0 引言
【研究意义】分子标记开发对基因定位和分子标记辅助育种有着重要意义,在玉米中采用SLAF-Seq可以快速开发出大量的多态性标记;果皮是玉米籽粒重要的组成部分之一,与玉米籽粒的抗性、脱水速度等性状相关。对甜玉米而言,果皮柔嫩度是甜玉米食用品质的决定因素之一,影响适口性这一重要的品质性状。果皮的柔嫩度作为衡量甜玉米口感品质的重要性状指标,其与果皮厚度和果皮纤维素含量有着密切的关系。鉴于果皮纤维素含量是玉米果皮柔嫩度的主要化学形成因子,对玉米纤维素含量调控基因区段的定位和候选基因的确定对玉米品质改良将具有重要意义。【前人研究进展】中国玉米种植面积和产量均居世界第二位,仅次于美国,是中国的主要粮食作物之一[1]。尽管中国玉米生产约占世界总产量的20%,但仍需进口大量玉米来满足国内所需。其中,饲料用玉米占总需求的70%以上,其余用作食用,工业等。近年来,中国特用玉米正大力发展,包括甜玉米、糯玉米、爆裂玉米等。特用玉米具有较高的经济价值、营养价值和加工价值[2]。甜玉米粮经果菜兼用,是重要的特用玉米之一[3]。其不仅含有丰富的蛋白质、维生素以及硒、镁、铁等抗癌微量元素,还含有大量的膳食纤维,具有消渴、利尿、解毒,增加肠胃蠕动,治疗便秘和防止结肠癌功效[4,5]。近年来,玉米栽培面积逐年上升,市场对优质、高产甜玉米的需求量不断增加。同时,消费者对甜玉米的品质要求也不断提高[6]。因此,甜度高、口感鲜脆、风味独特是甜玉米育种中除了丰产性好、抗病性强之外的重要目标[7,8,9,10]。目前,一些甜玉米以及超甜玉米品种如金银898[11]、沪甜1号[12]、闽甜6855[13]等品种的培育都朝着改善甜玉米的口感上发展。但是,中国的甜玉米品种与国外特别是美国相比尚有较大的差距,最大的不足是果皮柔嫩度差导致渣多、口感粗糙等问题,这也是现阶段中国甜玉米品质改良所面临的瓶颈。张士龙等[14]采用硬度计法测定了甜玉米籽粒发育过程中的果皮柔嫩度值,发现甜玉米的果皮柔嫩度受到发育时期,种植环境的影响,但果皮柔嫩度的好坏本质上仍决定于材料本身。另有研究表明,玉米果皮柔嫩度与籽粒的果皮厚度呈负相关[15],口感与果皮粗纤维含量的负相关也达到显著水平[16]。果皮厚度是玉米果皮柔嫩度的主要物理形成因子,而果皮纤维素含量则是玉米果皮柔嫩度的主要化学形成因子[14]。一般情况下,甜玉米的粗纤维含量均较高[17],因此,降低果皮纤维素含量对于甜玉米品质改良至关重要。浙江省农业科学院旱杂粮研究室的研究也表明甜玉米果皮细胞层数和果皮纤维素含量是影响果皮柔嫩性的关键因素,通过降低果皮细胞层数和果皮纤维素含量可以提高甜玉米的柔嫩性(王美兴等,未发表数据)。因此,甜玉米柔嫩度的改良需要在减少果皮细胞层数的同时降低果皮纤维素的含量,在果皮细胞层数无法减少的情况下,降低果皮纤维素含量则是玉米柔嫩度改良的唯一途径。【本研究切入点】尽管中国已育成了多个甜玉米品种[11,12,13],但是,甜玉米柔嫩度品质改良的研究还比较少,甜玉米果皮纤维素含量调控的研究也未见报道,控制此性状的QTL位点和调控基因也尚未定位。【拟解决的关键问题】本研究通过对玉米果皮纤维素含量差异的RILs群体中纤维素含量高、低材料和亲本进行SLAF测序,通过生物信息学分析的方法开发出多态性的SLAF标记,在此基础上,通过BSA的分析方法,定位调控玉米纤维素含量的染色体区段,将此区段的基因映射到拟南芥基因组,通过对比拟南芥基因功能,确定调控玉米果皮纤维素含量的候选基因。为玉米果皮纤维素含量品质改良提供理论依据和基因资源信息。1 材料与方法
1.1 植物材料及RILs群体的构建
以果皮纤维素含量显著差异的E327和G5-1为亲本,构建重组自交系(RILs)。E327为母本,其纤维素含量为17.62%;G5-1为父本,纤维素含量为11.34%。在138份F6代重组自交系中选择28个纤维素含量高的植株、22个纤维素含量低的植株进行关联分析。1.2 纤维素含量测定
