0 引言
【研究意义】紫色小麦是一种特殊的小麦种质,其籽粒呈现出的紫色与紫色种皮中的黄酮类化合物花青苷有关,而花青苷由花青素和糖基结合而成[1],花青素在许多植物中起重要作用,不仅作为防止UV-B照射的保护剂,还表现出抗氧化活性,因此具有在人类健康中用作潜在的抗癌和抗动脉硬化化合物的功能。此外,花青素构成生物活性代谢物,还参与调节植物与昆虫,哺乳动物和鸟类等的种间关系[2,3]。国家小麦改良中心贵州分中心于2004年用自育品系贵紫99-4小麦作母本,与自育品系贵农03-7作父本杂交后,采用系谱法于2010年选育得到小麦品系贵紫麦1号,并于2015年6月经贵州品种审定委员会通过品种审定,审定编号为黔审麦2015003号。贵紫麦1号籽粒不仅整粒为紫色,而且在产量、品质、抗病性(白粉病、条锈病、赤霉病等)、抗旱耐寒、耐瘠薄等方面均表现出较强的优势,同时与其他一些紫黑色小麦相比,其含有丰富的蛋白质、维生素和多种微量元素。分析贵紫麦1号小麦籽粒花青素合成的关键基因和关键酶,探寻其花青素代谢合成途径,可为小麦籽粒色素相关基因的鉴定、功能分析以及特异性表达研究提供参考。【前人研究进展】国内外****对小麦紫色籽粒的研究大多从遗传性状入手,由于不同品种小麦遗传背景不同,得出的结论也有差异。徐丙元等[4]确定漯珍一号的紫粒受到分别位于2A和3A染色体的2个独立基因的控制,且多酚氧化酶活性最高;DOBROVOLSKAYA等[5]认为Purple Feed和Purple与Saratovskaya杂交后的紫粒受2对互补的显性基因控制;黄碧光[6]以C75与紫粒小麦03初3为材料,认为紫粒呈2对显性互补基因控制的母性遗传;宗学凤等[7]认为PAL参与促进花青素的合成;李杏普等[8]认为多酚氧化酶活性影响小麦籽粒颜色,与小麦抗病性相关。除遗传性状外,环境因子和调控因子对花青素的合成代谢皆有影响。低温诱导相关基因表达,高温加剧植物分解代谢,影响酶活性,从而影响花青素的稳定性及含量[9,10,11,12]。强光诱导结构基因及调节基因的表达,调节转录因子表达模式,提高花青素累积量[13,14],不同光质调控相应光受体的表达,影响花青素的合成量[15]。植物激素如茉莉酸、赤霉素、脱落酸等可在花青素的合成途径中诱导或调控相关基因的表达[1,16-18]。前人对小麦紫色籽粒的研究大多从籽粒品质、发育形态和胁迫机制等方面入手,而在籽粒颜色形成的差异表达研究方面少见有报道,仅在其他一些作物上有相关研究,如周姚[19]发现水稻中OsCl调控OsDFR、OsANS突变后,可影响花青素的合成;BOVY等[20]将玉米中的转录因子Lc、C1转入番茄中,类黄酮总量提高,但不影响花青素的含量;LI等[21]将Lc转入水稻品种chao2-10中,虽然花青素含量明显提高,但会引起水稻不育;张长青等[22]以越橘为试材,发现PAL、C4H、CHS等酶基因在越橘幼果和/或成熟果中上调。【本研究切入点】通过对贵紫麦1号籽粒的初步横切和纵切观察,发现其中具有果皮、种皮、糊粉层均为紫色的籽粒。近年来,人们愈加重视紫色小麦的营养价值及生理生化指标,但对紫色小麦颜色相关的调控基因研究尚未完全明确。【拟解决的关键问题】对贵紫麦1号同株籽粒变紫前和变紫后2个灌浆时期的转录组测序,进行差异表达分析,对所找到的差异表达基因进行注释,并在富集和代谢中找到调控花青素合成的相关基因,进一步揭示紫色小麦颜色相关的基因调控途径,为贵紫麦1号在小麦育种研究中的应用打下分子基础。1 材料与方法
1.1 植物材料与处理
贵紫麦1号种植于贵州大学教学实验场国家小麦改良中心贵州分中心科研基地(26º23′N,106º40′E,海拔1 121 m),每小区面积30 m2,长×宽=6 m×5 m,小麦播幅33 cm,采用随机区组实验设计,每小区重复3次。田间管理浇水、除草和施肥等按统一方式进行。贵紫麦1号2016年4月6日左右抽穗,于4月10日开花期选取开花一致的麦穗进行挂牌标记并连续观察记载。本次试验以开花后12日籽粒未变紫作为变紫前的样品,以开花后25日整个籽粒2/3变紫作为变紫后的样品(图1),每日各取不同植株的3个麦穗籽粒进行样品混合,液氮速冻后置于-80℃超低温冰箱保存,用于总RNA提取和转录组测序。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1贵紫麦1号籽粒变紫前后比较
-->Fig. 1Observation of grains of Guizimai No.1 before and after purple-changing
-->
1.2 RNA提取及总RNA纯度与完整性检查
