 ,中国医学科学院血液病医院(中国医学科学院血液学研究所),实验血液学国家重点实验室,国家血液系统疾病临床医学研究中心,天津300020
,中国医学科学院血液病医院(中国医学科学院血液学研究所),实验血液学国家重点实验室,国家血液系统疾病临床医学研究中心,天津300020Zebrafish blood disease models and applications
Jiani Guo, Fan Liu, Lu Wang,State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases, Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin 300020, China通讯作者: 王璐,博士,研究员,研究方向:干细胞与再生医学。E-mail:wanglu1@ihcams.ac.cn
编委: 刘峰
收稿日期:2020-05-25修回日期:2020-06-27网络出版日期:2020-08-20
| 基金资助: |
Received:2020-05-25Revised:2020-06-27Online:2020-08-20
| Fund supported: |
作者简介 About authors
郭佳妮,在读硕士研究生,专业方向:干细胞与再生医学。E-mail:

摘要
血液发育是一个复杂有序且保守的过程,由多种转录因子和信号通路协同调控,任何环节的失调都可能引起血液系统发育或功能缺陷,导致血液疾病的发生。斑马鱼(Danio rerio)造血过程及分子调控机制与哺乳动物高度保守。应用斑马鱼模拟致病因子的异常变化构建相关血液疾病模型,为探究疾病发生机制、肿瘤发生发展可视化研究及高通量化学筛选提供了有力的工具。本文概述了斑马鱼血液疾病模型及其应用,这些疾病模型不仅有助于完善对血液系统病理生理学、血液疾病发生分子机制的认识,也为临床相关恶性血液疾病的治疗提供了新思路。
关键词:
Abstract
Hematopoiesis is a complex, orderly and conserved developmental process, coordinated by multiple factors including transcription factors and signaling pathways. Dysregulation of any of these factors may cause developmental or functional defects in the blood system, leading to the pathogenesis of blood diseases. Zebrafish hematopoiesis and the underlying molecular mechanisms are highly conserved with those in mammals. The use of zebrafish to recapitulate abnormal changes in pathogenic factors can build models of related blood diseases, thus providing powerful tools for exploring the molecular mechanisms of pathogenesis and progression, visualization of tumorigenesis and high-throughput chemical screening. In this review, we summarize the zebrafish models of blood diseases and their applications. These disease models not only help to improve our understanding of the pathophysiology of the blood system and the molecular mechanisms on pathogeneses of blood diseases, but also provide new ideas for the treatment of clinically relevant hematological malignancies.
Keywords:
PDF (703KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
郭佳妮, 刘帆, 王璐. 斑马鱼血液疾病模型及应用. 遗传[J], 2020, 42(8): 725-738 doi:10.16288/j.yczz.20-148
Jiani Guo.
自20世纪80年代美国俄勒冈大学George Streisinger首次使用斑马鱼(Danio rerio)作为模式生物以来[1],越来越多的实验室开始使用斑马鱼进行发育生物学及人类疾病的相关研究[2,3]。斑马鱼是一种淡水硬骨鱼,具有许多独特的生物学优势:体外受精与胚胎透明,便于观察与操作;繁殖能力强且生长迅速,有利于大规模筛选等[4]。此外,硬骨鱼类和哺乳动物拥有相当数量的同源基因,如82%的人类已知致病基因在斑马鱼中存在同源基因[5],且针对这些基因构建的斑马鱼突变体可作为人类相关疾病研究的动物模型[6,7]。目前,日渐成熟的基因编辑技术使得斑马鱼疾病模型的构建更加简便快捷。通过ZFN(zinc finger nucleases)、TALEN(transcription activator-like effector nuclease)以及CRISPR/Cas9(clustered regularly interspaced short palindromic repeats/CRISPR associated 9)技术进行基因编辑,科研人员获取斑马鱼特定基因缺失突变体,进行表型及功能的研究[8,9,10]。中国科研人员Sun等[11]应用CRISPR/Cas9技术对斑马鱼1号染色体基因进行敲除,获得了大量突变体,其中约有1/4突变体可以模拟人类相关疾病。基于自身的独特优势以及先进的技术方法,斑马鱼已逐渐成为研究胚胎发育与人类疾病发生机制的重要模型。
1 斑马鱼血液发育过程
血液系统是维持机体生命活动重要的系统之一,为机体提供氧气和营养物质,通过物质交换维持内环境的稳态,同时提供免疫防御与保护。血液系统包含红系细胞、髓系细胞以及淋系细胞等多种成熟血细胞,而这些血细胞均由造血过程产生。造血过程是造血干细胞(hematopoietic stem cells, HSCs)及各类血细胞产生、分化及发育成熟的过程,开始于胚胎发育早期并贯穿整个生命过程[12]。脊椎动物造血发育是高度保守的过程,从胚胎发育早期到成体,造血过程的阶段性是一致的,分为初级造血和次级造血两个阶段[13,14]。初级造血为早期胚胎发育提供必要的髓系细胞和红细胞。斑马鱼的初级造血发生在两个解剖位置—前侧板中胚层以及由后侧板中胚层发育形成的中间细胞团(intermediate cell mass, ICM),分别生成初级髓系细胞和初级红系细胞[4]。scl、lmo2、fli1、flk1和tif1γ等基因对初级造血与血管的发生至关重要[15,16,17];gata1和spi1(也被称为pu.1)分别促进红系和髓系的分化[18,19]。初级造血是一个瞬时过程,很快被次级造血过程替代。次级造血过程始于造血干细胞的产生,位于主动脉-性腺-中肾(aorta-gonad-mesenephros, AGM)区域背主动脉腹侧壁的生血内皮细胞,经过内皮造血转化过程生成HSCs[20,21]。随后HSCs迁移至尾部造血组织(caudal hematopoietic tissue, CHT)(相当于哺乳动物胎肝),经过短暂扩增之后,大部分HSCs迁移并定植于肾髓(相当于哺乳动物骨髓),一部分HSCs则迁移到胸腺分化为T淋巴前体细胞[22]。HSC命运决定的关键基因包括runx1、scl以及gata2等[23,24,25,26]。与初级造血相似,gata1和tif1γ驱动红细胞生成[15,18],spi1和c/ebp1促进髓系细胞分化[19,27]。此外,rag1、rag2、ikaros、lck、gata3、foxn1、irf4a和rac2等关键基因影响淋系细胞的分化[28,29,30,31,32,33]。
脊椎动物血液发育是维持机体正常生命活动的动态过程,受多种关键因子和信号通路的精细协同调控,多个造血组织和器官参与其中。在此过程中,任何环节的失调都可能引起血液系统发育或功能缺陷,最终导致血液疾病的发生。因此系统了解并深入探究血液发育的调控机理有助于解析疾病发生机制。
2 斑马鱼血液疾病模型及其应用
血液病是原发于血液系统,或影响造血系统伴发血液异常的一类疾病。血液病种类繁多,且多数为难治性或恶性疾患,近年来发病率有逐渐增高的趋势,其中白血病的发病率和死亡率更是居于我国恶性肿瘤前10位。因此,针对疾病临床需求深入开展基础研究,解析疾病发病机制具有重要意义。应用模式动物,针对关键致病因子构建疾病模型,对于人类疾病机制研究、治疗及药物评价至关重要。斑马鱼造血过程及调控机制与哺乳动物高度保守,结合体外受精、早期胚胎透明、丰富的转基因鱼系等独特优势,斑马鱼已成为造血发育以及人类疾病研究的常用模式动物之一。在斑马鱼体内模拟致病因子异常变化构建相关血液疾病模型,可用于肿瘤发生发展可视化研究及高通量化学筛选。本文系统总结了斑马鱼白血病模型和其他多种血液疾病模型的构建,及其在疾病研究中的应用。2.1 白血病模型
白血病是一类造血干祖细胞恶性克隆性疾病,按细胞类型和生长速度主要分为4类:急性淋巴细胞白血病(acute lymphoblastic leukemia, ALL)、慢性淋巴细胞白血病(chronic lymphocytic leukemia, CLL)、急性髓系白血病(acute myeloid leukemia, AML)和慢性髓系白血病(chronic myeloid leukemia, CML)。其中ALL可以分为急性T淋巴细胞白血病(acute T-lymphocytic leukemia, T-ALL)和急性B淋巴细胞白血病(acute B-lymphocytic leukemia, B-ALL)。2.1.1 T-ALL模型
T细胞恶性肿瘤是一类分子异质性疾病,由复杂的遗传变化所驱动[34]。淋巴细胞恶性转化的原因主要是异常的染色体易位和异常激活的信号通路[35,36]。在斑马鱼体内模拟这些改变可以构建T-ALL相关转基因斑马鱼系。
原癌基因MYC影响细胞生长和增殖,在人类癌症发生过程中发挥重要作用[37]。第一个T-ALL转基因斑马鱼模型是应用斑马鱼rag2启动子驱动EGFP-mMyc融合基因表达,借助该模型美国哈佛大学Thomas Look团队首次实时观察EGFP标记白血病细胞的发生发展过程[38]。然而,该模型的大多数后代在性成熟前便发展为晚期白血病,无法自然扩繁,需要通过体外受精进行繁殖,操作繁琐。为了解决这一问题,Thomas Look团队构建了条件性转基因鱼系Tg(rag2-loxP-dsRED2-loxP-EGFP-mMyc)。正常状态下该品系不发生白血病,Cre表达后才可能发生T-ALL,然而诱导率仅有13%[39]。为了突破低疾病外显率的限制,该团队引入热休克启动子hsp70驱动Cre表达。在受精后3天进行热休克处理,81%的转基因鱼发生T淋巴母细胞性淋巴瘤(T-lymphoblastic lymphoma, T-LBL),并快速进展为T-ALL,该模型大大提高了诱导T-ALL的效率[40]。近期,Jiang等[41]应用此疾病模型证明蛋白激酶Aurora B能够结合并磷酸化MYC,形成AURKB-MYC正反馈调控轴,加快T-ALL发生发展。
T-ALL病例中也常见到PTEN/PI3K/Akt通路发生遗传改变[42]。Thomas Look团队构建了诱导型T-ALL转基因斑马鱼模型,由4-羟基他莫昔芬诱导,激活原癌基因MYC导致疾病发生。撤除4-羟基他莫昔芬引起肿瘤消退,而斑马鱼pten基因功能缺失突变或持续激活型Akt2表达则可促进肿瘤继续发展。这些发现说明,即使MYC癌基因驱动的信号丢失,Akt通路激活也足以维持肿瘤生存[43]。T细胞前体向T-ALL转化的另一个关键基因是NOTCH1,约65%的T-ALL患者NOTCH1基因激活[44]。应用rag2启动子驱动notch1胞内段表达,成功构建了T-ALL模型。同时证明了Notch激活与抗凋亡分子Bcl2过表达相结合进一步加速了T-ALL发生,提示二者之间的协同作用[45]。
除了针对已知遗传学改变构建疾病模型,表型驱动的正向遗传筛选方法也得到广泛应用,美国犹他大学Nikolaus Trede团队应用N-乙基-N-亚硝基脲(N-ethyl-N-nitrosourea,ENU)化学诱变,筛选并鉴定了具有遗传倾向的3个T细胞恶性肿瘤鱼系—shrek(srk)、hulk(hlk)和oscar the grouch(otg)[46]。srk和hlk突变体作为家族性白血病和淋巴瘤的模型已被多次报道[47,48,49,50]。利用这些突变体进行重叠修饰因子筛选(superimposed modifier screens),识别影响疾病表型的协同基因。具有低外显率和长潜伏期的杂合突变体适合于筛选肿瘤发生促进因子;高外显率且早期发病的纯合突变体srk是筛选肿瘤抑制途径的最佳选择。此外,这些鱼系的肿瘤临床症状和分子特征与人类T-ALL和T-LBL相似,其中胸腺肿瘤症状类似于哺乳动物淋巴瘤模型和人类患者中的纵隔肿块[46]。从胸腺肿瘤、其他部位的播散性克隆或连续移植后的高度侵袭性肿瘤中分别纯化肿瘤细胞,比较分析疾病进展各个阶段的细胞特征,有助于揭示肿瘤转化的分子事件[46]。这些突变体作为T细胞恶性肿瘤的理想模型,为该疾病的研究提供了新的工具。
2.1.2 B-ALL模型
染色体易位t(12;21)是儿童B-ALL中最常见的易位形式,其导致的TEL-AML1(又称ETV6-RUNX1)融合基因存在于25%的患儿中[51,52]。然而,在小鼠(Mus musculus)中建立该融合基因B-ALL模型并不成功[53]。后续研究中应用不同的启动子,建立了泛表达或淋系祖细胞特异表达人TEL-AML1的多种斑马鱼模型,但其中只有少数斑马鱼发生了B-ALL[54]。近期研究发现,T-ALL模型Tg(rag2:hMYC)会同时发生B-ALL,借助淋系特异标记转基因品系Tg(lck:EGFP),发现T-ALL细胞呈现强绿色荧光,而B-ALL细胞则呈现较弱绿色荧光;此外,igic1s1与rag1基因分别表达于B-ALL与T-ALL,可以区分两种细胞簇。基于以上特征,T-ALL模型Tg(rag2:hMYC)可用于B-ALL疾病的研究[55]。
2.1.3 AML模型
染色体重排后产生的致癌融合基因可驱动急性髓系白血病的发生,如inv(8)(p11;q13)导致MYST3-NCOA2融合基因[56],t(8;21)q(21;22)导致AML1-ETO融合基因[57],以及t(7;11)(p15;p15)导致NUP98-HOXA9 (NHA9)融合基因[58]。在斑马鱼中表达常见的致癌融合基因可以构建AML疾病模型。
最早的AML模型是在斑马鱼胚胎中短暂表达人融合癌基因,然而这些模型均存在早期致死现象,无法在成体期进行研究[59,60]。第一个成功的非胚胎致死AML模型是用spi1启动子驱动MYST3-NCOA2融合基因表达,该模型中髓系前体细胞广泛侵袭斑马鱼肾脏,然而AML发病率低、潜伏期长[61]。在时间上控制致癌基因的表达可以有效解决胚胎死亡问题。在斑马鱼胚胎中热激处理诱导AML1-ETO融合基因表达可模拟人AML部分症状,机制研究表明AML1-ETO通过scl影响红-髓系祖细胞分化,促进粒细胞产生。同时用组蛋白去乙酰酶抑制剂Trichostatin A处理可以恢复scl和gata1的表达,AML1-ETO引起的粒细胞聚集也得以改善[62]。应用该模型开展的生物活性化合物筛选,结果显示环氧合酶-2(COX-2)选择性抑制剂—尼美舒利能拮抗AML1-ETO导致的异常造血分化[63]。而将热激活驱动与Cre/loxP系统结合,则可以实现在特定时间和空间位置诱导癌基因表达。应用转基因鱼系Tg(spi1-loxP-EGFP-loxP-NUP98-HOXA9)与Tg(hsp70:Cre)杂交,可以特异性地在髓系细胞中诱导致癌基因表达。胚胎期过表达NHA9可干扰早期造血发育,导致髓系前体占优势;而在成体期过表达NHA9可导致23%的转基因鱼在19~23个月时出现骨髓增殖性肿瘤(myeloproliferative neoplasms, MPN)[64]。利用此模型,加拿大戴尔豪斯大学Jason Berman团队发现DNA甲基转移酶抑制剂与组蛋白去乙酰化酶抑制剂结合可恢复NHA9过表达胚胎的正常血液发育[65],该发现揭示了NHA9与表观遗传调控之间的联系,展示了协同药物组合在NHA9诱导的髓系疾病中的治疗潜力。
除了染色体重排,基因表达水平的改变也与AML发生密切相关。癌基因MYCN(N-Myc)在AML患者体内过度表达,可作为AML不良预后的标志[66]。Shen等[67]构建了由热休克元件(heat shock elements, HSE)驱动小鼠n-Myc基因表达的AML模型。n-Myc通过上调scl与lmo2增强初级造血,并通过诱导spi1和mpo促进髓系细胞扩增,导致外周血髓系前体细胞积累。此外,细胞周期进程改变、糖代谢异常、MAPK/Ras及p53信号途径均参与n-Myc导致的血细胞恶性转化过程。该模型AML发病率高且潜伏期短,为研究MYCN致癌作用的分子调控网络建立了有力工具。
2.2其他血液肿瘤模型
2.2.1骨髓增殖性肿瘤模型骨髓增殖性肿瘤(MPN)起源于造血干细胞,表现为骨髓一系或多系血细胞过度增殖,包括真性红细胞增多症(polycythemia vera, PV)、原发性血小板增多症(essential thrombocythemia, ET)和原发性骨髓纤维化(primary myelofibrosis, PMF)[68]。JAK2、CALR、RAS、CBL等基因突变常见于骨髓增殖性肿瘤[69,70,71],其中最高频的突变是JAK2基因V617F的功能获得突变[72]。
斑马鱼jak2aV581F突变体可以模拟人JAK2V617F功能获得突变,表现为与人类PV高度相似的红细胞扩增[70]。在斑马鱼胚胎表达突变型人CALR基因会导致胚胎出现类似于ET患者的血小板生成增加表型[73]。然而,以上两种模型均是在斑马鱼胚胎瞬时过表达突变基因,并不能稳定遗传。
在内皮细胞诱导RAS突变基因表达,模拟MPN表型,表现为CHT区域红系、髓系祖细胞显著扩增,肾髓中血细胞分化受阻,以及外周血中红系、髓系祖细胞聚积[74]。斑马鱼irf8突变体呈现MPN类似表型,具体表现为髓系前体细胞扩增[75]。通过正向遗传筛选获得的LDD731鱼系,表现为红系、髓系细胞扩增,且该表型依赖Flt3信号途径[76]。这些模型模拟了人类MPN表型并初步阐释了其发病机制。
2.2.2骨髓增生异常综合征模型
骨髓增生异常综合征(myelodysplastic syndromes, MDS)是异质性造血干细胞疾病,表现为病态造血或无效造血,以及高风险向AML转化[77]。近年来大量基因组分析显示,MDS某些亚型与剪接体(spliceosome)或表观遗传因子的突变密切相关[78,79,80,81]。
剪接体突变是MDS发病的关键因素,约60%的MDS患者中会发生不同形式的剪接体突变[79]。剪接因子3B亚单位1(splicing factor 3B subunit 1, SF3B1)是MDS中最易突变的剪接体因子之一[79,81]。斑马鱼sf3b1功能缺失突变体的初级造血受损,髓系和红系细胞分化与增殖障碍,造血干祖细胞产生减少[82],该表型与MDS患者症状类似[82]。正向遗传筛选得到的cephalophonus(cph)突变体具有HSC产生正常、而髓系和红系细胞发育缺陷的表型,图位克隆分析鉴定出剪接因子prpf8是其突变基因[83]。上述剪接因子突变体具有类似表型,但也有各自独特的表现,与临床观察一致,即不同剪接因子突变的患者有共同的疾病特点,也有个体化的特征,斑马鱼模型的使用将有助于个体化精准医疗的实施。
TET2功能缺失突变常见于髓系恶性肿瘤患者(约30%的MDS以及约10%的再发AML病例)[84]。Gjini等[85]研究表明,纯合tet2突变体的胚胎期造血正常,但随着年龄的增长会出现进行性克隆性骨髓增生异常、贫血和髓系祖细胞扩增;受精后24个月,它们呈现出更严重的MDS表型,如外周血红细胞异常增生。
原癌基因c-myb是造血细胞增殖和分化的重要调控因子,其异常表达通常与多种血液疾病相关[86,87,88]。转基因斑马鱼Tg(c-myb:GFP)中c-myb基因过度表达,髓系细胞显著扩增,类似于人类MDS表型,且部分成鱼会发展为AML和ALL。这是由于该模型中过度表达的c-myb基因影响细胞周期相关基因的表达,导致造血前体细胞过度增殖。c-myb靶向药物flavopiridol可缓解c-mybhyper胚胎和成鱼的MDS样症状。该模型可用于MDS发生分子机制的探究以及治疗药物的筛选[89]。
2.3 斑马鱼血液疾病模型的应用
2.3.1移植评价实验应用斑马鱼进行血液肿瘤细胞移植可以用来定义和量化白血病增殖细胞(leukemia-propagating cells,LPCs)以及探究其启动白血病的潜能[90]。斑马鱼的移植评价实验具有诸多优势:使用受精后4周内的斑马鱼胚胎进行移植不需要免疫抑制,因为该时期斑马鱼缺乏成熟的适应性免疫系统[91];透明的胚胎或成体鱼Casper与多种荧光转基因品系相结合,促进了活体成像技术在肿瘤发生发展研究中的应用[92]。
首次移植实验是应用T-ALL模型Tg(rag2-EGFP-mMyc),将EGFP标记的白血病细胞移植到辐射后野生型成年斑马鱼腹腔内,这些细胞在腹腔注射后14天开始扩散,14~26天归巢于胸腺[38]。连续移植后诱发疾病的能力是肿瘤细胞自我更新的标志。T-ALL细胞连续移植揭示了大部分T-ALL细胞具有启动白血病的潜能。此外,大规模的单细胞移植实验则证实了这一启动潜能呈现差异性,即0.1%~15.9%甚至更少的白血病细胞有能力重建肿瘤[90]。
