 ,1,2,3
,1,2,3Assessing abundance and specificity of different types of sgRNA targeting miRNA precursors
Hailong Liu1, Yang Shen1, Yang Gao1, Ling Zhou2, Xiaosong Han1, Changzhi Zhao1, Gaojuan Yang1, Yilong Chen1, Hui Yang1, Shengsong Xie,1,2,3第一联系人:
编委: 李明洲
收稿日期:2018-01-23修回日期:2018-04-27网络出版日期:2018-07-20
| 基金资助: |
Received:2018-01-23Revised:2018-04-27Online:2018-07-20
| Fund supported: |
作者简介 About authors
刘海龙,硕士研究生,专业方向:动物遗传育种与繁殖E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1794KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘海龙, 谌阳, 高杨, 周玲, 韩晓松, 赵长志, 杨高娟, 陈毅龙, 杨慧, 谢胜松. 靶向miRNA前体不同类型sgRNA的丰度及特异性评估. 遗传[J], 2018, 40(7): 561-571 doi:10.16288/j.yczz.17-417
Hailong Liu, Yang Shen, Yang Gao, Ling Zhou, Xiaosong Han, Changzhi Zhao, Gaojuan Yang, Yilong Chen, Hui Yang, Shengsong Xie.
miRNAs是一类长约18~22个核苷酸的小分子非编码RNA,能调控转录后的靶基因表达[1, 2]。目前,miRBase数据库(v21)共收录来自223个物种28 645条miRNAs[3]。这些miRNAs在基因组上以单拷贝、多拷贝或基因簇的形式存在[4]。miRNA在加工成熟后,与相关蛋白组成的RNA诱导沉默复合体(RNA-induced silencing complex,RISC)共同参与抑制靶基因的翻译或mRNA降解[5]。miRNAs参与机体的多种生物学进程,如miR-291-3p、miR-294、miR-295和miR-106a/363等维持干细胞分化与增殖;miR-302/367提高诱导性多能干细胞(induced pluripotent stem cells,iPSCs)的制备效率[6,7,8,9];miR-146a调控免疫细胞的炎症反应[10, 11];miR-1、miR-133和miR-206等调节骨骼肌增殖与分化[12];附睾中的miR-29a参与调控雄激素信号通路[13]。目前,已有多种实验方法用于miRNA的功能研究,如采用化学法合成miRNA模拟物和抑制剂(miRNA mimics & inhibitors);在难转染的细胞中用慢病毒或腺病毒过表达特定的miRNA,也可过表达“封闭海绵(sponges封闭)”,竞争性抑制miRNA的靶基因;还可采用常规转基因或基因敲除策略,构建特定miRNA过表达或敲除的动物模型。
CRISPR/Cas9是第三代基因组定点编辑技术,可实现基因敲除、敲入、点突变和单碱基编辑等。目前,已有多篇利用CRISPR/Cas9技术靶向编辑miRNA的研究报道[14,15,16]。随着基因组编辑技术的发展,已发现多种识别不同PAM (protospacer adjacent motif)的Cas9变异体蛋白和新型核酸内切酶,如来自化脓链球菌的Cas9酶 (SpCas9)识别的PAM为“NGG”,其变异体识别的PAM为“NGAN”、“NGCG”和“NGCG”等;Cpf1识别的PAM为“TTTN”。目前,设计sgRNA(small guide RNAs)的软件较多,如sgRNAcas9[17]、CRISPRscan[18]、sgRNA Scorer 2.0[19]和CRISPRpred[20]等。除了采用软件设计特异性高的sgRNA来降低脱靶风险,还可通过Cas9单切口酶和成对gRNAs(paired-gRNAs)相结合的手段来提高基因编辑的特异性[17]。值得一提的是,针对CRISPR/ Cas系统不同核酸内切酶,本课题组前期开发了一个用户可以自定义PAM的sgRNA设计软件CRISPR- offinder[21]。为了研究miRNA前体中是否存在特异性高的sgRNAs靶点,本文利用CRISPR-offinder软件对靶向miRNA前体不同类型sgRNA的丰度及特异性特征进行了分析。此外,还利用CRISPR/Cas9慢病毒技术成功构建了猪miR-302/367基因簇敲除细胞系,为利用CRISPR/Cas技术靶向敲除miRNA提供了理论和实践参考。
1 材料和方法
1.1 数据来源
本文数据主要为miRNA前体序列,其中28 645条miRNA前体序列下载自miRBase数据库(http:// www.mirbase.org/index.shtml),808条猪前体序列分别来自Ensemble (http://www.Ensemble.org/index.html)和miRBase数据库。用于评估靶向猪miRNA前体的sgRNA脱靶效应的全基因组序列(Sus_scrofa. Sscrofa build 11.1)来自Ensemble数据库。通过将人miR-302/367基因簇与猪的基因组序列进行BLAST比对,获得猪的miR-302/367基因簇序列。1.2 sgRNA设计与脱靶评估
不同细菌来源或人工改造的基因编辑核酸内切酶识别的PAM不同,如来自化脓性链球菌(Streptococcus pyogenes)的SpCas9及其突变体SpCas9- VQR、SpCas9-EQR和SpCas9-VRER,来自金黄色葡萄球菌(Staphylococcus aureus)的SaCas9及突变体SaCas9-KKH,来自新凶手弗朗西丝菌(Francisella novicida)的FnCas9及突变体FnCas9-RHA,还有来自嗜热链球菌(Streptococcus thermophiles)的St1Cas9和St3Cas9、脑膜炎奈瑟菌(Neisseria meningitides)的NmCas9、氨基酸球菌(Acidaminococcus)的AsCpf1和毛螺科菌(Lachnospiraceae bacterium)的LbCpf1等。它们识别的PAM序列有:NGA、NGAG、NGCG、NGG、NGGNG、NNAGAAW、NNGRRT、NNNNGATT、NNNRRT、TTTN和YG,其中N代表A或T或C或G、R代表G或A、Y代表T或C、W代表A或T。因此,本文利用本课题组前期开发的CRISPR-offinder (v1.2)软件(网址:https://sourceforge. net/projects/crispr-offinder-v1-2/)设计靶向miRNA前体不同类型的sgRNA,该软件可自定义PAM及3°或5°的位置。利用sgRNAcas9 (网址:http://www. biootools.com/,版本号:v3.05)软件设计靶向猪miRNA前体的sgRNA,其脱靶评估最大允许5个碱基错配。本文进一步对sgRNAcas9软件设计的sgRNA脱靶评估结果进行了分类:(1)脱靶不严谨 型,即sgRNA靶点在基因组上唯一存在,脱靶位点不考虑错配碱基数量多少;(2)脱靶严谨型,即不仅考察sgRNA靶点在基因组上唯一存在,而且其脱靶位点碱基错配数量需要大于或等于3。1.3 引物设计和sgRNA慢病毒表达载体构建