籽粒浸泡24 h,剥取果皮,将果皮在70℃鼓风干燥箱内烘干,然后完全粉碎。取样品测定水分含量(w);G4玻璃砂芯漏斗于500℃灼烧至质量恒定;精确称取干燥样品1.00—1.05 g(m0),放入250 mL洁净干燥的锥形瓶中,加入25 mL硝酸-乙醇混合液[18],装上回流冷凝管,沸水浴加热1 h,用G4玻璃砂芯漏斗抽滤去除溶剂,重复上述操作3—5次,直至纤维变白。用10 mL硝酸-乙醇混合液洗涤残渣,再用热水洗涤至洗涤液用甲基橙试验不呈酸性反应为止,最后用无水乙醇洗涤2次,抽干滤液,将盛有残渣的玻璃砂芯漏斗移入烘箱,于105℃烘干至质量恒定,称质量(m1),然后置于坩埚中500℃灼烧至质量恒定,称取质量(m2)。纤维素含量C=$\frac{\text{m}1\text{-m}2}{\text{m}0(1\text{-w})}$×100%
1.3 SALF文库构建及高通量测序
对参与混池的各单株取等量的叶片,混合得到高纤维素混池样品(28单株)和低纤维素混池样品(22单株),采用CTAB法提取亲本E327、G5-1和两混池的DNA。对玉米参考基因组进行电子酶切,选择HaeⅢ和Hpy166Ⅱ酶对DNA样品进行酶切并回收414—464 bp的酶切片段,对回收得到的酶切片段(SLAF标签)进行3′端加A处理、连接Dual-index测序接头[19]、PCR扩增、纯化、混样、切胶选取目的片段,文库质检合格后用Illumina HiSeqTM 2500进行测序。1.4 多态性SLAF标签的鉴定及分布
通过水稻对照数据评估HaeⅢ+Hpy166Ⅱ的酶切效率,以此判断实验过程的准确性和有效性。根据序列相似性将每个样本测序产生的reads进行聚类,聚类到一起的reads来源于同一个SLAF片段(SLAF标签)。一个SLAF标签在不同样品间序列有差异(包括SNP和InDel),即定义为多态性SLAF标签[20]。统计玉米各染色体上SLAF标签和多态性SLAF标签的分布并作图。1.5 BAS关联分析
根据多态性SLAF标记的信息,使用GTAK对亲本与子代混池进行SNP开发。在进行BSA分析前,过滤任一混池中read支持度小于3的位点和与相应亲本的SNP类型差异的位点,得到高质量的可信SNP位点,并在此基础上识别两混池间差异的位点。采用ED(euclidean distance)算法[21]计算与目的基因连锁的区域。该算法利用混池间差异SNP的频率的差异,计算两混池间的SNP的ED值,ED值越大表明该SNP在两混池间的差异越大。根据连锁的原理,真实的关联区域附近的SNP位点会倾向于在两混池间表现出显著性差异,因此,目标区域内的ED值较大,通过作图,可以在目标关联区域附近观察到较明显的峰。为消除背景噪音,对原始ED值利用LOESS算法[21]进行拟合回归曲线操作,确定显著性阈值,并通过拟合曲线确定最终关联区域。公式如下:ED=$\sqrt{{{({{A_{高}}_{}}\text{-}{{A_{低}}_{}})}^{2}}+{{({{T_{高}}_{}}\text{-}{{T_{低}}_{}})}^{2}}+{{({{C_{高}}_{}}\text{-}{{C_{低}}_{}})}^{2}}+{{({{G_{高}}_{}}\text{-}{{G_{低}}_{}})}^{2}}}$
式中,A高/T高/C高/G高:在SNP位点上A/T/C/G碱基高纤维素混池测序reads中出现频率;A低/T低/C低/G低:在SNP位点上A/T/C/G碱基低纤维素混池测序reads中出现频率。
2 结果
2.1 RILs分离群体的建立
纤维素含量受多基因共同调控。以果皮纤维素含量较高的甜玉米自交系E327和果皮纤维素含量较低的甜玉米自交系G05-1为亲本,构建了纤维素含量变化的RILs群体。在第六代RILs群体中,纤维素含量呈现连续变化,最高的纤维含量为21.3%,而最低为9.3%(电子附表1)。为了开发玉米果皮纤维素含量的分子标记和定位相关调控基因,在RILs群体中挑选28个高纤维含量(17.3%—21.3%)和22个低纤维素含量(9.3%—14.0%)的两组植株,进行BSA(bulked segregate analysis)分析。2.2 SLAF文库的构建及评价
