 ,, 范元婵,, 王杰, 蒋海宾, 祝智威, 范小雪, 陈华枝, 杜宇, 周紫彧, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿,福建农林大学动物科学学院(蜂学学院),福州 350002
,, 范元婵,, 王杰, 蒋海宾, 祝智威, 范小雪, 陈华枝, 杜宇, 周紫彧, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿,福建农林大学动物科学学院(蜂学学院),福州 350002Regulatory Function of Long Non-Coding RNAs in Ascosphaera apis
ZHOU DingDing,, FAN YuanChan,, WANG Jie, JIANG HaiBin, ZHU ZhiWei, FAN XiaoXue, CHEN HuaZhi, DU Yu, ZHOU ZiYu, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, CHEN DaFu, GUO Rui,College of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002通讯作者:
责任编辑: 岳梅
收稿日期:2020-01-3接受日期:2020-01-20网络出版日期:2021-01-01
| 基金资助: |
Received:2020-01-3Accepted:2020-01-20Online:2021-01-01
作者简介 About authors
周丁丁,E-mail:
范元婵,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (6524KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
周丁丁, 范元婵, 王杰, 蒋海宾, 祝智威, 范小雪, 陈华枝, 杜宇, 周紫彧, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿. 蜜蜂球囊菌中长链非编码RNA的调控作用[J]. 中国农业科学, 2021, 54(1): 224-238 doi:10.3864/j.issn.0578-1752.2021.01.017
ZHOU DingDing, FAN YuanChan, WANG Jie, JIANG HaiBin, ZHU ZhiWei, FAN XiaoXue, CHEN HuaZhi, DU Yu, ZHOU ZiYu, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, CHEN DaFu, GUO Rui.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】蜜蜂球囊菌(Ascosphaera apis,简称球囊菌)是一种专性侵染蜜蜂幼虫的真菌性病原[1],对西方蜜蜂(Apis mellifera)幼虫、中华蜜蜂(Apis cerana cerana,简称中蜂)雄蜂和工蜂幼虫以及成年熊蜂均具有侵染性[2,3]。球囊菌可导致蜜蜂幼虫罹患白垩病,能够引起成年蜜蜂数量和蜂群生产力大幅下降[2,3,4,5]。长链非编码RNA(long non-coding RNA,lncRNA)是一类主要由RNA聚合酶II转录,不具备蛋白编码能力,缺少完整开放阅读框(ORF)且长度>200 nt的转录本[6],已被证明在真核生物的基因组印记和细胞周期等诸多生物学过程中扮演关键角色[7,8]。然而,球囊菌的lncRNA研究十分滞后。本研究基于前期获得的球囊菌菌丝和孢子混合样品的高质量lncRNA组学数据开展相关生物信息学分析和分子生物学验证,对球囊菌lncRNA的顺式(cis)作用、反义lncRNA(antisense lncRNA)作用和竞争性内源RNA(competing endogenous RNA,ceRNA)作用进行全面分析和深入探讨。研究结果可丰富球囊菌的lncRNA信息,为其他真菌的lncRNA提供参考,并揭示lncRNA在球囊菌中的潜在调控功能。【前人研究进展】LncRNA可通过顺式作用调控同一染色体上邻近蛋白编码基因的表达[9];通过反式作用调控距离较远基因的转录激活及表达[9,10];含有微小RNA反应元件(microRNA response element,MRE)的lncRNA还能够通过发挥ceRNA作用吸附结合miRNA,从而间接调节下游靶mRNA的表达[11];还能与特定蛋白质结合形成核酸蛋白复合物,调控蛋白活性或改变蛋白在细胞中的定位,以及作为某些小RNA的前体分子[12,13]。VAN WERVEN等[14]研究发现在酿酒酵母(Saccharomyces cerevisiae)的孢子形成过程中,lncRNA IRT1通过顺式作用抑制转录因子Rme1与启动子IME1的结合,从而抑制IME1的表达;孢子形成过程中另一个lncRNA IME4-AS的表达能够抑制IME4的表达。杜庆国[15]通过对正常磷水培和低磷水培2 d和8 d的地下和地上的玉米组织进行链特异性建库的RNA-seq测序,基于测序数据预测出65 099个lncRNA,进一步利用psRobot软件预测6 787个差异表达lncRNA(differentially expressed lncRNA,DElncRNA)可靶向结合miR399,推测这些DElncRNA通过影响miR399的表达介导玉米的低磷胁迫响应。SHAO等[16]通过对生理盐水和吗啡耐受两组小鼠的比较分析,鉴定到136个DElncRNA,进而通过构建和分析DElncRNA-miRNA-mRNA调控网络发现部分DElncRNA可作为潜在的ceRNA竞争性结合miRNA,从而间接影响泛素化途径、GRCP和TLR信号通路。真菌lncRNA的研究起步较晚且进展缓慢,有限的lncRNA研究主要集中于芽殖酵母(Schizosaccharomyces cerevisiae)、裂殖酵母(Schizosaccharomyces pombe)和粗糙脉孢菌(Neurospora crassa)等少数真菌物种[14,17-19],昆虫病原真菌的lncRNA研究未见报道。球囊菌的基因组序列信息早在2006年就已公布[20];但其完整的基因序列和功能注释信息直到2016年才正式公布[21]。因此,球囊菌的分子生物学及组学研究进展缓慢。前期研究中,笔者团队组装和注释了球囊菌的参考转录组[22];分析和探讨了侵染意大利蜜蜂(Apis mellifera ligustica,简称意蜂)幼虫和中蜂幼虫的球囊菌的转录组动态和致病机理[23,24];近期又利用PacBio单分子实时测序技术(SMRT)对实验室条件下纯培养的球囊菌进行了测序,重新组装和注释了更高质量的全长转录组,包含394 142条高可信度的全长转录本以及11 623条参考基因组未注释的全长转录本[25],可为参考基因组的完善、新基因的全长克隆以及可变剪接和可变腺苷酸化的研究提供数据基础。【本研究切入点】笔者团队前期利用链特异性建库的lncRNA-seq技术分别对纯化的球囊菌菌丝和孢子的混合样品进行了深度测序,对球囊菌lncRNA的数量、种类和保守性进行了分析,并全面比较了lncRNA和mRNA的结构特点[26]。但上述lncRNA在球囊菌中的作用方式及潜在调控功能尚不清楚。【拟解决的关键问题】基于已获得的高质量lncRNA组学数据对球囊菌lncRNA的顺式作用、反义lncRNA作用和ceRNA作用进行深入细致的分析和探讨,以期丰富球囊菌lncRNA的相关信息,并在组学水平揭示球囊菌lncRNA的潜在调控功能。1 材料与方法
试验于2019年在福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室完成。1.1 生物材料
球囊菌菌株由福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室分离、培养和保存[22,23,24]。1.2 LncRNA组学数据来源
笔者团队前期已利用链特异性建库的lncRNA- seq技术对球囊菌菌丝和孢子混合样品进行测序,共获得了70 612 299条原始读段(raw reads),质控后共获得48 268 696条有效读段(clean reads),平均Q30达到90.21%,测序数据质量良好[26];利用软件CNCI、CPC、Pfam和CAPT分别预测出578、1 848、1 460和1 662条lncRNA,四者的交集为379条lncRNA,即高可信度的球囊菌lncRNA[26]。高质量的lncRNA组学数据可用于本研究中lncRNA顺式作用、反义lncRNA作用和ceRNA作用的生物信息学分析。原始数据已上传NCBI SRA数据库,BioProject号:PRJNA395108。1.3 sRNA组学数据来源
笔者团队前期已利用small RNA-seq(sRNA-seq)技术对球囊菌菌丝和孢子的混合样品进行测序,获得了70 612 299条raw reads,质控后得到48 268 696条clean reads,平均Q30达到98.75%[27]。高质量的sRNA组学数据为本研究中lncRNA与miRNA以及miRNA和mRNA靶向结合关系的预测提供数据支撑。原始数据已上传NCBI SRA数据库,BioProject号:PRJNA560456。1.4 LncRNA上下游基因的功能及通路注释
LncRNA位于编码蛋白基因上下游,可能与启动子、增强子和可诱导元件等产生部分重叠,在转录或转录后水平对其邻近蛋白编码基因的表达具有调控作用[8,9]。参照周丁丁等[28]的方法,将lncRNA上下游100 kb范围内的邻近基因作为其调控的靶基因,利用Blast软件(采用默认参数)将lncRNA的上下游基因比对到GO数据库(1.5 LncRNA序列互补的mRNA预测及分析
LncRNA序列可以与对应的靶mRNA序列碱基互补配对,在配对的过程中,可以携带某些作用因子到靶mRNA,进而调控相关编码蛋白基因的表达[11]。参照周丁丁等[28]的方法,利用LncTar软件[29]对lncRNA的靶mRNA进行预测,基于lncRNA和mRNA之间的互补序列,计算配对位点自由能和标准化自由能,标准化自由能阈值以下的则认为是lncRNA的靶mRNA。利用Blast软件(采用默认参数)将lncRNA的互补序列基因比对到eggNOG数据库(1.6 LncRNA的ceRNA调控网络构建及分析
参照郭睿等[30]的方法,利用Target Finder软件[31]预测球囊菌lncRNA靶向结合的miRNA以及miRNA靶向结合的mRNA,利用三者之间的靶向结合关系构建lncRNA-miRNA及lncRNA-miRNA-mRNA调控网络,并通过Cytoscape v 3.7.1软件[28,31]对上述调控网络进行可视化。1.7 LncRNA和mRNA的RT-PCR验证
根据1.6中靶向预测的结果,随机选取9个调控网络中的lncRNA(MSTRG.6443.1、MSTRG.2135.1、MSTRG.2134.1、MSTRG.3422.1、MSTRG.2302.1、MSTRG.5023.1、MSTRG.5584.1、MSTRG.1870.1和MSTRG.1614.1)和8个靶mRNA(gene2674、gene4126、gene4125、gene1602、gene5384、gene1986、gene989、和gene1970)进行RT-PCR验证。根据上述lncRNA和mRNA的核酸序列,利用Primer Primer 5.0软件设计相应的特异性引物,委托上海生工生物工程有限公司合成引物(表1)。选取actin(gene6001)作为lncRNA和mRNA的内参基因(阳性对照)。将9对lncRNA(或8对mRNA)特异性上游引物(1 μL/种)和下游引物(1 μL/种)混合,以无菌水为模板进行RT-PCR,作为阴性对照。利用RNA抽提试剂盒(TaKaRa公司,日本)分别提取球囊菌菌丝的总RNA和孢子的总RNA,将1 μg菌丝RNA与1 μg孢子RNA等量混合,作为模板进行反转录,得到的cDNA作为模板进行PCR。PCR反应体系(20 μL):cDNA模板1 μL,上游引物1 μL,下游引物1 μL,Mixture 10 μL,无菌水7 μL。PCR程序:95℃预变性5 min;95℃变性30 s,55℃退火30 s,72℃延伸30 s,共34个循环;72℃再延伸5 min。PCR产物经1.5%琼脂糖凝胶电泳和凝胶成像仪(上海培清,中国)检测。Table 1
表1
表1本研究使用的引物
Table 1
| 核酸ID Nucleic acid ID | 引物名称 Primer name | 引物序列 Primer sequence (5′-3′) | 产物大小 Product size (bp) |
|---|---|---|---|
| MSTRG.6443.1 | F | AAAATGAAAAGGCAAATC | 210 |
| R | AAGGTCAAGAAGCACAAG | ||
| MSTRG.2135.1 | F | AGAAGCAGCAAGGAAGTCG | 128 |
| R | GGCAGGGCAATAACAAAAC | ||
| MSTRG.2134.1 | F | ACTCACTCTCTGCCCCTC | 238 |
| R | CCCATTTATTTGCTACCG | ||
| MSTRG.3422.1 | F | AACCGAAAAACTCAAGGA | 160 |
| R | TCGCAATCAGACATCAAA | ||
| MSTRG.2302.1 | F | TGTCTGTCCGTTCGTCCTT | 139 |
| R | CAGCGTAGCGTTGTGTAGT | ||
| MSTRG.5023.1 | F | CAAACCCAGATTTATTCC | 110 |
| R | TACCTTTCCTCCTTACGA | ||
| MSTRG.5584.1 | F | GAAGACATTCAATCCAACAC | 184 |
| R | AGCCACACACTTATCCACTA | ||
| MSTRG.1870.1 | F | GCCTCTTTTCGGTTTGCT | 158 |
