0 引言
【研究意义】基因组编辑技术是现代生命科学领域的一项重要革新性技术,在功能基因组学研究、经济物种的遗传改良、生物医药、基因治疗等领域发挥了重要的作用。自2013年CRISPR/Cas9技术成功地在人和小鼠细胞中实现基因组编辑[1,2,3]以来,该技术在生命科学领域的应用呈现出井喷的态势,目前已被广泛应用于动物、植物、微生物和人类医学等领域。猪胎儿成纤维细胞(porcine fetal fibroblasts, PFF)是研制转基因克隆猪的重要供体细胞,然而常规化学转染法对PFF细胞的转染效率较低[4,5]。慢病毒对分裂和非分裂细胞均有感染能力,且能够有效地感染较难转染的细胞[6],因此慢病毒介导的CRISPR/Cas9技术用于PFF细胞基因组编辑,可以更好地克服其难转染的问题。骨形态发生蛋白(bone morphogenetic proteins, BMPs)是转化生长因子β亚基(transforming growth factor-β, TGF-β)的一个亚家族,此家族所编码的多功能蛋白在各种组织中具有广泛的生物活性,在多种细胞中起调节生长和分化的作用。BMPR-IB基因属于BMP家族I型受体,在将BMPs通路信号从细胞表面传导至细胞核过程中起着至关重要的作用[7]。【前人研究进展】目前鲜见慢病毒介导的CRISPR/Cas9技术用于PFF细胞的报道。LIU等[8]和LAI等[9]利用慢病毒介导的CRISPR/Cas9技术分别将Cas9基因和Oct4-tdTomato报告基因敲入PFF细胞中,该两项研究均使用了超速离心机浓缩病毒。研究表明,BMPR-IB基因在成骨细胞[10,11,12]和卵泡颗粒细胞[13]的分化和凋亡过程中均起重要作用。此外,羊的BMPR-IB A746G突变位点被证明是影响部分绵羊品种多羔性状的主效突变位点[14,15,16]。ZHAO等[13]通过siRNA干扰的方式将猪卵泡颗粒细胞中的BMPR-IB基因沉默后,会显著抑制颗粒细胞的增殖和促进颗粒细胞的凋亡,并且会影响CylinD2、Cdk2、Bcl2、Cyp19a1等基因的表达[13]。【本研究切入点】目前利用慢病毒介导的CRISPR/Cas9技术针对PFF细胞开展基因组编辑的研究报道较少,且已有的研究往往利用昂贵的超速离心机对慢病毒进行了浓缩[8,9];此外,之前暂无BMPR-IB基因对PFF细胞增殖或凋亡的影响的相关报道。【拟解决的关键问题】本研究拟针对PFF细胞建立快速和高效的、慢病毒介导的CRISPR/Cas9基因组编辑技术体系;并分析BMPR-IB基因编辑后对PFF细胞BMP信号通路中重要功能基因表达及细胞增殖能力影响。1 材料与方法
本研究于2016—2017年在江西农业大学猪遗传改良与养殖技术省部共建国家重点试验室完成。1.1 试验材料
1.1.1 细胞系和质粒 猪胎儿成纤维细胞系由江西农业大学猪遗传改良与养殖技术省部共建国家重点实验室前期所建立;293T细胞购自中科院细胞库(http://www.cellbank.org.cn/);PX458(Addgene: #48138)、lentiCRISPR v2(Addgene: #52961)、psPAX2(Addgene: #12260)、pCMV-VSV-G(Addgene: #8454)均购自Addgene质粒库(http://www.addgene.org/);Trans 5α感受态细胞购自北京全式金生物技术公司。1.1.2 试验试剂 聚凝胺(polybrene)、嘌呤霉素为Sigma公司产品;胰酶(Trypsin-EDTA solution)、磷酸盐缓冲液(PBS)、高糖DMEM(11995-065)、胎牛血清(FBS)为Gibco公司产品;质粒小提试剂盒(QIA prep spin Mini prep kit)、胶回收试剂盒(QIAquick Gel Extraction Kit(50))为QIAGEN公司产品;Lipofectamine 3000 Transfection Reagent为Invitrogen 公司产品;T7核酸内切酶Ι(T7E1)、T4连接酶和限制性内切酶BsmB Ι购自NEB公司;琼脂糖为Biowest公司产品;细胞基因组DNA提取试剂盒(E.Z.N.A. Tissue DNA Kit)为Omega公司产品;r-Taq酶为Takara公司产品;TRIzol试剂(Invitrogen);PrimeScript RT reagent Kit with gDNA Eraser反转录试剂盒(TaKaRa);SYBR Premix ExTaq™ RT-PCR试剂盒((TaKaRa);RIPA裂解液(强)、BCA蛋白浓度测定试剂盒、超敏ECL化学发光试剂购自碧云天公司;兔源BMPR-IB一抗(ab97051)、鼠源β-actin一抗(mAbcam 8226)、BMPR-IB二抗(辣根过氧化物酶标记的羊抗兔IgG,ab97051)、β-actin二抗(辣根过氧化物酶标记的羊抗鼠IgG,ab6789)均购自abcam公司;Cell Counting Kit-8(CCK-8)试剂盒购自日本Dojindo公司。