从JGI网站(https://jgi.doe.gov/)下载玉米参考基因组序列,其大小为2.05 GB,GC含量为46.89%。对玉米的参考基因组进行电子酶切,最终确定选用HaeⅢ和Hpy166Ⅱ限制性内切酶对进行SLAF文库构建,酶切片段长度在414—464 bp的序列定义为SLAF标签,预测可得到246 254个SLAF标签(电子附表2),这些标签在玉米基因组上分布均匀(电子附表3),能满足基因定位和进一步开发分子标记的要求。对亲本E327,G05-1和高、低纤维素混池4个样品用HaeⅢ和Hpy166Ⅱ进行双酶切,Illumina建库,并筛选insert为400—500 bp的片段进行Illumnia HiSeq 2500测序。共获得39.77 M reads数据,测序平均Q30为87.57%,平均GC含量为43.59%。亲本和2个混池的read数和数据质量如表1所示。测序质量评估和碱基分布检查表明NGS(next-generation sequencing)测序获得的数据合格。双端比对效率为 95.76%,符合实验要求。根据Control测序pair-end mapped reads在基因组中的位置计算SLAF标签的实际长度,绘制Control reads插入片段的长度分布图,并估测实际片段选择范围(电子附表3),与预期待相符。通过对玉米NGS数据和Control数据的分析结果表明,所获得的玉米NGS数据符合要求,可以进一步进行SLAF标记开发和基于SLAF标记的BSA分析。
Table 1
表1
表1各样品测序数据统计表
Table 1The statistic of sequencing data
| 样品编号 Sample ID | 试验编号 Experiment ID | 总Reads数 Total reads | Q30百分数 Q30 Percentage (%) | GC百分含量 GC percentage (%) |
|---|---|---|---|---|
| G5-1 | P | 7981729 | 87.46 | 43.87 |
| E327 | M | 8306768 | 87.8 | 43.86 |
| 低纤维素材料池 Low cellulose content bulk | L | 10959916 | 88.26 | 43.35 |
| 高纤维素材料池 High cellulose content bulk | H | 12526139 | 86.74 | 43.29 |
| SD | 对照Control | 635758 | 87.59 | 40.7 |
新窗口打开
2.3 SLAF标签的确定及其在玉米染色体上的分布
通过SOAP软件[22]将SLAF定位到玉米(B73)的基因组(下载地址:https://www.ncbi.nlm.nih.gov/ assembly/GCF_000005005.1/)上,共获得268 524个SLAF标签,其中父本的测序深度为22.57×,母本的测序深度为22.64×,低纤维素混池(L)平均测序深度为26.86×,高纤维素混池(H)平均测序深度为31.05×(电子附表4)。不同染色体上的SLAF标签在染色体上均匀分布(图1-A)。通过两亲本SLAF标签的对比,找到有序列差异的SLAF标签73 786个,即为多态性SLAF标签。总体上,多态性SLAF标签占总SLAF标签数目的27.48%。SLAF标签及多态性SLAF标签的在玉米染色体上的统计见电子附表5。这些多态性标签在染色体上均匀分布(图1-B),满足作为开发分子标记的条件。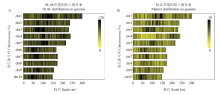 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1SLAF标签(A)和多态性SLAF(B)在染色体上的分布
-->Fig. 1The distribution of SLAF tags (A) and polymorphic SLAF (B) tags on maize genome