采用天根RNAsimple植物总RNA提取试剂盒提取总RNA,通过琼脂糖凝胶电泳检测RNA污染情况。使用Nanodrop分光光度计检测样品OD260/230值,继而对RNA进行纯度分析;Agitent2100生物分析仪检测样品浓度、RIN值以及28S/18S值。1.3 文库构建及测序
将样品送至北京诺禾致源生物信息科技有限公司进行建库测序,用带有Oligo(dT)的磁珠富集真核生物Mrna,随后加入fragmentation buffer将mRNA打断成短片段,以mRNA为模板,用六碱基随机引物(random hexamers)合成一链cDNA,加入缓冲液、dNTPs、DNA polymerase I和RNase H合成二链cDNA,AMPure XP beads纯化双链cDNA。纯化的双链cDNA先进行末端修复、加A尾并连接测序接头,再用AMPure XP beads进行片段大小选择。最后进行PCR扩增,并用AMPure XP beads纯化PCR产物,得到最终的文库。库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行Illumina HiSeq测序。1.4 原始数据过滤及转录本拼接
原始数据过滤流程如下:(1)去除带接头(adapter)的reads;(2)去除N(N表示无法确定碱基信息)比例>10%的reads;(3)去除低质量reads。为了保证信息分析质量,需对raw reads过滤,得到clean reads,后续分析都基于clean reads结果。由于贵紫麦1号转录组为无参转录组,获得clean reads后,诺禾致源采用Trinity对clean reads进行拼接,以获取后续分析的参考序列[23,24]。1.5 差异基因表达筛选分析
基因差异表达的输入数据为基因表达水平分析中得到的read count数据。首先采用TMM对read count数据进行标准化处理,再用DEGseq进行差异分析,筛选阈值q-value<0.005且|log2 (fold change)| >1。对于差异基因,如果基因的log2 (fold change) >0,则认为该差异基因为上调,反之,若log2 (fold change) <0,认为该差异基因为下调[25]。1.6 差异基因GO功能注释、分类及KEGG分析
本研究对于GO富集分析方法为GOseq,该法基于Wallenius non-central hyper-geometric distribution。首先将所有差异表达基因Gene Ontology数据库与各个term 映射,计算每个term的基因数目,随后与整个基因组背景相比,在差异表达基因中显著富集[26]。KEGG(Kyoto encyclopedia of genes and genomes)是有关Pathway的主要公共数据库。Pathway显著性富集分析以KEGG Pathway为单位,应用超几何检验,找出差异基因相对于所有有注释的基因显著富集的pathway。使用KOBAS(2.0),设置参数--fdr为BH(即使用BH校正)进行Pathway富集分析。FDR≤0.05的Pathway,即定义为在差异表达基因中显著富集的Pathway[27]。
1.7 差异表达基因荧光定量PCR(qRT-PCR)验证
以β-Actin作为内参基因,待验证基因为CHS和ANS,特异性引物序列见表1。使用TransScript II All-in-One First-Strand cDNA Synthesis SuperMix for qPCR(北京全式金)试剂盒对1.2中提取的模板RNA进行逆转录,SYBRGreen染料法进行qRT-PCR,每个样品3次重复。差异倍数采用2-△△Ct法进行计算。Table 1
表1
表1用于荧光定量PCR验证的基因及引物
Table 1The qRT-PCR primers for the three genes
| 基因名称 Gene name | 引物序列 Primer sequence (5′-3′) | 基因ID Gene ID |
|---|---|---|
| β-Actin | F: CCAAGGCGGAGTACGATGAGTCT | |
| R: TTCATACAGCAGGCAAGCACCAT | ||
| CHS | F: CAACCGAGCTTTTACAG | c61642_g2 |
| R: ATCACCATTTGCTTTCC | ||
| ANS | F: GACGGGAAGAGGGAGTGG | c57850_g1 |
| R: GAGGGAGAGGATGGCGAG |
新窗口打开
2 结果
2.1 测序产量统计及转录本拼接