1.1.3 引物合成 sgRNA链和检测引物从Invitrogen公司订购。
1.1.4 测序 普通测序及TA-克隆测序由华大基因武汉分公司完成。
1.2 试验方法
1.2.1 sgRNA 的设计和引物合成 针对猪BMPR-IB基因的第8个外显子,利用麻省理工学院张锋(Feng Zhang)实验室的在线网站(http://crispr.mit.edu)设计sgRNA。该软件针对所设计的sgRNA序列的潜在脱靶效应给予评分,分值越高(满分为100分)说明潜在脱靶效应越低。挑选评分最高的sgRNA序列:5′-GATTGGAAAAGGTCGCTATG-3′,在sgRNA链的5′端添加CACCG,在sgRNA互补链的5′端添加AAAC、3′端添加碱基C,以使双链sgRNA序列与线性化的lentiCRISPR v2质粒连接。表1中的P 1引物对用于检测sgRNA序列是否与lentiCRISPR v2质粒正确连接;P 2和P 3引物用于检测是否成功打靶;其他引物均用于RT-PCR分析。Table 1
表1
表1载体构建、打靶验证和RT-PCR引物
Table 1Primers used for validation of vector construction, targeting and RT-PCR
| 引物名称 Primer name | 序列(5′-3′) Sequence | 片段大小 Amplicon(bp) |
|---|---|---|
| P1 | FP:GCAGACAAATGGCAGTATTCA | 436 |
| RP:AAACCATAGCGACCTTTTCCAATCC | ||
| P2 | FP:CAAATCTGTCGTGCTGTGAATAC | 443 |
| RP:GCCTTAGTCATTCCTCCTGTGA | ||
| P3 | FP:CAAATCTGTCGTGCTGTGAA | 181 |
| RP:CCTCCGTGGTGAAGAACACT | ||
| BMPR-IB | FP:AACATTTTGGGCTTCATTGCT RP:ATCATAGAGGGAGCCGTTTTC | 99 |
| CyclinD2 | FP:GACTTCATCGAGCACATTCTTC RP:ACGTCGGTGTTGGTGATCCT | 248 |
| Cdk2 | FP:ATGGATGCCTCTGCTCTCAC RP:GGTTTAAGGTCTCGGTGCAA | 119 |
| Bcl2 | FP:ACTTTGCCGAGATGTCCAG RP:GTTGACGCTCTCCACACAC | 158 |
| Cyp19a1 | FP:GTCCTCAAGTGTGTTCCATGT RP:GCCGGACAGAGCTTTCATAA | 116 |
| GAPDH | FP:CACTCTTCCACTTTTGATGCTG RP:CCTGTTGCTGTAGCCAAATTC | 99 |
新窗口打开
1.2.2 CRISPR-sgRNA/Cas9打靶DNA的制备 利用20单位BsmB I酶切1 μg lentiCRISPR v2质粒DNA,电泳后通过QIAquick Gel Extraction Kit(50)试剂盒割胶回收片段为10 kb的条带。将sgRNA正向链和反向链按1﹕1比例于0.5 mL EP管中混匀后,将EP管置于装有沸水的烧杯,沸水于室温下冷却过夜,使单链退火形成双链。将50 ng线性化的lentiCRISPR v2质粒DNA与1 μL(50 nmol)sgRNA双链通过T 4连接酶相连接。连接产物转化于Trans 5α感受态细胞,经氨苄抗性平板筛选、菌落PCR(引物对为P 1)及测序,最后利用QIA Prep Spin Mini Prep Kit提取打靶质粒DNA。菌落PCR反应体系为:25 μL 体系中含1× buffer,0.5 mmol·L-1 Mg2+,0.2 mmol·L-1 dNTP(Takara),1 × Q-solution(QIAGEN),0.4 μmol·L-1上游和下游引物,2.5 U r-Taq酶(Takara),1 μL菌液模板。PCR反应条件:94℃ 5 min;(94℃ 30 s,65℃(-0.5℃/循环)30 s,72℃ 1 min)26个循环;(94℃ 30 s,55℃ 30 s,72℃ 1 min)14个循环;72℃ 10 min。