-->
2.4 SNP标记的开发
利用多态性SLAF标签信息,采用GTAK对亲本与子代混池进行SNP标记开发,一共得到SNP标记523 395个。过滤掉低可信度的标记后,在父本E327、母本G05-1、低纤维素混池、高纤维素混池样本中分别得到414 466、411 851、459 632和464 746个SNP标记。这些SNP在4个样本中的杂合度分别位17.37%、16.62%、44.46%和40.05%(表2),杂合度的分布符合亲本及BSA混池的特征。表明基于SLAF标签开发的SNP标记可以用来进行进一步的关联分析。同时,所得到的SNP位点的变异信息(电子附表6),可以用于进一步开发SNP分子标记引物,用于玉米的基因定位或分子标记辅助育种。Table 2
表2
表2本研究所检测到的SNP情况
Table 2The overview of SNPs identified in this study
| 样品编号Sample ID | SNP数目 SNP number | 过滤掉的数目 Deficiency | 杂合位点数 Heter Loci Num | 纯和位点数 Homo Loci Num | 杂合位点比例 Het loci ratio (%) | 有效率 Integrity ratio(%) |
|---|---|---|---|---|---|---|
| M | 414466 | 108929 | 72009 | 342457 | 17.37 | 79.18 |
| P | 411851 | 111544 | 68473 | 343378 | 16.62 | 78.68 |
| L | 459632 | 63763 | 204385 | 255247 | 44.46 | 87.81 |
| H | 464746 | 58649 | 186156 | 278590 | 40.05 | 88.79 |
| 总Total | 523395 |
新窗口打开
2.5 BSA关联分析
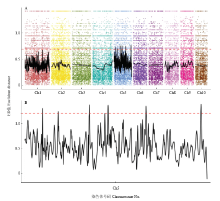
进一步对2个亲本和2个混池的SNP进行分析,为了保证实验的准确性,首先过滤任一混池中read支持度小于3的位点265 983个,然后过滤与相应亲本的SNP类型差异的位点132 740个,最终得到高质量的可信SNP 位点344 441个。其中,280 414位点在2个混池间是一致的,64 027个SNP位点在混间具有差异。针对这些混池间有差异的SNP位点,使用ED算法[21]计算每个SNP位点的ED值,ED值越大表明该SNP在两混池间的差异越大,差异SNP位点ED值在染色体上的分布如图2所示。为消除背景噪音,对原始ED值利用LOESS算法进行拟合回归曲线操作[21],取所有位点ED的第998百分位数(0.69)为阈值,关联到6个区段,均位于第5染色体上,标记为Region1-Region6。表3显示Region1-Region6在玉米基因组上的位置及区段长度,这6个区段一共有47个基因,Region1-Region6所对应的基因数分别为1、1、12、5、3和25。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2BSA分析结果
A:玉米不同染色体上BSA分析得到的ED值;B:玉米第5染色体的ED值。红线显示的是ED分析的阈值
-->Fig. 2The results from bulk segregant analysis
A: Shows the ED value along the 10 chromosomes of maize; B: Shows the ED value along the chromosome 5 of maize. The red line showed the threshold of ED analysis
-->
Table 3
表3
表3关联区域信息统计表
Table 3The statistic of associated regions.
