 ,, 仇雨歌, 陈胜男, 王晓萌, 王春生,东北林业大学生命科学学院,哈尔滨 150040
,, 仇雨歌, 陈胜男, 王晓萌, 王春生,东北林业大学生命科学学院,哈尔滨 150040CRISPR/Cas9 Mediated Exogenous Gene Knock-in at ROSA26 Locus in Sheep Umbilical Cord Mesenchymal Stem Cells
LI SongMei,, QIU YuGe, CHEN ShengNan, WANG XiaoMeng, WANG ChunSheng,College of Life Science,Northeast Forestry University, Harbin 150040通讯作者:
责任编辑: 林鉴非
收稿日期:2020-02-23接受日期:2020-05-13网络出版日期:2021-01-16
| 基金资助: |
Received:2020-02-23Accepted:2020-05-13Online:2021-01-16
作者简介 About authors
李松美,E-mail:songmei.

摘要
关键词:
Abstract
Keywords:
PDF (1150KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李松美, 仇雨歌, 陈胜男, 王晓萌, 王春生. CRISPR/Cas9介导的绵羊示踪脐带间充质干细胞系的建立[J]. 中国农业科学, 2021, 54(2): 400-411 doi:10.3864/j.issn.0578-1752.2021.02.015
LI SongMei, QIU YuGe, CHEN ShengNan, WANG XiaoMeng, WANG ChunSheng.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】间充质干细胞(mesenchymal stem cell,MSC)是一种来源于中胚层的具有自我复制和多向分化潜能的成体干细胞。由于被移植到体内的MSC具有可以向特定或病损的组织或器官定向迁移并分化为特定细胞来修复损伤的特点,其在临床治疗应用方面具有巨大潜力。但MSCs移植至宿主体内时存在时间非常短暂,难于检测到其作用轨迹和分化渠道,无法研究MSCs解决损伤的临床前和临床机制,如何实现MSCs位置标记成为开展此研究的关键点。新的技术如CRISPR/Cas9系统对DNA分子具有靶向切割的特性已经实现了外源基因向特定细胞基因组中的有效插入,为此开辟了一条新路径。利用CRISPR/ Cas9技术将外源GFP基因敲入sUMSCs,建立稳定的细胞系,通过检测GFP示踪MSCs去向和分化为何种细胞,此举为揭示MSCs在损伤修复中的作用奠定基础。【前人研究进展】从2010年至今,陆续出现的锌指核酸酶(ZFNs)、类转录激活因子效应物核酸酶(TALENs),及规律成簇的间隔短回文重复(CRISPR)等新型基因组编辑技术不仅大大降低了对动物进行基因敲除、修饰的难度,而且使动物转基因技术由传统的随机整合发展到高度精确的基因组定向删除、突变或插入等精确修饰改造[1]。CRISPR系统,通过Cas蛋白与CRISPR转录出来的RNA形成蛋白质核酸复合物行使免疫功能。CRISPR/Cas9系统是II型CRISPR/Cas系统,具有核酸内切酶功能,能选择性切割DNA序列,是一种基因编辑的革命性技术[2]。利用CRISPR/Cas9系统可以进行精确的基因操作实现特定细胞的遗传操作[3],此技术主要利用sgRNA引导内切酶定点切割DNA双链(DSBs),并在DNA断裂处将供体DNA上的序列与DSB两侧同源通过同源重组和非同源重组方式进行修复将目的序列引入基因组,实现基因敲入,既避免了细胞DNA降解,又维护了细胞的正常功能[4]。相较于ZFNs、TALENs,CRISPR/Cas9系统,因其具有可以定点打靶基因、编辑效率高、操作方便等特点已广泛应用于大小鼠、斑马鱼、猴和猪等转基因动物制备和基因功能等研究领域 [5,6,7,8]。选择“ROSA26”位点用于GFP基因安全敲入,是因为该位点适应于外源基因良性插入并能保证转入基因的正常稳定表达,该位点的基因敲入对细胞功能及动物的健康无副作用,且在胚胎和其他成体组织中都有所表达。该位点现已成为外源基因插入的重要位点[9]。间充质干细胞具有低免疫原性、多向分化、向病损组织定向迁移和免疫调节等优点,其来源广泛、获取简单且无伦理性争议,决定了其在疾病治疗和组织损伤修复等方面具有广阔的应用前景[10]。从临床数据可知,MSCs在骨与软骨疾病、脊髓损伤、移植排斥、肝脏疾病、自身免疫病和心血管疾病等方面具有一定的临床治疗效果[10]。例如,MSCs可以通过旁分泌营养介质,可以抑制缺血导致的凋亡[11]、抑制瘢痕形成、刺激血管新生和维持血管稳定性[12],对心脑血管疾病方面具有一定的改善作用。MSCs容易受趋化因子和生长因子的介导,向损伤或炎症部位定向迁移, 促进组织损伤修复 [13]。MSCs还具有直接分化为成骨和软骨等能力,能直接替代损伤细胞从而在骨与软骨损伤的修复中发挥重要作用[14]。MSCs在动物试验中显现出显著的神经保护作用,能减少神经细胞凋亡,改善新生儿缺氧缺血性脑病带来的损伤[15]。MSCs可对参与特异性和非特异性免疫应答的多种细胞发挥免疫调节作用。自体MSCs可通过影响促炎性细胞因子抑制淋巴细胞在有丝分裂原或抗体活化反应中的增殖 [16],也可通过抑制淋巴细胞分泌炎症因子调节免疫[17]。利用异体的MSCs与常规药物相结合的治疗方法,可明显改善类风湿性关节炎(rheumatoid arthritis, RA)患者的临床指标[18]和难治性系统性红斑狼疮(systemic lupus erythematosus, SLE)患者的肾功能,使临床缓解率显著上升[19],可见MSCs对自身免疫性疾病有一定的改善作用。MSCs还可以介导宿主的防御反应以抵抗病毒和寄生虫等微生物的感染,有望降低感染者的发病率和死亡率[20]。【本研究切入点】迄今尚未见到CRISPR/Cas9介导同源重组外源报告基因GFP于sUMSCs 的ROSA26位点来建立绵羊示踪脐带MSCs细胞系的报道。本研究以CRISPR/Cas9技术为基础,针对绵羊ROSA26位点设计sgRNA,构建了sgRNA/Cas9同源打靶载体,用脂质体转染该载体定点打靶绵羊MSCs,建立CRISPR/Cas9介导外源GFP基因敲入绵羊MSCs细胞系,获得具有绿色荧光的阳性克隆细胞系以供后续研究。【拟解决的关键问题】构建针对绵羊ROSA26位点的sgRNA/Cas9表达载体,转染检测活性,连接左右同源臂以获得绵羊同源打靶载体,然后利用脂质体转染绵羊MSCs,在绵羊MSCs中建立利用CRISPR/Cas9技术敲入外源GFP基因的示踪细胞系,为进一步研究绵羊间充质干细胞的功能和分化机制奠定基础。1 材料与方法
1.1 试验材料
试验于2016年9月至2019年5月在东北林业大学动物胚胎发育实验室完成。pMD19-T simple购自宝生物工程(大连)有限公司,sUMSCs和载体px330由东北林业大学生命科学学院动物胚胎发育实验室鉴定与保存,试验过程所用引物与测序分析由苏州金唯智生物科技有限公司完成。1.2 绵羊ROSA26基因sgRNA/Cas9表达载体的构建
