 ,1, 程苗苗1, 宋昉,1, 瞿宇晋1, 白晋丽1, 金煜炜1, 王红11
,1, 程苗苗1, 宋昉,1, 瞿宇晋1, 白晋丽1, 金煜炜1, 王红11 Familial study of spinal muscular atrophy carriers with SMN1 (2+0) genotype
Cao Yanyan,1, Cheng Miaomiao1, Song Fang,1, Qu Yujin1, Bai Jinli1, Wang Hong11 通讯作者: 通讯作者: 宋昉,硕士,研究员,研究方向:儿童罕见病机制研究。E-mail:songf_558@263.net
第一联系人:
收稿日期:2020-10-17
| 基金资助: |
Received:2020-10-17
| Fund supported: |

摘要
脊髓性肌萎缩症(spinal muscular atrophy, SMA)是一种儿童时期较为常见的神经肌肉病,属于常染色体隐性遗传。绝大多数SMA由运动神经元存活基因1 (survival motor neuron 1,SMN1)的纯合缺失突变所致。而SMN1的2+0基因型个体作为一种特殊的SMA携带者,给携带者筛查以及家系的遗传咨询带来了巨大的挑战。已有研究表明,g.27134T>G和g.27706_27707delAT多态位点变异对于Ashkenazi犹太人群中的2+0基因型个体具有提示作用。为进一步探究这两个多态位点是否在中国人群也具有特异性,本研究纳入了44例家系成员和204例已知SMN1基因拷贝数的对照样本。44例家系成员来自于9个无关的SMN1基因纯合缺失的SMA家系,先证者双亲之一疑似为2+0基因型携带者。利用多重连接探针扩增(multiplex ligation-dependent probe amplification, MLPA)和短串联重复(short tandem repeat, STR)连锁分析进行基因型的鉴定以及多态位点的筛查,最终通过对家系三代成员或多子女家系两代成员的分析确定了9个家系中的10例个体为2+0基因型携带者,多态位点筛查显示1例携带3拷贝SMN1基因的个体同时存在g.27134T>G和g.27706_27707delAT多态位点的变异。因此,本研究通过对2+0基因型携带者的鉴定,为家系遗传病的诊断提供了精准的遗传咨询。g.27134T>G和g.27706_27707delAT多态位点可能与中国人群2+0基因型个体的关联度较低,尚需寻找中国人群特异的多态位点以提高2+0基因型携带者的检出率。
关键词:
Abstract
Keywords:
PDF (1730KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
曹延延, 程苗苗, 宋昉, 瞿宇晋, 白晋丽, 金煜炜, 王红. 脊髓性肌萎缩症SMN1基因 2+0基因型携带者的家系研究 . 遗传[J], 2021, 43(2): 160-168 doi:10.16288/j.yczz.20-319
Cao Yanyan.
脊髓性肌萎缩症(spinal muscular atrophy, SMA)是儿童时期较为常见的神经肌肉病,呈常染色体隐性遗传。运动神经元存活基因1 (survival motor neuron 1,SMN1)是SMA的致病基因,绝大部分的SMA患儿是由于SMN1基因的纯合缺失突变所致。SMN2基因与SMN1基因高度同源,是SMA的表型修饰基因。此外,SMN1基因和SMN2基因均定位于5q13.2,SMN1基因靠近端粒,而SMN2基因靠近着丝粒,二者成镜像排列。这些特征使得SMN1基因易于发生缺失、重复以及与SMN2之间的基因转换,造成人群中SMN1基因拷贝数变异较大[1]。
正常情况下,每条染色体携带1拷贝SMN1基因和1拷贝SMN2基因,即每个个体的SMN1基因和SMN2基因的拷贝数均为2。对于SMN1基因纯合缺失的先证者(拷贝数为0),其双亲SMN1基因的拷贝数通常为1,也就是一条染色体上的SMN1基因丢失,即经典的SMA携带者。但是SMA还有一种特殊类型的携带者,这类携带者的SMN1基因拷贝数虽然为2,但是却位于同一条染色体(incis),而另一条染色体上的SMN1基因丢失,即2+0型携带者。有效识别2+0型携带者对于提高SMA携带者检出率以及为家庭提供精准的遗传咨询至关重要。