在猪miR-302/367基因簇的上游和下游区域设计了3条特异性的sgRNAs,分别为sgRNA-1、sgRNA-2和sgRNA-3 (表1)。sgRNA慢病毒载体骨架为Lenti-sgRNA-EGFP (由上海科技大学的黄行许教授实验室馈赠),对应该载体的克隆位点设计并合成寡核苷酸,分别为sgRNA-1-F和sgRNA-1-R,sgRNA-2-F和sgRNA-2-R,sgRNA-3-F和sgRNA-3-R (表1)。利用sgRNAcas9软件包中的“extract_ targetSeq.pl”脚本,根据sgRNA的位置信息,从猪基因组中提取长为1500~2000 bp且包含sgRNA靶点和miR-302/367基因簇在内的目标序列。利用Premier 5.0软件设计检测基因组编辑效率的PCR引物对ssc-miR-302/367-F和ssc-miR-302/367-R (表1)。引物由生工生物工程(上海)股份有限公司合成。Table 1
表1
表1 本研究使用的sgRNA和引物序列
Table 1
| 名称 | 序列(5°→3°) | 扩增产物长度(bp) | 用途 |
|---|---|---|---|
| sgRNA-1 | GTATTATCGAAGTAAGTCAGCGG | sgRNA序列 | |
| sgRNA-2 | GCGGCAAAACACGCTCCCGTCGG | ||
| sgRNA-3 | TTCTGGGATTACAGTTTCCTAGG | ||
| sgRNA-1-F | caccGTATTATCGAAGTAAGTCAG | 构建表达载体引物对 | |
| sgRNA-1-R | aaacCTGACTTACTTCGATAATAC | ||
| sgRNA-2-F | caccGCGGCAAAACACGCTCCCGT | ||
| sgRNA-2-R | aaacACGGGAGCGTGTTTTGCCGC | ||
| sgRNA-3-F | caccgTTCTGGGATTACAGTTTCCT | ||
| sgRNA-3-R | aaacAGGAAACTGTAATCCCAGAAc | ||
| ssc-miR-302/367-F | AGCTGAAGTGTCTGGCTTACC | 1379 | 检测基因编辑PCR引物 |
| ssc-miR-302/367-R | CAGACCCACCCAGGACCATA | ||
| U6seqF | ACTATCATATGCTTACCGTAAC | 测序引物 |
新窗口打开|下载CSV
sgRNA表达载体构建流程:取浓度为10 μmol/L的sgRNA-1-F和sgRNA-1-R寡核苷酸各5 μL,进行寡核苷酸链复性。反应条件为:95℃加热变性10 min,65℃复性1 h。根据DNA Ligation Kit (宝生物工程(大连)有限公司)说明书,取5 μL Ligation mix,1 ng左右的sgRNA-1-F和sgRNA-1-R寡核苷酸复性产物,50 ng由BbsⅠ酶切线性化后的Lenti-sgRNA- EGFP载体,最后补水至10 μL。混匀,置于16℃连接2 h。取2 μL连接产物,转化到Trans-T1感受态细胞(北京全式金生物技术有限公司),挑选PCR鉴定为阳性的菌液,由武汉奥科生物技术有限公司测序 (测序引物见表1)。使用去内毒素质粒提取试剂盒(Endo-free Plasmid DNA Mini KitⅡ,Omega Biotek,美国)抽提质粒并测定浓度。
1.4 慢病毒包装
将3 × 106个HEK 293T 细胞(中国科学院细胞库)接种于10 cm细胞培养皿,培养液为DMEM+ 10% FBS,培养条件为37℃、5% CO2。待第二天细胞汇合度达到70%,使用JetPRIME (Polyplus- transfection SA)转染试剂盒对三质粒慢病毒包装系统(包括目标质粒、psPAX2和pMD2.G)进行转染。转染体系为:sgRNA表达质粒12 μg,psPAX2质粒 8 μg,pMD2.G质粒 4 μg,转染条件参见JetPRIME 说明书。转染5 h后,更换新鲜的DMEM+10% FBS培养基。转染48 h后,收取含有慢病毒颗粒的细胞培养液,30 000 r/min超速冷冻离心2.5 h,弃上清液,加PBS溶液溶解过夜。用1.5 mL离心管收集病毒溶解液,于-80℃超低温冰箱中保存。1.5 猪miR-302/367基因簇敲除细胞系的构建与鉴定
用制备好的sgRNA慢病毒感染Cas9稳定表达的PK-15细胞系(未发表)。感染2天后,将细胞接种到10 cm细胞培养皿,使其形成单个细胞,且单个细胞之间保持一定的间距。继续培养10天左右,待形成单个细胞克隆时,挑取单克隆至24孔细胞培养板中扩大培养。待细胞长满,收集细胞,按照基因组DNA小量抽提试剂盒(天根生化科技(北京)有限公司)操作说明书抽提DNA。然后利用高保真DNA聚合酶(宝生物工程(大连)有限公司)扩增目标区域,引物对为ssc-miR-302/367-F和ssc-miR-302/ 367-R (表1)。PCR扩增程序为:94℃ 5 min;94℃ 30 s,60℃ 30 s,72℃ 45 s,32个循环;最后再72℃ 7 min。扩增的PCR产物用2%的琼脂糖凝胶电泳检测。将目标条带用PCR产物试剂盒纯化回收后,用TA克隆的方法连接、转化并挑选单菌落扩大培养,由武汉奥科生物技术有限公司测序。1.6 数据分析与作图
数据分析主要采用EXCEL软件和PERL语言编写的脚本,相关性分析使用Pearson函数;利用Sigma plot软件(https://systatsoftware.com/products/sigmaplot/)绘制柱形图。2 结果与分析
2.1 靶向miRNA前体不同类型sgRNA的丰度特征
针对miRBase数据库中28 645条miRNA序列,本文首先分析了靶向miRNA前体不同类型sgRNA丰度特征。结果表明,sgRNA长度为20~24nt (图1),GC%含量为20%~80% (附表1)。通过对sgRNA靶点PAM的数量进行统计(表2),发现靶点数量最多的PAM为“YG”,为345 025条,平均每条miRNA前体含12.04个sgRNA靶点;PAM为“NGG”的靶点数量适中,为169 517条,平均每个miRNA前体中有5.92个sgRNA靶点。本文对靶向miRNA前体不同类型sgRNA丰度进行分析时发现,当PAM为“NGA”、“NGG”、“NNNRRT”和“YG”时,miRNA前体中有大于95%的概率存在对应的sgRNA靶点(表2)。此外,通过比较靶向miRNA前体不同类型sgRNA数量,发现有24%的miRNA前体含有8种不同类型的sgRNA靶点(表3)。有少数miRNA (0.07%)前体中不含任何类型的sgRNA靶点,而含有所有11种不同类型sgRNA靶点的miRNA仅占1.86%。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1靶向miRNA前体不同类型sgRNA特征示意图
N代表A或T或C或G,R代表A或G,Y代表T或C,W代表A或T。红线条表示成熟miRNA。
Fig. 1Illustrations of the different types of sgRNAs that target miRNA precursors
Table 2
表2
表2 靶向miRNA前体不同类型sgRNA的丰度特征
Table 2
| PAM类型 | CRISPR核酸酶 | 靶向miRNA不同类型sgRNA的数量(条) | 靶向miRNA不同类型sgRNA的平均数(条) | 含有sgRNA靶点的miRNA前体数量(个) | 含有sgRNA靶点的miRNA前体百分比(%) |
|---|---|---|---|---|---|
| NGA | SpCas9 VQR变异体 | 198 751 | 6.94 | 28 379 | 99.07 |
| NGAG | SpCas9 EQR变异体 | 51 170 | 1.79 | 20 567 | 71.80 |
| NGCG | SpCas9 VRER变异体 | 20 395 | 0.71 | 10 833 | 37.82 |
| NGG | SpCas9和FnCas9 | 169 517 | 5.92 | 27 372 | 95.56 |
| NGGNG | St3Cas9 | 37 284 | 1.30 | 16 531 | 57.71 |
| NNAGAAW | St1Cas9 | 8 943 | 0.31 | 6 797 | 23.73 |