1.2.3 慢病毒包装 慢病毒包装前一天,在两个10 cm培养皿中分别接种约2.0×106 293T细胞,于37℃、5% CO2箱中培养,培养液为含10%胎牛血清的DMEM。第二天待细胞密度达到80%—90%时,通过Lipofectamine 3000转染慢病毒包装体系于一个10 cm培养皿中,该体系含有5 μg lentiCRISPR v2-sgRNA、4 μg psPAX2和1 μg pCMV-VSV-G。为评估转染效率,将携带绿色荧光蛋白(green fluorescent protein,GFP)的PX458质粒,转染于另一个10 cm培养皿中。转染6 h后更换新鲜的DMEM培养液;转染后48 h和72 h分别回收病毒上清液,上清液经0.45 μm的滤膜过滤后、分装后置于-80℃冰箱中备用。
1.2.4 慢病毒感染PFF细胞 在慢病毒感染前一天,将3个不同血缘的第三(P3)代PFF细胞接种至6孔板中培养,培养液为含10%胎牛血清和1%双抗的DMEM。当细胞汇合度约50%时,将6孔板中的培养液替换为病毒上清液与新鲜培养液的1﹕1混匀液,加入polybrene至终浓度为6 μg·mL-1。6孔板于1 000 rcf (relative centrifugal force)转速、32℃下离心1 h后,置于培养箱中继续培养6 h,然后更换成正常的培养液。在另一个6孔板中设置对应的3个不同血缘的P3代细胞作为对照组(野生型),对照组不加病毒上清、不离心,其他条件与试验组相同。
1.2.5 筛选打靶细胞 lentiCRISPR v2质粒含有嘌
呤霉素抗性基因,慢病毒感染前,用P3代PFF细胞开展嘌呤霉素的抗性试验。设置1.5、2.0、2.5、3.0、3.5和4.0 μg·mL-1的嘌呤霉素浓度梯度,记录细胞在不同浓度下抗药时间,选择4 d能杀死所有细胞的浓度(3.5 μg·mL-1)为后续细胞筛选的浓度。慢病毒感染72 h后,在细胞培养液中加入嘌呤霉素至3.5 μg·mL-1,加入嘌呤霉素后的前两天,细胞每天换液一次,之后隔天换液一次。待对照组细胞全部死亡之后,试验组细胞在含2.0 μg·mL-1嘌呤霉素的培养液中继续培养4 d。
1.2.6 细胞基因组DNA的提取 胰酶消化并收集打靶细胞(抗嘌呤霉素)和对照组细胞,用E.Z.N.A. Tissue DNA Kit提取DNA。
1.2.7 打靶效率的检测 为检测是否成功打靶目标基因,首先通过P 2引物对(表1)扩增细胞DNA,PCR反应体系及扩增循环与菌落PCR相同。PCR产物经琼脂糖凝胶电泳检测其扩增特异性后,再通过T7E1酶检测是否产生突变体。然后利用P 3引物对(表1)扩增打靶和对照组细胞DNA并送测序,PCR反应体系及扩增循环与菌落PCR相同。为分析打靶效率,将打靶细胞的PCR产物进行TA克隆测序,测序结果与对照组序列通过SeqMan软件比对分析。
1.2.8 脱靶效应检测 在线软件http://crispr.mit.edu/在设计出sgRNA序列的同时,还针对该sgRNA序列在整个基因组的同源性提供潜在的脱靶位点(off-target sites, OTS),并按脱靶可能性的高低给予评分。本试验针对20个可能性最高的潜在脱靶位点设计引物(表2),PCR扩增后送TA克隆测序,PCR反应体系和扩增循环与菌落PCR相同,对照组细胞的PCR产物送普通测序,测序结果通过SeqMan软件比对分析。
Table 2
表2
表220个潜在脱靶位点及其检测引物
Table 2Primers for the detection of twenty potential off-target sites
| 潜在脱靶位点 Potential off-target site | 脱靶位点所在位置 Position of off-target site | 引物序列(5′-3′) Primer sequence | 片段大小 Amplicon (bp) |
|---|---|---|---|
| OTS-1 | 4:99998654-99998676 | F:AAGTGAAGCCCTCGGGGAAATGT | 561 |
| R:CACTGGCTTCTTGATTGCTTTGG | |||
| OTS-2 | 6:65387020-65387042 | F:TTGGTAGCGGGGAGGCAGTAGAG | 623 |
