 ,1,2
,1,2The identification of enhancers and its application in cancer studies
Qian Liu1, Chunyan Li,1,2通讯作者: 李春燕,博士,特聘副研究员,研究方向:肿瘤基因组学。E-mail:lichunyan@buaa.edu.cn
责任编辑: 孙玉洁
收稿日期:2020-04-9修回日期:2020-07-23网络出版日期:2020-09-20
| 基金资助: |
Received:2020-04-9Revised:2020-07-23Online:2020-09-20
| Fund supported: |
作者简介 About authors
刘倩,在读硕士研究生,专业方向:生物医学工程。E-mail:

摘要
增强子是一类增强靶基因转录活性的DNA顺式作用元件。但是增强子与靶基因的方向和距离不确定,大大增加了研究增强子调控的靶基因及其作用机制的困难。已有大量研究显示,增强子的突变或功能异常与疾病发生发展相关;仅有少量研究报道增强子通过促进靶基因的表达,引发癌症或产生抗药性。目前与癌症发生发展和在癌症治疗过程中抗药性产生相关的增强子尚未得到充分鉴定,这些增强子的调控机制也未得到充分解析。本文对目前可在全基因组水平上预测和鉴定增强子以及解析增强子调控机制的方法进行总结和对比,并对近几年增强子在肿瘤诊断、治疗和发生发展机制中的研究进展进行综述。期望本文为筛选与癌症发生发展相关的增强子和解析这些增强子的调控机制提供参考,为提高癌症的诊断和制定癌症的治疗策略提供新的视角。
关键词:
Abstract
Enhancers are a type of cis-acting DNA elements that enhance transcriptional activity of target genes. However, the uncertainty in the orientation and distance between enhancers and target genes could post significant difficulties in identifying the target genes and the regulatory mechanisms of the enhancers. Numerous studies have shown that the mutations and/or abnormalities in the functions of enhancers are associated with development of diseases. A few studies have reported that enhancers could activate cancer development or drug resistance by promoting the expression of target genes. At present, enhancers involved in carcinogenesis and drug resistance have not been fully identified, and the underlying mechanism are still largely unknown. This paper summarizes the main methods used in identifying and characterizing enhancers and analyzing the regulatory mechanism at the genome-wide level. It further reviews the recent research progress of enhancers in cancer diagnosis, treatment, and the underlying mechanism during carcinogenesis, thereby providing a reference for the screening of these enhancers involved in carcinogenesis and drug resistance and exploring their regulatory mechanisms of target genes. It also provides a new perspective for improving the diagnosis of cancer and insights for formulating cancer therapeutic strategies.
Keywords:
PDF (827KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘倩, 李春燕. 增强子的鉴定及其在肿瘤研究中的应用. 遗传[J], 2020, 42(9): 817-831 doi:10.16288/j.yczz.20-097
Qian Liu.
增强子是一种通过与靶基因启动子相互作用来增强靶基因转录的DNA调控元件。增强子与调控的靶基因的位置不确定,有些位于靶基因的5?端,有些位于靶基因的3?端,甚至位于基因的内含子中。增强子与靶基因的距离也不确定,甚至远离靶基因达几 Mb仍可增强靶基因的转录。猿猴空泡病毒40 (simian virus 40, SV40)的增强子与其早期启动子相距3000 bp以上,并且该增强子无论是在启动子上游还是下游均能对转录起增强作用。在小鼠(Mus musculus)肢芽组织中,sonic hedgehog (shh)基因与增强子相距1 Mb以上,但是此增强子仍然可以通过促进shh的表达,参与器官的模式形成[1]。尽管在DNA序列上,增强子距离靶基因有很远的距离,但在染色质三维空间结构上,增强子区域和靶基因启动子区域形成三维环状结构(3D-loop)直接相互作用,促进靶基因的转录表达[1,2]。