复发T-ALL患者中经常发生克隆进化,导致肿瘤更具侵袭性,这一现象与LPCs的遗传多样性及其增强的白血病启动潜能相关[93,94]。研究表明,Notch信号能促进T-ALL癌前T细胞克隆扩增,提高积累必要突变并完全转化为LPCs的可能性和速度[95]。大规模细胞移植筛选实验显示单个克隆之间存在功能变异,少数克隆随着时间的推移提高了生长速度和扩增潜力。Blackburn等[96]研究证明,克隆进化激活Akt信号通路促进T-ALL的生长,同时可促使肿瘤细胞对地塞米松产生耐药性。以上研究提示,克隆进化促使T-ALL对化疗产生耐药性,而且这可能发生于药物暴露之前。
斑马鱼移植受体的免疫系统可被地塞米松或γ射线短暂抑制,却无法进行长期移植;且仅可用于同源移植,限制了移植模型的广泛应用[97]。为了解决这些问题,美国哈佛大学David Langenau团队构建了免疫缺陷斑马鱼模型—rag2E450fs突变体。该突变体中功能性T细胞和B细胞数量减少,但仍能存活和繁殖,并且可进行多种组织与癌细胞长期稳定移植。但是该模型纯合子斑马鱼不能繁殖,以及个体间B细胞缺陷差异极大,可能影响植入潜能[98]。免疫缺陷斑马鱼模型jak3P369fs突变体和prkdcD3612fs突变体,分别导致T细胞与NK细胞、成熟T细胞和B细胞的缺失,两种突变体均具有植入能力,但只有prkdc纯合突变体可以繁殖,并且在细胞移植后存活[97]。斑马鱼prkdc-/-,il2rg-/-双突变体中T细胞、B细胞和NK细胞缺陷,David Langenau团队应用该免疫缺陷突变体建立并评估了肿瘤移植模型,能够重现多种病人来源的肿瘤生长迁移等情况,并可在单细胞水平进行活体实时研究[99]。
2.3.2化学药物筛选
斑马鱼模型是进行高通量药物筛选的理想选择,主要基于两个优点:(1)整体动物模型,可以针对特定生物学事件发现活性化合物和药物靶标;(2)全面评估化合物的活性和副作用,排除具有明显毒副作用的化合物,缩短药物研发周期[100,101,102]。
研究表明前列腺素E2(prostaglandin E2, PGE2)的代谢活性衍生物二甲基前列腺素E2(16,16-dimethyl-PGE2, dmPGE2)可以增加HSCs数量,同时也可促进辐射损伤成体鱼中肾髓的恢复[103]。此外,dmPGE2可提升小鼠骨髓HSCs移植重建能力[103]。临床前分析显示,dmPGE2能显著促进体外人造血细胞集落形成,提高异种移植后人脐血干细胞(human cord blood, hCB)的植入效率[104]。人类脐带血移植的临床研究显示,经dmPGE2处理的脐带血细胞具有持久、多系重建潜能,且安全性高,移植后患者中性粒细胞的恢复速度也大大加快[105]。美国犹他大学Nikolaus Trede团队应用淋系转基因斑马鱼进行药物筛选,确定小分子化合物Lenaldekar(LDK)可以有效消除不成熟的T细胞而不影响正常细胞的细胞周期,且可延长大部分T-ALL成鱼的生存期[106]。在小鼠模型中,LDK也表现出了减缓疾病进展的功效。同时对于直接取自临床原发性白血病(包括难治性B-ALL和慢性粒细胞白血病)患者的样本,LDK可以杀死其中大部分的白血病细胞[106]。这项工作证明了使用斑马鱼筛选抗肿瘤药物的实用性,同时LDK的发现也为白血病靶向治疗提供了新方向[106]。
接受异种移植的斑马鱼也是药物筛选的一个重要模型。将人类白血病细胞移植到斑马鱼胚胎中用于筛选非致畸的白血病治疗药物,结果显示伊马替尼和奥沙福林可消除白血病细胞,且对受体胚胎无毒性;而全反式维甲酸和4EGI-1表现出致畸作用,不能作为抗白血病药物[107]。
3 结语与展望
斑马鱼血液疾病模型的应用为人们了解疾病的病理生理学、基因型与表型的相关性以及探索治疗方案提供了新的途径。应用正反向遗传学方法获得的斑马鱼血液疾病模型,可用于化学筛选靶向治疗药物,或进行异体移植研究白血病增殖细胞的功能等(图1)。然而斑马鱼模型的应用通常缺乏对肿瘤发病的时空控制,这限制了其在肿瘤进展和转移研究中的应用。最近一项研究开发了一种成年斑马鱼转基因电穿孔(transgene electroporation in adult zebrafish, TEAZ)技术,可以在成鱼体内定时定点导入特定的DNA,如原癌基因、CRISPR/Cas9基因编辑组件以及特定启动子驱动荧光蛋白的DNA载体[108],可以实现在特定组织和发育时期引入遗传改变。斑马鱼模型应用的另一个限制是缺乏可靠的细胞表面标记及抗体,目前大多数造血细胞分选都是基于转基因标记,极大地限制了特定谱系亚群的分选[109]。此外,人们对斑马鱼造血微环境的了解仍然很少,造血细胞之间的相互作用、与微环境细胞的相互作用及其与血液疾病的关系值得进一步探究。图1
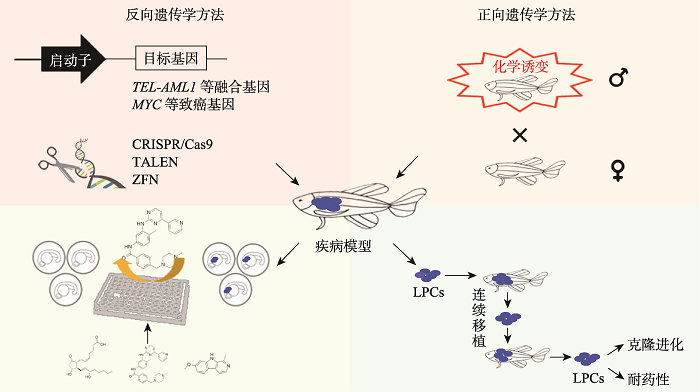 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1斑马鱼血液疾病模型构建与应用
应用正反向遗传学方法构建斑马鱼血液疾病模型,可用于化学药物筛选或肿瘤细胞移植评价研究。
Fig. 1Generation and application of zebrafish blood disease models
异种移植模型极大地加深了人们对白血病发生和干细胞生物学的理解。患者来源的异种移植物保持了人类癌症固有的克隆异质性,提供了优于体外系统的微环境,这对药物的开发与临床转化有重要意义[110,111]。虽然斑马鱼异种移植平台已初步建成[99,112,113],但是人类造血干祖细胞在斑马鱼体内仅能短暂地存活[114]。最近,一种人源化斑马鱼模型可以表达多种人类造血特异性的细胞因子,从而促进了受体中造血干祖细胞的存活、自我更新和多向分化,且移植白血病细胞表现出向造血组织的归巢,更准确地模拟了人类白血病的行为[115]。除了血液系统肿瘤,在其他人源肿瘤的移植方面斑马鱼也显示出独特的优势:活体成像可以对肿瘤生长、转移、血管生成以及肿瘤起始细胞进行动态分析,斑马鱼也为高通量筛选抗癌药物提供了一个经济有效的平台[99,116,117]。
疾病模型的应用有助于探索个体化精准医疗。最近,研究人员利用斑马鱼拯救了一位患有淋巴管疾病的12岁男孩的生命。全外显子测序发现该患儿X染色体上ARAF基因发生错义突变,过表达携带这种突变的人源基因导致斑马鱼淋巴管的过度生长,化学筛选证明MEK抑制剂对该表型有回救作用;这不仅明确了ARAF基因突变导致该患儿发生疾病,也寻找到了有效的治疗药物[118]。这是通过斑马鱼实现精准医疗的成功案例,但是由于生命活动的关联性与复杂性,基因的改变往往会造成级联反应,如何将斑马鱼的研究成果过渡到人类医疗中仍然需要持续的探索。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/291293a0URLPMID:7248006 [本文引用: 1]

Homozygous diploid zebra fish have been produced on a large scale by the application of simple physical treatments. Clones of homozygous fish have been produced from individual homozygotes. These clones and associated genetic methods will facilitate genetic analyses of this vertebrate.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:27018965 [本文引用: 2]
DOI:10.1038/nature12111URLPMID:23594743 [本文引用: 1]
Zebrafish have become a popular organism for the study of vertebrate gene function. The virtually transparent embryos of this species, and the ability to accelerate genetic studies by gene knockdown or overexpression, have led to the widespread use of zebrafish in the detailed investigation of vertebrate gene function and increasingly, the study of human genetic disease. However, for effective modelling of human genetic disease it is important to understand the extent to which zebrafish genes and gene structures are related to orthologous human genes. To examine this, we generated a high-quality sequence assembly of the zebrafish genome, made up of an overlapping set of completely sequenced large-insert clones that were ordered and oriented using a high-resolution high-density meiotic map. Detailed automatic and manual annotation provides evidence of more than 26,000 protein-coding genes, the largest gene set of any vertebrate so far sequenced. Comparison to the human reference genome shows that approximately 70% of human genes have at least one obvious zebrafish orthologue. In addition, the high quality of this genome assembly provides a clearer understanding of key genomic features such as a unique repeat content, a scarcity of pseudogenes, an enrichment of zebrafish-specific genes on chromosome 4 and chromosomal regions that influence sex determination.
DOI:10.1038/3041URLPMID:9806541 [本文引用: 1]
Defects in the enzymes involved in the haem biosynthetic pathway can lead to a group of human diseases known as the porphyrias. yquem (yqe(tp61)) is a zebrafish mutant with a photosensitive porphyria syndrome. Here we show that the porphyric phenotype is due to an inherited homozygous mutation in the gene encoding uroporphyrinogen decarboxylase (UROD); a homozygous deficiency of this enzyme causes hepatoerythropoietic porphyria (HEP) in humans. The zebrafish mutant represents the first genetically 'accurate' animal model of HEP, and should be useful for studying the pathogenesis of UROD deficiency and evaluating gene therapy vectors. We rescued the mutant phenotype by transient and germline expression of the wild-type allele.
DOI:10.1038/3049URLPMID:9806542 [本文引用: 1]
Many human anaemias are caused by defects in haemoglobin synthesis. The zebrafish mutant sauternes (sau) has a microcytic, hypochromic anaemia, suggesting that haemoglobin production is perturbed. During embryogenesis, sau mutants have delayed erythroid maturation and abnormal globin gene expression. Using positional cloning techniques, we show that sau encodes the erythroid-specific isoform of delta-aminolevulinate synthase (ALAS2; also known as ALAS-E), the enzyme required for the first step in haem biosynthesis. As mutations in ALAS2 cause congenital sideroblastic anaemia (CSA) in humans, sau represents the first animal model of this disease.
DOI:10.1016/j.tig.2016.10.005URLPMID:27836208 [本文引用: 1]
Geneticists have long sought the ability to manipulate vertebrate genomes by directly altering the information encoded in specific genes. The recently discovered clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 endonuclease has the ability to bind single loci within vertebrate genomes and generate double-strand breaks (DSBs) at those sites. These DSBs induce an endogenous DSB repair response that results in small insertions or deletions at the targeted site. Alternatively, a template can be supplied, in which case homology-directed repair results in the generation of engineered alleles at the break site. These changes alter the function of the targeted gene facilitating the analysis of gene function. This tool has been widely adopted in the zebrafish model; we discuss the development of this system in the zebrafish and how it can be manipulated to facilitate genome engineering.
URLPMID:28364234 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:18295580 [本文引用: 1]
DOI:10.1016/j.stem.2016.05.016URLPMID:27257760 [本文引用: 1]
Cell engineering has brought us tantalizingly close to the goal of deriving patient-specific hematopoietic stem cells (HSCs). While directed differentiation and transcription factor-mediated conversion strategies have generated progenitor cells with multilineage potential, to date, therapy-grade engineered HSCs remain elusive due to insufficient long-term self-renewal and inadequate differentiated progeny functionality. A cross-species approach involving zebrafish and mammalian systems offers complementary methodologies to improve understanding of native HSCs. Here, we discuss the role of conserved developmental timing processes in vertebrate hematopoiesis, highlighting how identification and manipulation of stage-specific factors that specify HSC developmental state must be harnessed to engineer HSCs for therapy.
URLPMID:23715539 [本文引用: 1]
DOI:10.1371/journal.pbio.0020237URLPMID:15314655 [本文引用: 2]
Hematopoiesis is precisely orchestrated by lineage-specific DNA-binding proteins that regulate transcription in concert with coactivators and corepressors. Mutations in the zebrafish moonshine (mon) gene specifically disrupt both embryonic and adult hematopoiesis, resulting in severe red blood cell aplasia. We report that mon encodes the zebrafish ortholog of mammalian transcriptional intermediary factor 1gamma (TIF1gamma) (or TRIM33), a member of the TIF1 family of coactivators and corepressors. During development, hematopoietic progenitor cells in mon mutants fail to express normal levels of hematopoietic transcription factors, including gata1, and undergo apoptosis. Three different mon mutant alleles each encode premature stop codons, and enforced expression of wild-type tif1gamma mRNA rescues embryonic hematopoiesis in homozygous mon mutants. Surprisingly, a high level of zygotic tif1gamma mRNA expression delineates ventral mesoderm during hematopoietic stem cell and progenitor formation prior to gata1 expression. Transplantation studies reveal that tif1gamma functions in a cell-autonomous manner during the differentiation of erythroid precursors. Studies in murine erythroid cell lines demonstrate that Tif1gamma protein is localized within novel nuclear foci, and expression decreases during erythroid cell maturation. Our results establish a major role for this transcriptional intermediary factor in the differentiation of hematopoietic cells in vertebrates.