已有研究显示在Ashkenazi犹太人群[2],大部分携带有SMN1重复等位基因的个体(SMN1拷贝数大于等于3)可存在一种由g.27134T>G和g.27706_ 27707delAT多态位点构成的SMN1单倍型。由此推测在特定人群中,这两个多态变异的存在对于2+0基因型携带者具有提示作用。
本研究纳入9个无关的中国SMA家系,先证者双亲之一疑似2+0型携带者。通过SMN基因(SMN1和SMN2基因)剂量分析结合短串联重复(short tandem repeat, STR)连锁分析明确疑似2+0携带者的基因型,并初步探讨g.27134T>G和g.27706-27707delAT多态位点是否适于中国人群2+0基因型的预测。
1 对象与方法
1.1 研究对象
本研究纳入了9个SMN1基因纯合缺失的SMA家系,共44例家系成员。其中,SMA先证者9例,先证者同胞2例,先证者双亲18例,先证者祖父母/外祖父母15例。所有先证者均为在首都儿科研究所遗传室加入“中国罕见病注册登记研究—SMA注册登记”项目(编号:2016YFC0901505)的患者,且所有参与家庭均签署知情同意书。本研究经首都儿科研究所伦理委员会批准(批准号:SHERLL2017007)。此外,另加入204例已知SMN1基因拷贝数的本研究室贮存DNA对照样本用于多态位点的筛查。1.2 拷贝数分析
先证者及家系成员留取EDTA抗凝血3 mL,采用EasyPure?Blood Genomic DNA Kit (北京全式金生物技术有限公司)提取基因组DNA。应用多重连接探针扩增(multiplex ligation-dependent probe amplification, MLPA)技术(MRC,荷兰)进行基因拷贝数和多态位点的分析。其中,P060-B2 SMA Kit用于测定SMN1基因和SMN2基因的拷贝数,P460-A1 SMA Kit用于多态位点g.27134T>G和g.27706-27707delAT的检测。1.3 STR连锁分析
选取12个STR位点应用毛细管电泳的方法进行连锁分析,其中6个位点(UHM2、UHM3、UHM4、UHM5、UHM7和UHM8)位于SMN2基因上游2 Mb范围内,其余6个位点(DHM1、DHM2、DHM4、DHM6、DHM7和DHM8)位于SMN1基因下游2Mb范围内。家系的STR连锁分析实验及数据处理由上海五色石医学研究股份有限公司完成。2 结果与分析
2.1 SMN1基因拷贝数分布
9例SMA先证者的SMN1基因拷贝数均为0,即为SMN1基因纯合缺失。先证者的双亲之一均携带2拷贝SMN1基因(3例父亲;6例母亲),其配偶SMN1基因的拷贝数均为1,即为SMN1基因杂合缺失。先证者的15例祖父母或外祖父母中,8例个体携带3拷贝SMN1基因,除1例仅有单方样本外,其余7例的配偶均为SMN1基因杂合缺失。最后,在2例SMA先证者的同胞中SMN1基因拷贝数分别为2和3 (表1)。2.2 SMN1基因型的分布
通常情况下,正常个体每条染色体携带1拷贝SMN1基因,即1+1基因型。0+0基因型表示两条染色体上的SMN1基因均丢失,即纯合缺失突变,为SMA患者。1+0基因型表示一条染色体上SMN1基因拷贝数为1,另一条染色体上的SMN1基因丢失,即经典的SMA携带者;2+0基因型表示2拷贝的SMN1基因位于同一条染色体,另一条染色体上SMN1基因丢失,即为特殊类型的SMA携带者。2+1基因型表示一条染色体携带2拷贝的SMN1基因,而另一条染色体上携带1拷贝的SMN1基因,SMN1基因的总拷贝数为3。2+0基因型和2+1基因型个体为SMN1重复等位基因的携带者,二者表型正常。进一步分析SMN1基因的基因型分布(表1),9例先证者均为0+0基因型,即SMN1基因纯合缺失。先证者的双亲中,50%(9/18)为经典型SMA携带者(1+0基因型),同时另一方(50%,9/18)均为疑似2+0型的携带者。同样,在3对祖父母和4对外祖父母中,一方(50%, 7/14)为经典1+0携带者,另一方(50%, 7/14)为2+1基因型。另有1例外祖父(仅有单方样本)为2+1基因型。2例先证者同胞中,SMN1基因型分别为2+0和2+1。