2006年,Chapel-Fernandes等[3]发现人PSA (prostate specific antigen)增强子可以增强PSA启动子驱动的绿色荧光蛋白在前列腺癌中的表达。近年来,越来越多研究表明增强子通过增强启动子的转录活性,促进癌基因的转录表达[4]。癌基因的表达水平上调是肿瘤形成的重要机制,经过基因重组或基因突变的增强子,通常可促进癌基因的表达[5,6]。近年来,汤柳笛等[5]发现,在基因重组过程中增强子位置变化与肿瘤发生相关,基因重组使原本距癌基因较远的增强子出现在癌基因附近,进而增强子可以调控附近癌基因的表达[5]。2017年,Abraham等[6]发现插入突变的增强子在肿瘤发生中起着重要作用,并在致癌基因附近的增强子序列中发现了多种插入突变,如插入突变造成调控原癌基因LMO2 (LIM only protein 2)的活性增强子形成,使正常体细胞癌变为原发性白血病肿瘤细胞。因此,对增强子位置、活性及功能的鉴定对研究癌症的发生机制具有重要意义,可提供全新的癌症治疗策略。本文主要总结了增强子和超级增强子的特点、鉴定和预测调控机制的方法及其在肿瘤研究中的应用。
1 增强子及其特性
增强子最早被发现于20世纪80年代初期。1980年,Grosschedl等[7]发现位于组蛋白H2A基因转录起始位点上游大于100 bp处的一段DNA序列能促进H2A基因有效转录。1981年,Benoist等[8]在SV40早期基因的上游发现了首个增强子序列,该序列的缺失会使SV40基因的转录效率降低,将增强子序列反转后放置在SV40基因的下游依然可以增强转录,由此将这种不依赖位置和方向增强基因转录的顺式元件命名为增强子[9]。随着增强子研究的深入,其作用特点以及生物学功能进一步被发现。近40年的研究证明增强子具有以下特征:(1)增强子是增强靶基因转录的顺式作用元件。但不能根据方向和距离预测增强子调控的靶基因,因为增强子可以位于靶基因的上游,也可位于下游,甚至可以位于基因内;而且两者的距离可远可近[10]。(2)增强子位于染色质开放区域。(3)增强子的活性与转录因子的结合、DNA序列的甲基化修饰及核小体中组蛋白的修饰有关,如增强子与转录因子结合可促进基因的转录[10];增强子DNA序列高甲基化会导致eRNA表达下降,相反,低甲基化可促进eRNA的表达[11];H3K4me1 (H3 lysine 4 monomethylation)修饰常常促进增强子序列转录表达,而H3K27me3 (H3 lysine 27 trimethylation)修饰会使增强子沉默无法转录表达;H3K27ac (H3 lysine 27 acetylation)修饰使增强子序列转录活跃[12,13]。(4)增强子区域需要和启动子区域形成三维环状结构直接相互作用才能发挥功能,增强子和启动子的相互作用由多种蛋白介导,如Mediator复合体和cohesin等[14,15]。
2 超级增强子及其特性
2013年首个超级增强子(super enhancers, SEs)由美国怀特黑德生物医学研究所Richard A. Young实验室在胚胎干细胞中发现,该实验室定义超级增强子是多个具有转录活性增强子串联而成的长约8~20 kb的片段,可以强力驱动相关基因的表达[16]。与增强子相比,超级增强子与重要转录因子的结合密度更高,调控转录的强度和敏感度更高[17]。随着对超级增强子研究的深入,研究人员提出了一些更为明确、普遍的超级增强子特性。目前,超级增强子区别于增强子的功能特性有:(1)超级增强子结合更多的转录因子以及转录辅助因子[14,15];(2)超级增强子具有更多的转录活性相关组蛋白修饰,如:H3K27ac和H3K4me1修饰;(3)超级增强子的DNA序列具有较低的甲基化修饰[11];(4)组成超级增强子的单个增强子同样具有增强子激活靶基因转录的功能[9];(5)超级增强子活性对转录因子表达水平的变化更敏感[18];(6)超级增强子驱动的基因表达量更高[9]。增强子与超级增强子结构特征和功能对比如图1所示。
3 增强子的研究方法
由于增强子多位于基因的非编码区,不同增强子相对于靶基因的位置不固定,而且增强子可能只在特定组织细胞或特殊生理情况下才具有活性[19]。因此,增强子的发现和功能注释变得更为复杂和具有挑战性。比较基因组方法(comparative genomics)和转录因子基序法(transcription factor motif matches)可以进行增强子的预测。在已知各物种基因组序列的前提下,比较基因组法是通过不同物种基因组的比较,进而发现在不同物种间高度保守的DNA序列。但与编码基因序列比较,增强子的保守性较差,因此基于保守性预测增强子的效率较低。转录因子基序法鉴定增强子的原理基于增强子中包含充当转录因子结合位点的DNA基序(一般只有6~10 bp),利用结合位点的保守性,在全基因组范围搜索与已知转录因子基序匹配的序列。较短的DNA序列在整个基因组中出现的频率较高,影响增强子鉴定的准确率[17]。此外,通过保守的基因组和基序,无法预测增强子的活性状态。因此需要新的方法进行增强子的预测和鉴定。随着高通量测序技术的快速发展,在全基因组水平上研究增强子方法和工具呈现多样化,如ChIP-Seq (chromatin immunoprecipitation followed by sequencing)、DNase-Seq (DNase I hypersensitive sites sequencing)、ATAC-Seq (assay for transposase-accessible chromatin with high throughput sequencing)、RNA-Seq (RNA sequencing)、基因编辑(gene editing)技术、亚硫酸氢盐测序法(bisulfite sequencing PCR, BSP)和染色质构象捕获(chromosome conformation capture, 3C)技术及其衍生技术,如环形染色质构象捕获(circular chromosome conformation capture, 4C)、染色质构象捕获碳拷贝(chromosome conformation capture carbon copy, 5C)和高通量染色体构象捕获(high- through chromosome conformation capture, Hi-C)等。3.1 基于ChIP-Seq技术鉴定增强子
1997年,Orlando等[20]创立了染色质免疫共沉淀(chromatin immunoprecipitation, ChIP)技术,该技术也称结合位点分析法,其原理是在活细胞状态下用甲醛固定蛋白质-DNA复合物,然后利用抗原抗体免疫结合将含目的蛋白的复合物沉淀下来,然后从中提取DNA进行PCR分析和序列测定,从而获得与目的蛋白互作的DNA信息。ChIP是在全基因组范围内检测DNA与蛋白质体内相互作用的方法[21]。ChIP-Seq技术是将ChIP技术与二代测序技术相结合在全基因组范围内分析蛋白质与DNA互作信息的方法。首先利用ChIP技术富集与目的蛋白特异性结合的DNA片段,然后利用二代测序技术对DNA片段进行高通量测序,从而在全基因组范围内获得与转录因子、辅助转录因子、或不同修饰的组蛋白结合的DNA信息。根据已知增强子的特征(特异性结合的转录因子和辅助转录因子或组蛋白修饰特征等),可利用ChIP-Seq技术在全基因组范围内寻找转录因子和辅助转录因子结合位点、组蛋白修饰区段,进而进行增强子的预测和鉴定[22]。3.1.1 基于转录因子和转录辅助因子结合位点鉴定增强子