DOI:10.1101/gad.12.5.621URLPMID:9499398 [本文引用: 1]
SCL/Tal-1 is a transcription factor necessary for hematopoietic stem cell differentiation. Although SCL is also expressed in endothelial and neural progenitors, SCL function in these cells remains unknown. In the zebrafish mutant cloche (clo), SCL expression is nearly abolished in hematopoietic and vascular tissues. Correspondingly, it was shown previously that clo fails to differentiate blood and angioblasts. Genetic analysis demonstrates that the clo mutation is not linked to the SCL locus. Forced expression of SCL in clo embryos rescues the blood and vascular defects, suggesting that SCL acts downstream of clo to specify hematopoietic and vascular differentiation.
DOI:10.1006/dbio.1998.8887URLPMID:9630750 [本文引用: 1]
In vertebrates, hematopoietic and vascular progenitors develop from ventral mesoderm. The first primitive wave of hematopoiesis yields embryonic red blood cells, whereas progenitor cells of subsequent definitive waves form all hematopoietic cell lineages. In this report we examine the development of hematopoietic and vasculogenic cells in normal zebrafish and characterize defects in cloche and spadetail mutant embryos. The zebrafish homologs of lmo2, c-myb, fli1, flk1, and flt4 have been cloned and characterized in this study. Expression of these genes identifies embryonic regions that contain hematopoietic and vascular progenitor cells. The expression of c-myb also identifies definitive hematopoietic cells in the ventral wall of the dorsal aorta. Analysis of b316 mutant embryos that carry a deletion of the c-myb gene demonstrates that c-myb is not required for primitive erythropoiesis in zebrafish even though it is expressed in these cells. Both cloche and spadetail mutant embryos have defects in primitive hematopoiesis and definitive hematopoiesis. The cloche mutants also have significant decreases in vascular gene expression, whereas spadetail mutants expressed normal levels of these genes. These studies demonstrate that the molecular mechanisms that regulate hematopoiesis and vasculogenesis have been conserved throughout vertebrate evolution and the clo and spt genes are key regulators of these programs.
DOI:10.1182/blood.v99.9.3089URLPMID:11964270 [本文引用: 2]
URLPMID:12051816 [本文引用: 2]
URLPMID:20154732 [本文引用: 1]
DOI:10.1038/nature08738URLPMID:20154733 [本文引用: 1]
A major goal of regenerative medicine is to instruct formation of multipotent, tissue-specific stem cells from induced pluripotent stem cells (iPSCs) for cell replacement therapies. Generation of haematopoietic stem cells (HSCs) from iPSCs or embryonic stem cells (ESCs) is not currently possible, however, necessitating a better understanding of how HSCs normally arise during embryonic development. We previously showed that haematopoiesis occurs through four distinct waves during zebrafish development, with HSCs arising in the final wave in close association with the dorsal aorta. Recent reports have suggested that murine HSCs derive from haemogenic endothelial cells (ECs) lining the aortic floor. Additional in vitro studies have similarly indicated that the haematopoietic progeny of ESCs arise through intermediates with endothelial potential. Here we have used the unique strengths of the zebrafish embryo to image directly the generation of HSCs from the ventral wall of the dorsal aorta. Using combinations of fluorescent reporter transgenes, confocal time-lapse microscopy and flow cytometry, we have identified and isolated the stepwise intermediates as aortic haemogenic endothelium transitions to nascent HSCs. Finally, using a permanent lineage tracing strategy, we demonstrate that the HSCs generated from haemogenic endothelium are the lineal founders of the adult haematopoietic system.
DOI:10.1016/j.bcmd.2013.07.009URLPMID:23916372 [本文引用: 1]
The zebrafish has become a commonly used model for studying hematopoiesis as a result of its unique attributes. Zebrafish are highly suitable for large-scale genetic and chemical screens compared to other vertebrate systems. It is now possible to analyze hematopoietic lineages in zebrafish and validate cell function via transplantation assays. Here, we review advancements over the past decade in forward genetic screens, chemical screens, fluorescence-activated cell sorting analysis, and transplantation assays. Integrating these approaches enables new chemical and genetic screens that assay cell function within the hematopoietic system. Studies in zebrafish will continue to contribute and expand our knowledge about hematopoiesis, and develop novel treatments for clinical applications.
DOI:10.1016/s0301-472x(02)00955-4URLPMID:12482499 [本文引用: 1]
OBJECTIVE: The AML/RUNX family of transcription factors plays important roles in hematopoiesis, neurogenesis, bone development, and segmentation in vertebrate embryos. The aim of this study was to isolate runt-related genes in a genetically and embryologically exploitable system, the zebrafish, and characterize their function during hematopoietic development. MATERIALS AND METHODS: Two runt-related genes were isolated by degenerate PCR and standard library screening, and a radiation hybrid panel, T51 RH, was used to resolve their chromosomal localization. In situ hybridization demonstrated their expression whereas their transcriptional activity was assessed using an AML1-responsive reporter gene in the MLA 144 T-cell line. RESULTS: We isolated the zebrafish runxa and runxb cDNAs, which encode proteins highly homologous to the human and murine Runx1 (AML1) and Runx3 (AML2) proteins. In contrast to a recent report, we detected runxa expression in both hematopoietic and neural tissues of the developing zebrafish. runxa transcripts first appear during segmentation in bilateral mesodermal cells that coexpress one of the earliest blood and endothelial cell markers, scl/tal-1. By 24 hours postfertilization (hpf), runxa transcripts are seen in the ventral wall of the dorsal aorta. Hematopoietic runxa expression is lost in cloche mutants, which are defective in blood and endothelial cell formation. runxb transcripts are seen in nonhematopoietic domains. Both Runxa and Runxb transactivate an AML1-responsive human promoter in hematopoietic cells. Genomic localization studies demonstrate that runxa is located on linkage group 1 (LG1), and the runxb gene is located on LG13. CONCLUSIONS: Our gene expression analysis strongly suggests that both the functional and spatial aorta-gonad-mesonephros (AGM) region has been conserved throughout evolution. Our runxa spatiotemporal expression data shed light on the role of vertebrate Runx1/AML1 in primitive vs definitive hematopoietic development.
DOI:10.1182/blood-2009-09-244640URLPMID:20185582 [本文引用: 1]
Recent lineage studies suggest that hematopoietic stem cells (HSCs) may be derived from endothelial cells. However, the genetic hierarchy governing the emergence of HSCs remains elusive. We report here that zebrafish ets1-related protein (etsrp), which is essential for vascular endothelial development, also plays a critical role in the initiation of definitive hematopoiesis by controlling the expression of 2 stem cell leukemia (scl) isoforms (scl-alpha and scl-beta) in angioblasts. In etsrp morphants, which are deficient in endothelial and HSC development, scl-alpha alone partially rescues angioblast specification, arterial-venous differentiation, and the expression of HSC markers, runx1 and c-myb, whereas scl-beta requires angioblast rescue by fli1a to restore runx1 expression. Interestingly, when vascular endothelial growth factor (Vegf) signaling is inhibited, HSC marker expression can still be restored by scl-alpha in etsrp morphants, whereas the rescue of arterial ephrinb2a expression is blocked. Furthermore, both scl isoforms partially rescue runx1 but not ephrinb2a expression in embryos deficient in Vegf signaling. Our data suggest that downstream of etsrp, scl-alpha and fli1a specify the angioblasts, whereas scl-beta further initiates HSC specification from this angioblast population, and that Vegf signaling acts upstream of scl-beta during definitive hematopoiesis.
DOI:10.1242/dev.097071URLPMID:24046317 [本文引用: 1]
Recent studies have shown that nascent hematopoietic stem cells (HSCs) derive directly from the ventral aortic endothelium (VAE) via endothelial to hematopoietic transition (EHT). However, whether EHT initiates from a random or predetermined subpopulation of VAE, as well as the molecular mechanism underlying this process, remain unclear. We previously reported that different zebrafish stem cell leukemia (scl) isoforms are differentially required for HSC formation in the ventral wall of the dorsal aorta. However, the exact stage at which these isoforms impact HSC development was not defined. Here, using in vivo time-lapse imaging of scl isoform-specific reporter transgenic zebrafish lines, we show that prior to EHT scl-beta is selectively expressed in hemogenic endothelial cells, a unique subset of VAE cells possessing hemogenic potential, whereas scl-alpha is expressed later in nascent HSCs as they egress from VAE cells. In accordance with their expression, loss-of-function studies coupled with in vivo imaging analysis reveal that scl-beta acts earlier to specify hemogenic endothelium, which is later transformed by runx1 into HSCs. Our results also reveal a previously unexpected role of scl-alpha in maintaining newly born HSCs in the aorta-gonads-mesonephros. Thus, our data suggest that a defined hemogenic endothelial population preset by scl-beta supports the deterministic emergence of HSCs, and unravel the cellular mechanisms by which scl isoforms regulate HSC development.
DOI:10.1084/jem.20130751URLPMID:24297996 [本文引用: 1]
Knowledge of the key transcription factors that drive hematopoietic stem cell (HSC) generation is of particular importance for current hematopoietic regenerative approaches and reprogramming strategies. Whereas GATA2 has long been implicated as a hematopoietic transcription factor and its dysregulated expression is associated with human immunodeficiency syndromes and vascular integrity, it is as yet unknown how GATA2 functions in the generation of HSCs. HSCs are generated from endothelial cells of the major embryonic vasculature (aorta, vitelline, and umbilical arteries) and are found in intra-aortic hematopoietic clusters. In this study, we find that GATA2 function is essential for the generation of HSCs during the stage of endothelial-to-hematopoietic cell transition. Specific deletion of Gata2 in Vec (Vascular Endothelial Cadherin)-expressing endothelial cells results in a deficiency of long-term repopulating HSCs and intra-aortic cluster cells. By specific deletion of Gata2 in Vav-expressing hematopoietic cells (after HSC generation), we further show that GATA2 is essential for HSC survival. This is in contrast to the known activity of the RUNX1 transcription factor, which functions only in the generation of HSCs, and highlights the unique requirement for GATA2 function in HSCs throughout all developmental stages.
URLPMID:11313249 [本文引用: 1]
DOI:10.1007/s002510050221URLPMID:9089097 [本文引用: 1]
The closely linked recombination activating genes, rag1 and rag2, encode components of the recombinase involved in V(D)J recombination of the immunoglobulin and T-cell receptor genes. These genes are expressed together exclusively in immature lymphocytes and are useful markers for following the development of lymphoid tissues. We cloned the rag locus of the zebrafish Danio rerio and sequenced the open reading frames of the rag1 and rag2 genes. Although the gene organization is similar to that in other species, the rag1 gene is unusual in possessing two introns within the coding region. In another teleost, the rainbow trout, the rag1 gene is interrupted by a single intron. Introns are not present in the rag1 gene of any other species examined to date. Expression of both rag1 and rag2 begins late in embryonic development, on day 4, by northern RNA blot analysis. Expression of rag1 was detected in the adult zebrafish thymus, pronephros, mesonephros, and ovary. This pattern of expression is consistent with previous histological studies of adult teleosts, which implicate the kidney as the major site of hematopoiesis and the thymus as the major lymphocyte-containing organ.
DOI:10.1002/dvdy.1223URLPMID:11748838 [本文引用: 1]
The Ikaros gene encodes a transcription factor that, in mice, has been shown to be essential for the correct differentiation of B and T lymphocytes and is expressed in all cells of the lymphoid lineage, including pluripotent hematopoietic stem cells. During development in zebrafish, Ikaros expression begins in lateral mesoderm, and continues in the intermediate cell mass (ICM), which is derived from lateral mesoderm and has been shown to generate primitive hematopoietic precursors. Cells expressing Ikaros were then seen on the ventral side of the dorsal aorta, known to be a location of definitive hematopoietic precursors in birds and mammals. Ikaros-expressing cells were also found in the pharyngeal region, near the forming thymus. Later, such cells were seen in the pronephros, the site of hematopoiesis in adult fish. The timing of appearance of Ikaros-expressing cells suggests that, similar to other vertebrates, lymphocytes in the thymus arise from hematopoietic tissue located near the dorsal aorta or in the ICM.
DOI:10.1073/pnas.0402248101URLPMID:15123839 [本文引用: 1]
Transgenic zebrafish that express GFP under control of the T cell-specific tyrosine kinase (lck) promoter were used to analyze critical aspects of the immune system, including patterns of T cell development and T cell homing after transplant. GFP-labeled T cells could be ablated in larvae by either irradiation or dexamethasone added to the water, illustrating that T cells have evolutionarily conserved responses to chemical and radiation ablation. In transplant experiments, thymocytes from lck-GFP fish repopulated the thymus of irradiated wild-type fish only transiently, suggesting that the thymus contains only short-term thymic repopulating cells. By contrast, whole kidney marrow permanently reconstituted the T lymphoid compartment of irradiated wild-type fish, suggesting that long-term thymic repopulating cells reside in the kidney.
URLPMID:23213226 [本文引用: 1]
DOI:10.1016/j.devcel.2015.07.011URLPMID:26300447 [本文引用: 1]
T lymphoid-primed progenitors are hematopoietic progenitors destined to enter the thymus. The in vivo characterization of these embryonic progenitors is challenging, however, due to the intrauterine development of mouse embryos. Thus, how the fate of these cells is determined has not been fully defined in mammals. Here we use zebrafish embryos to show that the homing of T lymphoid-primed progenitors to the thymus is impaired, concomitant with a decrease in ccr9a expression, in the absence of irf4a. Strikingly, fate mapping assays at the single-cell level showed a fate change of irf4a-deficient T lymphoid-primed progenitors to myeloid cells, accompanied by an increase in Pu.1 expression. These data indicate that in addition to regulating ccr9a expression, Irf4a is essential in T lymphoid-primed progenitors for repressing Pu.1 expression to prevent an alternate fate. Our findings provide insight into the fate determination mechanism of T lymphoid-primed progenitors.
DOI:10.4049/jimmunol.1901494URLPMID:32198141 [本文引用: 1]
The caudal hematopoietic tissue in zebrafish, the equivalent to the fetal liver in mammals, is an intermediate hematopoietic niche for the maintenance and differentiation of hematopoietic stem and progenitor cells before homing to the thymus and kidney marrow. As one of the ultimate hematopoietic organs, the thymus sustains T lymphopoiesis, which is essential for adaptive immune system. However, the mechanism of prethymic T lymphoid progenitors migrating to the thymus remains elusive. In this study, we identify an Rho GTPase Rac2 as a modulator of T lymphoid progenitor homing to the thymus in zebrafish. rac2-Deficient embryos show the inability of T lymphoid progenitors homing to the thymus because of defective cell-autonomous motility. Mechanistically, we demonstrate that Rac2 regulates homing of T lymphoid progenitor through Pak1-mediated AKT pathway. Taken together, our work reveals an important function of Rac2 in directing T lymphoid progenitor migration to the thymus during zebrafish embryogenesis.
DOI:10.1016/s1535-6108(02)00018-1URLPMID:12086890 [本文引用: 1]
Human T cell leukemias can arise from oncogenes activated by specific chromosomal translocations involving the T cell receptor genes. Here we show that five different T cell oncogenes (HOX11, TAL1, LYL1, LMO1, and LMO2) are often aberrantly expressed in the absence of chromosomal abnormalities. Using oligonucleotide microarrays, we identified several gene expression signatures that were indicative of leukemic arrest at specific stages of normal thymocyte development: LYL1+ signature (pro-T), HOX11+ (early cortical thymocyte), and TAL1+ (late cortical thymocyte). Hierarchical clustering analysis of gene expression signatures grouped samples according to their shared oncogenic pathways and identified HOX11L2 activation as a novel event in T cell leukemogenesis. These findings have clinical importance, since HOX11 activation is significantly associated with a favorable prognosis, while expression of TAL1, LYL1, or, surprisingly, HOX11L2 confers a much worse response to treatment. Our results illustrate the power of gene expression profiles to elucidate transformation pathways relevant to human leukemia.
URLPMID:16155013 [本文引用: 1]
URLPMID:15071128 [本文引用: 1]
DOI:10.1016/s0003-9861(03)00294-7URLPMID:12893289 [本文引用: 1]
The proto-oncogene c-MYC is implicated in various physiological processes-cell growth, proliferation, loss of differentiation, and cell death (apoptosis). Oncogenic c-MYC implies constitutive or deregulated expression of c-MYC and is associated with many human cancers often with poor prognosis. Recently, c-MYC has been implicated in the loss and dysfunction of insulin-producing beta cells in diabetes. Intriguingly, this raises the possibility that c-Myc may be a key contributor to disease, not only by deregulating cell proliferation, which is well established, but also by virtue of its opposing role in engendering apoptosis. However, given the fact that human diseases at diagnosis are generally advanced and pathologically complex, it is generally difficult to attribute a specific pathogenic role to c-MYC, or indeed any given single factor, or to assess the potential of therapies targeting individual such factors. Regulatable transgenic mouse models have shed light on these issues, have influenced our thinking about cancer, and have provided encouragement for the future development of cancer therapies based on targeting individual oncogenes such as c-MYC. Although still in its infancy, encouraging results have been reported for several approaches using gene targeting to interfere with c-MYC expression or activity both in vitro and in vivo.
