0 引言
【研究意义】提高家禽的抗病力,进行抗病育种是家禽育种工作者研究的热点。随着分子标记技术的发展,已鉴定出了影响鸡经济性状的诸多QTL区域,对抗病力的研究也在持续进行。脾脏是家禽重要的免疫器官,是机体对外界抗原起免疫反应的主要场所,常用于衡量家禽免疫机能的重要指标[1],动物随着年龄和机体免疫状态脾脏大小也有很大的变化。2010年伊莎公司提出了“100周龄500个蛋”的计划[2],目前在下韦尔特一个农场让这一计划成为现实,每只母鸡100周龄产蛋数达500.5个[3]。蛋鸡产蛋周期的延长可以降低生产成本,提高生产效益,而产蛋后期母鸡的健康状况将是影响这一目标实现的重要因素[4,5]。因此研究母鸡产蛋后期的免疫器官分子标记,对蛋鸡产蛋持久性具有间接的影响作用,可为产蛋后期母鸡的生存状况提供重要的理论指导。【前人研究进展】张磊等[6]利用60K芯片技术对北京油鸡100日龄脾脏进行GWAS分析,发现16个SNP位点与脾脏重显著相关,分布于8条染色体上,并将鉴定到JAK1、ZDHHC8、VAV3、SATB1候选基因。TERČIČ等[7]利用QTL作图对由体重双向选择构建的资源群体的F3代55日龄脾脏重进行研究,结果发现影响脾脏重的区域位于5号染色体13 cM位置。【本研究切入点】不同研究者对鸡脾脏重的研究多采用青年鸡,目前尚无对成年鸡产蛋后期脾脏重的GWAS进行分析。【拟解决的关键问题】基于1 501只由东乡绿壳蛋鸡和白来航蛋鸡构建的F2代资源群体,利用600 K SNP芯片进行基因型检测,对72周龄母鸡脾脏重进行全基因组关联分析,鉴定影响脾脏重的QTL区域,并基于基因芯片分析脾脏重的遗传参数,以期为提高母鸡鸡产蛋后期的健康状况提供参考依据。1 材料与方法
试验在中国农业科学院江苏省家禽科学研究所进行,血液DNA的提取在中国农业大学进行,600 K SNP基因芯片分析在Affymetrix公司进行。F2代母鸡群体饲养至72周时屠宰取脾脏称重。1.1 试验动物
以扬州翔龙禽业发展有限公司饲养的东乡绿壳蛋鸡和单冠白来航鸡为亲本构建的F2代母鸡群体为试验素材,资源群体构建及来源参见文献[8]。构建方式主要为以下方式:F0代由白来航公鸡(WL)和东乡绿壳蛋鸡母鸡(DX)进行正反交杂交产生F1代,F1代共计1 581只,其中正交(DX♂×WL♀)后代552只,反交(WL♂×DX♀)后代1 029只。以F1代为基础组建49个家系,其中25个家系以父系半同胞方式组建,交配的公鸡和母鸡间为半同胞关系并避免全同胞,分别有12个和13个家系来自于正交群体和反交群体;另外24个家系要求与配公母无血缘关系,来自于正交群体和反交群体的家系分别为11个和13个。同批出雏F2代3 749只,其中公鸡1 856只,母鸡1 893只。各代均有详细的系谱记录,三代共统计有2 447个个体,其中用于本试验统计的F2代群体母鸡为1 501只。试验过程中饲养管理条件一致,自由采食和饮水,产蛋期单笼饲养,按常规程序免疫,健康状况良好。采翅下静脉血ACD抗凝用于DNA提取。72周龄统一屠宰,取脾脏后称重。1.2 方法
1.2.1 性状表型值处理 进行GWAS分析的数据需要符合正态分布,因此首先对脾脏重利用SAS的univariate模块进行Shapiro-Wilk正态性检验。1.2.2 基因组DNA提取及质控 经翅静脉采集所有F2代个体血液,ACD抗凝后利用酚-氯仿法提取基因组DNA。基因组DNA利用 600 K Affymetrix Axiom Chicken Genotyping Array[9]进行基因分型。利用APT软件(http://affymetrix.com/)对数据筛选进行下一步分析,进行分型前的质量控制分析,采用axiom_dishqc_DQC进行质控,保留DQC≥0.82进行后续SNP分型;删除call rate小于97%的个体。SNP初步质控后,然后利用PLINK1.90[10]软件包进行数据前处理分析,剔除检出率<95%,次等位基因频率<0.01、偏离哈代温伯格P<10-6的SNP标记。BEAGLE v4.0 [11]进行基因型填充分析,选择R2>0.5的SNPs进入填充。最终得到1 501只个体和435 867个SNPs进行后续分析。
1.2.3 全基因组关联分析 为了消除群体分层可能导致的假阳性出现,在全基因组关联分析前,首先利用PLINK通过独立配对法对所有SNPs进行多维度主成分分析。利用PLINK软件计算成对SNP的r2值,高于0.2则剔除1个标记,计算内部成对SNP的r2值,并以5个SNPs为单位进行步移检测,以前5个主成分作为协变量加入到全基因组关联分析模型中。利用R脚本程序“simpleM”方法[12]计算由于各SNPs位点独立检验估计,共得到59 308个indepSNPs,然后进行bonferroni校正,计算出基因组显著水平和潜在显著水平阈值分别为8.43×10-7(1/59308)和1.69×10-5(0.05/59308)。