转录因子是可以直接结合在增强子的DNA序列上调控基因表达的蛋白质,可利用ChIP-Seq技术在全基因组内寻找增强子DNA序列与转录因子(蛋白质)的结合位点进行增强子的预测。转录辅助因子由转录因子募集到结合位点行使生物学功能,协助RNA聚合酶与靶基因启动子的结合,促进增强子RNA的转录。因此,也可利用ChIP-Seq技术检测辅助转录因子与DNA的结合位点,从而对增强子进行预测。如广泛表达的通用辅助转录激活因子组蛋白乙酰转移酶P300,参与多种转录因子的转录调控[10]。Visel等[23]曾在小鼠模型中进行转录辅助因子P300的ChIP-Seq实验,发现数千个P300结合位点,然后通过转基因技术在小鼠体内进行验证,结果表明绝大部分预测的结合位点都显示了增强子的调控活性。基因组上增强子与转录因子和转录辅助因子的结合机制为增强子的鉴定提供了可行方法。
3.1.2 基于组蛋白修饰鉴定增强子
缠绕在组蛋白上的DNA处于高度压缩状态,基因很难表达,为了提高基因的表达,需要改变DNA与组蛋白的紧密结合程度。组蛋白修饰是表观遗传学的重要部分,组蛋白修饰的改变可导致组蛋白与DNA结合程度的改变,从而实现表观修饰对基因表达的调控作用。组蛋白修饰主要包括甲基化、磷酸化、乙酰化和泛素化。目前,关于增强子研究较多的组蛋白修饰是组蛋白甲基化和乙酰化。不同的组蛋白甲基化修饰可能会激活也可能会抑制增强子的转录表达,如H3K4me1修饰会促进增强子转录,而H3K27me3修饰会抑制增强子转录。组蛋白乙酰化修饰可以减弱增强子DNA序列与组蛋白的结合程度,让部分DNA序列暴露,进而可以与转录因子结合促进增强子转录表达,如H3K27ac修饰使增强子序列转录活跃;相反,组蛋白去乙酰化会使增强子DNA序列与组蛋白的结合更加紧密,抑制增强子的转录表达[24]。目前H3K4me1和H3K27ac常用来作为鉴定增强子的组蛋白修饰特征。
Nathaniel等[25]利用ChIP-Seq技术在人类基因组中发现高密度的H3K4me1修饰存在于活性增强子区域[25]。研究者发现H3K27ac修饰是区分活性增强子和非活性增强子的重要标记,非活性增强子只存在H3K4me1的修饰;而活性增强子存在H3K4me1、H3K27ac等的修饰,位于转录基因的近端;存在H3K27me3和H3K4me1修饰却不存在H3K27ac修饰,不参与基因的转录的增强子为平衡增强子[26,27]。此外,有活性的增强子可以有多种组蛋白修饰方式,如H3K79me3 (H3 lysine 79 trimethylation)、H4K16ac (H3 lysine 16 acetylation)和H3K122ac (H3 lysine 122 acetylation)修饰等[27,28,29]。因此,利用ChIP-Seq技术在全基因组水平寻找富集多种不同的组蛋白修饰的位点进行增强子预测和活性鉴定。
3.2 基于DNA甲基化检测技术鉴定有活性的增强子
DNA甲基化(DNA methylation)是指在DNA甲基转移酶的催化下DNA序列特定碱基共价结合一个甲基基团,DNA甲基化通常发生在CpG (胞嘧啶-磷酸-鸟嘌呤)位点,胞嘧啶在DNA甲基转移酶的催化下结合一个甲基基团转化为5-甲基胞嘧啶。通过增强子DNA甲基化修饰,可以在不改变增强子DNA序列的情况下影响增强子的活性。增强子DNA序列高甲基化会导致eRNA表达下降,相反,低甲基化可促进eRNA的表达,因此,增强子序列CpG位点的甲基化成为增强子活性预测的标志物[30,31]。常用的DNA甲基化检测方法为BSP直接测序法,首先用重亚硫酸氢盐处理DNA,然后设计引物进行PCR扩增,直接对PCR产物测序进而判断CpG位点是否甲基化[32]。增强子序列的甲基化水平与增强子的转录活性成反比,通过BSP直接测序法检测已知增强子序列CpG位点是否甲基化,进而预测增强子的活性。
3.3 基于DNase-Seq、ATAC-Seq、RNA-Seq技术鉴定有活性的增强子
核小体是由DNA和组蛋白形成的染色质基本结构单位,一连串的核小体呈螺旋状排列构成染色质。常染色质状态下的DNA压缩包装比约为1000,在细胞有丝分裂前期染色质高度螺旋化成染色体,此染色体状态下的DNA压缩包装比最高可达8400,染色体长度约为伸展状态的万分之一,核小体的高度压缩使DNA序列不被暴露。非活性增强子通常由未修饰的核小体紧密包裹,因此它不能与转录因子或聚合酶结合。当增强子被激活时,它的局部染色质首先被修饰(如H3K4me1)变得松散,可以与转录因子和RNA聚合酶结合[10,33]。当增强子与一些转录因子如CBP(CREB- binding protein)结合,被充分激活,增强子将去除核小体结构,局部染色质完全开放,使其具有可接近性(亦称为染色质可及性,chromatin accessibility)。可以利用脱氧核糖核酸酶I超敏位点测序(DNase-Seq)技术或ATAC-Seq技术检测染色质开放区域。DNase-Seq技术利用脱氧核糖核酸酶I (DNase I)识别并切割染色质开放区域的DNA片段,然后对切出的DNA片段测序,与已知基因组序列进行比对,在全基因组范围内确定染色质开放的区域[33]。ATAC-Seq技术利用Tn5转座酶将已知序列标签插入到染色质开放区域,利用已知序列标签进行测序,捕获全基因组范围内完整的染色质开放区域。与DNase-Seq相比,ATAC-Seq不仅操作简单、可重复性强,并且可以在全基因组范围内捕获完整的染色质开放区域[33]。由于增强子位于染色质开放区域,利用DNase-Seq或ATAC-Seq在全基因组范围内检测染色质开放区域,可进行增强子的预测。
当增强子区域与RNA聚合酶结合启动转录,转录出增强子RNA。通过检测增强子表达出的eRNAs (如通过分析RNA-Seq数据),鉴定增强子的活性。Kim等[34]通过 RNA-Seq数据分析,在神经元细胞中发现了活跃增强子的广泛转录模式。2018年,Chen等[35]利用TCGA的RNA-Seq数据对33种癌症类型近9000例癌症患者样本中的增强子表达进行系统分析,发现癌症中广泛存在已激活的增强子。2019年,美国德州大学健康科学中心韩冷实验室利用RNA-Seq技术在全基因组范围内鉴定了9108种人类癌症中可检测到的eRNAs,构建了目前最完整的癌症eRNA图谱,充分展现了癌症中活性增强子的分布[36]。利用RNA-Seq在全基因组范围内检测eRNA的表达,判断已知增强子的转录活性。