| NNGRRT | SaCas9 | 39 959 | 1.39 | 19 528 | 68.17 |
| NNNNGATT | NmCas9 | 9 840 | 0.34 | 7 391 | 25.80 |
| NNNRRT | SaCas9 KKH变异体 | 175 605 | 6.13 | 28 155 | 98.29 |
| TTTN | AsCpf1和LbCpf1 | 68 334 | 2.39 | 19 907 | 69.50 |
| YG | FnCas9 RHA变异体 | 345 025 | 12.04 | 28 602 | 99.85 |
新窗口打开|下载CSV
Table 3
表3
表3 靶向miRNA前体不同类型sgRNA的数量及频率
Table 3
| 靶向miRNA前体的sgRNA类型数量(个) | 单个sgRNA靶点 | 成对gRNAs靶点 | ||
|---|---|---|---|---|
| miRNA前体(个) | 频率(%) | miRNA前体(个) | 频率(%) | |
| 0 | 21 | 0.07 | 6842 | 23.89 |
| 1 | 15 | 0.05 | 8523 | 29.75 |
| 2 | 31 | 0.11 | 6963 | 24.31 |
| 3 | 165 | 0.58 | 3863 | 13.49 |
| 4 | 833 | 2.91 | 1646 | 5.75 |
| 5 | 2079 | 7.26 | 593 | 2.07 |
| 6 | 4393 | 15.34 | 164 | 0.57 |
| 7 | 6480 | 22.62 | 35 | 0.12 |
| 8 | 6893 | 24.06 | 15 | 0.05 |
| 9 | 4983 | 17.40 | 1 | 0 |
| 10 | 2218 | 7.74 | 0 | 0 |
| 11 | 534 | 1.86 | 0 | 0 |
新窗口打开|下载CSV
2.2 miRNA前体中成对gRNAs靶点丰度
为了利用成对gRNAs技术编辑miRNA前体,本文分析了靶向miRNA前体不同类型成对gRNAs丰度特征(表4)。采用sgRNAcas9软件的默认参数设计“paired-gRNA”,即限定两个sgRNA之间的距离最小值为-2 bp,最大值为32 bp。由表4可见,miRNA前体序列中,不含任何类型成对gRNAs靶点的比例约占20%,且绝大部分前体中仅存在1~3个成对gRNAs靶点。当PAM为“NGA”时,miRNA前体中存在的成对gRNAs靶点数最多,占总数的47.75%;当PAM为“NNNNGATT”时,miRNA前体中不存在对应的成对gRNAs靶点。对于常用的PAM为“NGG”时,仅有33.52%的miRNA前体中存在成对gRNAs靶点。Table 4
表4
表4 靶向miRNA前体的不同类型成对gRNAs丰度
Table 4
| PAM类型 | CRISPR核酸酶 | 靶向miRNA不同类型成对gRNAs数量(条) | 靶向miRNA不同类型成对gRNAs平均数(条) | 含成对gRNAs靶点的miRNA前体数量(个) | 含成对gRNAs靶点的miRNA前体百分比(%) |
|---|---|---|---|---|---|
| NGA | SpCas9 VQR变异体 | 56 077 | 1.96 | 13 679 | 47.75 |
| NGAG | SpCas9 EQR变异体 | 3 210 | 0.11 | 1839 | 6.42 |
| NGCG | SpCas9 VRER变异体 | 961 | 0.03 | 598 | 2.09 |
| NGG | SpCas9和FnCas9 | 44 397 | 1.55 | 9603 | 33.52 |
| NGGNG | St3Cas9 | 1 988 | 0.07 | 1041 | 3.63 |
| NNAGAAW | St1Cas9 | 67 | 0.002 | 62 | 0.22 |
| NNGRRT | SaCas9 | 1 067 | 0.04 | 782 | 2.73 |
| NNNNGATT | NmCas9 | 0 | 0 | 0 | 0 |
| NNNRRT | SaCas9 KKH变异体 | 18 085 | 0.63 | 7558 | 26.39 |
| TTTN | AsCpf1和LbCpf1 | 2 172 | 0.08 | 672 | 2.35 |
| YG | FnCas9 RHA变异体 | 65 535 | 2.29 | 9111 | 31.81 |
新窗口打开|下载CSV
2.3 miRNA前体序列特征与不同类型sgRNA丰度的相关性
本文利用Pearson相关系数评估了28 645条miRNA前体长度、GC%含量与不同类型sgRNA丰度的关系(附表1)。miRNA前体长度集中在50~120nt之间,miRNA前体的GC%含量最低仅为8.1%,最高为93%。本文进一步对miRNA前体长度和GC%含量与不同类型sgRNA的丰度特征进行Pearson相关性分析,结果见表5。由表5可知,miRNA前体长度与sgRNA靶点数量的多少呈正相关(相关系数为0.89)。可见,miRNA前体序列越长,sgRNA的数量越多,而miRNA前体的GC%含量仅影响特定类型的sgRNA数量(表5)。Table 5
表5
表5 miRNA前体长度、GC%含量与不同类型sgRNA丰度相关性分析
Table 5
| PAM类型 | CRISPR核酸酶 | 与miRNA前体长度的相关性 (Pearson相关系数) | 与GC%含量的相关性 (Pearson相关系数) |
|---|---|---|---|
| NGA | SpCas9 VQR变异体 | 0.80 | -0.08 |
| NGAG | SpCas9 EQR变异体 | 0.49 | 0.12 |
| NGCG | SpCas9 VRER变异体 | 0.25 | 0.30 |
| NGG | SpCas9和FnCas9 | 0.59 | 0.35 |
| NGGNG | St3Cas9 | 0.36 | 0.31 |
| NNAGAAW | St1Cas9 | 0.39 | -0.15 |
| NNGRRT | SaCas9 | 0.58 | -0.10 |
| NNNNGATT | NmCas9 | 0.39 | -0.19 |
| NNNRRT | SaCas9 KKH变异体 | 0.83 | -0.32 |
| TTTN | AsCpf1和LbCpf1 | 0.65 | -0.38 |
| YG | FnCas9 RHA变异体 | 0.80 | 0.12 |
新窗口打开|下载CSV
2.4 靶向猪miRNA前体的sgRNA丰度及脱靶效应评估
针对核酸内切酶为SpCas9的CRISPR/Cas9系统,本文设计了靶向808条猪miRNA前体的sgRNA,并评估了其脱靶效应(图2A)。分析发现,预测每条sgRNA的脱靶位点总数在10~311 656个之间(图2B)。由表6可见,在不考虑脱靶的条件下,有679个miRNA前体中含有5362个sgRNA靶点,即CRISPR/Cas9系统可靶向编辑猪的miRNA前体占总数的84.3%。在脱靶不严谨型评估条件下,有28%的miRNA前体中存在唯一的sgRNA靶点。如果采用严谨型脱靶评估条件,仅有18.2%的miRNA含有特异性高的sgRNA靶点。通过进一步分析sgRNA在基因组上完全匹配的位点,发现有多达3192条sgRNA在基因组上无完全匹配靶点,基因组上存在唯一匹配靶点的sgRNA仅有1314条,而多数sgRNA在基因组上存在大于或等于2个的完全匹配位点(图2C)。总之,本文评估了靶向猪miRNA前体sgRNA的脱靶效应,发现在不考虑脱靶情况下,每个miRNA前体中存在5~6个靶点。如若评估其脱靶效应,发现大多数miRNA前体中仅含有1个特异性较高的sgRNA靶点。图2
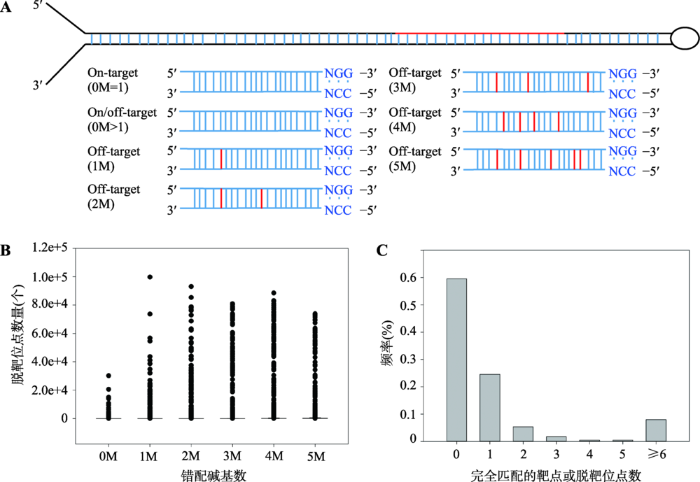 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2猪miRNA前体中sgRNA靶点丰度及脱靶效应
