 ,, 孔祥阳,昆明理工大学医学院疾病与药物遗传实验室,昆明 650500
,, 孔祥阳,昆明理工大学医学院疾病与药物遗传实验室,昆明 650500Lung microbiome mediates the progression from chronic obstructive pulmonary disease to lung cancer through inflammation
Yajie Wang, Shuangshuang Wu, Jiang Chu,, Xiangyang Kong,Genetics and Pharmacogenomics Laboratory, Medical School, Kunming University of Science and Technology, Kunming 650500, China通讯作者: 孔祥阳,博士,教授,研究方向:慢性阻塞性肺病和肺癌的基因组学研究。E-mail:kxy2772@yahoo.com储江,博士,讲师,研究方向:慢性阻塞性肺病和肺癌的代谢研究。E-mail:chujiang2015@126.com
编委: 姜长涛
收稿日期:2020-11-24修回日期:2020-12-28网络出版日期:2021-01-20
| 基金资助: |
Received:2020-11-24Revised:2020-12-28Online:2021-01-20
| Fund supported: |
作者简介 About authors
王娅洁,在读硕士研究生,专业方向:遗传学。E-mail:

摘要
肺部微生物组存在于呼吸道和实质组织中,通过菌群紊乱、代谢产物、炎症反应、免疫反应、基因毒性等方面介导肺部损伤。随着肺部微生物组的深入研究,发现肺部微生物组的相关活动与慢性阻塞性肺疾病(chronic obstructive pulmonary disease, COPD)和肺癌息息相关,能够促使COPD向肺癌的转变。本文主要介绍了肺部微生物组稳态及其通过炎症反应导致COPD和肺癌,重点探讨了肺部微生物组如何通过炎症反应介导COPD转化为肺癌,以期为COPD和肺癌的临床预防、优化治疗以及新型药物设计提供新的理论依据。
关键词:
Abstract
Lung microbiome exists in the respiratory tract and parenchymal tissues. It mediates lung injury through a variety of mechanisms, including bacterial disturbance, metabolites, inflammatory response, immune response, and genotoxicity. Accumulating evidences suggest that changes in lung microbiome correlates with chronic obstructive pulmonary disease (COPD) and lung cancer, and the microbiome promotes the progression from COPD to lung cancer. In this review, we mainly introduce the impairment of the homeostasis of the lung microbiome and its inflammation that leads to COPD and lung cancer, then focus on how the microbiome mediates the progression from COPD to lung cancer through inflammatory response. The review may provide a new theoretical basis for clinical prevention, optimal treatment strategy and design of new drugs for COPD and lung cancer.
Keywords:
PDF (628KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王娅洁, 吴爽爽, 储江, 孔祥阳. 肺部微生物组通过炎症反应介导慢性阻塞性肺疾病转化为肺癌的研究进展. 遗传[J], 2021, 43(1): 30-39 doi:10.16288/j.yczz.20-315
Yajie Wang.
随着16S/18S/基因间隔序列(intergenic transcribed spacer, ITS)扩增子测序、宏基因组测序、微生物全基因组测序等微生物组测序技术在微生物组检测中的应用[1],人们逐渐意识到下呼吸道实际上含有多种细菌,从而为肺部相关疾病研究提供了新的方向[2]。微生物组的稳态与宿主健康息息相关,“失调”的微生物菌群可能作为病原体,通过引起持续性炎症影响人体呼吸道对各种疾病的易感性[3]。
肺部微生物组对肺部的损害也逐渐得到证实,并且与慢性阻塞性肺疾病(chronic obstructive pulmonary disease, COPD)、肺癌、哮喘、囊性纤维化等多种肺部疾病相关[4,5]。其中COPD和肺癌是世界范围内导致死亡的重要原因。COPD作为一个主要的公共卫生问题,影响了全球约2.51亿人[6,7],是一种以不完全可逆性气流受限为特征的慢性肺疾病,与有毒颗粒和气体引起的炎症反应增强有关[8,9]。烟草烟雾、空气污染、遗传因素、表观遗传修饰及微生物组等被认为是 COPD的高危因素,但其根本病因尚不明确,临床无法根治[3]。同样,肺癌是癌症相关死亡的主要原因,几乎占全球癌症死亡人数的1/5[10]。据估计,到2030年,每年死于肺癌的人数将上升到1000万人[11]。肺癌是一种由致癌基因突变引起的,导致正常细胞在遗传和表观遗传学改变的影响下逐渐转化为恶性衍生物的复杂的异质性疾病[12]。根据其细胞起源和表型,肺癌可分为两个亚型:非小细胞肺癌(non-small cell lung cancer, NSCLC)和小细胞肺癌(small cell lung cancer, SCLC)[12]。NSCLC起源于上皮细胞,占大多数病例(80%),可进一步划分为腺癌、鳞状细胞癌和大细胞癌[12]。越来越多的证据显示,慢性炎症、表观遗传、遗传易感性和环境因素等都对COPD转化为肺癌有推动作用[3,13]。
肺部微生物组感染COPD患者呼吸道,并在气道中持续存在,导致气道上皮损伤和慢性炎症,进而引发病情加重[14]。而肺部微生物组在肿瘤发展中的作用机制也成为一个迅速发展的研究领域,研究发现细菌性肺部病原体可以通过调节肿瘤相关炎症以及直接促进癌细胞增殖来促进肺癌的发展[15]。因此肺部微生物组可能通过慢性炎症成为COPD和肺癌之间的“中介”,为COPD转化为肺癌提供一个新的角度。本文将从肺部微生物组如何通过炎症反应介导COPD转化为肺癌进行阐述,以期为COPD转化为肺癌研究提供参考,从而为临床预防、优化治疗以及新型药物设计提供新的理论依据。
1 肺部微生物组的多样性与COPD和肺癌
1.1 肺部微生物组
最近使用微生物免培养检测技术已经证实,无论是健康人群的还是疾病患者的肺部,都有不同的微生物菌群[16,17]。这一发现挑战了许多关于呼吸系统及肺部相关疾病长期存在的致病机制假设。肺部微生物组包含了存在于呼吸道和实质组织中的细菌、病毒和真菌,参与呼吸道免疫的成熟与稳态[18]。健康人的肺部微生物组具有多样性与稳定性,并与机体环境形成一种动态平衡,有利于机体新陈代谢、免疫系统发育和抵抗病原体[19]。然而肺部细菌负荷程度低,短暂的微生物组紊乱也会对其多样性造成显著的影响进而因“生态失调”而导致严重炎症反应[20]。因此如果肺部微生物组与宿主的动态平衡被打破,出现某种优势菌群从而使微生物组多样性改变,可能会导致气道失调和免疫紊乱,产生严重的肺部损伤[16]。
1.2 肺部微生物组与COPD
已有研究表明慢性呼吸道疾病患者下呼吸道的微生物组成与健康人群不同[21,22,23],COPD患者的微生物菌群由健康人群的拟杆菌门(Bacteriodetes)和厚壁菌门(Firmicutes)转变为流感嗜血杆菌(Haemophilus influenza)、肺炎链球菌(Streptococcus pneumoniae)和卡他莫拉菌(Moraxella Catarrhalis)以及恶化期的铜绿假单胞菌(Pseudomonas aeruginosa)等[24,25]。值得注意的是,可能由于采样部位的不同,也有许多研究认为硬壁菌门(Firmicutes)也是中重度COPD患者中主要细菌[3]。Garcia-Nu?ez等[26]对17名COPD患者的痰标本微生物组分析显示,支气管微生物组多样性与COPD严重程度密切相关,尤其是在疾病晚期,细菌的多样性显著下降。在患者中主要的微生物菌群包含变形菌门(Proteobacteria) (44%)、厚壁菌门(16%)和放线菌门(Actinobacteria) (13%)[27]。其中变形菌门相对丰度明显增高,包括大部分通常被视为潜在致病微生物的细菌,同时硬壁菌门中微生物相对丰度下降[26],这种微生物组成的变化破坏了从口咽到支气管微生物群落模式的连续性,与疾病的严重程度平行[28]。Galiana等[29]也发现与轻/中度患者相比,重度COPD患者微生物组多样性更低。随着对肺部微生物组的深入了解,也有人认为COPD的进展与恶化可能不仅仅与单个致病菌有关,而是与整个微生物菌群的紊乱相关[30]。
1.3 肺部微生物组与肺癌
肺部微生物组异常已经被证明与一系列肺部疾病相关,例如哮喘、COPD和囊性纤维化[31]。虽然与这些疾病相比,关于肺癌微生物组的研究仍比较少,但已经有研究显示肺癌与肺部微生物组的菌群多样性相关[31]。通过对肺癌患者微生物组成分析,发现微生物组的菌群多样性改变不仅与肺癌患者的疾病进展密切相关,也能反应不同的肺癌类型,因此特定的微生物组变化也许可以成为检测以及区分不同类型肺癌的生物标记物[32]。研究人员对来自165个癌症患者的非恶性肺组织样本中微生物组进行分析,发现变形菌属(Proteus Hauser)为主要微生物菌群,在晚期(IIIB, IV)患者的组织中栖热菌属(Thermus)更丰富,军团菌属(Legionella)在发生转移的患者中更多[33]。同时与非恶性肺组织的微生物菌群相比,肿瘤具有更高的微生物α多样性[33]。Lee等[34]通过对28名肺癌患者与良性肿块样病变的支气管肺泡灌洗液微生物组进行比较,结果显示肺癌患者与良性肿物样病变患者的微生物菌群存在差异,肺癌患者的厚壁菌门和TM7门相对丰度显著增加。肺癌患者中韦荣氏球菌属(Veillonella)和巨球形菌属(Megasphaera)也相对丰富,并且其组合预测肺癌的曲线下面积为0.888,暗示可以作为肺癌的诊断标记物[34]。
对一组从未吸烟的中国肺癌女性人群痰样本微生物组检测显示,与健康对照组相比营养缺陷菌属(Abiotrophia)、链球菌属(Streptococcus)及颗粒链菌属(Granulicatella)显著富集,同时微生物组多样性显著降低[35]。Mur等[31]的实验也得出了一致的结果,颗粒链菌属在肺癌患者中显著富集。Yan等[36]通过对腺癌、鳞状细胞癌患者和健康对照组唾液样本微生物组比较发现,在肺癌患者中微生物组多样性改变,二氧化碳噬纤维菌属(Capnocytophaga)、月形单胞菌属(Selenomonas)和韦荣氏球菌属显著富集而奈瑟菌属(Neisseria)丰度显著降低,其中二氧化碳噬纤维菌属联合韦荣氏球菌属作为生物标志物可有效区分腺癌或鳞状细胞癌与对照组,在肺癌的筛查中具有一定的应用价值。
2 肺部微生物组相关炎症与COPD和肺癌
2.1 肺部微生物组相关炎症
共生菌和致病性微生物,都是宿主免疫系统的关键调节者,也是炎症的调节者。共生菌通过抗菌肽和其他分子来抑制病原菌的生长[37],维持菌群的稳态,但当菌群紊乱时,优势菌群过度增殖可能会引发炎症,而炎症反应作为机体重要的一种环境应激反应,会加剧微生物组的紊乱,从而形成恶性循环[38]。Mayhew等[39,40]对COPD患者中的肺部微生物组纵向研究结果也表明,与稳定期的患者相比,个体内的恶化表现出更高的微生物群变异性,频繁恶化患者更有可能经历肺微生物组模式的显著变化。模式识别受体(pattern-recognition receptor, PRR)包括Toll样受体(Toll-like receptors, TLRs)、NOD样受体(NOD-like receptors, NLRs)、RIG样受体(RIG-I like receptors, RLRs)和C型凝集素受体(C-type lectin receptors, CLRs),是一类表达于固有免疫细胞表面可与微生物相关分子模式(microbe-associated molecular pattern, MAMP)结合的识别分子[41,42]。它们通过感应微生物细胞壁、细菌运动鞭毛蛋白、微生物核酸或应激分子来识别各种微生物感染[41]。
紊乱的肺部微生物组可以通过MAMP-PRR的相互作用,激活核转录因子κB (NF-κB)通路、信号转导与转录激活因子3 (signal transducer and activator of transcription 3, STAT3)等释放炎症细胞因子调节固有免疫和适应性免疫,引起炎症反应[4,43]。Th17和γδT炎症相关途径已经在许多COPD和肺癌患者中发现,通过分泌IL-6和IL-17等炎症因子促进炎症反应[20,44]。
在非肿瘤性的情况下,炎症可以诱发增生性病变,但是如果这些病变不受控制地增殖,它们可以转变并适应肿瘤和非典型腺瘤的外观,获得侵袭性、血管生成性和转移性,最终形成肿瘤[3]。
2.2 肺部微生物组相关炎症对COPD的影响
细菌容易感染COPD患者呼吸道并在气道中持续存在,导致气道上皮损伤和慢性炎症,进一步损伤肺防御系统和导致更严重的肺部微生物组紊乱,从而形成恶性循环并伴随严重后果[45]。病原微生物组常常定植于稳定的COPD患者,并通过放大肺部炎症来促进COPD的发展[46]。Yadava等[47]利用与人类COPD炎症表型与气道异常相似的小鼠模型进行研究,发现肺部微生物组失调,导致IL-17A表达增加。通过对比处理后无菌的小鼠,发现微生物组可以促进γδ+ T细胞产生IL-17A。从LPS/弹性蛋白酶处理的动物体内转移富集的微生物菌群,并在抗生素治疗的小鼠中同时激发LPS/弹性蛋白酶,显示IL-17A免疫表型的上调。通过这些对比实验,可以认为肺部微生物组的稳态与炎症反应密切相关。
Wang等[48]通过对87例COPD患者476份痰样本的16S核糖体RNA纵向调查显示,与稳定期患者相比,COPD加重期的患者肺部微生物组也发生了变化,卡他莫拉菌显著增加。同时观察到卡他莫拉菌和中性粒细胞百分比之间存在正相关关系,并与疾病的严重程度显著相关,这可能与宿主的免疫反应有关。以及分析得到痰液中CXCL8/IL-8与菌群微生物组多样性呈显著负相关,并通过募集中性粒细胞和上调气道粘蛋白基因来诱导气道炎症,从而产生粘液进一步导致慢性炎症[49]。而且有研究表明,气道炎症标志物如IL-6、IL-8、IL-1β、TNF-α、白三烯B4(LTB4)、基质金属蛋白酶(MMP)、髓过氧化物酶(MPO)和中性粒细胞弹性蛋白酶(NE)在稳定的COPD患者中的水平高与无致病菌定植的患者,强烈暗示细菌定植会产生有害的炎症反应,这种持续性炎症与症状恶化和死亡率有关[13,44,50]。
2.3 肺部微生物组相关炎症对肺癌的影响
已经有研究提出并验证了介导微生物组致癌的潜在机制,结果表明,微生物菌群的生态失调在多个水平上调控了恶性肿瘤的易感性,包括免疫反应、促炎反应、毒力增强和代谢改变。肺部微生物组多样性改变造成的紊乱,会使炎症诱导细菌增多,从而多层次诱发癌变[4]。MAMP和PRR系统,不仅可以识别病原微生物,引发炎症,还可以在一定的情况下触发上皮细胞的增殖和存活,从而促进癌症的发展[4]。TLRs通过激活NF-κB和STAT3通路引发炎症,诱导癌症发生[41]。NOD2 (nucleotide-binding oligomerization domain 2)同样在微生物组活动中起着重要的调节作用,被敲除NOD2基因的小鼠会出现细菌增多和炎症反应增加[51]。而且多项肺癌转移模型和K-ras诱导的肺癌模型显示,细菌引起的肺部炎症明显加速了肺癌的生长,同时实验证明无菌或抗生素治疗的小鼠对K-ras突变和p53缺失诱发的肺癌有明显的保护作用[15,52,53]。
另外,研究发现微生物组在髓系细胞中刺激髓样分化因子(MyD88)产生的IL-1β和IL-23,诱导Vγ6+Vδ1+γδ T细胞增殖和活化,释放IL-17和其他效应分子,会促进炎症和肿瘤细胞增殖[54]。Jungnickel等[55]将IL-17C (IL-17C-/-)缺陷小鼠和Toll样受体2和4 (TLR-2/4-/-)双缺陷小鼠置于转移性肺癌模型中,实验结果表明IL-17C不仅能促进中性粒细胞向肿瘤微环境的募集,而且能通过诱导肿瘤细胞源性趋化因子将中性粒细胞导向肿瘤组织。这些结果有力地表明了肺部微生物组可以通过引起局部炎症促进肺癌的发展。