3.4 基于基因编辑技术研究增强子
基因编辑技术可以在活细胞内对增强子进行靶向插入、敲除或修饰,进而探究增强子的功能和作用机制。基因编辑技术主要通过基因工程改造的核酸酶在基因组特定位点进行切割,使DNA双链断裂,通过同源重组(homologous recombinetion, HR)或非同源末端连接(non-homologous end joining, NHEJ)进行修复,从而导致基因的插入、删除或替换。基因编辑技术包括ZFN (zinc finger nucleases)技术、TALEN (transcription activator-like effector nucleases)技术以及CRISPR (clustered regularly interspaced short palindromic repeats)技术。与CRISPR相比,ZFN操作繁琐、价格昂贵,而TALEN的基因切割效率较低,因此CRISPR的应用更为广泛[37]。利用CRISPR靶向识别切割基因的特性对增强子的活性进行靶向调控,进而研究增强子的功能。研究者基于CRISPR开发出CRISPR-Cas (CRISPR-associated)系统,融合了向导RNA (guide RNA, gRNA),提高了基因编辑的靶向性和精确度,含有Cas9蛋白的CRISPR-Cas9在基因编辑中使用较为广泛。在CRISPR-Cas9系统的基础上开发出CRISPR-dCas9 (nuclease-deficient cas9, dCas9)系统,dCas9蛋白与效应器融合可以特定位点进行基因调控且不损伤DNA[4,38]。下面将分别列举 CRISPR-Cas9和 CRISPR-dCas9这两个工具在增强子功能研究中的应用。3.4.1 CRISPR-Cas9系统在增强子研究中的应用
目前最常用的基因编辑工具是CRISPR-Cas9,Cas9 核酸内切酶与gRNA融合,gRNA可将Cas9蛋白靶向到目标位点进行切割[4]。基于基因编辑技术构建靶向人增强子关键区的CRISPR-Cas9重组质粒,可实现目标增强子序列的定向敲除,便于增强子功能的预测和深入研究。郭晓龙等[37]人利用该技术构建靶向人ezrin增强子的CRISPR- Cas9载体并检测其基因敲除功能。该实验室设计了2个gRNA靶位点,分别靶向人ezrin增强子关键区的上、下游,并对食管癌EC109细胞进行传代培养,在CRISPR-Cas9重组质粒瞬时转染至食管癌EC109细胞群48h后,提取细胞基因组DNA,进行PCR扩增和亚克隆测序分析。结果表明,CRISPR-Cas9重组质粒构建正确,在共转染重组质粒的细胞基因组DNA中检测到ezrin增强子关键区的缺失,ezrin增强子的缺失会抑制癌细胞的增殖和迁移,进而为食管癌的治疗提供新的靶点(图2,黑色框所示)。莫文慧等[39]发现前列腺癌的独立风险因子位于一个增强子内部,并且该增强子与HOXA基因存在相互作用。为研究该增强子的功能,利用CRISPR-Cas9技术敲除该增强子,结果显示癌细胞的增殖和迁移能力降低,该增强子的缺失可以抑制前列腺癌细胞的增殖和迁移(图2,黑色框所示)。张晓燕等[40]利用CRISPR- Cas9技术敲除肺癌细胞中位于MYC基因3?端下游约450 kb的增强子,MYC基因表达降低,癌细胞的增殖和迁移能力降低。各种调控癌基因表达的增强子成为肿瘤治疗的新靶点。
图2
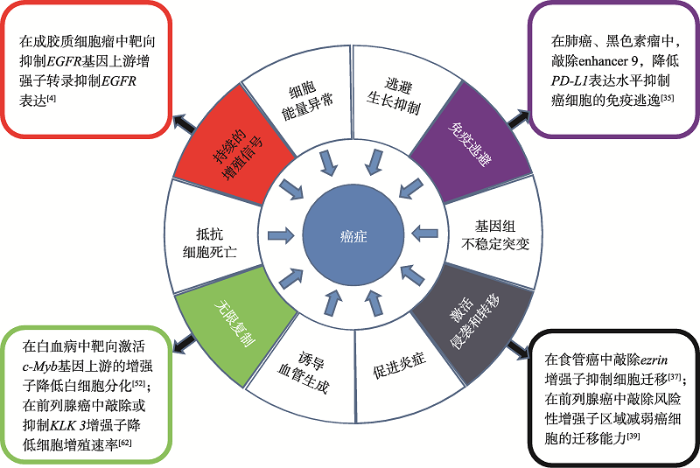 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2增强子对不同标志肿瘤的治疗应用
Fig. 2Application of enhancers in the treatment of tumors with different hallmarks
癌细胞的10大标志性特征如饼图所示;针对癌细胞的某标志性特征,与增强子相关的治疗方法分别列举在外围的4个方框中。
3.4.2 CRISPR-dCas9系统在增强子研究中的应用
CRISPR-dCas9系统是在CRISPR-Cas9的基础上对Cas9蛋白进行改造,使其失去内切酶活性,从而成为dCas9蛋白。在gRNA的引导下,dCas9蛋白只结合到目标序列上并不进行切割[41]。Qi等[42]利用CRISPR-dCas9系统,将抑制因子或激活因子与dCas9融合,靶向调控特定增强子的活性,调节相关靶基因的表达。抑制因子与dCas9融合后可抑制目标基因的转录,目前最常用的抑制因子是KRAB (kruppel-associated box, KRAB),KRAB通过组蛋白甲基化和去乙酰化修饰招募抑制转录的辅助因子,抑制基因的转录表达[41,43]。Gilbert等[4]利用dCas9-KRAB靶向已知的EGFR增强子,抑制增强子对EGFR基因转录的调控作用(图2,红色框所示)。dCas9-KRAB可通过靶向增强子来抑制其表达,而激活因子VP64和P300融合到dCas9中可增加目标基因的表达。Gao等[44]将VP64与dCas9蛋白融合,靶向激活远端增强子,增强子被激活表现出增强转录的功能[44]。徐丽等[45]将P300与dCas9融合,通过组蛋白乙酰化来招募促进转录的转录因子,靶向激活增强子的活性。利用CRISPR-dCas9系统靶向增强或抑制增强子进而影响靶基因的表达,该系统常用于已知增强子的功能研究。表1为研究增强子的常用方法描述及其优缺点总结。
Table 1
表1
表1增强子预测和功能解析的常用方法
Table 1
| 方法 | 描述 | 优势 | 局限性 |
|---|---|---|---|
| ChIP-Seq | 一种将染色质免疫沉淀与高通量测序技术相结合所产生的技术 | 高分辨率、低噪、高覆盖率、样本需求较少 | 与ChIP芯片技术相比成本较高,数据质量依赖抗体质量 |
| DNase-Seq | 一种结合高通量测序技术鉴定全基因组内DNase I超敏位点的方法,进行全基因组假定增强子等调控区域的预测 | 提供的信息比ChIP-Seq,更广泛 | 样本需求较大,重复性较差;DNase I对DNA的切割具有序列依赖性,测序误差较大;不能保证切割后的结果,就完全是蛋白质覆盖的区域 |