| R:GACCTTCTCACCCCTGACCCCTG | |||
| OTS-3 | 9:100625580-100625602 | F:CCAGCCACCAAGTTCTATGGGACC | 757 |
| R:GTGGTCCTTCCTGAATGTGCGGT | |||
| OTS-4 | 9:7148186-7148208 | F:TTGCTCTGATTTCTAGGTTATGG | 591 |
| R:GAATAGAAAATCTGAACAGCCCT | |||
| OTS-5 | 10:72263072-72263094 | F:GAGGTGAAAGGAAGAAGGAGAAT | 896 |
| R:TAACGAATCCGACTAGGAACCAT | |||
| OTS-6 | 11:11707678-11707700 | F:TAAACGGGATTTCTACACTCTGG | 516 |
| R:ATAGTCATCAAGGTTGAGGACGC | |||
| OTS-7 | X:16345186-16345208 | F:AGACAGAATGACTAACTCCCCTTTG | 469 |
| R:GGTGGGGTTATGAAATGATTATG | |||
| OTS-8 | 5:53711211-53711233 | F:AAGGCTTTACAGATTTCCTTGCGTTTA | 313 |
| R:GATTCCTTGACAACACTCAGGTGC | |||
| OTS-9 | 7:81706164-81706186 | F:AGTAGGTAATCTCTGAGCGTGGA | 897 |
| R:AGACTTCTGTTTGCTAATGGGTG | |||
| OTS-10 | 7:4955651-4955673 | F:TGGATTGTCAGTCCTATCAGCAT | 701 |
| R:GAATGGTTGCCACAATAGGTATG | |||
| OTS-11 | 13:147542203-147542225 | F:AGAATGAGAATGAACAGCGAGTG | 659 |
| R:GAAATACTGG ACACGGGTAACTC | |||
| OTS-12 | JH118494:77147-77169 | F:CGCTACTGCCGACTTCAGGACTT | 957 |
| R:GGGGTGAGGAAGGTCAAGGTCAG | |||
| OTS-13 | 3:54009134-54009156 | F:GCCCAAGAAATAGCAACAATAAC | 743 |
| R:TCAGATACGATATTGCTATGGCT | |||
| OTS-14 | 6:24160453-24160475 | F:GACGCTGACCTTTCTGCTACTAC | 799 |
| R:TGGTCCGTGTAATGCGATGTTTA | |||
| OTS-15 | 14:152741273-152741295 | F:GCGGGTCCCCCAGTTCCTGTTCC | 622 |
| R:GCTGTCTGGGAGCAGGGCTCTTG | |||
| OTS-16 | 14:55413707-55413729 | F:TCTCAGGTTGGTGTCAGGGAAAA | 291 |
| R:GGCTAAGTATTCATCC TTCCTTT | |||
| OTS-17 | 3:61277435-61277457 | F:GATTCGGCTTCCTCTCGGGTGCT | 515 |
| R:GACACAGCCACAGCAACACCAGA | |||
| OTS-18 | 7:13078393-13078415 | F:CCAAAACTCAGCCAGAAACAATC | 635 |
| R:AGCCCTTATTCCAAAAGCACTGT | |||
| OTS-19 | 3:5166589-5166611 | F:TTGGAACAGAGATGGAGCGAGCG | 432 |
| R:CCAGCATCTTGCTGAATCAAGAG | |||
| OTS-20 | 7:16444910-16444932 | F:GAAAAAAGGACAGAGAAATACCA | 559 |
| R:ATGAATAATGAGGTCGTTTTTGT |
新窗口打开