A:靶向猪miRNA前体sgRNA打靶或脱靶位点碱基错配示意图;B:预测的脱靶位点不同碱基错配数量分布;C:存在多个完全匹配结合位点的sgRNA频率分布。0M代表完全匹配的靶点,1M代表存在一个碱基错配的脱靶位点,2M代表存在2个碱基错配的脱靶位点,3M代表存在3个碱基错配的脱靶位点,4M代表存在4个碱基错配的脱靶位点,5M代表存在5个碱基错配的脱靶位点。On-target代表打靶位点,Off-target代表脱靶位点。红色竖线代表发生碱基错配。
Fig. 2The abundance of sgRNA target and its off-target effect in pig miRNA precursors
Table 6
表6
表6 不同脱靶效应评估条件下猪miRNA前体及sgRNA靶点丰度特征
Table 6
| 筛选类型 | 含sgRNA靶点的miRNA前体数量(个) | sgRNA的数量(个) |
|---|---|---|
| 脱靶不严谨型 | 225 (28%) | 1314 (24.5%) |
| 脱靶严谨型 | 147 (18.2%) | 676 (12.6%) |
新窗口打开|下载CSV
2.5 利用CRISPR/Cas9慢病毒技术构建猪miR-302/367基因簇敲除细胞系
为了检测用CRISPR/Cas9慢病毒技术构建miRNA敲除细胞系的效率,本文利用该技术构建了猪miR-302/367基因簇敲除细胞系。靶向miR-302/ 367基因簇上游和下游区域的3条sgRNAs位置见图3A。构建sgRNA载体并包装慢病毒,将3种sgRNA慢病毒等量混合并感染Cas9稳定表达的PK-15细胞。随机挑取和培养10个单克隆细胞株,提取DNA并进行PCR扩增。结果发现,有5株细胞能检测到小片段缺失,特别是7#无野生型扩增片段,表明其为纯合子突变(图3,B和C)。将PCR产物纯化后通过TA克隆测序,测序结果与野生型比对,发现第1#、3#、4#和7#单克隆细胞株成功敲除了miR-302/ 367基因簇(其中5#单克隆细胞株靶标序列TA克隆测序失败),而被敲除的片段分别为851 bp、797 bp (+35 bp)、815 bp、828 bp (+11 bp,7-up),937 bp (+10 bp,7-down)。据此推算,采用CRISPR/Cas9慢病毒策略构建miRNA基因簇敲除细胞系的效率达到40% (图3,C和D)。可见,利用CRISPR/Cas9技术可以实现高效靶向编辑miR-302/367基因簇。图3
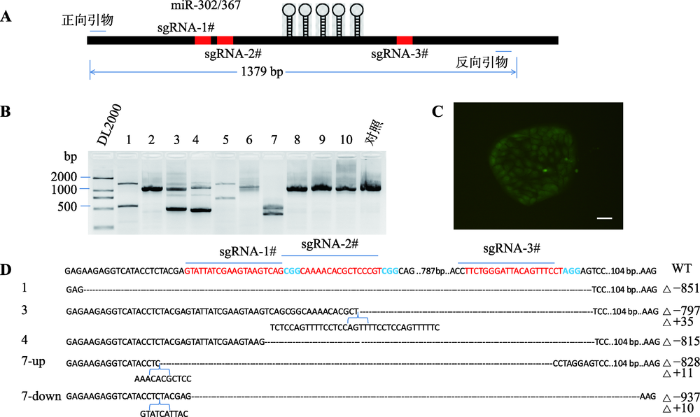 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3利用CRISPR/Cas9慢病毒技术构建猪miR-302/367基因簇敲除的PK-15细胞系
A:采用CRISPR/Cas9慢病毒技术敲除miR-302/367基因簇示意图;B:PCR扩增和凝胶电泳检测基因组编辑单克隆细胞株的基因型;C:miR-302/367基因簇敲除的PK-15单克隆细胞GFP荧光蛋白检测(标尺:100 μm);D:TA克隆测序鉴定基因组编辑细胞株的基因型。WT代表野生型序列。
Fig. 3Construction of porcine miR-302/367 cluster knockout PK-15 cell line using CRISPR/Cas9 technology
3 讨 论
miRNA是一类小分子非编码RNA,其先转录形成miRNA前体,进而加工成长度约为22nt的成熟体miRNA。通过基因组编辑技术靶向修饰miRNA前体,可干扰miRNA的加工成熟或破坏miRNA成熟体。目前采用CRISPR或CRISPRi策略[22]在小鼠或猪的细胞中可有效抑制miRNA表达。Bassett等[23]通过CRISPR/Cas9技术敲除miRNA靶基因3° UTR区的结合位点,能抑制miRNA与靶基因的结合。研究发现,在miRNA的5°端通过CRISPR/Cas9诱导的小片段插入/缺失(indels)突变会引起miRNA的消耗及RNaseⅢ (Drosha)加工的迟缓[24]。在RAW264.7细胞中,利用CRISPR/Cas9技术敲除miR-155会抑制炎症因子的产生[25]。此外,CRISPR/Cas9技术可有效在体内外水平抑制miRNA的表达[26]。通过全基因组水平CRISPR/Cas9文库敲除技术,研究人员发现了一些参与白血病细胞生长所必需的miRNAs[27]。利用CRISPR/Cas9技术也可有效敲除miRNA家 族[28]。通过CRISPR/Cas9技术构建的miR-26a-1/ miR-26a-2双敲除小鼠能影响肺泡表面活性物质的生成[29]。利用CRISPR/Cas9技术可快速构建miR- 301a和miR-29b1敲除小鼠模型[30,31]。总之,CRISPR/ Cas9技术能有效敲除或抑制miRNA的表达。但在miRNA前体序列中,是否含有适用于不同基因组编辑核酸内切酶且特异性高的sgRNA还未被系统研究。由此,本文采用生物信息学方法,全面分析了靶向miRNA前体不同类型sgRNA的丰度特征。结果表明,由于miRNA前体序列长度较短,并非所有miRNA前体中均含有sgRNA的靶点。因此,在使用CRISPR/Cas系统开展miRNA靶向编辑前,可利用CRISPR-offinder软件设计不同类型的sgRNA以增加靶点数量。通过实验,本文还发现采用CRISPR/ Cas9慢病毒技术可高效构建miRNA基因簇敲除的细胞系。脱靶风险是CRISPR/Cas9技术存在的主要问题之一,设计和筛选高效且特异的sgRNA是实验成功与否的关键因素。本实验室前期利用sgRNAcas9软件,针对miR-206前体设计了sgRNA并进行活性和脱靶评估。对13个预测的脱靶位点进行实验检测,发现其中2个预测的位点被证实的确发生了脱靶切割[32]。鉴于此,本研究针对808个猪miRNA前体设计了sgRNA并系统评估了其脱靶效应。结果发现,多数靶向猪miRNA前体的sgRNA在基因组上存在多个完全匹配的位点,可能存在较为严重的脱靶风险。而仅有18.2%的前体中含有特异性相对较高的sgRNA靶点。可见,利用CRISPR/Cas系统靶向编辑miRNA时,除了受sgRNA靶点类型及数量少等因素制约,还存在较大的脱靶风险。另外,本文发现miRNA前体中存在约8种不同类型的sgRNA靶点,且总数为20~37个。而对猪的sgRNA进行脱靶效应分析后,发现靶向miRNA前体的特异性sgRNA仅为18.2%。鉴于此,在利用CRISPR/Cas9技术靶向编辑miRNA时,需要选择合适的软件设计和评估sgRNA的特异性。综述所述,可结合不同基因组编辑酶及相关sgRNA的特征,并结合多种敲除手段,如表达单个、成对gRNAs或多个sgRNAs来实现靶向敲除miRNA前体。
附录:
附表1见文章电子版www.chinagene.cn。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]
URLPMID:11896390 [本文引用: 1]

Abstract Micro RNAs are a large family of noncoding RNAs of 21-22 nucleotides whose functions are generally unknown. Here a large subset of Drosophila micro RNAs is shown to be perfectly complementary to several classes of sequence motif previously demonstrated to mediate negative post-transcriptional regulation. These findings suggest a more general role for micro RNAs in gene regulation through the formation of RNA duplexes.