3 肺部微生物组与COPD和肺癌之间联系
COPD和肺癌是世界范围内与肺部疾病相关的高死亡率的主要原因[3]。流行病学证据表明表观遗传、免疫功能紊乱、遗传易感性、氧化应激和慢性炎症等机制都可以驱动COPD转化为肺癌[12]。同时COPD也是肺癌发生的独立因素,增加肺癌的发病率与死亡率,并对肺癌治疗预后带来负面效果,临床上常用广谱抗炎药物、支气管扩张剂等药物通过控制COPD症状降低肺癌发病率[56]。对肺部微生物组深入研究也为肺部相关疾病带来了新的解释。研究表明,在由微生物组导致的相关疾病中,微生物组通常通过破坏器官粘膜或上皮组织的完整性、破坏细胞,导致组织损伤,触发局部慢性炎症反应,并因此引起持续不断的微生物组紊乱[57]。同样根据微生物组、炎症与癌症进展的多项相关研究,Francescone等[57]认为,微生物组通常通过诱导炎症刺激肿瘤生长,而不是直接作用于癌细胞。因此,微生物组、宿主免疫系统和炎症之间的微妙平衡对癌症的发展或预防至关重要。
慢性炎症在肿瘤发生中起重要作用,增加患癌风险,高达10%~20%的癌症可归因于慢性炎症与感染,与肿瘤发生的各个过程相关,包括细胞转化、促进、存活、增殖、侵袭、血管生成和转移等[58]。许多慢性炎症性肺部疾病如特发性肺纤维化(idiopathic pulmonary fibrosis, IPF)、肺结核(tuberculosis, TB)、COPD等都是肺癌的危险因素,与肺癌的发生、发展密切相关[59]。在IPF中,持续存在的慢性炎症使支气管上皮细胞不断发生损伤和修复,最终导致DNA损伤,进而转变为肺癌,同时在TB中,可通过诱发慢性炎症和组织修复为肺癌发生提供环境[59]。COPD以慢性炎症为主要特征,涉及复杂的病理过程包括破坏肺部结构、改变肺部微环境甚至促进恶性肿瘤发展[56],相较于其他肺部慢性疾病,以慢性炎症为主要特征的COPD可以使肺癌的风险增加4~6倍,是导致肺癌的重要原因[13]。我们在前文中也已经阐述了肺部微生物组多样性及其紊乱引起的炎症反应对COPD和肺癌患者的影响,肺部微生物组和慢性炎症作为其共同致病因素,可能是介导COPD转化为肺癌的一个重要原因。
如前文所述,肺部微生物组的多样性在COPD和肺癌患者中不仅会发生改变,而且与COPD和肺癌的疾病进展密切相关。流感嗜血杆菌和硬壁菌门在COPD和肺癌患者中都是优势菌群[22]。同样是肺癌患者致病菌的TM7在COPD患者中也有数量增加的研究结果[22]。而吸烟作为COPD和肺癌的共同致病因素,可以通过破坏肺上皮细胞,诱发细菌感染和炎症反应,促进COPD转化为肺癌[34]。研究表明,吸烟的肺癌患者的硬壁菌门与拟杆菌门的比值明显比不吸烟的肺癌患者更高[34]。多个肺癌转移模型和K-ras诱导的肺癌模型均显示细菌引起的肺部炎症明显加速了肺癌的恶化[15,53,60]。这些结果都表明了COPD和肺癌在微生物组方面有着密切联系,肺部微生物组紊乱和共同的致病菌增加了COPD患者转变为肺癌的风险。
已经有多个动物模型表明不可分型流感嗜血杆菌(nontypeable Haemophilus influenza, NTHi)可能在COPD样气道炎症和肺癌促发中起到因果关系。NTHi是一种小的不可分型的缺乏荚膜的革兰氏阴性球杆菌,主要作为粘膜病原体,定植在约75%的正常成人的上呼吸道[61]。与大多数其他细菌感染一样,NTHi感染可通过显著释放细胞因子和趋化因子来诱导炎症。NTHi可以激活模式识别受体如NLRs和TLRs,通过NF-κB通路,显著增加促炎介质的表达和释放,包括IL-6、IL-8和TNF,气道上皮细胞能够通过先天免疫机制感知和响应炎症刺激从而导致肺部炎症[62]。
有研究发现NTHi在30%的慢性阻塞性肺病患者中及87%的急性加重期患者的支气管组织中定植,可通过诱导中性粒细胞进入气道[61]。中性粒细胞坏死并释放中性粒细胞弹性蛋白酶和其他基质金属蛋白酶以及产生氧自由基来促进COPD的进展[61,63]。同时也在肺癌中观察到NTHi通过肿瘤相关炎症促进肿瘤恶化,例如可通过NF-κB途径调节炎症介质(TNF、IL-6等)表达,从而促进肿瘤增殖[15]。
Moghaddam等[53]通过将携带LSL-K-rasG12D等位基因的小鼠与插入Clara细胞分泌蛋白(CCSP)位点的Cre重组酶的小鼠杂交,建立了两种新的肺癌小鼠模型(分别为CCSPCre-Neo/LSL-K-rasG12D和CCSPCre/LSL-K-rasG12D)。将CCSPCre/LSL-K-rasG12D小鼠暴露于NTHi裂解物中,形成COPD样气道炎症,最终结果显示肺表面肿瘤数量增加3.2倍[53]。
Barta等[64]在维甲酸诱导的G蛋白偶联受体敲除小鼠模型(Gprc5a-/-小鼠)中,分别研究了细菌诱导的COPD样炎症和烟草致癌物促进肿瘤发生/炎症的作用。结果表明细菌单独诱导的慢性外源性肺部炎症可增强Gprc5a-/-小鼠的肺癌发生,NTHi暴露可使增生灶数目增加6倍,肿瘤多样性增加2倍。NTHi暴露后,NF-κB通过激活HIF-1α(hypoxia inducible factor 1alpha)通路及其下游血管生成信号,促进Gprc5a-/-小鼠对细菌性炎症的反应,同时产生细胞因子和趋化因子(如IL-6和IL-17)[65],吸引白细胞,从而促进肿瘤进展、癌细胞生长和扩散、血管生成、侵袭和肿瘤免疫抑制[66]。
多项研究结果证明,NTHi诱导的COPD样气道炎症提供了一个有利于肺部肿瘤促进和发展的肿瘤微环境,表明了肺部微生物组可以通过炎症反应促进COPD转变为肺癌。
为了进一步探究COPD样炎症促进肺癌的机制,在细菌诱导的COPD样炎症小鼠模型中,IL-6基因敲除型及IL-17C-/-和TLR-2/4-/-双缺陷型小鼠均能抑制COPD炎症及肿瘤进展[67]。COPD样炎症促进肺癌的过程不仅与NF-κB和STAT3通路激活、炎症细胞因子的增加有关,而且与骨髓细胞反应的增强(M2型巨噬细胞极化及中性粒细胞和髓源性抑制细胞的积累)有关[67]。综上所述,这些结果都表明了肺部微生物组及相关炎症在促进COPD转化为肺癌方面的重要性。
4 结语与展望
COPD和肺癌是世界范围内导致死亡的重要原因,有共同的致病机制,而COPD在各种因素作用下可以发展为肺癌[3]。随着微生物组测序技术的发展,肺部微生物组在COPD和肺癌的影响也逐渐被大家认识,成为新的研究热点。人们的注意力也从初始的对肺部微生物组多样性的了解转移到特定的微生物菌群的定植与持续存在即紊乱,以及相关的炎症途径及导致的功能改变[68,69]。而肺部微生物组介导的慢性炎症作为COPD和肺癌的共同致病因素,也为COPD转变为肺癌提供了新的解释。肺部微生物组的改变与机体之间是双向关系,呼吸道的任何炎症源都会引发一系列的宿主反应,打破肺部微生物组与宿主之间的动态平衡,从而改变微生物组生长条件致使微生物菌群紊乱[16]。紊乱的微生物菌群通过MAMP-PPR的相互作用,激活NF-κB及STAT3等相关通路释放炎症细胞因子[43],介导慢性炎症以及刺激细胞增殖、血管生成、组织重塑或转移进而导致癌变[54],这会导致肺部微环境的进一步紊乱,从而形成一个恶性循环加剧疾病进展(图1)。因此,一个自我放大的反馈回路使呼吸道炎症以及推动炎症的紊乱微生物菌群持续存在,可能在COPD转化为肺癌中发挥重要作用。
图1
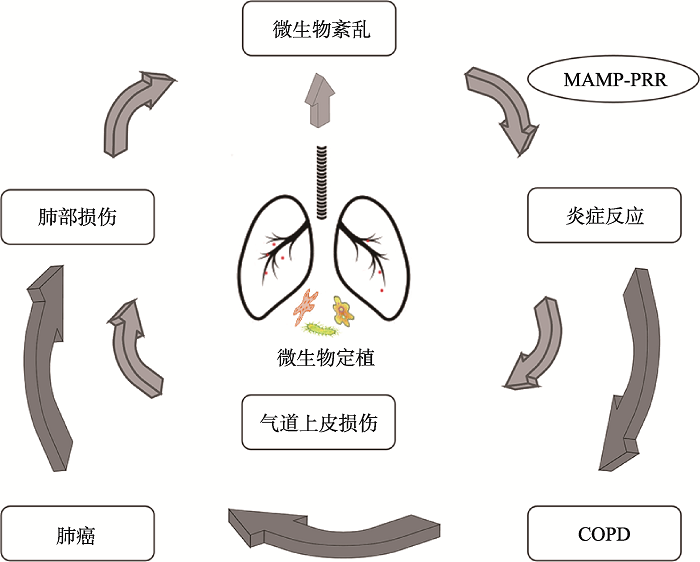 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1肺部微生物组与COPD、肺癌之间联系
Fig. 1The link between lung microbiomes and COPD and lung cancer
目前,肺部微生物组学的研究缺乏对采样类型、环境污染等因素的标准化质控以及大型实验研究,例如不同的样本类型如肺部组织、痰、支气管肺泡灌洗液及支气管镜标本可能会有不同的结果,取样偏差也会导致类似的问题[4]。对于肺部微生物组研究存在的问题,国际研究人员可以共同建立标准化样本类型及质控标准,以便于更深入的开展对肺部微生物组与疾病之间关系的研究。本实验室也正在大量收集云南省宣威市COPD和肺癌患者痰样本,针对肺部微生物组、COPD和肺癌之间联系进行研究,并希望可以为肺部微生物组的研究做出贡献。
虽然目前由于采样类型、环境污染及样本量等原因的影响,肺部微生物组学在COPD和肺癌中的研究仍然充满挑战。但从肺部微生物组角度研究COPD与肺癌之间的关系,有助于提供COPD和肺癌患者提临床预防和优化治疗的新方向,也为新型药物设计提供了新的靶点。有研究表明在NTHi诱导的COPD样炎症转化为肺癌的研究模型中,IL-6阻断剂不仅对肿瘤细胞有直接的内在抑制作用,而且可以通过改变肿瘤细胞与抗肿瘤免疫细胞的相对比例,使肺部微环境向抗肿瘤表型转化,可以作为预防和治疗K-ras突变型肺肿瘤的潜在药物靶点[67]。同样抗生素已被用作治疗COPD恶化的标准管理方法,但其在这方面的价值仍不确定,而如何更加有效的使用抗生素是一个巨大的挑战[70]。也许通过对肺部微生物组组成变化的深入了解,可以为抗生素的优化使用带来新的理论依据,也为COPD和肺癌的防治带来新的契机。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/emm.2017.7URLPMID:28408748 [本文引用: 1]

Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory disease, and bacterial infection plays a role in its pathogenesis. Bacteria secrete nanometer-sized extracellular vesicles (EVs), which may induce more immune dysfunction and inflammation than the bacteria themselves. We hypothesized that the microbiome of lung EVs might have distinct characteristics depending on the presence of COPD and smoking status. We analyzed and compared the microbiomes of 13 nonsmokers with normal spirometry, 13 smokers with normal spirometry (healthy smokers) and 13 patients with COPD by using 16S ribosomal RNA gene sequencing of surgical lung tissue and lung EVs. Subjects were matched for age and sex in all groups and for smoking levels in the COPD and healthy smoker groups. Each group included 12 men and 1 woman with the same mean age of 65.5 years. In all groups, EVs consistently showed more operational taxonomic units (OTUs) than lung tissue. In the healthy smoker and COPD groups, EVs had a higher Shannon index and a lower Simpson index than lung tissue and this trend was more prominent in the COPD group. Principal component analysis (PCA) showed clusters based on sample type rather than participants' clinical characteristics. Stenotrophomonas, Propionibacterium and Alicyclobacillus were the most commonly found genera. Firmicutes were highly present in the EVs of the COPD group compared with other samples or groups. Our analysis of the lung microbiome revealed that the bacterial communities present in the EVs and in the COPD group possessed distinct characteristics with differences in the OTUs, diversity indexes and PCA clustering.
DOI:10.21037/jtd.2019.10.54URLPMID:31737343 [本文引用: 7]
Chronic obstructive pulmonary disease (COPD) and lung cancer comprise the leading causes of lung disease-related mortality worldwide. Exposure to tobacco smoke is a mutual aetiology underlying the two diseases, accounting for almost 90% of cases. There is accumulating evidence supporting the role of immune dysfunction, the lung microbiome, extracellular vesicles and underlying genetic susceptibility in the development of COPD and lung cancer. Further, epigenetic factors, involving DNA methylation and microRNA expression, have been implicated in both diseases. Chronic inflammation is a key feature of COPD and could be a potential driver of lung cancer development. Using next generation technologies, further studies investigating the genomics, epigenetics and gene-environment interaction in key molecular pathways will continue to elucidate the pathogenic mechanisms underlying the development of COPD and lung cancer, and contribute to the development of novel diagnostic and prognostic tools for early intervention and personalised therapeutic strategies.
DOI:10.1016/j.canlet.2017.11.036URLPMID:29197615 [本文引用: 5]
