 ,2, 周钢桥,1,2
,2, 周钢桥,1,2Prognostic and predictive value of a DNA methylation-driven transcriptional signature in hepatocellular carcinoma
Hongbo Luo1, Pengbo Cao,2, Gangqiao Zhou,1,2通讯作者: 曹鹏博,博士,副研究员,研究方向: 医学遗传与基因组学。E-mail:birchcpb@163.com周钢桥,博士,研究员,研究方向:医学遗传与基因组学。E-mail:zhougq114@126.com
编委: 方向东
收稿日期:2020-05-18修回日期:2020-07-10网络出版日期:2020-08-20
| 基金资助: |
Received:2020-05-18Revised:2020-07-10Online:2020-08-20
| Fund supported: |
作者简介 About authors
骆红波,硕士研究生,专业方向:肝癌转录组学。E-mail:

摘要
肝细胞癌(hepatocellular carcinoma,简称肝癌)是最常见的恶性肿瘤之一。DNA甲基化的异常是恶性肿瘤的特征之一,并被发现在肝癌等肿瘤的发生发展中发挥重要作用。为了能为肝癌患者提供新的临床预后预测标志物,本研究首先采用整合组学分析策略在全基因组范围内鉴定与肝癌患者预后相关的DNA甲基化驱动的差异表达基因;然后,采用LASSO (least absolute shrinkage and selection operator)分析建立了10个最优基因组合的预后预测模型。Cox比例风险回归分析显示,在校正临床特征参数后,此预测模型高风险评分与患者不良预后显著相关,表明该模型具有潜在的独立预后价值。受试者工作特征(receiver operating characteristic, ROC)曲线分析显示该风险评分模型在预测患者短期和长期预后方面优于其他已被报道的肝癌预后预测模型。基因集富集分析(gene set enrichment analysis, GSEA)表明,高风险评分与细胞周期和DNA损伤修复通路相关。以上结果表明,本研究构建了一个基于10个DNA甲基化驱动基因的预后风险评分模型,该模型可作为肝癌患者的潜在预后生物标志物,有助于肝癌患者的生存预后评估和治疗策略的指导。
关键词:
Abstract
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide. DNA methylation alterations are frequently observed in malignant tumours and play critical roles in the development of cancers, including HCC. To provide novel clinical prognosis biomarkers for HCC patients, we first performed a comprehensive analysis and identified a collection of prognosis-associated genes with DNA methylation-driven expression dysregulation in HCCs. Then, we optimally established a 10-gene prognostic risk score model using the least absolute shrinkage and selection operator (LASSO) analysis. Cox's proportional hazards regression analysis revealed that the high-risk score is significantly associated with poor prognosis after being adjusted by clinical parameters, indicating its potential prognostic value. The receiver operating characteristic curve (ROC) analysis showed that this 10-gene prognostic risk score model outperformed several other publicly available models in predicting both short- and long-term prognosis. Gene set enrichment analysis revealed that the high-risk score is relevantly associated with pathways involved in cell cycle and DNA damage repair. The above results indicate that we have constructed a 10-DNA-methylation-driven-gene prognostic risk score model, which might serve as a potential prognostic biomarker for HCC patients and guide treatment decisions for patients at high risk of tumour progression.
Keywords:
PDF (1991KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
骆红波, 曹鹏博, 周钢桥. DNA甲基化驱动的转录表达特征作为肝癌预后预测标志物的价值. 遗传[J], 2020, 42(8): 775-787 doi:10.16288/j.yczz.20-139
Hongbo Luo.
肝细胞癌(hepatocellular carcinoma;简称肝癌)是最常见的原发性肝癌,也是全球范围内与癌症相关死亡的主要原因之一[1]。尽管现代医学的发展和多种治疗策略的结合较好地改善了肝癌患者的临床预后,但由于肝癌的高转移或高复发率,这些患者的长期预后状况仍然较差[2]。鉴定新的与肝癌复发及生存相关的分子,能为肝癌患者的临床预后预测提供新的候选标志物。
DNA甲基化(methylation)是最早被发现的DNA修饰类型之一。已有研究表明DNA甲基化能够引起染色质结构和DNA稳定性等发生改变,从而调控基因的表达[3]。位于启动子区域的异常DNA甲基化通常导致抑癌基因的转录沉默或癌基因的高表达,从而促进肿瘤的进展[4]。因此,DNA甲基化异常在肝癌等肿瘤的发生发展中发挥重要作用[5]。
为建立肝癌相关异常DNA甲基化所调控基因的预后预测模型,本研究通过肝癌组织的转录组和表观基因组的整合分析,鉴定出一系列与肝癌预后相关的DNA甲基化驱动的差异表达基因,并建立了一个高置信的肝癌预后预测模型,为肝癌患者的预后风险分层、预后评估及治疗策略的选择提供了新的参考指标,具有一定的潜在应用价值。
1 材料与方法
1.1 研究对象
本研究主要分为3个阶段:候选DNA甲基化驱动的差异表达基因的发掘阶段、模型训练阶段和模型验证阶段,共包括8个肝癌队列(1042个肝癌临床组织样本;表1,图1A)。对于涉及患者临床资料的分析,去除生存时间未知和TNM (tumour node metastasis)分期未知的患者。Table 1
表1
表1本研究中涉及的所有肝癌队列
Table 1