利用高通量测序技术,对贵紫麦1号籽粒灌浆期变紫前和变紫后2个时期进行转录本测序(表2)。贵紫麦1号籽粒变紫后获得原始序列片段114 058 286条,筛选过滤后,获得106 906 108条clean reads,占原始序列的93.73%。贵紫麦1号籽粒变紫前获得原始序列片段数量为107 009 534条,筛选过滤后,获得101 547 534条clean reads,占原始序列的94.90%。结果显示2个转录组文库测序质量较好,准确度高,满足后续数据分析需求。Table 2
表2
表2测序数据评估统计
Table 2The statistical results of sequencing data
| 名称Name | Raw reads | Clean reads | Clean bases (G) | 错误率Error rate (%) | Q20 (%) | Q30 (%) | GC含量 GC content (%) |
|---|---|---|---|---|---|---|---|
| WS1 WS2 | 114058286 107009534 | 106906108 101547534 | 13.36 12.69 | 0.03 0.03 | 94.16 95.01 | 88.98 90.19 | 51.66 53.08 |
新窗口打开
通过Trinity软件对所得clean reads进行拼接,整合并取每条基因中最长的转录本作为Unigene,共获得170 396条转录本,长度为119 020 625。贵紫麦1号clean reads获得119 572条70 339 868长的Unigenes。平均长度为588 bp,长度为200—500 bp的Unigenes数量最多占总数的66.44%。Unigenes N50和N90长度分别为869和250 bp。
2.2 贵紫麦1号转录组基因功能注释
为获得全面的基因功能信息,对获得的119 572条Unigenes进行了Nr、Nt、Pfam、KOG/COG、Swiss- prot、KEGG、GO 7大数据库的基因注释。119 572个高质量独特序列中,在BLAST搜索中,有86 004条(71.92%)Unigenes与现有基因模型具有至少1个显著匹配。通过将本研究中发现的DEG与NCBI数据库进行比较,鉴定了至少5种具有与来自节节麦(Aegilops tauschii)、乌拉尔图小麦(Triticum urartu)、大麦(Hordeum vulgare)、二穗短柄草(Brachypodium distachyon)、小麦(Triticum aestivum)已知基因同一性且序列相似性高的Unigenes(图2)。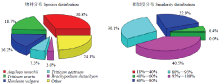 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2Nr库比对上的物种分布和相似度分布图
-->Fig. 2Species distribution and similarity distribution maps of Nr library
-->
2.3 KOG转录因子注释
将组装得到的20 255条Unigenes在KOG数据库进行比对,KOG分为26个group,将注释成功的基因按KOG进行分类,转录因子注释结果表明一般功能基因,蛋白质翻译后修饰与转运、分子伴侣以及翻译、核糖体结构与生物合成所占比例较大,分别为15.79%、14.51%和10.54%(图3)。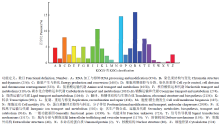 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3KOG功能分类图
-->Fig. 3Diagram of KOG functional classification
-->
2.4 RNA-seq整体质量评估
样品间基因表达水平相关性是检验试验可靠性和样本选择是否合理的重要指标,使用R语言进行Pearson相关系数计算后发现,2个材料相似系数为0.601,说明整体质量较好。2.5 差异基因分析
通过将贵紫麦1号变紫前和变紫后2个时期转录组进行比较,并将存在差异表达的基因进行统计分析发现,共找到差异表达基因643个,其中上调基因236个,下调基因407个,分别占差异基因数量的36.7%、63.3%(图4)。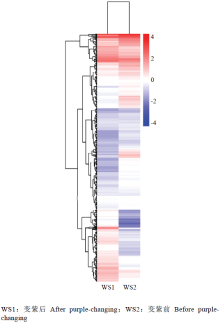 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4基因差异表达分析热图
-->Fig. 4Cluster analysis of DEGs
-->
2.6 GO注释及富集