根据绵羊Rosa26的基因序列分别设计3条正向引物和通用反向引物,利用点突变法(KOD-Plus- Mutagenesis Kit,Toyobo Life Science Department)构建绵羊ROSA26基因sgRNA/Cas9表达载体:以px330为模板,以KOD-R,sROSA26-sgRNA1、2、3(表1)为引物分别进行PCR扩增,用DpnⅠ对PCR产物进行消化(去除质粒DNA),随后进行PCR产物自身连接,Trans5α转化并挑取单克隆,碱裂解法小提质粒(高纯质粒小量制备试剂盒 BioTeke)。对突变质粒克隆载体进行BbsⅠ酶切鉴定及测序分析,测序正确的重组质粒分别命名为px330-sgRNA1/2/3,并利用试剂盒(Endo-free Plasmid Mini KitⅡ, OMEGA)提取无内毒素质粒用于后续试验。Table 1
表1
表1sgRNA和引物及其目的片段大小
Table 1
| sgRNA/引物 sgRNA/Primers | 序列 Oligonucleotide sequences | 片段 Length (bp) |
|---|---|---|
| KOD-R | 5′-GGTGTTTCGTCCTTTCCAC | 19 |
| px330-sgRNA-1 | 5′-CCAGCAGGTATAAGATTTAGGTTTTAGAGCTAGAATAGCAGGT | 43 |
| px330-sgRNA-2 | 5′-CCTCTAAATCTTATACCTGCGTTTTAGAGCTAGAATAGCAGGT | 43 |
| px330-sgRNA-3 | 5′-TGTCCTGCAGTGGATCCAGCGTTTTAGAGCTAGAATAGCAGGT | 43 |
| px330-TP-F | 5′-GCTGCCTGAAGGACAAGACTA | 21 |
| px330-TP-R | 5′-GGCAACACCTGGGACTGATTT | 21 |
| sROSA26-LHA-F | 5′-ACTAGTTACGCTGAAAGGGAAAGAGG | 26 |
| sROSA26-LHA-R | 5′-ACGCGTAGGACAACGCCCAGGATT | 24 |
| sROSA26-RHA-F | 5′-TGTACACCCAATTTCTTTATCTTCCC | 26 |
| sROSA26-RHA-R | 5′-ACATGTTCCTGTCAGTAGTTACCACCC | 27 |
| SRL-F | 5′-AAGAGGCTGTGCTCTGGG | 18 |
| SRL-R | 5′-CGTGAGTCAAACCGCTATCC | 20 |
| SRR-F | 5′-TCCCTAAAGAAACAGTGGC | 19 |
| SRR-R | 5′-CACGTTTGTGATGATGGAAT | 20 |
新窗口打开|下载CSV
1.3 绵羊ROSA26基因sgRNA/Cas9表达载体的活性检测
利用Thermo GeneJET Genomic DNA Purification Kit试剂盒提取绵羊脐带间充质干细胞(sUMSCs)全基因组:培养收集适量sUMSCs,裂解细胞,去除蛋白,洗脱获得纯化基因组,以sUMSCs全基因组作为对照,用px330-sgRNA1/2/3分别转染bMSCs,转染48—72 h后,提取其全基因组。根据绵羊ROSA26基因打靶位点和sgRNA序列在1 402 bp和1 910 bp处设计PCR上下游引物px330-TP-F和px330-TP-R,利用LA酶以绵羊基因组或转染后UMSC基因组DNA为模板进行PCR扩增。对PCR胶回收产物(康为世纪Gel Extraction Kit试剂盒)进行变性退火杂交,T7E1酶切后进行琼脂糖凝胶电泳分析。1.4 绵羊ROSA26同源打靶载体的构建
提取绵羊全基因组,根据sgRNA序列在绵羊ROSA26靶位点的上下游设计并合成左同源臂扩增引物sROSA26-LHA-F、sROSA26-LHA-R(长度1 093 bp,具有SpeⅠ、MluⅠ酶切位点)和右同源臂扩增引物sROSA26-RHA-F、sROSA26-RHA-R(长度928bp,具有BsrGⅠ、PciⅠ酶切位点)。以绵羊基因组DNA为模板,左臂以sROSA26-LHA-F和sROSA26-LHA-R为引物,右臂以sROSA26-RHA-F和sROSA26-RHA-R为引物,使用Ex Taq酶进行大量扩增,反应条件为:预变性94℃ 5min;94℃ 30s,52℃、55℃、58℃ 各1min,72℃ 7min,33循环;72℃延伸7min。PCR产物胶回收后与pMD19-Simple克隆载体连接。转化DH-5α、挑取单克隆、保存菌种、质粒小提。对右臂重组克隆载体进行PciⅠ、BsrGⅠ双酶切鉴定;对左臂重组克隆载体进行PstⅠ酶切鉴定。经酶切鉴定正确的左、右同源臂重组质粒且测序分析正确命名为sROSA26-LHA -pMD19和sROSA26-RHA-pMD19。将sROSA26- LHA-pMD19-T和Donor表达载体DC-DON-SH02 ROSA26分别进行SpeⅠ和MluⅠ大量酶切并进行胶回收,连接所需目的基因片段、转化、挑取单克隆、保存菌种及质粒小提,对左臂重组表达载体进行SmaⅠ单酶双酶切鉴定,酶切鉴定准确的左臂重组表达载体测序对比分析。准确的重组表达载体命名为sROSA26-LHA。将sROSA26- RHA-pMD19-T和sROSA26-LHA分别进行SpeⅠ和MluⅠ大量酶切并进行胶回收来获取目的片段。T4连接左右同源臂、转化、挑取单克隆、保存菌种及质粒小提,对重组表达载体进行HindⅢ单酶双酶切鉴定。对酶切鉴定正确的载体进行测序分析。正确的打靶载体命名为sROSA26-HA,无内毒素质粒提取用于后续试验。1.5 阳性细胞克隆筛选和同源重组检测
取生长良好的sUMSCs,观察加入不同浓度的嘌呤霉素的细胞存活时间,确定最佳抗性筛选浓度。利用Lipofectamine2000(Lipofectamine? 2000 Transfection Reagent,Thermo Fisher)转染sUMSCs,24 h后,根据sROSA26-HA表达的绿色荧光确定转染效率并收集细胞。根据sROSA26-HA载体携带有嘌呤霉素抗性基因的特点,转染48h后进行嘌呤霉素抗性筛选,在对照组细胞几乎全部死亡时,更换无嘌呤霉素的DMEM/F12合成培养基培养以扩大培养筛选得到的阳性细胞。提取其全基因组,根据绵羊ROSA26位点插入的片段序列设计同源重组检测引物,以提取纯化后的阳性细胞基因组为模板,上游以SRL-F和SRL-R为引物,下游以SRR-F和SRR-R为引物,利用Ex Taq酶进行同源重组PCR鉴定,反应条件为:预变性98℃ 5 min;98℃ 10s,58℃ 30s,72℃ 1 min,33循环;72℃延伸7 min。PCR产物进行琼脂糖凝胶电泳分析。2 结果
2.1 px330-sgRNA载体的构建与切割活性检测
