 ,, 祝智威,, 蒋海宾, 王杰, 范元婵, 范小雪, 万洁琦, 卢家轩, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿,福建农林大学动物科学学院(蜂学学院),福州 350002
,, 祝智威,, 蒋海宾, 王杰, 范元婵, 范小雪, 万洁琦, 卢家轩, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿,福建农林大学动物科学学院(蜂学学院),福州 350002Comparative Analysis of MicroRNAs and Corresponding Target mRNAs in Ascosphaera apis Mycelium and Spore
CHEN HuaZhi,, ZHU ZhiWei,, JIANG HaiBin, WANG Jie, FAN YuanChan, FAN XiaoXue, WAN JieQi, LU JiaXuan, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, CHEN DaFu, GUO Rui,College of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002通讯作者:
责任编辑: 岳梅
收稿日期:2019-12-25接受日期:2020-02-4网络出版日期:2020-09-01
| 基金资助: |
Received:2019-12-25Accepted:2020-02-4Online:2020-09-01
作者简介 About authors
陈华枝,E-mail:
祝智威,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (2720KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
陈华枝, 祝智威, 蒋海宾, 王杰, 范元婵, 范小雪, 万洁琦, 卢家轩, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿. 蜜蜂球囊菌菌丝和孢子中微小RNA及其靶mRNA的比较分析[J]. 中国农业科学, 2020, 53(17): 3606-3619 doi:10.3864/j.issn.0578-1752.2020.17.017
CHEN HuaZhi, ZHU ZhiWei, JIANG HaiBin, WANG Jie, FAN YuanChan, FAN XiaoXue, WAN JieQi, LU JiaXuan, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, CHEN DaFu, GUO Rui.
0 引言
【研究意义】蜜蜂球囊菌(Ascosphaera apis,球囊菌)是一种特异性侵染蜜蜂幼虫的致死性真菌病原,能够使蜜蜂幼虫罹患白垩病,可导致成年蜜蜂数量和群势的急剧下降[1]。近期的研究表明,球囊菌对中华蜜蜂(Apis cerana cerana,中蜂)幼虫和成虫、成年熊蜂也具有侵染性[2]。食物连同球囊菌孢子被蜜蜂幼虫摄入后进入中肠,幼虫期中肠与后肠之间存在一层隔膜,至预蛹期隔膜消失,孢子随食物残渣涌入后肠,在O2的刺激下孢子剧烈萌发、菌丝大量生长,先穿透肠壁再从尾部穿透体壁,进而包裹整个虫尸[3]。前人在球囊菌的分类鉴定[4,5]、增殖方式[6]和病理学[7,8]等方面开展了较多研究,但其分子生物学及组学研究较少。球囊菌微小RNA(microRNA,miRNA)等非编码RNA的相关研究尤为滞后。利用small RNA-seq(sRNA-seq)技术和生物信息学方法对球囊菌菌丝和孢子分别进行深度测序和组学分析,明确二者miRNA的数量和结构特征差异,揭示miRNA与菌丝生长、孢子萌发、杂交产孢、毒力因子合成与分泌等生物学过程的关系,可为明晰相关分子机理提供参考信息和数据基础。【前人研究进展】MiRNA是一类长度约为23 nt的细胞内源性小RNA分子,在真核生物中广泛存在且高度保守[9],可通过切割靶mRNA或阻止蛋白质的翻译过程来调控基因表达[10],从而参与调控生长发育及信号转导等相关进程[11]。自从在秀丽隐杆线虫(Caenorhabditis elegans)中发现第一个miRNA以来[12],人们已在动物[13]、植物[14]和微生物中[15,16]陆续发现了大量miRNA,但真菌miRNA的相关研究总体仍较为滞后。前期研究中,笔者团队已在mRNA组学水平对球囊菌侵染意大利蜜蜂(Apis mellifera ligustica,意蜂)幼虫和中蜂幼虫过程的宿主与病原相互作用进行了系统探究[17,18,19,20,21],组装并注释了球囊菌的参考转录组[21];基于球囊菌菌丝和孢子混合样品的高质量组学数据鉴定出379个长链非编码RNA和551个环状RNA,并通过分子生物学手段验证了它们的真实表达[22,23]。【本研究切入点】笔者团队前期基于球囊菌菌丝和孢子混合样品的sRNA组学数据鉴定出118个miRNA,并预测和分析了miRNA的靶mRNA及调控网络[24]。然而,球囊菌菌丝和孢子miRNA的数量、结构特征、表达谱差异仍未明确,miRNA与菌丝和孢子生长、发育、生殖和致病性的关系还不清楚。【拟解决的关键问题】明确球囊菌菌丝和孢子miRNA的数量、结构特征和表达谱差异,揭示共有miRNA、特有miRNA和差异表达miRNA(differentially expressed miRNA,DEmiRNA)与菌丝和孢子生长、发育、生殖及致病性的潜在关系。1 材料与方法
试验于2019年在福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室完成。1.1 供试真菌
球囊菌菌株由福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室分离和保存[22,23,24]。1.2 球囊菌培养及测序样品制备
参照笔者团队前期已建立的方法[19-20,22-23],将实验室保存的球囊菌孢子接种于10块马铃薯葡萄糖琼脂(potato dextrose agar,PDA)固体培养基上,置于33℃生化培养箱中培养,培养7 d的PDA固体培养基上层为蓬松的白色菌丝,覆盖着下层的黑色孢子囊,为避免菌丝和孢子囊之间的交叉污染,首先在超净台中用干净的接种环刮取最上层的白色菌丝,避免接触与孢子囊接触的菌丝,将刮取的菌丝(AaM)集中于一个RNA Free的EP管,经液氮速冻后迅速移至-80℃超低温冰箱保存备用;在超净台中通过无菌操作将覆盖在孢子囊上的薄层菌丝小心刮去,再用另一个干净的接种环将孢子囊刮至一个RNA Free的EP管;继而按照JENSEN等[25]的方法进行差速离心,球囊菌孢子密度较大,沉淀在EP管底部,弃去上清;重复上述差速离心3次,弃去上清,保留孢子沉淀;取少量孢子制备悬液,取少量悬液进行显微制片及观察,视野中可见较多的游离孢子,未见菌丝,因而得到的孢子沉淀即为球囊菌的纯净孢子(AaS);将上述EP管投入液氮速冻,然后迅速移至-80℃超低温冰箱保存备用。1.3 cDNA文库构建及深度测序
(1)用Trizol法分别提取AaM和AaS的总RNA,琼脂糖凝胶电泳切胶选择18—30 nt的片段,分别连接3′接头和5′接头;(2)对连接了两侧接头的small RNA进行反转录和PCR扩增;(3)琼脂糖凝胶电泳回收并纯化约140 bp的条带,完成文库构建。委托广州基迪奥生物科技有限公司对上述样品进行单端测序,测序平台为Illumina MiSeq。测序数据已上传到NCBI SRA数据库,BioProject号:PRJNA560456。1.4 测序数据质控
按照前期已建立的方法[24,26-27]对下机的原始读段(raw reads)进行质量控制:(1)过滤掉质量值低于20的碱基数超过1个的reads;(2)过滤除掉含有未知碱基(N)的reads;(3)过滤3′或5′接头的reads,并去除长度<18 bp的reads;(4)过滤包含poly A的reads。过滤后得到的clean reads用于后续分析。1.5 菌丝和孢子miRNA的预测与Venn分析