URLPMID:24275495 [本文引用: 1]
We describe an update of the miRBase database (http://www.mirbase.org/), the primary microRNA sequence repository. The latest miRBase release (v20, June 2013) contains 24 521 microRNA loci from 206 species, processed to produce 30 424 mature microRNA products. The rate of deposition of novel microRNAs and the number of researchers involved in their discovery continue to increase, driven largely by small RNA deep sequencing experiments. In the face of these increases, and a range of microRNA annotation methods and criteria, maintaining the quality of the microRNA sequence data set is a significant challenge. Here, we describe recent developments of the miRBase database to address this issue. In particular, we describe the collation and use of deep sequencing data sets to assign levels of confidence to miRBase entries. We now provide a high confidence subset of miRBase entries, based on the pattern of mapped reads. The high confidence microRNA data set is available alongside the complete microRNA collection at http://www.mirbase.org/. We also describe embedding microRNA-specific Wikipedia pages on the miRBase website to encourage the microRNA community to contribute and share textual and functional information.
URLPMID:11679671 [本文引用: 1]
Abstract Two small temporal RNAs (stRNAs), lin-4 and let-7, control developmental timing in Caenorhabditis elegans. We find that these two regulatory RNAs are members of a large class of 21- to 24-nucleotide noncoding RNAs, called microRNAs (miRNAs). We report on 55 previously unknown miRNAs in C. elegans. The miRNAs have diverse expression patterns during development: a let-7 paralog is temporally coexpressed with let-7; miRNAs encoded in a single genomic cluster are coexpressed during embryogenesis; and still other miRNAs are expressed constitutively throughout development. Potential orthologs of several of these miRNA genes were identified in Drosophila and human genomes. The abundance of these tiny RNAs, their expression patterns, and their evolutionary conservation imply that, as a class, miRNAs have broad regulatory functions in animals.
URL [本文引用: 1]
URLPMID:28666145 [本文引用: 1]
Transgene-free induced pluripotent stem cells (iPSCs) are valuable for both basic research and potential clinical applications. We previously reported that a replication-defective and persistent Sendai virus (SeVdp) vector harboring four reprogramming factors (SeVdp-iPS) can efficiently induce generation of transgene-free iPSCs. This vector can express all four factors stably and simultaneously without chromosomal integration and can be eliminated completely from reprogrammed cells by suppressing vector-derived RNA-dependent RNA polymerase. Here, we describe an improved SeVdp-iPS vector (SeVdp(KOSM)302L) that is automatically erased in response to microRNA-302 (miR-302), uniquely expressed in pluripotent stem cells (PSCs). Gene expression and genome replication of the SeVdp-302L vector, which contains miRNA-302a target sequences at the 3 untranslated region of L mRNA, are strongly suppressed in PSCs. Consequently, SeVdp(KOSM)302L induces expression of reprogramming factors in somatic cells, while it is automatically erased from cells successfully reprogrammed to express miR-302. As this vector can reprogram somatic cells into transgene-free iPSCs without the aid of exogenous short interfering RNA (siRNA), the results we present here demonstrate that this vector may become an invaluable tool for the generation of human iPSCs for future clinical applications.
URLPMID:19176999 [本文引用: 1]
Increasing experimental evidence suggests an important role of miRNAs in embryonic stem cell (ESC) biology. The miR-302-367 cluster is exclusively expressed at high levels in ESCs but not in either somatic stem cells or adult/embryonic differentiated cells. The human miR-302-367 gene structure has been recently described and its promoter has been identified, characterized and functionally validated in human stem cells. The miR-302-367 promoter activity depends on the ontogeny and hierarchical cellular stage. The miR-302-367 promoter is transcriptionally regulated by the ESC-specific transcription factors Oct3/4, Sox2 and Nanog and, its activity restricted to the ESC compartment. Functionally, this cluster regulates cell cycle in ESCs promoting self-renewal and pluripotency, therefore representing a master regulator in the maintenance of hESC stemness. We envision this data may open up new avenues to investigate the transcriptional regulators upstream miR-302-367 cluster and to dissect the complex interplay by which this miR-302-367 cluster integrates in the molecular network conferring pluripotency to ESCs. In this perspective, we summarize recent progress in the genomic and functional characterization of the miR-302-367 cluster and discuss its potential as a stemness determinant.
URLPMID:3035461 [本文引用: 1]
Global demethylation is required for early zygote development to establish stem cell pluripotency, yet our findings reiterate this epigenetic reprogramming event in somatic cells through ectopic introduction of mir-302 function. Here, we report that induced mir-302 expression beyond 1.3-fold of the concentration in human embryonic stem (hES) H1 and H9 cells led to reprogramming of human hair follicle cells (hHFCs) to induced pluripotent stem (iPS) cells. This reprogramming mechanism functioned through mir-302-targeted co-suppression of four epigenetic regulators, AOF2 (also known as KDM1 or LSD1), AOF1, MECP1-p66 and MECP2. Silencing AOF2 also caused DNMT1 deficiency and further enhanced global demethylation during somatic cell reprogramming (SCR) of hHFCs. Re-supplementing AOF2 in iPS cells disrupted such global demethylation and induced cell differentiation. Given that both hES and iPS cells highly express mir-302, our findings suggest a novel link between zygotic reprogramming and SCR, providing a regulatory mechanism responsible for global demethylation in both events. As the mechanism of conventional iPS cell induction methods remains largely unknown, understanding this microRNA (miRNA)-mediated SCR mechanism may shed light on the improvements of iPS cell generation.
URLPMID:21285944 [本文引用: 1]
Somatic cells can be reprogrammed to an ES-like state to create induced pluripotent stem cells (iPSCs) by ectopic expression of four transcription factors, Oct4, Sox2, Klf4 and cMyc. Here, we show that cellular microRNAs (miRNAs) regulate iPSC generation. Knock-down of key microRNA pathway proteins resulted in significant decreases in reprogramming efficiency. Three miRNA clusters, miR-1709080492, miR-106b09080425 and miR-106a090804363, were shown to be highly induced during early reprogramming stages. Several miRNAs, including miR-93 and miR-106b, which have very similar seed regions, greatly enhanced iPSC induction and modulated mesenchymal-to-epithelial transition step in the initiation stage of reprogramming, and inhibiting these miRNAs significantly decreased reprogramming efficiency. Moreover, miR-iPSC clones reached the fully reprogrammed state. Further analysis revealed that Tgfbr2 and p21 are directly targeted by these miRNAs and that siRNA knock-down of both genes indeed enhanced iPSC induction. Here, for the first time, we demonstrate that miR-93 and its family members directly target TGF-0205 receptor II to enhance iPSC generation. Overall, we demonstrate that miRNAs function in the reprogramming process and that iPSC induction efficiency can be greatly enhanced by modulating miRNA levels in cells.