| ATAC-Seq | 利用转座酶研究染色质可进入性的高通量测序技术,利用DNA转座酶技术实现染色质可及性分析 | 样本需求量少,灵敏度高,操作简单,耗时短,实验重复性好,能同时揭示开放染色质的基因组位置,DNA结合蛋白,转录结合位点的相互作用 | 有一半DNA片段无法利用,无法进行PCR富集,DNA剪切效果仍需优化 |
| RNA-Seq | 一种全基因组水平的基因表达差异研究,用于分析基因表达水平以了解细胞在不同状态下的基因表达差异 | 高通量,高灵敏度,可重复性高,检测范围广 | 检测细胞内累积的RNA,包括来自核糖体线粒体的RNA,影响RNA表达水平的准确性 |
| 3C | 一种结合免疫共沉淀和PCR扩增研究两个位点之间的相互作用的技术 | — | 覆盖范围通常小于1 Mb;只能研究点对点的互作方式 |
| 4C | 在全基因组范围研究一个特定基因与所有互作基因之间的相互作用的高通量测序技术 | 覆盖全基因组范围 | 只能研究与一个特定基因互作的所有基因 |
| 5C | 研究多基因与多基因之间的相互作用高通量测序技术 | 可以研究多个位点与其相互作用的所有位点之间的相互作用 | 覆盖范围有限,通常小于1 Mb |
| Hi-C | 在全基因组范围研究所有染色体和染色体外的相互作用的高通量测序技术 | 覆盖全基因组范围,操作时间短,花费少 | 分辨率低,噪声高 |
| ChIA-PET | 一种整合了免疫共沉淀、染色质铰链、双末端标签及高通量测序的技术,可以在全基因组范围研究染色质的相互作用 | 能够确定蛋白质结合位点间的相互作用;使用超声打断DNA-蛋白质复合体,避免使用限制性内切酶引入染色质随机连接 | 无法检测出不依赖蛋白质因子的染色体相互作用;抗体的纯度、质量、特异性要求较高 |
| CRISPR | 将Cas9核酸酶与向导RNA(gRNA)结合,可在基因组的特定位点切割,从而实现基因的移除、添加或替换 | 在内源性环境中研究增强子,对靶向位点进行修饰,操作简单、快速 | 易脱靶,有效切割效率低 |
新窗口打开|下载CSV
4 超级增强子的研究方法
超级增强子是由多个具有转录活性的增强子串联而成,超级增强子的预测和功能鉴定是在已鉴定出的活性增强子基础上进行的(第3节为增强子的预测和功能鉴定方法)。与增强子相比,超级增强子序列结合更多的转录因子、辅助转录因子和RNA聚合酶Ⅱ,同时也有更高的转录活性,转录出更多的eRNA (图1所示)[17]。结合超级增强子的上述特征,在增强子基础上进一步鉴定出超级增强子。目前超级增强子的预测和鉴定是借助ChIP-Seq技术和RNA-Seq技术基于转录因子和辅助转录因子的结合特征、组蛋白修饰特征及eRNA的转录水平进行的[17]。图1
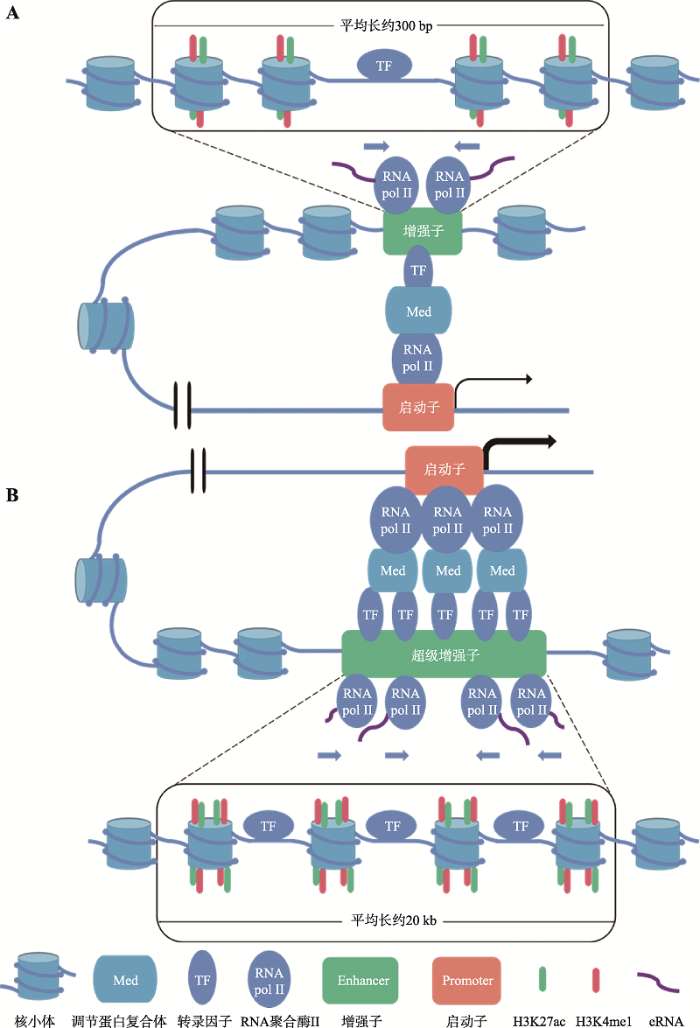 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1增强子与超级增强子的结构特征和功能
A:增强子的结构特征和功能。增强子位于染色质疏松的区域,平均长约300 bp。增强子区域的组蛋白富集H3K4me1和H3K27ac修饰,暴露的DNA序列招募并结合转录因子,转录因子招募Mediator复合体介导增强子与RNA聚合酶Ⅱ的相互作用。增强子在RNA聚合酶II的介导下双向转录eRNA。增强子区域和启动子区域形成三维环状结构相互作用,增强靶基因的转录水平。B:超级增强子的结构特征与功能。超级增强子是由多个具有转录活性的增强子串联而成,平均长约20 kb。与增强子相比,超级增强子区域组蛋白的H3K4me1和H3K27ac修饰更加富集,暴露的DNA序列结合更多的转录因子,转录因子招募更多的Mediator复合体介导增强子与RNA聚合酶Ⅱ的相互作用。暴露的超级增强子序列通过结合RNA聚合酶II双向转录出超级增强子RNA。超级增强子区域与启动子区域同样形成三维环状结构相互作用,但促进靶基因转录的效果更为显著(与A图比较,启动子下游的线更粗)。
Fig. 1Structural features and functions of enhancers and super enhancers
与增强子相比,超级增强子与转录因子和辅助转录因子的结合密度更高、组蛋白修饰也更加富集。因此可以借助ChIP-Seq技术在全基因组范围内鉴定出与转录因子或辅助转录因子结合或具有特定组蛋白修饰的增强子,再进一步根据转录因子和辅助转录因子的结合密度或组蛋白修饰的富集程度,筛选和鉴定出超级增强子[13,17,46]。Whyte等[16]利用ChIP-Seq技术在小鼠胚胎细胞中检测出8000多个增强子,发现其中231增强子的转录辅助因子结合密度远高于其余增强子,故将这些增强子命名为超级增强子。
此外,与增强子相比,超级增强子序列结合更多的RNA聚合酶II,因此超级增强子转录出的eRNA的水平更高。在已知的活性增强子基础上,利用RNA-Seq技术检测活性增强子的eRNA转录水平,进而判断该活性增强是否为超级增强子[17]。
5 增强子的应用方向
5.1 增强子在肿瘤诊断治疗上的应用潜力
肿瘤诊断是肿瘤治疗的前提,传统的癌症诊断依赖于影像学检查或活体组织检查(活检)。但是目前影像学检查的分辨率需要进一步提高;由于肿瘤有很强的异质性,活检组织的代表性越来越受质疑[47]。为了实现肿瘤的早期诊断,同时提高诊断的特异性和代表性,肿瘤的液体活检技术日益兴起,如血浆中循环游离DNA (circulating free DNA, cfDNA)的检测[31,48]。通过测序分析cfDNA中的增强子,根据增强子的肿瘤特异性诊断出癌基因的来源,肿瘤的早期诊断为肿瘤治疗提供了更大的成功率[48,49,50]。
癌细胞内的增强子通过与启动子相互作用促进癌基因的转录表达,进而导致肿瘤的发生,增强子的活性可以影响相关癌基因的表达。研究表明,免疫逃逸是肿瘤发生发展的重要机制。在肺癌和黑色素瘤中,PD-1/PD-L1通路的激活在肿瘤免疫逃逸过程中起着关键作用,T细胞表面的PD-1 (programmed cell death-1,程序性死亡受体-1)与肿瘤细胞表面的PD-L1 (programmed cell death 1 ligand 1,程序性死亡配体1)结合后传递抑制性信号,T细胞的增殖和活化受到阻碍,从而促进肿瘤细胞的免疫逃逸。抑制肿瘤细胞PD-L1的表达有利于进一步阻断PD-1/PD-L1信号通路的活化,使免疫治疗效果得以改善,因此,PD-L1成为了免疫治疗的主要靶点[51]。Chen等[35]利用TCGA的RNA-Seq数据对多种肿瘤样本分析发现,PD-L1 mRNA水平与PD-L1下游140 kb的增强子有很强的共表达关系。通过对Hi-C数据的再分析,进一步证实了增强子直接结合到PD-L1的下游。ChIP-Seq数据显示NF-κB结合在该增强子以及PD-L1启动子的p65结合基序上,表明NF-κB参与增强子与PD-L1的相互作用。利用CRISPR- Cas9技术敲除此增强子后,PD-L1在mRNA和蛋白水平上的表达均显著降低,并且很大程度上抑制了NF-κB对PD-L1表达的诱导效应,癌细胞免疫逃逸得到有效抑制(图2,紫色框所示)。这是一个增强子-启动子相互作用的模型,表明增强子还可作为预测癌症治疗反应的标志物。