| R | CGTTCTTGCTCGTGTCGT | ||
| MSTRG.1614.1 | F | AAATCGGAACGGTGAGGGA | 115 |
| R | GGCAGTGACCAAAGGCAAA | ||
| gene1602 | F | AGCCCCGCTACCAAACTC | 110 |
| R | CTGTCCCTCATCGCCATA | ||
| gene1970 | F | AGTTCTCTTACTGGCGTCTTTG | 100 |
| R | CTTTCACTTGCTGGGCTTTCTC | ||
| gene2674 | F | TGGCTTCCTACACAAACCT | 233 |
| R | TTCCTTATTCACCCGCTCC | ||
| gene4126 | F | ATGAACAATGTCAAGGAAGC | 320 |
| R | TCTGGAAAGAGTGTGGGGAG | ||
| gene4125 | F | CCAAACAGACAAATGGCTAA | 120 |
| R | TCGGGAAAGATGATTGAGAA | ||
| gene5384 | F | TATGGTTATGCCTATGGTTT | 122 |
| R | GATGTGGGGTATCTCGTGTT | ||
| gene1986 | F | CATTGATACCGATACAGAGAC | 237 |
| R | GAGATAACATTGACAACGCCT | ||
| gene989 | F | CCTCTTCCCACACTCTCACT | 236 |
| R | AGACTCCAAAACCTGCTCCT | ||
| miR-281-y | L | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCAAAGAGA | 72 |
| F | ACACTCCAGCTGGGTGTCATGGAGTTGC | ||
| miR-13-y | L | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAGCTCATC | 73 |
| F | ACACTCCAGCTGGGTATCACAGCCATTTT | ||
| miR-11980-y | L | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCTGCCAA | 68 |
| F | ACACTCCAGCTGGGGGGAACGGGC | ||
| miR-6057-y | L | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCATTTGTT | 72 |
| F | ACACTCCAGCTGGGTTTGTGACTGTAAC | ||
| miR-1-z | L | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGATACATAC | 72 |
| F | ACACTCCAGCTGGGTGGAATGTAAAGAA | ||
| miR-9-z | L | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCATACAG | 73 |
| F | ACACTCCAGCTGGGTCTTTGGTTATCTAG | ||
| miR-13-x | L | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCATTCCAC | 72 |
| F | ACACTCCAGCTGGGACATCAAATTGGTT | ||
| actin (gene6001) | F | GCTACTTCCCATCATTCGTC | 92 |
| R | CCCAATCTGTGACAATCCC | ||
| - | Universal R | CTCAACTGGTGTCGTGGA |
新窗口打开|下载CSV
1.8 MiRNA的Stem-loop RT-PCR验证
根据1.6中靶向预测的结果,随机选取7个调控网络中的靶miRNA(miR-281-y、miR-13-y、miR-11980-y、miR-6057-y、miR-1-z、miR-9-z和miR-13-x)参照郭睿等[27]和杜宇等[32]的方法,利用DNAMAN软件(Lynnon Biosoft 公司,美国)设计miRNA的Stem-loop引物、特异性上游引物和通用下游引物,委托上海生工生物工程有限公司合成引物(表1)。选取actin(gene6001)作为miRNA的内参基因(阳性对照)。将7对miRNA特异性上游引物(1 μL/种)和通用下游引物(1 μL/种)混合,以无菌水为模板进行RT-PCR,作为阴性对照。利用RNA抽提试剂盒(TaKaRa公司,日本)分别提取球囊菌菌丝的总RNA和孢子的总RNA,将1 μg菌丝RNA与1 μg孢子RNA等量混合作为反转录的模板。利用Stem-loop引物反转录得到cDNA,作为模板进行 PCR扩增。PCR 体系(20 μL):上下游引物(1.67 mol·L-1)各1 μL,cDNA模板1 μL,PCR mix 10 μL,无菌水7 μL。PCR程序:95℃预变性5 min;95℃变性30 s,梯度退火30 s,72℃延伸30 s,共34个循环;72℃再延伸5 min。PCR产物经1.5%琼脂糖凝胶电泳和凝胶成像仪(上海培清,中国)检测。2 结果
2.1 球囊菌lncRNA的顺式作用分析
球囊菌的371个lncRNA共预测出5 852个上下游基因。GO数据库注释结果显示,这些上下游基因可注释到48个功能条目,涉及生物学进程、分子功能和细胞组分3大类;其中生物学进程相关的前5个条目分别是细胞进程(1 707)、代谢进程(1 705)、单组织进程(1 231)、定位(473)和生物学调控(325);细胞组分相关的前5个条目分别是细胞(1 163)、细胞组件(1 163)、细胞器(795)、细胞膜(532)和大分子复合物(501);分子功能相关的前5个条目分别是催化活性(1 796)、结合(1 388)、转运活性(180)、分子结构活性(106)和核酸结合转化因素(40)(图1)。括号内的数字表示注释到该条目的上下游基因数。KEGG数据库注释结果显示,这些上下游基因可注释到121条通路,注释基因数最多的前10位分别为新陈代谢途径(564)、次生代谢产物的生物合成(233)、抗生素的生物合成(176)、氨基酸的生物合成(89)、碳代谢(81)、核糖体(78)、嘌呤代谢(75)、剪接体(66)、RNA转运(65)和细胞周期-酵母(62)。括号内的数字表示注释到该通路的上下游基因数。图1
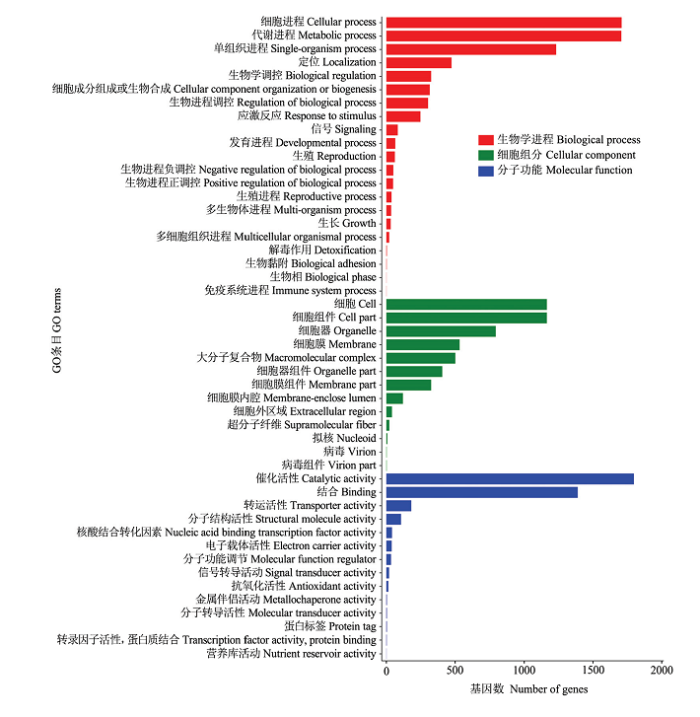 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1球囊菌lncRNA上下游基因的GO数据库注释
Fig. 1GO database annotation of upstream and downstream genes of A. apis lncRNAs
2.2 球囊菌中反义lncRNA分析
共预测出球囊菌的7个lncRNA与7个mRNA序列互补,它们的结合关系如图2所示。其中,有1个靶mRNA(gene3444)在KEGG数据库注释为核孔复合体蛋白An-Nup120和假定蛋白(K14303),另有1个靶mRNA(gene1554)注释为假定蛋白(K07117);共有5个靶基因(gene1316、gene1194、gene4650、gene5233和gene1554)在eggNOG数据库中注释为假定蛋白(表2)。图2
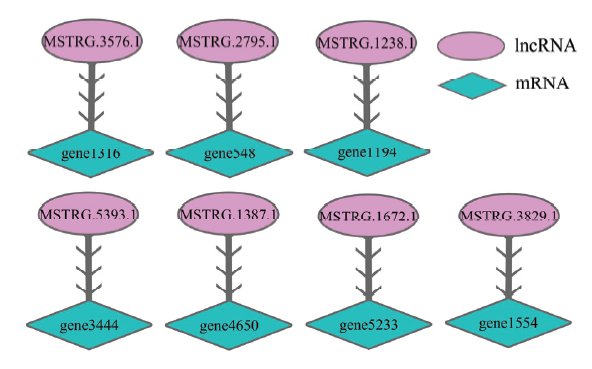 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2球囊菌中反义lncRNA及其靶标的结合关系
Fig. 2Binding relationship between antisense lncRNAs and their targets in A. apis
Table 2
表2
表2球囊菌中与lncRNA序列互补靶标的功能注释
Table 2
| LncRNA ID | LncRNA大小 LncRNA size (bp) | 靶mRNA ID Target mRNA ID | 靶mRNA大小 Target mRNA size (bp) | eggNOG数据库注释 Annotation in eggNOG database |
|---|---|---|---|---|
| MSTRG.3576.1 | 285 | gene1316 | 1851 | 保守性假定蛋白(土曲霉NIH2624) Conserved hypothetical protein (Aspergillus terreus NIH2624) |
| MSTRG.2795.1 | 253 | gene548 | 1407 | — |
| MSTRG.1238.1 | 294 | gene1194 | 1569 | 保守性假定蛋白(土曲霉NIH2624) Conserved hypothetical protein (Aspergillus terreus NIH2624) |
| MSTRG.5393.1 | 238 | gene3444 | 3831 | 核孔复合体蛋白An-Nup120(皮炎芽生菌ATCC 18188) Nuclear pore complex protein An-Nup120 (Blastomyces dermatitidis ATCC 18188) |
| MSTRG.1387.1 | 206 | gene4650 | 3186 | 假定蛋白AN4929.2(构巢曲霉FGSC A4) Hypothetical protein AN4929.2 (Aspergillus nidulans FGSC A4) |
| MSTRG.1672.1 | 221 | gene5233 | 7065 | 假定蛋白PABG_05261(巴西副球孢子菌Pb03) Hypothetical protein PABG_05261 (Paracoccidioides brasiliensis Pb03) |
| MSTRG.3829.1 | 296 | gene1554 | 3036 | 假定蛋白CIMG_07970(粗球孢子菌RS) Hypothetical protein CIMG_07970 (Coccidioides immitis RS) |
新窗口打开|下载CSV
2.3 球囊菌lncRNA的ceRNA作用分析
共预测出227个lncRNA靶向结合73个miRNA,二者之间形成较为复杂的调控网络,其中MSTRG.2597.1、MSTRG.4497.1和MSTRG.1789.9靶向结合的miRNA数量较多,分别为7、7和6个;但多数lncRNA(79.02%)仅能结合1—2个miRNA;部分miRNA可靶向多个lncRNA,其中miR-34-x、miR-10-x和miR-375-y结合的lncRNA数量最多,分别为31、27和24个(图3);但多数miRNA(80.82%)可靶向的lncRNA数少于10个。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3球囊菌中的lncRNA-miRNA调控网络
Fig. 3LncRNAs-miRNAs regulatory networks in A. apis
2.4 氧化磷酸化和MAPK信号通路相关的ceRNA调控网络
根据lncRNA与miRNA、miRNA与mRNA的靶向结合关系构建lncRNA-miRNA-mRNA调控网络,发现三者之间具有更为复杂的调控关系,227个lncRNA靶向73个miRNA,这些miRNA可结合50个靶mRNA。对上述靶mRNA进行KEGG数据库注释,结果显示它们共注释到28条通路,注释最多的通路是新陈代谢途径(9),其次为次生代谢产物的生物合成(4)、自噬-其他真核生物(3)、线粒体自噬-酵母(3)和自噬-酵母(3)(表3)。进一步分析发现有10个靶mRNA可注释到9条物质能量代谢通路,分别为谷胱甘肽代谢(2)、脂肪酸延长(1)、不饱和脂肪酸的生物合成(1)、鞘脂类代谢(1)、脂肪酸代谢(1)、碳代谢(1)、氧化磷酸化(1)、丙酸代谢(1)、糖酵解和糖质生(1)。括号内的数字代表注释在该通路的靶mRNA数。Table 3
表3
表3球囊菌ceRNA网络中靶mRNA注释数前12位的KEGG通路
Table 3
| 通路 Pathway | 通路ID Pathway ID | mRNA数 Number of mRNAs | P值 P value |
|---|---|---|---|
| 新陈代谢途径Metabolic pathway | ko01100 | 9 | 0.2834603 |
| 次生代谢物的生物合成Biosynthesis of secondary metabolite | ko01110 | 4 | 0.3784708 |
| 自噬-其他真核生物Autophagy-other eukaryote | ko04136 | 3 | 0.0012982 |
| 线粒体自噬-酵母Mitophagy-yeast | ko04139 | 3 | 0.0059399 |
| 自噬-酵母Autophagy-yeast | ko04138 | 3 | 0.0165266 |
| 抗生素生物合成Biosynthesis of antibiotics | ko01130 | 3 | 0.4179210 |
| 谷胱甘肽代谢Glutathione metabolism | ko00480 | 2 | 0.0271424 |
| 寿命调节途径-多物种Longevity regulating pathway-multiple species | ko04213 | 2 | 0.0302527 |
| 内质网蛋白加工Protein processing in endoplasmic reticulum | ko04141 | 2 | 0.1919356 |
| 减数分裂-酵母Meiosis-yeast | ko04113 | 2 | 0.2092014 |