Gene Ontology(基因本位,GO)是全面描述生物体中基因及其产物的属性的分类系统。对获得的基因进行GO注释之后,按照基因参与的生物过程(biological process)、所处的细胞组分(cellular component)及具有的分子功能(molecular function)的下一层级进行分类GO注释,结果显示(图5)共44个分类。注释成功的39 906条基因中基因参与的生物过程有19组下级分类,大部分注释在代谢过程(GO:0008152,21 253条)、细胞过程(GO:0009987,20 960条)及单生物过程(GO:0044699,15 921条)。其次与所处的细胞组分有关,共14组下级分类,注释较多的分别是细胞(GO:0005623,11 749条)和细胞部分(GO:004464,11 739条)。最少的是与具有的分子功能有关的差异表达基因,共11组下级分类,20 953和17 973条基因被注释在结合(GO:0005488)和催化活性(GO:0003824)类别下。在DAG图上将不同富集程度的GO标上不同的颜色,可清楚地展示研究的生物学意义(图6)。GO注释的433个差异基因富集在生物过程和分子功能上,从富集情况来看,富集在生物过程的差异基因较为显著的有碳水化合物代谢过程(GO:0005975,16.03%)、应激反应(GO:0006950,10.83%)、防御反应(GO:0006952,7.32%)等,富集在分子功能的差异基因主要有水解酶活性分子功能(GO:0016787,34.84%),酯键(GO:0016788,11.53%)、肽酶活性(GO:0008233,9.41%)。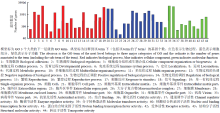 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5差异表达基因GO功能分类图
-->Fig. 5Diagram of GO functional classification of DEGs
-->
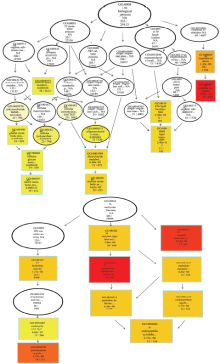 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图6topGO有向无环图
每个方框或圆圈代表一个GO term,方框中内容从上到下代表的含义依次为GO term的id、GO的描述、GO富集的corrected P-value、该GO下差异基因的数目/该GO下背景基因的数目
-->Fig. 6Directed acyclic graph of topGO
Each box or circle represents a GO term, the contents of the box from top to bottom on behalf of the meaning of id to GO term, GO description, GO enriched corrected P-value, the number of differential genes/the number of background genes under the GO
-->
2.7 差异基因KEGG分析
类黄酮生物合成代谢途径(ko00941)共66个相关的基因(表3),排在上调差异基因富集前20的通路中(图7)。KO00941含有2条相关的上调差异Unigenes。矫正后的P-value为0.6577,基因ID为c61642_g2、c57850_g1,涉及ANS[EC:1.14.11.19 ]和CHS[EC:2.3.1.74 ],log2 (fold change)分别为3.4164和2.1258。由通路可知,贵紫麦1号合成的花青素主要为天竺葵色素、矢车菊色素和飞燕草色素(图8)。Table 3
表3
表3类黄酮生物合成途径相关基因
Table 3Flavonoid biosynthesis pathway related genes
| 基因ID Gene ID | 基因NR描述 Gene NR description | log2 (fold change) | q-value |
|---|---|---|---|
| c8011_g1 | — | NA | NA |
| c60031_g3 | Unnamed protein product | 0.12908 | 0.99989 |
| c20668_g1 | Chalcone synthase 1 | 0.030594 | 0.99989 |
| c80569_g1 | Chalcone synthase A | 0.49357 | 0.99989 |
| c6641_g1 | — | NA | NA |