c-Myb是造血过程中重要的转录因子,在造血祖细胞中c-Myb表达量很高,但随着细胞分化,c-Myb表达量会逐渐降低甚至不表达。c-Myb的异常表达会引发各种癌症,如白血病,乳腺癌,结肠癌。为研究c-Myb基因表达的调控机制,陈连香等[52]使用4C技术在人的白血病细胞系K562中发现,在c-Myb基因的上游有两个远端增强子与启动子互作较强。为进一步验证增强子的功能,利用dCas9-P300靶向激活增强子,发现c-Myb基因的表达增强,在一定程度上降低了k562细胞的分化,通过验证两个上游增强子对c-Myb的影响从而为白血病的治疗提拱了一个新的突破点(图2,绿色框所示)[52]。此外,还有一些增强子用于肿瘤特异性治疗研究,如在结肠癌细胞中敲除位于MYC基因上游335 kb处的肿瘤特异性增强子,癌细胞的生长和增殖受到抑制,而对正常肠道细胞没有任何影响[53];在急性T淋巴细胞白血病(T-cell acute lymphoblastic leukemia, T-ALL)的癌细胞中敲除位于TAL1 (T-cell acute lymphocytic leukemia 1)基因上游8 kb处的癌症特异性增强子的缺失,导致TAL1基因沉默进而癌细胞存活率降低[54]等。增强子的敲除可以使用CRISPR-Cas9技术,对增强子的抑制不仅可以使用CRISPR-dCas9技术,还可以利用化合物干扰转录因子与增强子的结合,进而达到抑制增强子的目的,如通过构建HBV (hepatitis B virus)人工转录因子靶向抑制HBV增强子活性[55]。
近年来研究发现,癌基因的高倍数扩增多发生于大规模DNA重组,在重组过程中发生的调控元件位置变化与肿瘤发生发展相关。这类重组包括了一种以染色体外环状DNA (extrachromosomal circular DNA, eccDNA)形式存在的特殊扩增体[56,57]。近年来研究表明,eccDNA几乎不存在于正常细胞中,而存在于近一半的人类癌症细胞中,且其富集处癌基因扩增明显,表明了这类DNA对于肿瘤细胞进化可能存在的重要意义[58]。2019年Mischel等[59]研究发现,在人类肿瘤细胞中发现的大量eccDNA,eccDNA内癌基因与增强子空间位置的变化,改变了癌基因的表达方式,从而促进了癌细胞的侵袭性,并在肿瘤快速进化和抵御威胁的能力(如化疗、放疗和其他治疗)中发挥了关键作用[59]。同年,Morton等[60]发现癌基因及其邻近增强子通过eccDNA的形式进行扩增从而增强已有联系或建立新的联系、促进癌症发展的全新分子机制。他们发现在非EGFR扩增的肿瘤细胞中,EGFR启动子仅与其上游的两个染色体内增强子具有显著联系;而在EGFR扩增的肿瘤细胞中,EGFR启动子尽管仍保持了与其上游两个增强子的联系,但同时也获得了大量新的互作对象。包括两个上游邻近增强子在内的多个于扩增后新获得的互作增强子都具有对EGFR扩增肿瘤细胞活性很强的正向效应。该研究表明增强子在由癌基因环化扩增介导的促癌效应中所发挥的重要作用,展现了超越通常规定的基因边界的基因调控网络。环状染色体外DNA上增强子新的互作模式为癌基因的高倍数扩增提供了依据,也为肿瘤的有效抑制提供新的方向。
Hanahan等[61]总结了目前癌细胞的十大标志性特征,如持续不断的增殖信号、逃避生长抑制因子、逃避免疫破坏,遗传信息的持续复制、促进新血管生成、基因组的不稳定性、抵抗细胞死亡、激活入侵和转移等。针对部分癌细胞的标志性特征,已有增强子相关的对应治疗方法(图2)。
5.2 eRNA在肿瘤诊断治疗上的应用潜力
eRNA在调控多种肿瘤信号通路中起着重要作用,介导靶基因的激活,调节基因表达。KLK3-eRNA (kallikrein related peptidase 3, KLK3)是人类前列腺癌激肽释放酶3 (KLK3)的增强子区域转录生成的,抑制KLK3-eRNA的合成可下调KLK3基因的表达,降低前列腺癌细胞增殖速率(图2,绿色框所示)[62]。因此,可通过分析肿瘤相关eRNAs来评价肿瘤的治疗效果。2019年,Zhang等[36]鉴定了9108种人类癌症中可检测到的eRNAs,并将这些可检测的eRNAs分为3组:652个普遍存在的eRNAs(在10种以上的癌症类型中表达);3124个中间特异性eRNAs (在2~9种癌症类型中表达);5332个癌症特异性eRNAs (只在一种癌症类型中表达)。表明大量eRNAs可能在特定的特异性癌症类型中具有生物标记作用。研究者通过分析癌细胞系的eRNAs表达水平与抗癌药物敏感性之间的相关性,发现无论是在直接目标通路内还是间接交叉通路,eRNAs和抗癌药物之间都有很强的相关性[36]。
5.3 超级增强子在肿瘤研究中的进展
5.3.1 超级增强子与肿瘤的关系近年研究发现,超级增强子所驱动的异常基因转录对维持肿瘤细胞特性至关重要。2013年美国怀特黑德生物医学研究所Richard A. Young实验室首次在多发性骨髓瘤细胞中发现了超级增强子通过调控 MYC、IRF4、PRDMI和XBPI 等基因,促进多发性骨髓瘤的发生和发展[63]。研究发现,超级增强子通过促进mRNA的生成、microRNA (miRNA)的转录以及成熟和lncRNA (long non-coding RNA)的转录生成对靶基因实现调控[9]。2018年Jiang等[64]在食道鳞状细胞癌(esophageal squamous cell carcinoma, ESCC)细胞中发现了主要转录因子TP63和SOX2,通过激活超级增强子和启动子协同调控长链非编码RNA CCAT1的表达,CCAT1促进细胞的增殖,SOX2或TP63基因的过表达可促进细胞增殖和肿瘤发生,CCAT1与TP63和SOX2形成复合物,通过与EGFR的超级增强子结合调控EGFR表达,激活EGFR下游信号通路,从而促进食道鳞状细胞癌发生。同年,Peng等[65]在肝癌(hepatocellular carcinoma, HCC)细胞中发现一种超级增强子驱动的长链非编码RNA HCCL5,可促进HCC细胞的存活、迁移和侵袭,促进肿瘤发生。