The human microbiome confers benefits or disease susceptibility to the human body through multiple pathways. Disruption of the symbiotic balance of the human microbiome is commonly found in systematic diseases such as diabetes, obesity, and chronic gastric diseases. Emerging evidence has suggested that dysbiosis of the microbiota may also play vital roles in carcinogenesis at multiple levels, e.g., by affecting metabolic, inflammatory, or immune pathways. Although the impact of the gut microbiome on the digestive cancer has been widely explored, few studies have investigated the interplay between the microbiome and lung cancer. Some recent studies have shown that certain microbes and microbiota dysbiosis are correlated with development of lung cancer. In this mini-review, we briefly summarize current research findings describing the relationship between the lung microbiome and lung cancer. We further discuss the potential mechanisms through which the lung microbiome may play a role in lung carcinogenesis and impact lung cancer treatment. A better knowledge of the interplay between the lung microbiome and lung cancer may promote the development of innovative strategies for early prevention and personalized treatment in lung cancer.
DOI:10.1002/iub.1969URLPMID:30466159 [本文引用: 1]
It is now well appreciated that the human microbiome plays a significant role in a number of processes in the body, significantly affecting its metabolic, inflammatory, and immune homeostasis. Recent research has revealed that almost every mucosal surface in the human body is associated with a resident commensal microbiome of its own. While the gut microbiome and its role in regulation of host metabolism along with its alteration in a disease state has been well studied, there is a lacuna in understanding the resident microbiota of other mucosal surfaces. Among these, the scientific information on the role of lung microbiota in pulmonary diseases is currently severely limited. Historically, lungs have been considered to be sterile and lung diseases have only been studied in the context of bacterial pathogenesis. Recently however, studies have revealed a resilient microbiome in the upper and lower respiratory tracts and there is increased evidence on its central role in respiratory diseases. Knowledge of lung microbiome and its metabolic fallout (local and systemic) is still in its nascent stages and attracting immense interest in recent times. In this review, we will provide a perspective on lung-associated metabolic disorders defined for lung diseases (e.g., chronic obstructive pulmonary disease, asthma, and respiratory depression due to infection) and correlate it with lung microbial perturbation. Such perturbations may be due to altered biochemical or metabolic stress as well. Finally, we will draw evidence from microbiome and classical microbiology literature to demonstrate how specific lung morbidities associate with specific metabolic characteristics of the disease, and with the role of microbiome in this context. (c) 2018 IUBMB Life, 71(1):152-165, 2019.
[本文引用: 1]
[本文引用: 1]
DOI:10.1017/gheg.2018.1URLPMID:29868229 [本文引用: 1]
DOI:10.1007/s10565-019-09473-9URLPMID:31119467 [本文引用: 1]
The functions of body gradually decrease as the age increases, leading to a higher frequency of incidence of age-related diseases. Diseases associated with aging in the respiratory system include chronic obstructive pulmonary disease (COPD), IPF (idiopathic pulmonary fibrosis), asthma, lung cancer, and so on. The mitochondrial dysfunction is not only a sign of aging, but also is a disease trigger. This article aims to explain mitochondrial dysfunction as an aging marker, and its role in aging diseases of lung. We also discuss whether the mitochondria can be used as a target for the treatment of aging lung disease.
[本文引用: 1]
[本文引用: 1]
DOI:10.3322/caac.21387URLPMID:28055103 [本文引用: 1]
Each year, the American Cancer Society estimates the numbers of new cancer cases and deaths that will occur in the United States in the current year and compiles the most recent data on cancer incidence, mortality, and survival. Incidence data were collected by the Surveillance, Epidemiology, and End Results Program; the National Program of Cancer Registries; and the North American Association of Central Cancer Registries. Mortality data were collected by the National Center for Health Statistics. In 2017, 1,688,780 new cancer cases and 600,920 cancer deaths are projected to occur in the United States. For all sites combined, the cancer incidence rate is 20% higher in men than in women, while the cancer death rate is 40% higher. However, sex disparities vary by cancer type. For example, thyroid cancer incidence rates are 3-fold higher in women than in men (21 vs 7 per 100,000 population), despite equivalent death rates (0.5 per 100,000 population), largely reflecting sex differences in the
DOI:10.3322/caac.21262URLPMID:25651787 [本文引用: 1]
Cancer constitutes an enormous burden on society in more and less economically developed countries alike. The occurrence of cancer is increasing because of the growth and aging of the population, as well as an increasing prevalence of established risk factors such as smoking, overweight, physical inactivity, and changing reproductive patterns associated with urbanization and economic development. Based on GLOBOCAN estimates, about 14.1 million new cancer cases and 8.2 million deaths occurred in 2012 worldwide. Over the years, the burden has shifted to less developed countries, which currently account for about 57% of cases and 65% of cancer deaths worldwide. Lung cancer is the leading cause of cancer death among males in both more and less developed countries, and has surpassed breast cancer as the leading cause of cancer death among females in more developed countries; breast cancer remains the leading cause of cancer death among females in less developed countries. Other leading causes of cancer death in more developed countries include colorectal cancer among males and females and prostate cancer among males. In less developed countries, liver and stomach cancer among males and cervical cancer among females are also leading causes of cancer death. Although incidence rates for all cancers combined are nearly twice as high in more developed than in less developed countries in both males and females, mortality rates are only 8% to 15% higher in more developed countries. This disparity reflects regional differences in the mix of cancers, which is affected by risk factors and detection practices, and/or the availability of treatment. Risk factors associated with the leading causes of cancer death include tobacco use (lung, colorectal, stomach, and liver cancer), overweight/obesity and physical inactivity (breast and colorectal cancer), and infection (liver, stomach, and cervical cancer). A substantial portion of cancer cases and deaths could be prevented by broadly applying effective prevention measures, such as tobacco control, vaccination, and the use of early detection tests.
DOI:10.1038/nrc3477URLPMID:23467302 [本文引用: 4]
Numerous epidemiological studies have consistently linked the presence of chronic obstructive pulmonary disease (COPD) to the development of lung cancer, independently of cigarette smoking dosage. The mechanistic explanation for this remains poorly understood. Progress towards uncovering this link has been hampered by the heterogeneous nature of the two disorders: each is characterized by multiple sub-phenotypes of disease. In this Review, I discuss the nature of the link between the two diseases and consider specific mechanisms that operate in both COPD and lung cancer, some of which might represent either chemopreventive or chemotherapeutic targets.