1.2.9 RT-PCR检测 以GAPDH为内参基因,针对打靶和对照组细胞中BMPR-IB、CylinD2、Cdk2、Bcl2、Cyp19a1基因进行RT-PCR检测。RT-PCR采用10.0 μL反应体系,体系包括:1× SYBR Premix ExTaq,1× ROX References Dye,0.2 μmol·L-1上游和下游引物,cDNA模板1.0 μL;反应条件为:50℃ 2 min;95℃ 5 min;(95℃ 15 s,60℃ 50 s)40个循环;95℃ 15 s;60℃ 15 s;95℃ 15 s。mRNA相对表达量计算采用LIVAK等的2ˉΔΔCt法[17]。RT-PCR反应在7900HT Fast Real-Time PCR System(ABI公司)上完成,采用SPSS17.0统计软件进行t检验,以P<0.05表示差异显著,以P<0.01表示差异极显著。
1.2.10 Western blotting检测BMPR-IB蛋白表达量 使用RIPA裂解液(强)分别提取打靶和对照组PFF细胞的总蛋白。利用BCA蛋白浓度测定试剂盒检测2种细胞的蛋白浓度,各取40 μg蛋白上样(每种细胞设置一个重复样),进行SDS-PAGE凝胶电泳。电泳结束后,以30 mA 的恒定电流冰浴湿转3 h ,然后将凝胶上的蛋白转移至PVDF膜上。于含5%脱脂奶粉的TBST、4℃封闭过夜,然后用TBST洗膜3次,再分别加入1﹕800 稀释的兔源BMPR-IB一抗和1﹕1 000 稀释的鼠源β-actin一抗,室温下孵育3 h;用TBST洗膜3次,之后分别加入1﹕10 000稀释的目标基因及内参基因二抗,在室温条件下孵育2 h后用TBST洗膜3次。最后用超敏ECL化学发光试剂显色,采取Quantity-One软件分析条带光密度值。
1.2.11 CCK-8法检测细胞增殖 本试验经打靶药筛的细胞为P4代,取1.2.4所述的对照组(3个样本)和试验组(3个样本)的P4代细胞,在6孔板中分别传代至P6和P8代,待CCK-8检测的细胞经计数后接种到24孔板中培养(即为P5、P7和P9代),每孔的细胞数量约为7.5×104个。本试验设置3个组:野生型细胞,打靶细胞,加嘌呤霉素的打靶细胞;不含细胞的培养液为空白对照。依据CCK-8试剂盒说明书完成相应的细胞处理,每个样本设置1个重复。最后用全自动酶标仪(TECAN,infinite M200 PRO)测定吸光度。通过SPSS17.0统计软件对数据进行t检验,以P<0.05表示差异显著,以P<0.01表示差异极显著。
2 结果
2.1 打靶载体的构建及慢病毒包装
本试验针对猪BMPR-IB基因相对较长的外显子8(196 bp),并通过http://crispr.mit.edu/软件共设计出21条sgRNA序列,得分最高的sgRNA成功连接在lentiCRISPR v2载体上。利用Lipofectamine 3000试剂将病毒包装体系转染293T细胞,最终过滤回收到靶向BMPR-IB基因的慢病毒溶液;对照PX458质粒的转染效率达90%以上(图片未展示)。
2.2 打靶细胞筛选及打靶效率分析
本试验在1.5—4.0 μg·mL-1的浓度范围内,按0.5 μg·mL-1的间隔设置了6个浓度梯度开展筛选试验,最终选择3.5 μg·mL-1的嘌呤霉素浓度筛选打靶细胞。筛选至第4天,对照组细胞全部死亡(图1-d),试验组(T25,P4代)中发现10余个细胞群,每个细胞群中含有数量不等的抗嘌呤霉素细胞,较大细胞群中的细胞数多于100个(图1-b)。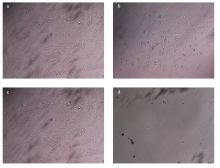 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1嘌呤霉素筛选BMPR-IB打靶细胞
a:试验组未经药筛的细胞;b:试验组经药筛4 d的细胞群;c:对照组未经药筛的细胞;d:对照组细胞经药筛4 d后(全部死亡)
-->Fig. 1Puromycin selected cells targeting BMPR-IB
a: Cells of experiment group without selection; b: Cell population of experiment group with selection for 4 days; c: Cells of control group without selection; d: Cells of control group with selection for 4 days (all were dead)
-->
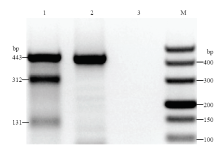