针对绵羊Rosa26位点设计3条正向PCR引物px330-sgRNA-1/2/3和1条通用反向引物分别进行PCR扩增。利用DpnⅠ酶切去除质粒,琼脂糖凝胶电泳检测显示在大于8 000 bp处看到单一条带,获得PCR产物。利用PCR产物自身环化反应得到重组突变表达载体,因为与原载体相比,重组质粒的BbsⅠ酶切位点发生突变,不能被切开。使用BbsⅠ进行酶切鉴定,跑胶验证未被切开的重组质粒初步断定为正确的sgRNA/Cas9表达载体(图1),测序结果正确的载体命名为sROSA26-sgRNA1。(sROSA26- sgRNA1效率最高,故本试验只表述sROSA26-sgRNA1结果)。图1
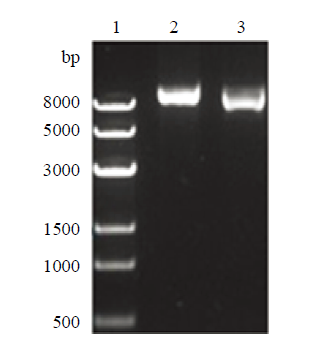 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1sROSA26-sgRNA-1 BbsⅠ酶切鉴定
1:Trans8k DNA Marker;2:PX330质粒;3:重组质粒BbsⅠ酶切鉴定
Fig. 1The BbsⅠenzyme identification of sROSA26-sgRNA-1
1: Trans8k DNA Marker; 2: Plasmid PX330; 3: Enzyme identification of restructuring plasmid
sUMSCs汇合度达到70%—80%时(图2-A),脂质体转染sROSA26-sgRNA-1/2/3载体,对照组转染pAcGFP1-N1载体,48 h后,观察细胞状态和荧光强度(图2-B),根据对照组,转染效率为40%左右(图2-C)。以px330-TP-F和px330-TP-R为引物对转染后细胞基因组PCR后,电泳检测,对照组和转染组均在约500 bp处检测到单一条带。对该PCR产物分别进行T7E1酶切检测碱基错配情况。结果表明,与对照组相比,sg1转染组的PCR产物能够被T7E1切割为大小132 bp和377 bp两条带(图3)。根据公式突变率=突变型/(野生型+突变型)计算编辑效率约为20%,但是sg2和3的PCR产物未被T7E1切开。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2转染效率
A:sUMSCs;B:转染后sUMSCs;C:对照组sUMSCs(50×)
(A:在sUMSCs细胞系中转染空载作为阳性对照;B、C:在sUMSCs细胞系中trim away技术,转入带有GFP的sROSA26-sgRNA-1/2/3载体,转染48h后的亮视野和暗视野,根据观察到的荧光强度确定转染效率)
Fig. 2Transfection efficiency
A:sUMSCs;B:sUMSCs After transfecion;C:Control group sUMSCs(50×)
(A: the sUMSCs cell line after transfecting empty plasmid as a positive control; B, C:Transfer sROSA26-sgRN-1/2/3 vectors with GFP into sUMSCs cell lines using Trim Away technique, capture the bright and dark field after transfection for 48h, and the transfection efficiency was determined according to the observed fluorescence intensity.)
图3
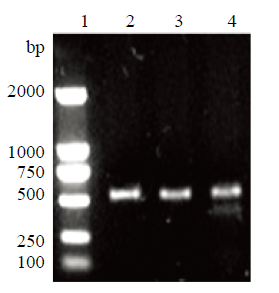 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3T7E1酶切鉴定
1:Trans2K DNA Marker;2:野生型PCR产物;3:野生型T7E1酶切产物;4:突变型T7E1酶切产物
Fig. 3T7E1 digestion identification
1: Trans2K DNA Marker; 2: Wild type PCR product; 3: Wild type T7E1 digestion product; 4: Mutated T7E1 digestion product
2.2 绵羊ROSA26左右同源臂表达载体构建
以上述编辑效率最高的sgRNA1的切割位点为中心,设计敲入载体的左、右同源臂引物,以绵羊基因组为模板分别扩增。电泳结果显示,在约1 100 bp和900 bp处检测到特异性单一条带,与理论值相符(图4、5)。将上述PCR产物分别与克隆载体pMD19- Simple连接,利用PCR和酶切对重组质粒进行鉴定。PCR结果显示,左、右同源臂分别在约1 100 bp和900 bp处检测到特异单一条带。含有同源左臂的重组质粒经PstⅠ酶切后,检测到与理论值相符的两个条带,大小为732 bp和3 200 bp(图6);右臂克隆载体经PciⅠ和BsrGⅠ双酶切后,检测到与理论值相符的两个条带,大小为928 bp和1 693 bp(图7)。经测序进一步鉴定后分别命名为pMD19- LHA和pMD19-RHA。随后经一系列分子生物学步骤将左右同源臂先后连接到表达载体,对左同源臂重组表达载体和重组表达载体进行PCR和SmaⅠ、Hind Ⅲ酶切鉴定。PCR鉴定电泳检测显示:分别在约为1 100 bp和900 bp大小处检测到特异性单一条带,SmaⅠ酶切检测到大小约为700 bp和8 200 bp的两个片段(图8);HindⅢ酶切割后,检测到大小约为2 400 bp和5 800 bp的两个片段,均与理论相符,表明成功将左右同源臂连入表达载体,将测序正确的同源重组载体命名sROSA26-HA(图9)(构建过程和所用载体图谱如图10、11所示)。图4
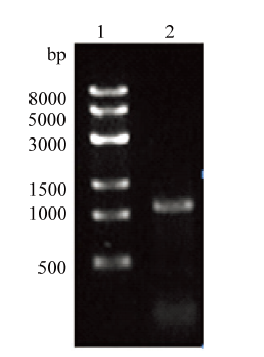 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4左同源臂PCR扩增
1:左臂PCR产物;2:Trans8K DNA Marker
Fig. 4Left homology arm PCR amplification
1: Left arm PCR product; 2: Trans8K DNA Marker
图5
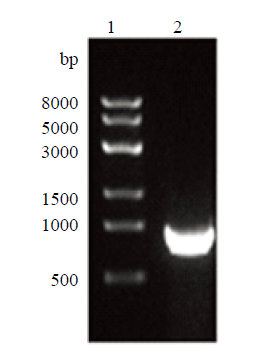 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5右同源臂PCR扩增
1:右臂PCR产物;2:Trans8K DNA Marker
Fig. 5Right homology arm PCR amplification
1: Right arm PCR product; 2: Trans8K DNA Marker
图6
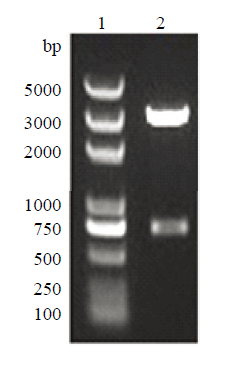 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6左臂克隆载体酶切鉴定
1:D2000Plus DNA Marker;2:左臂克隆载体PstⅠ酶切鉴定