将严格质控后得到的small RNA有效标签序列(clean tags)分别比对到GenBank数据库(http://www. ncbi.nlm.nih.gov/Web/Genbank/)、Rfam数据库(http://rfam.xfam.org/)、核糖体数据库(http://oberon. fvms.ugent.be:8080/rRNA/lsu/index.html)和球囊菌参考基因组(assembly AAP 1.0,www.ncbi.nlm.nih.gov/ genome/656?genome_assembly_id=274809),以去除可能的rRNA、scRNA、snoRNA、snRNA和tRNA,以及比对上的基因组外显子区域、内含子区域和重复序列区域的clean tags,将未比对上的clean tags与miRBase数据库(www.mirbase.org/)中的miRNA前体序列进行比对,从而鉴定已知miRNA序列。利用Mireap_V 0.2软件将剩余的未比对上的clean tags比对球囊菌参考基因组(assembly AAP 1.0),得到可能的前体序列,根据clean tags在前体序列的分布信息和前体结构能量信息,采用贝叶斯模型打分预测新miRNA。按照TPM=T×106/N(T表示miRNA的tags,N表示总miRNA的tags)算法对AaM和AaS中每个miRNA的表达量进行归一化处理。利用基迪奥在线工具集合(www.omicshare.com)对AaM和AaS的miRNA进行Venn分析,采用默认参数,分别筛选出AaM和AaS的共有miRNA及特有miRNA。1.6 共有与特有miRNA的靶mRNA的GO和KEGG数据库注释分析
采用RNA hybrid(v2.1.2)+svm_light(v6.01)、Miranda(v3.3a)和TargetScan(Version 7.0)软件[28,29,30,31]分别预测共有miRNA和特有miRNA的靶mRNA,将预测结果的交集作为可信度高的靶标集合。利用BLAST工具将靶mRNA比对到GO数据库(http:// geneontology.org/)和KEGG数据库(https://www. kegg.jp/),获得相应的功能和通路注释信息,并统计比对上各功能条目或通路(pathway)的靶mRNA数量。1.7 DEmiRNA的筛选和靶mRNA的预测及分析
以|log2 fold change (FC)|≥1且P≤0.05为标准,利用edgeR软件[32]筛选AaM vs AaS的DEmiRNA。前期已利用链特异性建库的RNA-seq技术对球囊菌菌丝和孢子分别进行测序,其中高质量的mRNA组学数据可作为本研究中靶mRNA的数据来源(未发表数据)。分别采用RNA hybrid+svm_light、Miranda和TargetScan软件[28,29,30,31]预测DEmiRNA的靶mRNA,将预测结果的交集作为可信度高的靶标集合。利用BLAST工具将DEmiRNA的靶mRNA比对到GO和KEGG数据库,获得相应的功能和通路注释信息,并统计比对上的功能条目和通路的靶mRNA数量。根据上述DEmiRNA与mRNA的靶向结合关系,按照自由能<-40 kcal·mol-1和P<0.05的阈值筛选靶向结合关系并构建调控网络,通过Cytoscape软件进行调控网络的可视化。1.8 DEmiRNA的Stem-loop RT-qPCR验证
随机选取5个DEmiRNA(miR5658-x、miR-10285- y、miR-3245-y、miR4404-x、miR-9-z)进行Stem-loop RT-qPCR验证。参照CHEN等[33]和郭睿等[34]的方法,利用DNAMAN软件(Lynnon Biosoft公司,美国)设计上述DEmiRNA的Stem-loop引物、特异性上游引物和通用下游引物。委托上海生工生物工程股份有限公司合成引物(表1)。利用总RNA抽提试剂盒(Promega公司,中国)分别抽提菌丝和孢子的总RNA,然后用RNase-free DNase I去除各自基因组DNA残留。吸取1 μL RNA,用NanoDrop One(Thermo Scientific公司,美国)分别对菌丝、孢子的RNA浓度进行测定。利用Stem-loop引物,按照cDNA第1链合成试剂盒(TaKaRa公司,日本)说明书进行总RNA的反转录,得到的cDNA作为模板进行qPCR。选用球囊菌的actin(5417)作为内参基因。反应体系为20 μL:SYBR Green Dye 10 μL,特异性上游引物1 μL,通用下游引物1 μL,cDNA模板1 μL,RNA-Free H2O 7 μL。每个反应进行3次技术重复。采用2-ΔΔCt法计算DEmiRNA的相对表达量,然后利用GraphPad Prism 7软件进行Student’s t-test检验及绘图。Table 1
表1
表1本研究使用的引物
Table 1
| 引物名称 Primer name | 引物序列Primer sequence (5′-3′) |
|---|---|
| Loop-miR5658-x | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCATCATC |
| Loop-miR-10285-y | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACAATTGG |
| Loop-miR-3245-y | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCCCCGGAC |
| Loop-miR4404-x | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGCCAGTCT |
| Loop-miR-9-z | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCATACAG |
| miR5658-x-F | ACACTCCAGCTGGGCGATGATGAT |
| miR-10285-y-F | ACACTCCAGCTGGGTCATTTGTGGT |
| miR-3245-y-F | ACACTCCAGCTGGGCTTGGGAGAG |
| miR4404-x-F | ACACTCCAGCTGGGAATACGTAGA |
| miR-9-z-F | ACACTCCAGCTGGGTCTTTGGTTATCTAG |
| Universal-R | CTCAACTGGTGTCGTGGA |
| actin-F | CAGGAAAGGCTATGTTCGC |
| actin-R | ATTACCGAGGAGCAAGACG |
新窗口打开|下载CSV
2 结果
2.1 测序数据的质控与评估
AaM和AaS分别测得12 982 320和12 708 832条raw reads,经严格过滤和质控后分别得到10 800 101和9 888 848条clean tags,占raw reads的比例分别为84.34%和78.83%;此外,比对上参考基因组的clean tags比例分别为80.59%和71.60%(表2)。上述结果表明本研究的sRNA-seq数据质量良好,可满足后续分析。Table 2
表2
表2sRNA-seq数据概览
Table 2
| 样品 Sample | 原始读段 Raw reads | 有效标签序列 Clean tags | 比对上参考基因组的clean tags Clean tags mapped to reference genome |
|---|---|---|---|
| AaM | 12982320 | 10800101 (84.34%) | 8703440 (80.59%) |
| AaS | 12708832 | 9888848 (78.83%) | 7080369 (71.60%) |
新窗口打开|下载CSV
2.2 球囊菌菌丝和孢子miRNA的预测及比较分析
基于AaM和AaS的高质量数据分别预测出193和275个miRNA。其中,AaM中miRNA的长度介于18—26 nt(图1-A),AaS中miRNA的长度介于18—24 nt(图1-C);AaM和AaS中首位碱基偏向于U的miRNA数量最多,占比分别达到30.77%和45.73%(图1-B、1-D)。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1AaM和AaS中miRNA的结构特征比较
A:AaM中miRNA的长度分布 Length distribution of miRNAs in AaM;B:AaM中miRNA的首位碱基偏向性 First base bias of miRNAs in AaM;C:AaS中miRNA的长度分布 Length distribution of miRNAs in AaS;D:AaS中miRNA的首位碱基偏向性 First base bias of miRNAs in AaS
Fig. 1Comparison of structural properties of miRNAs between AaM and AaS
AaM中表达量最高的是miR6478-x、miR10516-x和miR482-x,TPM值分别达到294 585.5787、200 923.7875和99 563.767;AaS中表达量最高的同样为miR6478-x、miR482-x和miR10516-x,TPM值分别为209 814.8692、71 995.2983和71 407.5815。AaM和AaS中表达量最高的前10位miRNA的详细信息如表3和表4所示。
