cytosine DNA immunoprecipitation followed by Illumina sequencing (MeDIP-seq), we mapped a total of 129,360,151 36- or 32-mer reads to the P. trichocarpa reference genome. We validated MeDIP-seq results by bisulfite sequencing, and compared methylation and gene expression using published microarray data. Qualitative DNA methylation differences among tissues were obvious on a chromosome scale. Methylated genes had lower expression than unmethylated genes, but genes with methylation in transcribed regions (
DOI:10.1007/s00412-018-0664-yURLPMID:29520650 [本文引用: 1]
Dioecious species accounted for 6% of all plant species, including a number of crops and economically important species, such as poplar. However, sex determination and sex chromosome evolution have been studied only in few dioecious species. In poplar, the sex-determining locus was mapped to chromosome 19. Interestingly, this locus was mapped to either a peritelomeric or a centromeric region among different poplar species. We developed an oligonucleotide (oligo)-based chromosome painting probe based on the sequence of chromosome 19 from Populus trichocarpa. We performed chromosome painting in P. tomentosa and P. deltoides. Surprisingly, the distal end on the short arm of chromosome 19, which corresponds to the location of the sex-determining locus reported in several species, was not painted in both species. Thus, the DNA sequences associated with this region have not been anchored to the current chromosome 19 pseudomolecule, which was confirmed by painting of somatic metaphase chromosome 19 of P. trichocarpa. Interestingly, the unpainted distal ends of the two chromosome 19 did not pair at the pachytene stage in 22-24% of the meiotic cells in the two species, suggest that these regions from the sex chromosomes have structurally diverged from each other, resulting in the reduced pairing frequency. These results shed light on divergence of a pair of young sex chromosomes in poplar.
[本文引用: 3]
DOI:10.7554/eLife.01426URLPMID:24347547 [本文引用: 1]
Knowledge of the exact distribution of meiotic crossovers (COs) and gene conversions (GCs) is essential for understanding many aspects of population genetics and evolution, from haplotype structure and long-distance genetic linkage to the generation of new allelic variants of genes. To this end, we resequenced the four products of 13 meiotic tetrads along with 10 doubled haploids derived from Arabidopsis thaliana hybrids. GC detection through short reads has previously been confounded by genomic rearrangements. Rigid filtering for misaligned reads allowed GC identification at high accuracy and revealed an approximately 80-kb transposition, which undergoes copy-number changes mediated by meiotic recombination. Non-crossover associated GCs were extremely rare most likely due to their short average length of approximately 25-50 bp, which is significantly shorter than the length of CO-associated GCs. Overall, recombination preferentially targeted non-methylated nucleosome-free regions at gene promoters, which showed significant enrichment of two sequence motifs. DOI: http://dx.doi.org/10.7554/eLife.01426.001.
URLPMID:23017241 [本文引用: 1]
DOI:10.1007/s11427-015-4811-xURLPMID:25651968 [本文引用: 1]
Meiotic recombination is a deeply conserved process within eukaryotes that has a profound effect on patterns of natural genetic variation. During meiosis homologous chromosomes pair and undergo DNA double strand breaks generated by the Spo11 endonuclease. These breaks can be repaired as crossovers that result in reciprocal exchange between chromosomes. The frequency of recombination along chromosomes is highly variable, for example, crossovers are rarely observed in heterochromatin and the centromeric regions. Recent work in plants has shown that crossover hotspots occur in gene promoters and are associated with specific chromatin modifications, including H2A.Z. Meiotic chromosomes are also organized in loop-base arrays connected to an underlying chromosome axis, which likely interacts with chromatin to organize patterns of recombination. Therefore, epigenetic information exerts a major influence on patterns of meiotic recombination along chromosomes, genetic variation within populations and evolution of plant genomes.
DOI:10.1016/j.tig.2003.09.004URLPMID:14585614 [本文引用: 1]
DOI:10.1007/s00412-016-0624-3URLPMID:28101670 [本文引用: 1]
Homologous chromosomes exchange genetic information through recombination during meiosis, a process that increases genetic diversity, and is fundamental to sexual reproduction. In an attempt to shed light on the dynamics of mammalian recombination and its implications for genome organization, we have studied the recombination characteristics of 112 individuals belonging to 28 different species in the family Bovidae. In particular, we analyzed the distribution of RAD51 and MLH1 foci during the meiotic prophase I that serve, respectively, as proxies for double-strand breaks (DSBs) which form in early stages of meiosis and for crossovers. In addition, synaptonemal complex length and meiotic DNA loop size were estimated to explore how genome organization determines DSBs and crossover patterns. We show that although the number of meiotic DSBs per cell and recombination rates observed vary between individuals of the same species, these are correlated with diploid number as well as with synaptonemal complex and DNA loop sizes. Our results illustrate that genome packaging, DSB frequencies, and crossover rates tend to be correlated, while meiotic chromosomal axis length and DNA loop size are inversely correlated in mammals. Moreover, axis length, DSB frequency, and crossover frequencies all covary, suggesting that these correlations are established in the early stages of meiosis.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/0014-4827(85)90544-0URLPMID:3967684 [本文引用: 1]
There appears to be only a weak correlation between genome size and the corresponding total length of a complete set of synaptonemal complexes (SCs) based on published evidence for several fungal, plant, and animal species. This result is unexpected, considering the strong positive correlations between genome size (DNA amount) and total chromosome length and volume and between relative lengths of chromosomes and SCs. Because the observed weak correlation was based on limited data, we systematically investigated the relationship between genome size and SC length, using ten higher plant species. Two-dimensional spreads of SCs from primary microsporocytes at pachytene were prepared using a hypotonic bursting technique. The SC spreads were examined either by light or electron microscopy, and the lengths of at least ten complete sets of SCs were measured for each of the ten species. Additionally, the genome size of each species was determined from pollen tetrad protoplasts using flow cytometry. A strong correlation (r = 0.97) between total SC length and genome size was observed for higher plants, indicating a constant amount of DNA is associated with a given length of SC, at least when averaged over the whole genome.