DOI:10.1080/14728222.2019.1615884URLPMID:31079559 [本文引用: 3]
INTRODUCTION: COPD and lung cancer are leading causes of morbidity and mortality worldwide, and they share a common environmental risk factor in cigarette smoke exposure and a genetic predisposition represented by their incidence in only a fraction of smokers. This reflects the ability of cigarette smoke to induce an inflammatory response in the airways of susceptible smokers. Moreover, COPD could be a driving factor in lung cancer, by increasing oxidative stress and the resulting DNA damage and repression of the DNA repair mechanisms, chronic exposure to pro-inflammatory cytokines, repression of innate immunity and increased cellular proliferation. Areas covered: We have focused our review on the potential pathogenic molecular links between tobacco smoking-related COPD and lung cancer and the potential molecular targets for new drug development by understanding the common signaling pathways involved in COPD and lung cancer. Expert commentary: Research in this field is mostly limited to animal models or small clinical trials. Large clinical trials are needed but mostly combined models of COPD and lung cancer are necessary to investigate the processes caused by chronic inflammation, including genetic and epigenetic alteration, and the expression of inflammatory mediators that link COPD and lung cancer, to identify new molecular therapeutic targets.
DOI:10.21037/jtd.2018.06.143URLPMID:30123556 [本文引用: 1]
DOI:10.1152/ajplung.00116.2015URLPMID:26209273 [本文引用: 4]
Microorganisms have an important role in tumorgenesis by the induction of inflammation and by a direct impact on tumor cells. Chronic obstructive pulmonary disease (COPD) is associated with an increased risk for lung cancer and microbial colonization. We asked whether bacterial pathogens act as tumor promoters during CS-induced pulmonary inflammation. In a metastatic lung cancer (LC) model, Lewis lung carcinoma (LLC) cells were injected in mice to initiate the growth of tumors in the lung. Exposure to the combination of cigarette smoke (CS) and nontypeable Haemophilus influenzae (NTHi) synergistically increased metastatic growth. Lung levels of albumin and LDH, translocation of bacterial factors into tumor tissue, tumor inflammation, and tumor proliferation were significantly increased in mice exposed to CS in combination with NTHi. Bacterial pathogens increased the proliferation of cultured LLC cells and human cancer cell lines. Metastatic growth induced by the exposure to CS in combination with NTHi was reduced in mice deficient for IL-17. Our data provide evidence that CS-induced loss of pulmonary barrier integrity allows bacterial factors to translocate into tumor tissue and to regulate tumor-associated inflammation and tumor proliferation. Translocation of bacterial factors in tumor tissue links CS-induced inflammation with tumor proliferation.
DOI:10.1146/annurev-physiol-021115-105238URLPMID:26527186 [本文引用: 3]
Although the notion that
DOI:10.1164/rccm.201502-0223OCURLPMID:25945594 [本文引用: 1]
RATIONALE: The relatively sparse but diverse microbiome in human lungs may become less diverse in chronic obstructive pulmonary disease (COPD). This article examines the relationship of this microbiome to emphysematous tissue destruction, number of terminal bronchioles, infiltrating inflammatory cells, and host gene expression. METHODS: Culture-independent pyrosequencing microbiome analysis was used to examine the V3-V5 regions of bacterial 16S ribosomal DNA in 40 samples of lung from 5 patients with COPD (Global Initiative for Chronic Obstructive Lung Disease [GOLD] stage 4) and 28 samples from 4 donors (controls). A second protocol based on the V1-V3 regions was used to verify the bacterial microbiome results. Within lung tissue samples the microbiome was compared with results of micro-computed tomography, infiltrating inflammatory cells measured by quantitative histology, and host gene expression. MEASUREMENTS AND MAIN RESULTS: Ten operational taxonomic units (OTUs) was found sufficient to discriminate between control and GOLD stage 4 lung tissue, which included known pathogens such as Haemophilus influenzae. We also observed a decline in microbial diversity that was associated with emphysematous destruction, remodeling of the bronchiolar and alveolar tissue, and the infiltration of the tissue by CD4(+) T cells. Specific OTUs were also associated with neutrophils, eosinophils, and B-cell infiltration (P < 0.05). The expression profiles of 859 genes and 235 genes were associated with either enrichment or reductions of Firmicutes and Proteobacteria, respectively, at a false discovery rate cutoff of less than 0.1. CONCLUSIONS: These results support the hypothesis that there is a host immune response to microorganisms within the lung microbiome that appears to contribute to the pathogenesis of COPD.
DOI:10.1016/S2213-2600(18)30510-1URLPMID:30975495 [本文引用: 1]
The composition of the lung microbiome is increasingly well characterised, with changes in microbial diversity or abundance observed in association with several chronic respiratory diseases such as asthma, cystic fibrosis, bronchiectasis, and chronic obstructive pulmonary disease. However, the precise effects of the microbiome on pulmonary health and the functional mechanisms by which it regulates host immunity are only now beginning to be elucidated. Bacteria, viruses, and fungi from both the upper and lower respiratory tract produce structural ligands and metabolites that interact with the host and alter the development and progression of chronic respiratory diseases. Here, we review recent advances in our understanding of the composition of the lung microbiome, including the virome and mycobiome, the mechanisms by which these microbes interact with host immunity, and their functional effects on the pathogenesis, exacerbations, and comorbidities of chronic respiratory diseases. We also describe the present understanding of how respiratory microbiota can influence the efficacy of common therapies for chronic respiratory disease, and the potential of manipulation of the microbiome as a therapeutic strategy. Finally, we highlight some of the limitations in the field and propose how these could be addressed in future research.
DOI:10.1055/s-0037-1617441URLPMID:29579771 [本文引用: 1]
Once considered a sterile site below the larynx, the tracheobronchial tree and parenchyma of the lungs are now known to harbor a rich diversity of microbial species including bacteria, viruses, fungi, and archaea. Many of these organisms, particularly the viruses which comprise the human respiratory virome, have not been identified, so their true role is unknown. It seems logical to conclude that a
DOI:10.1128/microbiolspec.FUNK-0030-2016URLPMID:28597810 [本文引用: 2]
In this article, we review some of the best-studied fungi used as food sources, in particular, the cheese fungi, the truffles, and the fungi used for drink fermentation such as beer, wine, and sake. We discuss their history of consumption by humans and the genomic mechanisms of adaptation during artificial selection.
[本文引用: 1]
DOI:10.1164/rccm.201111-2075OCURLPMID:22427533 [本文引用: 3]
RATIONALE: Based on surface brushings and bronchoalveolar lavage fluid, Hilty and coworkers demonstrated microbiomes in the human lung characteristic of asthma and chronic obstructive pulmonary disease (COPD), which have now been confirmed by others. OBJECTIVES: To extend these findings to human lung tissue samples. METHODS: DNA from lung tissue samples was obtained from nonsmokers (n = 8); smokers without COPD (n = 8); patients with very severe COPD (Global Initiative for COPD [GOLD] 4) (n = 8); and patients with cystic fibrosis (CF) (n = 8). The latter served as a positive control, with sterile water as a negative control. All bacterial community analyses were based on polymerase chain reaction amplifying 16S rRNA gene fragments. Total bacterial populations were measured by quantitative polymerase chain reaction and bacterial community composition was assessed by terminal restriction fragment length polymorphism analysis and pyrotag sequencing. MEASUREMENT AND MAIN RESULTS: Total bacterial populations within lung tissue were small (20-1,252 bacterial cells per 1,000 human cells) but greater in all four sample groups versus the negative control group (P < 0.001). Terminal restriction fragment length polymorphism analysis and sequencing distinguished three distinct bacterial community compositions: one common to the nonsmoker and smoker groups, a second to the GOLD 4 group, and the third to the CF-positive control group. Pyrotag sequencing identified greater than 1,400 unique bacterial sequences and showed an increase in the Firmicutes phylum in GOLD 4 patients versus all other groups (P < 0.003) attributable to an increase in the Lactobacillus genus (P < 0.0007). CONCLUSIONS: There is a detectable bacterial community within human lung tissue that changes in patients with very severe COPD.
DOI:10.1183/13993003.02086-2016URLPMID:28404649 [本文引用: 1]
The healthy lung has previously been considered to be a sterile organ because standard microbiological culture techniques consistently yield negative results. However, culture-independent techniques report that large numbers of microorganisms coexist in the lung. There are many unknown aspects in the field, but available reports show that the lower respiratory tract microbiota: 1) is similar in healthy subjects to the oropharyngeal microbiota and dominated by members of the Firmicutes, Bacteroidetes and Proteobacteria phyla; 2) shows changes in smokers and well-defined differences in chronic respiratory diseases, although the temporal and spatial kinetics of these changes are only partially known; and 3) shows relatively abundant non-cultivable bacteria in chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis, cystic fibrosis and bronchiectasis, with specific patterns for each disease. In all of these diseases, a loss of diversity, paralleled by an over-representation of Proteobacteria (dysbiosis), has been related to disease severity and exacerbations. However, it is unknown whether dysbiosis is a cause or a consequence of the damage to bronchoalveolar surfaces.Finally, little is known about bacterial functionality and the interactions between viruses, fungi and bacteria. It is expected that future research in bacterial gene expressions, metagenomics longitudinal analysis and host-microbiome animal models will help to move towards targeted microbiome interventions in respiratory diseases.
DOI:10.1016/j.rmed.2019.08.012URLPMID:31450162 [本文引用: 1]
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory disorder with a course that is not uniform for all COPD patients. Although smoking is considered as the major cause of the disease, persistent or recurrent infections seem to play a particular role in the disease establishment and progression. COPD is characterized by dysregulated immunity that has been associated with the bacterial colonization and infections. The establishment of culture-independent techniques has shed new light on the relationships between bacterial ecology and health status and expanded our knowledge on the lung microbiome. Interactions between the host and lung microbiome result in inflammation and activation of resident cells. The lung microbiome contains populations of symbionts and pathobionts in balance which lose their equilibrium and disturb the balance of T-helper and regulatory T-cells (Treg) upon infection, or lung disease. In COPD factors such as disease severity, exacerbations, degree of inflammation, and type of treatment used (e.g inhaled or systemic steroids and antibiotics) affect the composition of lung microbiota. Recent data indicate that the presence of specific bacterial taxa in the airways has the potential to influence the host immune response and possibly to interfere with disease phenotype. Although, there is a growing body of evidence for the role of microbiome in COPD several unanswered questions still exist for its clinical relevance.
DOI:10.3389/fcimb.2020.00213URLPMID:32477966 [本文引用: 1]
Chronic respiratory diseases including chronic rhinosinusitis, otitis media, asthma, cystic fibrosis, non-CF bronchiectasis, and chronic obstructive pulmonary disease are a major public health burden. Patients suffering from chronic respiratory disease are prone to persistent, debilitating respiratory infections due to the decreased ability to clear pathogens from the respiratory tract. Such infections often develop into chronic, life-long complications that are difficult to treat with antibiotics due to the formation of recalcitrant biofilms. The microbial communities present in the upper and lower respiratory tracts change as these respiratory diseases progress, often becoming less diverse and dysbiotic, correlating with worsening patient morbidity. Those with chronic respiratory disease are commonly infected with a shared group of respiratory pathogens including Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, Pseudomonas aeruginosa, and Moraxella catarrhalis, among others. In order to understand the microbial landscape of the respiratory tract during chronic disease, we review the known inter-species interactions among these organisms and other common respiratory flora. We consider both the balance between cooperative and competitive interactions in relation to microbial community structure. By reviewing the major causes of chronic respiratory disease, we identify common features across disease states and signals that might contribute to community shifts. As microbiome shifts have been associated with respiratory disease progression, worsening morbidity, and increased mortality, these underlying community interactions likely have an impact on respiratory disease state.