DOI:10.1126/science.1080280URLPMID:12574629 [本文引用: 2]
The zebrafish is an attractive model organism for studying cancer development because of its genetic accessibility. Here we describe the induction of clonally derived T cell acute lymphoblastic leukemia in transgenic zebrafish expressing mouse c-myc under control of the zebrafish Rag2 promoter. Visualization of leukemic cells expressing a chimeric transgene encoding Myc fused to green fluorescent protein (GFP) revealed that leukemias arose in the thymus, spread locally into gill arches and retro-orbital soft tissue, and then disseminated into skeletal muscle and abdominal organs. Leukemic cells homed back to the thymus in irradiated fish transplanted with GFP-labeled leukemic lymphoblasts. This transgenic model provides a platform for drug screens and for genetic screens aimed at identifying mutations that suppress or enhance c-myc- induced carcinogenesis.
DOI:10.1073/pnas.0408708102URLPMID:15827121 [本文引用: 1]
We have created a stable transgenic rag2-EGFP-mMyc zebrafish line that develops GFP-labeled T cell acute lymphoblastic leukemia (T-ALL), allowing visualization of the onset and spread of this disease. Here, we show that leukemias from this transgenic line are highly penetrant and render animals moribund by 80.7 +/- 17.6 days of life (+/-1 SD, range = 50-158 days). These T cell leukemias are clonally aneuploid, can be transplanted into irradiated recipient fish, and express the zebrafish orthologues of the human T-ALL oncogenes tal1/scl and lmo2, thus providing an animal model for the most prevalent molecular subgroup of human T-ALL. Because T-ALL develops very rapidly in rag2-EGFP-mMyc transgenic fish (in which
DOI:10.1111/j.1365-2141.2007.06625.xURLPMID:17593023 [本文引用: 1]
The zebrafish is an ideal vertebrate model system to investigate the complex genetic basis of cancer because it has the capacity for in vivo tumour-cell imaging and forward genetic screens, and the molecular mechanisms regulating malignancy are remarkably conserved when compared with human. Our laboratory has previously generated transgenic zebrafish models that overexpress the mouse c-Myc gene fused to enhanced green fluorescent protein (EGFP) and develop T-cell acute lymphoblastic leukaemia (T-ALL) that recapitulates the human disease both molecularly and pathologically. Our previous models have been limited by disease onset prior to sexual maturity and by the low disease penetrance when conditional transgenic embryos are injected with Cre RNA. Here, we report a novel system in which compound transgenic fish expressed both Cre controlled by the heat-shock promoter and a rag2-promoter-regulated lox-dsRED2-lox-EGFP-mMyc cassette rag2-LDL-EMyc in developing T cells. After heat-shock treatment at 3 d postfertilisation (dpf) for 45 min at 37 degrees C, 81% of compound transgenic fish developed T-lymphoblastic lymphoma (T-LBL, mean latency 120 +/- 43 (standard deviation) days of life), which rapidly progressed to T-ALL. Heat-shock-regulated transgenic technology in zebrafish provides the missing link necessary to exploit the powerful genetic capacity of this organism to probe the multi-step molecular pathogenesis of leukaemia.
DOI:10.1016/j.ccell.2020.01.001URLPMID:32049046 [本文引用: 1]
Deregulation of MYC plays an essential role in T cell acute lymphoblastic leukemia (T-ALL), yet the mechanisms underlying its deregulation remain elusive. Herein, we identify a molecular mechanism responsible for reciprocal activation between Aurora B kinase (AURKB) and MYC. AURKB directly phosphorylates MYC at serine 67, counteracting GSK3beta-directed threonine 58 phosphorylation and subsequent FBXW7-mediated proteasomal degradation. Stabilized MYC, in concert with T cell acute lymphoblastic leukemia 1 (TAL1), directly activates AURKB transcription, constituting a positive feedforward loop that reinforces MYC-regulated oncogenic programs. Therefore, inhibitors of AURKB induce prominent MYC degradation concomitant with robust leukemia cell death. These findings reveal an AURKB-MYC regulatory circuit that underlies T cell leukemogenesis, and provide a rationale for therapeutic targeting of oncogenic MYC via AURKB inhibition.
URLPMID:19458356 [本文引用: 1]
DOI:10.1084/jem.20101691URL [本文引用: 1]
The MYC oncogenic transcription factor is overexpressed in most human cases of T cell acute lymphoblastic leukemia (T-ALL), often downstream of mutational NOTCH1 activation. Genetic alterations in the PTEN-PI3K-AKT pathway are also common in T-ALL. We generated a conditional zebrafish model of T-ALL in which 4-hydroxytamoxifen (4HT) treatment induces MYC activation and disease, and withdrawal of 4HT results in T-ALL apoptosis and tumor regression. However, we found that loss-of-function mutations in zebrafish pten genes, or expression of a constitutively active Akt2 transgene, rendered tumors independent of the MYC oncogene and promoted disease progression after 4HT withdrawal. Moreover, MYC suppresses pten mRNA levels, suggesting that Akt pathway activation downstream of MYC promotes tumor progression. Our findings indicate that Akt pathway activation is sufficient for tumor maintenance in this model, even after loss of survival signals driven by the MYC oncogene.
DOI:10.1126/science.1102160URLPMID:15472075 [本文引用: 1]
Very rare cases of human T cell acute lymphoblastic leukemia (T-ALL) harbor chromosomal translocations that involve NOTCH1, a gene encoding a transmembrane receptor that regulates normal T cell development. Here, we report that more than 50% of human T-ALLs, including tumors from all major molecular oncogenic subtypes, have activating mutations that involve the extracellular heterodimerization domain and/or the C-terminal PEST domain of NOTCH1. These findings greatly expand the role of activated NOTCH1 in the molecular pathogenesis of human T-ALL and provide a strong rationale for targeted therapies that interfere with NOTCH signaling.
DOI:10.1038/sj.leu.2404546URLPMID:17252014 [本文引用: 1]
Activating mutations in the NOTCH1 gene have been found in about 60% of patients with T-cell acute lymphoblastic leukemia (T-ALL). In order to study the molecular mechanisms by which altered Notch signaling induces leukemia, a zebrafish model of human NOTCH1-induced T-cell leukemia was generated. Seven of sixteen mosaic fish developed a T-cell lymphoproliferative disease at about 5 months. These neoplastic cells extensively invaded tissues throughout the fish and caused an aggressive and lethal leukemia when transplanted into irradiated recipient fish. However, stable transgenic fish exhibited a longer latency for leukemia onset. When the stable transgenic line was crossed with another line overexpressing the zebrafish bcl2 gene, the leukemia onset was dramatically accelerated, indicating synergy between the Notch pathway and the bcl2-mediated antiapoptotic pathway. Reverse transcription-polymerase chain reaction analysis showed that Notch target genes such as her6 and her9 were highly expressed in NOTCH1-induced leukemias. The ability of this model to detect a strong interaction between NOTCH1 and bcl2 suggests that genetic modifier screens have a high likelihood of revealing other genes that can cooperate with NOTCH1 to induce T-ALL.
DOI:10.1038/leu.2009.116URLPMID:19516274 [本文引用: 3]
T-cell neoplasias are common in pediatric oncology, and include acute lymphoblastic leukemia (T-ALL) and lymphoblastic lymphoma (T-LBL). These cancers have worse prognoses than their B-cell counterparts, and their treatments carry significant morbidity. Although many pediatric malignancies have characteristic translocations, most T-lymphocyte-derived diseases lack cytogenetic hallmarks. Lacking these informative lesions, insight into their molecular pathogenesis is less complete. Although dysregulation of the NOTCH1 pathway occurs in a substantial fraction of cases, many other genetic lesions of T-cell malignancy have not yet been determined. To address this deficiency, we pioneered a phenotype-driven forward-genetic screen in zebrafish (Danio rerio). Using transgenic fish with T-lymphocyte-specific expression of enhanced green fluorescent protein (EGFP), we performed chemical mutagenesis, screened animals for GFP(+) tumors, and identified multiple lines with a heritable predisposition to T-cell malignancy. In each line, the patterns of infiltration and morphological appearance resembled human T-ALL and T-LBL. T-cell receptor analyses confirmed their clonality. Malignancies were transplantable and contained leukemia-initiating cells, like their human correlates. In summary, we have identified multiple zebrafish mutants that recapitulate human T-cell neoplasia and show heritable transmission. These vertebrate models provide new genetic platforms for the study of these important human cancers.
DOI:10.1111/j.1600-0609.1975.tb01063.xURLPMID:1188315 [本文引用: 1]
A family survey was conducted among 909 patients with leukaemia of all types, with the purpose of establishing the incidence of further cases of leukaemia among relatives. Among a total of 41,807 relatives 8,349 were deceased, and the cause of death was objectively confirmed in 5,011. 72 patients had one or more relatives with leukaemia. First degree relatives with leukaemia were much more frequent in families of patients with chronic lymphocytic than in those of patients with chronic granulocytic leukaemia. The incidence of leukaemia among first degree relatives was established to be 2.8-3.0 times, among more distant relatives about 2.3 times, and overall about 2.5 times that expected. This excess is of the order of that observed in relatives of patients with certain solid tumors. Genetic factors may have accounted for much of the excess incidence in chronic lymphocytic and acute leukaemia, but there was little evidence for a genetic background in chronic granulocytic leukaemia. With the possible exception of one family with multiple cases, a simple Mendelian mechanism did not appear to be involved in the leukaemia families investigated. It appeared more likely that a polygenic mechanism led to a heightened susceptibility to the disease in these families.
URLPMID:7932824 [本文引用: 1]
DOI:10.1038/sj.leu.2400707URLPMID:9264391 [本文引用: 1]
Familial leukemia is rare, but, as is the case with other cancer family syndromes, its study is likely to lead to the identification of genes causative of the far more common, sporadic cases. I review the clinical and, what is known of the molecular genetic features of familial leukemia. I propose a nosology based on whether the leukemia is a component of a medical syndrome or exists as a solitary disease, the apparent mode of inheritance, and the distribution of leukemia types and subtypes in affected family members. I review the recent findings from my group that leukemia is inherited with 'anticipation', in the form of a declining age of onset with each passing generation. I consider two models of leukemia genesis that can potentially account for anticipation in familial cases and incorporate epidemiological observations made in sporadic cases. The first model is analogous to trinucleotide repeat expansion in Huntington disease, myotonic dystrophy, and other inherited neurodegenerative illness demonstrating anticipation. The second model considers evidence that anticipation may be common to multiple types of familial cancer and is based on the intergenerational inheritance of multiple downstream mutations resulting from a defect in a single DNA repair gene.
DOI:10.1016/j.bcmd.2003.10.005URLPMID:14757442 [本文引用: 1]
We have reviewed the world's literature that addresses familial leukemia, lymphoma, and myeloma. We have catalogued the phenotypic abnormalities associated with an increased risk of developing a hematological malignancy. These syndromes, such as Fanconi anemia or familial platelet syndrome, have been well characterized and in many cases the gene responsible for the predisposition has been defined. We have focused, however, on reports of a familial incidence of hematological malignancy in which no prior predisposing syndrome was reported. In this circumstance, so-called pure familial leukemia, lymphoma, or myeloma, the intergenerational incidence of disease occurred in ostensibly healthy persons. These families have been grouped into sets in which (a) anticipation, (b) immune abnormalities, (c) linkage to HLA phenotypes, (d) linkage to chromosome abnormalities, or (e) gene abnormalities have been reported. They have also been grouped by type of leukemia. Purely descriptive reports, not accompanied by some information on pathogenesis, have not been included. They are catalogued in some of the references cited in this paper. Anticipation is a prominent feature of familial leukemia, lymphoma, and myeloma, supporting the concept of germline transmission of a susceptibility gene. Although linkage to an HLA phenotype occurs in some families, no consistent intrafamilial pattern has emerged. Deletion of chromosome 7 is associated with familial acute myelogenous leukemia, but no other recurring localization has been established. Although putative susceptibility genes have been identified in some families, the likelihood is that the mode of inheritance is different in different families and different genes are involved even within a specific Mendelian pattern. Although as yet not reported, the frequency of familial CLL and the intensity of its study indicates that the gene or genes involved in that familial disorder(s) should be identified conclusively soon if sufficient families for study can be assembled through international cooperation.
URLPMID:7780150 [本文引用: 1]
URLPMID:28809761 [本文引用: 1]
Pediatric B-cell acute lymphoblastic leukemia (B-ALL) is the most common hematological malignancy in children, and the t(12;21)(p13;q22) occurs in approximately 25% of these cases, making it is the most prevalent chromosomal abnormality. The t(12;21) which disrupts hematopoietic differentiation and proliferation, and can be present as a sole abnormality or within the context of a complex karyotype characterized by three or more chromosomal abnormalities. The prognosis of t(12;21) within a complex karyotype is extensively debated. In this review, we discuss the literature regarding t(12;21) and summarize the cytogenetic features found in 363 pediatric cases compiled from the Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. Cytogenetically, most of the cases had secondary chromosomal abnormalities, about half of which were in the context of a complex karyotype. Trisomy 21 was found to be the most common numerical abnormality in almost one-fifth of the cases, and deletions on chromosome 12 and 6 occurred in 16.9% and 12.5% of cases, respectively. In general, t(12;21) in B-ALL is associated with a favorable prognosis. Herein, we found no significant difference in survival outcome of t(12;21) with a on-complex or complex karyotype.
DOI:10.1016/s0165-4608(01)00518-0URLPMID:11675129 [本文引用: 1]
ETV6/CBFA2 (TEL/AML1) is the most frequent genetic abnormality associated with acute lymphoblastic leukemias in children, and is associated with a favorable prognosis. To investigate the influence of ETV6/CBFA2 on cellular transformation, the fusion gene was cloned into a murine ecotropic retroviral vector and transduced into IL-3-dependent Ba/F3 and 32Dcl.3 and IL-7-dependent IxN/2b murine hematopoietic cell lines. Different variants of ETV6/CBFA2, corresponding to CBFA2 alternatively spliced variants, and the reciprocal product CBFA2/ETV6, were stably expressed in each of these cell lines. However, although Western blot analysis demonstrated expression of each variant, none of the stable cell lines expressing CBFA2/ETV6 or the variants conferred factor-independent growth. We further investigated the effect of ETV6/CBFA2 expression in vivo by generating transgenic mice in which expression of the fusion was directed to lymphoid cells using the immunoglobulin heavy chain enhancer/promoter. Four founder mice were identified showing transmission and expression of the chimeric product. The mice were bred for five generations and followed for more than 24 months. The mice did not develop a malignant hematologic disorder, nor did they display histopathologic, morphologic, or immunophenotypic abnormalities, although ETV6/CBFA2 expression was confirmed in each line. We conclude that the expression of ETV6/CBFA2 alone is not sufficient for induction of growth factor independence in hematopoietic cell lines or hematologic disease in transgenic mice.
DOI:10.1073/pnas.0603349103URLPMID:17015828 [本文引用: 1]
Acute lymphoblastic leukemia (ALL) is a clonal disease that evolves through the accrual of genetic rearrangements and/or mutations within the dominant clone. The TEL-AML1 (ETV6-RUNX1) fusion in precursor-B (pre-B) ALL is the most common genetic rearrangement in childhood cancer; however, the cellular origin and the molecular pathogenesis of TEL-AML1-induced leukemia have not been identified. To study the origin of TEL-AML1-induced ALL, we generated transgenic zebrafish expressing TEL-AML1 either ubiquitously or in lymphoid progenitors. TEL-AML1 expression in all lineages, but not lymphoid-restricted expression, led to progenitor cell expansion that evolved into oligoclonal B-lineage ALL in 3% of the transgenic zebrafish. This leukemia was transplantable to conditioned wild-type recipients. We demonstrate that TEL-AML1 induces a B cell differentiation arrest, and that leukemia development is associated with loss of TEL expression and elevated Bcl2/Bax ratio. The TEL-AML1 transgenic zebrafish models human pre-B ALL, identifies the molecular pathways associated with leukemia development, and serves as the foundation for subsequent genetic screens to identify modifiers and leukemia therapeutic targets.
URLPMID:30111845 [本文引用: 1]
URLPMID:9731070 [本文引用: 1]
Chromosomal abnormalities in acute leukemia have led to the discovery of many genes involved in normal hematopoiesis and in malignant transformation. We have identified the fusion partners in an inv(8)(p11q13) from a patient with acute mixed lineage leukemia. We show by fluorescence in situ hybridization (FISH) analysis, Southern blotting, and reverse transcriptase-polymerase chain reaction (RT-PCR) that the genes for MOZ, monocytic leukemia zinc finger protein, and TIF2, transcriptional intermediary factor 2, are involved in the inv(8)(p11q13). We demonstrate that the inversion creates a fusion between the 5' end of MOZ mRNA and the 3' end of TIF2 mRNA maintaining the translational frame of the protein. The predicted fusion protein contains the zinc finger domains, the nuclear localization domains, the histone acetyltransferase (HAT) domain, and a portion of the acidic domain of MOZ, coupled to the CREB-binding protein (CBP) interaction domain and the activation domains of TIF2. The breakpoint is distinct from the breakpoint in the t(8;16)(p11;p13) translocation in acute monocytic leukemia with erythrophagocytosis that fuses MOZ with CBP. The reciprocal TIF2-MOZ fusion gene is not expressed, perhaps as a result of a deletion near the chromosome 8 centromere. The MOZ-TIF2 fusion is one of a new family of chromosomal rearrangements that associate HAT activity, transcriptional coactivation, and acute leukemia.