抽提细胞DNA,PCR扩增跨sgRNA序列的区域,利用T7E1酶切PCR产物,打靶细胞中出现了312 bp和131 bp的产物,而对照组中仅有443 bp的条带(图2)。这初步说明CRISPR -sgRNA/Cas9质粒成功打靶目标基因。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2试验组和对照组细胞PCR产物的T7E1酶切结果
1:试验组;2:对照组;3:水;M:marker
-->Fig. 2T7E1 nuclease assay of PCR products of experiment and control groups
1: The experiment group; 2: The control group; 3: ddH2O; M: marker
-->
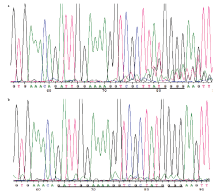
为进一步确认细胞打靶成功,本试验通过PCR测序获得了打靶细胞sgRNA区域的测序结果(图3)。从图中可见,试验组PAM(GGG)前3—4个碱基处出现明显的重叠峰,而对照组中未出现重叠峰。这说明打靶基因PAM附近出现了碱基缺失或(和)插入突变、导致重叠峰的出现,从而确证了成功编辑目标基因。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3试验组和对照组细胞sgRNA区域测序图
a:试验组;b:对照组。长直线标注为sgRNA序列,短直线标注为PAM序列
-->Fig. 3Sequencing results of the sgRNA region in cells from experiment and control groups
a: The experiment group; b: The control group. The long straight lines indicate the sgRNA sequence; The short straight lines indicate the PAM sequence
-->
本试验通过TA-克隆测序获得了随机20个克隆的sgRNA区域的靶向位点序列,分析了其打靶效率(图4)。结果表明,20个克隆中出现了1个15 bp和3 bp的缺失突变,1个克隆为CT插入突变,11个克隆为1个T碱基的插入突变,而6个克隆与野生型细胞的序列一致(图4)。整体打靶效率为70%。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4打靶细胞sgRNA区域20个单克隆的测序结果
#1至#20为sgRNA区域20个单克隆测序结果。斜体序列为sgRNA序列,单下划线标注的为PAM序列,虚线表示碱基缺失,双下划线表示为插入突变。WT,野生型细胞
-->Fig. 4Sequencing results of the sgRNA region in 20 clones from targeting cells
#1 to # 20: Sequencing results of the sgRNA region in 20 clones. The italic sequences indicate sgRNA, the single underlines indicate the PAM sequence, the dotted lines indicate deletions, and the double underlines indicate insertions. WT: Wild type cells
-->
针对sgRNA设计软件所预测的脱靶效应最高的20个潜在脱靶位点,本试验通过PCR-TA克隆测序分别获得了每个位点的20个克隆的序列。通过与对照组序列的比对,发现仅在OTS-2中出现了两种突变,分别为1 bp和2 bp的插入突变(图5)。针对OTS-2的脱靶率为10%(2/20),而针对20个OTS的脱靶率为0.5%(2/400)。
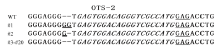 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5打靶细胞OTS-2的TA克隆测序结果
#1-#20:OTS-2的20个单克隆测序结果。斜体表示为sgRNA序列,单下划线表示为PAM序列,双下划线表示为插入突变。WT,野生型细胞
-->Fig.5The TA clone and sequencing results of OTS-2 from targeting cell.