Table 3
表3
表3AaM中表达量最高的前10位miRNA
Table 3
| miRNA ID | miRNA序列 miRNA sequence | TPM值 TPM value |
|---|---|---|
| miR6478-x | GCGACTTTAGCTCAGTTGG | 294585.5787 |
| miR10516-x | GATCCTCTGCAGACGACTGA | 200923.7875 |
| miR482-x | TTGGAGTGGGTGGGTTGGGAA | 99563.767 |
| miR-8440-y | GTTCGTTTCTGGGTCAGG | 84423.9158 |
| miR5658-x | CGATGATGATGATGATGA | 30792.9176 |
| miR-11987-x | AGGAAACTCTGGTGGAGGT | 27713.6259 |
| miR-10285-y | TCATTTGTGGTCCAATTGT | 11547.3441 |
| miR-3245-y | CTTGGGAGAGGTCCGGGG | 10264.3059 |
| miR-1332-y | CAGTTGGTTAGAGCTGGT | 9494.4829 |
新窗口打开|下载CSV
Table 4
表4
表4AaS中表达量最高的前10位miRNA
Table 4
| miRNA ID | miRNA序列 miRNA sequence | TPM值 TPM value |
|---|---|---|
| miR6478-x | GCGACTTTAGCTCAGTTGG | 209814.8692 |
| miR482-x | TTGGAGTGGGTGGGTTGGGAA | 71995.2983 |
| miR10516-x | GATCCTCTGCAGACGACTGA | 71407.5815 |
| miR-21-x | TAGCTTATCAGACTGATGTTGA | 32324.4196 |
| miR-8440-y | GTTCGTTTCTGGGTCAGG | 31736.7029 |
| miR-143-y | TGAGATGAAGCACTGTAGCTCT | 29973.5527 |
| miR-11987-x | AGGAAACTCTGGTGGAGGT | 26741.1108 |
| let-7-x | TGAGGTAGTAGGTTGTATAGTT | 19688.5101 |
| novel-m0040-3p | TCTTGAACTGAGAGATGGGGC | 16456.0682 |
| miR159-y | TCTTGGGGTGAAGGGCGG | 15574.4931 |
新窗口打开|下载CSV
2.3 球囊菌菌丝和孢子共有和特有miRNA的筛选及靶mRNA分析
Venn分析结果显示,AaM和AaS的共有miRNA为76个(19.4%),特有miRNA分别为117个(29.8%)和199个(50.8%)。AaM和AaS的共有miRNA可靶向5 946个mRNA,二者的特有miRNA可分别靶向6 141和6 346个mRNA。GO数据库注释结果显示,上述共有miRNA的靶mRNA可注释到42个功能条目,其中细胞组分大类注释靶mRNA数最多的条目是细胞组件(1 027)、细胞(1 027)和细胞器(692),分子功能大类注释靶mRNA数最多的条目是催化活性(1 199)、结合(871)和转运活性(129),生物学进程大类注释靶mRNA数最多的条目是代谢进程(1 303)、细胞进程(1 301)和单一组织进程(975);AaM的特有miRNA的靶mRNA可注释到42个功能条目,其中细胞组分大类注释靶mRNA数最多的条目是细胞组件(1 095)、细胞(1 095)和细胞器(736),分子功能大类注释靶mRNA数最多的条目是催化活性(1 241)、结合(900)和转运活性(128),生物学进程大类注释靶mRNA数最多的条目是代谢进程(1 362)、细胞进程(1 350)和单一组织进程(1 021);AaS的特有miRNA的靶mRNA可注释到42个功能条目,其中细胞组分大类注释靶mRNA数最多的条目是细胞组件(1 121)、细胞(1 121)和细胞器(762),分子功能大类注释靶mRNA数最多的条目是催化活性(1 256)、结合(915)和转运活性(126),生物学进程大类注释靶mRNA数最多的条目是代谢进程(1 383)、细胞进程(1 379)和单一组织进程(1 039)。括号内的数字表示注释到该条目的靶mRNA数。KEGG数据库注释结果显示,上述共有miRNA和二者特有miRNA的靶mRNA均可注释到120条通路,涉及代谢、遗传信息处理、环境信息处理和细胞进程4个大类。其中,共有miRNA的靶mRNA注释数量最多的通路是新陈代谢总览(690)、翻译(264)、碳水化合物代谢(232)、折叠、分类与降解(223)、运输与代谢(186)、传染性疾病(156)、信号转导(138)、能量代谢(133)、细胞生长与死亡(133)和脂质代谢(124);AaM的特有miRNA的靶mRNA注释数量最多的是新陈代谢总览(712)、翻译(283)、碳水化合物代谢(232)、折叠与分类降解(231)、氨基酸代谢(202)、运输与代谢(189)、传染性疾病(164)、信号转导(144)、能量代谢(137)和细胞生长与死亡(134);AaS的特有miRNA的靶mRNA注释数量最多的是新陈代谢总览(725)、翻译(292)、碳水化合物代谢(236)、折叠、分类与降解(236),氨基酸代谢(209)、运输与代谢(194)、传染性疾病(170)、信号转导(148)、细胞生长与死亡(138)和能量代谢(135)。进一步分析发现,AaM的116个特有miRNA靶向的287个mRNA可注释到次级代谢物的生物合成通路;AaS的197个特有miRNA靶向的288个mRNA可注释到次级代谢通路的生物合成通路;44个共有miRNA靶向的14个mRNA可注释到自噬通路;56个共有miRNA靶向的6个mRNA,以及AaM的95个特有miRNA靶向的28个mRNA可注释到MAPK信号通路。括号内的数字表示注释到该通路的靶mRNA数。
2.4 球囊菌菌丝和孢子中miRNA的差异表达分析
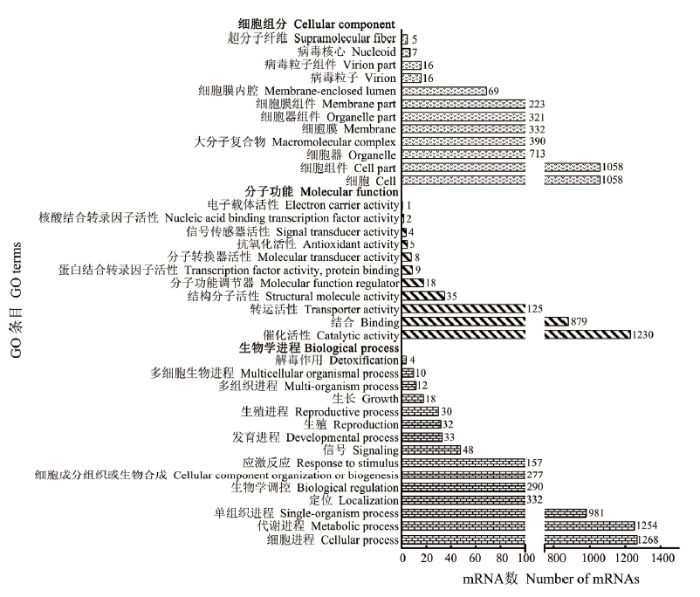
从AaM vs AaS中筛选出93个DEmiRNA,包括65个上调miRNA和28个下调miRNA;其中上调幅度最大的是novel-m0040-3p(log2FC=20.65019,P=5.98E-17),其次是novel-m0016-3p(log2FC= 20.45754,P=3.77E-15)和miR319-y(log2FC=20.23515,P=4.75E-13);下调幅度最大的为miR-4028-y(log2FC= -19.6472765,P=4.77E-10),其次为miR-4171-x(log2FC=-18.2322391,P=0.00049)和miR7787-y(log2FC=-18.1067082,P=0.000979)(表5)。上述DEmiRNA能靶向结合6 090个mRNA。DEmiRNA的靶mRNA可注释到38个功能条目,包括细胞进程(1 268)、代谢进程(1 254)、单一组织进程(981)、定位(332)和生物学调控(290)等15条生物学进程相关条目;催化活性(1 230)、结合(879)、转运活性(125)、结构分子活性(35)及分子功能调节器(18)等11条分子功能相关条目;细胞(1058)、细胞组件(1 058)、细胞器(713)、大分子复合物(390)和细胞膜(332)等12条细胞组分相关条目(图2)。此外,这些靶mRNA可注释到120条通路,注释数量最多的是新陈代谢通路(652)、次级代谢物的生物合成(285)、抗生素的生物合成(212)、微生物在不同环境中的代谢(172)、氨基酸的生物合成(115)、碳代谢(101)、嘌呤代谢(81)、核糖体(78)、剪接体(77)及RNA转运(76)(表6)。括号内的数字代表注释到该条目(通路)的靶mRNA数。
Table 5
表5
表5AaM vs AaS比较组中前10位上调和下调miRNA
Table 5
| 差异表达miRNA ID DEmiRNA ID | AaM的TPM值 TPM value in AaM | AaS的TPM值 TPM value in AaS | Log2差异倍数 Log2FC | P 值 P value |
|---|---|---|---|---|
| 上调miRNA Up-regulated miRNAs | ||||