Fig. 6Left arm cloning vector digestion identification
1: D2000Plus DNA Marker; 2: Identification of the left arm cloning
图7
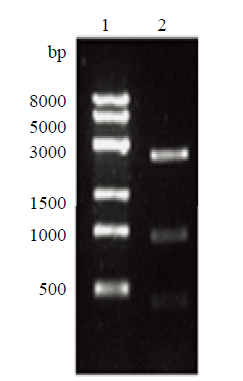 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7右臂克隆载体酶切鉴定
1:右臂克隆载体双酶切鉴定;2:Trans8K DNA Marker
Fig7.Right arm cloning vector digestion identification
1: Identification of right arm cloning; 2: Trans8K DNA Marker
图8
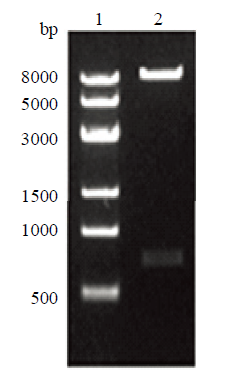 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8左臂表达载体酶切鉴定
1:Trans8K DNA Marker;2:左臂表达载体酶切鉴定
Fig. 8Left arm expression vector enzyme digestion identification
1: Trans8K DNA Marker; 2: Left arm expression vector enzyme digestion identification
图9
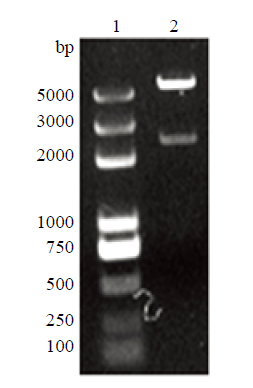 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9sROSA26-HA酶切鉴定
1:D2000Plus DNA Marker;2:sROSA26-HA酶切鉴定
Fig. 9sROSA26-HA enzyme digestion identification
1: D2000Plus DNA Marker; 2: sROSA26-HA enzyme digestion identification
图10
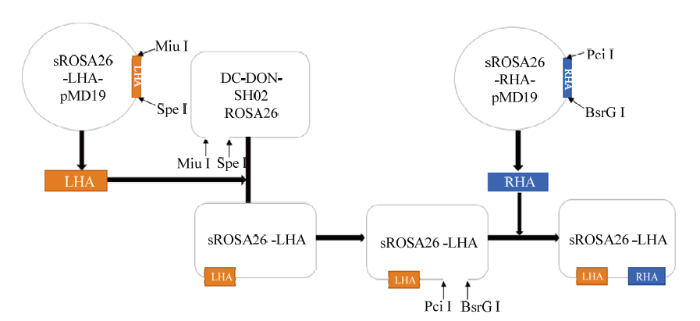 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10donor载体构建流程图
Fig. 10Donor vector construction flowchart
图11
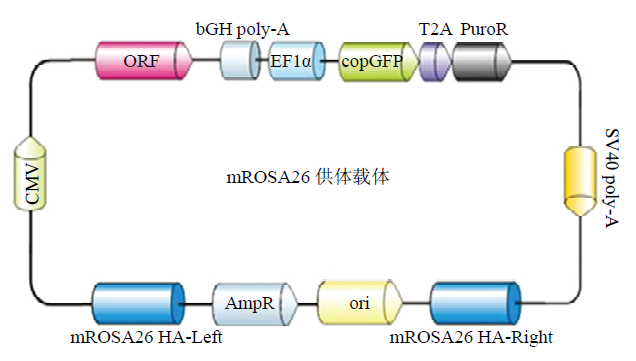 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11DC-DON-SH02 ROSA26供体载体图谱
Fig. 11Construction of the DC-DON-SH02 ROSA26 vector
2.3 细胞转染与筛选
解冻并扩大培养sUMSCs,铺满6孔板, sUMSCs呈长梭形并涡旋样生长(图12-A)。使用1.0、1.5、2.0和2.5 μg·mL-1浓度梯度的嘌呤霉素加压筛选最适浓度,24h后,除1.0 μg·mL-1组外的其余3组均出现大量死亡现象(图12-B),1.0 μg·mL-1浓度组每天死亡数量较少,在筛选14d后,仍有20%的细胞存活;2.0和2.5 μg·mL-1浓度组在第6天和第4天绝大多数死亡,对细胞伤害较大(图13);1.5 μg·mL-1组在第12天细胞死光,既有筛选效果又作用温和,后续试验选其浓度进行(图12-C)。使用脂质体Lipofectamine2000共转染sROSA26-sgRNA1和sROSA26-HA到sUMSCs中,处理24 h后可表达绿色荧光。48 h后,对转染组和对照组同时进行1.5 μg·mL-1浓度嘌呤霉素加压筛选至对照组细胞全部死亡,更换无嘌呤霉素的新鲜培养基,可观察到转染组剩余细胞出现大量绿色荧光,成功获得阳性克隆(图14、15)。图12
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图12筛选最佳嘌呤霉素浓度
A:sUMSCs(50×);B:1.5μg·mL-1 嘌呤霉素筛选48h后sUMSCs(50×);C:嘌呤霉素筛选结束时sUMSCs(50×)
(在汇合度为70%-80%的sUMSCs中加入抗性,记录细胞几乎全部死亡的时间和浓度作为后续抗性筛选依据)
Fig. 12Select the best puromycin concentration
A: sUMSCs(50×); B: After 1.5μg·mL-1 puromycin screening for 48h sUMSCs(50×); C: At the end of puromycin screening sUMSCs(50×)
(Add puromycin to sUMSCs when the confluence was 70%-80%, and record the time and concentration of almost all cell death as the foundation for subsequent resistance screening. )
图13
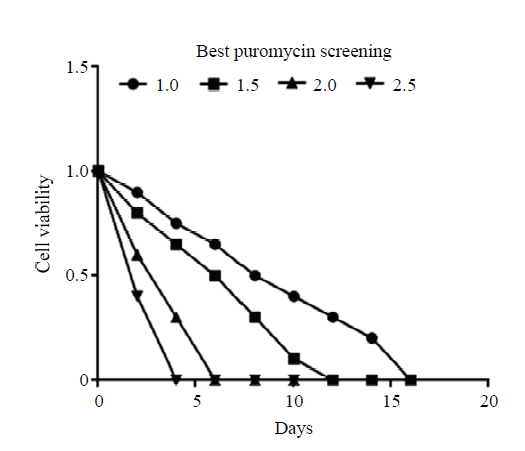 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图13筛选最佳嘌呤霉素浓度统计图
Fig. 13Screening of best puromycin concentration statistics
图14
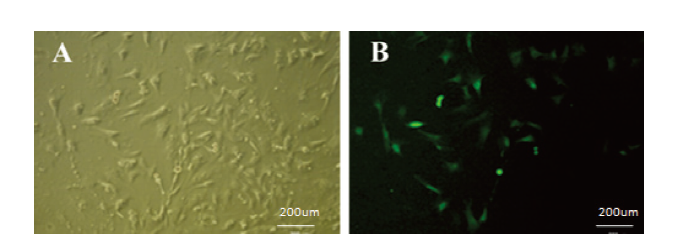 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图14阳性细胞克隆筛选
A:嘌呤霉素筛选16d后sUMSC-1(明视野50×);B:嘌呤霉素筛选16d后sUMSC(暗视野50×)
(在sUMSCs细胞系中转入带有GFP的sROSA26-HA载体,转染24h后加压筛选16天后获得的细胞多数表达绿色荧光。之后,换为普通培养基)
Fig. 14Cloning and screening of positive cells
A: After puromycin screening for 16d sUMSC-1(Bright field 50×); B: After puromycin screening for 16d sUMSC(Dark field 50×)
(Transfect the sROSA26-HA vector with GFP into the sUMSCs cell line, add puromycin after transfecion 24h, most cells express green fluorescence after 16 days of screening.After that, change to normal medium.)