#1-#20: Sequencing results of 20 clones of OTS-2. The italic sequences indicate sgRNA and the single underlines indicate the PAM sequence, the double underlines indicate insertions. WT: Wild type cells
-->
2.3 RT-PCR和Western blotting结果
针对BMPR-IB、CylinD2、Cdk2、Bcl2、Cyp19a1基因,分别在试验组和对照组细胞中开展了RT-PCR试验。结果显示,打靶细胞的BMPR-IB、CylinD2、Cdk2、Bcl2基因表达量极显著下调(P<0.01,图6),但Cyp19a1基因的表达量无显著性差异(P>0.05,图6)。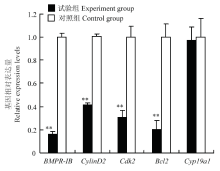 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图6试验组和对照组细胞中5个基因mRNA表达量分析
猪的GAPDH基因作为内参基因,每个反应3个重复。** 差异极显著(P<0.01)
-->Fig. 6mRNA expression levels of five genes in cells from experiment and control groups
The porcine GAPDH gene was used as an internal control, each reaction was performed in triplicate. **Highly significant difference (P<0.01)
-->
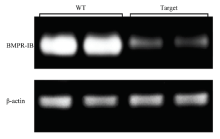
Western blotting结果表明,正常未编辑细胞的BMPR-IB蛋白表达量是打靶细胞的2.6倍(图7),打靶细胞BMPR-IB蛋白量降低62%。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图7BMPR-IB蛋白的Western blotting结果
WT:野生型细胞,Target:打靶细胞
-->Fig. 7Western blotting results of BMPR-IB
WT: Wild type cells, Target: Targeting cells
-->
2.4 细胞增殖检测结果
CCK-8试验检测结果表明,在同一代细胞中,野生型细胞的增殖能力极显著高于打靶细胞(P<0.01),而添加嘌呤霉素与不加嘌呤霉素实验组之间细胞的增殖能力无显著差异(P>0.05)(图8-A)。野生型细胞不同代次之间的细胞增殖能力无显著性差异(P>0.05),而打靶细胞增殖水平随着代数的增加极显著下降(P<0.01)(图8-B)。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图8细胞增殖能力比较分析
WT:野生型细胞;Target+P:加嘌呤霉素的打靶细胞;Target:打靶细胞。每个试验组3个样本,每个样本一次重复。**. 差异极显著(P<0.01)
-->Fig. 8Comparative analysis of cell proliferation
WT: Wild type cells; Target +P: Targeting cells with puromycin; Target: Targeting cells. Each experiment group had three samples, each sample was performed in duplicate. **Highly significant difference(P<0.01)
-->
3 讨论
sgRNA设计是CRISPR/Cas9编辑技术的一个重要环节,高特异性的sgRNA可有效提高打靶效率和降低脱靶效应。目前已有多种软件可用来设计sgRNA[18],这些软件针对参数设置、脱靶位点预测、sgRNA序列评分等具有不同特点。本试验利用麻省理工学院张锋(Feng Zhang)实验室建立的CRISPR设计软件(http://crispr.mit.edu/),针对BMPR-IB基因的第8个外显子,共设计出21条sgRNA序列,得分最低(特异性最差)的为30分,得分最高的为90分,得分80分以上的有5条(具体结果未显示)。本试验选择得分最高的sgRNA用于打靶载体的构建,最终针对PFF细胞获得了70%的打靶效率。http://crispr.mit.edu/软件在设计sgRNA序列的同时,还针对每一条sgRNA序列预测潜在脱靶位点,并按脱靶效应的高低给予评分,分值越高则脱靶效应越强。针对本研究的sgRNA,该软件共预测了50个可能的脱靶位点,评分最高的为0.6分(满分为100分),最低分为0.1分(具体信息未显示)。本试验选择了脱靶效应排在前20的开展PCR-TA克隆测序分析,结果在OTS-2(评分为0.5分)的20个TA克隆中发现了2个突变体。这说明即使软件给予的脱靶效应评分较低,也不排除脱靶发生的可能性。众所周知,存在脱靶效应是CRISPR/Cas9技术目前较明显的一个缺点。