| c53082_g1 | Leucoanthocyanidin reductase | 0.3125 | 0.99989 |
| c61642_g2 | Chalcone synthase 1 | 2.1258 | 1.7513e-05 |
| c56224_g1 | — | -4.7918 | 0.99989 |
| c60035_g2 | Naringenin, 2-oxoglutarate 3-dioxygenase | -0.10587 | 0.99989 |
| c104728_g1 | — | -1.6764 | 0.99989 |
| c61459_g2 | Predicted protein | 0.40503 | 0.99989 |
| c58846_g2 | — | NA | NA |
| c90641_g1 | — | NA | NA |
| c43136_g1 | Predicted protein | -0.67636 | 0.99989 |
| c89181_g1 | Anthocyanidin reductase | 0.49357 | 0.99989 |
| c63309_g4 | Trans-cinnamate 4-monooxygenase | 1.0785 | 0.99989 |
| c43204_g1 | — | -4.1503 | 0.99989 |
| c52115_g1 | Anthranilate N-benzoyltransferase protein 1 | 0.33007 | 0.99989 |
| c28690_g1 | Predicted protein | 0.71596 | 0.99989 |
| c60822_g1 | Unnamed protein product | -0.18387 | 0.99989 |
| c52097_g1 | Chalcone flavanone isomerase | 0.40066 | 0.99989 |
| c35628_g1 | — | NA | NA |
| c84548_g1 | Leucoanthocyanidin reductase | 0.49357 | 0.99989 |
| c102988_g1 | Anthocyanidin reductase | -0.50643 | 0.99989 |
| c60035_g1 | Flavanone 3-hydroxylase | 0.24375 | 0.99989 |
| c79617_g1 | o-methyltransferase, putative | 0.49357 | 0.99989 |
| c49339_g1 | Predicted protein | -0.29872 | 0.79408 |
| c52205_g1 | Predicted protein | 0.083343 | 0.99989 |
| c61459_g1 | Anthranilate N-benzoyltransferase protein 1 | 1.2209 | 0.31027 |
| c22834_g1 | Cytochrome P450 | -0.50643 | 0.99989 |
| c34686_g1 | Predicted protein | 0.13993 | 0.99989 |
| c66443_g1 | Anthocyanidin reductase | -1.284 | 0.99989 |
| c57850_g1 | Anthocyanidin synthase | 3.4164 | 3.2221e-26 |
| c56260_g1 | Chalcone synthase 1 | 0.72183 | 0.99989 |
| c62495_g1 | Predicted protein | -0.80578 | 0.99989 |
| c38330_g1 | Putative caffeoyl-CoA O-methyltransferase | NA | NA |
| c61459_g3 | Predicted protein | -0.060974 | 0.99989 |
| c52129_g1 | — | -5.3644 | 0.99989 |
| c58160_g2 | Predicted protein | -0.72242 | 0.99989 |
| c116744_g1 | — | 1.4936 | 0.99989 |
| c87169_g1 | — | NA | NA |
| c109587_g1 | Putative caffeoyl-CoA O-methyltransferase | 2.4936 | 0.99989 |