| 染色体编号Chromosome ID | 区域编号 Region ID | 关联区域起始位置 Start(bp) | 关联区域终止位置 End(bp) | 关联区域大小 Size (Mb) | 关联区域内的基因数量 Gene number |
|---|---|---|---|---|---|
| Chr.5 | 区段1 Region1 | 21439635 | 21445702 | 0.01 | 1 |
| Chr.5 | 区段2 Region2 | 40114342 | 40229347 | 0.12 | 1 |
| Chr.5 | 区段3 Region3 | 77344879 | 77758436 | 0.41 | 12 |
| Chr.5 | 区段4 Region4 | 99585215 | 100008471 | 0.42 | 5 |
| Chr.5 | 区段5 Region5 | 145155300 | 145429145 | 0.27 | 3 |
| Chr.5 | 区段6 Region6 | 210394169 | 210982131 | 0.59 | 25 |
| 总计Total | - | - | - | 1.82 | 47 |
新窗口打开
使用BLAST软件将关联区域内的47个基因分别与NR、SwissProt、GO、COG、KEGG数据库比对(电子附表7和电子附表8)。
2.6 候选基因的分析
在这6个关联区域中,Region1和Region2分别只对应一个基因,分别为GRMZM2G413544和GRMZM2G413544,这两个基因可以作为2个候选基因。其他区域分别对应多个基因,但是在每个区域中,可能只有少数基因真正调控纤维素含量变化。因此,需要搜索相关文献对这些基因已经报道的功能进行查阅,进一步缩小候选基因的范围。然而,在玉米中这些基因功能的研究尚未见报道,而模式植物拟南芥的基因功能研究较其他植物更为深入,因为不同植物物种中,同源基因一般具有相同或类似的功能,通过同源比对的方法把这47个基因映射到拟南芥基因组,找到它们对应的拟南芥基因及其注释信息(电子附表8),并查阅拟南芥相关基因的研究工作,推测这些玉米基因的功能。如果某一玉米基因对应的拟南芥同源基因直接或间接影响纤维素的代谢,那么这个玉米基因就确定为可能的候选基因。通过这种方法,最终在Region3、Region4、Region5、Region6分别各确定了2个基因作为候选基因(表4)。Table 4
表4
表4各关联区段的候选基因分析
Table 4The identification of candidate genes located in Region 1-6
| 关联区域 Associated region | 基因数目 Number of gene | 候选基因数 Number of candidate gene | 玉米基因 Maize gene | 拟南芥基因 Gene loci in Arabidopsis | 基因名称 Gene name | 拟南芥基因注释 Gene annotation in Arabidopsis | 支持文献 Reference |
|---|---|---|---|---|---|---|---|
| 区段1 Region 1 | 1 | 1 | GRMZM2G413544 | AT4G10730 | 蛋白激酶超蛋白家族蛋白 Protein kinase superfamily protein | ||
| 区段2 Region 2 | 1 | 1 | GRMZM2G016878 | AT3G63220 | 半乳糖氧化酶/kelch重复超家族蛋白 Galactose oxidase/kelch repeat superfamily protein | ||
| 区段3 Region 3 | 12 | 1 | GRMZM5G805485 | AT1G12040 | LRX1 | 富亮氨酸重复/伸展蛋白1 leucine-rich repeat/extensin 1 | [24-25] |
| 区段4 Region 4 | 5 | 2 | GRMZM2G103937 | AT2G04570 AT2G42990 AT4G26790 | 类GDSL脂肪酶/酰基水解酶超家族蛋白 GDSL-like Lipase/Acylhydrolase superfamily protein | [26] | |
| GRMZM2G137507 | AT2G04570 AT2G42990 AT4G26790 | 类GDSL脂肪酶/酰基水解酶超家族蛋白 GDSL-like Lipase/Acylhydrolase superfamily protein | |||||