本试验对20个OTS进行验证,仅发现一个脱靶位点,但由于未针对所有预测的OTS进行检测,因此也不排除存在其它脱靶位点的可能性。随着CRISPR/Cas9技术的发展,目前已建立起多种捕获脱靶位点的方法,如CHIP-seq[19,20,21,22]、GUIDE-seq[23]、IDLV[24]、digenome-seq[25]等。随着重测序成本的逐步降低,针对单克隆细胞的全基因组测序可以较准确地获得脱靶位点的信息。本试验选择了携带嘌呤霉素的lentiCRISPR v2质粒用于编辑PFF细胞的目标基因。相比传统的新霉素,嘌呤霉素可以更快速地用于哺乳动物细胞筛选;前者往往需要10—14 d全部杀死没有抗性的细胞[26,27],而后者一般需要3—5 d即可全部杀死不带抗性基因的细胞。本试验选择了4 d可杀死所有PFF细胞的浓度(3.5 μg·mL-1)用于筛选打靶细胞,用8 d即获得了具有嘌呤霉素稳定抗性的细胞群,合并计算载体构建、慢病毒包装等工作,仅用两星期左右的时间即获得了打靶细胞。在转染慢病毒包装体系的过程中,携带GFP基因的PX458质粒同步转染于另一盘细胞中,以有效监测转染效率。HOTTA等[28]在包装CRISPR-sgRNA/ Cas9慢病毒试验中,同样以带GFP的质粒作为包装转染效率的质控,并认为当293T细胞的发光比率达90%以上,慢病毒的包装效率是有保障的[28]。本试验选择PX458(9.3 kb)作为病毒包装时评估转染效率的参照质粒,虽然PX458的质粒结构与lentiCRISPR v2的较相似,但质粒大小有一定的差距(lentiCRISPR v2-sgRNA大小为12.9 kb),而质粒大小对转染效率有影响。因此,今后在开展类似的试验时,应采用片段大小更接近的质粒作为对照,如lentiCRISPR v2GFP(Addgene编号:82416)片段大小为13.1 kb,更适用于本试验监控病毒包装时的转染效率。慢病毒离心法感染宿主细胞被证实可以提高感染效率5倍以上[29],该方法无需通过价值昂贵的超速离心机浓缩病毒,无需测定慢病毒的滴度,直接取病毒溶液过滤后通过离心法感染细胞,使操作过程更简便,只需一台普通的温控离心机即可开展。本试验采用离心法也获得了较好的感染效率,在一个T25培养瓶中找到了10余个克隆群。慢病毒针对不同细胞的感染效率有较大不同[30]。本试验也尝试用病毒上清液与细胞培养液1﹕1混合后(含6.0 μg·mL-1的polybrene)直接感染293T细胞和PFF细胞,293T细胞经嘌呤霉素筛选3—4 d细胞可长满,而PFF细胞无法获得细胞群,这说明慢病毒对PFF细胞的感染效率远低于293T细胞(具体结果未展示)。
本试验还利用RT-PCR检测了打靶和对照组PFF细胞中的BMPR-IB、CylinD2、Cdk2、Bcl2、Cyp19a1基因表达情况,结果显示BMPR-IB基因被编辑之后,其表达量极显著下调(检测引物位于靶位点下游),同时CylinD2、Cdk2、Bcl2基因的表达量也极显著下调,但Cyp19a1基因的表达量无显著性差异。BMPR-IB基因的Western blotting结果显示,打靶细胞BMPR-IB蛋白表达量下降62%。众所周知,细胞的增殖主要取决于细胞周期,尤其是哺乳动物细胞,G1/S、G2/M期在其增值过程中有着关键性的作用[31]。在哺乳动物细胞周期中,调节G1期的因子主要是D型的细胞周期蛋白(Cyclins)和细胞周期蛋白依赖激酶(Cdks)。Cdk2在细胞周期起始以及从G1至S期均具有重要的调节作用[32]。BMPR-IB基因是BMPs的I型受体,其位于BMP-Smad信号传导通路II型受体的下游,在配体结合后,I型受体被磷酸化激活,将信号传导给下游信号载体蛋白,有利于细胞增殖信号的传导。当BMPR-IB基因被编辑后,BMP信号通路被阻断,抑制了细胞增殖,从而引起了CylinD2、Cdk2基因的表达极显著下调[13]。研究表明,Bcl-2基因家族在卵泡发育过程中对卵巢颗粒细胞凋亡的调节具有重要作用,ZHAO等[13]发现BMPR-IB基因被编辑后会促进颗粒细胞的凋亡,使线粒体功能发生障碍,进而极显著下调Bcl-2基因的表达。Cyp19a1是芳香化酶的编码基因,ZHAO等[13]发现BMPR-IB基因被编辑后会极显著下调Cyp19a1基因的表达,而本试验中该基因的表达并未出现显著性改变(P>0.05),推测是由于不同细胞的BMP信号通路表达调控差异所造成。猪胎儿成纤维细胞中BMPR-IB基因被编辑使得CylinD2、Cdk2、Bcl2基因表达量极显著下调,可能严重影响了猪胎儿成纤维细胞的增殖和凋亡,在试验过程中发现,编辑 BMPR-IB基因的PFF细胞传代2—3次后,明显出现细胞增殖缓慢、细胞形态变差的现象。CCK-8检测结果也表明,打靶细胞随着代数的增加其增殖水平极显著下降(P<0.01,图8)。
4 结论
利用慢病毒介导的CRISPR/Cas9技术快速地获得了编辑BMPR-IB基因的PFF细胞,打靶细胞突变率为70%;在所检测的20个最可能的脱靶位点中,仅一个存在脱靶现象。研究显示,BMPR-IB基因可能对BMPs信号通路中重要功能基因的表达和PFF细胞的增殖有重要的调节作用。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}