URLPMID:28825144 [本文引用: 1]
Autophagy is a self-degrading process that is triggered by diverse stimuli including starvation,accumulation of misfolded proteins and infection conditions.The role of autophagy in diseases,such as cancer and infections,has been elucidated.However,the signaling pathways regulating autophagy still remain obscure.Recently,other groups reported that several microRNAs(miRNAs),such as miR-376b and miR-181a,are novel and potent modulators of the autophagic activity.Here,we found that miR-146a is a new autophagy-regulating miRNA.We showed that overexpression of miR-146a abrogated DENV2-,rapamycin-and LPS-induced autophagy.Consistently,LNA-mediated silencing of endogenous miRNA activity promoted autophagy.Furthermore,we demonstrated that silence of TRAF6,one of miR-146a targets,decreased DENV2-induced IFN- expression,and then abrogated autophagy.And co-overexpression of TRAF6 reversed the inhibition role of miR-146a on autophagy.Therefore,miR-146a is a novel and important regulator of autophagy and TRAF6 is the direct target in this effect.Moreover,our data showed that in A549 cells,autophagy promoted DENV2-induced TNF-伪and IL-6 production.These findings consummated the role of miR-146a in DENV infection,which might be vital in the pathogenesis of DENV diseases.
URLPMID:28784800 [本文引用: 1]
microRNA-146a (miR-146a) has been previously implicated as an essential molecular brake, preventing immune overreaction and malignant transformation by attenuating NF-κB signaling, putatively via repression of the Traf6 and Irak1 genes. The exact contribution of miR-146a–mediated silencing of these genes to the control of immune activation is currently unknown. Therefore, we...
URLPMID:26708096 [本文引用: 1]
61Muscle-specific microRNAs are called myomiRs.61MyomiRs involve miR-1, miR-133a/b, miR-206, miR-208a/b, miR-486 and miR-499.61MyomiRs play a significant role in virtually every stage of skeletal muscle development.
URLPMID:23960076 [本文引用: 1]
MicroRNAs are involved in a number of cellular processes; thus, their deregulation is usually apt to the occurrence of diverse diseases. Previous studies indicate that abnormally up-regulated miR-29a is associated with several diseases, such as human acute myeloid leukemia and diabetes; therefore, the proper level of miR-29a is critical for homeostasis. Herein, we observed that miR-29a was repressed by androgen/androgen receptor signaling in mouse epididymis by targeting a conserved androgen response element located 8 kb upstream of miR-29b1a loci. It is well known that multiple regulatory programs often form a complicated network. Here, we found that miR-29a reversibly suppressed androgen receptor and its target genes by targeting IGF1 and p53 pathways. miR-29b1a-overexpressing transgenic mice displayed epididymis hypoplasia partially similar to the phenotype of those mice with an impaired androgen-androgen receptor signal system. Taken together, the results demonstrated that there is a regulatory circuitry between the androgen signaling pathway and miR-29a in mouse epididymis that may be vital for epididymal development and functions.
URLPMID:23792628 [本文引用: 1]
Abstract Clustered, regularly interspaced, short palindromic repeat (CRISPR) RNA-guided nucleases (RGNs) have rapidly emerged as a facile and efficient platform for genome editing. Here, we use a human cell-based reporter assay to characterize off-target cleavage of CRISPR-associated (Cas)9-based RGNs. We find that single and double mismatches are tolerated to varying degrees depending on their position along the guide RNA (gRNA)-DNA interface. We also readily detected off-target alterations induced by four out of six RGNs targeted to endogenous loci in human cells by examination of partially mismatched sites. The off-target sites we identified harbored up to five mismatches and many were mutagenized with frequencies comparable to (or higher than) those observed at the intended on-target site. Our work demonstrates that RGNs can be highly active even with imperfectly matched RNA-DNA interfaces in human cells, a finding that might confound their use in research and therapeutic applications.
.
URL [本文引用: 1]
基于CRISPR/Cas9系统介导的第三代基因组编辑技术,已成功应用于动物、植物和微生物等诸多物种的基因组改造。如何提高CRISPR/Cas9技术的基因组编辑效率和最大限度降低脱靶风险一直是本领域的研究热点,而使用高效且特异的sg RNA(Small guide RNA)是基因组改造成功的关键性因素之一。目前,已有多款针对CRISPR/Cas9技术的sg RNA设计和/或脱靶效应评估软件,但不同的软件各有优缺点。本文重点对16款sg RNA设计和脱靶效应评估在线和单机版软件的特点进行了阐述,通过制定38项评估指标对不同软件进行了比较分析,最后对11种用于检测基因组编辑效率和脱靶的实验方法,以及如何筛选高效且特异的sg RNA进行了归纳总结。
URL [本文引用: 1]
基于CRISPR/Cas9系统介导的第三代基因组编辑技术,已成功应用于动物、植物和微生物等诸多物种的基因组改造。如何提高CRISPR/Cas9技术的基因组编辑效率和最大限度降低脱靶风险一直是本领域的研究热点,而使用高效且特异的sg RNA(Small guide RNA)是基因组改造成功的关键性因素之一。目前,已有多款针对CRISPR/Cas9技术的sg RNA设计和/或脱靶效应评估软件,但不同的软件各有优缺点。本文重点对16款sg RNA设计和脱靶效应评估在线和单机版软件的特点进行了阐述,通过制定38项评估指标对不同软件进行了比较分析,最后对11种用于检测基因组编辑效率和脱靶的实验方法,以及如何筛选高效且特异的sg RNA进行了归纳总结。
Magsci [本文引用: 1]
<p>CRISPR/Cas系统广泛存在于细菌及古生菌中, 是机体长期进化形成的RNA指导的降解入侵病毒或噬菌体DNA的适应性免疫系统。对Ⅱ型CRISPR/Cas系统的改造使其成为继锌指核酸酶(ZFNs)和TALE核酸酶(TALENs)以来的另一种对基因组进行高效定点修饰的新技术, 与ZFNs和TALENs相比, CRISPR/Cas系统更简单, 并且更容易操作。文章重点介绍了Ⅱ型CRISPR/Cas系统的基本结构、作用原理及这一技术在基因组定点修饰中的应用, 剖析了该技术可能存在的问题, 展望了CRISPR/Cas系统的应用前景, 为开展这一领域的研究工作提供参考。</p>
Magsci [本文引用: 1]
<p>CRISPR/Cas系统广泛存在于细菌及古生菌中, 是机体长期进化形成的RNA指导的降解入侵病毒或噬菌体DNA的适应性免疫系统。对Ⅱ型CRISPR/Cas系统的改造使其成为继锌指核酸酶(ZFNs)和TALE核酸酶(TALENs)以来的另一种对基因组进行高效定点修饰的新技术, 与ZFNs和TALENs相比, CRISPR/Cas系统更简单, 并且更容易操作。文章重点介绍了Ⅱ型CRISPR/Cas系统的基本结构、作用原理及这一技术在基因组定点修饰中的应用, 剖析了该技术可能存在的问题, 展望了CRISPR/Cas系统的应用前景, 为开展这一领域的研究工作提供参考。</p>
URLPMID:24956386 [本文引用: 2]
Although the CRISPR/Cas9/sgRNA system efficiently cleaves intracellular DNA at desired target sites, major concerns remain on potential 0904off-target0909 cleavage that may occur throughout the whole genome. In order to improve CRISPR-Cas9 specificity for targeted genome editing and transcriptional control, we describe a bioinformatics tool 0904sgRNAcas90909, which is a software package developed for fast design of CRISPR sgRNA with minimized off-target effects. This package consists of programs to perform a search for CRISPR target sites (protospacers) with user-defined parameters, predict genome-wide Cas9 potential off-target cleavage sites (POT), classify the POT into three categories, batch-design oligonucleotides for constructing 20-nt (nucleotides) or truncated sgRNA expression vectors, extract desired length nucleotide sequences flanking the on- or off-target cleavage sites for designing PCR primer pairs to validate the mutations by T7E1 cleavage assay. Importantly, by identifying potential off-target sites in silico, the sgRNAcas9 allows the selection of more specific target sites and aids the identification of bona fide off-target sites, significantly facilitating the design of sgRNA for genome editing applications. sgRNAcas9 software package is publicly available at BiooTools website (www.biootools.com) under the terms of the GNU General Public License.