利用GEMMA软件[13]进行全基因组关联分析。利用单变量混合线性模型对脾脏重进行全基因组关联分析,计算公式如下:
y=Wα+xβ+u+ε (1)
式(1)中,y代表所有个体的表型性状的n×1向量值;W指协变量矩阵(指包括一列向量1和5个主成分的固定效应),α为包括截距在内对应系数的一列向量,Wα代表群体结构效应;x为标记基因型向量,β标记位点效应的大小,xβ代表SNP的效应;u为个体的随机效应向量,u~N(0, KVg),K代表由SNP标记计算出的已知的n×n遗传关系矩阵,Vg多基因加方差;ε代表n×1随机误差向量,符合ε~N(0, IVe)分布,I代表n×n身份矩阵,Ve代表残差组分。
本次研究中主要利用Wald测验作为GWAS分析显著统计指标。
为了判断有无假阳性的出现,利用R软件包GenABEL[14]计算基因膨胀系数λ。由于λ随着群体样本规模的增加而增大,因此通过样本规模1000来计算校正的λ值。
$\lambda =\frac{Median(T_{i}^{2})}{0.455}$ (2)
${{\lambda }_{1000}}=1+\frac{\lambda \text{-}1}{n}\times 1000$ (3)
式(2)中Ti2代表Wald估计值在零假设下渐近符合自由度为1的卡方分布,0.455是在零假设下假设没有关联的期望中位数。
式(3)中n代表总样本量,λ代表总样本量的估计值。
全基因组关联分析曼哈顿图和QQ图R软件中由“gap”和“qqman”并修改相应参数绘制完成[15]。
1.2.4 连锁不平衡分析 由于基因间的连锁关系导致在一个区间内许多SNPs在全基因组关联分析中被检测均具有显著效应。为了区分由于连锁不平衡造成的假阳性,将候选SNP位点的基因型作为协变量,重复1.2.3中GWAS分析。经条件分析后,原先显著的SNP位点均不显著时即将该位点作为候选位点筛选候选基因。
1.2.5 基于SNP数据的遗传参数计算 将质控后的SNPs数据利用PLINK处理成GCTA v1.24[16]处理格式。然后利用GCTA v1.24软件的单性状REML方法估计脾脏重的遗传参数。同时计算候选SNP位点对性状表型方差的贡献率(CPV)以及各染色体对脾脏重解释的表型方差。
1.2.6 基因注释 将分析得到的显著SNP位点查找Ensembl和NCRI上Galgal4 assembly序列,按照SNP突变位点所在区间选择候选基因,以“有义突变”、“cds区”、“外显子区”、“UTR”、“内含子区”、“下游\上游”的原则顺序选择候选基因进行分析[17,18]。
2 结果
2.1 脾脏重表型值和遗传力统计
脾脏重的型值和基于芯片计算的遗传力见表1。由表知脾脏重的平均值在1.64 g,其变异系数为48.36%,说明在72周龄老鸡的脾脏重存在很大的差异。其遗传力为0.236,为低遗传力性状。Table 1
表1
表1脾脏重表型数据和遗传参数
Table 1Descriptive statistics and hertibilty for spleen weight in the F2 population
| 指标 Index | 值 Value |
|---|---|
| 均值±标准差(g)Mean±SD | 1.64±0.79 |
| 变异系数CV (%) | 48.36 |
| 遗传力hsnp2 | 0.236 |
新窗口打开
2.2 全基因组关联分析
对脾脏重进行全基因组关联分析后发现达到基因组显著水平的SNP位点共412个,见表2和图1。分别有383个和29个位于1号和28号染色体上,同时这两条染色体上分别有242个和34个潜在性显著水平。在4号和16号染色体上分别有3个和2个潜在性SNP位点。λ是判断群体是否存在分层的参数,一般在1.05以上说明群体存在分层,不适宜进行全基因组关联分析。由本研究图1-B的QQ图知,群体的λ值为1.042,说明所有个体均匀分布,不存在群体分层现象。Table 2
表2
表2与脾脏重显著和潜在显著关联SNP位点信息
Table 2Number and distribution of significant and suggestive SNPs association with spleen weight
| 染色体 GGA | 显著位点 Sig. | 潜在显著位点 Sug. | 区域 Region(bp) |
|---|---|---|---|
| 1 | 383 | 242 | 161105630-174178625 |
| 28 | 29 | 34 | 470414-1267646 |
| 4 | 3 | 75912399-76073771 | |
| 16 | 2 | 175263-175325 | |
| 合计 Total | 412 | 281 |
新窗口打开
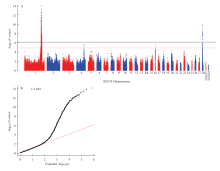 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1脾脏重全基因组关联分析曼哈顿图和QQ图