| novel-m0040-3p | 0.01 | 16456.0682 | 20.65019 | 5.98E-17 |
| novel-m0016-3p | 0.01 | 14399.0597 | 20.45754 | 3.77E-15 |
| miR319-y | 0.01 | 12342.0511 | 20.23515 | 4.75E-13 |
| novel-m0010-3p | 0.01 | 8521.8924 | 19.70081 | 3.80E-09 |
| novel-m0028-3p | 0.01 | 7346.459 | 19.48669 | 6.05E-08 |
| miR-1546-x | 0.01 | 5583.3088 | 19.09076 | 3.85E-06 |
| miR-6882-x | 0.01 | 5289.4505 | 19.01276 | 7.69E-06 |
| novel-m0001-3p | 0.01 | 4995.5921 | 18.9303 | 1.54E-05 |
| novel-m0017-5p | 0.01 | 4701.7338 | 18.84283 | 3.07E-05 |
| miR-992-x | 0.01 | 4701.7338 | 18.84283 | 3.07E-05 |
| 下调miRNA Down-regulated miRNAs | ||||
| miR-4028-y | 8211.4447 | 0.01 | -19.6472765 | 4.77E-10 |
| miR-4171-x | 3079.2918 | 0.01 | -18.2322391 | 0.00049 |
| miR7787-y | 2822.6841 | 0.01 | -18.1067082 | 0.000979 |
| miR-92-x | 2566.0765 | 0.01 | -17.9692047 | 0.0020 |
| novel-m0011-3p | 2309.4688 | 0.01 | -17.8172015 | 0.0040 |
| miR5782-y | 2052.861 | 0.01 | -17.6473 | 0.0078 |
| novel-m0013-5p | 2052.861 | 0.01 | -17.6473 | 0.0078 |
| novel-m0031-3p | 2052.861 | 0.01 | -17.6473 | 0.0078 |
| novel-m0036-5p | 2052.861 | 0.01 | -17.6473 | 0.0078 |
| miR-3533-y | 1796.254 | 0.01 | -17.4546 | 0.0156 |
| novel-m0003-5p | 1796.254 | 0.01 | -17.4546 | 0.0156 |
| novel-m0006-5p | 1796.254 | 0.01 | -17.4546 | 0.0156 |
新窗口打开|下载CSV
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2AaM vs AaS比较组中DEmiRNA的靶mRNA的GO数据库注释
Fig. 2GO database annotation of target mRNAs of DEmiRNAs in AaM vs AaS comparison group
Table 6
表6
表6AaM vs AaS比较组中DEmiRNA的靶mRNA注释数前10位通路
Table 6
| 通路名称 Pathway name | 通路 ID Pathway ID | 靶mRNA数 Number of target mRNAs | P 值 P value |
|---|---|---|---|
| 新陈代谢通路Metabolic pathway | ko01100 | 652 | 0.174 |
| 次级代谢物的生物合成Biosynthesis of secondary metabolites | ko01110 | 285 | 5.63E-05 |
| 抗生素的生物合成Biosynthesis of antibiotics | ko01130 | 212 | 0.002 |
| 微生物在不同环境中的代谢Microbial metabolism in diverse environments | ko01120 | 172 | 0.001 |
| 氨基酸的生物合成Biosynthesis of amino acids | ko01230 | 115 | 0.012 |
| 碳代谢Carbon metabolism | ko01200 | 101 | 0.025 |
| 嘌呤代谢Purine metabolism | ko00230 | 81 | 0.916 |
| 核糖体Ribosome | ko03010 | 78 | 1.000 |
| 剪接体Spliceosome | ko03040 | 77 | 0.861 |
| RNA转运RNA transport | ko03013 | 76 | 0.936 |
新窗口打开|下载CSV
2.5 DEmiRNA的调控网络构建及分析
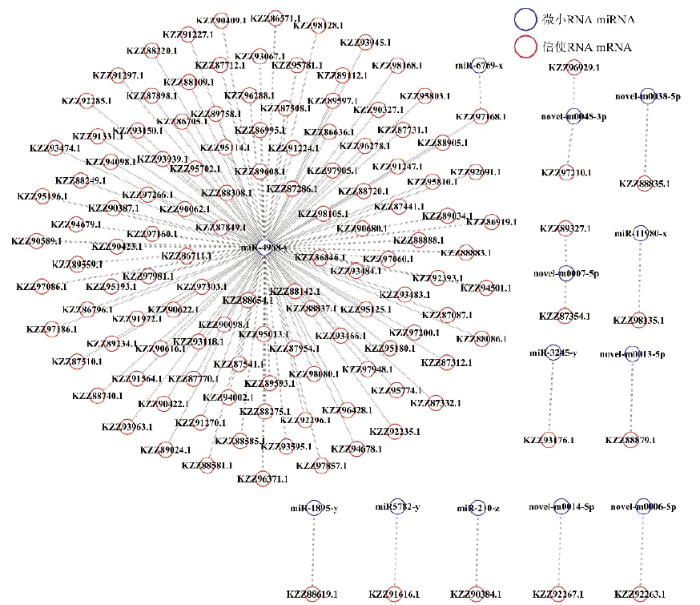
根据靶向结合关系构建调控网络,分析结果显示13个DEmiRNA可靶向结合131个mRNA;同一个DEmiRNA可靶向结合多个mRNA,如miR-4968-y可靶向结合多达118个mRNA;同时,部分mRNA可靶向结合1—2个DEmiRNA,如KZZ97168.1分别靶向结合miR-4968-y和miR-6769-y,KZZ91616靶向结合miR5782-y(图3)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3DEmiRNA-mRNA的调控网络
Fig. 3Regulatory network of DEmiRNA-mRNA
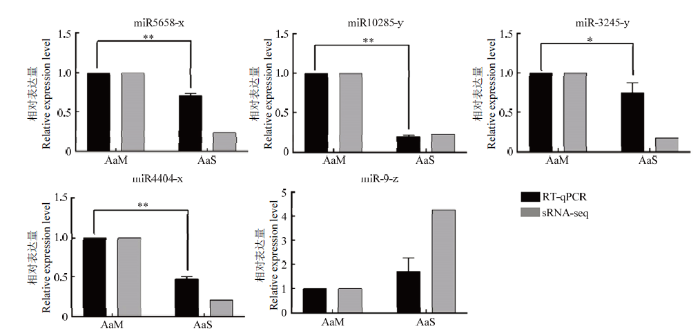
2.6 DEmiRNA的RT-qPCR验证
RT-qPCR结果显示,miR5658-x、miR-10285-y、miR-3245-y、miR4404-x和miR-9-z的表达变化趋势与测序数据相符(图4),证实了本研究中sRNA-seq数据的可靠性。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4DEmiRNA的RT-qPCR验证
RT-qPCR 组中, *表示 P<0.05, **表示 P<0.01 In RT-qPCR group, * indicates P<0.05, ** indicates P<0.01
Fig. 4RT-qPCR verification of DEmiRNAs
3 讨论
MiRNA已被证实能够参与调控真菌的菌丝生长和孢子形成过程[35,36,37,38]。例如,SHAO等通过比较分析筛选出冬虫夏草(Cordyceps militaris)无性生殖阶段和有性生殖阶段的19个DEmiRNA,进而通过过表达和敲除实验证实milR4和milR16参与了菌丝生长过程的调控[36]。前期研究中,为最大限度鉴定miRNA,笔者利用sRNA-seq技术对球囊菌菌丝和孢子的混合样品进行测序,利用miRDeep2软件鉴定到118个novel miRNA,这是关于球囊菌miRNA的首例报道[24]。然而,由于测序得到的混合数据无法区分来源于菌丝的数据和来源于孢子的数据,导致难以进一步对菌丝和孢子中的miRNA进行数量、结构特征、表达谱、靶mRNA及调控网络的比较分析和深入挖掘。因此,本研究首先在实验室条件下获得纯培养的球囊菌,利用sRNA-seq技术对纯净的球囊菌菌丝样品、孢子样品分别进行测序,基于二者的高质量sRNA组学数据分别鉴定到193和275个miRNA,它们的长度分别介于18—26和18—24 nt。分布在18 nt长度的miRNA数量最多,且首位碱基主要偏向U,其结构特征与灰盖鬼伞菌(coprinopsis cinerea)[37]、新月弯孢(Curvularia lunata)[39]和马尔尼菲青霉(Penicillium marneffei)[40]等真菌以及蜜蜂和棉花等动植物[41,42]的miRNA结构高度相似。本研究中,有76个miRNA同时在球囊菌菌丝和孢子中表达,占二者全部miRNA的比例分别为39.38%和27.64%,推测这些共有miRNA在球囊菌的不同生长发育时期均具有一定的调控功能;此外,分别有117和199个miRNA在球囊菌菌丝和孢子中特异性表达,鉴于孢子是球囊菌的休眠态,新陈代谢等生命活动较之菌丝更低,该结果一定程度说明较多的miRNA通过在孢子中特异性表达发挥更强的基因表达抑制(或降解)作用。共有miRNA的靶mRNA可注释到代谢进程、生殖、生殖进程、生长等42个功能条目,以及代谢途径、次级代谢物的生物合成、不同环境下微生物的代谢、氨基酸的生物合成、碳代谢及嘌呤代谢和氧化磷酸化等120条通路,表明共有miRNA在球囊菌菌丝和孢子的生长发育、物质和能量代谢、环境适应等方面具有广泛的调控功能。菌丝的特有miRNA可靶向结合6 141个mRNA,这些靶标可注释到42个功能条目和120条通路;孢子的特有miRNA可靶向结合6 346个mRNA,同样可注释到42个功能条目和120条通路。对于真菌孢子中是否存在转录和翻译等生命活动,相关研究报道很少。笔者团队前期通过分子生物学和组学手段证实另一种广泛存在的蜜蜂真菌病原东方蜜蜂微孢子虫(Nosema ceranae)的孢子中存在基因转录[43]。本研究中,在球囊菌孢子中鉴定到199个特有miRNA,上述结果一定程度表明球囊菌孢子中同样存在基因转录以及miRNA介导的基因表达调控现象。此外,共有93个miRNA在菌丝和孢子中差异表达,其中上调miRNA(65)的数量明显多于下调miRNA(28),进一步说明对于球囊菌孢子,除了具有较多的特异性表达miRNA外,其部分miRNA还能通过上调表达量增强对靶基因的抑制(或降解)作用,从而维持较低的新陈代谢水平。推测这对于球囊菌孢子抵抗外界不良环境及长期存活具有重要意义。