| 嘌呤代谢Purine metabolism | ko00230 | 2 | 0.2678463 |
| 细胞周期-酵母Cell cycle-yeast | ko04111 | 2 | 0.3210316 |
新窗口打开|下载CSV
根据靶mRNA对应的Nr数据库蛋白注释信息,筛选与氧化磷酸化通路和MAPK信号通路相关的靶mRNA及与上述靶mRNA存在靶向结合关系的miRNA及相应lncRNA,分析发现氧化磷酸化通路相关调控网络包含222个lncRNA、78个靶miRNA和50个靶mRNA,其中MSTRG.2597.1、MSTRG.4497.1及MSTRG.1789.9结合的miRNA数最多,分别为7、7和6个(图4-A);MAPK信号通路相关调控网络包含222个lncRNA、76个靶miRNA和46个靶mRNA,其中MSTRG.4497.1、MSTRG.2597.1及MSTRG.1789.9结合的miRNA数最多,分别为7、7和6个(图4-B);共有217个lncRNA和66个miRNA同时涉及上述两条通路的调控网络。
图4
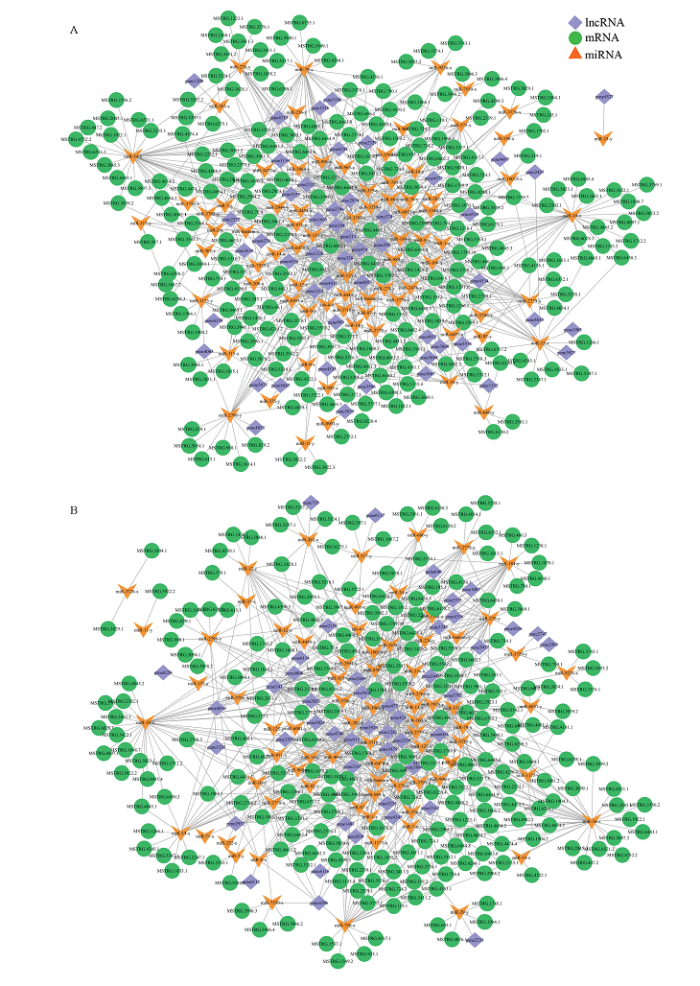 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4球囊菌中氧化磷酸化(A)和MAPK信号通路(B)相关的lncRNA-miRNA-mRNA调控网络
Fig. 4LncRNAs-miRNAs-mRNAs regulatory networks involved in oxidative phosphorylation (A) and MAPK signaling pathway (B) in A. apis
2.5 球囊菌ceRNA调控网络中lncRNA、miRNA和mRNA的表达验证
为验证ceRNA网络中lncRNA、靶miRNA和靶mRNA的表达,通过RT-PCR对上述ceRNA网络中lncRNA和靶mRNA的进行扩增,扩增产物的电泳结果显示9个lncRNA均能扩增出预期大小的目的条带,阳性对照能扩增出目的片段而阴性对照未扩增出片段;8个靶mRNA能扩增出预期片段,阳性对照能扩增出目的片段而阴性对照未扩增出片段;上述结果证实了lncRNA和靶mRNA的真实表达。利用Stem-loop RT-PCR对7个miRNA进行检测,电泳结果显示它们均能扩增出预期大小的目的片段,阳性对照能扩增出目的片段而阴性对照未扩增出片段(图5),证实了miRNA的真实表达。上述结果表明球囊菌中lncRNA、靶miRNA和靶mRNA真实表达。图5
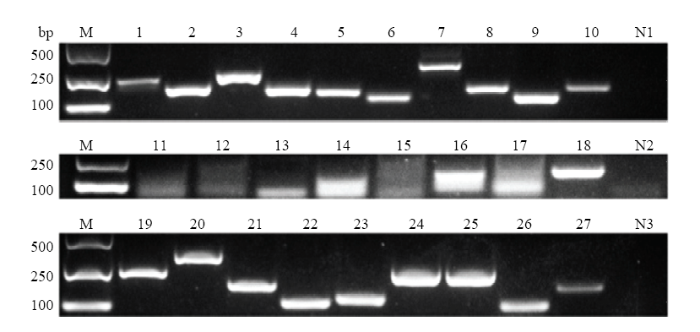 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5球囊菌中lncRNA、靶miRNA和靶mRNA的RT-PCR验证
Fig. 5RT-PCR verification of lncRNAs, target miRNAs and target mRNAs in A. apis
M: DNA marker; 1: MSTRG.6443.1; 2: MSTRG.2135.1; 3: MSTRG.2134.1; 4: MSTRG.3422.1; 5: MSTRG.2302.1; 6: MSTRG.5023.1; 7: MSTRG.5584.1; 8: MSTRG.1870.1; 9: MSTRG.1614.1; 11: miR-281-y; 12: miR-13-y; 13: miR-11980-x; 14: miR-6057-x; 15: miR-1-z; 16: miR-9-z; 17: miR-13-x; 19: gene2674; 20: gene4126; 21: gene4125; 22: gene1602; 23: gene5384; 24: gene1986; 25: gene989; 26: gene1970; 10, 18, 27: actin (positive control); N1, N2, N3: Sterile water (negative control)
3 讨论
球囊菌侵染蜜蜂幼虫导致白垩病,该病危害蜜蜂健康并长期困扰养蜂生产。相对于酵母等模式真菌,球囊菌的分子生物学及组学研究较为滞后,miRNA和lncRNA等ncRNA的相关研究更为匮乏。目前,较多的研究表明lncRNA在人类、黑腹果蝇(Drosophila melanogaster)、秀丽隐杆线虫(Caenorhabditis elegans)、拟南芥(Arabidopsis thaliana)和酵母(Saccharomyces)等模式生物中发挥特殊而重要的调控功能[8,10-14,33-35]。笔者团队前期已对球囊菌的miRNA进行了全基因组鉴定、分析和验证[27];并对侵染中华蜜蜂6日龄幼虫的球囊菌的miRNA差异表达谱及调控网络进行了解析[36]。近期,为最大限度鉴定球囊菌的lncRNA,笔者团队利用链特异性建库的lncRNA-seq技术对球囊菌菌丝和孢子混合样品进行了深度测序,通过生物信息学方法对lncRNA的数量、种类、结构特征进行了初步分析,鉴定到379个lncRNA,包括242个反义lncRNA、123个基因间区lncRNA(lincRNA)、13个正义链lncRNA(sense lncRNA)和1个内含子lncRNA(intronic transcript lncRNA)[26];并对部分lncRNA的真实表达进行了分子验证[26];此为球囊菌lncRNA的首次研究报道。基于已获得的高质量lncRNA组学数据,本研究进一步探究球囊菌lncRNA的顺式作用、反义lncRNA作用和ceRNA作用,发现lncRNA在球囊菌的物质和能量代谢、遗传信息传递、环境刺激应答、信号通路、细胞生长和细胞周期等生物学过程中具有潜在的调控功能。3.1 LncRNA对球囊菌生长发育以及物质能量代谢具有潜在的顺式调控作用
LncRNA发挥顺式作用主要是通过影响同一染色体上的邻近蛋白编码基因的表达发挥调控功能[9]。CAI等[37]利用RNAi技术和细胞实验对7周龄的兴化雌鸡各组织和器官进行研究,发现位于Six1基因上游432 bp处的lncRNA-Six1可编码一个约7.26 kD的小肽,进而通过顺式作用影响Six1的表达以促进鸡肉细胞增殖及诱导细胞分裂。FENG等[38]利用lncRNA-seq技术对长牡蛎(Crassostrea gigas)的白色、黑色、金色和正常外壳进行深度测序,预测出1 157个lncRNA及潜在调控的24 057个上下游基因,进一步分析结果显示427个DElncRNA可靶向2 088个上下游基因,这些上下游基因涉及RNA甲基转移酶和tRNA甲基转移酶活性,以及ECM-受体相互作用、Jak-STAT信号通路和Notch信号通路,作者推测相关DElncRNA通过顺式作用调节牡蛎壳的色素沉淀。本研究中,371个lncRNA共预测出5 852个上下游基因,其中分别有1 705、64、31和61个上下游基因注释到代谢进程、发育进程、生长和繁殖。还发现分别有1 163、1 163和795个上下游基因注释到细胞、细胞组件和细胞器,表明lncRNA可能通过顺式作用调节球囊菌的细胞生命活动。碳代谢在真菌生长和发育中扮演关键角色,其中一碳代谢在机体代谢活动发挥重要作用;而中心碳代谢是生物体所需能量的主要来源,并为其他代谢提供前体反应物,糖酵解途径和磷酸戊糖途径都属于中心碳代谢[39]。WANG等[40]利用转录组测序技术和分子生物学手段对野生型和突变型的谷氨酸棒杆菌(Corynebacterium glutamicum)进行深入分析,通过13C代谢通量分析,发现两个转录因子gntR1-E70K和ramA-A52V可促进突变型谷氨酸棒杆菌的糖酵解和磷酸戊糖途径相关基因上调表达,说明这两个转录因子加快了谷氨酸棒杆菌的中心碳代谢。本研究中,分别有81、39和21个上下游基因注释到碳代谢、糖酵解/糖异生途径和磷酸戊糖途径,表明相应的lncRNA可能通过顺式作用调控球囊菌的一碳代谢和中心碳代谢。此外,分别有57、21和11个上下游基因注释到氧化磷酸化、甲烷代谢和硫代谢等能量代谢途径,说明相应的lncRNA可能通过顺式作用调控上述能量代谢通路。
3.2 LncRNA对球囊菌次级代谢产物的合成与降解以及过氧化物酶体具有潜在的顺式调控作用
生物体能够以糖类、氨基酸和核苷酸等初级代谢产物为前体,合成一些对于生长发育不可或缺的次级代谢产物,例如苯丙素类、萜类和黄酮类化合物[41,42]。本研究中,分别有30、27、20、16、13、12和7个上下游基因涉及氨基糖和核苷糖代谢、缬氨酸和亮氨酸及异亮氨酸的降解、果糖和甘露糖代谢、烟酸和烟酰胺代谢、赖氨酸降解、鞘脂类代谢和维生素B6代谢,表明相应的lncRNA通过调控上述初级代谢产物的加工,合成必要的次级代谢产物供球囊菌的生长和发育;此外,分别有233、15、7、4、1和1个lncRNA上下游基因涉及次级代谢物生物合成、萜类化合物生物合成、泛醌和其他烯萜类生物合成、酮体的合成与降解、碳青霉烯类生物合成和倍半萜三萜类生物合成,再次说明相关lncRNA通过调控次级代谢产物的合成与降解参与对球囊菌生长和发育的调节。HUARTE-BONNET等[43]利用含烷烃培养基培养球孢白僵菌(Beauveria bassiana),发现过氧化物酶体生物合成途径相关的Bbhyd1和Bbhyd2两个基因被激活,在过氧化物酶活性高的菌丝团形成菌丝球过程中,球孢白僵菌表现出过氧化物酶体增多和表面明显增厚的现象。本研究中,有32个上下游基因注释到过氧化物酶体,说明相关lncRNA(MSTRG.4299.3、MSTRG.6298.3和MSTRG.6220.4等)对该通路具有潜在的顺式调控作用。3.3 LncRNA对球囊菌的环境适应及细胞生化反应具有潜在的顺式调控作用
应激反应是生物体对外界刺激的一种适应性机制,真菌在面对营养物质缺乏、温度胁迫、物理化学因子刺激和机械损伤时可在一定环境压力范围内通过自身应激调节来适应和生存[44]。金属伴侣蛋白是一类胞内可溶性金属结合蛋白,可以将金属离子准确地交付给目标蛋白,促使机体激活金属酶,同时也保证细胞免受金属危害[45]。本研究发现,分别有1 796、247、106、19、3和3个上下游基因涉及催化活性、应激反应、结构性分子活性、信号传感器活性、分子传感器活性和金属伴侣蛋白活性,说明相应的lncRNA可能通过顺式作用参与调控球囊菌的环境适应及细胞内的生化反应,以及保护细胞抵御外界不良因素的影响。3.4 球囊菌反义lncRNA对核孔复合体蛋白的合成等生物学过程具有潜在调控作用
有些lncRNA能够与一些距离较远的基因序列互补配对,也可以携带某些调控因子到互补的靶mRNA上,通过影响基因的表达发挥调控功能[9,28]。CHACKO等[46]基于Northern blot、单分子荧光原位杂交和RNA-seq等技术对新型隐球酵母(Cryptococcus neoformans)进行深入研究,发现lncRNA RZE1影响新型隐球酵母的关键基因ZNF2的转录,RZE1可以调控新型隐球酵母菌丝的形成。本研究预测到7个lncRNA(MSTRG.3576.1、MSTRG.2795.1、MSTRG.1238.1、MSTRG.5393.1、MSTRG.1387.1、MSTRG.1672.1和MSTRG.3829.1)与7个靶mRNA(gene1316、gene548、gene1194、gene3444、gene4650、gene5233和gene1554)之间存在序列互补;分析发现其中5个靶mRNA对应的蛋白在eggNOG数据库中注释为假定蛋白(表2),说明球囊菌的基因注释信息尚不完善,基因组质量偏低和分子生物学研究滞后是主要原因。仅有gene3444对应的蛋白注释为核孔复合体蛋白(表2),表明相应的lncRNA(MSTRG.5393.1)通过与相应mRNA序列互补调控球囊菌核孔复合体蛋白的合成,可能会影响核苷酸、酶和转录因子进出细胞核,以及遗传信息的传递。然而,本研究并未发现可以作为miRNA前体的lncRNA。鉴于本研究的组学数据来源于实验室条件下的球囊菌纯培养,而lncRNA的表达具有细胞、组织、发育阶段和胁迫时期特异性[47],因此对于球囊菌lncRNA是否能通过作为miRNA前体发挥更为广泛而灵活的调控作用,仍需要进一步研究。下一步将利用链特异性建库的lncRNA-seq技术对球囊菌侵染的不同日龄蜜蜂幼虫肠道进行测序,通过连续比对核糖体数据库、宿主基因组和球囊菌基因组筛滤得到纯净的病原lncRNA数据,进而开展处于侵染过程的球囊菌的lncRNA调控作用研究。3.5 球囊菌lncRNA对物质能量代谢以及生长发育具有潜在的ceRNA调控作用
ceRNA假说认为,具有MRE的不同种类RNA如lncRNA、circRNA和假基因等,可通过竞争性结合miRNA间接调节基因表达[48]。目前,该假说已被越来越多的研究证实。周丁丁等[28]研究发现东方蜜蜂微孢子虫(Nosema ceranae)的lncRNA通过ceRNA作用调控孢子中的物质和能量代谢等基本生命活动。笔者团队前期系统分析和探讨了意蜂工蜂中肠发育过程的lncRNA差异表达谱及调控网络[30]、意蜂工蜂中肠响应东方蜜蜂微孢子虫胁迫的lncRNA差异表达谱及调控网络[49],发现相关DElncRNA可能通过ceRNA作用参与意蜂工蜂中肠、意蜂幼虫肠道的发育及真菌病原胁迫应答过程。氧化磷酸化是提供真菌生命活动所需能量的基础代谢途径[50]。MAPK信号通路在酵母等真菌的信号转导、生长发育、胁迫应答和致病性等方面起到关键作用[51]。本研究中,球囊菌的227个lncRNA与92个miRNA及96个mRNA之间形成的调控网络十分复杂,因此针对氧化磷酸化通路和MAPK信号通路相关的lncRNA、靶miRNA和靶mRNA构建调控网络,分析发现MSTRG.2597.1、MSTRG.4497.1和MSTRG.1789.9等球囊菌lncRNA靶向结合miRNA数量较多;共有217个lncRNA(MSTRG.1135.4、MSTRG.2904.2和MSTRG.5757.1等)同时参与上述两个通路的调控;表明相关的球囊菌lncRNA可能作为ceRNA间接调节靶mRNA的表达。进一步分析发现,调控网络中的靶mRNA可注释到碳代谢和脂肪酸代谢等18条物质能量代谢通路,以及细胞周期和减数分裂。上述结果说明相应的球囊菌lncRNA可能通过ceRNA作用对物质和能量代谢以及生殖进行调控。本研究中,MSTRG.3576.1、MSTRG.1238.1和MSTRG.3829.1同时参与了顺式作用、反义lncRNA作用和ceRNA作用,说明这3个lncRNA作为多功能的调控因子,可通过多种方式对球囊菌的生物学过程进行灵活调控。4 结论