图A为全基因组关联分析曼哈顿图,横坐标为染色体号和两个连锁群(LGE64和LGE32),黑色和绿色水平线分别代表全基因组关联分析显著性(8.43 × 10-7)和潜在显著性(1.69 × 10-5)性阈值;图B为群体分层QQ检验,横坐标代表由-log10转换后的P值,λ为基因膨胀系数
-->Fig. 1Manhattan plot and QQ-plot of genome-wide association analysis for spleen weight
Plot A is the Manhattan plot, which shows the -log10 (observed P values) for association of SNPs (y-axis) plotted against their chromosomal positions on each chromosome (x-axis), and the horizontal black and green lines depict the genome-wide significant (8.43 × 10-7) and suggestive significant (1.69 × 10-5) thresholds, respectively. Plot B is the QQ test for population structure, the x-axis indicates the expected -log10-transformed P values, and the y-axis shows the observed -log10-transformed P values. λ represents the genomic inflation factor
-->
染色体1号和28号上鉴定出的较多SNP位点可能是由于连锁不平衡造成,而通常有义突变更容易造成较大的表型改变,因此首先以SNP位点在CDS区间作为候选基因,通过查找glagal4参考基因组序列后,显著和潜在显著的SNPs没有位于编码区序列。而UTR区域的突变可以改变顺式调控元件,从而影响mRNA翻译和转录效率的稳定性[19],本研究因此选择了1号和28号染色体上位于UTR-3区域的SNPs作为候选位点在1号染色体上发现有4个SNPs位点位于UTR-3区(表3),分别位于基因CCDC122、KCTD4、SLC25A30和SUCLA2区间内。28号染色体为1个SNP位点位于PCASP2基因的UTR-3区,因此选择这5个SNP位点作为候选位点进行初步分析。同时将4号和28号的潜在显著性SNPs参照参考基因组,筛选距离最近的基因作为候选基因。
Table 3
表3
表3脾脏重显著和潜在性SNP位点信息
Table 3Information of significant and suggestive mutations association with spleen weight
| 突变点 Snp | 染色体 Chr | 物理位置 Position (bp) | EA/AA | MAF | beta | se | P值 P value | 基因区间 Location | 基因注释 Gene symbol | CPV (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| rs314975697 | 1 | 166476909 | A/T | 0.329 | -0.173 | 0.035 | 7.54E-07 | UTR-3 | CCDC122 | 2.83 |
| rs314001986 | 1 | 166994822 | A/G | 0.488 | -0.244 | 0.033 | 3.51E-13 | UTR-3/intron | KCTD4/GTF2F2 | 5.91 |
| rs316299009 | 1 | 167087710 | C/G | 0.458 | -0.231 | 0.033 | 7.83E-12 | UTR-3 | SLC25A30 | 5.33 |
| rs317751826 | 1 | 167942690 | A/G | 0.338 | 0.190 | 0.036 | 2.26E-07 | UTR-3 | SUCLA2 | 3.20 |
| rs312729296 | 28 | 977835 | A/G | 0.115 | 0.257 | 0.046 | 2.82E-08 | UTR-3 | PCASP2 | 2.52 |
| rs315695199 | 4 | 75912399 | A/G | 0.286 | 0.153 | 0.033 | 3.21E-06 | intron | LDB2 | 1.81 |
| rs315270535 | 4 | 75955611 | C/T | 0.139 | 0.205 | 0.045 | 5.42E-06 | intron | LDB2 | 1.91 |