| 研究队列 | 数据集 | 样本量 | 数据类型 | 数据来源 |
|---|---|---|---|---|
| 发掘队列 | SRP069212 | 20例配对的癌和癌旁组织 | mRNA表达 | GEO |
| SRP118972 | 12例癌组织样本和8例癌旁组织 | mRNA表达 | GEO | |
| GSE89852 | 33例配对的癌和癌旁组织 | DNA甲基化 | GEO | |
| GSE54503 | 66例配对的癌和癌旁组织 | DNA甲基化 | GEO | |
| 模型训练队列 | TCGA-LIHC | 371例癌组织和50例癌旁组织 | mRNA表达和DNA甲基化 | TCGA |
| ICGC-LIRI-JP | 203例癌组织 | mRNA表达 | ICGC | |
| 模型验证队列 | GSE76427 | 115例癌组织 | 基因表达 | GEO |
| GSE84005 | 37例癌组织 | 基因表达 | GEO |
新窗口打开|下载CSV
发掘阶段:从基因表达汇编数据库(gene expression omnibus, GEO)下载获得2个肝癌组织及癌旁配对组织的DNA甲基化数据集(GEO: GSE89852和GSE54503),以及2个肝癌组织及癌旁配对组织的转录组RNA测序数据集(GEO: SRP069212和SRP118972)。
模型建立阶段:从癌症基因组图谱-肝细胞癌项目(the cancer genome atlas-liver hepatocellular carcinoma, TCGA-LIHC)下载获得level 3转录组RNA测序数据和DNA甲基化数据以及临床信息数据。该队列用于验证发掘阶段所鉴定出的DNA甲基化驱动的差异表达基因及建立和训练预后预测模型。
模型验证阶段:包括3个独立的肝癌队列。从国际癌症基因组联盟(international cancer genome consortium, ICGC)获得的日本肝癌项目(liver cancer- RIKEN of JP project)的转录组基因表达谱数据集作为第1个模型验证队列,从GEO下载的GSE76427以及GSE84005表达谱数据集分别作为第2和3个模型验证队列。
1.2 差异表达基因及差异甲基化基因的鉴定
针对GEO下载的RNA测序数据,采用STAR软件[6]以hg19基因组为参考进行比对,然后采用HTseq-count软件[7]进行基因表达的定量。采用DESeq2软件[8]进行差异表达分析,差异表达基因(differently expressed gene, DEG)的筛选标准为|log2[fold change]|≥1.5且错误发现率(false discovery rate,FDR)<0.05。
采用ChAMP软件[9]处理DNA甲基化数据,通过limma软件包[10]对肝癌和癌旁间每个CpG位点甲基化水平(β)进行t检验。差异甲基化的CpG位点定义为:|Δβ|>0.25且FDR<0.05。对于匹配到多个DNA甲基化探针的基因,选择甲基化水平倍数变化显著的作为代表[11]。
利用维恩图获得上述发掘的2个候选基因集合(即差异表达基因和差异甲基化基因)间的重叠基因,即可得到肝癌组织中相对癌旁组织“高甲基化-低表达”和“低甲基化-高表达”的基因,作为后续LASSO (least absolute shrinkage and selection operator)回归分析的候选基因。
1.3 预后预测评分模型的建立
基于发掘的候选基因,采用两个步骤来构建预后预测评分模型。首先,通过单因素Cox回归分析获得与模型训练队列中患者总体生存期(overall survival)相关的候选基因(P<0.05),然后通过具有Cox比例风险模型的LASSO回归根据最佳惩罚因子(λ)对候选基因进行降维筛选[12]。LASSO回归是一种根据λ缩小回归系数的方法,一些系数可能会缩小为0,然后从模型中删除。将具有Cox比例风险模型的LASSO回归应用于模型训练队列,然后基于10倍交叉验证(10-fold cross validation)确定最佳惩罚因子,进而估计模型系数。如果系数为0,则删除对应的基因,然后将保留系数不为0的基因用于构建预后预测评分模型。每位患者的风险分数可以通过以下公式计算得出:风险评分(Risk score)=(EXPgene1×βgene1)+(EXPgene2×βgene2)+…+(EXPgenen×βgenen)
其中EXPgene代表某一特定基因的表达水平,βgene代表LASSO回归系数。
以患者总体风险评分的中位数为界,将队列中的患者划分为高风险组和低风险组。采用单因素和多因素Cox回归分析计算该风险模型对总体生存期的风险比例(hazard ratios, HR)。采用时间依赖的受试者工作特征曲线(time-dependent receiver operating characteristic curve, time-dependent ROC)评估该风险模型在预测患者预后方面的性能[13]。
1.4 列线图的建立
用多因素Cox回归构建列线图(nomogram)来量化患者的风险评估,进而预测患者的临床预后。分析中纳入模型训练队列和模型验证队列共有的临床参数,包括:性别、年龄、肿瘤分期和基于预测模型计算的风险评分。列线图通过rms工具包构建,同时采用校准曲线(calibration curves)用来评价该模型在预测患者生存的准确性[14]。1.5 基因集富集分析
基于KEGG基因集,采用基因富集分析(gene set enrichment analysis, GSEA)对高、低风险组(基于中位数分组)进行富集分析,鉴定与生存风险相关的信号通路,参考基因集为MsigDB (6.2版本),样本置换检验1000次,FDR<0.05作为差异显著性的评价标准。1.6 统计分析
采用R 3.4.4软件进行所有的统计学分析。采用survival工具包绘制Kaplan-Meier生存曲线,采用Log-rank检验计算生存率差异显著性。采用χ2检验计算组间患者临床特征分布差异。对于所有的假设检验,P<0.05被认为有统计学意义。2 结果与分析
2.1 鉴定肝癌组织中DNA甲基化驱动的差异表达基因
为了鉴定肝癌中DNA甲基化变化驱动的差异表达基因,本研究在发掘队列肝癌组织和配对癌旁组织的数据集中进行了全基因组层面的差异表达基因及差异DNA甲基化基因的整合分析。通过差异表达分析共鉴定到547个基因在肝癌组织中上调表达和291个基因在肝癌组织中下调表达(图1B);通过差异甲基化分析共鉴定到788个基因在肝癌组织中高甲基化和5126个基因在肝癌组织中低甲基化(图1B)。通过取这两组基因集合的交集,在发掘队列中共鉴定出197个“低甲基化-高表达”基因和18个“高甲基化-低表达”基因(图1B)。在这215个基因中,有163个基因(占75.8%)在模型训练队列中被成功地重复,其中包括153个“低甲基化-高表达”基因和10个“高甲基化-低表达”基因。热图显示这163个候选基因在发掘队列和模型训练队列中DNA甲基化水平和表达水平高度一致(图1C)。图1
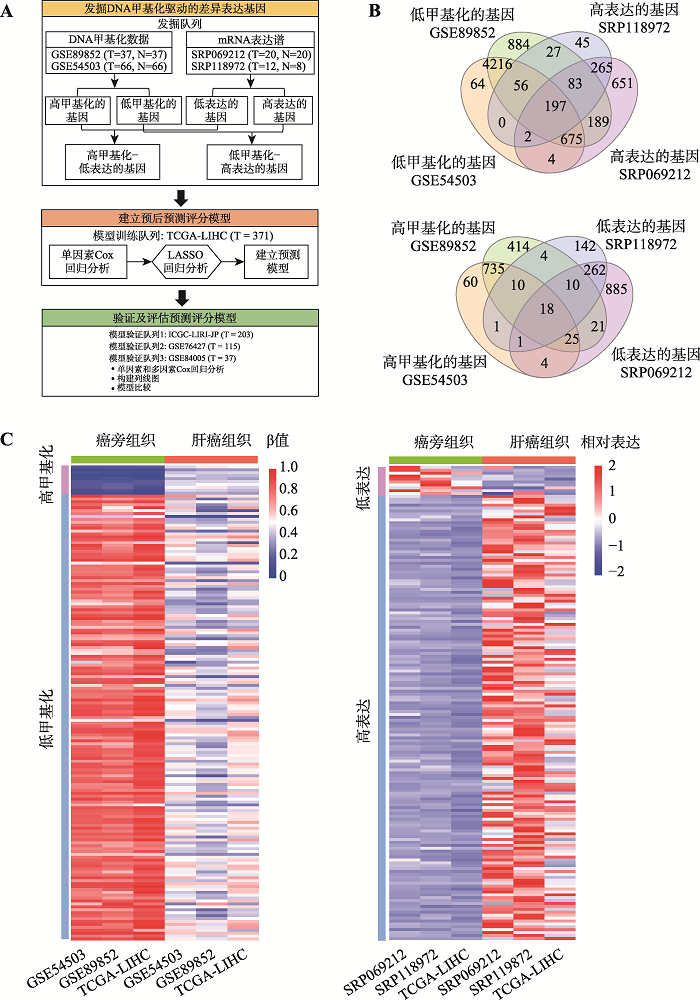 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1肝癌中DNA甲基化驱动的差异表达基因的鉴定
A:研究技术路线图。主要包括候选DNA甲基化驱动的差异表达基因的发掘阶段、模型训练阶段和模型验证和评估阶段。B:在发掘队列中鉴定出的DNA甲基化驱动的差异表达基因数量。上图为鉴定出的“低甲基化-高表达”基因数量,下图为鉴定出“高甲基化-低表达”基因数量。C:在发掘队列和模型训练队列中DNA甲基化驱动的差异表达基因的热图。左图为基因DNA甲基化水平热图(平均甲基化水平),右图为基因表达热图(平均表达水平)。ICGC-LIRI-JP:国际癌症基因组联盟日本肝癌项目(international cancer genome consortium liver cancer-RIKEN of JP project);N:肝癌癌旁组织数量;T:肝癌组织数量;TCGA-LIHC:癌症基因组图谱-肝细胞癌项目(the cancer genome atlas-liver hepatocellular carcinoma);β:基因的DNA甲基化水平。