DOI:10.1128/JCM.01967-14URL [本文引用: 2]
Bronchial colonization by potentially pathogenic microorganisms (PPMs) is often demonstrated in chronic obstructive pulmonary disease (COPD), but culture-based techniques identify only a portion of the bacteria in mucosal surfaces. The aim of the study was to determine changes in the bronchial microbiome of COPD associated with the severity of the disease. The bronchial microbiome of COPD patients was analyzed by 16S rRNA gene amplification and pyrosequencing in sputum samples obtained during stable disease. Seventeen COPD patients were studied (forced expiratory volume in the first second expressed as a percentage of the forced vital capacity [FEV1%] median, 35.0%; interquartile range [IQR], 31.5 to 52.0), providing a mean of 4,493 (standard deviation [SD], 2,598) sequences corresponding to 47 operational taxonomic units (OTUs) (SD, 17) at a 97% identity level. Patients were dichotomized according to their lung function as moderate to severe when their FEV1% values were over the median and as advanced when FEV1% values were lower. The most prevalent phyla in sputum were Proteobacteria (44%) and Firmicutes (16%), followed by Actinobacteria (13%). A greater microbial diversity was found in patients with moderate-to-severe disease, and alpha diversity showed a statistically significant decrease in patients with advanced disease when assessed by Shannon (rho = 0.528; P = 0.029, Spearman correlation coefficient) and Chao1 (rho = 0.53; P = 0.028, Spearman correlation coefficient) alpha-diversity indexes. The higher severity that characterizes advanced COPD is paralleled by a decrease in the diversity of the bronchial microbiome, with a loss of part of the resident flora that is replaced by a more restricted microbiota that includes PPMs.
DOI:10.1186/s40168-017-0381-4URLPMID:29316977 [本文引用: 1]
BACKGROUND: Oral taxa are often found in the chronic obstructive pulmonary disease (COPD) lung microbiota, but it is not clear if this is due to a physiologic process such as aspiration or experimental contamination at the time of specimen collection. METHODS: Microbiota samples were obtained from nine subjects with mild or moderate COPD by swabbing lung tissue and upper airway sites during lung lobectomy. Lung specimens were not contaminated with upper airway taxa since they were obtained surgically. The microbiota were analyzed with 16S rRNA gene qPCR and 16S rRNA gene hypervariable region 3 (V3) sequencing. Data analyses were performed using QIIME, SourceTracker, and R. RESULTS: Streptococcus was the most common genus in the oral, bronchial, and lung tissue samples, and multiple other taxa were present in both the upper and lower airways. Each subject's own bronchial and lung tissue microbiota were more similar to each other than were the bronchial and lung tissue microbiota of two different subjects (permutation test, p = 0.0139), indicating more within-subject similarity than between-subject similarity at these two lung sites. Principal coordinate analysis of all subject samples revealed clustering by anatomic sampling site (PERMANOVA, p = 0.001), but not by subject. SourceTracker analysis found that the sources of the lung tissue microbiota were 21.1% (mean) oral microbiota, 8.7% nasal microbiota, and 70.1% unknown. An analysis using the neutral theory of community ecology revealed that the lung tissue microbiota closely reflects the bronchial, oral, and nasal microbiota (immigration parameter estimates 0.69, 0.62, and 0.74, respectively), with some evidence of ecologic drift occurring in the lung tissue. CONCLUSION: This is the first study to evaluate the mild-moderate COPD lung tissue microbiota without potential for upper airway contamination of the lung samples. In our small study of subjects with COPD, we found oral and nasal bacteria in the lung tissue microbiota, confirming that aspiration is a source of the COPD lung microbiota.
DOI:10.21037/atm.2017.04.20URLPMID:28706919 [本文引用: 1]
The introduction of culture-independent techniques for the microbiological analysis of respiratory samples has confirmed that the respiratory system hosts a large number of microorganisms, which include a wide range of bacteria. The regular exposure to tobacco smoke changes the microbiome in healthy smokers, first in the oropharynx, increasing the presence of a restricted number of genera which attain high relative abundance, a pattern that may be considered as dysbiosis. In chronic obstructive pulmonary disease (COPD), microbiome analyses of sputum samples have demonstrated an important decline in bacterial diversity, with a change to a restricted flora with an overrepresentation of the Proteobacteria phylum, which include most of the bacteria commonly considered as potentially pathogenic microorganisms, paralleled by a decline in the relative abundance of microorganisms part of the Firmicutes phylum. In exacerbations, specific bacteria overrepresented in microbiome analyses and potentially causal of the acute episode may not be recovered by sputum culture, while colonizing microorganisms grow easily, in spite that their relative abundance have not changed from previous stability. This situation has been described in patients showing chronic colonization by Pseudomonas aeruginosa, who suffer from exacerbations that in most cases are due to other PPMs, in spite of the persistence of positive cultures for the colonizing Pseudomonas strains. Interaction between different microorganisms can be addressed through microbiome analyses, and functional metagenomics, that describes the genomic potential of the community, has shown that, in spite that the bronchial microbiome as a whole may not change significantly, clear changes in carbohydrate metabolism, cancer, cell growth and death, transport and catabolism pathways often appear during exacerbations. These functional changes may be important because through them the resident community as a whole show its power to modify important metabolic patterns.
DOI:10.1183/09031936.00191513URLPMID:24311775 [本文引用: 1]
DOI:10.1371/journal.pone.0144448URLPMID:26632844 [本文引用: 1]
The course of chronic obstructive pulmonary disease (COPD) is frequently aggravated by exacerbations, and changes in the composition and activity of the microbiome may be implicated in their appearance. The aim of this study was to analyse the composition and the gene content of the microbial community in bronchial secretions of COPD patients in both stability and exacerbation. Taxonomic data were obtained by 16S rRNA gene amplification and pyrosequencing, and metabolic information through shotgun metagenomics, using the Metagenomics RAST server (MG-RAST), and the PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) programme, which predict metagenomes from 16S data. Eight severe COPD patients provided good quality sputum samples, and no significant differences in the relative abundance of any phyla and genera were found between stability and exacerbation. Bacterial biodiversity (Chao1 and Shannon indexes) did not show statistical differences and beta-diversity analysis (Bray-Curtis dissimilarity index) showed a similar microbial composition in the two clinical situations. Four functional categories showed statistically significant differences with MG-RAST at KEGG level 2: in exacerbation, Cell growth and Death and Transport and Catabolism decreased in abundance [1.6 (0.2-2.3) vs 3.6 (3.3-6.9), p = 0.012; and 1.8 (0-3.3) vs 3.6 (1.8-5.1), p = 0.025 respectively], while Cancer and Carbohydrate Metabolism increased [0.8 (0-1.5) vs 0 (0-0.5), p = 0.043; and 7 (6.4-9) vs 5.9 (6.3-6.1), p = 0.012 respectively]. In conclusion, the bronchial microbiome as a whole is not significantly modified when exacerbation symptoms appear in severe COPD patients, but its functional metabolic capabilities show significant changes in several pathways.
DOI:10.3332/ecancer.2018.866URLPMID:30263057 [本文引用: 3]
The lung microbiome has been shown to reflect a range of pulmonary diseases-for example: asthma, chronic obstructive pulmonary disease (COPD) and cystic fibrosis. Studies have now begun to show microbiological changes in the lung that correlate with lung cancer (LC) which could provide new insights into lung carcinogenesis and new biomarkers for disease screening. Clinical studies have suggested that infections with tuberculosis or pneumonia increased the risk of LC possibly through inflammatory or immunological changes. These have now been superseded by genomic-based microbiome sequencing studies based on bronchoalveolar lavage, sputum or saliva samples. Although some discrepancies exist, many have suggested changes in particular bacterial genera in LC samples particularly, Granulicatella, Streptococcus and Veillonella. Granulicatella is of particular interest, as it appeared to show LC stage-specific increases in abundance. We propose that these microbial community changes are likely to reflect biochemical changes in the LC lung, linked to an increase in anaerobic environmental niches and altered pyridoxal/polyamine/nitrogenous metabolism to which Granulicatella could be particularly responsive. These are clearly preliminary observations and many more expansive studies are required to develop our understanding of the LC microbiome.
[本文引用: 1]
DOI:10.1186/s13059-016-1021-1URLPMID:27468850 [本文引用: 2]
BACKGROUND: The human lung tissue microbiota remains largely uncharacterized, although a number of studies based on airway samples suggest the existence of a viable human lung microbiota. Here we characterized the taxonomic and derived functional profiles of lung microbiota in 165 non-malignant lung tissue samples from cancer patients. RESULTS: We show that the lung microbiota is distinct from the microbial communities in oral, nasal, stool, skin, and vagina, with Proteobacteria as the dominant phylum (60 %). Microbiota taxonomic alpha diversity increases with environmental exposures, such as air particulates, residence in low to high population density areas, and pack-years of tobacco smoking and decreases in subjects with history of chronic bronchitis. Genus Thermus is more abundant in tissue from advanced stage (IIIB, IV) patients, while Legionella is higher in patients who develop metastases. Moreover, the non-malignant lung tissues have higher microbiota alpha diversity than the paired tumors. CONCLUSIONS: Our results provide insights into the human lung microbiota composition and function and their link to human lifestyle and clinical outcomes. Studies among subjects without lung cancer are needed to confirm our findings.
DOI:10.1016/j.lungcan.2016.10.016URLPMID:27987594 [本文引用: 4]
OBJECTIVES: Disruption in the stability of respiratory microbiota is known to be associated with many chronic respiratory diseases. However, only few studies have examined microbiomes in lung cancer. Therefore, we characterized and compared the microbiomes of patients with lung cancer and those with benign mass-like lesions. MATERIALS AND METHODS: Bronchoalveolar fluid was collected prospectively to evaluate lung masses in patients who had undergone bronchoscopies from May to September 2015. Twenty-eight patients (20 male, 8 female) were enrolled: 20 diagnosed with lung cancer and 8 diagnosed with benign diseases. Samples were analysed by 16S rRNA-based next-generation sequencing. RESULTS: The participants' mean age was 64+/-11years. Bacterial operational taxonomic units were classified into 26 phyla, 44 classes, 81 orders, 153 families, 288 genera, and 797 species. The relative abundance of two phyla (Firmicutes and TM7) was significantly increased in patients with lung cancer (p=0.037 and 0.035, respectively). Furthermore, two genera (Veillonella and Megasphaera) were relatively more abundant in lung cancer patients (p=0.003 and 0.022, respectively). The area under the curve of a combination of these two genera used to predict lung cancer was 0.888 (sensitivity=95.0%, specificity=75.0% and sensitivity=70.0%, specificity=100.0%; p=0.002). CONCLUSION: The results indicate that differences exist in the bacterial communities of patients with lung cancer and those with benign mass-like lesions. The genera Veillonella and Megasphaera showed the potential to serve as biomarkers to predict lung cancer. Thus, the lung microbiota may change the environment in patients with lung cancer.
DOI:10.1002/ijc.31098URLPMID:29023689 [本文引用: 1]
The functional role of respiratory microbiota has attracted an accumulating attention recently. However, the role of respiratory microbiome in lung carcinogenesis is mostly unknown. Our study aimed to characterize and compare bilateral lower airway microbiome of lung cancer patients with unilateral lobar masses and control subjects. Protected bronchial specimen brushing samples were collected from 24 lung cancer patients with unilateral lobar masses (paired samples from cancerous site and the contralateral noncancerous site) and 18 healthy controls undergoing bronchoscopies and further analyzed by 16S rRNA amplicon sequencing. As results, significant decreases in microbial diversity were observed in patients with lung cancer in comparison to the controls, alpha diversity steadily declined from healthy site to noncancerous to cancerous site. Genus Streptococcus was significantly more abundant in cancer cases than the controls, while Staphylococcus was more abundant in the controls. The area under the curve of genus Streptococcus used to predict lung cancer was 0.693 (sensitivity = 87.5%, specificity = 55.6%). The abundance of genus Streptococcus and Neisseria displayed an increasing trend whereas Staphylococcus and Dialister gradually declined from healthy to noncancerous to cancerous site. Collectively, lung cancer-associated microbiota profile is distinct from that found in healthy controls, and the altered cancer-associated microbiota is not restricted to tumor tissue. The genus Streptococcus was abundant in lung cancer patients and exhibited moderate classification potential. The gradual microbiota profile shift from healthy site to noncancerous to paired cancerous site suggested a change of the microenvironment associated with the development of lung cancer.