与增强子相比,大多数致癌基因的表达异常是由超级增强子驱动的,癌细胞通过构建驱动致癌基因过表达的超级增强子,显著促进多种癌基因表达,从而增强肿瘤细胞的增殖、侵袭和转移的能力;抑制超级增强子的活性,则显著抑制肿瘤细胞的生长和存活[17,66]。目前已在多种实体肿瘤细胞中鉴定出处于异常激活状态的超级增强子,如发病率较高的乳腺癌、结肠癌、小细胞肺癌、T细胞急性淋巴细胞白血病等[13]。研究并揭示肿瘤细胞中处于异常激活状态的超级增强子的构建机制和激活路径,为基因水平上抑制癌基因的过表达提供了新的思路,进一步改善肿瘤的临床治疗效果。
5.3.2 超级增强子在肿瘤治疗中的应用潜力
在超级增强子驱动致癌基因表达而导致的肿瘤细胞中,因超级增强子区域过大(平均在20 kb以上)不易敲除,为了精确抑制癌基因的表达,需要抑制超级增强子对癌基因的转录调控作用[49,67]。超级增强子调控癌基因转录的关键调节点可作为靶点特异性地抑制癌基因表达[13]。
超级增强子调控转录的关键调节点有:Mediator复合体、BRD4 (bromodomain containing 4)和关键的CDK (cyclin-dependent kinase),这些关键调节点基因表达水平降低均会抑制超级增强子调控的相关基因表达,因此,通过靶向关键调节点可以影响超级增强子调控的癌基因转录[13]。2016年Jiang等[46]发现了一种治疗食管鳞状细胞癌的特异性CDK7抑制剂THZ1,可抑制多种致癌基因转录表达。目前通过靶向抑制上述关键调节点进而针对超级增强子的治疗药物主要有:(1)BRD家族蛋白抑制剂或降解剂如:针对BRD4治疗多发性骨髓瘤的抑制剂JQ1;针对BDR4治疗白血病的抑制剂 iBET151;针对BRD2、BRD3、BRD4治疗成神经细胞瘤的抑制剂OTX051等。(2)CDK7抑制剂如:针对CDK7治疗食管鳞状细胞癌的抑制剂THZ1;针对CDK7治疗成人晚期实体瘤的抑制剂SY-1365等。(3)其他类型抑制剂如:针对RARα (retinoic acid receptor alpha, RARα)治疗急性髓样细胞样白血病的抑制剂SY-1425等[13]。
6 结语与展望
增强子在人类的生命过程中扮演着十分重要的角色,了解和研究增强子有助于精细解读人类基因组的功能、转录调控、细胞分化以及个体发育的机制。随着基因组水平鉴定技术的成熟和高通量测序技术的不断完善以及第三代测序技术的应用,增强子的定位、功能、鉴定会有较大突破,为肿瘤治疗提供新的有效的靶点。多个具有转录活性且作用方向相关的增强子构成超级增强子。研究显示,在多种肿瘤细胞中超级增强子处于激活状态,对癌基因的表达有促进作用。目前对超级增强子的组成、超级增强子内部每个增强子的活性以及超级增强子内部多个增强子的协作机制知之甚少。为了更好地治疗肿瘤,人们需要探索超级增强子内各个组分的作用机制,探索怎样利用药物同时抑制超级增强子内的多个增强子活性,从而抑制癌基因的表达。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 2]
DOI:10.1016/j.physbeh.2017.03.040URL [本文引用: 1]
URLPMID:16741521 [本文引用: 1]
URLPMID:25307932 [本文引用: 4]
[本文引用: 3]
[本文引用: 3]
DOI:10.1038/s41467-016-0009-6URL [本文引用: 2]
URLPMID:6938957 [本文引用: 1]
DOI:10.1038/290304a0URL [本文引用: 1]
[本文引用: 4]
[本文引用: 4]
[本文引用: 4]
[本文引用: 4]
[本文引用: 2]
[本文引用: 2]
URLPMID:21295696 [本文引用: 1]
[本文引用: 6]
[本文引用: 6]
[本文引用: 2]
DOI:10.1101/gr.184986.114URL [本文引用: 2]
DOI:10.1016/j.cell.2013.03.035URL [本文引用: 2]

Master transcription factors Oct4, Sox2, and Nanog bind enhancer elements and recruit Mediator to activate much of the gene expression program of pluripotent embryonic stem cells (ESCs). We report here that the ESC master transcription factors form unusual enhancer domains at most genes that control the pluripotent state. These domains, which we call super-enhancers, consist of clusters of enhancers that are densely occupied by the master regulators and Mediator. Super-enhancers differ from typical enhancers in size, transcription factor density and content, ability to activate transcription, and sensitivity to perturbation. Reduced levels of Oct4 or Mediator cause preferential loss of expression of super-enhancer-associated genes relative to other genes, suggesting how changes in gene expression programs might be accomplished during development. In other more differentiated cells, super-enhancers containing cell-type-specific master transcription factors are also found at genes that define cell identity. Super-enhancers thus play key roles in the control of mammalian cell identity.