Fig. 1Identification of the DNA methylation-driven differentially expressed genes in HCC
2.2 采用LASSO回归分析建立由10个基因组合的肝癌预后预测评分模型
针对这163个基因,首先在模型训练队列中进行了单因素Cox比例风险回归分析,共鉴定出51个与肝癌患者预后显著相关的候选基因(均P<0.05)。随后,LASSO回归进一步降维筛选出其中10个有显著性的候选基因组合用于风险模型的构建(MAEL、PRC1、TTC39A、SFN、LPL、STC2、PBK、CDCA8、MYO18B和MAPT;表2,图2A)。除此之外,在模型训练队列中对这10个基因的基因组变异频率进行分析,结果显示它们均表现出低频(≤10%)的点突变或拷贝数变异(表2),表明表观遗传的改变可能是导致这些基因在肝癌中发生表达异常的主要原因。Table 2
表2
表210个最优基因的Cox比例风险回归分析结果、LASSO回归系数和基因组变异频率
Table 2
| 基因 | HR (95% CI) | P值 | LASSO系数 | 基因组变异频率(%) |
|---|---|---|---|---|
| CDCA8 | 2.21 (1.54~3.01) | <0.0001 | 0.1194 | 0.0 |
| PRC1 | 1.85 (1.29~2.54) | 0.0005 | 0.08869 | 0.3 |
| MAPT | 1.72 (1.21~2.46) | 0.0021 | 0.2597 | 1.7 |
| SFN | 1.72 (1.21~2.45) | 0.0021 | 0.001652 | 0.3 |
| STC2 | 1.73 (1.22~2.43) | 0.0021 | 0.03600 | 0.8 |
| MYO18B | 1.69 (1.19~2.37) | 0.0031 | 0.1932 | 4.0 |
| PBK | 1.67 (1.17~2.35) | 0.0036 | 0.06524 | 6.0 |
| MAEL | 1.55 (1.09~2.17) | 0.013 | -0.1766 | 10.0 |
| TTC39A | 1.55 (1.09~2.17) | 0.013 | -0.01347 | 1.4 |
| LPL | 1.55 (1.10~2.18) | 0.014 | 0.003126 | 7.0 |
新窗口打开|下载CSV
基于这10个基因的表达水平及其对应的LASSO回归分析的系数,在模型训练队列中建立了预后评分模型:风险评分(Risk score)=(-0.1766× EXPMAEL)+(0.08869×EXPPRC1)+(0.01347×EXPTTC39A)+(0.001652×EXPSFN)+(0.003126×EXPLPL)+(0.03600× EXPSTC2)+(0.06524×EXPPBK)+(0.1194×EXPCDCA8)+ (0.1932×EXPMYO18B)+(0.2597×EXPMAPT)。根据风险评分的中位数,将肝癌患者分为高风险组和低风险组(表3)。单因素Cox比例风险回归分析显示,高风险评分与患者较短的总体生存率显著相关(P<0.0001;图2B)。进一步采用多因素Cox分析校正患者的肿瘤分期,结果显示高风险评分仍然是肝癌患者总体生存期的独立风险因素(P<0.0001;表3)。此外,该风险评分预测患者1年、2年、3年、4年和5年的生存率对应曲线下面积AUC(areas under the curve)分别为0.82、0.74、0.72、0.68和0.66 (图2B),提示该模型能较好预测模型队列中患者预后情况。
2.3 基于10个基因的肝癌预后预测评分模型在验证队列中表现出良好的预测能力
随后,在另外3个独立肝癌队列中验证此预测评分模型的性能。在验证队列1(包含203例肝癌患者)中,高风险评分与肝癌患者较短的总体生存期显著相关(P=0.0015,图2C);在校正患者的肿瘤分期后,高风险评分仍然显示为肝癌患者总体生存期的独立风险因素(P=0.025;表3);ROC分析显示,虽然预测模型在验证队列1中的预测能力略差于发掘队列,但该模型仍具有良好的预测性能,预测模型在此队列中预测患者1年、2年、3年和4年生存率的AUC分别为0.67、0.75、0.68和0.63 (图2C)。在验证队列2(包含115例肝癌患者)中,虽然没有临床特征参数表现出显著的预后价值(表3),但高风险评分也与肝癌患者较短的总体生存期显著相关(P= 0.0014;图2C);风险评分预测1年、2年、3年、4年和5年生存率的AUC分别为0.63、0.60、0.63、0.69和0.69 (图2C)。在验证队列3 (包含37例肝癌患者)中,虽然没有临床特征参数表现出显著的预后价值,但是风险评分仍然与肝癌患者的总体生存期显著相关(P=0.0016;图2C) (表3);且其预测肝癌患者1年、2年、3年和4年生存率的AUC分别达到0.79、0.71、0.66和0.57 (图2C)。综上,本研究建立的预测评分模型可以显著地将肝癌患者分为预后高风险组和低风险组。图2
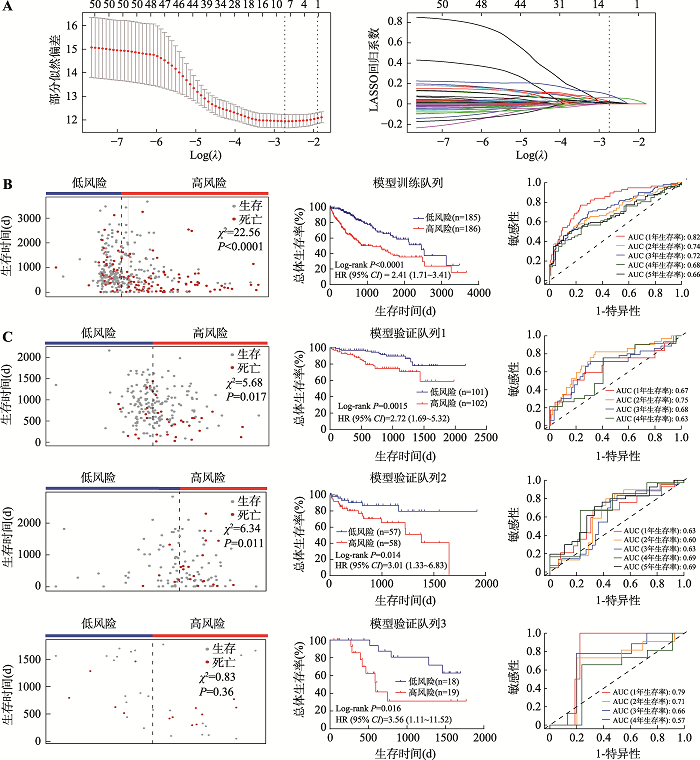 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2建立和验证10个基因的预后评分模型
A:使用LASSO回归分析和10倍交叉验证构建预后预测评分模型。左图为基于最小原则(minimum criteria)采用10倍交叉验证对LASSO模型进行调参,通过LASSO回归交叉验证计算的部分似然偏差(partial likelihood deviance)被绘制为log(λ)的函数。y轴表示部分似然偏差,x轴表示log(λ),沿x轴上方的数字表示预测变量的平均数量,红点表示具有给定λ的每个模型的平均偏差值,穿过红点的竖线表示偏差的上限值和下限值,垂直虚线分别表示最小误差的λ值和最大λ值。右图为51个预后基因的LASSO系数分布,垂直虚线表示采用10倍交叉验证选取的基因数,当基因数为10时,部分似然偏差为最小值,对应最小λ值。B:基于LASSO系数和基因表达在模型训练队列建立预后模型。左图为模型训练队列中肝癌患者的风险评分及生存时间的散点图,中间图为模型训练队列中不同风险评分组(中位数分组)患者的生存曲线图,右图为采用ROC曲线分析评估预测模型对训练队列中患者生存率的预测性能。C:在模型验证队列中验证预后模型。左图为模型验证队列中肝癌患者的风险评分及生存时间的散点图,中间图为模型验证队列中不同风险评分组(中位数分组)患者的生存曲线图,右图为ROC曲线分析评估预测模型对验证队列中患者生存率的预测性能。由于验证队列1和3中患者达到5年生存的数量较少,所以并未对患者5年生存率进行ROC分析。卡方检验(χ2)用于评价组间患者生存分布差异;组间生存差异采用Log-rank方法进行比较;ROC分析用于评估模型预测性能。AUC:曲线下面积(area under curve);CI:置信区间(confidence interval);HR:风险比例(hazard ratios);LASSO:最小绝对值收敛和选择算子(least absolute shrinkage and selection operator);ROC:受试者工作特征曲线(receiver operating characteristic curve)。