图15
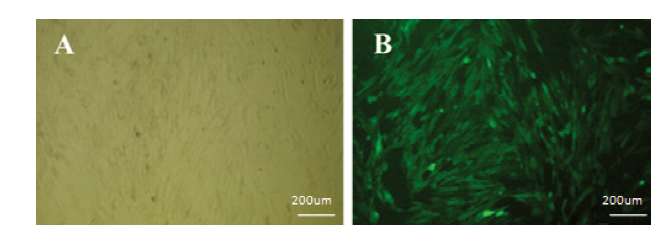 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图15阳性细胞扩大培养
A:嘌呤霉素筛选16d后sUMSCs(明视野50×);B:嘌呤霉素筛选16d后sUMSCs(暗视野50×)。
(将筛选后的阳性克隆扩大培养以获得足够的细胞数进行后续实验)
Fig. 15Positive cells were further cultured
A: After puromycin screening for 16d sUMSCs(Bright field 50×); B: After puromycin screening for 16d sUMSCs(Dark field 50×)
(Subculture the screened positive clones to obtain sufficient cell numbers for subsequent experimental studies)
2.4 阳性细胞克隆的同源重组鉴定
以提取阳性克隆细胞的全基因组为模板,针对插入外源片段后的绵羊ROSA26位点序列,分别设计上游引物SRL-F、SRL-R和下游引物SRR-F、SRR-R,PCR检测阳性细胞的同源重组情况,经琼脂糖凝胶电泳检测,分别在约1 400 bp和1 000 bp处检测到特异性条带,且野生型则没有条带(图16、17),与理论大小相符,表明利用CRISPR/Cas9技术对sUMSCs在ROSA26位点实现外源基因的定点导入。图16
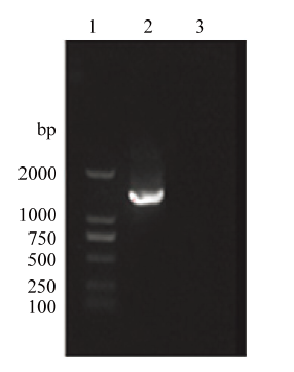 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图16同源重组上游PCR鉴定
1:D2000 DNA Marker;2:同源重组上游PCR产物;3:野生型PCR产物
Fig. 16Upstream PCR identification of homologous recombination
1: D2000 DNA Marker; 2: Homologous recombination upstream PCR product; 3: Wild type PCR product
图17
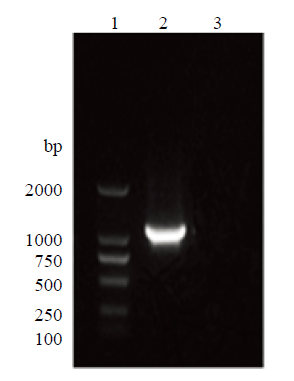 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图17同源重组下游PCR鉴定
1:D2000 DNA Marker;2:同源重组下游PCR产物;3:野生型PCR产物
Fig. 17Downstream PCR identification of homologous recombination
1: D2000 DNA Marker; 2: Homologous recombination downstream PCR product; 3: Wild type PCR product
3 讨论
3.1 MSCs的临床应用
MSCs是来源于中胚层的成体干细胞, 形态类似于成纤维细胞,具有成骨、成脂及成软骨等细胞类型的分化潜能,能自我更新和高度增殖[21]。已有研究证明,MSC的营养支持作用与免疫调节功能在组织损伤修复与疾病治疗中起到重要作用,移植到体内的MSC受到不同趋化因子和生长因子的影响向特定或病损的组织或器官定向迁移,可分化成特定细胞替代病损的组织器官修复损伤[10];同时MSCs已经被证明可以调节内源组织和免疫细胞。在同种异体移植时缺乏诱导T细胞活化的CD80等共刺激因子,因此不易发生排斥反应,既能通过增强内源修复进程解决损伤又具有较低的免疫原性,在骨与软骨等损伤修复中具有良好的治疗作用[22]。但MSC移植至宿主体内时存在时间非常短暂,难于检测到其作用轨迹和渠道。建立利用CRISPR/Cas9系统介导敲入报告基因的绵羊示踪脐带间充质干细胞系,通过检测GFP靶向定位MSCs,此举对探索MSCs临床治疗过程与机制具有重要意义。3.2 sgRNA的设计与筛选
根据王楚端等[9]发表的绵羊ROSA26的基因序列,针对绵羊ROSA26位点选取3条评分较高的sgRNA并合成引物。由于基因组可能出现与sgRNA识别位点相似的序列错误引导Cas9蛋白切割DNA出现脱靶效应[23],以及生理状态下染色质精密的螺旋结构和多种状态共存的复杂情况,因此理论设计的sgRNA仍需要在细胞内进行验证以确定其编辑效率[24,25,26]。利用KOD-Plus-Mutagenesis Kit点突变法构建sgRNA/Cas9真核表达载体px330-sgRNA-1/2/3。将经过BbsⅠ酶切(图1)和测序鉴定正确后的表达质粒利用Lipofectamine2000转染至sUMSCs,T7E1酶切鉴定显示只有sgRNA1构建的载体PCR产物有3条特异性条带(图3),表明px330-sgRNA-1具有打靶活性,经计算编辑效率约为20%,可以用于后续试验。3.3 ROSA26 safe harbor的选择
选择适合用于外源基因良性插入并稳定表达的基因组位点很重要,ROSA26 safe harbor概念来源于在ROSAβgeo26品系的小鼠被随机插入单个基因,结果在所有组织中均检测到高表达的β半乳糖苷酶且纯合子幼崽能正常发育并繁殖。试验发现插入位点位于6号染色体,该位点表达一个编码转录本和两个非编码的转录本,只有非编码转录本的序列受到外源插入的干扰。因此“ROSA26”位点能保证转入基因的正常稳定表达并对细胞及小鼠的健康无副作用[27]。而后,赖良学等在猪基因组中也找到了一个特殊基因位点ROSA26,成功地构建了世界上第一个由TALEN介导的ROSA26定点敲入Cre重组酶报告基因的大动物模型[28]。在此基础上,在ROSA26位点还引入一对异源loxp位点,利用Cre/loxP重组酶系统,可以将任意靶基因敲入到ROSA26位点,实现目的基因在大动物所有组织中的无差异表达,从而解决了一直困扰转基因猪研究领域效率低下、表型不确定、携带抗性基因的问题[29]。ROSA26基因可以在几乎所有的生物体中编码一种非必需的核RNA,因而成为了外源基因插入的一个热点,因此本试验根据绵羊Rosa26基因序列设计合成sgRNA,构建含同源序列和目的序列的载体,利用CRISPR-Cas9特异靶向ROSA26 位点生成 DNA 双链断裂,触发细胞的 DNA 修复机制,诱导基因组与其供体克隆之间发生同源重组(HR),将靶基因整合到基因组上的 ROSA26 safe harbor 位点[30,31],实现在sUMSCs中稳定高效表达绿色荧光。