[本文引用: 7]
[本文引用: 7]
DOI:10.1016/j.cell.2013.09.005URL [本文引用: 1]
DOI:10.1038/nature12787URL [本文引用: 1]
Enhancers control the correct temporal and cell-type-specific activation of gene expression in multicellular eukaryotes. Knowing their properties, regulatory activity and targets is crucial to understand the regulation of differentiation and homeostasis. Here we use the FANTOM5 panel of samples, covering the majority of human tissues and cell types, to produce an atlas of active, in vivo-transcribed enhancers. We show that enhancers share properties with CpG-poor messenger RNA promoters but produce bidirectional, exosome-sensitive, relatively short unspliced RNAs, the generation of which is strongly related to enhancer activity. The atlas is used to compare regulatory programs between different cells at unprecedented depth, to identify disease-associated regulatory single nucleotide polymorphisms, and to classify cell-type-specific and ubiquitous enhancers. We further explore the utility of enhancer redundancy, which explains gene expression strength rather than expression patterns. The online FANTOM5 enhancer atlas represents a unique resource for studies on cell-type-specific enhancers and gene regulation.
DOI:10.1006/meth.1996.0407URLPMID:8993033 [本文引用: 1]
Recent advances leave no doubt that higher order chromatin structures play a fundamental role in many developmentally important mechanisms of gene regulation. In particular analyses in genetic model systems like yeast and Drosophila uncovered novel proteins that are involved in the regulation of chromatin structures. Many of these proteins do not bind directly to DNA but interact in large multimeric complexes. To identify the DNA elements regulated by these multiprotein complexes, alternative approaches to the standard methods of DNA-protein analysis had to be devised. Here we present a method that preserves the architecture of the higher order chromatin structures by cross-linking cells in vivo with formaldehyde. An immunoprecipitation strategy is then used to identify the DNA targets of chromosomal proteins of interest. This method can be applied to study the distribution of proteins at high resolution over extended chromosomal regions.
URLPMID:25827895 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nature07730URLPMID:19212405 [本文引用: 1]
A major yet unresolved quest in decoding the human genome is the identification of the regulatory sequences that control the spatial and temporal expression of genes. Distant-acting transcriptional enhancers are particularly challenging to uncover because they are scattered among the vast non-coding portion of the genome. Evolutionary sequence constraint can facilitate the discovery of enhancers, but fails to predict when and where they are active in vivo. Here we present the results of chromatin immunoprecipitation with the enhancer-associated protein p300 followed by massively parallel sequencing, and map several thousand in vivo binding sites of p300 in mouse embryonic forebrain, midbrain and limb tissue. We tested 86 of these sequences in a transgenic mouse assay, which in nearly all cases demonstrated reproducible enhancer activity in the tissues that were predicted by p300 binding. Our results indicate that in vivo mapping of p300 binding is a highly accurate means for identifying enhancers and their associated activities, and suggest that such data sets will be useful to study the role of tissue-specific enhancers in human biology and disease on a genome-wide scale.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/ng1966URLPMID:17277777 [本文引用: 2]
Eukaryotic gene transcription is accompanied by acetylation and methylation of nucleosomes near promoters, but the locations and roles of histone modifications elsewhere in the genome remain unclear. We determined the chromatin modification states in high resolution along 30 Mb of the human genome and found that active promoters are marked by trimethylation of Lys4 of histone H3 (H3K4), whereas enhancers are marked by monomethylation, but not trimethylation, of H3K4. We developed computational algorithms using these distinct chromatin signatures to identify new regulatory elements, predicting over 200 promoters and 400 enhancers within the 30-Mb region. This approach accurately predicted the location and function of independently identified regulatory elements with high sensitivity and specificity and uncovered a novel functional enhancer for the carnitine transporter SLC22A5 (OCTN2). Our results give insight into the connections between chromatin modifications and transcriptional regulatory activity and provide a new tool for the functional annotation of the human genome.
URLPMID:21106759 [本文引用: 1]
URLPMID:22231485 [本文引用: 2]
DOI:10.1101/gr.155028.113URL [本文引用: 1]
Compared with histone H3, acetylation of H4 tails has not been well studied, especially in mammalian cells. Yet, H4K16 acetylation is of particular interest because of its ability to decompact nucleosomes in vitro and its involvement in dosage compensation in flies. Here we show that, surprisingly, loss of H4K16 acetylation does not alter higher-order chromatin compaction in vivo in mouse embryonic stem cells (ESCs). As well as peaks of acetylated H4K16 and KAT8 histone acetyltransferase at the transcription start sites of expressed genes, we report that acetylation of H4K16 is a new marker of active enhancers in ESCs and that some enhancers are marked by H3K4me1, KAT8, and H4K16ac, but not by acetylated H3K27 or EP300, suggesting that they are novel EP300 independent regulatory elements. Our data suggest a broad role for different histone acetylation marks and for different histone acetyltransferases in long-range gene regulation.
URLPMID:27089178 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.immuni.2010.02.008URL [本文引用: 3]
Summary
Enhancers determine tissue-specific gene expression programs. Enhancers are marked by high histone H3 lysine 4 mono-methylation (H3K4me1) and by theacetyl-transferase p300, which has allowed genome-wide enhancer identification. However, the regulatory principles by which subsets of enhancers become active in specific developmental and/or environmental contexts are unknown. We exploited inducible p300 binding to chromatin to identify, and then mechanistically dissect, enhancers controlling endotoxin-stimulated gene expression in macrophages. In these enhancers, binding sites for the lineage-restricted and constitutive Ets protein PU.1 coexisted with those for ubiquitous stress-inducible transcription factors such as NF-κB, IRF, and AP-1. PU.1 was required for maintaining H3K4me1 at macrophage-specific enhancers. Reciprocally, ectopic expression of PU.1 reactivated these enhancers in fibroblasts. Thus, the combinatorial assembly of tissue- and signal-specific transcription factors determines the activity of a distinct group of enhancers. Wesuggest that this may represent a general paradigm in tissue-restricted and stimulus-responsive gene regulation.Highlights
? LPS-inducible binding of p300 to chromatin reveals inflammatory gene enhancers ? These enhancers combine sites for tissue-specific and inducible transcription factors ? Combination of tissue- and signal-specific TFs adapts the response to the context[本文引用: 1]
[本文引用: 2]
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}