球囊菌的371个lncRNA通过顺式作用潜在调控5 852个上下游基因的表达,227个lncRNA可通过92个miRNA间接调控96个靶mRNA的表达,部分lncRNA可能通过顺式作用和ceRNA作用调节球囊菌的生长发育及物质能量代谢;球囊菌的7个lncRNA可作为反义RNA调控7个互补配对的mRNA的表达,MSTRG.5393.1潜在参与调控球囊菌的核孔复合体的蛋白合成。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1371/journal.pone.0124868URLPMID:25885679 [本文引用: 1]

Fungi in the genus Ascosphaera (Ascomycota: Eurotiomycetes: Ascosphaerales) cause chalkbrood disease in larvae of bees. Here, we report the first-ever detection of the fungus in adult bumble bees that were raised in captivity for studies on colony development. Wild queens of Bombus griseocollis, B. nevadensis and B. vosnesenskii were collected and maintained for establishment of nests. Queens that died during rearing or that did not lay eggs within one month of capture were dissected, and tissues were examined microscopically for the presence of pathogens. Filamentous fungi that were detected were plated on artificial media containing broad spectrum antibiotics for isolation and identification. Based on morphological characters, the fungus was identified as Ascosphaera apis (Maasen ex Claussen) Olive and Spiltoir, a species that has been reported earlier only from larvae of the European honey bee, Apis mellifera, the Asian honey bee, Apis cerana, and the carpenter bee Xylocopa californica arizonensis. The identity of the fungus was confirmed using molecular markers and phylogenetic analysis. Ascosphaera apis was detected in queens of all three bumble bee species examined. Of 150 queens dissected, 12 (8%) contained vegetative and reproductive stages of the fungus. Both fungal stages were also detected in two workers collected from colonies with Ascosphaera-infected B. nevadensis queens. In this study, wild bees could have been infected prior to capture for rearing, or, the A. apis infection could have originated via contaminated European honey bee pollen fed to the bumble bees in captivity. Thus, the discovery of A. apis in adult bumble bees in the current study has important implications for commercial production of bumble bee colonies and highlights potential risks to native bees via pathogen spillover from infected bees and infected pollen.
DOI:10.1051/apido:19960401URL [本文引用: 2]
DOI:10.1051/apido:19940604URL [本文引用: 2]
DOI:10.1080/00218839.1987.11100758URL [本文引用: 1]
DOI:10.1016/0022-2011(88)90141-3URL [本文引用: 1]
URL [本文引用: 1]
Researches in lncRNAs have been booming for the past few years, but only a few studies about lncRNAs in infectious diseases have been reported. After pathogen infected a host, on the one hand, changes of its own lncRNAs may lead to a host of pathogenic; on the other hand, the pathogen may cause adaptive changes in host lncRNAs, which may play a role in immune defense.
URL [本文引用: 1]
Researches in lncRNAs have been booming for the past few years, but only a few studies about lncRNAs in infectious diseases have been reported. After pathogen infected a host, on the one hand, changes of its own lncRNAs may lead to a host of pathogenic; on the other hand, the pathogen may cause adaptive changes in host lncRNAs, which may play a role in immune defense.
URLPMID:23498938 [本文引用: 1]
DOI:10.4161/rna.7.5.13216URLPMID:20930520 [本文引用: 3]
Long noncoding RNAs (lncRNAs) are pervasively transcribed and critical regulators of the epigenome[1, 2]. These long, polyadenylated RNAs do not code for proteins, but function directly as RNAs, recruiting chromatin modifiers to mediate transcriptional changes in processes ranging from X-inactivation (XIST) to imprinting (H19)[3]. The recent discovery that lncRNA HOTAIR can link chromatin changes to cancer metastasis[4] furthers the relevance of lncRNAs to human disease. Here, we discuss lncRNAs as regulatory modules and explore the implications for disease pathogenesis. Although large-scale analyses of mammalian transcriptomes have revealed that more than 50% of transcripts have no protein coding potential[2, 5, 6], the functions of these putative transcripts are largely unknown. A subset of these noncoding transcripts are termed long noncoding RNAs (lncRNAs), based on an arbitrary minimum length of 200 nucleotides. LncRNAs are roughly classified based on their position relative to protein-coding genes: intergenic (between genes), intragenic/intronic (within genes), and antisense[2]. Initial efforts to characterize these molecules demonstrated that they function in cis, regulating their immediate genomic neighbors. Examples include AIR, XIST, and Kcnq1ot (reviewed in [1, 7, 8]), which recruit chromatin modifying complexes to silence adjacent sites. The scope of lncRNAs in gene regulation was advanced with the finding that lncRNA HOTAIR exhibited trans regulatory capacities. HOTAIR is transcribed at the intersection of opposing chromatin domains in the HOXC locus, but targets Polycomb Repressive Complex 2 (PRC2) to silence 40 kilobases of HOXD[9], a locus involved in developmental patterning. A subsequent study revealed that HOTAIR is overexpressed in approximately one quarter of human breast cancers, directing PRC2 to approximately 800 ectopic sites in the genome, which leads to histone H3 lysine 27 trimethylation and changes in gene expression[4]. The impacts of lncRNA-mediated chromatin changes are noteworthy: not only did HOTAIR drive metastasis in a mouse model, but HOTAIR expression in human breast cancer was found to be an independent prognostic marker for death and metastasis[4]. The fact that HOTAIR drives chromatin reprogramming genome-wide suggests that long-range regulation by lncRNAs may be a widespread mechanism. This is supported by a study showing that > 20% of tested lncRNAs are bound by PRC2 and other chromatin modifiers[10]. Furthermore, this is an underestimate of the total RNAs involved in chromatin modification, as PRC2 target genes also transcribe smaller 50-200 nt RNAs that interact with SUZ12 to mediate gene repression[11]. These findings provoke questions regarding the initial triggers for HOTAIR overexpression and whether understanding of lncRNA mechanics may have clinical relevance.