Fig. 2Construction and validation of the 10-gene prognostic risk score model
Tbale 3
Tbale 3Univariate and multivariate survival analyses of variables associated with overall survival in HCC patients
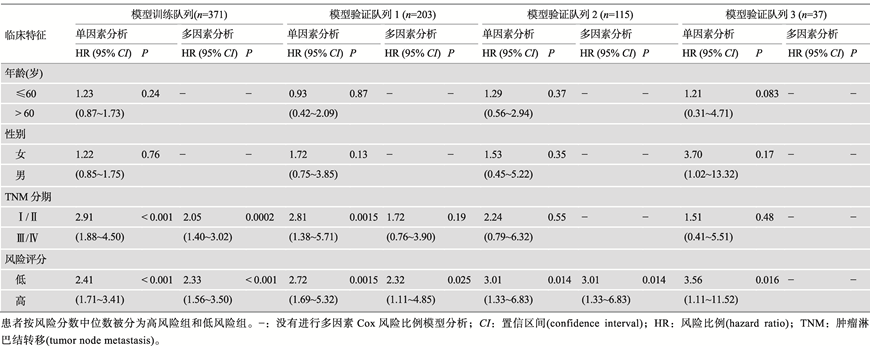 |
新窗口打开|下载CSV
2.4 整合的临床参数与预后预测评分模型建立列线图
为了进一步建立可应用于预测肝癌患者总体生存率预测的直接定量方法,在模型训练队列中,将预测模型评分和临床特征参数进行多因素Cox比例风险回归分析构建列线图(图3A)。列线图显示,相比较其他临床特征参数,预后预测评分模型贡献了最大的风险值(范围为0~100) (图3A),提示此预测模型在所有列线图变量中的作用最为显著。由于本研究中有2个验证队列生存超过4年的样本例数较少,为了避免结果的偏倚,校正曲线图只计算3年生存期预测的准确性。结果可见,通过列线图构建的模型能够较好地预测肝癌患者的3年生存状态(图3B)。图3
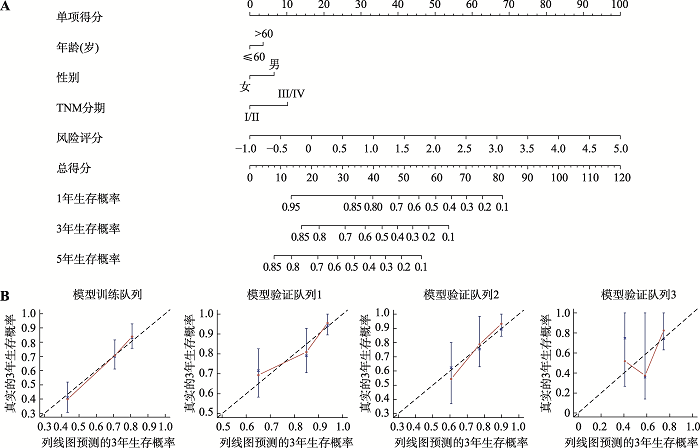 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3基于列线图评估肝癌患者的预后
A:列线图预测模型训练队列中患者1年、3年和5年的生存情况;B:校正曲线描绘列线图预测患者3年生存与实际情况之间的一致性。粉红色实线表示列线图的预测性能,45°斜线表示一种理想的校正模型。TNM:肿瘤淋巴结转移(tumor node metastasis)。
Fig. 3The nomogram for predicting the overall survival of HCC patients
2.5 基于10个基因的肝癌预后预测评分模型优于其他模型
为了进一步评估此模型的预测性能,本研究比较了该预测模型与上述建立的列线图、肿瘤分期系统以及3个已被报道的肝癌预后模型[Zheng等[15]的4基因模型(SPINK1、TXNRD1、LCAT、PZP)、Yang等[16]的3基因模型(SPP2、CDC37L1、ECHDC2)和Long等[17]的2基因模型(SPP1、LCAT)]的预测性能。有趣的是,无论是在模型训练队列还是在3个验证队列中,本研究建立的10个基因的预后预测模型与列线图对患者总体生存期的预测性能大致相同(图4)。经对比发现,此10个基因的预后预测模型和列线图在模型训练队列中比其他3个已被报道的模型和肿瘤分期系统表现出更好的预测性能(图4A)。在3个验证队列中,本研究的模型预测1年总体生存期(验证队列1~3的AUC分别为0.67、0.63和0.79),3年总体生存期(验证队列1~3的AUC分别为0.68、0.63和0.66)和5年总体生存期(验证队列2的AUC为0.69)的结果表明10个基因的预后预测模型在预测短期和长期生存期方面均表现出良好的性能(图4B)。综上所述,本研究建立的预后评分模型在预测肝癌患者总体生存期方面显示出与整合临床特征参数的列线图相似的能力,且优于传统的肿瘤分期系统和其他已被报道的模型。图4
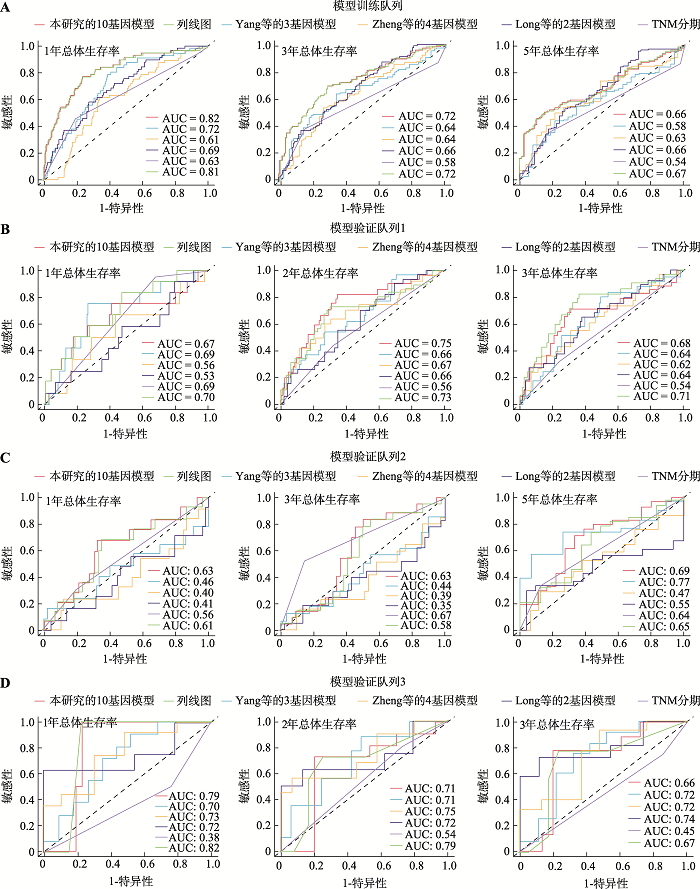 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4基于10个基因的肝癌预后预测评分模型的预测性能优于其他模型
A:模型训练队列中预测性能的比较;B:模型验证队列中预测性能的比较。采用ROC曲线分析比较本研究的10个基因预测模型与其他5个模型(列线图模型、TNM阶段、Zheng等[15]的4基因模型、Yang等[16]的3基因模型和Long等[17]的2基因模型)的预测性能。AUC:曲线下面积(area under curve);ROC:受试者工作特征曲线(receiver operating characteristic curve)。
Fig. 4Performance comparison of our 10-gene prognostic risk score model with the other models
2.6 不同的风险评分组具有生物学差异
采用基因集富集分析(gene set enrichment analysis, GSEA)探索与高风险评分或低风险评分相关的生物学信号通路(图5A),结果显示高风险组显著富集于与DNA复制、细胞周期和DNA修复相关的信号通路(图5B),表明高风险评分反映了增殖信号和DNA修复信号传导的异常,这些通路的异常已经被报道是肿瘤发生发展的驱动力[18]。相反地,低风险组显著富集于与补体和凝血级联、脂肪酸、药物和丙酸酯代谢等肝代谢途径(图5B),提示低风险评分的患者保留了相对正常的肝功能,这可以保护肝癌患者免受各种药物引起的持续药物毒性。图5
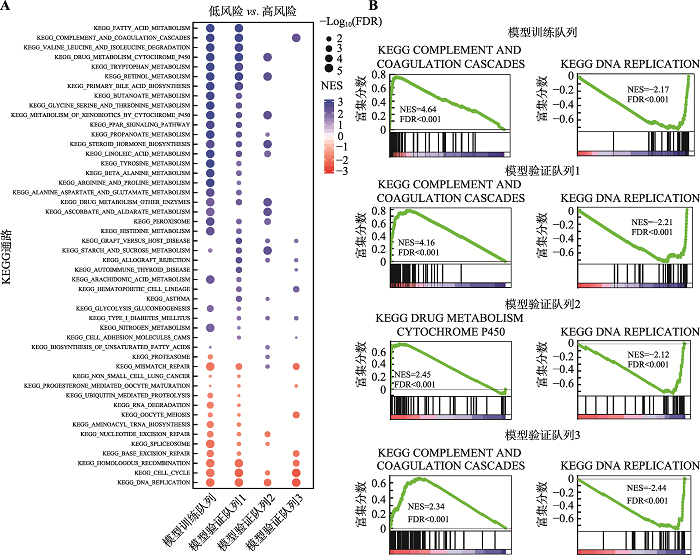 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5模型训练队列和验证列中的基因集富集分析
A:在模型训练队列和验证队列中的基因GSEA结果展示。蓝色和红色点分别表示在低风险组和高风险组(中位数分组)中显著富集的基因集,点的大小表示为P值,基因集至少在2个队列中显著富集(FDR<0.05)才被绘制。B:在模型训练队列和验证队列中最显著富集基因集的GSEA图。FDR:错误发现率(false discovery rate);GSEA:基因集富集分析(gene set enrichment analysis);NES:标准化富集分数(normalized enrichment score)。
Fig. 5Gene set enrichment analysis in model training cohort and validation cohort
3 讨论
缺乏有效和可靠的预后生物标志物或预测模型仍然是改善肝癌患者临床结局的主要挑战。已有研究表明,表观遗传异常和基因组异常变化是导致肝癌发生发展的重要原因[19]。因此,迫切需要有效和可靠的表观遗传生物标志物作为肝癌患者预后预测指标和治疗靶标。本研究基于肝癌组织的转录组与表观基因组,采用整合组学的分析策略,发现了一系列与肝癌预后相关的DNA甲基化驱动的差异表达基因,并建立了一个高置信的肝癌预后预测模型,为肝癌患者的生存预后评估提供了候选参考标准,将有助于指导临床治疗策略的选择。生物标志物不仅可以作为肿瘤患者预后预测工具,而且对测量治疗反应,监测肿瘤复发以及指导临床决策具有重要意义。近来,已经提出了几个基于DNA甲基化驱动的基因建立的肝癌预后模型,包括Zheng等[15]的4基因模型、Yang等[16]的3基因模型和Long等[17]的2基因模型。但是,这些模型大多数是在小型队列中建立或缺乏独立队列验证。本研究使用大规模的模型训练队列(即 TCGA-LIHC,371 例肝癌组织样本)建立了10个基因的预后预测评分模型,并在两个公共的独立大规模验证队列(即 ICGC-LIRI-JP,203例肝癌组织样本;GSE76427,115例肝癌组织样本)中进一步验证了该模型。与那些已报道的预测模型和传统的TNM分期系统相比,本研究的10个基因预后预测评分模型显示出更优的预测能力。此外,本研究的10个基因预后预测评分模型的预测能力与整合有风险评分模型和临床特征的列线图预测能力相似。因此,本研究建立的10个基因预后预测评分模型为预测肝癌患者的预后提供一种简单而准确的方法。