3.4 同源重组方式敲入标记基因GFP
为了探讨MSCs在患者体内的作用分化途径,明确其黏附定植的靶组织,必须构建标记特性和生物学特性稳定的标记细胞系,以示踪细胞在体内的治疗过程,而利用基因工程技术构建标记MSCs的关键是选择合适的标记物和表达载体。由于GFP具有对细胞无毒害、抗光漂白能力强、不需要任何反应底物和辅助因子、荧光强度高效、稳定持久等特点[32]。因此,本试验利用GFP 基因标记间充质干细胞,以实现在患者体内实时跟踪细胞治疗的动态过程。研究证实,sgRNA/Cas9 在目的位点产生双链断裂后,可通过同源重组方式进行修复[26],将外源导入的DNA作为同源重组模板,最终将外源 DNA插入到基因组中。共转染供体同源臂质粒和sgRNA/Cas9质粒,sgRNA/Cas9质粒能使基因组发生DSB, 同源臂质粒定点整合,已成功在斑马鱼中实现外源基因的整合重组[33,34]。至今尚未见到利用CRISPR/Cas9技术同源重组外源报告基因于绵羊间充质干细胞ROSA26位点建立sUMSCs细胞系的报道。本研究旨在sUMSCs中利用CRISPR/Cas9系统在ROSA26位点敲入报告基因GFP追踪细胞动向,本试验的技术难点在于如何克服knoch-in的编辑效率低下和脐带间充质干细胞转染困难、抗性筛选后细胞增殖困难等问题,成功获得表达绿色荧光的阳性细胞。试验过程中首先进行预试验明晰sUMSCs的转染试剂种类、试剂和质粒配比、转染时间等条件,之后构建针对sROSA26的donor载体sROSA26-HA(图4—9)和gRNA质粒px330-sgRNA-1,两者共转入sUMSCs中,在载体上具有嘌呤霉素标签,为了避免嘌呤霉素浓度过高将导致转染的阳性细胞被杀死,浓度过低将造成正常细胞仍然可以存活,干扰结果。用不同浓度嘌呤霉素处理间充质干细胞,得到 1.5μg·mL-1的最小致死浓度,在转染后的细胞加入嘌呤霉素筛选,以未转染组细胞全部死亡作为终止嘌呤霉素筛选时间点,获得表达绿色荧光的阳性细胞(图14、15),并利用PCR检测其在ROSA26位点发生了GFP同源重组(图16、17),建立了利用CRISPR/ Cas9介导的绵羊示踪脐带MSC系,并为绵羊MSC功能研究和临床应用奠定基础。尽管我们多次尝试采用有限稀释法尝试获取表达GFP的单细胞克隆,但均未成功。其原因可能在于sUMSCs转染比较困难,试验过程中转染试剂和嘌呤霉素可能对细胞产生了一定损伤,挑的单个细胞传代几次就会发生增殖停滞或细胞凋亡,因而难以形成细胞集落。不过,分别以位于基因组和载体上的引物,提取获得的阳性细胞基因组进行PCR鉴定,电泳结果显示可扩增到特异条带,表明载体已经整合到基因组中,而且在显微镜下可以观察到阳性细胞绝大部分可以表达绿色荧光。因此我们判断发生整合的细胞占数量优势,可以用于肌肉损伤等动物模型,注射制备的阳性sUMSCs到损伤部位,追踪sUMSCs的去向和对在组织修复中的贡献度。
4 结论
利用突变法根据sgRNA序列打靶位点与PX330载体引入部位序列结合成功构建重组表达载体。转染检测其sgRNA/Cas9酶切效率约为20%。再构建sROSA26的同源打靶载体,利用脂质体共转双质粒在sUMSCs的ROSA26位点敲入报告基因GFP, 抗性筛选阳性克隆, PCR验证同源重组情况,成功建立CRISPR/Cas9介导sUMSCs定点敲入外源基因GFP的绵羊示踪脐带间充质干细胞系。本研究结果为间充质干细胞在活体动物模型中的分化去向研究和转基因绵羊的制备提供了科学依据。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/nrg3686URLPMID:24690881 [本文引用: 1]

Programmable nucleases - including zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and RNA-guided engineered nucleases (RGENs) derived from the bacterial clustered regularly interspaced short palindromic repeat (CRISPR)-Cas (CRISPR-associated) system - enable targeted genetic modifications in cultured cells, as well as in whole animals and plants. The value of these enzymes in research, medicine and biotechnology arises from their ability to induce site-specific DNA cleavage in the genome, the repair (through endogenous mechanisms) of which allows high-precision genome editing. However, these nucleases differ in several respects, including their composition, targetable sites, specificities and mutation signatures, among other characteristics. Knowledge of nuclease-specific features, as well as of their pros and cons, is essential for researchers to choose the most appropriate tool for a range of applications.