| 区段5 Region 5 | 3 | 2 | GRMZM2G129543 | AT5G05340 | PRX52 | 过氧化物酶超家族蛋白 Peroxidase superfamily protein | [31-32] |
| GRMZM2G035506 | AT5G05340 | PRX52 | 过氧化物酶超家族蛋白 Peroxidase superfamily protein | ||||
| 区段6 Region 6 | 25 | 2 | GRMZM2G154332 | AT2G46690 | 类SAUR生长素应答蛋白家族 SAUR-like auxin-responsive protein family | [38] | |
| GRMZM5G894568 | AT1G68150 | WRKY9 | WRKY DNA结合蛋白 WRKY DNA-binding protein 9 | [39-40] |
新窗口打开
3 讨论
SLAF-seq(specific-locus amplified fragment sequencing)技术是利用二代高通量测序发展而来的一套简化基因组测序技术。通过特定内切酶酶切基因组并筛选特定大小的片段建库测序,从而选择性地得到目标基因组的片段序列。对不同样本SLAF片段进行多样性分析可以得到多态性SLAF标签,用于BSA关联分析。SLAF-seq技术结合BSA关联分析具有通量高、准确性高、成本低、周期短等优势。另外,基于多态性SLAF标签上变异位点,可以开发出大量的SNP标记,用于后续的分子标记辅助育种[20]。目前,基于SLAF-seq的分子标记技术已经在水稻、小麦、大豆、油菜、棉花等作物中得到应用。在玉米中,SLAF标记已成功用于鉴定fea*-9LB030突变体的突变基因[23]。但是,利用SLAF-seq技术开发玉米分子标记尚未见报道。本研究利用果皮纤维素含量差异的两亲本E327、G05-1构建RILs分离群体,并对亲本和纤维素含量高、低的2个混池进行SLAF测序,得到了73 786个在亲本中存在多态性的标签。另外,与参考基因组进行比对,在4个样本中分别得到了414 466、411 851、459 632和464 746个SNP位点,这些位点的信息(电子附表6)可以用于进一步开发SNP分子标记,用于玉米基因定位和分子辅助育种。结合BSA分析的方法,本研究一共得到6个与果皮纤维素含量差异性状关联的染色体区段,在这6个区段中一共注释到47个基因。作为模式植物拟南芥,其基因功能的研究远远领先于其他植物。鉴于此,以拟南芥基因组作为参考,根据这47个基因在拟南芥中的同源基因是否直接或间接影响纤维素的代谢来确定候选基因。在关联到的染色体区段1和区段2,虽然没有报道表明其对应的拟南芥基因与纤维素的代谢相关,但由于这两个区段各只有一个基因,因此,这两个基因(GRMZM2G413544和GRMZM2G016878)将作为候选基因进行后续的功能验证分析;第3区段有12个基因,其中GRMZM5G805485在拟南芥中的同源基因是LRX1(AT1G12040)。LRX1是拟南芥根毛特异表达基因[24],其与LRX2功能冗余,在拟南芥根毛形成中能够促进细胞壁的合成[25],表明LRX1可能间接的影响纤维素的合成。因此,GRMZM5G805485被确定为区段3的候选基因;区段4有5个基因,分别为GRMZM2G103937、GRMZM2G300672、GRMZM2G103011、GRMZM2G137507和GRMZM2G071626。其中GRMZM2G103937和GRMZM2G137507在拟南芥中有共同的同源基因AT2G04570、AT2G42990和AT4G26790,它们同属于GDSL-like Lipase/ Acylhydrolase superfamily protein基因家族。这个基因家族在拟南芥中还没有参与或影响纤维素含量的报道,但最近在棉花中的一项研究表明:棉花中的同源基因GhGDSL位于GhMYB1转录因子的下游,参与棉花中纤维的合成[26]。因此,GRMZM2G103937和GRMZM2G137507被确定此区段的候选基因;染色体区段5有3个基因,其中GRMZM2G129543和GRMZM2G035506均与拟南芥的AT5G05340(PRX52)同源。PRX52属于过氧化物酶超级家族蛋白(peroxidase superfamily protein),是一个Ⅲ类过氧化物酶,在细胞壁中表达,调节次生壁的合成[27]。过氧化物酶(peroxidase,POD)可以在细胞中催化超氧化物(O2·--)和过氧化氢(H2O2)的产生,使细胞壁松弛,促进细胞壁的合成[28]。有报道表明,PRX52的表达下调,可以抑制细胞的伸长和根的生长[29,30];在拟南芥prx52突变体中,当植物受到铝胁迫时,纤维素合成基因CesA7和CesA8的表达显著下调[31]。