| c47275_g2 | Chalcone synthase 2 | 2.0203 | 0.99989 |
| c63309_g1 | Predicted protein | 0.18904 | 0.99989 |
| c64042_g2 | Hypothetical protein OsI_07154 | -0.50643 | 0.99989 |
| c96559_g1 | Flavonoid 3’, 5’-hydroxylase 2 | 1.8155 | 0.99989 |
| c58846_g1 | Unnamed protein product | -1.5379 | 0.99989 |
| c46502_g1 | Predicted protein | 0.62001 | 0.99989 |
| c60031_g1 | Cytochrome P450 | -0.10847 | 0.99989 |
| c75680_g1 | Anthranilate N-benzoyltransferase protein 2 | -0.41332 | 0.99989 |
| c39099_g1 | Chalcone synthase 2 | 0.87208 | 0.99989 |
| c47275_g1 | Chalcone synthase 1 | 0.86444 | 0.99989 |
| c58160_g1 | Leucoanthocyanidin reductase | -1.0783 | 0.99989 |
| c22352_g1 | Anthocyanidin reductase | -0.50643 | 0.99989 |
| c61792_g1 | Predicted protein | 1.1501 | 0.99989 |
| c63309_g2 | Hypothetical protein OsI_04230 | 1.4619 | 0.99989 |
| c51756_g1 | Flavonol synthase/flavanone 3-hydroxylase | -0.76947 | 0.60692 |
| c46706_g1 | Unnamed protein product | 0.33934 | 0.99989 |
| c62495_g2 | Predicted protein | 0.54527 | 0.99989 |
| c11599_g1 | — | -2.3138 | 0.99989 |
| c108668_g1 | — | NA | NA |
| c11518_g1 | Cytochrome P450 98A1 | 0.49357 | 0.99989 |
| c45760_g1 | Flavonoid 3’, 5’-hydroxylase | 0.046107 | 0.99989 |
| c8267_g1 | — | NA | NA |
| c42665_g1 | — | NA | NA |
| c47131_g1 | — | NA | NA |
新窗口打开
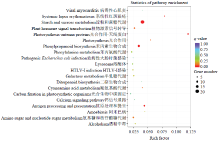 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图7KEGG通路富集程度散点图
-->Fig. 7Scatter diagram of KEGG pathway enrichment
-->
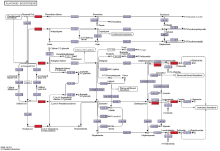 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图8KEGG数据库中类黄酮生物合成途径
-->Fig. 8Flavonoid biosynthesis pathway in KEGG database
-->
2.8 差异表达基因的qRT-PCR验证
为验证基因差异表达的可靠性,选取CHS(c61642_g2)、ANS(c57850_g1)2个目的基因进行qRT-PCR验证(图9)。结果发现随着籽粒变紫,目的基因表达水平变化趋势相同均表现上调,表明通过转录组测序技术分析出的差异基因结果真实可靠。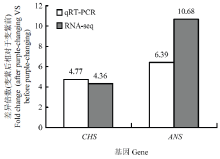 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图9差异表达基因qRT-PCR与RNA-seq结果对比
-->Fig. 9Comparison of DEGs between qRT-PCR and RNA-seq
-->
3 讨论