肝癌是一种高异质性的肿瘤,探索与肝癌进展有关的失调基因可能有助于改善治疗策略和预后。在本研究的预后预测评分模型中,这10个特征基因在肝癌组织中均呈现低DNA甲基化引起的高表达。GSEA分析表明,高风险评分与DNA复制、细胞增殖和DNA修复等通路改变有关。在这10个基因中,其中8个已被报道在肝癌组织中高表达。在这8个基因中,MALE、LPL、MYO18B和CDCA8已被报道与DNA损伤响应和细胞周期检查点密切相关[20,21,22];同时,PRC1、STC2、SFN和PBK被发现参与调控多条与肿瘤进展相关的激酶通路[23,24,25]。其他两个基因MAPT和TTC39A尚未有与肿瘤相关的研究报道。但是,MAPT作为微管相关蛋白可以参与细胞的迁移[26];TTC39A可以通过泛素化降解胆固醇受体LXR参与调节高密度脂蛋白的代谢[27]。因此,本研究首次鉴定发现MAPT和TTC39A可以作为肝癌患者的预后标志物,它们可能在肝癌发生发展中发挥重要作用,具有成为新候选靶标的潜力。
综上所述,本研究通过表观基因组和转录组整合分析建立的预后预测评分模型为肝癌患者预后评估提供了一种简单而准确的方法,为患者生存的预测和治疗策略的选择提供指导。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1056/NEJMra1713263URLPMID:30970190 [本文引用: 1]
DOI:10.4254/wjh.v9.i2.80URLPMID:28144389 [本文引用: 1]

Many patients with hepatocellular carcinoma (HCC) are diagnosed in an advanced stage, so they cannot be offered the option of curative treatments. The results of systemic chemotherapy are unsatisfactory and this has led to molecular targeted approaches. HCC develops in chronically damaged tissue due to cirrhosis in most patients. Several different cell types and molecules constitute a unique microenvironment in the liver, which has significant implications in tumor development and invasion. This, together with genome instability, contributes to a significant heterogeneity which is further enhanced by the molecular differences of the underlying causes. New classifications based on genetic characteristics of the tissue microenvironment have been proposed and key carcinogenic signaling pathways have been described. Tumor and adjacent tissue profiling seem biologically promising, but have not yet been translated into clinical settings. The encouraging first results with molecular - genetic signatures should be validated and clinically applicable. A more personalized approach to modern management of HCC is urgently needed.
DOI:10.16288/j.yczz.19-077URLPMID:31307967 [本文引用: 1]
Cancer is a complex disease caused by the malignant cellular proliferation and metastasis. Elucidating its pathogenic mechanism is one of the major challenges that we face currently. Epigenetic mechanisms are essential for maintaining specific patterns of gene expression and normal development and growth of living individuals. Disorders of epigenetic markers, such as histone modification, DNA/RNA methylation, and changes in the three-dimensional conformation of chromatin, can interfere with gene expression to some extent, and result in cancers. This review provides a brief overview of epigenetics, focusing on their association with the genesis of cancers, and we look forward to the application of epigenetics in cancer clinical diagnosis and treatment.
DOI:10.16288/j.yczz.19-077URLPMID:31307967 [本文引用: 1]
Cancer is a complex disease caused by the malignant cellular proliferation and metastasis. Elucidating its pathogenic mechanism is one of the major challenges that we face currently. Epigenetic mechanisms are essential for maintaining specific patterns of gene expression and normal development and growth of living individuals. Disorders of epigenetic markers, such as histone modification, DNA/RNA methylation, and changes in the three-dimensional conformation of chromatin, can interfere with gene expression to some extent, and result in cancers. This review provides a brief overview of epigenetics, focusing on their association with the genesis of cancers, and we look forward to the application of epigenetics in cancer clinical diagnosis and treatment.