DOI:10.1046/j.1365-2141.1999.01377.xURLPMID:10460585 [本文引用: 1]
DOI:10.1038/ng0296-159URLPMID:8563754 [本文引用: 1]
The t(7;11)(p15;p15) translocation is a recurrent chromosomal abnormality associated primarily with acute myeloid leukaemia (FAB M2 and M4). We present here the molecular definition of this translocation. On chromosome 7 positional cloning revealed the consistent rearrangement of the HOXA9 gene, which encodes a class I homeodomain protein potentially involved in myeloid differentiation. On chromosome 11 the translocation targets the human homologue of NUP98, a member of the GLFG nucleoporin family. Chimaeric messages spliced over the breakpoint fuse the GLFG repeat domains of NUP98 in-frame to the HOXA9 homeobox. The predicted NUP98-HOXA9 fusion protein may promote leukaemogenesis through inhibition of HOXA9-mediated terminal differentiation and/or aberrant nucleocytoplasmic transport.
URLPMID:11934867 [本文引用: 1]
RUNX1/AML1/CBFA2 is essential for definitive hematopoiesis, and chromosomal translocations affecting RUNX1 are frequently involved in human leukemias. Consequently, the normal function of RUNX1 and its involvement in leukemogenesis remain subject to intensive research. To further elucidate the role of RUNX1 in hematopoiesis, we cloned the zebrafish ortholog (runx1) and analyzed its function using this model system. Zebrafish runx1 is expressed in hematopoietic and neuronal cells during early embryogenesis. runx1 expression in the lateral plate mesoderm co-localizes with the hematopoietic transcription factor scl, and expression of runx1 is markedly reduced in the zebrafish mutants spadetail and cloche. Transient expression of runx1 in cloche embryos resulted in partial rescue of the hematopoietic defect. Depletion of Runx1 with antisense morpholino oligonucleotides abrogated the development of both blood and vessels, as demonstrated by loss of circulation, incomplete development of vasculature and the accumulation of immature hematopoietic precursors. The block in definitive hematopoiesis is similar to that observed in Runx1 knockout mice, implying that zebrafish Runx1 has a function equivalent to that in mammals. Our data suggest that zebrafish Runx1 functions in both blood and vessel development at the hemangioblast level, and contributes to both primitive and definitive hematopoiesis. Depletion of Runx1 also caused aberrant axonogenesis and abnormal distribution of Rohon-Beard cells, providing the first functional evidence of a role for vertebrate Runx1 in neuropoiesis. To provide a base for examining the role of Runx1 in leukemogenesis, we investigated the effects of transient expression of a human RUNX1-CBF2T1 transgene [product of the t(8;21) translocation in acute myeloid leukemia] in zebrafish embryos. Expression of RUNX1-CBF2T1 caused disruption of normal hematopoiesis, aberrant circulation, internal hemorrhages and cellular dysplasia. These defects reproduce those observed in Runx1-depleted zebrafish embryos and RUNX1-CBF2T1 knock-in mice. The phenotype obtained with transient expression of RUNX1-CBF2T1 validates the zebrafish as a model system to study t(8;21)-mediated leukemogenesis.
DOI:10.1155/2018/6705842URLPMID:30003105 [本文引用: 1]
The 11q23 of the mixed lineage leukemia 1 (MLL1) gene plays a crucial role in early embryonic development and hematopoiesis. The MLL-AF9 fusion gene, resulting from chromosomal translocation, often leads to acute myeloid leukemia with poor prognosis. Here, we generated a zebrafish model expressing the human MLL-AF9 fusion gene. Microinjection of human MLL-AF9 mRNA into zebrafish embryos resulted in enhanced hematopoiesis and the activation of downstream genes such as meis1 and hox cluster genes. Embryonic MLL-AF9 expression upregulated HSPC and myeloid lineage markers. Doxorubicin and MI-2 (a menin inhibitor) treatments significantly restored normal hematopoiesis in MLL-AF9-expressing animals. This study provides insight into the role of MLL-AF9 in zebrafish hematopoiesis and establishes a robust and efficient in vivo model for high-throughput drug screening.
DOI:10.1111/j.1365-2141.2008.07362.xURLPMID:18729850 [本文引用: 1]
The inv(8)(p11q13) chromosomal abnormality, described in acute myeloid leukaemias (AML), fuses the histone acetyl-transferase (HAT) MYST3 (MOZ) gene with another HAT gene, NCOA2 (TIF2). We generated a transgenic zebrafish in which the MYST3/NCOA2 fusion gene was expressed under control of the spi1 promoter. An AML developed in 2 of 180 MYST3/NCOA2-EGFP-expressing embryos, 14 and 26 months after injection of the fusion gene in a one-cell embryo, respectively. This leukaemia was characterised by an extensive invasion of kidneys by myeloid blast cells. This model, which is the first zebrafish model of AML, demonstrates the oncogenic potency of MYST3/NCOA2 fusion gene.
DOI:10.1242/dev.008904URLPMID:18156164 [本文引用: 1]
AML1-ETO is one of the most common chromosomal translocation products associated with acute myelogenous leukemia (AML). Patients carrying the AML1-ETO fusion gene exhibit an accumulation of granulocyte precursors in the bone marrow and the blood. Here, we describe a transgenic zebrafish line that enables inducible expression of the human AML1-ETO oncogene. Induced AML1-ETO expression in embryonic zebrafish causes a phenotype that recapitulates some aspects of human AML. Using this highly tractable model, we show that AML1-ETO redirects myeloerythroid progenitor cells that are developmentally programmed to adopt the erythroid cell fate into the granulocytic cell fate. This fate change is characterized by a loss of gata1 expression and an increase in pu.1 expression in myeloerythroid progenitor cells. Moreover, we identify scl as an early and essential mediator of the effect of AML1-ETO on hematopoietic cell fate. AML1-ETO quickly shuts off scl expression, and restoration of scl expression rescues the effects of AML1-ETO on myeloerythroid progenitor cell fate. These results demonstrate that scl is an important mediator of the ability of AML1-ETO to reprogram hematopoietic cell fate decisions, suggesting that scl may be an important contributor to AML1-ETO-associated leukemia. In addition, treatment of AML1-ETO transgenic zebrafish embryos with a histone deacetylase inhibitor, Trichostatin A, restores scl and gata1 expression, and ameliorates the accumulation of granulocytic cells caused by AML1-ETO. Thus, this zebrafish model facilitates in vivo dissection of AML1-ETO-mediated signaling, and will enable large-scale chemical screens to identify suppressors of the in vivo effects of AML1-ETO.
DOI:10.1038/nchembio.147URLPMID:19172146 [本文引用: 1]
It has been proposed that inhibitors of an oncogene's effects on multipotent hematopoietic progenitor cell differentiation may change the properties of the leukemic stem cells and complement the clinical use of cytotoxic drugs. Using zebrafish, we developed a robust in vivo hematopoietic differentiation assay that reflects the activity of the oncogene AML1-ETO. Screening for modifiers of AML1-ETO-mediated hematopoietic dysregulation uncovered unexpected roles of COX-2- and beta-catenin-dependent pathways in AML1-ETO function. This approach may open doors for developing therapeutics targeting oncogene function within leukemic stem cells.
DOI:10.1111/j.1365-2141.2011.08810.xURLPMID:21810091 [本文引用: 1]
NUP98-HOXA9 [t(7;11) (p15;p15)] is associated with inferior prognosis in de novo and treatment-related acute myeloid leukaemia (AML) and contributes to blast crisis in chronic myeloid leukaemia (CML). We have engineered an inducible transgenic zebrafish harbouring human NUP98-HOXA9 under the zebrafish spi1(pu.1) promoter. NUP98-HOXA9 perturbed zebrafish embryonic haematopoiesis, with upregulated spi1 expression at the expense of gata1a. Markers associated with more differentiated myeloid cells, lcp1, lyz, and mpx were also elevated, but to a lesser extent than spi1, suggesting differentiation of early myeloid progenitors may be impaired by NUP98-HOXA9. Following irradiation, NUP98-HOXA9-expressing embryos showed increased numbers of cells in G2-M transition compared to controls and absence of a normal apoptotic response, which may result from an upregulation of bcl2. These data suggest NUP98-HOXA9-induced oncogenesis may result from a combination of defects in haematopoiesis and an aberrant response to DNA damage. Importantly, 23% of adult NUP98-HOXA9-transgenic fish developed a myeloproliferative neoplasm (MPN) at 19-23 months of age. In summary, we have identified an embryonic haematopoietic phenotype in a transgenic zebrafish line that subsequently develops MPN. This tool provides a unique opportunity for high-throughput in vivo chemical modifier screens to identify novel therapeutic agents in high risk AML.
DOI:10.1038/leu.2015.126URLPMID:26017032 [本文引用: 1]
Acute myeloid leukemia (AML) occurs when multiple genetic aberrations alter white blood cell development, leading to hyperproliferation and arrest of cell differentiation. Pertinent animal models link in vitro studies with the use of new agents in clinical trials. We generated a transgenic zebrafish expressing human NUP98-HOXA9 (NHA9), a fusion oncogene found in high-risk AML. Embryos developed a preleukemic state with anemia and myeloid cell expansion, and adult fish developed a myeloproliferative neoplasm (MPN). We leveraged this model to show that NHA9 increases the number of hematopoietic stem cells, and that oncogenic function of NHA9 depends on downstream activation of meis1, the PTGS/COX pathway and genome hypermethylation through the DNA methyltransferase, dnmt1. We restored normal hematopoiesis in NHA9 embryos with knockdown of meis1 or dnmt1, as well as pharmacologic treatment with DNA (cytosine-5)-methyltransferase (DNMT) inhibitors or cyclo-oxygenase (COX) inhibitors. DNMT inhibitors reduced genome methylation to near normal levels. Strikingly, we discovered synergy when we combined sub-monotherapeutic doses of a histone deacetylase inhibitor plus either a DNMT inhibitor or COX inhibitor to block the effects of NHA9 on zebrafish blood development. Our work proposes novel drug targets in NHA9-induced myeloid disease, and suggests rational therapies by combining minimal doses of known bioactive compounds.
URLPMID:1954386 [本文引用: 1]
The myc proto-oncogenes encode nuclear phosphoproteins, which are believed to participate in the control of cell proliferation and differentiation. Deregulated expression of c-myc has been implicated in several human hematopoietic malignancies. We have studied the expression and mRNA processing of human L-myc, N-myc, and c-myc genes in a panel of human leukemias, leukemia cell lines, and normal hematopoietic cells. L-myc mRNA was expressed in three acute myeloid leukemias (AML) studied and in several myeloid leukemia cell lines. Only low expression levels were observed in adult bone marrow and in fetal spleen and thymus. The K562 and Dami leukemia cell lines showed a unique pattern of L-myc mRNA processing, with approximately 40% of L-myc mRNA lacking exon III and intron I. N-myc was expressed in five of six AML cases studied, in one of nine acute lymphocytic leukemia (ALL) cases, and in several leukemia cell lines, while c-myc mRNA was detected in all leukemias and leukemia cell lines studied. Coexpression of all three myc genes was observed in Dami and MOLT-4 cell lines and in two AMLs, and either L-myc or N-myc was coexpressed with c-myc in several other cases. These results show that in addition to c-myc, the L-myc and N-myc genes are expressed in some human leukemias and leukemia cell lines, and suggest a lack of mutually exclusive cross-regulation of the myc genes in human leukemia cells.
DOI:10.1371/journal.pone.0059070URLPMID:23554972 [本文引用: 1]
BACKGROUND: Amplification of MYCN (N-Myc) oncogene has been reported as a frequent event and a poor prognostic marker in human acute myeloid leukemia (AML). The molecular mechanisms and transcriptional networks by which MYCN exerts its influence in AML are largely unknown. METHODOLOGY/PRINCIPAL FINDINGS: We introduced murine MYCN gene into embryonic zebrafish through a heat-shock promoter and established the stable germline Tg(MYCN:HSE:EGFP) zebrafish. N-Myc downstream regulated gene 1 (NDRG1), negatively controlled by MYCN in human and functionally involved in neutrophil maturation, was significantly under-expressed in this model. Using peripheral blood smear detection, histological section and flow cytometric analysis of single cell suspension from kidney and spleen, we found that MYCN overexpression promoted cell proliferation, enhanced the repopulating activity of myeloid cells and the accumulation of immature hematopoietic blast cells. MYCN enhanced primitive hematopoiesis by upregulating scl and lmo2 expression and promoted myelopoiesis by inhibiting gata1 expression and inducing pu.1, mpo expression. Microarray analysis identified that cell cycle, glycolysis/gluconeogenesis, MAPK/Ras, and p53-mediated apoptosis pathways were upregulated. In addition, mismatch repair, transforming and growth factor beta (TGFbeta) were downregulated in MYCN-overexpressing blood cells (p<0.01). All of these signaling pathways are critical in the proliferation and malignant transformation of blood cells. CONCLUSION/SIGNIFICANCE: The above results induced by overexpression of MYCN closely resemble the main aspects of human AML, suggesting that MYCN plays a role in the etiology of AML. MYCN reprograms hematopoietic cell fate by regulating NDRG1 and several lineage-specific hematopoietic transcription factors. Therefore, this MYCN transgenic zebrafish model facilitates dissection of MYCN-mediated signaling in vivo, and enables high-throughput scale screens to identify the potential therapeutic targets.
DOI:10.1016/j.jbior.2018.11.007URLPMID:30528537 [本文引用: 1]
Myeloproliferative neoplasms (MPNs) are haematopoietic stem cell-derived clonal disorders characterised by proliferation of some or all myeloid lineages, depending on the subtype. MPNs are classically categorized into three disease subgroups; essential thrombocythaemia (ET), polycythaemia vera (PV) and primary myelofibrosis (PMF). The majority (>85%) of patients carry a disease-initiating or driver mutation, the most prevalent occurring in the janus kinase 2 gene (JAK2 V617F), followed by calreticulin (CALR) and myeloproliferative leukaemia virus (MPL) genes. Although these diseases are characterised by shared clinical, pathological and molecular features, one of the most challenging aspects of these disorders is the diverse clinical features which occur in each disease type, with marked variability in risks of disease complications and progression to leukaemia. A remarkable aspect of MPN biology is that the JAK2 V617F mutation, often occurring in the absence of additional mutations, generates a spectrum of phenotypes from asymptomatic ET through to aggressive MF, associated with a poor outcome. The mechanisms promoting MPN heterogeneity remain incompletely understood, but contributing factors are broad and include patient characteristics (gender, age, comorbidities and environmental exposures), additional somatic mutations, target disease-initiating cell, bone marrow microenvironment and germline genetic associations. In this review, we will address these in detail and discuss their role in heterogeneity of MPN disease phenotypes. Tailoring patient management according to the multiple different factors that influence disease phenotype may prove to be the most effective approach to modify the natural history of the disease and ultimately improve outcomes for patients.
DOI:10.1038/sj.leu.2401240URLPMID:10049057 [本文引用: 1]
Juvenile myelomonocytic leukemia (JMML) is a malignant hematopoietic disorder of early childhood with excessive proliferation of the myeloid and monocytic lineage. Deregulation of the RAS signal transduction pathway is thought to play a key role in its pathogenesis. We examined peripheral blood or bone marrow cells of 36 children with JMML for activating point mutations in codons 12, 13 and 61 of the NRAS and KRAS proto-oncogenes by allele-specific restriction assay, single-strand conformation polymorphism and/or direct sequencing. Codons 12, 13 and 61 of HRAS were examined in 26 of these patients. We detected RAS mutations in six cases (17%) located at N12 (n = 2), N13 (n = 3) and K13 (n = 1). In addition, we performed clonality studies on different cell lineages in four of these patients applying the RAS mutation, the karyotype and X-chromosome inactivation patterns as clonal markers. Erythroid cells carried mutant RAS, indicating clonal origin. In EBV B cell lines, one of three patients studied harbored a RAS mutation, while the other two patients had polyclonal B cells with wild-type RAS. T lymphocytes were examined in one patient; they were polyclonal and had wild-type RAS. It is likely that JMML is a heterogeneous disease with respect to clonal involvement of different lineages.
DOI:10.1182/blood-2009-01-198416URLPMID:19571318 [本文引用: 2]
Juvenile myelomonocytic leukemia is an aggressive myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Seventy-five percent of patients harbor mutations in the NF1, NRAS, KRAS, or PTPN11 genes, which encode components of Ras signaling networks. Using single nucleotide polymorphism arrays, we identified a region of 11q isodisomy that contains the CBL gene in several JMML samples, and subsequently identified CBL mutations in 27 of 159 JMML samples. Thirteen of these mutations alter codon Y371. In this report, we also demonstrate that CBL and RAS/PTPN11 mutations were mutually exclusive in these patients. Moreover, the exclusivity of CBL mutations with respect to other Ras pathway-associated mutations indicates that CBL may have a role in deregulating this key pathway in JMML.