Table 1
表1
表1SMN1拷贝数和基因型的分布
Table 1
| SMN1拷贝数 | SMN1基因型 | 先证者(%) | 先证者父亲(%) | 先证者母亲(%) | 先证者祖父/母(%) | 先证者外祖父/母(%) | 先证者同胞(%) |
|---|---|---|---|---|---|---|---|
| 0 | 0+0 | 9 (100) | 0 | 0 | 0 | 0 | 0 |
| 1 | 1+0 | 0 | 6 (66.7) | 3 (33.3) | 3 (50) | 4 (44.4) | 0 |
| 2 | 2+0 | 0 | 3(33.3) | 6 (66.7) | 0 | 0 | 1 (50) |
| 3 | 2+1 | 0 | 0 | 0 | 3 (50) | 5 (55.6) a | 1 (50) |
| 合计 | - | 9 | 9 | 9 | 6 | 9 | 2 |
新窗口打开|下载CSV
2.3 SMN1基因拷贝数的家系分布
9个家系均进行了包括先证者、同胞、父母、祖父母或外祖父母在内的SMN1和SMN2基因拷贝数检测以及STR连锁分析,以明确先证者携带2拷贝SMN1基因双亲的基因型(表2)。Table 2
表2
表2SMN1基因拷贝数在家系中的分布
Table 2
| 家系 | I (祖父/祖母或 外祖父/外祖母) | II (父亲/母亲) | III (先证者/同胞) |
|---|---|---|---|
| #1 | 1/3 | 2/1 | 0 |
| #2 | 3/1 | 1/2 | 0 |
| #3 | 1/3 | 1/2 | 0 |
| #4 | 3/1 | 2/1 | 0 |
| #5 | 1/3 | 1/2 | 0 |
| #6 | 1/3 | 1/2 | 0 |
| #7 | 1/3 | 2/1 | 0 |
| #8 | 3/NA | 1/2 | 0/2 |
| #9 | NA | 1/2 | 0/3 |
| 合计 | 8 | 9 | 9 |
新窗口打开|下载CSV
以#7号家系为例,先证者SMN1基因纯合缺失,母亲为SMN1基因的杂合缺失,父亲SMN1基因的拷贝数为2 (图1)。因此,先证者父亲疑似为2+0基因型。此外,先证者的祖父和祖母SMN1基因的拷贝数分别为1和3。结合STR连锁分析显示,父亲携带黑色0拷贝的SMN1基因遗传自祖父,携带绿色2拷贝的SMN1基因遗传自祖母。随后父亲将黑色0拷贝的SMN1基因传递给先证者,同时母亲也将橙色0拷贝的SMN1基因传递给先证者,导致先证者发生SMA。因此,该家系通过三代SMN1基因拷贝数结合STR连锁分析,基本确定了先证者父亲SMN1基因为2+0型。同时也基本排除了先证者SMN1基因的新生变异。
图1
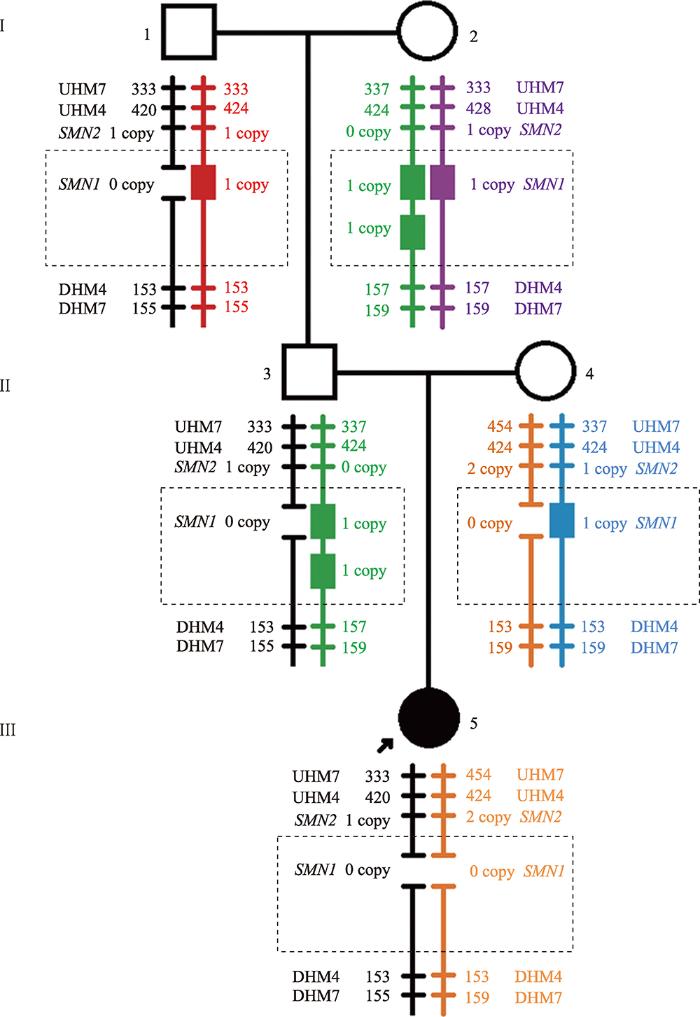 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1#7家系 SMN基因拷贝数与STR连锁分析模式图
I:祖父/祖母;II:父亲/母亲;III:先证者;正方形:男性;圆形:女性;黑色圆形:女性先证者;不同颜色的竖线:不同来源的染色体;短横线及数字:STR位点及其片段长度;实心长方形:1拷贝的SMN1基因;两短横线间空白:0拷贝的SMN1基因;虚线框:SMN1基因的总拷贝数;UHM7:上游单倍型标记7;UHM4:上游单倍型标记4;DHM4:下游单倍型标记4;DHM7:下游单倍型标记7。
Fig. 1Pattern graph for SMN copies and STR linkage analysis in #7 family
此外,值得注意的是#8和#9家系。#8家系中先证者的外祖母由于过世没有获得样本,该家系有两个孩子。如图2所示,先证者母亲为2+0基因型,其蓝色的2拷贝SMN1基因遗传自先证者的外祖父,推测橙色的0拷贝SMN1基因可能遗传自先证者的外祖母。随后母亲将橙色的0拷贝SMN1基因传递给先证者,加之另一个遗传自先证者父亲的黑色0拷贝SMN1基因,先证者发病。但是,先证者母亲将蓝色的2拷贝SMN1基因传递给了先证者的姐姐,而姐姐也同时继承了父亲黑色0拷贝SMN1基因。因此,姐姐的SMN1基因拷贝数虽然为2,但是她实际与母亲一样,为2+0携带者。