URLPMID:26693063 [本文引用: 1]
Microbes are residents in a number of body sites, including the oral and nasal cavities, which are connected to the lung via the pharynx. The associations between oral diseases and increased risk of lung cancer have been reported in previous prospective studies. In this study, we measured variations of salivary microbiota and evaluated their potential association with lung cancer, including squamous cell carcinoma (SCC) and adenocarcinoma (AC). A three-phase study was performed: First, we investigated the salivary microbiota from 20 lung cancer patients (10 SCC and 10 AC) and control subjects (n=10) using a deep sequencing analysis. Salivary Capnocytophaga, Selenomonas, Veillonella and Neisseria were found to be significantly altered in patients with SCC and AC when compared to that in control subjects. Second, we confirmed the significant changes of Capnocytophaga, Veillonella and Neisseria in the same lung cancer patients using quantitative PCR (qPCR). Finally, these bacterial species were further validated on new patient/control cohorts (n=56) with qPCR. The combination of two bacterial biomarkers, Capnocytophaga and Veillonella, yielded a receiver operating characteristic (ROC) value of 0.86 with an 84.6% sensitivity and 86.7% specificity in distinguishing patients with SCC from control subjects and a ROC value of 0.80 with a 78.6% sensitivity and 80.0% specificity in distinguishing patients with AC from control subjects. In conclusion, we have for the first time demonstrated the association of saliva microbiota with lung cancer. Particularly, the combination of the 16S sequencing discovery with qPCR validation studies revealed that the levels of Capnocytophaga and Veillonella were significantly higher in the saliva from lung cancer patients, which may serve as potential biomarkers for the disease detection/classification.
DOI:10.1016/j.chom.2015.01.012URLPMID:25674978 [本文引用: 1]
The mammalian immune system communicates with skin-resident microbes, and some of these microbes provide benefits to the host. In a recent paper in Nature, Naik et al. (2015) provide evidence that the murine epidermis permits S. epidermidis, a skin-specific bacterium, to shape the immune response.
[本文引用: 1]
DOI:10.1136/thoraxjnl-2017-210408URLPMID:29386298 [本文引用: 1]
BACKGROUND: Alterations in the composition of the lung microbiome associated with adverse clinical outcomes, known as dysbiosis, have been implicated with disease severity and exacerbations in COPD. OBJECTIVE: To characterise longitudinal changes in the lung microbiome in the AERIS study (Acute Exacerbation and Respiratory InfectionS in COPD) and their relationship with associated COPD outcomes. METHODS: We surveyed 584 sputum samples from 101 patients with COPD to analyse the lung microbiome at both stable and exacerbation time points over 1 year using high-throughput sequencing of the 16S ribosomal RNA gene. We incorporated additional lung microbiology, blood markers and in-depth clinical assessments to classify COPD phenotypes. RESULTS: The stability of the lung microbiome over time was more likely to be decreased in exacerbations and within individuals with higher exacerbation frequencies. Analysis of exacerbation phenotypes using a Markov chain model revealed that bacterial and eosinophilic exacerbations were more likely to be repeated in subsequent exacerbations within a subject, whereas viral exacerbations were not more likely to be repeated. We also confirmed the association of bacterial genera, including Haemophilus and Moraxella, with disease severity, exacerbation events and bronchiectasis. CONCLUSIONS: Subtypes of COPD have distinct bacterial compositions and stabilities over time. Some exacerbation subtypes have non-random probabilities of repeating those subtypes in the future. This study provides insights pertaining to the identification of bacterial targets in the lung and biomarkers to classify COPD subtypes and to determine appropriate treatments for the patient. TRIAL REGISTRATION NUMBER: Results, NCT01360398.
DOI:10.1136/thoraxjnl-2017-210741URLPMID:29269441 [本文引用: 1]
BACKGROUND: Recent studies suggest that lung microbiome dysbiosis, the disease associated disruption of the lung microbial community, might play a key role in chronic obstructive pulmonary disease (COPD) exacerbations. However, characterising temporal variability of the microbiome from large longitudinal COPD cohorts is needed to better understand this phenomenon. METHODS: We performed a 16S ribosomal RNA survey of microbiome on 716 sputum samples collected longitudinally at baseline and exacerbations from 281 subjects with COPD at three UK clinical centres as part of the COPDMAP consortium. RESULTS: The microbiome composition was similar among centres and between stable and exacerbations except for a small significant decrease of Veillonella at exacerbations. The abundance of Moraxella was negatively associated with bacterial alpha diversity. Microbiomes were distinct between exacerbations associated with bacteria versus eosinophilic airway inflammation. Dysbiosis at exacerbations, measured as significant within subject deviation of microbial composition relative to baseline, was present in 41% of exacerbations. Dysbiosis was associated with increased exacerbation severity indicated by a greater fall in forced expiratory volume in one second, forced vital capacity and a greater increase in CAT score, particularly in exacerbations with concurrent eosinophilic inflammation. There was a significant difference of temporal variability of microbial alpha and beta diversity among centres. The variation of beta diversity significantly decreased in those subjects with frequent historical exacerbations. CONCLUSIONS: Microbial dysbiosis is a feature of some exacerbations and its presence, especially in concert with eosinophilic inflammation, is associated with more severe exacerbations indicated by a greater fall in lung function. TRIAL REGISTRATION NUMBER: Results, NCT01620645.
DOI:10.1016/j.lfs.2019.116671URLPMID:31336122 [本文引用: 3]
Toll-like receptors (TLRs) comprise a clan of proteins involved in identification and triggering a suitable response against pathogenic attacks. As lung is steadily exposed to multiple infectious agents, antigens and host-derived danger signals, the inhabiting stromal and myeloid cells of the lung express an aggregate of TLRs which perceive the endogenously derived damage-associated molecular patterns (DAMPs) along with pathogen associated molecular patterns (PAMPs) and trigger the TLR-associated signalling events involved in host defence. Thus, they form an imperative component of host defence activation in case of microbial infections as well as non-infectious pulmonary disorders such as interstitial lung disease, acute lung injury and airways disease, such as COPD and asthma. They also play an equally important role in lung cancer. Targeting the TLR signalling network would pave ways to the design of more reliable and effective vaccines against infectious agents and control deadly infections, desensitize allergens and reduce inflammation. Moreover, TLR agonists may act as adjuvants by increasing the efficiency of cancer vaccines, thereby contributing their role in treatment of lung cancer too. Overall, TLRs present a compelling and expeditiously bolstered area of research and addressing their signalling events would be of significant use in pulmonary diseases.
DOI:10.3389/fimmu.2018.03082URLPMID:30692992 [本文引用: 1]
The detection of microbial pathogens relies on the recognition of highly conserved microbial structures by the membrane sensor Toll-like receptors (TLRs) and cytosolic sensor NOD-like receptors (NLRs). Upon detection, these sensors trigger innate immune responses to eradicate the invaded microbial pathogens. However, it is unclear whether TLR and NOD signaling are both critical for innate immunity to initiate inflammatory and antimicrobial responses against microbial infection. Here we report that activation of both TLR and NOD signaling resulted in an augmented inflammatory response and the crosstalk between TLR and NOD led to an amplified downstream NF-kappaB activation with increased nuclear transactivation of p65 at both TNF-alpha and IL-6 promoters. Furthermore, co-stimulation of macrophages with TLR and NOD agonists maximized antimicrobial activity with accelerated phagosome maturation. Importantly, administration of both TLR and NOD agonists protected mice against polymicrobial sepsis-associated lethality with increased serum levels of inflammatory cytokines and accelerated clearance of bacteria from the circulation and visceral organs. These results demonstrate that activation of both TLR and NOD signaling synergizes to induce efficient inflammatory and antimicrobial responses, thus conferring protection against microbial infection.
DOI:10.1038/mi.2016.108URLPMID:27966551 [本文引用: 2]
The lungs are not sterile or free from bacteria; rather, they harbor a distinct microbiome whose composition is driven by different ecological rules than for the gastrointestinal tract. During disease, there is often a shift in community composition towards Gammaproteobacteria, the bacterial class that contains many common lung-associated gram-negative
DOI:10.1186/s40168-019-0751-1URLPMID:31699146 [本文引用: 2]
BACKGROUND: Regulatory T cell (Treg) deficiency leads to IPEX syndrome, a lethal autoimmune disease, in Human and mice. Dysbiosis of the gut microbiota in Treg-deficient scurfy (SF) mice has been described, but to date, the role of the gut microbiota remains to be determined. RESULTS: To examine how antibiotic-modified microbiota can inhibit Treg deficiency-induced lethal inflammation in SF mice, Treg-deficient SF mice were treated with three different antibiotics. Different antibiotics resulted in distinct microbiota and metabolome changes and led to varied efficacy in prolonging lifespan and reducing inflammation in the liver and lung. Moreover, antibiotics altered plasma levels of several cytokines, especially IL-6. By analyzing gut microbiota and metabolome, we determined the microbial and metabolomic signatures which were associated with the antibiotics. Remarkably, antibiotic treatments restored the levels of several primary and secondary bile acids, which significantly reduced IL-6 expression in RAW macrophages in vitro. IL-6 blockade prolonged lifespan and inhibited inflammation in the liver and lung. By using IL-6 knockout mice, we further identified that IL-6 deletion provided a significant portion of the protection against inflammation induced by Treg dysfunction. CONCLUSION: Our results show that three antibiotics differentially prolong survival and inhibit lethal inflammation in association with a microbiota-IL-6 axis. This pathway presents a potential avenue for treating Treg deficiency-mediated autoimmune disorders.
DOI:10.1111/resp.12732URLPMID:26852737 [本文引用: 1]
Traditional culture techniques confirm that bacteria have an important role in Chronic Obstructive Pulmonary Disease (COPD). In individuals with COPD, acquisition of novel bacterial strains is associated with onset of acute exacerbation of COPD, which leads to further lung dysfunction and enormous health-care costs. Recent study of the human microbiome, the total composite of the bacteria on the human body, posited the microbiome as the last human organ studied, as the microbiome performs a multitude of metabolic functions absent in the human genome. The largest project to study the human microbiome was the National Institutes of Health (NIH) human microbiome project (HMP) started in 2007 to understand the 'normal' microbiome. However due to the presumption that the healthy human lung was sterile, the respiratory tract was not included in that study. The advent of next-generation sequencing technologies has allowed the investigation of the human respiratory microbiome, which revealed that the healthy lung does have a robust microbiome. Subsequent studies in individuals with COPD revealed that the microbiome composition fluctuates with severity of COPD, composition of the individual aero-digestive tract microbiomes, age, during an acute exacerbation of COPD and with the use of steroids and/or antibiotics. Understanding the impact of the microbiome on COPD progression and risk of exacerbation will lead to directed therapies for prevention of COPD progression and exacerbation.
DOI:10.1186/s12931-015-0204-8URL [本文引用: 1]
DOI:10.1164/rccm.201504-0779OCURLPMID:26630356 [本文引用: 1]
RATIONALE: Changes in the pulmonary microbiota are associated with progressive respiratory diseases including chronic obstructive pulmonary disease (COPD). Whether there is a causal relationship between these changes and disease progression remains unknown. OBJECTIVES: To investigate the link between an altered microbiota and disease, we used a murine model of chronic lung inflammation that is characterized by key pathological features found in COPD and compared responses in specific pathogen-free (SPF) mice and mice depleted of microbiota by antibiotic treatment or devoid of a microbiota (axenic). METHODS: Mice were challenged with LPS/elastase intranasally over 4 weeks, resulting in a chronically inflamed and damaged lung. The ensuing cellular infiltration, histological damage, and decline in lung function were quantified. MEASUREMENTS AND MAIN RESULTS: Similar to human disease, the composition of the pulmonary microbiota was altered in diseased animals. We found that the microbiota richness and diversity were decreased in LPS/elastase-treated mice, with an increased representation of the genera Pseudomonas and Lactobacillus and a reduction in Prevotella. Moreover, the microbiota was implicated in disease development as mice depleted, or devoid, of microbiota exhibited an improvement in lung function, reduced inflammation, and lymphoid neogenesis. The absence of microbial cues markedly decreased the production of IL-17A, whereas intranasal transfer of fluid enriched with the pulmonary microbiota isolated from diseased mice enhanced IL-17A production in the lungs of antibiotic-treated or axenic recipients. Finally, in mice harboring a microbiota, neutralizing IL-17A dampened inflammation and restored lung function. CONCLUSIONS: Collectively, our data indicate that host-microbial cross-talk promotes inflammation and could underlie the chronicity of inflammatory lung diseases.