DOI:10.1016/j.gde.2017.07.009URLPMID:28843809 [本文引用: 5]
Pervasive transcription in mammalian genomes produces thousands of long noncoding RNA (lncRNA) transcripts. Although they have been implicated in diverse biological processes, the functional relevance of most lncRNAs remains unknown. Recent studies reveal the prevalence of lncRNA-mediated cis regulation on nearby transcription. In this review, we summarize cis- and trans-acting lncRNAs involved in stem cell pluripotency and reprogramming, highlighting the role of regulatory lncRNAs in providing an additional layer of complexity to the regulation of genes that govern cell fate during development.
DOI:10.1016/j.cub.2017.12.048URLPMID:29395921 [本文引用: 2]
The cell fate decision leading to gametogenesis requires the convergence of multiple signals on the promoter of a master regulator. In fission yeast, starvation-induced signaling leads to the transcriptional induction of the ste11 gene, which encodes the central inducer of mating and gametogenesis, known as sporulation. We find that the long intergenic non-coding (linc) RNA rse1 is transcribed divergently upstream of the ste11 gene. During vegetative growth, rse1 directly recruits a Mug187-Lid2-Set1 complex that mediates cis repression at the ste11 promoter through SET3C-dependent histone deacetylation. The absence of rse1 bypasses the starvation-induced signaling and induces gametogenesis in the presence of nutrients. Our data reveal that the remodeling of chromatin through ncRNA scaffolding of repressive complexes that is observed in higher eukaryotes is a conserved, likely very ancient mechanism for tight control of cell differentiation.
[本文引用: 2]
URL [本文引用: 2]
URLPMID:24091684 [本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.cell.2012.06.049URLPMID:22959267 [本文引用: 3]
The cell-fate decision leading to gametogenesis is essential for sexual reproduction. In S. cerevisiae, only diploid MATa/alpha but not haploid MATa or MATalpha cells undergo gametogenesis, known as sporulation. We find that transcription of two long noncoding RNAs (lncRNAs) mediates mating-type control of sporulation. In MATa or MATalpha haploids, expression of IME1, the central inducer of gametogenesis, is inhibited in cis by transcription of the lncRNA IRT1, located in the IME1 promoter. IRT1 transcription recruits the Set2 histone methyltransferase and the Set3 histone deacetylase complex to establish repressive chromatin at the IME1 promoter. Inhibiting expression of IRT1 and an antisense transcript that antagonizes the expression of the meiotic regulator IME4 allows cells expressing the haploid mating type to sporulate with kinetics that are indistinguishable from that of MATa/alpha diploids. Conversely, expression of the two lncRNAs abolishes sporulation in MATa/alpha diploids. Thus, transcription of two lncRNAs governs mating-type control of gametogenesis in yeast.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1186/s13041-018-0365-8URLPMID:29636075 [本文引用: 1]
Morphine tolerance is a challenging clinical problem that limits the use of morphine in pain treatment, but the mechanisms of morphine tolerance remain unclear. Recent research indicates that long noncoding RNAs (lncRNAs) might be a novel and promising target in the pathogeneses of diseases. Therefore, we hypothesized that lncRNAs might play a role in the development of morphine tolerance. Male Sprague-Dawley rats were intrathecally injected with 10 mug morphine twice daily for 7 consecutive days. The animals were then sacrificed for lncRNA microarray tests, and the results were validated by RT-qPCR. Next, functional predictions for the differentially expressed mRNAs (DEmRNAs) were made with the Gene Ontology/Kyoto Encyclopedia of Genes and Genomes (GO/KEGG), and predictions for the differentially expressed lncRNAs (DElncRNAs) were made based on competitive endogenous RNA (ceRNA) analyses. The rats successfully developed morphine tolerance. LncRNA microarray analysis revealed that, according to the criteria of a log2 (fold change) > 1.5 and a P-value < 0.05, 136 lncRNAs and 278 mRNAs were differentially expressed in the morphine tolerance group (MT) compared with the normal saline group (NS). The functions of the DEmRNAs likely involve in the processes of the ion channel transport, pain transmission and immune response. The ceRNA analysis indicated that several possible interacting networks existed, including (MRAK150340, MRAK161211)/miR-219b/Tollip.Further annotations of the potential target mRNAs of the miRNAs according to the gene database suggested that the possible functions of these mRNAs primarily involved the regulation of ubiquitylation, G protein-linked receptors, and Toll-like receptors, which play roles in the development of morphine tolerance. Our findings revealed the profiles of differentially expressed lncRNAs in morphine tolerance conditions, and among these lncRNAs, some DElncRNAs might be new therapeutic targets for morphine tolerance.
DOI:10.1038/srep19376URLPMID:26786024 [本文引用: 1]
Telomerase is a ribonucleoprotein that maintains the ends of linear chromosomes in most eukaryotes. Loss of telomerase activity results in shortening of telomeric DNA and eventually a specific G2/M cell-cycle arrest known as senescence. In humans, telomere shortening occurs during aging, while inappropriate activation of telomerase is associated with approximately 90% of cancers. Previous studies have identified several classes of noncoding RNAs (ncRNA) also associated with aging-related senescence and cancer, but whether ncRNAs are also involved in short-telomere-induced senescence in yeast is unknown. Here, we report 112 putative novel lncRNAs in the yeast Saccharomyces cerevisiae, 41 of which are only expressed in telomerase-negative yeast. Expression of approximately half of the lncRNAs is strongly correlated with that of adjacent genes, suggesting this subset may influence transcription of neighboring genes. Our results reveal a new potential mechanism governing adaptive changes in senescing and post-senescent survivor yeast cells.
DOI:10.1038/ncomms6576URL [本文引用: 1]
DOI:10.1111/j.1365-2583.2006.00694.xURLPMID:17069642 [本文引用: 1]
Genome sequences offer a broad view of host-pathogen interactions at the systems biology level. With the completion of the sequence of the honey bee, interest in the relevant pathogens is heightened. Here we report the genome sequences of two of the major pathogens of honey bees, the bacterium Paenibacillus larvae (causative agent for American foulbrood disease) and the fungus Ascosphaera apis. (causative agent for chalkbrood disease). Ongoing efforts to characterize the genomes of these species can be used to understand and mitigate the effects of two important pathogens, and will provide a contrast with pathogenic, benign and freeliving relatives.
DOI:10.1093/gbe/evw082URLPMID:27071652 [本文引用: 1]
Fungal pathogens of plants and animals have multifarious effects; they cause devastating damages to agricultures, lead to life-threatening diseases in humans, or induce beneficial effects by reducing insect pest populations. Many virulence factors have been determined in different fungal pathogens; however, the molecular determinants contributing to fungal host selection and adaptation are largely unknown. In this study, we sequenced the genomes of seven ascomycete insect pathogens and performed the genome-wide analyses of 33 species of filamentous ascomycete pathogenic fungi that infect insects (12 species), plants (12), and humans (9). Our results revealed that the genomes of plant pathogens encode more proteins and protein families than the insect and human pathogens. Unexpectedly, more common orthologous protein groups are shared between the insect and plant pathogens than between the two animal group pathogens. We also found that the pathogenicity of host-adapted fungi evolved multiple times, and that both divergent and convergent evolutions occurred during pathogen-host cospeciation thus resulting in protein families with similar features in each fungal group. However, the role of phylogenetic relatedness on the evolution of protein families and therefore pathotype formation could not be ruled out due to the effect of common ancestry. The evolutionary correlation analyses led to the identification of different protein families that correlated with alternate pathotypes. Particularly, the effector-like proteins identified in plant and animal pathogens were strongly linked to fungal host adaptation, suggesting the existence of similar gene-for-gene relationships in fungus-animal interactions that has not been established before. These results well advance our understanding of the evolution of fungal pathogenicity and the factors that contribute to fungal pathotype formation.
URL [本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
DOI:10.13343/j.cnki.wsxb.20160551URL [本文引用: 2]
[Objective] RNA-seq technology was used to sequence the larval guts of Apis cerana cerana under stress of Ascosphaera apis. Subsequently, trend was analyzed for differentially expressed genes (DEGs) to obtain significant gene expression patterns, followed by transcriptome analysis of A. apis stressing the larval gut.[Methods] Infected honeybee larval guts were sequenced at Illumina HiSeq 2500 platform and in-depth analyses were done using corresponding biological software. Finally, RT-qPCR was conducted to validate RNA-seq data.[Results] A total of 41133932 high-quality clean reads were obtained. Trend analysis result showed that 22865 DEGs were grouped into 8 gene expression patterns, among them 16769 DEGs were assigned to 4 significant expression patterns including 2 up-regulated trends and 2 down-regulated trends. GO enrichment analysis result showed that all DEGs within significant up-and down-regulated patterns were enriched in 40 and 37 GO terms, respectively, and the mostly enriched one is cellular process (2486 unigenes). KEGG enrichment analysis result displayed that the DEGs within significant up-and down-regulated trends were enriched in 119 and 112 pathways, respectively, and biosynthesis of amino acids (127 unigenes) and ribosome (98 unigenes) were mostly enriched. A. apis facilitated its proliferation through enhancing the biosynthesis and the host could fight A. apis by inhibiting the protein synthesis of the fungal pathogen during the stress process. Furthermore, expression levels of 11 DEGs enriched in the pathogen's MAPK signaling pathway decreased when the stressing time of A. apis was prolonged, suggesting that A. c. cerana larvae could constrain the pathogen's replication by disturbing this pathway.[Conclusion] This is the first report of transcriptome investigation of A. apis infecting A. c. cerana larvae. Our data provide gene expression profiles of A. apis stressing the larval gut of A. c. cerana, as well lay the foundation of unraveling molecular mechanisms regulating the pathogenesis of A. apis.
DOI:10.13343/j.cnki.wsxb.20160551URL [本文引用: 2]
[Objective] RNA-seq technology was used to sequence the larval guts of Apis cerana cerana under stress of Ascosphaera apis. Subsequently, trend was analyzed for differentially expressed genes (DEGs) to obtain significant gene expression patterns, followed by transcriptome analysis of A. apis stressing the larval gut.[Methods] Infected honeybee larval guts were sequenced at Illumina HiSeq 2500 platform and in-depth analyses were done using corresponding biological software. Finally, RT-qPCR was conducted to validate RNA-seq data.[Results] A total of 41133932 high-quality clean reads were obtained. Trend analysis result showed that 22865 DEGs were grouped into 8 gene expression patterns, among them 16769 DEGs were assigned to 4 significant expression patterns including 2 up-regulated trends and 2 down-regulated trends. GO enrichment analysis result showed that all DEGs within significant up-and down-regulated patterns were enriched in 40 and 37 GO terms, respectively, and the mostly enriched one is cellular process (2486 unigenes). KEGG enrichment analysis result displayed that the DEGs within significant up-and down-regulated trends were enriched in 119 and 112 pathways, respectively, and biosynthesis of amino acids (127 unigenes) and ribosome (98 unigenes) were mostly enriched. A. apis facilitated its proliferation through enhancing the biosynthesis and the host could fight A. apis by inhibiting the protein synthesis of the fungal pathogen during the stress process. Furthermore, expression levels of 11 DEGs enriched in the pathogen's MAPK signaling pathway decreased when the stressing time of A. apis was prolonged, suggesting that A. c. cerana larvae could constrain the pathogen's replication by disturbing this pathway.[Conclusion] This is the first report of transcriptome investigation of A. apis infecting A. c. cerana larvae. Our data provide gene expression profiles of A. apis stressing the larval gut of A. c. cerana, as well lay the foundation of unraveling molecular mechanisms regulating the pathogenesis of A. apis.