URLPMID:4589495 [本文引用: 1]
CRISPR/Cas9 technology provides a powerful system for genome engineering. However, variable activity across different single guide RNAs (sgRNAs) remains a significant limitation. We have analyzed the molecular features that influence sgRNA stability, activity and loading into Cas9in vivo. We observe that guanine enrichment and adenine depletion increase sgRNA stability and activity, while loading, nucleosome positioning and Cas9 off-target binding are not major determinants. We additionally identified truncated and 5′ mismatch-containing sgRNAs as efficient alternatives to canonical sgRNAs. Based on these results, we created a predictive sgRNA-scoring algorithm (CRISPRscan.org) that effectively captures the sequence features affecting Cas9/sgRNA activityin vivo. Finally, we show that targeting Cas9 to the germ line using a Cas9-nanos-3′-UTR fusion can generate maternal-zygotic mutants, increase viability and reduce somatic mutations. Together, these results provide novel insights into the determinants that influence Cas9 activity and a framework to identify highly efficient sgRNAs for genome targetingin vivo.
URLPMID:28146356 [本文引用: 1]
It has been possible to create tools to predict single guide RNA (sgRNA) activity in the CRISPR/Cas9 system derived from Streptococcus pyogenes due to the large amount of data that has been generated in sgRNA library screens. However, with the discovery of additional CRISPR systems from different bacteria, which show potent activity in eukaryotic cells, the approach of generating large data sets for each of these systems to predict their activity is not tractable. Here, we present a new guide RNA tool that can predict sgRNA activity across multiple CRISPR systems. In addition to predicting activity for Cas9 from S. pyogenes and Streptococcus thermophilus CRISPR1, we experimentally demonstrate that our algorithm can predict activity for Cas9 from Staphylococcus aureus and S. thermophilus CRISPR3. We also have made available a new version of our software, sgRNA Scorer 2.0, which will allow users to identify sgRNA sites for any PAM sequence of interest.
URLPMID:28767689 [本文引用: 1]
Abstract The CRISPR/Cas9-sgRNA system has recently become a popular tool for genome editing and a very hot topic in the field of medical research. In this system, Cas9 protein is directed to a desired location for gene engineering and cleaves target DNA sequence which is complementary to a 20-nucleotide guide sequence found within the sgRNA. A lot of experimental efforts, ranging from in vivo selection to in silico modeling, have been made for efficient designing of sgRNAs in CRISPR/Cas9 system. In this article, we present a novel tool, called CRISPRpred, for efficient in silico prediction of sgRNAs on-target activity which is based on the applications of Support Vector Machine (SVM) model. To conduct experiments, we have used a benchmark dataset of 17 genes and 5310 guide sequences where there are only 20% true values. CRISPRpred achieves Area Under Receiver Operating Characteristics Curve (AUROC-Curve), Area Under Precision Recall Curve (AUPR-Curve) and maximum Matthews Correlation Coefficient (MCC) as 0.85, 0.56 and 0.48, respectively. Our tool shows approximately 5% improvement in AUPR-Curve and after analyzing all evaluation metrics, we find that CRISPRpred is better than the current state-of-the-art. CRISPRpred is enough flexible to extract relevant features and use them in a learning algorithm. The source code of our entire software with relevant dataset can be found in the following link: https://github.com/khaled-buet/CRISPRpred.
URL [本文引用: 1]
Designing efficient and specific CRISPR single-guide RNAs (sgRNAs) is vital for the successful application of CRISPR technology. Currently, a growing number of new RNA-guided endonucleases with a different protospacer adjacent motif (PAM) have been discovered, suggesting the necessity to develop a versatile tool for designing sgRNA to meet the requirement of different RNA-guided DNA endonucleases. Here, we report the development of a flexible sgRNA design program named “CRISPR-offinder”. Support for user-defined PAM and sgRNA length was provided to increase the targeting range and specificity. Additionally, evaluation of on- and off-target scoring algorithms was integrated into the CRISPR-offinder. The CRISPR-offinder has provided the bench biologist a rapid and efficient tool for identification of high quality target sites, and it is freely available athttps://sourceforge.net/projects/crispr-offinder-v1-2/orhttp://www.biootools.com.
URLPMID:24487629 [本文引用: 1]
Here, we report a convenient and efficient miRNA inhibition strategy employing the CRISPR system. Using specifically designed gRNAs, miRNA gene has been cut at a single site by Cas9, resulting in knockdown of the miRNA in murine cells. Using a modified CRISPR interference system (CRISPRi), inactive Cas9 can reversibly prevent the expression of both monocistronic miRNAs and polycistronic miRNA clusters. Furthermore, CRISPR/CRISPRi is also capable of suppressing genes in porcine cells.
.
URLPMID:25135198 [本文引用: 1]
Abstract MicroRNA (miRNA) target recognition is largely dictated by short 'seed' sequences, and single miRNAs therefore have the potential to regulate a large number of genes. Understanding the contribution of specific miRNA-target interactions to the regulation of biological processes in vivo remains challenging. Here we use transcription activator-like effector nuclease (TALEN) and clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 technologies to interrogate the functional relevance of predicted miRNA response elements (MREs) to post-transcriptional silencing in zebrafish and Drosophila. We also demonstrate an effective strategy that uses CRISPR-mediated homology-directed repair with short oligonucleotide donors for the assessment of MRE activity in human cells. These methods facilitate analysis of the direct phenotypic consequences resulting from blocking specific miRNA-MRE interactions at any point during development.
URLPMID:4615719 [本文引用: 1]
MicroRNA knockout by genome editing technologies is promising. In order to extend the application of the technology and to investigate the function of a specific miRNA, we used CRISPR/Cas9 to deplete human miR-93 from a cluster by targeting its 5090005 region in HeLa cells. Various small indels were induced in the targeted region containing the Drosha processing site and seed sequences. Interestingly, we found that even a single nucleotide deletion led to complete knockout of the target miRNA with high specificity. Functional knockout was confirmed by phenotype analysis. Furthermore, de novo microRNAs were not found by RNA-seq. Nevertheless, expression of the pri-microRNAs was increased. When combined with structural analysis, the data indicated that biogenesis was impaired. Altogether, we showed that small indels in the 5090005 region of a microRNA result in sequence depletion as well as Drosha processing retard.
URLPMID:4677169 [本文引用: 1]
MicroRNA 155 (miR-155) is a key proinflammatory regulator in clinical and experimental rheumatoid arthritis (RA). Here we generated a miR-155 genome knockout (GKO) RAW264.7 macrophage cell line using the clustered regulatory interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (CAS9) technology. While upregulating the Src homology-2 domain-containing inositol 5-phosphatase 1 (SHIP1), the miR-155 GKO line is severely impaired in producing proinflammatory cytokines but slightly increased in osteoclastogenesis upon treatment with receptor activator of nuclear factor- B ligand (RANKL). Taken together, our results suggest that genome editing of miR-155 holds the potential as a therapeutic strategy in RA.