DOI:10.1016/j.exphem.2015.06.007URLPMID:26209551 [本文引用: 1]
Major progress has been recently made in understanding the molecular pathogenesis of myeloproliferative neoplasms (MPN). Mutations in one of four genes-JAK2, MPL, CALR, and CSF3R-can be found in the vast majority of patients with MPN and represent driver mutations that can induce the MPN phenotype. Hyperactive JAK/STAT signaling appears to be the common denominator of MPN, even in patients with CALR mutations and the so-called
DOI:10.1182/blood-2016-10-695940URLPMID:28028029 [本文引用: 1]
The genetic landscape of classical myeloproliferative neoplasm (MPN) is in large part elucidated. The MPN-restricted driver mutations, including those in JAK2, calreticulin (CALR), and myeloproliferative leukemia virus (MPL), abnormally activate the cytokine receptor/JAK2 pathway and their downstream effectors, more particularly the STATs. The most frequent mutation, JAK2V617F, activates the 3 main myeloid cytokine receptors (erythropoietin receptor, granulocyte colony-stimulating factor receptor, and MPL) whereas CALR or MPL mutants are restricted to MPL activation. This explains why JAK2V617F is associated with polycythemia vera, essential thrombocythemia (ET), and primary myelofibrosis (PMF) whereas CALR and MPL mutants are found in ET and PMF. Other mutations in genes involved in epigenetic regulation, splicing, and signaling cooperate with the 3 MPN drivers and play a key role in the PMF pathogenesis. Mutations in epigenetic regulators TET2 and DNMT3A are involved in disease initiation and may precede the acquisition of JAK2V617F. Other mutations in epigenetic regulators such as EZH2 and ASXL1 also play a role in disease initiation and disease progression. Mutations in the splicing machinery are predominantly found in PMF and are implicated in the development of anemia or pancytopenia. Both heterogeneity of classical MPNs and prognosis are determined by a specific genomic landscape, that is, type of MPN driver mutations, association with other mutations, and their order of acquisition. However, factors other than somatic mutations play an important role in disease initiation as well as disease progression such as germ line predisposition, inflammation, and aging. Delineation of these environmental factors will be important to better understand the precise pathogenesis of MPN.
DOI:10.1038/bcj.2016.83URLPMID:27716741 [本文引用: 1]
CALR mutations are identified in about 30% of JAK2/MPL-unmutated myeloproliferative neoplasms (MPNs) including essential thrombocythemia (ET) and primary myelofibrosis. Although the molecular pathogenesis of CALR mutations leading to MPNs has been studied using in vitro cell lines models, how mutant CALR may affect developmental hematopoiesis remains unknown. Here we took advantage of the zebrafish model to examine the effects of mutant CALR on early hematopoiesis and model human CALR-mutated MPNs. We identified three zebrafish genes orthologous to human CALR, referred to as calr, calr3a and calr3b. The expression of CALR-del52 and CALR-ins5 mutants caused an increase in the hematopoietic stem/progenitor cells followed by thrombocytosis without affecting normal angiogenesis. The expression of CALR mutants also perturbed early developmental hematopoiesis in zebrafish. Importantly, morpholino knockdown of mpl but not epor or csf3r could significantly attenuate the effects of mutant CALR. Furthermore, the expression of mutant CALR caused jak-stat signaling activation in zebrafish that could be blocked by JAK inhibitors (ruxolitinib and fedratinib). These findings showed that mutant CALR activates jak-stat signaling through an mpl-dependent mechanism to mediate pathogenic thrombopoiesis in zebrafish, and illustrated that the signaling machinery related to mutant CALR tumorigenesis are conserved between human and zebrafish.
DOI:10.1038/leu.2013.132URLPMID:23625115 [本文引用: 1]
Human oncogenes involved in the development of hematological malignancies have been widely used to model experimental leukemia. However, models of myeloid leukemia rarely reproduce the human disease in full, due to genetic complexity or to difficulties in targeting leukemia initiating cells. Here, we used a zebrafish genetic model to induce the expression of oncogenic RAS in endothelial cells, including the hemogenic endothelium of the dorsal aorta that generates hematopoietic cells, and observed the development of a myelo-erythroid proliferative disorder. In larvae, the phenotype is characterized by disruption of the vascular system and prominent expansion of the caudal hematopoietic tissue. In few surviving juveniles, increased number of immature hematopoietic cells and arrest of myeloid maturation was found in kidney marrow. Peripheral blood showed increased erythroblasts and myeloid progenitors. We found that the abnormal phenotype is associated with a downregulation of the Notch pathway, whereas overexpressing an activated form of Notch together with the oncogene prevents the expansion of the myelo-erythroid compartment. This study identifies the downregulation of the Notch pathway following an oncogenic event in the hemogenic endothelium as an important step in the pathogenesis of myelo-erythroid disorders and describes a number of potential effectors of this transformation.
DOI:10.1038/leu.2017.189URLPMID:28626217 [本文引用: 1]
Interferon regulatory factor (IRF)-8 is a critical transcription factor involved in the pathogenesis of myeloid neoplasia. However, the underlying mechanisms in vivo are not well known. Investigation of irf8-mutant zebrafish in this study indicated that Irf8 is evolutionarily conserved as an essential neoplastic suppressor through tight control of the proliferation and longevity of myeloid cells. Surviving irf8 mutants quickly developed a myeloproliferative neoplasm (MPN)-like disease with enhanced output of the myeloid precursors, which recurred after transplantation. Multiple molecules presented notable alteration and Mertk signaling was aberrantly activated in the hematopoietic cells in irf8 mutants. Transgenic mertk overexpression in Tg(coro1a:mertk) zebrafish recapitulated the myeloid neoplasia-like syndrome in irf8 mutants. Moreover, functional interference with Mertk, via morpholino knockdown or genetic disruption, attenuated the myeloid expansion phenotype caused by Irf8 deficiency. Therefore, Mertk signaling is a critical downstream player in the Irf8-mediated regulation of the progression of myeloid neoplasia. Our study extends the understanding of the mechanisms underlying leukemogenesis.
DOI:10.1038/leu.2015.154URLPMID:26104663 [本文引用: 1]
Controlled self-renewal and differentiation of hematopoietic stem/progenitor cells (HSPCs) are critical for vertebrate development and survival. These processes are tightly regulated by the transcription factors, signaling molecules and epigenetic factors. Impaired regulations of their function could result in hematological malignancies. Using a large-scale zebrafish N-ethyl-N-nitrosourea mutagenesis screening, we identified a line named LDD731, which presented significantly increased HSPCs in hematopoietic organs. Further analysis revealed that the cells of erythroid/myeloid lineages in definitive hematopoiesis were increased while the primitive hematopoiesis was not affected. The homozygous mutation was lethal with a median survival time around 14-15 days post fertilization. The causal mutation was located by positional cloning in the c-cbl gene, the human ortholog of which, c-CBL, is found frequently mutated in myeloproliferative neoplasms (MPN) or acute leukemia. Sequence analysis showed the mutation in LDD731 caused a histidine-to-tyrosine substitution of the amino acid codon 382 within the RING finger domain of c-Cbl. Moreover, the myeloproliferative phenotype in zebrafish seemed dependent on the Flt3 (fms-like tyrosine kinase 3) signaling, consistent with that observed in both mice and humans. Our study may shed new light on the pathogenesis of MPN and provide a useful in vivo vertebrate model of this syndrome for screening drugs.
URLPMID:2672599 [本文引用: 1]
The myelodysplastic syndromes are a heterogeneous group of hematopoietic stem cell disorders characterized by dysplastic and ineffective hematopoiesis and a varying risk of transformation to acute leukemia. Although the natural history of these syndromes is variable, several factors appear to be of prognostic importance, including the French-American-British classification, the karyotype, in vitro colony formation, and others. The pathogenesis of the myelodysplastic syndrome is not known, but recent evidence suggests that alterations of cellular oncogenes may be a causative factor. There is no standard therapy for myelodysplasia, and thus novel approaches to patient management are warranted.
DOI:10.1038/nature10496URLPMID:21909114 [本文引用: 1]
Myelodysplastic syndromes and related disorders (myelodysplasia) are a heterogeneous group of myeloid neoplasms showing deregulated blood cell production with evidence of myeloid dysplasia and a predisposition to acute myeloid leukaemia, whose pathogenesis is only incompletely understood. Here we report whole-exome sequencing of 29 myelodysplasia specimens, which unexpectedly revealed novel pathway mutations involving multiple components of the RNA splicing machinery, including U2AF35, ZRSR2, SRSF2 and SF3B1. In a large series analysis, these splicing pathway mutations were frequent ( approximately 45 to approximately 85%) in, and highly specific to, myeloid neoplasms showing features of myelodysplasia. Conspicuously, most of the mutations, which occurred in a mutually exclusive manner, affected genes involved in the 3'-splice site recognition during pre-mRNA processing, inducing abnormal RNA splicing and compromised haematopoiesis. Our results provide the first evidence indicating that genetic alterations of the major splicing components could be involved in human pathogenesis, also implicating a novel therapeutic possibility for myelodysplasia.
DOI:10.1038/leu.2013.336URLPMID:24220272 [本文引用: 3]
High-throughput DNA sequencing significantly contributed to diagnosis and prognostication in patients with myelodysplastic syndromes (MDS). We determined the biological and prognostic significance of genetic aberrations in MDS. In total, 944 patients with various MDS subtypes were screened for known/putative mutations/deletions in 104 genes using targeted deep sequencing and array-based genomic hybridization. In total, 845/944 patients (89.5%) harbored at least one mutation (median, 3 per patient; range, 0-12). Forty-seven genes were significantly mutated with TET2, SF3B1, ASXL1, SRSF2, DNMT3A, and RUNX1 mutated in >10% of cases. Many mutations were associated with higher risk groups and/or blast elevation. Survival was investigated in 875 patients. By univariate analysis, 25/48 genes (resulting from 47 genes tested significantly plus PRPF8) affected survival (P<0.05). The status of 14 genes combined with conventional factors revealed a novel prognostic model ('Model-1') separating patients into four risk groups ('low', 'intermediate', 'high', 'very high risk') with 3-year survival of 95.2, 69.3, 32.8, and 5.3% (P<0.001). Subsequently, a 'gene-only model' ('Model-2') was constructed based on 14 genes also yielding four significant risk groups (P<0.001). Both models were reproducible in the validation cohort (n=175 patients; P<0.001 each). Thus, large-scale genetic and molecular profiling of multiple target genes is invaluable for subclassification and prognostication in MDS patients.
DOI:10.1182/blood-2013-08-518886URLPMID:24030381 [本文引用: 1]
Myelodysplastic syndromes (MDS) are a heterogeneous group of chronic hematological malignancies characterized by dysplasia, ineffective hematopoiesis and a variable risk of progression to acute myeloid leukemia. Sequencing of MDS genomes has identified mutations in genes implicated in RNA splicing, DNA modification, chromatin regulation, and cell signaling. We sequenced 111 genes across 738 patients with MDS or closely related neoplasms (including chronic myelomonocytic leukemia and MDS-myeloproliferative neoplasms) to explore the role of acquired mutations in MDS biology and clinical phenotype. Seventy-eight percent of patients had 1 or more oncogenic mutations. We identify complex patterns of pairwise association between genes, indicative of epistatic interactions involving components of the spliceosome machinery and epigenetic modifiers. Coupled with inferences on subclonal mutations, these data suggest a hypothesis of genetic
DOI:10.1182/blood-2012-09-399725URLPMID:23160465 [本文引用: 2]
Precursor mRNA splicing is catalyzed by the spliceosome, a macromolecule composed of small nuclear RNAs associated with proteins. The SF3B1 gene encodes subunit 1 of the splicing factor 3b, which is important for anchoring the spliceosome to precursor mRNA. In 2011, whole-exome sequencing studies showed recurrent somatic mutations of SF3B1 and other genes of the RNA splicing machinery in patients with myelodysplastic syndrome or myelodysplastic/myeloproliferative neoplasm. SF3B1 mutations had a particularly high frequency among conditions characterized by ring sideroblasts, which is consistent with a causal relationship. SF3B1 mutants were also detected at a lower frequency in a variety of other tumor types. In chronic lymphocytic leukemia, SF3B1 was found to be the second most frequently mutated gene. In myelodysplastic syndromes, SF3B1 mutations appear to be founding genetic lesions and are associated with a low risk of leukemic evolution. In contrast, SF3B1 mutations have a lower incidence in early stages of chronic lymphocytic leukemia, are more common in advanced disease, and tend to be associated with poor prognosis, suggesting that they occur during clonal evolution of the disease. The assessment of SF3B1 mutation status may become innovative diagnostic and prognostic tools and the availability of spliceosome modulators opens novel therapeutic prospects.
DOI:10.1016/j.exphem.2016.05.012URLPMID:27260753 [本文引用: 2]
SF3B1 (Splicing factor 3b, subunit 1) is one of the most commonly mutated factors in myelodysplastic syndrome (MDS). Although the genetic correlation between SF3B1 mutations and MDS etiology are quite strong, no in vivo model currently exists to explore how SF3B1 loss alters blood cell development. Using zebrafish mutants, we show here that proper function of Sf3b1 is required for all hematopoietic lineages. As in MDS patients, zebrafish sf3b1 mutants develop a macrocytic-anemia-like phenotype due to a block in maturation at a late progenitor stage. The mutant embryos also develop neutropenia, because their primitive myeloid cells fail to mature and turn on differentiation markers such as l-plastin and myeloperoxidase. In contrast, production of definitive hematopoietic stem and progenitor cells (HSPCs) from hemogenic endothelial cells within the dorsal aorta is greatly diminished, whereas arterial endothelial cells are correctly fated. Notch signaling, imperative for the endothelial-to-hematopoietic transition, is also normal, indicating that HSPC induction is blocked in sf3b1 mutants downstream or independent of Notch signaling. The data demonstrate that Sf3b1 function is necessary during key differentiation fate decisions in multiple blood cell types. Zebrafish sf3b1 mutants offer a novel animal model with which to explore the role of splicing in hematopoietic development and provide an excellent in vivo system with which to delve into the question of why and how Sf3b1 dysfunction is detrimental to hematopoietic differentiation, which could improve MDS diagnosis and treatment.
DOI:10.1016/j.febslet.2013.05.030URLPMID:23714367 [本文引用: 1]
Mutated spliceosome components are recurrently being associated with perturbed tissue development and disease pathogenesis. Cephalophonus (cph), is a zebrafish mutant carrying an early premature STOP codon in the spliceosome component Prpf8 (pre-mRNA processing factor 8). Cph initially develops normally, but then develops widespread cell death, especially in neurons, and is embryonic lethal. Cph mutants accumulate aberrantly spliced transcripts retaining both U2- and U12-type introns. Within early haematopoiesis, myeloid differentiation is impaired, suggesting Prpf8 is required for haematopoietic development. Cph provides an animal model for zygotic PRPF8 dysfunction diseases and for evaluating therapeutic interventions.
DOI:10.1016/j.ccr.2011.06.001URLPMID:21723200 [本文引用: 1]
Somatic loss-of-function mutations in the ten-eleven translocation 2 (TET2) gene occur in a significant proportion of patients with myeloid malignancies. Although there are extensive genetic data implicating TET2 mutations in myeloid transformation, the consequences of Tet2 loss in hematopoietic development have not been delineated. We report here an animal model of conditional Tet2 loss in the hematopoietic compartment that leads to increased stem cell self-renewal in vivo as assessed by competitive transplant assays. Tet2 loss leads to a progressive enlargement of the hematopoietic stem cell compartment and eventual myeloproliferation in vivo, including splenomegaly, monocytosis, and extramedullary hematopoiesis. In addition, Tet2(+/-) mice also displayed increased stem cell self-renewal and extramedullary hematopoiesis, suggesting that Tet2 haploinsufficiency contributes to hematopoietic transformation in vivo.
DOI:10.1128/MCB.00971-14URLPMID:25512612 [本文引用: 1]
The ten-eleven translocation 2 gene (TET2) encodes a member of the TET family of DNA methylcytosine oxidases that converts 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) to initiate the demethylation of DNA within genomic CpG islands. Somatic loss-of-function mutations of TET2 are frequently observed in human myelodysplastic syndrome (MDS), which is a clonal malignancy characterized by dysplastic changes of developing blood cell progenitors, leading to ineffective hematopoiesis. We used genome-editing technology to disrupt the zebrafish Tet2 catalytic domain. tet2(m/m) (homozygous for the mutation) zebrafish exhibited normal embryonic and larval hematopoiesis but developed progressive clonal myelodysplasia as they aged, culminating in myelodysplastic syndromes (MDS) at 24 months of age, with dysplasia of myeloid progenitor cells and anemia with abnormal circulating erythrocytes. The resultant tet2(m/m) mutant zebrafish lines show decreased levels of 5hmC in hematopoietic cells of the kidney marrow but not in other cell types, most likely reflecting the ability of other Tet family members to provide this enzymatic activity in nonhematopoietic tissues but not in hematopoietic cells. tet2(m/m) zebrafish are viable and fertile, providing an ideal model to dissect altered pathways in hematopoietic cells and, for small-molecule screens in embryos, to identify compounds with specific activity against tet2 mutant cells.