本研究利用转录组测序技术,对贵紫麦1号同株麦粒变紫前和变紫后2个灌浆时期的转录组测序获得大量数据。将存在的差异表达基因进行统计分析,结果表明2个材料存在大量的差异基因,下调基因多于上调基因,这些上调或下调基因导致不同灌浆时期差异表现。通过将得到的差异基因与NCBI数据库进行比对,至少鉴定了5种与来自节节麦、乌拉图尔小麦、大麦、二穗短柄草和普通小麦的已知基因同一性且序列相似性高的Unigenes。将组装得到的Unigenes在KOG数据库比对后,得到26个group。GO注释结果表明,注释成功的基因按照参与的生物过程、所处的细胞组分及具有的分子功能的下一层级进行分类注释,共44个分类。433个差异基因富集较为显著的有碳水化合物代谢过程、应激反应、防御反应、水解酶活性分子功能等,这与前人在水稻[28,29]、玉米籽粒[30]转录组得到的结果一致,说明在小麦灌浆期间,大量相关调控基因参与到生物过程与分子过程中,影响着小麦生长发育和相关酶催化调节的相互作用,推测这些富集显著的差异基因是调控小麦灌浆过程中的关键基因。由KEGG注释结果来看,353个差异基因富集在碳代谢、氨基糖等代谢通路,富集在淀粉、蔗糖、苯丙素代谢通路上的基因呈下调趋势,这与李怀珠[31]所研究的籽粒发育形态建成时期前期的结果相反,但同时印证了在灌浆后期,储存物质合成已大致完成,调控形态建成的差异基因开始大量表达。小麦籽粒的类黄酮生物合成代谢途径共66个相关基因,排在上调差异基因显著性的前20个通路之中,2条相关的上调差异Unigenes皆富集在生物学途径的分子功能上,经过转录组测序结果分析及qRT-PCR验证分析表明,类黄酮代谢途径中,CHS、ANS呈明显上调趋势,推测在贵紫麦1号灌浆期间,由于这些生物合成酶的大量上调,导致在较短时间内籽粒颜色迅速变紫。由KEGG通路可知,贵紫麦1号合成的花青素主要为天竺葵色素、矢车菊色素、飞燕草色素、芍药素和锦葵色素,这与赵善仓等[32]测定紫繁3小麦含有矢车菊色素的结果一致,但紫繁3含有的芍药素、锦葵色素在贵紫麦1号中为天竺葵色素、矢车菊色素和燕草色素的次生代谢产物。
CHS催化4-香豆酸CoA与丙二酰CoA合成查尔酮,形成的查尔酮提供基本碳骨架。CHS在贵紫麦1号中处于花青素合成的第二阶段,合成矢车菊色素的前体物质,调控花青素的合成。由于CHS是花青素早期合成阶段重要的调控基因,依赖R2R3-MYB转录因子合成类黄酮,在后续研究中,可通过导入反义或敲除CHS,从不同材料中获得其他有色或无色的小麦品种,也可利用贵紫麦1号为桥梁亲本,通过杂交、回交转育,结合分子辅助选择,将与紫色控制相关基因转移到其他普通主栽小麦品种中,培育出适合不同生态型、品质优良、高产的特色小麦品种,从而提高紫色小麦的利用价值,促进特色小麦产业化发展。
ANS的表达具有一定的组织特异性且受外界环境因素的影响[33]。ANS在贵紫麦1号中的表达使果皮、种皮、糊粉层皆呈紫色,这也印证了在水稻NP突变体中,ANS过表达使原花青素前体物在果皮中生成花青素,而玉米a2突变体中,ANS的表达使花青素产生于糊粉层之中[34]。据研究MYB转录因子对ANS具有正、负调控的作用,且同类的MYB转录因子在不同植物中对ANS调控作用也不同[35]。由于ANS在贵紫麦1号中呈上调表达,推测MYB在小麦中对ANS起正调控的作用。符思路[36]鉴定的23个小麦bHLH Unigene中,13个bHLH小麦转录因子在拟南芥中存在同源基因,其他基因在小麦中发生扩增,没有发现小麦特有基因,但鉴定的组织仅为幼苗、根、花药小穗等小麦生育前期组织,并未涉及籽粒灌浆时期。NESI等[37]发现矮牵牛AN1和玉米IN1与拟南芥TT8较为相似,但TT8与AN1中的转录因子都可激活,IN1却在花青素的生物合成中体现抑制性,因此认为调控基因的调控框架与调控子的等级有一定的相关性,不能单靠同源性预测,贵紫麦1号是否具有bHLH特异基因还需进一步验证。分析黄酮类KEGG通路时发现,在合成异甘草素的代谢途径中,PKR和CHS在共同调控异甘草素的合成,CHS呈明显上调趋势,PKR既不上调也不下调。研究表明,PKR是病毒感染时诱导抗病毒应激颗粒的细胞浆体生产
的关键因素[38],尹祥佳[39]将PKR以农杆菌介导玉米茎尖中进行遗传转化,结果呈阳性且部分植株结实。如果敲除或导入PKR,观察贵紫麦1号中异黄酮生物合成和抗病性表现,或许可进一步明确PKR在小
麦中的作用。
4 结论
通过对贵紫麦1号籽粒2个灌浆时期转录组测序及分析,获得66个类黄酮生物合成代谢途径相关基因在贵紫麦1号中的表达信息,发现236个上调差异基因,407个下调差异基因。CHS、ANS在调控花青素形成过程中具有关键作用。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}