| rs14491507 | 4 | 76073771 | A/G | 0.051 | 0.302 | 0.067 | 6.03E-06 | D14057,U/72200 | TAPT1,FGFBP2 | 1.73 |
| rs314065899 | 16 | 175263 | C/T | 0.411 | -0.133 | 0.028 | 3.06E-06 | intron | HEP21 | 1.67 |
| rs314122647 | 16 | 175325 | C/T | 0.431 | -0.126 | 0.028 | 7.99E-06 | intron | HEP21 | 1.50 |
新窗口打开
对1号和28号的5个SNP位点以及4号和16号的潜在显著性位点进行位点CPV计算,结果见表3,由表知以1号染色体上显著位点解释的CPV均最大,最高的是位点rs314001986,解释了5.91%的表型方差,因此将rs314001986作为1号染色体的候选位点进行条件分析。同时对28号染色体上位点rs312729296进行条件分析。以判断在各位点之间的显著性是否由连锁不平衡造成,对1号和28号染色体以rs314001986和rs312729296候选位点进行条件分析后结果显示原先显著的位点均不显著(图2)。对4号和16号染色上潜在的显著位点进行连锁不平衡分析结果见图3,由图知4号和16号上潜在性显著位点均存在着较强的连锁不平衡状态。
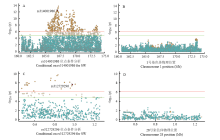 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图21号和28号染色体显著位点条件分析
A:在1号染色体上区间161.1-174.2 Mb所有SNPs的-log10 (P值),有383个为显著性SNPs(桔色),242个潜在性显著SNPs(灰色);B,将rs314001986基因型作为协变量进行条件分析后的SNPs的-log10 (P值),经条件分析后原先显著显著(桔色)和潜在(灰色)均不显著。C:在28号染色体上区间0.47-1.27 Mb所有SNPs的-log10 (P值),有29个为显著性SNPs(桔色),34个潜在性显著SNPs(灰色);D:将rs312729296基因型作为协变量进行条件分析后的SNPs的-log10 (P值),经条件分析后原先显著(桔色)和潜在显著(灰色)的点均不显著
-->Fig. 2Regional plot for conditional analysis about significant loci on GGA1 and GGA28
A: In the region 161.1 to 174.2Mb on GGA the -log10 (observed P values) of the SNPs (y-axis) are presented according to their chromosomal positions (x-axis). A number of 383 and 242 SNPs reached a genome-wide significance (8.43 × 10-7) and suggestive level (1.69× 10-5 ). B: The genotype of rs314001986 was placed into the univariate test as covariance for conditional analysis. After conditioning on rs314001986, the significant SNPs in plot A were all substantially attenuated below genome-wide significant level in plot B. C: In the region 0.44 to 1.3 Mb on GGA28 the -log10 (observed P values) of the SNPs (y-axis) are presented according to their chromosomal positions (x-axis). A number of 29 and 34 SNPs reached a genome-wide significance (8.43 × 10-7) and suggestive level (1.69× 10-5 ). D: The genotype of rs312729296 was placed into the univariate test as covariance for conditional analysis. After conditioning on rs312729296, the significant SNPs in plot C were all substantially attenuated below genome-wide significant level in plot D