真菌在侵染宿主时会分泌一些次级代谢物促进自身增殖并使宿主致死[44,45]。例如,球孢白僵菌(Beauveria bassiana)和卵孢白僵菌(Beauveria tenella)合成及分泌的草酸、类草酸晶体和柠檬酸等次级代谢物可协同致死宿主[44]。有研究表明真菌在菌丝状态产生的次级代谢物主要与真菌毒素有关[45]。本研究发现,球囊菌菌丝特有的116个miRNA(miR-11971-y、miR-14-x、miR-12227-y等)的287个靶mRNA(KZZ86592.1、KZZ86645.1和KZZ86652.1等)可注释到次级代谢物的生物合成通路,表明相应的菌丝特有miRNA参与调控次级代谢物的生物合成过程,进而影响球囊菌毒素的合成。真菌病原对昆虫寄主的侵染能力取决于蛋白酶、几丁质酶及脂酶等毒力因子的协同作用,以及菌丝对围食膜、肠壁和表皮的机械破坏力[46]。CORNMAN等[46]通过同源性比较发现球囊菌包含61个脂酶基因、51个蛋白酶基因和4个几丁质酶基因。本研究分别有16个(miR-14-x、miR11173-y、miR-1002-x等)和9个(miR-12227-y、miR-4171-x、miR-1002-x等)菌丝特有miRNA靶向几丁质酶合成相关的mRNA(KZZ93915.1和KZZ93066.1),暗示这些miRNA参与调控几丁质酶合成,在球囊菌突破宿主几丁质体表过程中发挥重要调控功能。次级代谢物还能影响真菌的孢子萌发[45,47],例如玉米赤霉烯酮和禾谷镰孢(Fusarium graminearum)的次级代谢物可诱导禾谷镰孢分生孢子和菌落的产生[47]。本研究发现,197个孢子特有miRNA调控的288个靶mRNA(KZZ86592.1、KZZ86645.1和KZZ86652.1等)注释到了次级代谢物的生物合成,笔者推测孢子特有miRNA可能通过调控次级代谢物合成的相关mRNA影响孢子的萌发。李琼等[48]研究发现,mro-miR-33负调控罗伯茨绿僵菌(Metarhizium robertsii)的产孢关键基因BrlA的表达,敲除mro-miR-33后BrlA的表达量明显上调,同时病原的产孢量增加。本研究中,miR-33-x与mro-miR-33高度同源且在孢子中特异性表达,可能参与调控球囊菌的杂交产孢。细胞自噬与丝状真菌的产孢、程序性细胞死亡及致病力紧密相关[49]。在米曲霉(Aspergillus oryzae)中,Aoatg4、Aoatg13及Aoatg15等自噬基因的功能缺失将影响其产孢过程[50]。此外,有研究表明烟曲霉(Aspergillus fumigatus)也依赖自噬来调控孢子的形成[51]。本研究中,菌丝和孢子的44个共有miRNA的14个靶mRNA(KZZ87046.1、KZZ87260.1和KZZ87338.1等)可注释到自噬通路,推测这些共有miRNA可通过负调控相关靶mRNA调节球囊菌的杂交产孢。
丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)级联反应参与真菌的交配繁殖、致病性、孢子形成以及毒力水平的调控[52,53],其依赖的MAPK激酶可协助病原真菌侵染宿主[54]。前期研究发现,对于侵染意蜂幼虫的球囊菌,有48个差异表达基因富集在MAPK信号通路且表达水平随着球囊菌胁迫时间的延长而显著增强[19];但对于侵染中蜂幼虫的球囊菌,富集在MAPK信号通路的11个差异表达基因均表现为下调趋势[20]。本研究中,菌丝和孢子的56个共有miRNA(let-7-x、miR-10285-y和miR-11980-x等)的靶mRNA,以及菌丝的95个特有miRNA(miR-11971-y、miR-12227-y和miR-14-x等)的28个靶mRNA均可注释到MAPK信号通路,表明上述miRNA与MAPK信号通路具有潜在的调控关系。目前,笔者团队已利用sRNA-seq技术对正常及球囊菌侵染的意蜂幼虫肠道、中蜂幼虫肠道进行测序,下一步将过滤得到侵染两种蜜蜂幼虫的球囊菌的sRNA组学数据,并结合本研究中的球囊菌孢子的sRNA组学数据进行比较分析,更深入地探讨球囊菌对不同抗性蜜蜂幼虫的侵染机制。
本研究中,DEmiRNA与mRNA之间存在较为复杂的调控关系,miR-4968-y可同时被118个mRNA同时靶向结合,表明miR-4968-y处于调控网络的核心位置,可能在球囊菌菌丝和孢子的生长和发育过程发挥关键调控功能,值得进一步深入研究。根癌农杆菌介导(AtMT)的真菌遗传转化体系已成功用于球孢白僵菌、金龟子绿僵菌(Metarhizium anisopliae)和蜡蚧轮枝菌(Lecanicillium lecanii)等昆虫病原真菌的基因功能研究[55,56,57]。目前,球囊菌的基因功能研究未见报道。利用AtMT技术对本研究筛选出的菌丝和孢子的关键miRNA进行转基因操作,进而探究其在菌丝和孢子生长发育以及病原致病性方面的功能是下一步的工作重点。
4 结论
分别在球囊菌菌丝和孢子中鉴定出193和275个miRNA,二者中特异性表达的miRNA分别为117和199个;菌丝和孢子中的miRNA具有类似的结构特征,但表达谱表现出明显差异;菌丝和孢子可能通过特异性表达和差异表达部分miRNA对其生长、发育和生殖进行调控。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/j.jip.2009.06.018URL [本文引用: 1]
URLPMID:25885679 [本文引用: 1]
DOI:10.1016/j.cois.2015.04.015URLPMID:29588016 [本文引用: 1]

Chalkbrood is a fungal brood disease of the honey bee, Apis mellifera, caused by the parasite Ascosphaera apis. Considered as a stress-related disease, the severity of chalkbrood outbreaks depend on a multitude of interacting factors. The specific relationship between host and parasite in this disease is interesting because the parasite is both heterothallic and semelparous. Recent studies highlight that this specific host-parasite relationship is influenced by factors such as interactions with other parasite strains or species, and environmental perturbations. To understand how to protect pollinators most effectively, it is crucial that future research takes a more ecologically relevant approach by studying the basic biology of the host-parasite relationship in the context of the multi-factorial processes that influence it.