DOI:10.1183/13993003.01406-2015URLPMID:26917613 [本文引用: 1]
Increasing evidence suggests that the lung microbiome plays an important role in chronic obstructive pulmonary disease (COPD) severity. However, the dynamics of the lung microbiome during COPD exacerbations and its potential role in disease aetiology remain poorly understood.We completed a longitudinal 16S ribosomal RNA survey of the lung microbiome on 476 sputum samples collected from 87 subjects with COPD at four visits defined as stable state, exacerbation, 2 weeks post-therapy and 6 weeks recovery.Our analysis revealed a dynamic lung microbiota where changes appeared to be associated with exacerbation events and indicative of specific exacerbation phenotypes. Antibiotic and steroid treatments appear to have differential effects on the lung microbiome. We depict a microbial interaction network for the lung microbiome and suggest that perturbation of a few bacterial operational taxonomic units, in particular Haemophilus spp., could greatly impact the overall microbial community structure. Furthermore, several serum and sputum biomarkers, in particular sputum interleukin-8, appear to be highly correlated with the structure and diversity of the microbiome.Our study furthers the understanding of lung microbiome dynamics in COPD patients and highlights its potential as a biomarker, and possibly a target, for future respiratory therapeutics.
DOI:10.4049/jimmunol.0803022URLPMID:19596978 [本文引用: 1]
Airway inflammation and mucus hypersecretion/overproduction/obstruction are pathophysiological characteristics of cystic fibrosis, asthma, and chronic obstructive pulmonary disease. Up-regulation of airway mucin genes by inflammatory/immune response mediators is one of the major contributors to mucin overproduction. IL-8, a potent proinflammatory mediator and neutrophil chemoattractant, is present at high levels in the airway secretions of such patients. In this study, the effects of IL-8 on expression of two major airway mucin genes, MUC5AC and MUC5B, were evaluated. IL-8 increased the mRNA abundance of both mucin genes in two human respiratory tract-derived cell lines (A549 and NCI-H292) in a time- and concentration-dependent manner. IL-8 also increased MUC5AC and MUC5B mRNA levels in primary normal differentiated human bronchial epithelial cells, with a high concentration of IL-8 required to increase MUC5B mRNA levels. IL-8 did not transcriptionally up-regulate MUC5AC gene expression, but rather increased the stability of the MUC5AC transcript, suggesting regulation at the posttranscriptional level. In addition, IL-8 altered the levels of RNA-binding proteins to specific domains in the 3'-untranslated region of the MUC5AC transcript. Taken together, these data indicate that the IL-8-induced binding of RNA-binding proteins to the 3'-untranslated region of MUC5AC is a potential mechanism for regulating MUC5AC gene expression at the posttranscriptional level, thus suggesting a new role whereby IL-8 sustains mucin gene expression in inflamed airways.
DOI:10.1111/imm.12760URLPMID:28542929 [本文引用: 1]
The microbiota plays a central role in human health and disease by shaping immune development, immune responses and metabolism, and by protecting from invading pathogens. Technical advances that allow comprehensive characterization of microbial communities by genetic sequencing have sparked the hunt for disease-modulating bacteria. Emerging studies in humans have linked the increased abundance of Prevotella species at mucosal sites to localized and systemic disease, including periodontitis, bacterial vaginosis, rheumatoid arthritis, metabolic disorders and low-grade systemic inflammation. Intriguingly, Prevotella abundance is reduced within the lung microbiota of patients with asthma and chronic obstructive pulmonary disease. Increased Prevotella abundance is associated with augmented T helper type 17 (Th17) -mediated mucosal inflammation, which is in line with the marked capacity of Prevotella in driving Th17 immune responses in vitro. Studies indicate that Prevotella predominantly activate Toll-like receptor 2, leading to production of Th17-polarizing cytokines by antigen-presenting cells, including interleukin-23 (IL-23) and IL-1. Furthermore, Prevotella stimulate epithelial cells to produce IL-8, IL-6 and CCL20, which can promote mucosal Th17 immune responses and neutrophil recruitment. Prevotella-mediated mucosal inflammation leads to systemic dissemination of inflammatory mediators, bacteria and bacterial products, which in turn may affect systemic disease outcomes. Studies in mice support a causal role of Prevotella as colonization experiments promote clinical and inflammatory features of human disease. When compared with strict commensal bacteria, Prevotella exhibit increased inflammatory properties, as demonstrated by augmented release of inflammatory mediators from immune cells and various stromal cells. These findings indicate that some Prevotella strains may be clinically important pathobionts that can participate in human disease by promoting chronic inflammation.
DOI:10.2147/JIR.S137606URLPMID:29483781 [本文引用: 1]
The nucleotide-binding oligomerization domain (NOD) protein, NOD2, belonging to the intracellular NOD-like receptor family, detects conserved motifs in bacterial peptidoglycan and promotes their clearance through activation of a proinflammatory transcriptional program and other innate immune pathways, including autophagy and endoplasmic reticulum stress. An inactive form due to mutations or a constitutive high expression of NOD2 is associated with several inflammatory diseases, suggesting that balanced NOD2 signaling is critical for the maintenance of immune homeostasis. In this review, we discuss recent developments about the pathway and mechanisms of regulation of NOD2 and illustrate the principal functions of the gene, with particular emphasis on its central role in maintaining the equilibrium between intestinal microbiota and host immune responses to control inflammation. Furthermore, we survey recent studies illustrating the role of NOD2 in several inflammatory diseases, in particular, inflammatory bowel disease, of which it is the main susceptibility gene.
DOI:10.1172/JCI62236URL [本文引用: 1]
Instability in the composition of gut bacterial communities (dysbiosis) has been linked to common human intestinal disorders, such as Crohn's disease and colorectal cancer. Here, we show that dysbiosis caused by Nod2 deficiency gives rise to a reversible, communicable risk of colitis and colitis-associated carcinogenesis in mice. Loss of either Nod2 or RIP2 resulted in a proinflammatory microenvironment that enhanced epithelial dysplasia following chemically induced injury. The condition could be improved by treatment with antibiotics or an anti-interleukin-6 receptor-neutralizing antibody. Genotype-dependent disease risk was communicable via maternally transmitted microbiota in both Nod2-deficient and WT hosts. Furthermore, reciprocal microbiota transplantation reduced disease risk in Nod2-deficient mice and led to long-term changes in intestinal microbial communities. Conversely, disease risk was enhanced in WT hosts that were recolonized with dysbiotic fecal microbiota from Nod2-deficient mice. Thus, we demonstrated that licensing of dysbiotic microbiota is a critical component of disease risk. Our results demonstrate that NOD2 has an unexpected role in shaping a protective assembly of gut bacterial communities and suggest that manipulation of dysbiosis is a potential therapeutic approach in the treatment of human intestinal disorders.
DOI:10.1165/rcmb.2008-0198OCURLPMID:18927348 [本文引用: 4]
Lung cancer is the leading cause of cancer deaths in the United States. In addition to genetic abnormalities induced by cigarette smoke, several epidemiologic studies have found that smokers with chronic obstructive pulmonary disease (COPD), an inflammatory disease of the lungs, have an increased risk of lung cancer (1.3- to 4.9-fold) compared to smokers without COPD. This suggests a link between chronic airway inflammation and lung carcinogenesis, independent of tobacco smoke exposure. We studied this association by assaying the inflammatory impact of products of nontypeable Haemophilus influenzae, which colonizes the airways of patients with COPD, on lung cancer promotion in mice with an activated K-ras mutation in their airway epithelium. Two new mouse models of lung cancer were generated by crossing mice harboring the LSL-K-ras(G12D) allele with mice containing Cre recombinase inserted into the Clara cell secretory protein (CCSP) locus, with or without the neomycin cassette excised (CCSP(Cre) and CCSP(Cre-Neo), respectively). Lung lesions in CCSP(Cre-Neo)/LSL-K-ras(G12D) and CCSP(Cre)/LSL-K-ras(G12D) mice appeared at 4 and 1 month of age, respectively, and were classified as epithelial hyperplasia of the bronchioles, adenoma, and adenocarcinoma. Weekly exposure of CCSP(Cre)/LSL-K-ras(G12D) mice to aerosolized nontypeable Haemophilus influenzae lysate from age 6-14 weeks resulted in neutrophil/macrophage/CD8 T-cell-associated COPD-like airway inflammation, a 3.2-fold increase in lung surface tumor number (156 +/- 9 versus 45 +/- 7), and an increase in total lung tumor burden. We conclude that COPD-like airway inflammation promotes lung carcinogenesis in a background of a G12D-activated K-ras allele in airway secretory cells.
DOI:10.1016/j.cell.2018.12.040URLPMID:30712876 [本文引用: 2]
Lung cancer is closely associated with chronic inflammation, but the causes of inflammation and the specific immune mediators have not been fully elucidated. The lung is a mucosal tissue colonized by a diverse bacterial community, and pulmonary infections commonly present in lung cancer patients are linked to clinical outcomes. Here, we provide evidence that local microbiota provoke inflammation associated with lung adenocarcinoma by activating lung-resident gammadelta T cells. Germ-free or antibiotic-treated mice were significantly protected from lung cancer development induced by Kras mutation and p53 loss. Mechanistically, commensal bacteria stimulated Myd88-dependent IL-1beta and IL-23 production from myeloid cells, inducing proliferation and activation of Vgamma6(+)Vdelta1(+) gammadelta T cells that produced IL-17 and other effector molecules to promote inflammation and tumor cell proliferation. Our findings clearly link local microbiota-immune crosstalk to lung tumor development and thereby define key cellular and molecular mediators that may serve as effective targets in lung cancer intervention.
DOI:10.1038/onc.2017.28URLPMID:28346430 [本文引用: 1]
Chronic obstructive pulmonary disease (COPD) is associated with an increased risk for lung cancer and an aberrant microbiota of the lung. Microbial colonization contributes to chronic neutrophilic inflammation in COPD. Nontypeable Haemophilus influenzae (NTHi) is frequently found in lungs of stable COPD patients and is the major pathogen triggering exacerbations. The epithelial cytokine interleukin-17C (IL-17C) promotes the recruitment of neutrophils into inflamed tissues. The purpose of this study was to investigate the function of IL-17C in the pulmonary tumor microenvironment. We subjected mice deficient for IL-17C (IL-17C(-/-)) and mice double deficient for Toll-like receptor 2 and 4 (TLR-2/4(-/-)) to a metastatic lung cancer model. Tumor proliferation and growth as well as the number of tumor-associated neutrophils was significantly decreased in IL-17C(-/-) and TLR-2/4(-/-) mice exposed to NTHi. The NTHi-induced pulmonary expression of IL-17C was dependent on TLR-2/4. In vitro, IL-17C increased the NTHi- and tumor necrosis factor-alpha-induced expression of the neutrophil chemokines keratinocyte-derived chemokine and macrophage inflammatory protein 2 in lung cancer cells but did not affect proliferation. Human lung cancer samples stained positive for IL-17C, and in non-small cell lung cancer patients with lymph node metastasis, IL-17C was identified as a negative prognostic factor. Our data indicate that epithelial IL-17C promotes neutrophilic inflammation in the tumor microenvironment and suggest that IL-17C links a pathologic microbiota, as present in COPD patients, with enhanced tumor growth.
[本文引用: 2]
[本文引用: 2]
URL [本文引用: 2]
Inflammation has long been suspected to play a major role in the pathogenesis of cancer. Only recently, however, have some mechanisms of its tumor promoting effects become known. Microbes, both commensal and pathogenic, are critical regulators of the host immune system and, ultimately, of inflammation. Consequently, microbes have the potential power to influence tumor progression as well, through a wide variety of routes, including chronic activation of inflammation, alteration of tumor microenvironment, induction of genotoxic responses, and metabolism. In this review, we will provide a general overview of commensal microbiota, inflammation, and cancer, as well as how microbes fit into this emerging field.