DOI:10.1186/1476-4598-10-41URLPMID:21501493 [本文引用: 1]
BACKGROUND: MicroRNAs are important regulators of transcription in hematopoiesis. Their expression deregulations were described in association with pathogenesis of some hematological malignancies. This study provides integrated microRNA expression profiling at different phases of chronic myeloid leukemia (CML) with the aim to identify microRNAs associated with CML pathogenesis. The functions of in silico filtered targets are in this report annotated and discussed in relation to CML pathogenesis. RESULTS: Using microarrays we identified differential expression profiles of 49 miRNAs in CML patients at diagnosis, in hematological relapse, therapy failure, blast crisis and major molecular response. The expression deregulation of miR-150, miR-20a, miR-17, miR-19a, miR-103, miR-144, miR-155, miR-181a, miR-221 and miR-222 in CML was confirmed by real-time quantitative PCR. In silico analyses identified targeted genes of these miRNAs encoding proteins that are involved in cell cycle and growth regulation as well as several key signaling pathways such as of mitogen activated kinase-like protein (MAPK), epidermal growth factor receptor (EGFR, ERBB), transforming growth factor beta (TGFB1) and tumor protein p53 that are all related to CML. Decreased levels of miR-150 were detected in patients at diagnosis, in blast crisis and 67% of hematological relapses and showed significant negative correlation with miR-150 proved target MYB and with BCR-ABL transcript level. CONCLUSIONS: This study uncovers microRNAs that are potentially involved in CML and the annotated functions of in silico filtered targets of selected miRNAs outline mechanisms whereby microRNAs may be involved in CML pathogenesis.
DOI:10.1182/blood-2006-12-064683URLPMID:17452517 [本文引用: 1]
The C-Myb transcription factor is essential for hematopoiesis, including in the T-cell lineage. The C-Myb locus is a common site of retroviral insertional mutagenesis, however no recurrent genomic involvement has been reported in human malignancies. Here, we identified 2 types of genomic alterations involving the C-MYB locus at 6q23 in human T-cell acute leukemia (T-ALL). First, we found a reciprocal translocation, t(6;7)(q23;q34), that juxtaposed the TCRB and C-MYB loci (n = 6 cases). Second, a genome-wide copy-number analysis by array-based comparative genomic hybridization (array-CGH) identified short somatic duplications that include C-MYB (MYB(dup), n = 13 cases of 84 T-ALL, 15%). Expression analysis, including allele-specific approaches, showed stronger C-MYB expression in the MYB-rearranged cases compared with other T-ALLs, and a dramatically skewed C-MYB allele expression in the TCRB-MYB cases, which suggests that a translocation-driven deregulated expression may overcome a cellular attempt to down-regulate C-MYB. Strikingly, profiling of the T-ALLs by clinical, genomic, and large-scale gene expression analyses shows that the TCRB-MYB translocation defines a new T-ALL subtype associated with a very young age for T-cell leukemia (median, 2.2 years) and with a proliferation/mitosis expression signature. By contrast, the MYB(dup) alteration was associated with the previously defined T-ALL subtypes.
DOI:10.1038/ng2025URLPMID:17435759 [本文引用: 1]
We identified a duplication of the MYB oncogene in 8.4% of individuals with T cell acute lymphoblastic leukemia (T-ALL) and in five T-ALL cell lines. The duplication is associated with a threefold increase in MYB expression, and knockdown of MYB expression initiates T cell differentiation. Our results identify duplication of MYB as an oncogenic event and suggest that MYB could be a therapeutic target in human T-ALL.
DOI:10.1038/leu.2016.170URLPMID:27457538 [本文引用: 1]
The c-MYB transcription factor is a key regulator of hematopoietic cell proliferation and differentiation, and dysregulation of c-MYB activity often associates with various hematological disorders. Yet, its pathogenic role remains largely unknown due to lack of suitable animal models. Here, we report a detail characterization of a c-myb-gfp transgenic zebrafish harboring c-Myb hyperactivity (named c-myb(hyper)). This line exhibits abnormal granulocyte expansion that resembles human myelodysplastic syndrome (MDS) from embryonic stage to adulthood. Strikingly, a small portion of c-myb(hyper) adult fish develops acute myeloid leukemia-like or acute lymphoid leukemia-like disorders with age. The myeloid and lymphoid malignancies in c-myb(hyper) adult fish are likely caused by the hyperactivity of c-myb, resulting in the dysregulation of a number of cell-cycle-related genes and hyperproliferation of hematopoietic precursor cells. Finally, treatment with c-myb target drug flavopiridol can relieve the MDS-like symptoms in both c-myb(hyper) embryos and adult fish. Our study establishes a zebrafish model for studying the cellular and molecular mechanisms underlying c-Myb-associated leukemogenesis as well as for anti-leukemic drug screening.
DOI:10.1182/blood-2009-10-246488URLPMID:20056790 [本文引用: 2]
Self-renewal is a feature of cancer and can be assessed by cell transplantation into immune-compromised or immune-matched animals. However, studies in zebrafish have been severely limited by lack of these reagents. Here, Myc-induced T-cell acute lymphoblastic leukemias (T-ALLs) have been made in syngeneic, clonal zebrafish and can be transplanted into sibling animals without the need for immune suppression. These studies show that self-renewing cells are abundant in T-ALL and comprise 0.1% to 15.9% of the T-ALL mass. Large-scale single-cell transplantation experiments established that T-ALLs can be initiated from a single cell and that leukemias exhibit wide differences in tumor-initiating potential. T-ALLs also can be introduced into clonal-outcrossed animals, and T-ALLs arising in mixed genetic backgrounds can be transplanted into clonal recipients without the need for major histocompatibility complex matching. Finally, high-throughput imaging methods are described that allow large numbers of fluorescent transgenic animals to be imaged simultaneously, facilitating the rapid screening of engrafted animals. Our experiments highlight the large numbers of zebrafish that can be experimentally assessed by cell transplantation and establish new high-throughput methods to functionally interrogate gene pathways involved in cancer self-renewal.
DOI:10.1007/978-1-4614-0106-3_15URLPMID:21948373 [本文引用: 1]
The zebrafish (Danio rerio) has been extensively used in biomedical research as a model to study vertebrate development and hematopoiesis and recently, it has been adopted into varied fields including immunology. After fertilization, larvae survive with only the innate immune responses because adaptive immune system is morphologically and functionally mature only after 4-6 weeks postfertilization. This temporal separation provides a suitable system to study the vertebrate innate immune response in vivo, independently from the adaptive immune response. The transparency of early life stages allows a useful real-time visualization. Adult zebrafish which have complete (innate and adaptative) immune systems offer also advantages over other vertebrate infection models: small size, relatively rapid life cycle, ease of breeding, and a growing list of molecular tools for the study of infectious diseases. In this review, we have tried to give some examples of the potential of zebrafish as a valuable model in innate immunity and inflammation studies.
DOI:10.1016/j.stem.2007.11.002URLPMID:18371439 [本文引用: 1]
Summary
The zebrafish is a useful model for understanding normal and cancer stem cells, but analysis has been limited to embryogenesis due to the opacity of the adult fish. To address this, we have created a transparent adult zebrafish in which we transplanted either hematopoietic stem/progenitor cells or tumor cells. In a hematopoiesis radiation recovery assay, transplantation of GFP-labeled marrow cells allowed for striking in vivo visual assessment of engraftment from 2 hr–5 weeks posttransplant. Using FACS analysis, both transparent and wild-type fish had equal engraftment, but this could only be visualized in the transparent recipient. In a tumor engraftment model, transplantation of RAS-melanoma cells allowed for visualization of tumor engraftment, proliferation, and distant metastases in as little as 5 days, which is not seen in wild-type recipients until 3 to 4 weeks. This transparent adult zebrafish serves as the ideal combination of both sensitivity and resolution for in vivo stem cell analyses.DOI:10.1084/jem.20110105URLPMID:21464223 [本文引用: 1]
Genomic studies in human acute lymphoblastic leukemia (ALL) have revealed clonal heterogeneity at diagnosis and clonal evolution at relapse. In this study, we used genome-wide profiling to compare human T cell ALL samples at the time of diagnosis and after engraftment (xenograft) into immunodeficient recipient mice. Compared with paired diagnosis samples, the xenograft leukemia often contained additional genomic lesions in established human oncogenes and/or tumor suppressor genes. Mimicking such genomic lesions by short hairpin RNA-mediated knockdown in diagnosis samples conferred a selective advantage in competitive engraftment experiments, demonstrating that additional lesions can be drivers of increased leukemia-initiating activity. In addition, the xenograft leukemias appeared to arise from minor subclones existing in the patient at diagnosis. Comparison of paired diagnosis and relapse samples showed that, with regard to genetic lesions, xenograft leukemias more frequently more closely resembled relapse samples than bulk diagnosis samples. Moreover, a cell cycle- and mitosis-associated gene expression signature was present in xenograft and relapse samples, and xenograft leukemia exhibited diminished sensitivity to drugs. Thus, the establishment of human leukemia in immunodeficient mice selects and expands a more aggressive malignancy, recapitulating the process of relapse in patients. These findings may contribute to the design of novel strategies to prevent or treat relapse.
DOI:10.1038/nature09733URLPMID:21248843 [本文引用: 1]
Many tumours are composed of genetically diverse cells; however, little is known about how diversity evolves or the impact that diversity has on functional properties. Here, using xenografting and DNA copy number alteration (CNA) profiling of human BCR-ABL1 lymphoblastic leukaemia, we demonstrate that genetic diversity occurs in functionally defined leukaemia-initiating cells and that many diagnostic patient samples contain multiple genetically distinct leukaemia-initiating cell subclones. Reconstructing the subclonal genetic ancestry of several samples by CNA profiling demonstrated a branching multi-clonal evolution model of leukaemogenesis, rather than linear succession. For some patient samples, the predominant diagnostic clone repopulated xenografts, whereas in others it was outcompeted by minor subclones. Reconstitution with the predominant diagnosis clone was associated with more aggressive growth properties in xenografts, deletion of CDKN2A and CDKN2B, and a trend towards poorer patient outcome. Our findings link clonal diversity with leukaemia-initiating-cell function and underscore the importance of developing therapies that eradicate all intratumoral subclones.
DOI:10.1038/leu.2012.116URLPMID:22538478 [本文引用: 1]
NOTCH1 pathway activation contributes to the pathogenesis of over 60% of T-cell acute lymphoblastic leukemia (T-ALL). While Notch is thought to exert the majority of its effects through transcriptional activation of Myc, it also likely has independent roles in T-ALL malignancy. Here, we utilized a zebrafish transgenic model of T-ALL, where Notch does not induce Myc transcription, to identify a novel Notch gene expression signature that is also found in human T-ALL and is regulated independently of Myc. Cross-species microarray comparisons between zebrafish and mammalian disease identified a common T-ALL gene signature, suggesting that conserved genetic pathways underlie T-ALL development. Functionally, Notch expression induced a significant expansion of pre-leukemic clones; however, a majority of these clones were not fully transformed and could not induce leukemia when transplanted into recipient animals. Limiting-dilution cell transplantation revealed that Notch signaling does not increase the overall frequency of leukemia-propagating cells (LPCs), either alone or in collaboration with Myc. Taken together, these data indicate that a primary role of Notch signaling in T-ALL is to expand a population of pre-malignant thymocytes, of which a subset acquire the necessary mutations to become fully transformed LPCs.
DOI:10.1016/j.ccr.2014.01.032URLPMID:24613413 [本文引用: 1]
Clonal evolution and intratumoral heterogeneity drive cancer progression through unknown molecular mechanisms. To address this issue, functional differences between single T cell acute lymphoblastic leukemia (T-ALL) clones were assessed using a zebrafish transgenic model. Functional variation was observed within individual clones, with a minority of clones enhancing growth rate and leukemia-propagating potential with time. Akt pathway activation was acquired in a subset of these evolved clones, which increased the number of leukemia-propagating cells through activating mTORC1, elevated growth rate likely by stabilizing the Myc protein, and rendered cells resistant to dexamethasone, which was reversed by combined treatment with an Akt inhibitor. Thus, T-ALL clones spontaneously and continuously evolve to drive leukemia progression even in the absence of therapy-induced selection.
DOI:10.1084/jem.20160378URLPMID:27810924 [本文引用: 2]
Cell transplantation into immunodeficient mice has revolutionized our understanding of regeneration, stem cell self-renewal, and cancer; yet models for direct imaging of engrafted cells has been limited. Here, we characterize zebrafish with mutations in recombination activating gene 2 (rag2), DNA-dependent protein kinase (prkdc), and janus kinase 3 (jak3). Histology, RNA sequencing, and single-cell transcriptional profiling of blood showed that rag2 hypomorphic mutant zebrafish lack T cells, whereas prkdc deficiency results in loss of mature T and B cells and jak3 in T and putative Natural Killer cells. Although all mutant lines engraft fluorescently labeled normal and malignant cells, only the prkdc mutant fish reproduced as homozygotes and also survived injury after cell transplantation. Engraftment into optically clear casper, prkdc-mutant zebrafish facilitated dynamic live cell imaging of muscle regeneration, repopulation of muscle stem cells within their endogenous niche, and muscle fiber fusion at single-cell resolution. Serial imaging approaches also uncovered stochasticity in fluorescently labeled leukemia regrowth after competitive cell transplantation into prkdc mutant fish, providing refined models to assess clonal dominance and progression in the zebrafish. Our experiments provide an optimized and facile transplantation model, the casper, prkdc mutant zebrafish, for efficient engraftment and direct visualization of fluorescently labeled normal and malignant cells at single-cell resolution.
DOI:10.1038/NMETH.3031URLPMID:25042784 [本文引用: 1]
Cell transplantation into adult zebrafish has lagged behind mouse models owing to the lack of immunocompromised strains. Here we have created rag2(E45fs) mutant zebrafish that have reduced numbers of functional T and B cells but are viable and fecund. Mutant fish engraft muscle, blood stem cells and various cancers. rag2(E45fs) mutant zebrafish are the first immunocompromised zebrafish model that permits robust, long-term engraftment of multiple tissues and cancer.
DOI:10.1016/j.cell.2019.04.004URLPMID:31031007 [本文引用: 3]
Xenograft cell transplantation into immunodeficient mice has become the gold standard for assessing pre-clinical efficacy of cancer drugs, yet direct visualization of single-cell phenotypes is difficult. Here, we report an optically-clear prkdc(-/-), il2rga(-/-) zebrafish that lacks adaptive and natural killer immune cells, can engraft a wide array of human cancers at 37 degrees C, and permits the dynamic visualization of single engrafted cells. For example, photoconversion cell-lineage tracing identified migratory and proliferative cell states in human rhabdomyosarcoma, a pediatric cancer of muscle. Additional experiments identified the preclinical efficacy of combination olaparib PARP inhibitor and temozolomide DNA-damaging agent as an effective therapy for rhabdomyosarcoma and visualized therapeutic responses using a four-color FUCCI cell-cycle fluorescent reporter. These experiments identified that combination treatment arrested rhabdomyosarcoma cells in the G2 cell cycle prior to induction of apoptosis. Finally, patient-derived xenografts could be engrafted into our model, opening new avenues for developing personalized therapeutic approaches in the future.
DOI:10.1038/nprot.2009.144URLPMID:19745824 [本文引用: 1]
Chemical genetic screening can be described as a discovery approach in which chemicals are assayed for their effects on a defined biological system. The zebrafish, Danio rerio, is a well-characterized and genetically tractable vertebrate model organism that produces large numbers of rapidly developing embryos that develop externally. These characteristics allow for flexible, rapid and scalable chemical screen design using the zebrafish. We describe a protocol for screening compounds from a chemical library for effects on early zebrafish development using an automated in situ based read-out. As screenings are carried out in the context of a complete, developing organism, this approach allows for a more comprehensive analysis of the range of a chemical's effects than that provided by, for example, a cell culture-based or in vitro biochemical assay. Using a 24-h chemical treatment, one can complete a round of screening in 6 d.
DOI:10.1016/j.chembiol.2003.10.003URLPMID:14583256 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nature05883URLPMID:17581586 [本文引用: 2]
Haematopoietic stem cell (HSC) homeostasis is tightly controlled by growth factors, signalling molecules and transcription factors. Definitive HSCs derived during embryogenesis in the aorta-gonad-mesonephros region subsequently colonize fetal and adult haematopoietic organs. To identify new modulators of HSC formation and homeostasis, a panel of biologically active compounds was screened for effects on stem cell induction in the zebrafish aorta-gonad-mesonephros region. Here, we show that chemicals that enhance prostaglandin (PG) E2 synthesis increased HSC numbers, and those that block prostaglandin synthesis decreased stem cell numbers. The cyclooxygenases responsible for PGE2 synthesis were required for HSC formation. A stable derivative of PGE2 improved kidney marrow recovery following irradiation injury in the adult zebrafish. In murine embryonic stem cell differentiation assays, PGE2 caused amplification of multipotent progenitors. Furthermore, ex vivo exposure to stabilized PGE2 enhanced spleen colony forming units at day 12 post transplant and increased the frequency of long-term repopulating HSCs present in murine bone marrow after limiting dilution competitive transplantation. The conserved role for PGE2 in the regulation of vertebrate HSC homeostasis indicates that modulation of the prostaglandin pathway may facilitate expansion of HSC number for therapeutic purposes.