图2
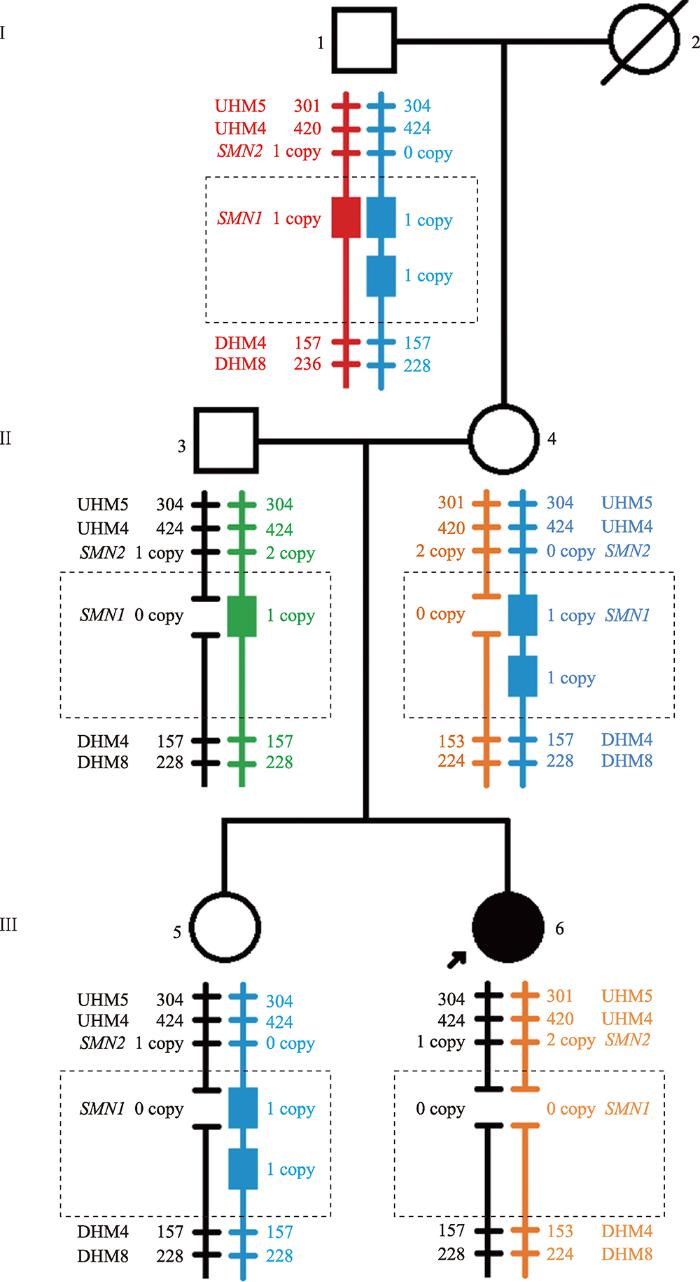 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2#8家系 SMN基因拷贝数与STR连锁分析模式图
I:外祖父/外祖母(外祖母已去世);II:父亲/母亲;III:先证者/同胞;正方形:男性;圆形:女性;黑色圆形:女性先证者;不同颜色的竖线:不同来源的染色体;短横线及数字:STR位点及其片段长度;实心长方形:1拷贝的SMN1基因;两短横线间空白:0拷贝的SMN1基因;虚线框:SMN1基因的总拷贝数;UHM5:上游单倍型标记5;UHM4:上游单倍型标记4;DHM4:下游单倍型标记4;DHM8:下游单倍型标记8。
Fig. 2Pattern graph for SMN copies and STR linkage analysis in #8 family
#9家系,该家系中先证者的外祖父母均已过世,家中有两个孩子。如图3所示,先证者为SMN1基因纯合缺失,父亲为SMN1基因杂合缺失,母亲的SMN1基因拷贝数为2。因为无法得到先证者外祖父和外祖母的样本,因而不能通过三代连锁分析确定母亲的基因型。母亲SMN1基因为2拷贝主要有两种可能:2+0基因型携带者或母亲为正常的1+1基因型。鉴于先证者哥哥SMN1基因的拷贝数为3,STR连锁分析显示,哥哥的绿色1拷贝SMN1基因遗传自父亲,另2拷贝的SMN1基因只能来自母亲,故而推测出另2拷贝的SMN1基因位于同一条染色体。因此,通过家系中的多子女连锁分析也可确定先证者母亲为2+0基因型。
图3
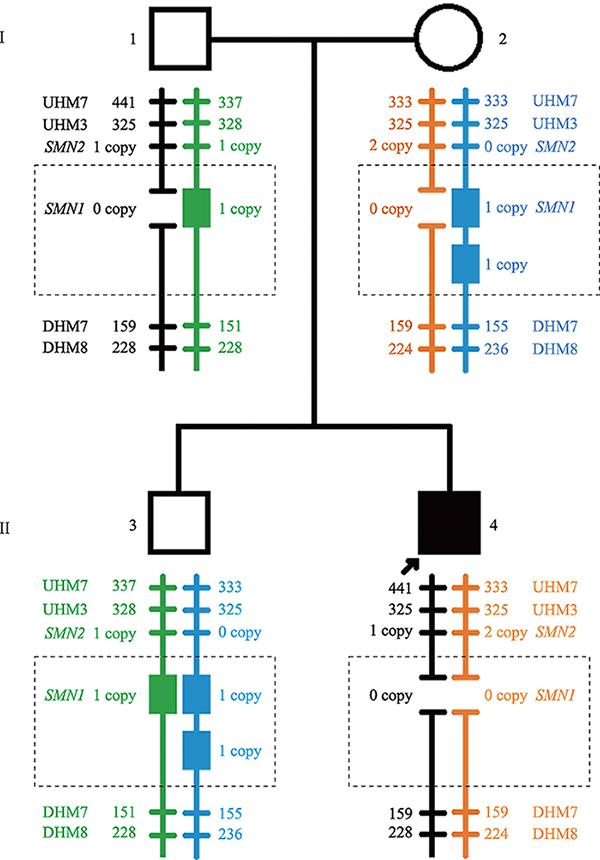 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3#9家系 SMN基因拷贝数与STR连锁分析模式图
I:父亲/母亲;II:先证者/同胞;正方形:男性;黑色正方形:男性先证者;圆形:女性;不同颜色的竖线:不同来源的染色体;短横线及数字:STR位点及其片段长度;实心长方形:1拷贝的SMN1基因;两短横线间空白:0拷贝的SMN1基因;虚线框:SMN1基因的总拷贝数;UHM7:上游单倍型标记7;UHM3:上游单倍型标记3;DHM7:下游单倍型标记7;DHM8:下游单倍型标记8。
Fig. 3Pattern graph for SMN copies and STR linkage analysis in #9 family
2.4 多态位点分析
为明确多态位点g.27134T>G和g.27706_ 27707delAT是否可用于中国人群2+0基因型的预测,本研究筛查了44例家系成员以及204例携带不同SMN1基因拷贝数的对照样本。本研究中的44例家系成员,SMN1基因型为0+0的有9例,1+0基因型16例,经实验确定的2+0基因型10例,还有2+1基因型9例。这些样本中均未发现g.27134T>G和g.27706_27707delAT多态位点变异。考虑到这些家系成员间存在亲缘关系,我们在204例无亲缘关系且已知SMN1基因拷贝数的对照样本中对这两个多态位点进行了筛查。对照样本中,187例携带2拷贝SMN1基因,其中包括18例疑似2+0基因型个体。这18例个体的子代均为SMN1基因纯合缺失的SMA患儿,配偶均为SMN1基因杂合缺失的SMA携带者。此外,对照样本中还包括16例携带3拷贝SMN1基因个体和1例携带4拷贝SMN1基因的个体。结果显示,有1例SMN1基因拷贝数为3的个体同时存在g.27134T>G和g.27706_ 27707delAT多态位点变异,而其余样本未发现携带有这两个位点的多态性改变。(表3)。
Table 3
表3
表3多态位点的分布
Table 3
| 多态位点 | SMA家系成员(SMN1基因型) | 对照组(SMN1拷贝数) | |||||
|---|---|---|---|---|---|---|---|
| 0+0 | 1+0 | 2+0 | 2+1 | 2拷贝 a | 3拷贝 | 4拷贝 | |
| g.27134T>G | 0 | 0 | 0 | 0 | 0 | 1 (6.3%) | 0 |