DOI:10.1002/ajb2.1955.42.issue-6URL [本文引用: 1]
DOI:10.1007/s13225-018-0394-8URL [本文引用: 1]
DOI:10.1007/s002530100721URL [本文引用: 1]
DOI:10.1016/j.vetmic.2005.03.005URL [本文引用: 1]
DOI:10.3852/mycologia.97.6.1171URLPMID:16722211 [本文引用: 1]
Chalkbrood in honeybees (Apis mellifera L. Himenoptera: Apidae) is a fungal disease caused by Ascosphaera apis (Maassen ex Claussen) Olive and Spiltoir. This disease requires the presence of fungal spores and a predisposing condition in the susceptible brood for the disease to develop. In this study we examined the role of pollen in the development of chalkbrood disease under two experimental conditions: (i) pollen combs were transferred from infected to uninfected beehives and (ii) colonies were deprived of adequate pollen supplies to feed the brood. The results of both treatments confirmed that pollen is an element that should be taken into account when controlling this honeybee disease.
DOI:10.1016/j.cell.2009.01.002URLPMID:19167326 [本文引用: 1]
MicroRNAs (miRNAs) are endogenous approximately 23 nt RNAs that play important gene-regulatory roles in animals and plants by pairing to the mRNAs of protein-coding genes to direct their posttranscriptional repression. This review outlines the current understanding of miRNA target recognition in animals and discusses the widespread impact of miRNAs on both the expression and evolution of protein-coding genes.
DOI:10.1007/s10142-014-0359-2URLPMID:24448659 [本文引用: 1]
MicroRNAs (miRNAs) are an extensive class of endogenous posttranscriptional gene regulators that function to mediate gene expression by cleaving target mRNAs or by preventing protein translation. Because of their importance in mediating gene regulation, identifying and elucidating the function of miRNAs have been the primary focus of many researchers. Now that many miRNAs have been identified and assessed for their functionality, the next step is to create expression profiles for miRNAs, so that gene expression studies can be further enhanced with knowledge of the basal expression levels of miRNAs and their targets. In a previous study, 259 putative miRNAs were identified in tobacco, in which 11 of them were confirmed. The primary goal of this study was to further expand on that study and create an expression profile for nine miRNAs and their targets in a tissue-specific manner in tobacco. We chose to study miRNAs that largely play a role in floral development and nutrient stress response. The results of our study show that all tested miRNAs and their targets were expressed in a differential manner. The results of our study also show that out of the tested miRNAs and their targets, miR159, miR157, miR167, miR172, and superoxide dismutase were expressed the highest, suggesting that these genes may play a vital role in the growth and development of tobacco. Corrected expression of miRNAs and their targets regulates floral development.
DOI:10.1038/nature02366URLPMID:14999284 [本文引用: 1]
Gene regulation by RNA interference requires the functions of the PAZ domain protein Argonaute. In plants, mutations in ARGONAUTE1 (AGO1) are associated with distinctive developmental defects that suggest a role for microRNA (miRNA) in organ polarity. Potential targets of miRNA regulation are the homeodomain/leucine zipper genes PHABULOSA (PHB) and PHAVOLUTA (PHV). These genes are expressed in a polar fashion in leaf primordia and are required for adaxial cell fate. Here we show that a 21-nucleotide miRNA that directs cleavage of PHB/PHV messenger RNA accumulates first in the embryonic meristem, and then in the abaxial domain of the developing leaf. miRNA distribution is disrupted by mutations in AGO1, indicating that AGO1 affects the regulation of miRNA. In addition, interactions between homeodomain/leucine zipper genes and an allelic series of ago1 indicate that miRNA acts as a signal to specify leaf polarity.
DOI:10.1016/0092-8674(93)90529-yURLPMID:8252621 [本文引用: 1]
lin-4 is essential for the normal temporal control of diverse postembryonic developmental events in C. elegans. lin-4 acts by negatively regulating the level of LIN-14 protein, creating a temporal decrease in LIN-14 protein starting in the first larval stage (L1). We have cloned the C. elegans lin-4 locus by chromosomal walking and transformation rescue. We used the C. elegans clone to isolate the gene from three other Caenorhabditis species; all four Caenorhabditis clones functionally rescue the lin-4 null allele of C. elegans. Comparison of the lin-4 genomic sequence from these four species and site-directed mutagenesis of potential open reading frames indicated that lin-4 does not encode a protein. Two small lin-4 transcripts of approximately 22 and 61 nt were identified in C. elegans and found to contain sequences complementary to a repeated sequence element in the 3' untranslated region (UTR) of lin-14 mRNA, suggesting that lin-4 regulates lin-14 translation via an antisense RNA-RNA interaction.
DOI:10.1038/nature07415URLPMID:18830242 [本文引用: 1]
In bilaterian animals, such as humans, flies and worms, hundreds of microRNAs (miRNAs), some conserved throughout bilaterian evolution, collectively regulate a substantial fraction of the transcriptome. In addition to miRNAs, other bilaterian small RNAs, known as Piwi-interacting RNAs (piRNAs), protect the genome from transposons. Here we identify small RNAs from animal phyla that diverged before the emergence of the Bilateria. The cnidarian Nematostella vectensis (starlet sea anemone), a close relative to the Bilateria, possesses an extensive repertoire of miRNA genes, two classes of piRNAs and a complement of proteins specific to small-RNA biology comparable to that of humans. The poriferan Amphimedon queenslandica (sponge), one of the simplest animals and a distant relative of the Bilateria, also possesses miRNAs, both classes of piRNAs and a full complement of the small-RNA machinery. Animal miRNA evolution seems to have been relatively dynamic, with precursor sizes and mature miRNA sequences differing greatly between poriferans, cnidarians and bilaterians. Nonetheless, miRNAs and piRNAs have been available as classes of riboregulators to shape gene expression throughout the evolution and radiation of animal phyla.