DOI:10.16288/j.yczz.14-443URLPMID:26351047 [本文引用: 1]
Liver cancer is a severe harmful disease. It is the fifth most frequently diagnosed cancer and second most frequent cause of cancer deaths worldwide. As the most popular histologic subtype of hepatocellular carcinoma (HCC), primary HCC is a heterogeneous disease whose management requires a multidisciplinary approach combining genetics, genomics and environmental toxicology. Although many molecular targeted therapies such as sorafenib have entered clinical application and proven effective, the cytotoxicity and other negative effects cannot be ignored. There is an urgent need to identify new therapeutic targets and drugs, which can kill HCC cells with high efficiency and specificity. Plenty of evidence suggests that occurrence and development of HCC is closely related with epigenetics. DNA methylation, histone modification, aberrant expression of miRNAs and dysregulated expression of many epigenetic regulatory genes are significantly altered in HCC. Epigenetic therapeutic drugs may reverse abnormal gene expression, thus controlling the occurrence and development of HCC. In this review, we summarize the latest research progresses in epigenetics and its therapeutic application in HCC,and the potential treatments to be used in the future.
DOI:10.16288/j.yczz.14-443URLPMID:26351047 [本文引用: 1]
Liver cancer is a severe harmful disease. It is the fifth most frequently diagnosed cancer and second most frequent cause of cancer deaths worldwide. As the most popular histologic subtype of hepatocellular carcinoma (HCC), primary HCC is a heterogeneous disease whose management requires a multidisciplinary approach combining genetics, genomics and environmental toxicology. Although many molecular targeted therapies such as sorafenib have entered clinical application and proven effective, the cytotoxicity and other negative effects cannot be ignored. There is an urgent need to identify new therapeutic targets and drugs, which can kill HCC cells with high efficiency and specificity. Plenty of evidence suggests that occurrence and development of HCC is closely related with epigenetics. DNA methylation, histone modification, aberrant expression of miRNAs and dysregulated expression of many epigenetic regulatory genes are significantly altered in HCC. Epigenetic therapeutic drugs may reverse abnormal gene expression, thus controlling the occurrence and development of HCC. In this review, we summarize the latest research progresses in epigenetics and its therapeutic application in HCC,and the potential treatments to be used in the future.
DOI:10.1016/j.molonc.2009.03.004URLPMID:19497796 [本文引用: 1]
We performed a global methylation profiling assay on 1505 CpG sites across 807 genes to characterize DNA methylation patterns in pancreatic cancer genome. We found 289 CpG sites that were differentially methylated in normal pancreas, pancreatic tumors and cancer cell lines. We identified 23 and 35 candidate genes that are regulated by hypermethylation and hypomethylation in pancreatic cancer, respectively. We also identified candidate methylation markers that alter the expression of genes critical to gemcitabine susceptibility in pancreatic cancer. These results indicate that aberrant DNA methylation is a frequent epigenetic event in pancreatic cancer; and by using global methylation profiling assay, it is possible to identify these markers for diagnostic and therapeutic purposes in this disease.
DOI:10.1093/bioinformatics/bts635URL [本文引用: 1]
Motivation: Accurate alignment of high-throughput RNA-seq data is a challenging and yet unsolved problem because of the non-contiguous transcript structure, relatively short read lengths and constantly increasing throughput of the sequencing technologies. Currently available RNA-seq aligners suffer from high mapping error rates, low mapping speed, read length limitation and mapping biases.
Results: To align our large (> 80 billon reads) ENCODE Transcriptome RNA-seq dataset, we developed the Spliced Transcripts Alignment to a Reference (STAR) software based on a previously undescribed RNA-seq alignment algorithm that uses sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure. STAR outperforms other aligners by a factor of > 50 in mapping speed, aligning to the human genome 550 million 2 x 76 bp paired-end reads per hour on a modest 12-core server, while at the same time improving alignment sensitivity and precision. In addition to unbiased de novo detection of canonical junctions, STAR can discover non-canonical splices and chimeric (fusion) transcripts, and is also capable of mapping full-length RNA sequences. Using Roche 454 sequencing of reverse transcription polymerase chain reaction amplicons, we experimentally validated 1960 novel intergenic splice junctions with an 80-90% success rate, corroborating the high precision of the STAR mapping strategy.
DOI:10.1038/nmeth.3317URLPMID:25751142 [本文引用: 1]
HISAT (hierarchical indexing for spliced alignment of transcripts) is a highly efficient system for aligning reads from RNA sequencing experiments. HISAT uses an indexing scheme based on the Burrows-Wheeler transform and the Ferragina-Manzini (FM) index, employing two types of indexes for alignment: a whole-genome FM index to anchor each alignment and numerous local FM indexes for very rapid extensions of these alignments. HISAT's hierarchical index for the human genome contains 48,000 local FM indexes, each representing a genomic region of approximately 64,000 bp. Tests on real and simulated data sets showed that HISAT is the fastest system currently available, with equal or better accuracy than any other method. Despite its large number of indexes, HISAT requires only 4.3 gigabytes of memory. HISAT supports genomes of any size, including those larger than 4 billion bases.
DOI:10.1186/s13059-014-0550-8URLPMID:25516281 [本文引用: 1]
In comparative high-throughput sequencing assays, a fundamental task is the analysis of count data, such as read counts per gene in RNA-seq, for evidence of systematic changes across experimental conditions. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. We present DESeq2, a method for differential analysis of count data, using shrinkage estimation for dispersions and fold changes to improve stability and interpretability of estimates. This enables a more quantitative analysis focused on the strength rather than the mere presence of differential expression. The DESeq2 package is available at http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html webcite.
DOI:10.1093/bioinformatics/btt684URL [本文引用: 1]
The Illumina Infinium HumanMethylation450 BeadChip is a new platform for high-throughput DNA methylation analysis. Several methods for normalization and processing of these data have been published recently. Here we present an integrated analysis pipeline offering a choice of the most popular normalization methods while also introducing new methods for calling differentially methylated regions and detecting copy number aberrations.
DOI:10.1093/nar/gkv007URLPMID:25605792 [本文引用: 1]
limma is an R/Bioconductor software package that provides an integrated solution for analysing data from gene expression experiments. It contains rich features for handling complex experimental designs and for information borrowing to overcome the problem of small sample sizes. Over the past decade, limma has been a popular choice for gene discovery through differential expression analyses of microarray and high-throughput PCR data. The package contains particularly strong facilities for reading, normalizing and exploring such data. Recently, the capabilities of limma have been significantly expanded in two important directions. First, the package can now perform both differential expression and differential splicing analyses of RNA sequencing (RNA-seq) data. All the downstream analysis tools previously restricted to microarray data are now available for RNA-seq as well. These capabilities allow users to analyse both RNA-seq and microarray data with very similar pipelines. Second, the package is now able to go past the traditional gene-wise expression analyses in a variety of ways, analysing expression profiles in terms of co-regulated sets of genes or in terms of higher-order expression signatures. This provides enhanced possibilities for biological interpretation of gene expression differences. This article reviews the philosophy and design of the limma package, summarizing both new and historical features, with an emphasis on recent enhancements and features that have not been previously described.