DOI:10.1016/j.jip.2020.107475URLPMID:32976816 [本文引用: 1]
Ascosphaera apis is a widespread fungal pathogen of honeybee larvae that results in chalkbrood disease, leading to heavy losses for the beekeeping industry in China and many other countries. This work was aimed at generating a full-length transcriptome of A. apis using PacBio single-molecule real-time (SMRT) sequencing. Here, more than 23.97 Gb of clean reads was generated from long-read sequencing of A. apis mycelia, including 464,043 circular consensus sequences (CCS) and 394,142 full-length non-chimeric (FLNC) reads. In total, we identified 174,095 high-confidence transcripts covering 5141 known genes with an average length of 2728 bp. We also discovered 2405 genic loci and 11,623 isoforms that have not been annotated yet within the current reference genome. Additionally, 16,049, 10,682, 4520 and 7253 of the discovered transcripts have annotations in the Non-redundant protein (Nr), Clusters of Eukaryotic Orthologous Groups (KOG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. Moreover, 1205 long non-coding RNAs (lncRNAs) were identified, which have less exons, shorter exon and intron lengths, shorter transcript lengths, lower GC percent, lower expression levels, and fewer alternative splicing (AS) evens, compared with protein-coding transcripts. A total of 253 members from 17 transcription factor (TF) families were identified from our transcript datasets. Finally, the expression of A. apis isoforms was validated using a molecular approach. Overall, this is the first report of a full-length transcriptome of entomogenous fungi including A. apis. Our data offer a comprehensive set of reference transcripts and hence contributes to improving the genome annotation and transcriptomic study of A. apis.
URLPMID:29894727 [本文引用: 5]
DOI:10.13343/j.cnki.wsxb.20170535URL [本文引用: 3]
[Objective] In this study, pure culture of A. apis was sequenced using sRNA-seq technology, followed by prediction, identification and analysis of A. apis microRNAs. The microRNAs-mRNAs regulation network was further constructed. [Methods] Illumina Hiseq Xten platform was used to sequence mycelium and spores of A. apis, and corresponding softwares were used to predict and analyze A. apis microRNAs, some of which were identified via Stem-loop PCR. Cytoskype software was used to construct A. apis microRNAs-mRNAs regulation network. [Results] A total of 48268696 clean reads were obtained, and 118 miRNAs of A. apis were predicted, whose length was distributed between 18 nt and 25 nt. The preference of the first base of miRNAs with different length was obviously various. Stem-loop PCR result showed target fragments with expected sizes were amplified from 10 microRNAs, implying most of the predicted microRNAs' true existence. In total, 6529 target genes of A. apis microRNAs were predicted, and among them 5725 could be annotated in Nr, Swissprot, KOG, GO and KEGG databases. Further investigation demonstrated 24 target genes were annotated in MAPK signaling pathway. Cytoskype software analysis suggested complicated regulation networks exist between microRNAs and mRNAs in A. apis, and majority of miRNAs inside the networks bind to several mRNAs. [Conclusion] Our findings enrich the understanding of A. apis microRNAs, provide beneficial supplement for basic biology information of A. apis, and lay some foundation for illustrating the molecular mechanism regulating the pathogenesis of this widespread fungal pathogen.
DOI:10.13343/j.cnki.wsxb.20170535URL [本文引用: 3]
[Objective] In this study, pure culture of A. apis was sequenced using sRNA-seq technology, followed by prediction, identification and analysis of A. apis microRNAs. The microRNAs-mRNAs regulation network was further constructed. [Methods] Illumina Hiseq Xten platform was used to sequence mycelium and spores of A. apis, and corresponding softwares were used to predict and analyze A. apis microRNAs, some of which were identified via Stem-loop PCR. Cytoskype software was used to construct A. apis microRNAs-mRNAs regulation network. [Results] A total of 48268696 clean reads were obtained, and 118 miRNAs of A. apis were predicted, whose length was distributed between 18 nt and 25 nt. The preference of the first base of miRNAs with different length was obviously various. Stem-loop PCR result showed target fragments with expected sizes were amplified from 10 microRNAs, implying most of the predicted microRNAs' true existence. In total, 6529 target genes of A. apis microRNAs were predicted, and among them 5725 could be annotated in Nr, Swissprot, KOG, GO and KEGG databases. Further investigation demonstrated 24 target genes were annotated in MAPK signaling pathway. Cytoskype software analysis suggested complicated regulation networks exist between microRNAs and mRNAs in A. apis, and majority of miRNAs inside the networks bind to several mRNAs. [Conclusion] Our findings enrich the understanding of A. apis microRNAs, provide beneficial supplement for basic biology information of A. apis, and lay some foundation for illustrating the molecular mechanism regulating the pathogenesis of this widespread fungal pathogen.
DOI:10.3864/j.issn.0578-1752.2020.10.018URL [本文引用: 5]
【Background】Long non-coding RNAs (lncRNAs) are a kind of non-coding transcripts with a length of more than 200 nt. LncRNAs can regulate gene expression at transcriptional level and post-transcriptional level via cis effect, trans effect or competing endogenous RNA (ceRNA) mechanism. LncRNAs have been proved to play pivotal roles in many biological processes such as growth and development.【Objective】The objective of this study is to perform detailed investigation and discussion of the regulatory manner of lncRNAs by combining small RNA (sRNA) omics dataset obtained in this work and previously gained lncRNA omics dataset, and to reveal the putative function of lncRNAs in Nosema ceranae spores.【Method】Here, purified spores of N. ceranae were sequenced using small RNA-seq (sRNA-seq), related bioinformatics software was used to conduct quality control of sequencing data. The upstream and downstream genes of N. ceranae lncRNAs were predicted based on the positional relationship between lncRNAs and mRNAs. Annotations of these upstream and downstream genes in GO and KEGG databases were carried out using Blast software. The lncRNA-miRNA and lncRNA-miRNA-mRNA regulatory networks were predicted using Target Finder software and then visualized with Cytoscape v3.7.2 software. The expression of partial lncRNAs, miRNAs and mRNAs within regulatory networks was validated with RT-PCR.【Result】In total, 16 597 883, 15 451 791 and 12 248 316 raw reads were obtained from sRNA-seq of N. ceranae spore samples, respectively, and after quality control, 15 608 370, 14 249 255 and 11 440 684 clean reads with a mean Q30 above 98.58% were gained, respectively. A total of 310 upstream and downstream genes of lncRNAs were predicted. These genes could be annotated to 35 functional terms associated with metabolic process, cell process, catalytic activity, binding and cells, etc. Additionally, these genes could be annotated to 56 metabolic pathways, including material metabolic pathways such as purine metabolism, carbon metabolism, pyruvate metabolism; and energy metabolic pathways including methane metabolism, oxidative phosphorylation, glycolysis/gluconeogenesis and so forth. The results indicated that corresponding lncRNAs could regulate the expression level of upstream and downstream genes through cis effect, thus participating in regulation of material and energy metabolisms as well as cell activities in N. ceranae spore. Furthermore, lncRNA-miRNA regulatory networks were constructed and analyzed, the result showed that MSTRG.3636.1, MSTRG.4498.1 and MSTRG.4883.1 could bind to four miRNAs including nce-miR-7729, nce-miR-7502, nce-miR-8639 and nce-miR-8565, suggesting that these three lncRNAs as ceRNAs could exert potential function in N. ceranae spore. LncRNA-miRNA-mRNA regulatory network analysis demonstrated that complex networks existed among them, miRNAs were located in the center, connecting lncRNAs and mRNAs, nce-miR-7502 and nce-miR-8639 could bind to the most mRNAs (28). MiRNAs targeting MSTRG.3636.1, MSTRG.4498.1 and MSTRG.4883.1 could target several mRNAs, indicating that these three lncRNAs might play a role via ceRNA mechanism, thus affecting the mechanism and vital activity in N. ceranae spore.【Conclusion】LncRNAs are likely to regulate the expression of upstream and downstream genes via cis effect, and indirectly affect the expression of target genes by absorbing miRNAs as ceRNAs, thus controlling basic activities in N. ceranae spore such as material metabolism and energy metabolism.
DOI:10.3864/j.issn.0578-1752.2020.10.018URL [本文引用: 5]
【Background】Long non-coding RNAs (lncRNAs) are a kind of non-coding transcripts with a length of more than 200 nt. LncRNAs can regulate gene expression at transcriptional level and post-transcriptional level via cis effect, trans effect or competing endogenous RNA (ceRNA) mechanism. LncRNAs have been proved to play pivotal roles in many biological processes such as growth and development.【Objective】The objective of this study is to perform detailed investigation and discussion of the regulatory manner of lncRNAs by combining small RNA (sRNA) omics dataset obtained in this work and previously gained lncRNA omics dataset, and to reveal the putative function of lncRNAs in Nosema ceranae spores.【Method】Here, purified spores of N. ceranae were sequenced using small RNA-seq (sRNA-seq), related bioinformatics software was used to conduct quality control of sequencing data. The upstream and downstream genes of N. ceranae lncRNAs were predicted based on the positional relationship between lncRNAs and mRNAs. Annotations of these upstream and downstream genes in GO and KEGG databases were carried out using Blast software. The lncRNA-miRNA and lncRNA-miRNA-mRNA regulatory networks were predicted using Target Finder software and then visualized with Cytoscape v3.7.2 software. The expression of partial lncRNAs, miRNAs and mRNAs within regulatory networks was validated with RT-PCR.【Result】In total, 16 597 883, 15 451 791 and 12 248 316 raw reads were obtained from sRNA-seq of N. ceranae spore samples, respectively, and after quality control, 15 608 370, 14 249 255 and 11 440 684 clean reads with a mean Q30 above 98.58% were gained, respectively. A total of 310 upstream and downstream genes of lncRNAs were predicted. These genes could be annotated to 35 functional terms associated with metabolic process, cell process, catalytic activity, binding and cells, etc. Additionally, these genes could be annotated to 56 metabolic pathways, including material metabolic pathways such as purine metabolism, carbon metabolism, pyruvate metabolism; and energy metabolic pathways including methane metabolism, oxidative phosphorylation, glycolysis/gluconeogenesis and so forth. The results indicated that corresponding lncRNAs could regulate the expression level of upstream and downstream genes through cis effect, thus participating in regulation of material and energy metabolisms as well as cell activities in N. ceranae spore. Furthermore, lncRNA-miRNA regulatory networks were constructed and analyzed, the result showed that MSTRG.3636.1, MSTRG.4498.1 and MSTRG.4883.1 could bind to four miRNAs including nce-miR-7729, nce-miR-7502, nce-miR-8639 and nce-miR-8565, suggesting that these three lncRNAs as ceRNAs could exert potential function in N. ceranae spore. LncRNA-miRNA-mRNA regulatory network analysis demonstrated that complex networks existed among them, miRNAs were located in the center, connecting lncRNAs and mRNAs, nce-miR-7502 and nce-miR-8639 could bind to the most mRNAs (28). MiRNAs targeting MSTRG.3636.1, MSTRG.4498.1 and MSTRG.4883.1 could target several mRNAs, indicating that these three lncRNAs might play a role via ceRNA mechanism, thus affecting the mechanism and vital activity in N. ceranae spore.【Conclusion】LncRNAs are likely to regulate the expression of upstream and downstream genes via cis effect, and indirectly affect the expression of target genes by absorbing miRNAs as ceRNAs, thus controlling basic activities in N. ceranae spore such as material metabolism and energy metabolism.