| g.27706_27707delAT | 0 | 0 | 0 | 0 | 0 | 1 (6.3%) | 0 |
| 筛查例数 | 9 | 16 | 10 | 9 | 187 | 16 | 1 |
新窗口打开|下载CSV
3 讨论
对于纯合缺失突变的SMA家系来说,当双亲一方为经典1+0携带者,而另一方SMN1基因拷贝数为2时,明确双亲的基因型对于家庭SMA再发风险的评估至关重要。若携带2拷贝SMN1基因的双亲一方为2+0基因型,那么他们再次生育,子代SMA的再发风险为25%;若为正常的1+1基因型,那么先证者可能发生了新生变异抑或是双亲之一为生殖腺嵌合体,因而该家系SMA的再发风险很低。本研究通过对家系三代成员,或多子女家系的两代成员进行SMN基因拷贝数检测,并结合SMN基因上下游STR位点的家系连锁分析最终确定了来自9个家系的10例个体为SMN1基因的2+0携带者。同时,也明确了这9个家系中的先证者非SMN1基因的新生变异所致,为该家系提供精准的遗传咨询奠定了基础。作为特殊类型的携带者,2+0基因型在一般人群中的携带率约为5%~8%[3],在肯定携带者中约占4%[4]。以MLPA、Real-time PCR为代表的基因定量检测技术虽然可以明确SMN1和SMN2基因的拷贝数,但是无法区分2+0基因型和正常的1+1基因型,需要辅以其他检测技术综合分析。Chen等[5]早在1999年就利用SMN1基因荧光定量结合单倍型分析的方法证实了2+0基因型的存在。2000年,Yan等[6]通过将人类细胞与小鼠细胞融合后进行选择性培养,可以分离出单个人类染色体。2001年Mailman等[7]利用上述技术成功检测出了2+0基因型,但是这项技术耗时且费力。近来也有报道利用单精子测序的方法辅助明确男性个体2+0基因型,但不适于女性个体[8]。因此,就目前而言,基因定量技术已经成熟,对于疑似2+0基因型个体,可以优先检测其双亲SMN1基因的拷贝数。当双亲一方为1+0携带者,另一方SMN1基因的拷贝数大于等于3则高度提示疑似个体为2+0基因型。
因为SMN1基因和SMN2基因的拷贝数变异很大程度上源自亲代遗传,并可以稳定地传递下去[9],所以2+0基因型携带者在人群中也会稳定存在(#8家系先证者姐姐)。而常规的检测方法不能直接确定2+0基因型,那么发现与SMN1重复等位基因连锁的单倍型可以有效提示2+0基因型的存在。Luo等[2]报道了在Ashkenazi犹太人中,由多态位点g.27134T> G和g.27706-27707delAT构成的单倍型仅存在于携带SMN1重复等位基因的个体中(SMN1拷贝数大于等于3),而不存在于对照个体(SMN1拷贝数为2)。提示这两个多态位点可能与SMN1重复等位基因呈现连锁不平衡。那么在一个SMN1基因拷贝数为2的个体中检测到这两个多态变异时,则高度提示这2个拷贝的SMN1基因位于同一条染色体,该个体为2+0携带者的风险增加。这一发现使得Ashkenazi犹太人群中SMA携带者检出率由90%提升至94%。但也并不是所有的2+0基因型个体都可以检测到这两个多态变异的存在。因此在特定人群中,当携带2拷贝SMN1基因的个体不存在g.27134T>G或g.27706_27707delAT多态变异中的任何一个,尚不能排除2+0携带者的可能,但是可以降低该个体为携带者的风险。
最近研究显示,非洲人群中携带3拷贝和4拷贝SMN1基因的个体所占比例分别为41.4%(373/902)和13.4% (121/902),显著高于东亚人群的5.6% (33/593)和0% (0/593)[10]。而在SMN1拷贝数同样为3或4的个体中,非洲人群g.27134T>G的检出率为86.4% (427/494),显著高于东亚人群的3.0% (1/33)。以上结果提示,SMN1基因拷贝数的分布以及g.27134T>G多态位点变异具有种族特异性。中国人群中携带3拷贝和4拷贝SMN1基因的个体所占比例分别为7.0% (1434/20403)和0.3% (60/20403)[11],尚无多态位点g.27134T>G和g.27706_27707delAT的相关数据。