DOI:10.1371/journal.pone.0104158URLPMID:25093504 [本文引用: 1]
Hepatocellular Carcinoma (HCC) is one of the leading causes of cancer-associated mortality worldwide. However, the role of epigenetic changes such as aberrant DNA methylation in hepatocarcinogenesis remains largely unclear. In this study, we examined the methylation profiles of 59 HCC patients. Using consensus hierarchical clustering with feature selection, we identified three tumor subgroups based on their methylation profiles and correlated these subgroups with clinicopathological parameters. Interestingly, one tumor subgroup is different from the other 2 subgroups and the methylation profile of this subgroup is the most distinctly different from the non-tumorous liver tissues. Significantly, this subgroup of patients was found to be associated with poor overall as well as disease-free survival. To further understand the pathways modulated by the deregulation of methylation in HCC patients, we integrated data from both the methylation as well as the gene expression profiles of these 59 HCC patients. In these patients, while 4416 CpG sites were differentially methylated between the tumors compared to the adjacent non-tumorous tissues, only 536 of these CpG sites were associated with differences in the expression of their associated genes. Pathway analysis revealed that forty-four percent of the most significant upstream regulators of these 536 genes were involved in inflammation-related NFkappaB pathway. These data suggest that inflammation via the NFkappaB pathway play an important role in modulating gene expression of HCC patients through methylation. Overall, our analysis provides an understanding on aberrant methylation profile in HCC patients.
[本文引用: 1]
DOI:10.1002/sim.5958URLPMID:24027076 [本文引用: 1]
The area under the time-dependent ROC curve (AUC) may be used to quantify the ability of a marker to predict the onset of a clinical outcome in the future. For survival analysis with competing risks, two alternative definitions of the specificity may be proposed depending of the way to deal with subjects who undergo the competing events. In this work, we propose nonparametric inverse probability of censoring weighting estimators of the AUC corresponding to these two definitions, and we study their asymptotic properties. We derive confidence intervals and test statistics for the equality of the AUCs obtained with two markers measured on the same subjects. A simulation study is performed to investigate the finite sample behaviour of the test and the confidence intervals. The method is applied to the French cohort PAQUID to compare the abilities of two psychometric tests to predict dementia onset in the elderly accounting for death without dementia competing risk. The 'timeROC' R package is provided to make the methodology easily usable.
DOI:10.1093/eurheartj/ehu207URL [本文引用: 1]
Clinical prediction models provide risk estimates for the presence of disease (diagnosis) or an event in the future course of disease (prognosis) for individual patients. Although publications that present and evaluate such models are becoming more frequent, the methodology is often suboptimal. We propose that seven steps should be considered in developing prediction models: (i) consideration of the research question and initial data inspection; (ii) coding of predictors; (iii) model specification; (iv) model estimation; (v) evaluation of model performance; (vi) internal validation; and (vii) model presentation. The validity of a prediction model is ideally assessed in fully independent data, where we propose four key measures to evaluate model performance: calibration-in-the-large, or the model intercept (A); calibration slope (B); discrimination, with a concordance statistic (C); and clinical usefulness, with decision-curve analysis (D). As an application, we develop and validate prediction models for 30-day mortality in patients with an acute myocardial infarction. This illustrates the usefulness of the proposed framework to strengthen the methodological rigour and quality for prediction models in cardiovascular research.
[本文引用: 3]
DOI:10.7150/jca.23762URLPMID:29896284 [本文引用: 3]
Integration of public genome-wide gene expression data together with Cox regression analysis is a powerful weapon to identify new prognostic gene signatures for cancer diagnosis and prognosis. Hepatitis B virus (HBV) is a major cause of hepatocellular carcinoma (HCC), however, it remains largely unknown about the specific gene prognostic signature of HBV-associated HCC. Using Robust Rank Aggreg (RRA) method to integrate seven whole genome expression datasets, we identified 82 up-regulated genes and 577 down-regulated genes in HBV-associated HCC patients. Combination of several enrichment analysis, univariate and multivariate Cox proportional hazards regression analysis, we revealed that a three-gene (SPP2, CDC37L1, and ECHDC2) prognostic signature could act as an independent prognostic indicator for HBV-associated HCC in both the discovery cohort and the internal testing cohort. Gene set enrichment analysis showed that the high-risk group with lower expression levels of the three genes was enriched in bladder cancer and cell cycle pathway, whereas the low-risk group with higher expression levels of the three genes was enriched in drug metabolism-cytochrome P450, PPAR signaling pathway, fatty acid and histidine metabolisms. This indicates that patients of HBV-associated HCC with higher expression of these three genes may preserve relatively good hepatic cellular metabolism and function, which may also protect HCC patients from persistent drug toxicity in response to various medication. Our findings suggest a three-gene prognostic model that serves as a specific prognostic signature for HBV-associated HCC.
DOI:10.7150/thno.31155URLPMID:31695766 [本文引用: 3]
In this study, we performed a comprehensively analysis of gene expression and DNA methylation data to establish diagnostic, prognostic, and recurrence models for hepatocellular carcinoma (HCC). Methods: We collected gene expression and DNA methylation datasets for over 1,200 clinical samples. Integrated analyses of RNA-sequencing and DNA methylation data were performed to identify DNA methylation-driven genes. These genes were utilized in univariate, least absolute shrinkage and selection operator (LASSO), and multivariate Cox regression analyses to build a prognostic model. Recurrence and diagnostic models for HCC were also constructed using the same genes. Results: A total of 123 DNA methylation-driven genes were identified. Two of these genes (SPP1 and LCAT) were chosen to construct the prognostic model. The high-risk group showed a markedly unfavorable prognosis compared to the low-risk group in both training (HR = 2.81; P < 0.001) and validation (HR = 3.06; P < 0.001) datasets. Multivariate Cox regression analysis indicated the prognostic model to be an independent predictor of prognosis (P < 0.05). Also, the recurrence model successfully distinguished the HCC recurrence rate between the high-risk and low-risk groups in both training (HR = 2.22; P < 0.001) and validation (HR = 2; P < 0.01) datasets. The two diagnostic models provided high accuracy for distinguishing HCC from normal samples and dysplastic nodules in the training and validation datasets, respectively. Conclusions: We identified and validated prognostic, recurrence, and diagnostic models that were constructed using two DNA methylation-driven genes in HCC. The results obtained by integrating multidimensional genomic data offer novel research directions for HCC biomarkers and new possibilities for individualized treatment of patients with HCC.
DOI:10.3389/fgene.2015.00157URLPMID:25954303 [本文引用: 1]
DNA damage has been long recognized as causal factor for cancer development. When erroneous DNA repair leads to mutations or chromosomal aberrations affecting oncogenes and tumor suppressor genes, cells undergo malignant transformation resulting in cancerous growth. Genetic defects can predispose to cancer: mutations in distinct DNA repair systems elevate the susceptibility to various cancer types. However, DNA damage not only comprises a root cause for cancer development but also continues to provide an important avenue for chemo- and radiotherapy. Since the beginning of cancer therapy, genotoxic agents that trigger DNA damage checkpoints have been applied to halt the growth and trigger the apoptotic demise of cancer cells. We provide an overview about the involvement of DNA repair systems in cancer prevention and the classes of genotoxins that are commonly used for the treatment of cancer. A better understanding of the roles and interactions of the highly complex DNA repair machineries will lead to important improvements in cancer therapy.
DOI:10.1016/j.canlet.2005.07.009URLPMID:16154256 [本文引用: 1]
Hepatocellular carcinoma (HCC) is one of the most frequent tumor types in the world, with short survival times and few treatment options. Hepatitis B virus (HBV) and hepatitis C virus (HCV) are major etiologic agents of HCC, although the associated mechanisms are incompletely understood. The available evidence suggests that both viruses promote tumorigenesis by up-regulating genes that promote hepatocellular growth and survival, and by down-regulating other genes that act as tumor suppressors and negative growth regulatory molecules. Significantly, a number of the pathways that are altered by these viruses are the same ones that accumulate genetic alterations during tumor progression. This suggests that the pathways that promote virus persistence and replication may also promote cell growth and survival. From the perspective of the virus, this promotes chronic infection, while from the perspective of the host, this promotes tumorigenesis.