DOI:10.1093/bib/bbu048URLPMID:25524864 [本文引用: 1]
Long noncoding RNAs (lncRNAs) represent a big category of noncoding RNA molecules, and increasing studies have shown that they play important roles in various critical biological processes. They show a diversity of functions through diverse mechanisms, among which regulating RNA molecules is one of the most popular ones. Given the big number of lncRNAs, it becomes urgent and important to predict the RNA targets of lncRNAs in a large scale for the comprehensive understanding of lncRNA functions and action mechanisms. Although several methods have been developed to predict RNA-RNA interactions, none of them can be used to predict the RNA targets of lncRNAs in a large scale. Here we presented a tool, LncTar, which shows the ability to efficiently predict the RNA targets of lncRNAs in a large scale. To test the accuracy of LncTar, we applied it to 10 experimentally supported lncRNA-mRNA interactions. As a result, LncTar successfully predicted 8 (80%) of the 10 lncRNA-mRNA pairs, suggesting that LncTar has a reliable accuracy. Finally, we believe that LncTar could be an efficient tool for the fast identification of the RNA targets of lncRNAs. LncTar is freely available at http://www.cuilab.cn/lnctar.
DOI:10.3864/j.issn.0578-1752.2018.18.016URL [本文引用: 2]
【Objective】Long non-coding RNA (lncRNA) plays an important role in regulation of gene expression, epigenetics and cell cycle in eukaryotes. The objective of this study is to investigate the expression profile and role of lncRNAs in the developmental process of Apis mellifera ligustica worker’s midgut. 【Method】In this study, 7- and 10-day-old worker’s midguts of A. m. ligustica (Am7, Am10) were sequenced using RNA-seq technology and strand-specific library construction method. Using Perl script, raw reads were filtered to obtain clean reads with high-quality. Bowtie tool was used to compare clean reads to the ribosome database, and TopHat2 software was employed to compare unmapped clean reads to the reference genome. CPC and CNCI softwares were utilized to predict coding capacity of the transcripts. RT-PCR was performed to identify partial lncRNAs. Investigation of differentially expressed lncRNAs (DElncRNAs) was carried out with edgeR, followed by prediction of upstream and downstream genes, for which GO and KEGG pathway enrichment analyses were performed. RNAhybrid, Miranda and TargetScan softwares were utilized together to predict target miRNAs of DElncRNAs and target genes of miRNAs, and DElncRNAs-miRNAs-mRNAs regulation networks were visualized via Cytoscape. Finally, RT-qPCR was conducted to verify reliability of the sequencing data.【Result】134 802 058 and 147 051 470 raw reads were gained from deep sequencing of Am7 and Am10, respectively, and after stringent filtration, 134 166 157 and 146 293 288 were obtained. In total, 6 353 lncRNAs were predicted, and 3 890 DElncRNAs were obtained based on expression calculation, including 2 005 up-regulated lncRNAs and 1 885 down-regulated lncRNAs. The result of RT-PCR suggested the expected signal bands could be amplified from 8 lncRNAs, implying their true existence. There were 1 793 upstream and downstream genes of DElncRNAs, which were involved in 42 GO terms, including metabolic processes, developmental processes, cellular processes, stress responses, immune system processes and so forth. They were also associated with 251 KEGG pathways, including material metabolism pathways such as carbon metabolism, purine metabolism and fatty acid biosynthesis; energy metabolism pathways such as sulfur metabolism, methane metabolism and oxidative phosphorylation; signaling pathways such as Hippo, Wnt and Notch signaling pathways; cellular immune pathways such as lysosome, endocytosis and ubiquitin mediated proteolysis; humoral immune pathways such as MAPK, Jak-STAT and NF-kappa B pathways, these results demonstrated the DElncRNAs were involved in the material and energy metabolism, cell life activity and immunity regulation in the developmental process of A. m. ligustica worker’s midgut. Further analysis showed TCONS_00020918 might play a regulatory part in the nutrient absorption and caste differentiation in the worker’s midgut. Analysis of regulation networks demonstrated that complex networks existed between DElncRNAs and target miRNAs and mRNAs, partial DElncRNAs lie in the central of the networks and link many miRNAs, and partial miRNAs could be bound by many DElncRNAs, which indicated that these DElncRNAs might play an important role during the developmental process of the worker’s midgut. Finally, 5 DElncRNAs were randomly selected for RT-qPCR assay, and the result proved the reliability of sequencing data in this study.【Conclusion】DElncRNA is widely involved in the metabolism, cellular activity and immune regulation of A. m. ligustica worker’s midgut, and plays a role as a competitive endogenous RNA (ceRNA). The results provide the necessary data support for the screening and functional study of key lncRNA.
DOI:10.3864/j.issn.0578-1752.2018.18.016URL [本文引用: 2]
【Objective】Long non-coding RNA (lncRNA) plays an important role in regulation of gene expression, epigenetics and cell cycle in eukaryotes. The objective of this study is to investigate the expression profile and role of lncRNAs in the developmental process of Apis mellifera ligustica worker’s midgut. 【Method】In this study, 7- and 10-day-old worker’s midguts of A. m. ligustica (Am7, Am10) were sequenced using RNA-seq technology and strand-specific library construction method. Using Perl script, raw reads were filtered to obtain clean reads with high-quality. Bowtie tool was used to compare clean reads to the ribosome database, and TopHat2 software was employed to compare unmapped clean reads to the reference genome. CPC and CNCI softwares were utilized to predict coding capacity of the transcripts. RT-PCR was performed to identify partial lncRNAs. Investigation of differentially expressed lncRNAs (DElncRNAs) was carried out with edgeR, followed by prediction of upstream and downstream genes, for which GO and KEGG pathway enrichment analyses were performed. RNAhybrid, Miranda and TargetScan softwares were utilized together to predict target miRNAs of DElncRNAs and target genes of miRNAs, and DElncRNAs-miRNAs-mRNAs regulation networks were visualized via Cytoscape. Finally, RT-qPCR was conducted to verify reliability of the sequencing data.【Result】134 802 058 and 147 051 470 raw reads were gained from deep sequencing of Am7 and Am10, respectively, and after stringent filtration, 134 166 157 and 146 293 288 were obtained. In total, 6 353 lncRNAs were predicted, and 3 890 DElncRNAs were obtained based on expression calculation, including 2 005 up-regulated lncRNAs and 1 885 down-regulated lncRNAs. The result of RT-PCR suggested the expected signal bands could be amplified from 8 lncRNAs, implying their true existence. There were 1 793 upstream and downstream genes of DElncRNAs, which were involved in 42 GO terms, including metabolic processes, developmental processes, cellular processes, stress responses, immune system processes and so forth. They were also associated with 251 KEGG pathways, including material metabolism pathways such as carbon metabolism, purine metabolism and fatty acid biosynthesis; energy metabolism pathways such as sulfur metabolism, methane metabolism and oxidative phosphorylation; signaling pathways such as Hippo, Wnt and Notch signaling pathways; cellular immune pathways such as lysosome, endocytosis and ubiquitin mediated proteolysis; humoral immune pathways such as MAPK, Jak-STAT and NF-kappa B pathways, these results demonstrated the DElncRNAs were involved in the material and energy metabolism, cell life activity and immunity regulation in the developmental process of A. m. ligustica worker’s midgut. Further analysis showed TCONS_00020918 might play a regulatory part in the nutrient absorption and caste differentiation in the worker’s midgut. Analysis of regulation networks demonstrated that complex networks existed between DElncRNAs and target miRNAs and mRNAs, partial DElncRNAs lie in the central of the networks and link many miRNAs, and partial miRNAs could be bound by many DElncRNAs, which indicated that these DElncRNAs might play an important role during the developmental process of the worker’s midgut. Finally, 5 DElncRNAs were randomly selected for RT-qPCR assay, and the result proved the reliability of sequencing data in this study.【Conclusion】DElncRNA is widely involved in the metabolism, cellular activity and immune regulation of A. m. ligustica worker’s midgut, and plays a role as a competitive endogenous RNA (ceRNA). The results provide the necessary data support for the screening and functional study of key lncRNA.
DOI:10.1093/bioinformatics/bti211URLPMID:15598838 [本文引用: 2]
UNLABELLED: TargetFinder is a PC/Windows program for interactive effective antisense oligonucleotide (AO) selection based on mRNA accessible site tagging (MAST) and secondary structures of target mRNA. To make MAST result intuitive, both the alignment result and tag frequency profile is illustrated. As theoretical reference, secondary structure and single strand probability profile of target mRNA is also represented. All of these sequences and profiles are displayed in aligned mode, which facilitates identification of the accessible sites in target mRNA. Graphical, user-friendly interface makes TargetFinder a useful tool in AO target site selection. AVAILABILITY: The software is freely available at http://www.bioit.org.cn/ao/targetfinder.htm CONTACT: sqwang@nic.bmi.ac.cn.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41467-017-00304-1URLPMID:28819095 [本文引用: 1]
Thousands of genes have been well demonstrated to play important roles in cancer progression. As genes do not function in isolation, they can be grouped into
DOI:10.18632/aging.101062URLPMID:27687893
Dietary restriction (DR) extends lifespan in many species which is a well-known phenomenon. Long non-coding RNAs (lncRNAs) play an important role in regulation of cell senescence and important age-related signaling pathways. Here, we profiled the lncRNA and mRNA transcriptome of fruit flies at 7 day and 42 day during DR and fully-fed conditions, respectively. In general, 102 differentially expressed lncRNAs and 1406 differentially expressed coding genes were identified. Most informatively we found a large number of differentially expressed lncRNAs and their targets enriched in GO and KEGG analysis. We discovered some new aging related signaling pathways during DR, such as hippo signaling pathway-fly, phototransduction-fly and protein processing in endoplasmic reticulum etc. Novel lncRNAs XLOC_092363 and XLOC_166557 are found to be located in 10 kb upstream sequences of hairy and ems promoters, respectively. Furthermore, tissue specificity of some novel lncRNAs had been analyzed at 7 day of DR in fly head, gut and fat body. Also the silencing of lncRNA XLOC_076307 resulted in altered expression level of its targets including Gadd45 (involved in FoxO signaling pathway). Together, the results implicated many lncRNAs closely associated with dietary restriction, which could provide a resource for lncRNA in aging and age-related disease field.
DOI:10.3390/genes8080198URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3389/fphys.2017.00230URLPMID:28473774 [本文引用: 1]
Long non-coding RNAs (lncRNAs) play important roles in epigenetic regulation of skeletal muscle development. In our previous RNA-seq study (accession number GSE58755), we found that lncRNA-Six1 is an lncRNA that is differentially expressed between White Recessive Rock (WRR) and Xinghua (XH) chicken. In this study, we have further demonstrated that lncRNA-Six1 is located 432 bp upstream of the gene encoding the protein Six homeobox 1 (Six1). A dual-luciferase reporter assay identified that lncRNA-Six1 overlaps the Six1 proximal promoter. In lncRNA-Six1, a micropeptide of about 7.26 kDa was found to play an important role in the lncRNA-Six1 in cis activity. Overexpression of lncRNA-Six1 promoted the mRNA and protein expression level of the Six1 gene, while knockdown of lncRNA-Six1 inhibited Six1 expression. Moreover, tissue expression profiles showed that both the lncRNA-Six1 and the Six1 mRNA were highly expressed in chicken breast tissue. LncRNA-Six1 overexpression promoted cell proliferation and induced cell division. Conversely, its loss of function inhibited cell proliferation and reduced cell viability. Similar effects were observed after overexpression or knockdown of the Six1 gene. In addition, overexpression or knockdown of Six1 promoted or inhibited, respectively, the expression levels of muscle-growth-related genes, such as MYOG, MYHC, MYOD, IGF1R, and INSR. Taken together, these data demonstrate that lncRNA-Six1 carries out cis-acting regulation of the protein-encoding Six1 gene, and encodes a micropeptide to activate Six1 gene, thus promoting cell proliferation and being involved in muscle growth.