本研究筛查了44例家系成员和204例携带不同SMN1基因拷贝数的对照样本,仅在对照组中发现1例SMN1基因拷贝数为3的个体同时携带g.27134T>G和g.27706_27707delAT多态变异。因此,g.27134T>G和g.27706_27707delAT在携带3拷贝SMN1基因个体中的检出率均为6.3% (1/16),g.27134T>G的检出率显著低于非洲人群(84.5%)[10]和西班牙人群(11.8%)[12],略高于东亚人群(3.0%)[10],g.27706_27707delAT的检出率显著低于西班牙人群(11.8%)[12]。而本研究在10例确定2+0基因型和18例疑似2+0基因型个体中均未发现这两个多态变异。以上结果提示多态位点g.27134T>G和g.27706_ 27707delAT在中国人群中的比例可能相对较低,对于2+0基因型的提示作用尚需进一步扩大样本进行验证。同时,也需要寻找中国人群特异性的单倍型或多态位点,以提升2+0携带者的检出率。但是,鉴于我国为多民族国家,是否存在普适的单倍型或多态位点还有待考究。
综上所述,本研究通过多代系SMN1基因定量结合STR连锁分析确定了纯合缺失SMA家系中10例2+0基因型个体,为家系遗传病的诊断提供了更为精准的遗传咨询。对于疑似2+0基因型个体,首先推荐检测其双亲SMN1基因的拷贝数。此外,本研究结果也表明g.27134T>G和g.27706_27707delAT多态位点可能与中国人群2+0基因型个体的关联度较低,尚需进一步探寻中国人群特异性的分子标记以提高2+0基因型携带者的检出率。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/ejhg.2010.54URLPMID:20442745 [本文引用: 1]

Screening for carriers of spinal muscular atrophy (SMA) is necessary for effective clinical/prenatal diagnosis and genetic counseling. However, a population-based study of SMA prevalence in mainland China has not yet been conducted. In this study, the copy number of survival motor neuron (SMN) genes was determined in 1712 newborn cord blood samples collected from southern China and from 25 core families, which included 26 SMA patients and 44 parents, to identify SMA carriers. The results presented 13 groups with different SMN1/SMN2 ratios among 1712 newborn individuals, which corresponded to 1535 subjects with two copies of SMN1, 119 with three copies of SMN1, 17 with four copies of SMN1, and 41 with a heterozygous deletion of SMN1 exon 7. Simultaneously, two '2+0' genotypes and two point mutations were found among the 44 obligate carriers in the core families, including a novel SMN1 splice-site mutation that was identified in the junction between intron 6 and exon 7 (c. 835-1G>A). These results indicated that the carrier frequency is 1/42 in the general Chinese population