-->
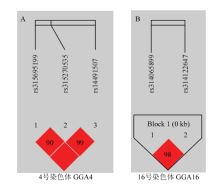 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图34号和16号染色体上潜在性显著位点LD分析
-->Fig. 3LD analysis about suggestive loci on GGA4 and GGA16
-->
综合上述分析,最终以rs314001986、rs312729296、rs315270535和rs314065899作为候选SNPs进行基因注释分析。根据参考基因组glagla4可知,这4个位点分别位于基因KCTD4(potassium channel tetramerisation domain containing 4,钾离子聚合结构域)、PCASP2(mucosa associated lymphoid tissue lymphoma translocation gene 1-like,黏膜相关性淋巴样组织)UTR-3区域内、以能LDB2(LIM domain binding 2,LIM结构域结合基因)和HEP21(hen egg protein 21,卵清组成蛋白)内含子区域内。
分别对1号和28号染色上候选SNPs位点rs314001986和 rs312729296在各个体上的基因型对应的表型分析见图4。由图4可知两个SNP的纯合子基因型G/G对脾脏重均具有增重效应。rs314001986位点3种基因型对应的表型之间存在极显著差异,rs312729296三种基因型为两个纯合子存在极显著差异。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图 4rs314001986和 rs312729296在各个体上基因型与表型关联分析
柱子上数字代表各基因型的基因型频率;柱子上字母上标不同代表表型值间极显著差异
-->Fig. 4Relationship between genotype and phenotype on rs314001986 and rs312729296
Decimals inside the bars of indicate genotype frequency; The phenotypic data with different letters means extremely significant difference.
-->
2.3 染色体遗传力
利用GCTA软件对染色体的遗传力和染色体长度进行分析结果见图5。每条染色体的遗传力和其长度的相关系数为R2 =0.386,为中等极显著相关(P= 1.50×10-4),1号染色体的遗传力最大为9.25%,其次为28号染色体为4.55%(图5-A)。将用于条件分析的位点SNPs作为协变量后重新计算染色体遗传力,1号染色体的遗传力变为4.03%,28号染色体变为2.05%(图5-B),而其他染色体的遗传力均几乎未变。
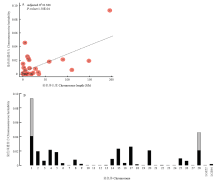 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5脾脏重各染色体的遗传力
A图染色体解释的遗传力散点图——横轴代表每条染色体的长度(bp),纵轴表示每条指的每条染色体解释的遗传力,其中圆圈上不同的号代表不同的染色体;B图是染色体解释的遗传力柱状图——其中灰色的柱子代表显著区域的遗传力
-->Fig. 5Genome partitioning for spleen weight by joint analysis
A. The estimated proportion of variance captured by each chromosome against its size. The characters in the circles are the chromosome numbers. B. Contributions of genome-wide association study (GWAS) SNPs partitioned by chromosome. The whole bars indicate the estimates of variance explained by each chromosome, in which the two grey bars represent the same values by two resulting loci
-->
3 讨论