DOI:10.1038/s41598-018-19950-6URLPMID:29362405 [本文引用: 1]
Long non-coding RNAs (lncRNAs) play crucial roles in diverse biological processes and have drawn extensive attention in the past few years. However, lncRNAs remain poorly understood about expression and roles in Crassostrea gigas, a potential model organism for marine molluscan studies. Here, we systematically identified lncRNAs in the mantles of C. gigas from four full-sib families characterized by white, black, golden, and partially pigmented shell. Using poly(A)-independent and strand-specific RNA-seq, a total of 441,205,852 clean reads and 12,243 lncRNA transcripts were obtained. LncRNA transcripts were relatively short with few exons and low levels of expression in comparison to protein coding mRNA transcripts. A total of 427 lncRNAs and 349 mRNAs were identified to differentially express among six pairwise groups, mainly involving in biomineralization and pigmentation through functional enrichment. Furthermore, a total of 6 mRNAs and their cis-acting lncRNAs were predicted to involve in synthesis of melanin, carotenoid, tetrapyrrole, or ommochrome. Of them, chorion peroxidase and its cis-acting lincRNA TCONS_00951105 are implicated in playing an essential role in the melanin synthetic pathway. Our studies provided the first systematic characterization of lncRNAs catalog expressed in oyster mantle, which may facilitate understanding the molecular regulation of shell colour diversity and provide new insights into future selective breeding of C. gigas for aquaculture.
DOI:10.13344/j.microbiol.china.140745URL [本文引用: 1]
【目的】利用Biolog-FF技术对4种不同金霉素降解真菌代谢95种碳源特征进行测定分析。【方法】测定不同时段4种真菌对95种碳源代谢的吸光值,并将4种真菌分别接种到金霉素药渣固废中,测定不同发酵时间药渣中的总有机碳含量。【结果】4种真菌利用碳源种类和活性差异较大,桔青霉LJ318、哈茨木霉LJ245、小刺青霉LJ236、草酸青霉LJ302能够利用碳源数量依次为41、39、15和14种;菌株LJ245和LJ318利用碳源的平均活性显著高于菌株LJ236和LJ302;4种真菌能够较好利用的碳源类型为糖类、氨基酸类、聚合物类等物质。【结论】菌株LJ245和LJ318代谢药渣中的碳源明显快于菌株LJ236和LJ302,这与Biolog方法测定结果趋势一致。Biolog-FF技术是一种快速测定真菌单菌落碳代谢特征的有效方法。研究为探讨真菌碳代谢特征与生物降解环境残留金霉素提供了科学依据。
DOI:10.13344/j.microbiol.china.140745URL [本文引用: 1]
【目的】利用Biolog-FF技术对4种不同金霉素降解真菌代谢95种碳源特征进行测定分析。【方法】测定不同时段4种真菌对95种碳源代谢的吸光值,并将4种真菌分别接种到金霉素药渣固废中,测定不同发酵时间药渣中的总有机碳含量。【结果】4种真菌利用碳源种类和活性差异较大,桔青霉LJ318、哈茨木霉LJ245、小刺青霉LJ236、草酸青霉LJ302能够利用碳源数量依次为41、39、15和14种;菌株LJ245和LJ318利用碳源的平均活性显著高于菌株LJ236和LJ302;4种真菌能够较好利用的碳源类型为糖类、氨基酸类、聚合物类等物质。【结论】菌株LJ245和LJ318代谢药渣中的碳源明显快于菌株LJ236和LJ302,这与Biolog方法测定结果趋势一致。Biolog-FF技术是一种快速测定真菌单菌落碳代谢特征的有效方法。研究为探讨真菌碳代谢特征与生物降解环境残留金霉素提供了科学依据。
DOI:10.1016/j.ymben.2018.05.004URLPMID:29753071 [本文引用: 1]
Evolution, i.e. the change in heritable characteristics of biological populations over successive generations, has created the diversity of life that exists today. In this study we have harnessed evolution to create faster growing mutants of Corynebacterium glutamicum, i.e. to debottleneck growth rate of this highly important industrial workhorse. After approximately 1500 generations of Adaptive Laboratory Evolution (ALE) in defined minimal medium with glucose, we obtained faster growing mutants with specific growth rate as high as 0.64h(-1) as compared with 0.45h(-1) for the wild type, and this 42% improvement is the highest reported for C. glutamicum to date. By genome resequencing and inverse metabolic engineering, we were able to pinpoint two mutations contributing to most of the growth improvement, and these resided in the transcriptional regulators GntR1 (gntR1-E70K) and RamA (ramA-A52V). We confirmed that the two mutations lead to alteration rather than elimination of function, and their introduction in the wild-type background resulted in a specific growth rate of 0.62h(-1). The glycolytic and pentose phosphate pathway fluxes had both increased significantly, and a transcriptomic analyses supported this to be associated with increased capacity. Interestingly, the observed fast growth phenotype was not restricted to glucose but was also observed on fructose, sucrose and xylose, however, the effect of the mutations could only be seen in minimal medium, and not rich BHI medium, where growth was already fast. We also found that the mutations could improve the performance of resting cells, under oxygen-deprived conditions, where an increase in sugar consumption rate of around 30% could be achieved. In conclusion, we have demonstrated that it is feasible to reprogram C. glutamicum into growing faster and thus enhance its industrial potential.
URL [本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.funbio.2017.09.003URLPMID:29801789 [本文引用: 1]
The entomopathogenic fungus Beauveria bassiana is able to grow on insect cuticle hydrocarbons, inducing alkane assimilation pathways and concomitantly increasing virulence against insect hosts. In this study, we describe some physiological and molecular processes implicated in growth, nutritional stress response, and cellular alterations found in alkane-grown fungi. The fungal cytology was investigated using light and transmission electron microscopy while the surface topography was examined using atomic force microscopy. Additionally, the expression pattern of several genes associated with oxidative stress, peroxisome biogenesis, and hydrophobicity were analysed by qPCR. We found a novel type of growth in alkane-cultured B. bassiana similar to mycelial pellets described in other alkane-free fungi, which were able to produce viable conidia and to be pathogenic against larvae of the beetles Tenebrio molitor and Tribolium castaneum. Mycelial pellets were formed by hyphae cumulates with high peroxidase activity, exhibiting peroxisome proliferation and an apparent surface thickening. Alkane-grown conidia appeared to be more hydrophobic and cell surfaces displayed different topography than glucose-grown cells. We also found a significant induction in several genes encoding for peroxins, catalases, superoxide dismutases, and hydrophobins. These results show that both morphological and metabolic changes are triggered in mycelial pellets derived from alkane-grown B. bassiana.
DOI:10.1128/mSphere.00219-19URLPMID:31118302 [本文引用: 1]
The majority of fungal species prefer the 12 degrees to 30 degrees C range, and relatively few species tolerate temperatures higher than 35 degrees C. Our understanding of the mechanisms underpinning the ability of some species to grow at higher temperatures is incomplete. Nosema ceranae is an obligate intracellular fungal parasite that infects honey bees and can cause individual mortality and contribute to colony collapse. Despite a reduced genome, this species is strikingly thermotolerant, growing optimally at the colony temperature of 35 degrees C. In characterizing the heat shock response (HSR) in N. ceranae, we found that this and other microsporidian species have lost the transcriptional regulator HSF and possess a reduced set of putative core HSF1-dependent HSR target genes. Despite these losses, N. ceranae demonstrates robust upregulation of the remaining HSR target genes after heat shock. In addition, thermal stress leads to alterations in genes involved in various metabolic pathways, ribosome biogenesis and translation, and DNA repair. These results provide important insight into the stress responses of microsporidia. Such a new understanding will allow new comparisons with other pathogenic fungi and potentially enable the discovery of novel treatment strategies for microsporidian infections affecting food production and human health.IMPORTANCE We do not fully understand why some fungal species are able to grow at temperatures approaching mammalian body temperature. Nosema ceranae, a microsporidium, is a type of fungal parasite that infects honey bees and grows optimally at the colony temperature of 35 degrees C despite possessing cellular machinery for responding to heat stress that is notably simpler than that of other fungi. We find that N. ceranae demonstrates a robust and broad response to heat shock. These results provide important insight into the stress responses of this type of fungus, allow new comparisons with other pathogenic fungi, and potentially enable the discovery of novel treatment strategies for this type of fungus.
DOI:10.7536/PC121007URL [本文引用: 1]
Biometals play important roles in biological process by different chemical actions. The biomembranes enable different metals to acquire different distribution mode among compartments in a biological system.Metalloproteins or metal chaperones are required to maintain cellular metal ions homeostasis. In cells and intracellular organelles there are many proteins whose metal binding sites consist of more metal ions and are not equivalent in thermodynamics. The functions of metalloproteins are mediated by the binding of metal ions, such as that of concanavalin A, Cu/Zn superoxide dismutase, centrin, and zinc fingers protein. That the binding of metal ions to proteins are investigated is of great significance for bioinorganic chemists to understand the roles of metal ions in mediating the functional changes of metalloproteins.
Contents
1 Introduction
2 Concanavalin A
2.1 Structure
2.2 Functional changes mediated by the binding of metal ions
3 Cu/Zn superoxide dismutase
3.1 Structure
3.2 Biological functions
3.3 Functional changes mediated by the binding of metal ions
4 Centrin
4.1 Biological functions
4.2 Structure
4.3 Functional changes mediated by the binding of metal ions
5 Zinc fingers protein
5.1 Structures of zinc fingers
5.2 The binding of metal ions
5.3 Regulation of biological functions by zinc ion
6 Conclusion and outlook
DOI:10.7536/PC121007URL [本文引用: 1]
Biometals play important roles in biological process by different chemical actions. The biomembranes enable different metals to acquire different distribution mode among compartments in a biological system.Metalloproteins or metal chaperones are required to maintain cellular metal ions homeostasis. In cells and intracellular organelles there are many proteins whose metal binding sites consist of more metal ions and are not equivalent in thermodynamics. The functions of metalloproteins are mediated by the binding of metal ions, such as that of concanavalin A, Cu/Zn superoxide dismutase, centrin, and zinc fingers protein. That the binding of metal ions to proteins are investigated is of great significance for bioinorganic chemists to understand the roles of metal ions in mediating the functional changes of metalloproteins.
Contents
1 Introduction
2 Concanavalin A
2.1 Structure
2.2 Functional changes mediated by the binding of metal ions
3 Cu/Zn superoxide dismutase
3.1 Structure
3.2 Biological functions
3.3 Functional changes mediated by the binding of metal ions
4 Centrin
4.1 Biological functions
4.2 Structure
4.3 Functional changes mediated by the binding of metal ions
5 Zinc fingers protein
5.1 Structures of zinc fingers
5.2 The binding of metal ions
5.3 Regulation of biological functions by zinc ion
6 Conclusion and outlook
DOI:10.1371/journal.pgen.1005692URLPMID:26588844 [本文引用: 1]
In the fungal pathogen Cryptococcus neoformans, the switch from yeast to hypha is an important morphological process preceding the meiotic events during sexual development. Morphotype is also known to be associated with cryptococcal virulence potential. Previous studies identified the regulator Znf2 as a key decision maker for hypha formation and as an anti-virulence factor. By a forward genetic screen, we discovered that a long non-coding RNA (lncRNA) RZE1 functions upstream of ZNF2 in regulating yeast-to-hypha transition. We demonstrate that RZE1 functions primarily in cis and less effectively in trans. Interestingly, RZE1's function is restricted to its native nucleus. Accordingly, RZE1 does not appear to directly affect Znf2 translation or the subcellular localization of Znf2 protein. Transcriptome analysis indicates that the loss of RZE1 reduces the transcript level of ZNF2 and Znf2's prominent downstream targets. In addition, microscopic examination using single molecule fluorescent in situ hybridization (smFISH) indicates that the loss of RZE1 increases the ratio of ZNF2 transcripts in the nucleus versus those in the cytoplasm. Taken together, this lncRNA controls Cryptococcus yeast-to-hypha transition through regulating the key morphogenesis regulator Znf2. This is the first functional characterization of a lncRNA in a human fungal pathogen. Given the potential large number of lncRNAs in the genomes of Cryptococcus and other fungal pathogens, the findings implicate lncRNAs as an additional layer of genetic regulation during fungal development that may well contribute to the complexity in these
[本文引用: 1]
URL [本文引用: 1]
DOI:10.1016/j.cell.2011.07.014URLPMID:21802130 [本文引用: 1]
Here, we present a unifying hypothesis about how messenger RNAs, transcribed pseudogenes, and long noncoding RNAs
DOI:10.3390/insects10080245URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}