DOI:10.1371/journal.pgen.1002354URLPMID:22072983 [本文引用: 1]
In most species, crossovers (COs) are essential for the accurate segregation of homologous chromosomes at the first meiotic division. Their number and location are tightly regulated. Here, we report a detailed, genome-wide characterization of the rate and localization of COs in Arabidopsis thaliana, in male and female meiosis. We observed dramatic differences between male and female meiosis which included: (i) genetic map length; 575 cM versus 332 cM respectively; (ii) CO distribution patterns: male CO rates were very high at both ends of each chromosome, whereas female CO rates were very low; (iii) correlations between CO rates and various chromosome features: female CO rates correlated strongly and negatively with GC content and gene density but positively with transposable elements (TEs) density, whereas male CO rates correlated positively with the CpG ratio. However, except for CpG, the correlations could be explained by the unequal repartition of these sequences along the Arabidopsis chromosome. For both male and female meiosis, the number of COs per chromosome correlates with chromosome size expressed either in base pairs or as synaptonemal complex length. Finally, we show that interference modulates the CO distribution both in male and female meiosis.
[本文引用: 1]
[本文引用: 1]
URLPMID:12907787 [本文引用: 1]
URLPMID:6545727 [本文引用: 1]
The question of recombination control has been the subject of intensive investigation over many years. This paper is restricted to an evaluation of the information obtainable from cytological studies of chiasmata, since other approaches are well represented in other papers in this volume. The advantages and disadvantages of chiasmata as means of studying recombination are discussed. Cytological observations reveal many constraints on the distribution of chiasmata, which are interpreted as outward expressions of controls operating at different levels, namely between cells, between bivalents and within bivalents. Detailed descriptions of these constraints are presented as a preliminary to discussing models and mechanisms of control. They include, at the between-bivalent level, over-dispersal of chiasma number per bivalent and the formation of an obligatory minimum of one chiasma per bivalent; and at the within-bivalent level, chiasma localization and chiasma interference. The genetical basis of these constraints is emphasized and evidence presented which suggests that chiasma distribution at different levels is subject to both separate and coordinate genetical controls. These observations lead to the consideration and evaluation of general stochastic models of chiasma control based on the modification of site probabilities for chiasma formation. While these models are a useful approach to thinking about the control of recombination, they do not give a fully satisfactory explanation of all aspects of chiasma control. Finally, specific mechanisms for chiasma distribution control are considered. In a few exceptional species, localized pairing and synaptonemal complexes lead to localized chiasma formation; details of two such cases are presented. In the majority of species, pairing is not localized or restricted and in these species the control of chiasma distribution is more conjectural. Arguments are presented which suggest that even in these cases of full pairing, the pattern of pairing initiation and progression and perhaps the timing of these events could be important factors controlling the distribution of chiasmata.
DOI:10.1016/j.tig.2017.03.003URLPMID:28359582 [本文引用: 1]
Rates of meiotic recombination are widely variable both within and among species. However, the functional significance of this variation remains largely unknown. Is the observed within-species variation in recombination rate adaptive? Recent work has revealed new insight into the scale and scope of population-level variation in recombination rate. These data indicate that the magnitude of within-population variation in recombination is similar among taxa. The apparent similarity of the variance in recombination rate among individuals between distantly related species suggests that the relative costs and benefits of recombination that establish the upper and lower bounds may be similar across species. Here we review the current data on intraspecific variation in recombination rate and discuss the molecular and evolutionary costs and benefits of recombination frequency. We place this variation in the context of adaptation and highlight the need for more empirical studies focused on the adaptive value of variation in recombination rate.
DOI:10.1371/journal.pgen.0030106URLPMID:17604455 [本文引用: 1]
In many species, sex-related differences in crossover (CO) rates have been described at chromosomal and regional levels. In this study, we determined the CO distribution along the entire Arabidopsis thaliana Chromosome 4 (18 Mb) in male and female meiosis, using high density genetic maps built on large backcross populations (44 markers, >1,300 plants). We observed dramatic differences between male and female map lengths that were calculated as 88 cM and 52 cM, respectively. This difference is remarkably parallel to that between the total synaptonemal complex lengths measured in male and female meiocytes by immunolabeling of ZYP1 (a component of the synaptonemal complex). Moreover, CO landscapes were clearly different: in particular, at both ends of the map, male CO rates were higher (up to 4-fold the mean value), whereas female CO rates were equal or even below the chromosomal average. This unique material gave us the opportunity to perform a detailed analysis of CO interference on Chromosome 4 in male and female meiosis. The number of COs per chromosome and the distances between them clearly departs from randomness. Strikingly, the interference level (measured by coincidence) varied significantly along the chromosome in male meiosis and was correlated to the physical distance between COs. The significance of this finding on the relevance of current CO interference models is discussed.
URLPMID:30770831 [本文引用: 1]
[Dissertation]. Beijing Forestry University,
[本文引用: 1]
[学位论文].
[本文引用: 1]
[本文引用: 1]
).
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}