and that duplicated SMN1 alleles and de novo deletion mutations are present in a small number of SMA carriers. In addition, we developed and validated a new alternative screening method using a reverse dot blot assay for rapid genotyping of deletional SMA. Our research elucidated the genetic load and SMN gene variants that are present in the Chinese population, and could serve as the basis for a nationwide program of genetic counseling and clinical/prenatal diagnosis to prevent SMA in China.
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
URLPMID:10405443 [本文引用: 1]
Approximately 95% of individuals with spinal muscular atrophy (SMA) lack both copies of the SMNt gene at 5q13. The presence of a nearly identical centromeric homolog of the SMNt gene, SMNc, necessitates a quantitative polymerase chain reaction approach to direct carrier testing. Adapting a radioactivity-based method described previously, multiplex polymerase chain reaction was performed using fluorescently labeled primers followed by analysis on an ABI 373a DNA sequencer. The SMNt copy number was calculated from ratios of peak areas using both internal and genomic standards. Samples from 60 presumed carriers (50 parents of affected individuals and 10 relatives implicated by linkage analysis) and 40 normal control individuals were tested. Normalized results (to the mean of five or more control samples harboring two copies of the SMNt gene) were consistently within the ranges of 0.4 to 0.6 for carriers (one copy) and 0.8 to 1.2 for normal controls (two copies), without overlap. Combining linkage analyses with direct carrier test results demonstrated de novo deletions associated with crossovers, unaffected individuals carrying two SMNt gene copies on one chromosome and zero SMNt gene copies on the other chromosome, and