DOI:10.1016/j.semcancer.2015.03.006URL [本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1073/pnas.1319051111URLPMID:24706787 [本文引用: 1]
Lung cancer development is associated with extensive pulmonary inflammation. In addition, the linkage between chronic obstructive pulmonary disease (COPD) and lung cancer has been demonstrated in population-based studies. IL-17-producing CD4 helper T cells (Th17 cells) play a critical role in promoting chronic tissue inflammation. Although Th17 cells are found in human COPD and lung cancer, their role is not understood. We have thus used a mouse model of lung cancer, in which an oncogenic form of K-ras (K-ras(G12D)), frequently found in human lung cancer, is restrictedly expressed in lung epithelial cells [via Clara cell secretory protein (CCSP(cre))]. In this model, Th17 and Treg but not Th1 cells were found enriched at the tumor tissues. When CCSP(cre)/K-ras(G12D) mice were weekly challenged with a lysate of nontypeable Haemophilus influenza (NTHi), which induces COPD-type inflammation and accelerates the tumor growth, they showed greatly enhanced Th17 cell infiltration in the lung tissues. Lack of IL-17, but not IL-17F, resulted in a significant reduction in lung tumor numbers in CCSP(cre)/K-ras(G12D) mice and also those treated with NTHi. Absence of IL-17 not only resulted in reduction of tumor cell proliferation and angiogenesis, but also decreased the expression of proinflammatory mediators and reduced recruitment of myeloid cells. Depletion of Gr-1(+)CD11b(+) myeloid cells in CCSP(cre)/K-ras(G12D) mice suppressed tumor growth in lung, indicating Gr-1(+)CD11b(+) myeloid cells recruited by IL-17 play a protumor role. Taken together, our data demonstrate a critical role for Th17 cell-mediated inflammation in lung tumorigenesis and suggest a novel way for prevention and treatment of this disease.
[本文引用: 3]
DOI:10.1186/s40248-018-0123-xURL [本文引用: 1]
DOI:10.1128/IAI.73.5.2728-2735.2005URLPMID:15845475 [本文引用: 1]
Interactions of nontypeable Haemophilus influenzae (NTHI) with human macrophages contribute to the pathogenesis of NTHI-induced infection in humans. However, the immunologic mechanisms that initiate and perpetuate NTHI-mediated macrophage responses have not been well explored. Outer membrane protein (OMP) P6 is a conserved lipoprotein expressed by NTHI in vivo that possesses a Pam(3)Cys terminal motif, characteristic of immunoactive bacterial lipoproteins associated with Toll-like receptor signaling. We theorized that OMP P6 is a potent immunomodulator of human macrophages. To test this hypothesis, we purified OMP P6 as well as OMP P2, the predominant NTHI outer membrane protein, and lipooligosaccharide (LOS), the specific endotoxin of NTHI, from NTHI strain 1479. Human blood monocyte-derived macrophages, purified from healthy donors, were incubated with each outer membrane constituent, and cytokine production of macrophage supernatants interleukin-1beta (IL-1beta), tumor necrosis factor alpha (TNF-alpha), IL-10, IL-12, and IL-8 was measured. OMP P6 selectively upregulated IL-10, TNF-alpha, and IL-8. While OMP P6 (0.1 mug/ml for 8 h) elicited slightly greater concentrations of IL-10, it resulted in over ninefold greater concentrations of TNF-alpha and over fourfold greater concentrations of IL-8 than did OMP P2. OMP P6 at doses as low as 10 pg/ml was still effective at induction of macrophage IL-8, while OMP P2 and LOS were not. OMP P6 of NTHI is a specific trigger of bacteria-induced human macrophage inflammatory events, with IL-8 and TNF-alpha as key effectors of P6-induced macrophage responses.
DOI:10.1186/1476-4598-11-4URLPMID:22239913 [本文引用: 1]
BACKGROUND: Although cigarette smoking is the principal cause of lung carcinogenesis, chronic obstructive pulmonary disease (COPD), an inflammatory disease of the lung, has been identified as an independent risk factor for lung cancer. Bacterial colonization, particularly with non-typeable Haemophilus influenzae (NTHi), has been implicated as a cause of airway inflammation in COPD besides cigarette smoke. Accordingly, we hypothesized that lung cancer promotion may occur in a chronic inflammatory environment in the absence of concurrent carcinogen exposure. RESULTS: Herein, we investigated the effects of bacterial-induced COPD-like inflammation and tobacco carcinogen-enhanced tumorigenesis/inflammation in the retinoic acid inducible G protein coupled receptor knock out mouse model (Gprc5a-/- mouse) characterized by late-onset, low multiplicity tumor formation. Three-month-old Gprc5a-/- mice received 4 intraperitoneal injections of the tobacco-specific carcinogen, NNK, followed by weekly exposure to aerosolized NTHi lysate for 6 months. The numbers of inflammatory cells in the lungs and levels of several inflammatory mediators were increased in Gprc5a-/- mice treated with NTHi alone, and even more so in mice pretreated with NNK followed by NTHi. The incidence of spontaneous lung lesions in the Gprc5a-/- mice was low, but NTHi exposure led to enhanced development of hyperplastic lesions. Gprc5a-/- mice exposed to NNK alone developed multiple lung tumors, while NTHi exposure increased the number of hyperplastic foci 6-fold and the tumor multiplicity 2-fold. This was associated with increased microvessel density and HIF-1alpha expression. CONCLUSION: We conclude that chronic extrinsic lung inflammation induced by bacteria alone or in combination with NNK enhances lung tumorigenesis in Gprc5a-/- mice.
DOI:10.1016/j.ccr.2004.08.012URL [本文引用: 1]
Abstract
We used an experimental murine cancer metastasis model in which a colon adenocarcinoma cell line generates lung metastases, whose growth is stimulated in response to injection of bacterial lipopolysaccharide (LPS), to investigate the role of NF-κB in inflammation-induced tumor growth. We found that LPS-induced metastatic growth response in this model depends on both TNFα production by host hematopoietic cells and NF-κB activation in tumor cells. Inhibition of NF-κB in both colon and mammary carcinoma cells converts the LPS-induced growth response to LPS-induced tumor regression. The latter response is TNFα-independent, but depends on another member of the TNF superfamily, TRAIL, whose receptor is induced in NF-κB-deficient cancer cells.DOI:10.1155/2019/1394191URLPMID:31485458 [本文引用: 1]
Recent research on cancer-associated microbial communities led to the accumulation of data on the interplay between bacteria, immune and tumor cells, the pathways of bacterial induction of carcinogenesis, and its meaningfulness for medicine. Microbial communities that have any kind of impact on tumor progression and microorganisms associated with tumors have been defined as oncobiome. Over the last decades, a number of studies were dedicated to Helicobacter pylori and its role in the progression of stomach tumors, so this correlation can be regarded as proven. Involvement of bacteria in the induction of lung cancer has been largely ignored for a long time, though some correlations between this type of cancer and lung microbiome were established. Despite the fact that in the present the microbial impact on lung cancer progression has many confirmations, the underlying mechanisms are poorly understood. Microorganisms can contribute to tumor initiation and progression through production of bacteriotoxins and other proinflammatory factors. The purpose of this review is to organize the available data on lung cancer microbiome and its role in malignant tumor progression.
DOI:10.1158/0008-5472.CAN-15-2840URLPMID:27197187 [本文引用: 3]
Activating mutations of K-ras are the most common oncogenic alterations found in lung cancer. Unfortunately, attempts to target K-ras-mutant lung tumors have thus far failed, clearly indicating the need for new approaches in patients with this molecular profile. We have previously shown NF-kappaB activation, release of IL6, and activation of its responsive transcription factor STAT3 in K-ras-mutant lung tumors, which was further amplified by the tumor-enhancing effect of chronic obstructive pulmonary disease (COPD)-type airway inflammation. These findings suggest an essential role for this inflammatory pathway in K-ras-mutant lung tumorigenesis and its enhancement by COPD. Therefore, here we blocked IL6 using a monoclonal anti-IL6 antibody in a K-ras-mutant mouse model of lung cancer in the absence or presence of COPD-type airway inflammation. IL6 blockade significantly inhibited lung cancer promotion, tumor cell-intrinsic STAT3 activation, tumor cell proliferation, and angiogenesis markers. Moreover, IL6 inhibition reduced expression of protumor type 2 molecules (arginase 1, Fizz 1, Mgl, and IDO), number of M2-type macrophages and granulocytic myeloid-derived suppressor cells, and protumor T-regulatory/Th17 cell responses. This was accompanied by increased expression of antitumor type 1 molecule (Nos2), and antitumor Th1/CD8 T-cell responses. Our study demonstrates that IL6 blockade not only has direct intrinsic inhibitory effect on tumor cells, but also reeducates the lung microenvironment toward an antitumor phenotype by altering the relative proportion between protumor and antitumor immune cells. This information introduces IL6 as a potential druggable target for prevention and treatment of K-ras-mutant lung tumors. Cancer Res; 76(11); 3189-99. (c)2016 AACR.
DOI:10.1038/nmicrobiol.2016.31URLPMID:27572644 [本文引用: 1]
Microaspiration is a common phenomenon in healthy subjects, but its frequency is increased in chronic inflammatory airway diseases, and its role in inflammatory and immune phenotypes is unclear. We have previously demonstrated that acellular bronchoalveolar lavage samples from half of the healthy people examined are enriched with oral taxa (here called pneumotypeSPT) and this finding is associated with increased numbers of lymphocytes and neutrophils in bronchoalveolar lavage. Here, we have characterized the inflammatory phenotype using a multi-omic approach. By evaluating both upper airway and acellular bronchoalveolar lavage samples from 49 subjects from three cohorts without known pulmonary disease, we observed that pneumotypeSPT was associated with a distinct metabolic profile, enhanced expression of inflammatory cytokines, a pro-inflammatory phenotype characterized by elevated Th-17 lymphocytes and, conversely, a blunted alveolar macrophage TLR4 response. The cellular immune responses observed in the lower airways of humans with pneumotypeSPT indicate a role for the aspiration-derived microbiota in regulating the basal inflammatory status at the pulmonary mucosal surface.
DOI:10.1016/j.jaci.2015.05.044URLPMID:26220531 [本文引用: 1]
BACKGROUND: Asthma is heterogeneous, and airway dysbiosis is associated with clinical features in patients with mild-to-moderate asthma. Whether similar relationships exist among patients with severe asthma is unknown. OBJECTIVE: We sought to evaluate relationships between the bronchial microbiome and features of severe asthma. METHODS: Bronchial brushings from 40 participants in the Bronchoscopic Exploratory Research Study of Biomarkers in Corticosteroid-refractory Asthma (BOBCAT) study were evaluated by using 16S ribosomal RNA-based methods. Relationships to clinical and inflammatory features were analyzed among microbiome-profiled subjects. Secondarily, bacterial compositional profiles were compared between patients with severe asthma and previously studied healthy control subjects (n = 7) and patients with mild-to-moderate asthma (n = 41). RESULTS: In patients with severe asthma, bronchial bacterial composition was associated with several disease-related features, including body mass index (P < .05, Bray-Curtis distance-based permutational multivariate analysis of variance; PERMANOVA), changes in Asthma Control Questionnaire (ACQ) scores (P < .01), sputum total leukocyte values (P = .06), and bronchial biopsy eosinophil values (per square millimeter, P = .07). Bacterial communities associated with worsening ACQ scores and sputum total leukocyte values (predominantly Proteobacteria) differed markedly from those associated with body mass index (Bacteroidetes/Firmicutes). In contrast, improving/stable ACQ scores and bronchial epithelial gene expression of FK506 binding protein (FKBP5), an indicator of steroid responsiveness, correlated with Actinobacteria. Mostly negative correlations were observed between biopsy eosinophil values and Proteobacteria. No taxa were associated with a TH2-related epithelial gene expression signature, but expression of TH17-related genes was associated with Proteobacteria. Patients with severe asthma compared with healthy control subjects or patients with mild-to-moderate asthma were significantly enriched in Actinobacteria, although the largest differences observed involved a Klebsiella genus member (7.8-fold increase in patients with severe asthma, adjusted P < .001). CONCLUSIONS: Specific microbiota are associated with and may modulate inflammatory processes in patients with severe asthma and related phenotypes. Airway dysbiosis in patients with severe asthma appears to differ from that observed in those with milder asthma in the setting of inhaled corticosteroid use.
[本文引用: 1]

{kind=link}
{kind=link}