此外,在玉米中也有研究发现POD的上调表达与细胞壁的合成和节间伸长相关[32]。基于这些研究,第5区段中的2个编码玉米过氧化物酶基因GRMZM2G129543和GRMZM2G035506被确定为候选基因;CesA是关键的纤维素合成酶,桉树中的一项研究表明,EgraCesA1、EgraCesA2和EgraCesA3的启动子上含有W-box,MYC识别为点(MYC recognition site),EEC元件(EEC element)、CCA1-结合位点(CCA1-binding site)以及ARR1-结合位点(ARR1-binding element)。其中,W-box是WRKY转录因子的识别位点[33,34,35,36,37],ARR1-结合原件是生长素信号途径转录因子ARR结合的位点。被子植物的树干和树枝在重力的刺激下会形成应拉木(tension wood,TW)。杨树的TW中富含纤维素,其TW和常规木(normal wood,NW)的转录组分析发现两者存在大量的差异基因。其中,属于WRKY基因家族的PU06356(对应拟南芥的WRKY20,基因位点为At4g26640)在富含纤维素的TW中的表达量是NW的3.47倍,暗示WRKY转录因子与纤维素合成存在着相关性;在同样一组数据中,研究者发现12个生长素应答基因的差异表达[38],这表明,生长素信号途径也可能间接影响纤维素的合成。也有报道表明拟南芥WRKY12和棉花的GhWRKY15参与调控细胞壁的形成[39,40]。这些研究暗示,纤维素的合成受到WRKY转录因子的调控,同时也受到生长素信号途径的调节。本研究利用BSA分析关联到的染色体区段6中一共有25个基因,其中,GRMZM5G894568编码WRKY转录因子,与拟南芥的WRKY9是同源基因;GRMZM2G154332在拟南芥中的同源基因是AT2G46690,编码一个类SUAR生长素应答蛋白(SAUR-like auxin-responsive protein)。基于这些研究,将这两个基因确定为候选基因(表4)。
4 结论
通过对玉米果皮纤维素含量差异的亲本和RILs群体混池进行SLAF测序,共获得73 786个SLAF多态性标签,并检测到523 395个SNP位点。获得6个控制玉米果皮纤维素含量的染色体区段,共确定9个候选基因。表明SLAF测序结合BSA分析的方法可以用于快速基因定位的分析。附表1 RILs群体纤维素含量及混池信息
Supplemental table 1 The cellulose content of RILs and the bulk information
附表2 电子酶切信息统计
Supplemental table 2 The statistical information of electrical enzyme digestion
附表3 SLAF 标签在各染色体上的数量统计
Supplemental table 3 SLAF number on different chromosome
附表4 SLAF 标签统计
Supplemental table 4 The statistical information of SLAF tags
附表5 SLAF标签和多态性SLAF标签染色体分布统计
Supplemental table 5 The statistic of SLAF tags and polymorphic SLAF tags on different chromosomes
附表6 亲本和高、低纤维素混中的多态性SNP
Supplemental table 6 The SNPs in parent lines and high/low cellulose content bulks
附表7 关联区域内基因注释统计表
Supplemental table 7 The statistic of annotated genes in associated regions
附表8 关联区域基因的注释
Supplemental table 8 Annotation of associated gene in associated regions
附表9 关联区域玉米基因对应的拟南芥基因的注释
Supplemental table 9 Annotation of Arabidopsis homological genes of Maize genes in associated regions
请根据该文DOI:10.3864/j.issn.0578-1752.2018.08.001或扫描右侧二维码,从相应网页“附件”处下载查阅以上信息(手机或不兼容)。

The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}