URLPMID:12119378 [本文引用: 1]
DOI:10.1038/nature05903URLPMID:17538623 [本文引用: 1]
MicroRNAs (miRNAs) in eukaryotes guide post-transcriptional regulation by means of targeted RNA degradation and translational arrest. They are released by a Dicer nuclease as a 21-24-nucleotide RNA duplex from a precursor in which an imperfectly matched inverted repeat forms a partly double-stranded region. One of the two strands is then recruited by an Argonaute nuclease that is the effector protein of the silencing mechanism. Short interfering RNAs (siRNAs), which are similar to miRNAs, are also produced by Dicer but the precursors are perfectly double-stranded RNA. These siRNAs guide post-transcriptional regulation, as with miRNAs, and epigenetic genome modification. Diverse eukaryotes including fungi, plants, protozoans and metazoans produce siRNAs but, until now, miRNAs have not been described in unicellular organisms and it has been suggested that they evolved together with multicellularity in separate plant and animal lineages. Here we show that the unicellular alga Chlamydomonas reinhardtii contains miRNAs, putative evolutionary precursors of miRNAs and species of siRNAs resembling those in higher plants. The common features of miRNAs and siRNAs in an alga and in higher plants indicate that complex RNA-silencing systems evolved before multicellularity and were a feature of primitive eukaryotic cells.
DOI:10.1101/gad.1543507URLPMID:17470535 [本文引用: 1]
Endogenous small RNAs function in RNA interference (RNAi) pathways to control gene expression through mRNA cleavage, translational repression, or chromatin modification. Plants and animals contain many microRNAs (miRNAs) that play vital roles in development, including helping to specify cell type and tissue identity. To date, no miRNAs have been reported in unicellular organisms. Here we show that Chlamydomonas reinhardtii, a unicellular green alga, encodes many miRNAs. We also show that a Chlamydomonas miRNA can direct the cleavage of its target mRNA in vivo and in vitro. We further show that the expression of some miRNAs/Candidates increases or decreases during Chlamydomonas gametogenesis. In addition to miRNAs, Chlamydomonas harbors other types of small RNAs including phased small interfering RNAs (siRNAs) that are reminiscent of plant trans-acting siRNAs, as well as siRNAs originating from protein-coding genes and transposons. Our findings suggest that the miRNA pathway and some siRNA pathways are ancient mechanisms of gene regulation that evolved prior to the emergence of multicellularity.
DOI:10.1016/j.gene.2017.04.022URLPMID:28427951 [本文引用: 1]
Honeybees are susceptible to a variety of diseases, including chalkbrood, which is capable of causing huge losses of both the number of bees and colony productivity. This research is designed to characterize the transcriptome profiles of Ascosphaera apis-treated and un-treated larval guts of Apis mellifera ligustica in an attempt to unravel the molecular mechanism underlying the immune responses of western honeybee larval guts to mycosis. In this study, 24, 296 and 2157 genes were observed to be differentially expressed in A. apis-treated Apis mellifera (4-, 5- and 6-day-old) compared with un-treated larval guts. Moreover, the expression patterns of differentially expressed genes (DEGs) were examined via trend analysis, and subsequently, gene ontology analysis and KEGG pathway enrichment analysis were conducted for DEGs involved in up- and down-regulated profiles. Immunity-related pathways were selected for further analysis, and our results demonstrated that a total of 13 and 50 DEGs were annotated in the humoral immune-related and cellular immune-related pathways, respectively. Additionally, we observed that many DEGs up-regulated in treated guts were part of cellular immune pathways, such as the lysosome, ubiquitin mediated proteolysis, and insect hormone biosynthesis pathways and were induced by A. apis invasion. However, more down-regulated DEGs were restrained. Surprisingly, a majority of DEGs within the Toll-like receptor signaling pathway, and the MAPK signaling pathway were up-regulated in treated guts, while all but two genes involved in the NF-kappaB signaling pathway were down-regulated, which suggested that most genes involved in humoral immune-related pathways were activated in response to the invasive fungal pathogen. This study's findings provide valuable information regarding the investigation of the molecular mechanism of immunity defenses of A. m. ligustica larval guts to infection with A. apis. Furthermore, these studies lay the groundwork for future researches on key genes controlling the susceptibility of A. m. ligustica larvae to chalkbrood.
DOI:10.1016/j.jip.2019.107210URLPMID:31211962 [本文引用: 1]
Chalkbrood is the most common fungal disease in honeybees. The objective of this study was to reveal immune responses in the Apis cerana cerana larval gut following Ascosphaera apis invasion. Combining a previously assembled transcriptome of A. c. cerana larval gut and the high-throughput sequencing data obtained in this study, 6152 differentially expressed genes (DEGs) were clustered into eight profiles. Trend analysis showed three significant up-regulated profiles (p
[本文引用: 3]
[本文引用: 3]
[本文引用: 3]
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
DOI:10.1016/j.jip.2018.06.001URLPMID:29894727 [本文引用: 3]
Ascospheara apis is a widespread fungal pathogen that exclusively invades honeybee larvae. Thus far, non-coding RNA in A. apis has not yet been documented. In this study, we sequenced A. apis using strand specific cDNA library construction and Illumina RNA sequencing methods, and identified 379 lncRNAs, including antisense lncRNAs, lincRNAs, intronic lncRNAs and sense lncRNAs. Additionally, these lncRNAs were found to be shorter in length and have fewer exons and transcript isoforms than protein-coding genes, similar to those identified in mammals and plants. Furthermore, the existence of 15 predicted lncRNAs of A. apis was confirmed using RT-PCR and expression levels of 11 were lower than those of adjacent protein-coding genes. Our findings not only enlarge the lncRNA database for fungi, but also lay a foundation for further investigation of potential lncRNA-mediated regulation of genes in A. apis.
DOI:10.1016/j.gene.2018.07.076URLPMID:30077766 [本文引用: 3]
Ascosphaera apis is a widespread fungal pathogen of honeybee larvae, which causes heavy losses in apiculture. To date, knowledge about non-coding RNA (ncRNA) including circular RNA (circRNA) in A. apis is lacking. In this study, A. apis mycelia and spores were sequenced using RNA-seq technology. A total of 551 circRNAs were predicted on the basis of bioinformatic analyses, and most of the circRNAs were 200-600bp in length, which were different from animal and plant circRNAs. In addition, the expression of six circRNAs in A. apis were confirmed using divergent and convergent PCR. Moreover, circRNA-microRNA regulation networks in A. apis were constructed, and further investigation showed that A. apis circRNAs could regulate gene expression by competitively binding miRNAs. GO and KEGG pathway enrichment analyses of the miRNAs target genes of circRNAs demonstrated that these A. apis circRNAs are likely to play key roles in metabolism, environmental response and gene expression.
DOI:10.13343/j.cnki.wsxb.20170535URL [本文引用: 4]
[Objective] In this study, pure culture of A. apis was sequenced using sRNA-seq technology, followed by prediction, identification and analysis of A. apis microRNAs. The microRNAs-mRNAs regulation network was further constructed. [Methods] Illumina Hiseq Xten platform was used to sequence mycelium and spores of A. apis, and corresponding softwares were used to predict and analyze A. apis microRNAs, some of which were identified via Stem-loop PCR. Cytoskype software was used to construct A. apis microRNAs-mRNAs regulation network. [Results] A total of 48268696 clean reads were obtained, and 118 miRNAs of A. apis were predicted, whose length was distributed between 18 nt and 25 nt. The preference of the first base of miRNAs with different length was obviously various. Stem-loop PCR result showed target fragments with expected sizes were amplified from 10 microRNAs, implying most of the predicted microRNAs' true existence. In total, 6529 target genes of A. apis microRNAs were predicted, and among them 5725 could be annotated in Nr, Swissprot, KOG, GO and KEGG databases. Further investigation demonstrated 24 target genes were annotated in MAPK signaling pathway. Cytoskype software analysis suggested complicated regulation networks exist between microRNAs and mRNAs in A. apis, and majority of miRNAs inside the networks bind to several mRNAs. [Conclusion] Our findings enrich the understanding of A. apis microRNAs, provide beneficial supplement for basic biology information of A. apis, and lay some foundation for illustrating the molecular mechanism regulating the pathogenesis of this widespread fungal pathogen.