DOI:10.1002/hep.26677URL [本文引用: 1]
Amplification of 1q is one of the most frequent chromosomal alterations in human hepatocellular carcinoma (HCC). In this study we identified and characterized a novel oncogene, Maelstrom (MAEL), at 1q24. Amplification and overexpression of MAEL was frequently detected in HCCs and significantly associated with HCC recurrence (P = 0.031) and poor outcome (P = 0.001). Functional study demonstrated that MAEL promoted cell growth, cell migration, and tumor formation in nude mice, all of which were effectively inhibited when MAEL was silenced with short hairpin RNA (shRNAs). Further study found that MAEL enhanced AKT activity with subsequent GSK-3 phosphorylation and Snail stabilization, finally inducing epithelial-mesenchymal transition (EMT) and promoting tumor invasion and metastasis. In addition, MAEL up-regulated various stemness-related genes, multidrug resistance genes, and cancer stem cell (CSC) surface markers at the messenger RNA (mRNA) level. Functional study demonstrated that overexpression of MAEL increased self-renewal, chemoresistance, and tumor metastasis. Conclusion: MAEL is an oncogene that plays an important role in the development and progression of HCC by inducing EMT and enhancing the stemness of HCC. (Hepatology 2014;59:531-543)
DOI:10.1186/s13000-018-0763-3URLPMID:30390677 [本文引用: 1]
BACKGROUND: MYO18B has been identified as a novel tumor suppressor gene in several cancers. However, its specific roles in the progression of hepatocellular carcinoma (HCC) has not been well defined. METHODS: We firstly identified the expression and prognostic values of MYO18B in HCC using TCGA cohort and our clinical data. Then, MYO18B knockdown by RNA inference was implemented to investigate the effects of MYO18B on HCC cells. Quantitative RT-PCR and Western blot were used to determine gene and protein expression levels. CCK-8 and colony formation assays were performed to examine cell proliferation capacity. Wound healing and transwell assays were used to evaluate the migration and invasion of HepG2 cells. RESULTS: MYO18B was overexpressed and correlated with poor prognosis in HCC. MYO18B expression was an independent risk factor for overall survival. Knockdown of MYO18B significantly inhibited the proliferation, migration and invasion of HepG2 cells. Meanwhile, MYO18B knockdown could effectively suppress the phosphorylation of PI3K, AKT, mTOR and P70S6K, suggesting that MYO18B might promote HCC progression by targeting PI3K/AKT/mTOR signaling pathway. CONCLUSIONS: MYO18B promoted tumor growth and migration via the activation of PI3K/AKT/mTOR signaling pathway. MYO18B might be a promising target for clinical intervention of HCC.
DOI:10.1111/liv.13183URLPMID:27264722 [本文引用: 1]
BACKGROUND & AIMS: Although it is well established that fatty acids (FA) are indispensable for the proliferation and survival of cancer cells in hepatocellular carcinoma (HCC), inhibition of Fatty Acid Synthase (FASN) cannot completely repress HCC cell growth in culture. Thus, we hypothesized that uptake of exogenous FA by cancer cells might play an important role in the development and progression of HCC. Lipoprotein lipase (LPL) is the enzyme that catalyses the hydrolysis of triglycerides into free fatty acids (FFA) and increases the cellular uptake of FA. METHODS: We used immunohistochemistry and quantitative reverse transcription real-time polymerase chain reaction to evaluate LPL expression in human and mouse HCC samples. Using lipoprotein-deficient medium as well as siRNAs against LPL and/or FASN, we investigated whether human HCC cells depend on both endogenous and exogenous fatty acids for survival in vitro. RESULTS: We found that LPL is upregulated in mouse and human HCC samples. High expression of LPL in human HCC samples is associated with poor prognosis. In HCC cell lines, silencing of FASN or LPL or culturing the cells in lipoprotein-deficient medium significantly decreased cell proliferation. Importantly, when FASN suppression was coupled to concomitant LPL depletion, the growth restraint of cell lines was further augmented. CONCLUSIONS: The present study strongly suggests that both de novo synthetized and exogenous FA play a major role along hepatocarcinogenesis. Thus, combined suppression of LPL and FASN might be highly beneficial for the treatment of human HCC.
DOI:10.1038/s41598-017-12855-wURLPMID:28978924 [本文引用: 1]
Angiogenesis plays an important role in hepatocellular carcinoma (HCC), the inhibition of which is explored for cancer prevention and treatment. The dietary phytochemical sulforaphane (SFN) is known for its anti-cancer properties in vitro and in vivo; but until now, no study has focused on the role of SFN in HCC tumor angiogenesis. In the present study, in vitro cell models using a HCC cell line, HepG2, and human endothelial cells, HUVECs, as well as ex vivo and in vivo models have been used to investigate the anti-tumor and anti-angiogenic effect of SFN. The results showed that SFN decreased HUVEC cell viability, migration and tube formation, all of which are important steps in angiogenesis. More importantly, SFN markedly supressed HepG2-stimulated HUVEC migration, adhesion and tube formation; which may be due to its inhibition on STAT3/HIF-1alpha/VEGF signalling in HepG2 cells. In addition, SFN significantly reduced HepG2 tumor growth in a modified chick embryo chorioallantoic membrane (CAM) assay, associated with a decrease of HIF-1alpha and VEGF expression within tumors. Collectively, these findings provide new insights into the inhibitory effect of SFN on HCC tumor angiogenesis as well as tumor growth, and indicate that SFN has potential for the prevention and treatment of HCC.
[本文引用: 1]
DOI:10.1136/gutjnl-2015-310625URLPMID:26941395 [本文引用: 1]
OBJECTIVES: Hepatocellular carcinoma (HCC) is the second leading cause of cancer mortality worldwide. Alterations in microtubule-associated proteins (MAPs) have been observed in HCC. However, the mechanisms underlying these alterations remain poorly understood. Our aim was to study the roles of the MAP protein regulator of cytokinesis 1 (PRC1) in hepatocarcinogenesis and early HCC recurrence. DESIGN: PRC1 expression in HCC samples was evaluated by microarray, immunoblotting and immunohistochemistry analysis. Molecular and cellular techniques including siRNA-mediated and lentiviral vector-mediated knockdown were used to elucidate the functions and mechanisms of PRC1. RESULTS: PRC1 expression was associated with early HCC recurrence and poor patient outcome. In HCC, PRC1 exerted an oncogenic effect by promoting cancer proliferation, stemness, metastasis and tumourigenesis. We further demonstrated that the expression and distribution of PRC1 is dynamically regulated by Wnt3a signalling. PRC1 knockdown impaired transcription factor (TCF) transcriptional activity, decreased Wnt target expression and reduced nuclear beta-catenin levels. Mechanistically, PRC1 interacts with the beta-catenin destruction complex, regulates Wnt3a-induced membrane sequestration of this destruction complex, inhibits adenomatous polyposis coli (APC) stability and promotes beta-catenin release from the APC complex. In vivo, high PRC1 expression correlated with nuclear beta-catenin and Wnt target expression. PRC1 acted as a master regulator of a set of 48 previously identified Wnt-regulated recurrence-associated genes (WRRAGs) in HCC. Thus, PRC1 controlled the expression and function of WRRAGs such as FANCI, SPC25, KIF11 and KIF23 via Wnt signalling. CONCLUSIONS: We identified PRC1 as a novel Wnt target that functions in a positive feedback loop that reinforces Wnt signalling to promote early HCC recurrence.
DOI:10.1146/annurev.cellbio.13.1.83URLPMID:9442869 [本文引用: 1]
The polymerization dynamics of microtubules are central to their biological functions. Polymerization dynamics allow microtubules to adopt spatial arrangements that can change rapidly in response to cellular needs and, in some cases, to perform mechanical work. Microtubules utilize the energy of GTP hydrolysis to fuel a unique polymerization mechanism termed dynamic instability. In this review, we first describe progress toward understanding the mechanism of dynamic instability of pure tubulin and then discuss the function and regulation of microtubule dynamic instability in living cells.
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}