URLPMID:23287722 [本文引用: 1]
DOI:10.1038/cr.2013.46URLPMID:23545779 [本文引用: 1]
DOI:10.1038/cr.2014.9URLPMID:24418757 [本文引用: 1]
DOI:10.1038/nbt.2661URLPMID:23929336 [本文引用: 1]
URLPMID:23360964 [本文引用: 1]
URLPMID:25859012 [本文引用: 1]
[本文引用: 1]
[本文引用: 2]
URL [本文引用: 3]
URL [本文引用: 3]
[本文引用: 1]
DOI:10.1155/2020/8857057URLPMID:33424980 [本文引用: 1]
A stably established population of mouse bone marrow stromal cells (BMSCs) with self-renewal and multilineage differentiation potential was expanded in vitro for more than 50 passages. These cells express high levels of mesenchymal stem cell markers and can be differentiated into adipogenic, chondrogenic, and osteogenic lineages in vitro. Subjected to basic fibroblast growth factor (bFGF) treatment, a typical neuronal phenotype was induced in these cells, as supported by neuronal morphology, induction of neuronal markers, and relevant electrophysiological excitability. To identify the genes regulating neuronal differentiation, cDNA microarray analysis was conducted using mRNAs isolated from cells differentiated for different time periods (0, 4, 24, and 72 h) after bFGF treatment. Various expression patterns of neuronal genes were stimulated by bFGF. These gene profiles were shown to be involved in developmental, functional, and structural integration of the nervous system. The expression of representative genes stimulated by bFGF in each group was verified by RT-PCR. Amongst proneural genes, the mammalian achate-schute homolog 1 (Mash-1), a basic helix-loop-helix transcriptional factor, was further demonstrated to be significantly upregulated. Overexpression of Mash-1 in mouse BMSCs was shown to induce the expression of neuronal specific enolase (NSE) and terminal neuronal morphology, suggesting that Mash-1 plays an important role in the induction of neuronal differentiation of mouse BMSCs.
DOI:10.1002/jcb.21172URLPMID:17295203 [本文引用: 1]
The recruitment of bone marrow CD34- mesenchymal stem- and progenitor cells (MSC) and their subsequent differentiation into distinct tissues is the precondition for in situ tissue engineering. The objective of this study was to determine the entire chemokine receptor expression profile of human MSC and to investigate their chemotactic response to the selected chemokines CCL2, CXCL8 and CXCL12. Human MSC were isolated from iliac crest bone marrow aspirates and showed a homogeneous population presenting a typical MSC-related cell surface antigen profile (CD14-, CD34-, CD44+, CD45-, CD166+, SH-2+). The expression profile of all 18 chemokine receptors was determined by real-time PCR and immunohistochemistry. Both methods consistently demonstrated that MSC express CC, CXC, C and CX(3)C receptors. Gene expression and immunohistochemical analysis documented that MSC express chemokine receptors CCR2, CCR8, CXCR1, CXCR2 and CXCR3. A dose-dependent chemotactic activity of CXCR4 and CXCR1/CXCR2 ligands CXCL12 and CXCL8 (interleukin-8) was demonstrated using a 96-well chemotaxis assay. In contrast, the CCR2 ligand CCL2 (monocyte chemoattractant protein-1, MCP-1) did not recruited human MSC. In conclusion, we report that the chemokine receptor expression profile of human MSC is much broader than known before. Furthermore, for the first time, we demonstrate that human MSC migrate upon stimulation with CXCL8 but not CCL2. In combination with already known data on MSC recruitment and differentiation these are promising results towards in situ regenerative medicine approaches based on guiding of MSC to sites of degenerated tissues.
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:21617697 [本文引用: 1]
DOI:10.1002/art.34401URLPMID:22275298 [本文引用: 1]
OBJECTIVE: Rheumatoid arthritis is characterized by persistent synovial inflammation and progressive joint destruction, which are mediated by innate and adaptive immune responses. Cytokine blockade successfully treats some patient subsets; however, approximately 50% do not respond to this approach. Targeting of pathogenic T lymphocytes is emerging as an effective alternative/complementary therapeutic strategy, yet the factors that control T cell activation in joint disease are not well understood. Tenascin-C is an arthritogenic extracellular matrix glycoprotein that is not expressed in healthy synovium but is elevated in the rheumatoid joint, where high levels are produced by myeloid cells. Among these cells, tenascin-C expression is most highly induced in activated dendritic cells (DCs). The aim of this study was to examine the role of tenascin-C in this cell type. METHODS: We systematically compared the phenotype of DCs isolated from wild-type mice or mice with a targeted deletion of tenascin-C by assessing cell maturation, cytokine synthesis, and T cell polarization. RESULTS: Dendritic cells derived from tenascin-C-null mice exhibited no defects in maturation; induction of the class II major histocompatibility complex and the costimulatory molecules CD40 and CD86 was unimpaired. Dendritic cells that did not express tenascin-C, however, produced lower levels of inflammatory cytokines than did cells from wild-type mice and exhibited specific defects in Th17 cell polarization. Moreover, tenascin-C-null mice displayed ablated levels of interleukin-17 in the joint during experimental arthritis. CONCLUSION: These data demonstrate that tenascin-C is important in DC-mediated polarization of Th17 lymphocytes during inflammation and suggest a key role for this endogenous danger signal in driving adaptive immunity in erosive joint disease.
DOI:10.1136/ard.2009.123463URLPMID:20650877 [本文引用: 1]
OBJECTIVE: To determine the safety and efficacy of allogeneic mesenchymal stem cell transplantation (MSCT) in refractory systemic lupus erythematosus (SLE). METHODS: A total of 15 patients with persistently active SLE underwent MSCT. Outcome was evaluated by changes in the SLE disease activity index (SLEDAI), serological features (anti-nuclear antibodies and anti-double-stranded DNA (anti-dsDNA)), renal function and percentage of peripheral blood regulatory T cells. RESULTS: From 11 March 2007 to 4 November 2008, 15 patients with persistently active SLE were enrolled and underwent MSCT. The mean follow-up period was 17.2+/-9.5 months. A total of 13 patients have been followed for more than 12 months. All patients clinically improved following treatment with mesenchymal stem cells with a marked decrease in the SLEDAI score and 24 h proteinuria. At 12-month follow-up, SLEDAI scores decreased from 12.2+/-3.3 to 3.2+/-2.8 and proteinuria decreased from 2505.0+/-1323.9 to 858.0+/-800.7 mg/24 h (all p<0.05, by paired t test, n=12). At 1-year follow-up in 13 patients, 2 had a relapse of proteinuria, while the other 11 continue to have decreased disease activity on minimal treatment. Anti-dsDNA levels decreased. Improvement in glomerular filtration rate was noted in two patients in which formal testing was performed. Non-renal-related manifestations also improved significantly. No serious adverse events were reported. CONCLUSION: Allogeneic MSCT in patients with refractory lupus resulted in amelioration of disease activity, improvement in serological markers and stabilisation of renal function. MSCT appears beneficial in treatment of patients with SLE refractory to conventional treatment options.