unaffected individuals with three copies of the SMNt gene. This report demonstrates that fluorescence-based carrier testing for SMA is accurate, reproducible, and useful for genetic risk assessment, and that carrier testing may need to be combined with linkage analysis in certain circumstances.
[本文引用: 1]
URLPMID:11281448 [本文引用: 1]
DOI:10.1038/ejhg.2009.198URLPMID:19904299 [本文引用: 1]
With the detection of a homozygous deletion of the survival motor neuron 1 gene (SMN1), prenatal and preimplantation genetic diagnosis (PGD) for spinal muscular atrophy has become feasible and widely applied. The finding of a de novo rearrangement, resulting in the loss of the SMN1 gene, reduces the recurrence risk from 25% to a lower percentage, the residual risk arising from recurrent de novo mutation or germline mosaicism. In a couple referred to our PGD center because their first child was affected with SMA, the male partner was shown to carry two SMN1 copies. An analysis of the SMN1 gene and two flanking markers was performed on 12 single spermatozoa, to determine whether the father carried a CIS duplication of the SMN1 gene on one chromosome and was a carrier, or if the deletion has occurred de novo. We showed that all spermatozoa that were carriers of the 'at-risk haplotype' were deleted for the SMN1 gene, confirming the carrier status of the father. We provide an original application of single germ cell studies to recessive disorders using coamplification of the gene and its linked markers. This efficient and easy procedure might be useful to elucidate complex genetic situations when samples from other family members are not available.
DOI:10.1038/s10038-020-0730-1URLPMID:32051521 [本文引用: 1]
URLPMID:32066871 [本文引用: 3]
URLPMID:33446828 [本文引用: 1]
[本文引用: 2]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}