前人利用关联分析或QTL定位对脾脏重进行研究,多以青年鸡为研究对象[6, 20]。随着“100周龄500个蛋”的计划实现,研究母鸡产蛋后期的健康状况尤为重要。本研究首次利用600 K高密度基因芯片对母鸡产蛋后期的脾脏重进行全基因组关联分析,研究发现4个QTL区域与脾脏重显著或潜在性关联。其中有412个显著性SNP位点和281个潜在性SNP位点,在1号、28号、4号和16号染色体上均有分布,其中以1号染色体上分布最多,且1号染色体的遗传力为9.23%。PARK等[20]研究对70日龄鸡脾脏重的QTL定位发现,10号和11号染色体对脾脏重解释遗传力在分别为3.9%和2.8%,均较本研究中4、16和28号染色体上区域要高,而低于本研究中1号染色体的显著区域的遗传力(图5-B)。1号染色体上与脾脏重显著关联的区域在165—173 Mb区间,在本研究群体发现也是影响蛋重、卵巢重和饲料消耗[8, 21-22]的重要区域,而在其他研究也发现与体重具有重要关系[23,24],说明1号染色体170 Mb左右的区间是影响鸡生长的重要QTL区域。张磊等[6]研究发现影响100日龄脾脏重的显著性SNP位点分布于1、2、3、4、13、18、19和25号染色体上。本研究中所定位到的关联区域与上述不同,代表了新的影响脾脏重的QTL区域或位点,然不同品系间以及不同时期可能会表现出不同的病原免疫应答反应[25],本研究中所得到的QTL区域需要进一步进行功能验证筛选。本研究中,1号染色体中共有383个SNPs位点达到基因组显著水平,对显著水平最高的SNP位点rs314001986进行条件分析后原显著的位点均不显著,以rs314001986作为候选位点,该位点位于KCTD基因UTR-3区域。KCTD蛋白参与离子运输过程[26],KCTD家族中含有26个成员,均具有保守的N端结构和钾通道四聚化结构域,参与了Wnt/beta-catenin、FGF、PPAR等信号通路,在许多生物过程和疾病中发挥作用,KCTD4在人神经母细胞瘤中上表达[27]。对鸡的生物学作用以及参与的免疫调控尚不清楚,根据上述170 Mb区间可能是影响鸡各性状“重量”的区域,KCTD4有可能是促进脾脏增殖等作用,其作用机理有待于进一步研究。
张磊等[6]发现影响脾脏重的4号染色体SNP位点在62.8 Mb区间,本研究得到的76 Mb区间与其研究不同,本研究对张磊研究中得到的SNP位点(rs14479254)与本研究中位点rs315270535进行连锁不平衡分析发现两个位点之间不存在连锁不平衡(D’ =0.5, R2=0.05),可能在4号染色上存在两个区域影响脾脏重。本研究中位点rs315270535位于LDB2基因的UTR-3区域,LDB2 基因能够与多种转录因子结合,在脑的发育和血管形成过程中发挥重要作用,与人的动脉瘤疾病相关。吴丹[28]对北就油鸡的全基因组关联分析发现LDB2基因下游108 kb的一个SNP位点与100日龄体重潜在关联,GU等[29]也发现LDB2与鸡7—12周体重显著关联并与6—12周平均日增重相关。在发生细胞免疫和体液免疫时,脾脏组织通过脾细胞增殖功能使淋巴小结增生或脾索内浆细胞及巨噬细胞显著增加进行免疫应答,综上,1号和4号染色体上发现的显著和潜在性显著SNPs以及相关候选基因可能均参与了免疫应答过程中的脾脏增殖作用。
本研究在16号和28号染色体得到的潜在和显著性QTL区域为首次发现。而其中与免疫系统密切相关的大部分主要组织相容性复合体(MHC)基因位于16号染色体上(http://www.ncbi.nlm.nih.gov/gene),可能16号的潜在显著区间参与了MHC在转录或转录后调控上发挥作用,以进一步增强免疫功能。其潜在性SNP位点rs314065899位于HEP21(hen egg protein 21)内含子区间,HEP21卵清组成蛋白[30],随着输卵管发育成熟而上调,HEP21同时参与胚胎孵化过程中的一些免疫反应[31],由此推测HEP21可能参与到脾脏的免疫应答过程。
28号染色上影响脾脏重的区域位于0.47—1.27 Mb区间内,筛选到的SNP位点rs312729296位于PCASP2基因UTR-3区域,PCASP2是MALT家族之一,MALT1(黏膜相关淋巴组织淋巴瘤转运蛋白1)基因在人上参与了先天免疫以及炎症反应,参与了NF-кB的激活过程。这个家族常见的有基因PCASP1、PCASP2和PCASP3,PCASP2在人类基因上已经消失,然在鸡基因组上仍起作用[32]。本研究推测在鸡上PCASP2可能发挥了与哺乳动物上MALT1一致的作用,具有参与炎症反应的功能。
4 结论
本研究利用高密度基因芯片,对蛋鸡72周龄脾脏重进行了全基因组关联分析,共发现了412和281个SNPs位点与脾脏重显著和潜在显著关联,分别位于1、28、4和16号染色体上,筛选到KCTD4、LDB2、HEP21和PCASP2候选基因。并利用生物信息学分析方法得到1号染色体解释的遗传力为9.25%,脾脏重的遗传力为0.236。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}