URLPMID:24388428 [本文引用: 1]
URLPMID:25329189 [本文引用: 1]
DOI:10.1016/j.stem.2008.03.002URLPMID:18397751 [本文引用: 1]
The concept of mesenchymal stem cells has gained wide popularity. Despite the rapid growth of the field, uncertainties remain with respect to the defining characteristics of these cells, including their potency and self-renewal. These uncertainties are reflected in a growing tendency to question the very use of the term. This commentary revisits the experimental origin of the concept of the population(s) referred to as mesenchymal stem cells and the experimental framework required to assess their stemness and function.
[本文引用: 1]
DOI:10.1186/s13059-017-1164-8URLPMID:28219395 [本文引用: 1]
BACKGROUND: Precise genome editing via homology-directed repair (HDR) after double-stranded DNA (dsDNA) cleavage facilitates functional genomic research and holds promise for gene therapy. However, HDR efficiency remains low in some cell types, including some of great research and clinical interest, such as human induced pluripotent stem cells (iPSCs). RESULTS: Here, we show that a double cut HDR donor, which is flanked by single guide RNA (sgRNA)-PAM sequences and is released after CRISPR/Cas9 cleavage, increases HDR efficiency by twofold to fivefold relative to circular plasmid donors at one genomic locus in 293 T cells and two distinct genomic loci in iPSCs. We find that a 600 bp homology in both arms leads to high-level genome knockin, with 97-100% of the donor insertion events being mediated by HDR. The combined use of CCND1, a cyclin that functions in G1/S transition, and nocodazole, a G2/M phase synchronizer, doubles HDR efficiency to up to 30% in iPSCs. CONCLUSIONS: Taken together, these findings provide guidance for the design of HDR donor vectors and the selection of HDR-enhancing factors for applications in genome research and precision medicine.
URLPMID:28524166 [本文引用: 1]
DOI:10.1126/science.1138140URLPMID:17379808 [本文引用: 1]
Clustered regularly interspaced short palindromic repeats (CRISPR) are a distinctive feature of the genomes of most Bacteria and Archaea and are thought to be involved in resistance to bacteriophages. We found that, after viral challenge, bacteria integrated new spacers derived from phage genomic sequences. Removal or addition of particular spacers modified the phage-resistance phenotype of the cell. Thus, CRISPR, together with associated cas genes, provided resistance against phages, and resistance specificity is determined by spacer-phage sequence similarity.
URLPMID:26524520 [本文引用: 2]
URLPMID:20508142 [本文引用: 1]
DOI:10.1038/cr.2014.15URLPMID:24503648 [本文引用: 1]
DOI:10.1371/journal.pone.0107945URLPMID:25232950 [本文引用: 1]
Genetically modified pigs have become a popular model system in fundamental research, agricultural and biomedical applications. However, random integration often result in unstable expression of transgene and unpredictable phenotypes. The Rosa26 locus has been widely used to produce genetic modified animals with high and consistent expressing of transgene in mouse, human and rat, as it can be targeted efficiently and is not subject to gene-silencing effects. Recently, the first case of reporter gene targeting pigs in porcine Rosa26 (pRosa26) locus was reported. In the study, full sequence of pRosa26 locus was further characterized, and the pRosa26 promoter (pR26) was cloned and we evidenced that the new porcine endogenous promoter is suitable for driving transgene expression in a high and stable manner by avoiding DNA methylation. Furthermore, elongation factor 1a promoter (EF1a) -driven GFP reporter and Myostatin promoter (MyoP)-driven Follistatin (Fst) were successfully targeted into the pRosa26 locusby traditional homologous recombination (HR) strategy. EF1a showed high activity and hypomethylation at the locus. And, muscle-specific promoter MyoP was activated strictly in muscle of the pRosa26 targeted pigs, indicating Rosa26 locus supports tissue-specific promoter driving transgene expression in its own manner. The study provided further demonstration on biomedical and agricultural applications of porcine Rosa26 promoter and locus.
DOI:10.1101/gad.5.9.1513URLPMID:1653172 [本文引用: 1]
A general strategy for selecting insertion mutations in mice has been devised. Constructs lacking a promoter and including a beta-galactosidase gene, or a reporter gene encoding a protein with both beta-galactosidase and neomycin phosphotransferase activity, were designed so that activation of the reporter gene depends on its insertion within an active transcription unit. Such insertion events create a mutation in the tagged gene and allow its expression to be followed by beta-galactosidase activity. Introduction of promoter trap constructs into embryonic stem (ES) cells by electroporation or retroviral infection has led to the derivation of transgenic lines that show a variety of beta-galactosidase expression patterns. Intercrossing of heterozygotes from 24 strains that express beta-galactosidase identified 9 strains in which homozygosity leads to an embryonic lethality. Because no overt phenotype was detected in the remaining strains, these results suggest that a substantial proportion of mammalian genes identified by this approach are not essential for development.
DOI:10.1016/j.cell.2014.09.014URLPMID:25263330 [本文引用: 1]
CRISPR-Cas9 is a versatile genome editing technology for studying the functions of genetic elements. To broadly enable the application of Cas9 in vivo, we established a Cre-dependent Cas9 knockin mouse. We demonstrated in vivo as well as ex vivo genome editing using adeno-associated virus (AAV)-, lentivirus-, or particle-mediated delivery of guide RNA in neurons, immune cells, and endothelial cells. Using these mice, we simultaneously modeled the dynamics of KRAS, p53, and LKB1, the top three significantly mutated genes in lung adenocarcinoma. Delivery of a single AAV vector in the lung generated loss-of-function mutations in p53 and Lkb1, as well as homology-directed repair-mediated Kras(G12D) mutations, leading to macroscopic tumors of adenocarcinoma pathology. Together, these results suggest that Cas9 mice empower a wide range of biological and disease modeling applications.
[本文引用: 1]
URLPMID:21048762 [本文引用: 1]
DOI:10.1038/nature13011URLPMID:24476820 [本文引用: 1]
The clustered regularly interspaced short palindromic repeats (CRISPR)-associated enzyme Cas9 is an RNA-guided endonuclease that uses RNA-DNA base-pairing to target foreign DNA in bacteria. Cas9-guide RNA complexes are also effective genome engineering agents in animals and plants. Here we use single-molecule and bulk biochemical experiments to determine how Cas9-RNA interrogates DNA to find specific cleavage sites. We show that both binding and cleavage of DNA by Cas9-RNA require recognition of a short trinucleotide protospacer adjacent motif (PAM). Non-target DNA binding affinity scales with PAM density, and sequences fully complementary to the guide RNA but lacking a nearby PAM are ignored by Cas9-RNA. Competition assays provide evidence that DNA strand separation and RNA-DNA heteroduplex formation initiate at the PAM and proceed directionally towards the distal end of the target sequence. Furthermore, PAM interactions trigger Cas9 catalytic activity. These results reveal how Cas9 uses PAM recognition to quickly identify potential target sites while scanning large DNA molecules, and to regulate scission of double-stranded DNA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}