DOI:10.1016/j.stem.2011.02.003URLPMID:21474107 [本文引用: 1]
Hematopoietic stem cells (HSCs) are used in transplantation therapy to reconstitute the hematopoietic system. Human cord blood (hCB) transplantation has emerged as an attractive alternative treatment option when traditional HSC sources are unavailable; however, the absolute number of hCB HSCs transplanted is significantly lower than bone marrow or mobilized peripheral blood stem cells (MPBSCs). We previously demonstrated that dimethyl-prostaglandin E2 (dmPGE2) increased HSCs in vertebrate models. Here, we describe preclinical analyses of the therapeutic potential of dmPGE2 treatment by using human and nonhuman primate HSCs. dmPGE2 significantly increased total human hematopoietic colony formation in vitro and enhanced engraftment of unfractionated and CD34(+) hCB after xenotransplantation. In nonhuman primate autologous transplantation, dmPGE2-treated CD34(+) MPBSCs showed stable multilineage engraftment over 1 year postinfusion. Together, our analyses indicated that dmPGE2 mediates conserved responses in HSCs from human and nonhuman primates and provided sufficient preclinical information to support proceeding to an FDA-approved phase 1 clinical trial.
DOI:10.1182/blood-2013-05-503177URLPMID:23996087 [本文引用: 1]
Umbilical cord blood (UCB) is a valuable source of hematopoietic stem cells (HSCs) for use in allogeneic transplantation. Key advantages of UCB are rapid availability and less stringent requirements for HLA matching. However, UCB contains an inherently limited HSC count, which is associated with delayed time to engraftment, high graft failure rates, and early mortality. 16,16-Dimethyl prostaglandin E2 (dmPGE2) was previously identified to be a critical regulator of HSC homeostasis, and we hypothesized that brief ex vivo modulation with dmPGE2 could improve patient outcomes by increasing the
DOI:10.1182/blood-2011-12-398818URLPMID:22490804 [本文引用: 3]
To detect targeted antileukemia agents we have designed a novel, high-content in vivo screen using genetically engineered, T-cell reporting zebrafish. We exploited the developmental similarities between normal and malignant T lymphoblasts to screen a small molecule library for activity against immature T cells with a simple visual readout in zebrafish larvae. After screening 26 400 molecules, we identified Lenaldekar (LDK), a compound that eliminates immature T cells in developing zebrafish without affecting the cell cycle in other cell types. LDK is well tolerated in vertebrates and induces long-term remission in adult zebrafish with cMYC-induced T-cell acute lymphoblastic leukemia (T-ALL). LDK causes dephosphorylation of members of the PI3 kinase/AKT/mTOR pathway and delays sensitive cells in late mitosis. Among human cancers, LDK selectively affects survival of hematopoietic malignancy lines and primary leukemias, including therapy-refractory B-ALL and chronic myelogenous leukemia samples, and inhibits growth of human T-ALL xenografts. This work demonstrates the utility of our method using zebrafish for antineoplastic candidate drug identification and suggests a new approach for targeted leukemia therapy. Although our efforts focused on leukemia therapy, this screening approach has broad implications as it can be translated to other cancer types involving malignant degeneration of developmentally arrested cells.
DOI:10.3324/haematol.2010.031401URLPMID:21228037 [本文引用: 1]
Zebrafish were proposed as an alternative to mammalian models to assess the efficacy and toxicity of antileukemic drugs. Due to the limited number of transgenic zebrafish leukemia models, we explored human leukemic cell xenograft in zebrafish embryos. Human leukemic cell lines and blast cells sorted from patients with acute myelogenous leukemia were injected 48 hours post-fertilization and remained in the circulation of zebrafish embryos for several days without affecting their development. Imatinib and oxaphorines did not demonstrate any toxicity on normal zebrafish embryos and decreased the leukemic burden in animals xenografted with sensitive leukemic cell lines. Two other molecules, all-trans retinoic acid and the translation inhibitor 4EGI-1, demonstrated teratogenic effects at concentrations shown to be efficient in vitro, which precluded investigation of their antileukemic activity in such models. Altogether, xenografted leukemic cells in zebrafish embryos are a pharmacologically relevant model for screening non-teratogenic drugs.
DOI:10.1242/dmm.031534URLPMID:30061196 [本文引用: 1]
Deletions of chromosome 1p36 are associated with a high incidence of congenital heart defects (CHDs). The arginine-glutamic acid dipeptide repeats gene (RERE) is located in a critical region for CHD on chromosome 1p36 and encodes a cardiac-expressed nuclear receptor co-regulator. Mutations affecting RERE cause atrial and ventricular septal defects (VSDs) in humans, and RERE-deficient mice also develop VSDs. During cardiac development, mesenchymal cells destined to form part of the atrioventricular (AV) septum are generated when endocardial cells in the AV canal undergo epithelial-to-mesenchymal transition (EMT) and migrate into the space between the endocardium and the myocardium. These newly generated mesenchymal cells then proliferate to fill the developing AV endocardial cushions. Here, we demonstrate that RERE-deficient mouse embryos have reduced numbers of mesenchymal cells in their AV endocardial cushions owing to decreased levels of EMT and mesenchymal cell proliferation. In the endocardium, RERE colocalizes with GATA4, a transcription factor required for normal levels of EMT and mesenchymal cell proliferation. Using a combination of in vivo and in vitro studies, we show that Rere and Gata4 interact genetically in the development of CHDs, RERE positively regulates transcription from the Gata4 promoter and GATA4 levels are reduced in the AV canals of RERE-deficient embryos. Tissue-specific ablation of Rere in the endocardium leads to hypocellularity of the AV endocardial cushions, defective EMT and VSDs, but does not result in decreased GATA4 expression. We conclude that RERE functions in the AV canal to positively regulate the expression of GATA4, and that deficiency of RERE leads to the development of VSDs through its effects on EMT and mesenchymal cell proliferation. However, the cell-autonomous role of RERE in promoting EMT in the endocardium must be mediated by its effects on the expression of proteins other than GATA4.This article has an associated First Person interview with the first author of the paper.
DOI:10.1002/wdev.312URLPMID:29436122 [本文引用: 1]
Hematopoiesis is a complex process with a variety of different signaling pathways influencing every step of blood cell formation from the earliest precursors to final differentiated blood cell types. Formation of blood cells is crucial for survival. Blood cells carry oxygen, promote organ development and protect organs in different pathological conditions. Hematopoietic stem and progenitor cells (HSPCs) are responsible for generating all adult differentiated blood cells. Defects in HSPCs or their downstream lineages can lead to anemia and other hematological disorders including leukemia. The zebrafish has recently emerged as a powerful vertebrate model system to study hematopoiesis. The developmental processes and molecular mechanisms involved in zebrafish hematopoiesis are conserved with higher vertebrates, and the genetic and experimental accessibility of the fish and the optical transparency of its embryos and larvae make it ideal for in vivo analysis of hematopoietic development. Defects in zebrafish hematopoiesis reliably phenocopy human blood disorders, making it a highly attractive model system to screen small molecules to design therapeutic strategies. In this review, we summarize the key developmental processes and molecular mechanisms of zebrafish hematopoiesis. We also discuss recent findings highlighting the strengths of zebrafish as a model system for drug discovery against hematopoietic disorders. This article is categorized under: Adult Stem Cells, Tissue Renewal, and Regeneration > Stem Cell Differentiation and Reversion Vertebrate Organogenesis > Musculoskeletal and Vascular Nervous System Development > Vertebrates: Regional Development Comparative Development and Evolution > Organ System Comparisons Between Species.
DOI:10.1158/2159-8290.CD-14-0001URLPMID:25185190 [本文引用: 1]
UNLABELLED: Recently, there has been an increasing interest in the development and characterization of patient-derived tumor xenograft (PDX) models for cancer research. PDX models mostly retain the principal histologic and genetic characteristics of their donor tumor and remain stable across passages. These models have been shown to be predictive of clinical outcomes and are being used for preclinical drug evaluation, biomarker identification, biologic studies, and personalized medicine strategies. This article summarizes the current state of the art in this field, including methodologic issues, available collections, practical applications, challenges and shortcomings, and future directions, and introduces a European consortium of PDX models. SIGNIFICANCE: PDX models are increasingly used in translational cancer research. These models are useful for drug screening, biomarker development, and the preclinical evaluation of personalized medicine strategies. This review provides a timely overview of the key characteristics of PDX models and a detailed discussion of future directions in the field.
DOI:10.1158/0008-5472.CAN-13-1069URLPMID:23733750 [本文引用: 1]
Tumor graft models (also known as patient-derived xenografts or PDX) are based on the transfer of primary tumors directly from the patient into an immunodeficient mouse. Because PDX mice are derived from human tumors, they offer a tool for developing anticancer therapies and personalized medicine for patients with cancer. In addition, these models can be used to study metastasis and tumor genetic evolution. This review examines the development, challenges, and broad use of these attractive preclinical models.
DOI:10.3324/haematol.2014.110742URLPMID:25281505 [本文引用: 1]
Cancer therapeutics is evolving to precision medicine, with the goal of matching targeted compounds with molecular aberrations underlying a patient's cancer. While murine models offer a pre-clinical tool, associated costs and time are not compatible with actionable patient-directed interventions. Using the paradigm of T-cell acute lymphoblastic leukemia, a high-risk disease with defined molecular underpinnings, we developed a zebrafish human cancer xenotransplantation model to inform therapeutic decisions. Using a focused chemical genomic approach, we demonstrate that xenografted cell lines harboring mutations in the NOTCH1 and PI3K/AKT pathways respond concordantly to their targeted therapies, patient-derived T-cell acute lymphoblastic leukemia can be successfully engrafted in zebrafish and specific drug responses can be quantitatively determined. Using this approach, we identified a mutation sensitive to gamma-secretase inhibition in a xenograft from a child with T-cell acute lymphoblastic leukemia, confirmed by Sanger sequencing and validated as a gain-of-function NOTCH1 mutation. The zebrafish xenotransplantation platform provides a novel cost-effective means of tailoring leukemia therapy in real time.
DOI:10.1111/j.1365-2141.2011.08661.xURLPMID:21517816 [本文引用: 1]
DOI:10.12688/f1000research.14507.2URLPMID:29946444 [本文引用: 1]
Haematopoietic stem cell (HSC) transplantation is a critical therapy for haematopoietic malignancies and immune disorders. Incomplete or delayed engraftment of HSCs in the host results in increased risk of infection and morbidity. The mechanisms of HSC engraftment are poorly understood and understanding these processes will increase transplantation success on many levels. Current animal models are immunocompromised 'humanised' mice transplanted with human HSCs. Harmful procedures include genetic manipulations and irradiation to ablate the mouse immune system, and opaque mouse tissues make visualisation of the early steps of HSC engraftment impossible. There is a need for new models to offer alternatives to humanised mice in the study of HSC transplantation. Here we described a detailed method for transplantation of human HSCs into zebrafish, before the onset of adaptive immunity. Human HSCs were purified from whole blood by enrichment of the CD34 cell population using a positive magnetic selection and further purified using an anti-CD34 antibody and cell sorting. Sorted CD34 cells were transplanted into the blood stream of 52 hour old zebrafish larvae. Human HSCs home into the zebrafish haematopoietic niche, where they engage with endothelial cells and undergo cell division. Our model offers the opportunities to image in vivo human HSC engraftment in a transparent organism, without the myeloablative strategies used in mice, and provides a unique system to understand the dynamic process of engraftment and replace current murine models. This technique can be applied to current engraftment protocols to validate the viability and efficiency of cryofrozen HSC grafts. This humanised zebrafish model will be instrumental to develop the 3Rs values in stem cell transplantation research and our detailed protocol will increase the chances of uptake of this zebrafish model by the mouse community.
URLPMID:32732367 [本文引用: 1]
Hemophagocytic lymphohistiocytosis (HLH) is an immune-regulatory disorder characterized by excessive production of inflammatory cytokines. The treatment recommendations of the HLH-1994 and HLH-2004 protocols have long been used in HLH therapy, but some patients still do not respond well to or have unacceptable side effects from conventional therapies. It is believed that cytokine-targeted strategies that directly target disease-driving pathways will be promising options for HLH. This prospective study aimed to investigate the efficacy and safety of ruxolitinib, a Janus kinase (JAK) 1/2 inhibitor, as a front-line therapy in children with secondary HLH. Twelve newly diagnosed patients without previous treatment were enrolled in this study with a median follow-up of 8.2 (7.1-12.0) months, including 8 cases of Epstein-Barr virus associated HLH (EBV-HLH), 2 cases of autoinflammatory disorder (AID)- associated HLH, and 2 cases of unknown etiology. Patients received oral ruxolitinib dosed on 2.5 mg, 5 mg or 10 mg twice daily depending on the body weight for 28 consecutive days. The overall response rate at the end of treatment (day 28) was 83.3% (10/12), with 66.7% (8/12) in complete response (CR), 8.3% (1/12) in partial response (PR), and 8.3% (1/12) in HLH improvement. Among the patients achieving CR, 87.5% (7/8) maintained CR condition for>6 months, and one patient with EBV-HLH relapsed following CR. For the EBV-HLH subgroup, all 8 patients responded to ruxolitinib, with a CR rate of 75% and a PR rate of 25%. Two patients with AID-associated HLH had quite different responses, with one showing reversal of the HLH abnormalities soon and the other showing no improvement, as did the two cases of unknown etiology. Patients who had no response or discontinued ruxolitinib all responded well to the subsequent HLH-1994 regimen. The expected 6-month event-free survival (EFS) rate was 58.3%+/-10.2%. No serious adverse effects were reported. Our study provides further support for the possibility of ruxolitinib targeted therapy for secondary HLH in children. This study was registered in the Chinese Clinical Trials Registry Platform (http://www.chictr.org.cn/) as ChiCTR2000029977.
DOI:10.1038/nprot.2010.8URLPMID:20203658 [本文引用: 1]
Transplantable tumors are an accepted gold standard in cancer studies in rodents. The progress of this model in zebrafish has long been constrained by the lack of true inbred lines in zebrafish. We have generated several lines of homozygous diploid clonal zebrafish lines, which allow serial transplantations of tumor cells from one fish to another without sublethal gamma-irradiation. The spectrum of transplantable tumors that were initially induced and maintained in inbred clonal zebrafish lines was limited to different types of spontaneous and diethylnitrosamine-induced hepatic tumors. However, this model can readily be extended to a broad range of extrahepatic tumors, transgenic tumors with defined mechanisms of induction and fluorescence-tagged tumor lines. These models will further facilitate in-depth analysis of invasive tumor growth, angiogenesis, metastasis and tumor-initiating cells by in vivo imaging and provide a cost-effective system for high-throughput (HTP) screening of anticancer therapeutics, including biological response modifiers. In addition, homozygous zebrafish lines are an indispensable tool for immunogenetics, mapping of quantitative trait loci and other genetic applications. The whole procedure, from generation of a gynogenetic female homozygous fish (a founder) to obtaining 3-4 consecutive passages of a syngeneic tumor, takes approximately 12-18 months. This time-frame largely depends on methods of tumor induction, tumor type and tumor growth rate.
DOI:10.1111/pcmr.12592URLPMID:28379616 [本文引用: 1]
Melanoma is the most aggressive and deadliest form of skin cancer. A detailed knowledge of the cellular, molecular, and genetic events underlying melanoma progression is highly relevant to diagnosis, prognosis and risk stratification, and the development of new therapies. In the last decade, zebrafish have emerged as a valuable model system for the study of melanoma. Pathway conservation, coupled with the availability of robust genetic, transgenic, and chemical tools, has made the zebrafish a powerful model for identifying novel disease genes, visualizing cancer initiation, interrogating tumor-microenvironment interactions, and discovering new therapeutics that regulate melanocyte and melanoma development. In this review, we will give an overview of these studies, and highlight recent advancements that will help unravel melanoma pathogenesis and impact human disease.
DOI:10.1038/s41591-019-0479-2URLPMID:31263281 [本文引用: 1]
The treatment of lymphatic anomaly, a rare devastating disease spectrum of mostly unknown etiologies, depends on the patient manifestations(1). Identifying the causal genes will allow for developing affordable therapies in keeping with precision medicine implementation(2). Here we identified a recurrent gain-of-function ARAF mutation (c.640T>C:p.S214P) in a 12-year-old boy with advanced anomalous lymphatic disease unresponsive to conventional sirolimus therapy and in another, unrelated, adult patient. The mutation led to loss of a conserved phosphorylation site. Cells transduced with ARAF-S214P showed elevated ERK1/2 activity, enhanced lymphangiogenic capacity, and disassembly of actin skeleton and VE-cadherin junctions, which were rescued using the MEK inhibitor trametinib. The functional relevance of the mutation was also validated by recreating a lymphatic phenotype in a zebrafish model, with rescue of the anomalous phenotype using a MEK inhibitor. Subsequent therapy of the lead proband with a MEK inhibitor led to dramatic clinical improvement, with remodeling of the patient's lymphatic system with resolution of the lymphatic edema, marked improvement in his pulmonary function tests, cessation of supplemental oxygen requirements and near normalization of daily activities. Our results provide a representative demonstration of how knowledge of genetic classification and mechanistic understanding guides biologically based medical treatments, which in our instance was life-saving.

{kind=link}
{kind=link}