.
[本文引用: 1]
URLPMID:26422227 [本文引用: 1]
Cpf1 is a RNA-guided DNA nuclease that provides immunity in bacteria and can be adapted for genome editing in mammalian cells.
URLPMID:27572667 [本文引用: 1]
A large number of microRNAs (miRNAs) are grouped into families derived from the same phylogenetic ancestors. miRNAs within a family often share the same physiological functions despite differences in their primary sequences, secondary structures, or chromosomal locations. Consequently, the generation of animal models to analyze the activity of miRNA families is extremely challenging. Using zebrafish as a model system, we successfully provide experimental evidence that a large number of miRNAs can be simultaneously mutated to abrogate the activity of an entire miRNA family. We show that injection of the Cas9 nuclease and two, four, ten, and up to twenty-four multiplexed single guide RNAs (sgRNAs) can induce mutations in 90% of the miRNA genomic sequences analyzed. We performed a survey of these 45 mutations in 10 miRNA genes, analyzing the impact of our mutagenesis strategy on the processing of each miRNA both computationally andin vivo. Our results offer an effective approach to mutate and study the activity of miRNA families and pave the way for further analysis on the function of complex miRNA families in higher multicellular organisms.
URLPMID:5340672 [本文引用: 1]
Abstract Pulmonary surfactant (PS), which is synthesized by type II alveolar epithelial cells (AECIIs), maintains alveolar integrity by reducing surface tension. Many premature neonates who lack adequate PS are predisposed to developing respiratory distress syndrome (RDS), one of the leading causes of neonatal morbidity and mortality. PS synthesis is influenced and regulated by various factors, including microRNAs. Previous in vitro studies have shown that PS synthesis is regulated by miR-26a in fetal rat AECIIs. This study aimed to investigate the role of miR-26a in PS synthesis in vivo. To obtain a miR-26a-1/miR-26a-2 double knockout mouse model, we used the clustered regularly interspaced short palindromic repeat/CRISPR-associated protein 9 (CRISPR/Cas9) system, an important genome editing technology. Real-time PCR was performed to determine the miR-26a levels in various organs, as well as the mRNA levels of surfactant-associated proteins. Moreover, AECIIs and surfactant-associated proteins in lung tissues were analyzed by hematoxylin-eosin staining and immunohistochemistry. Homozygous offspring of miR-26a-1/miR-26a-2 double knockout mice generated using the CRISPR/Cas9 system were successfully obtained, and PS synthesis and the number of AECIIs were significantly increased in the miR-26a knockout mice. These results indicate that miR-26a plays an important role in PS synthesis in AECIIs.
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
目的应用CRISPR/Cas9技术构建miRNA-29b1基因敲除小鼠。方法针对miRNA-29b1基因设计一段sgRNA,sgRNA和Cas9体外转录后显微注射至C57BL/6小鼠受精卵细胞。小鼠出生后取其基因组DNA进行测序以鉴定基因型,同时取小鼠心、肝、脾、肺、肾等脏器研磨后提取总RNA,通过real-time PCR分析miRNA-29b1在这些脏器中的表达。结果设计了20 bp的miRNA-29b1sgRNA并与Cas9一起进行了体外转录,显微注射小鼠受精卵细胞后获得miRNA-29b1基因突变小鼠。测序结果表明突变小鼠有两种基因型,一种为10 bp的缺失突变;另一种为22 bp的缺失突变,同时伴有3 bp的插入突变。与野生型小鼠相比,基因突变小鼠心、肝、脾、肺、肾等组织中miRNA-29b1表达量下降明显。结论应用CRISPR/Cas9技术成功构建miRNA-29b1基因敲除小鼠。
URL [本文引用: 1]
目的应用CRISPR/Cas9技术构建miRNA-29b1基因敲除小鼠。方法针对miRNA-29b1基因设计一段sgRNA,sgRNA和Cas9体外转录后显微注射至C57BL/6小鼠受精卵细胞。小鼠出生后取其基因组DNA进行测序以鉴定基因型,同时取小鼠心、肝、脾、肺、肾等脏器研磨后提取总RNA,通过real-time PCR分析miRNA-29b1在这些脏器中的表达。结果设计了20 bp的miRNA-29b1sgRNA并与Cas9一起进行了体外转录,显微注射小鼠受精卵细胞后获得miRNA-29b1基因突变小鼠。测序结果表明突变小鼠有两种基因型,一种为10 bp的缺失突变;另一种为22 bp的缺失突变,同时伴有3 bp的插入突变。与野生型小鼠相比,基因突变小鼠心、肝、脾、肺、肾等组织中miRNA-29b1表达量下降明显。结论应用CRISPR/Cas9技术成功构建miRNA-29b1基因敲除小鼠。
URL [本文引用: 1]
CRISPR/Cas9系统介导的基因组编辑技术是新一代功能强大的基因修饰技术。然而,脱靶效应(Off-target effects)是目前CRISPR/Cas9技术面临的最大问题。因此,设计脱靶风险低的sgRNA(single guide RNA)就成为关键。sgRNAcas9是一款专门用于sgRNA设计和评估脱靶效应的软件包。针对其核心运行程序,我们利用Java程序语言开发了其图形用户界面。此外,依据脱靶位点碱基数和 sgRNA 种子序列(seed sequences)的特异性,通过设置不同的风险等级对sgRNA的脱靶效应进行评估。随后,利用此软件设计了34124条靶向人、小鼠、大鼠、猪和鸡中共4691个microRNA(miRNA)前体的sgRNAs。此外,随机挑选了一个靶向人miR-206前体的sgRNA进行脱靶效应评估和验证。结果发现,sgRNAcas9软件人机交互界面友好,大多数miRNA前体可通过该软件寻找到 sgRNA,且他们的 GC%含量范围集中于40%~-60%。利用 sgRNAcas9软件设计的靶向 miR-206的 sgRNA,其基因组编辑活性和脱靶位点可被实验验证。本研究表明 sgRNAcas9图形用户界面软件能针对任意物种设计特异性的sgRNA,其可在BiooTools(http://www.biootools.com/)网站下载。
URL [本文引用: 1]
CRISPR/Cas9系统介导的基因组编辑技术是新一代功能强大的基因修饰技术。然而,脱靶效应(Off-target effects)是目前CRISPR/Cas9技术面临的最大问题。因此,设计脱靶风险低的sgRNA(single guide RNA)就成为关键。sgRNAcas9是一款专门用于sgRNA设计和评估脱靶效应的软件包。针对其核心运行程序,我们利用Java程序语言开发了其图形用户界面。此外,依据脱靶位点碱基数和 sgRNA 种子序列(seed sequences)的特异性,通过设置不同的风险等级对sgRNA的脱靶效应进行评估。随后,利用此软件设计了34124条靶向人、小鼠、大鼠、猪和鸡中共4691个microRNA(miRNA)前体的sgRNAs。此外,随机挑选了一个靶向人miR-206前体的sgRNA进行脱靶效应评估和验证。结果发现,sgRNAcas9软件人机交互界面友好,大多数miRNA前体可通过该软件寻找到 sgRNA,且他们的 GC%含量范围集中于40%~-60%。利用 sgRNAcas9软件设计的靶向 miR-206的 sgRNA,其基因组编辑活性和脱靶位点可被实验验证。本研究表明 sgRNAcas9图形用户界面软件能针对任意物种设计特异性的sgRNA,其可在BiooTools(http://www.biootools.com/)网站下载。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}