DOI:10.13343/j.cnki.wsxb.20170535URL [本文引用: 4]
[Objective] In this study, pure culture of A. apis was sequenced using sRNA-seq technology, followed by prediction, identification and analysis of A. apis microRNAs. The microRNAs-mRNAs regulation network was further constructed. [Methods] Illumina Hiseq Xten platform was used to sequence mycelium and spores of A. apis, and corresponding softwares were used to predict and analyze A. apis microRNAs, some of which were identified via Stem-loop PCR. Cytoskype software was used to construct A. apis microRNAs-mRNAs regulation network. [Results] A total of 48268696 clean reads were obtained, and 118 miRNAs of A. apis were predicted, whose length was distributed between 18 nt and 25 nt. The preference of the first base of miRNAs with different length was obviously various. Stem-loop PCR result showed target fragments with expected sizes were amplified from 10 microRNAs, implying most of the predicted microRNAs' true existence. In total, 6529 target genes of A. apis microRNAs were predicted, and among them 5725 could be annotated in Nr, Swissprot, KOG, GO and KEGG databases. Further investigation demonstrated 24 target genes were annotated in MAPK signaling pathway. Cytoskype software analysis suggested complicated regulation networks exist between microRNAs and mRNAs in A. apis, and majority of miRNAs inside the networks bind to several mRNAs. [Conclusion] Our findings enrich the understanding of A. apis microRNAs, provide beneficial supplement for basic biology information of A. apis, and lay some foundation for illustrating the molecular mechanism regulating the pathogenesis of this widespread fungal pathogen.
DOI:10.3896/IBRA.1.52.1.13URLPMID:24198438 [本文引用: 1]
Chalkbrood and stonebrood are two fungal diseases associated with honey bee brood. Chalkbrood, caused by Ascosphaera apis, is a common and widespread disease that can result in severe reduction of emerging worker bees and thus overall colony productivity. Stonebrood is caused by Aspergillus spp. that are rarely observed, so the impact on colony health is not very well understood. A major concern with the presence of Aspergillus in honey bees is the production of airborne conidia, which can lead to allergic bronchopulmonary aspergillosis, pulmonary aspergilloma, or even invasive aspergillosis in lung tissues upon inhalation by humans. In the current chapter we describe the honey bee disease symptoms of these fungal pathogens. In addition, we provide research methodologies and protocols for isolating and culturing, in vivo and in vitro assays that are commonly used to study these host pathogen interactions. We give guidelines on the preferred methods used in current research and the application of molecular techniques. We have added photographs, drawings and illustrations to assist bee-extension personnel and bee scientists in the control of these two diseases.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/978-1-4939-6539-7_13URLPMID:27807838 [本文引用: 2]
microRNAs are short RNAs that reduce gene expression by binding to their targets. Computational predictions indicate that all human genes may be regulated by microRNAs, with each microRNA possibly targeting thousands of genes. Commonly used software will produce a prohibitive number of predicted targets for each microRNA. Here I describe procedures that refine these predictions by integrating available software and expression data from experiments available online. These procedures are tailored to experiments where predicting true targets is more important than detecting all putative targets.
DOI:10.1261/rna.5248604URLPMID:15383676 [本文引用: 2]
MicroRNAs (miRNAs) are short RNAs that post-transcriptionally regulate the expression of target genes by binding to the target mRNAs. Although a large number of animal miRNAs has been defined, only a few targets are known. In contrast to plant miRNAs, which usually bind nearly perfectly to their targets, animal miRNAs bind less tightly, with a few nucleotides being unbound, thus producing more complex secondary structures of miRNA/target duplexes. Here, we present a program, RNA-hybrid, that predicts multiple potential binding sites of miRNAs in large target RNAs. In general, the program finds the energetically most favorable hybridization sites of a small RNA in a large RNA. Intramolecular hybridizations, that is, base pairings between target nucleotides or between miRNA nucleotides are not allowed. For large targets, the time complexity of the algorithm is linear in the target length, allowing many long targets to be searched in a short time. Statistical significance of predicted targets is assessed with an extreme value statistics of length normalized minimum free energies, a Poisson approximation of multiple binding sites, and the calculation of effective numbers of orthologous targets in comparative studies of multiple organisms. We applied our method to the prediction of Drosophila miRNA targets in 3'UTRs and coding sequence. RNAhybrid, with its accompanying programs RNAcalibrate and RNAeffective, is available for download and as a Web tool on the Bielefeld Bioinformatics Server (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/).
DOI:10.1093/nar/gkl243URLPMID:16845047 [本文引用: 2]
In the elucidation of the microRNA regulatory network, knowledge of potential targets is of highest importance. Among existing target prediction methods, RNAhybrid [M. Rehmsmeier, P. Steffen, M. Hochsmann and R. Giegerich (2004) RNA, 10, 1507-1517] is unique in offering a flexible online prediction. Recently, some useful features have been added, among these the possibility to disallow G:U base pairs in the seed region, and a seed-match speed-up, which accelerates the program by a factor of 8. In addition, the program can now be used as a webservice for remote calls from user-implemented programs. We demonstrate RNAhybrid's flexibility with the prediction of a non-canonical target site for Caenorhabditis elegans miR-241 in the 3'-untranslated region of lin-39. RNAhybrid is available at http://bibiserv.techfak.uni-bielefeld.de/rnahybrid.
DOI:10.1016/j.cell.2005.04.004URLPMID:15851028 [本文引用: 2]
Plants and animals use small RNAs (microRNAs [miRNAs] and siRNAs) as guides for posttranscriptional and epigenetic regulation. In plants, miRNAs and trans-acting (ta) siRNAs form through distinct biogenesis pathways, although they both interact with target transcripts and guide cleavage. An integrated approach to identify targets of Arabidopsis thaliana miRNAs and ta-siRNAs revealed several new classes of small RNA-regulated genes, including conventional genes such as Argonaute2 and an E2-ubiquitin conjugating enzyme. Surprisingly, five ta-siRNA-generating transcripts were identified as targets of miR173 or miR390. Rather than functioning as negative regulators, miR173- and miR390-guided cleavage was shown to set the 21-nucleotide phase for ta-siRNA precursor processing. These data support a model in which miRNA-guided formation of a 5' or 3' terminus within pre-ta-siRNA transcripts, followed by RDR6-dependent formation of dsRNA and Dicer-like processing, yields phased ta-siRNAs that negatively regulate other genes.
DOI:10.1093/bioinformatics/btp616URLPMID:19910308 [本文引用: 1]
SUMMARY: It is expected that emerging digital gene expression (DGE) technologies will overtake microarray technologies in the near future for many functional genomics applications. One of the fundamental data analysis tasks, especially for gene expression studies, involves determining whether there is evidence that counts for a transcript or exon are significantly different across experimental conditions. edgeR is a Bioconductor software package for examining differential expression of replicated count data. An overdispersed Poisson model is used to account for both biological and technical variability. Empirical Bayes methods are used to moderate the degree of overdispersion across transcripts, improving the reliability of inference. The methodology can be used even with the most minimal levels of replication, provided at least one phenotype or experimental condition is replicated. The software may have other applications beyond sequencing data, such as proteome peptide count data. AVAILABILITY: The package is freely available under the LGPL licence from the Bioconductor web site (http://bioconductor.org).
DOI:10.1093/nar/gni178URLPMID:16314309 [本文引用: 1]
A novel microRNA (miRNA) quantification method has been developed using stem-loop RT followed by TaqMan PCR analysis. Stem-loop RT primers are better than conventional ones in terms of RT efficiency and specificity. TaqMan miRNA assays are specific for mature miRNAs and discriminate among related miRNAs that differ by as little as one nucleotide. Furthermore, they are not affected by genomic DNA contamination. Precise quantification is achieved routinely with as little as 25 pg of total RNA for most miRNAs. In fact, the high sensitivity, specificity and precision of this method allows for direct analysis of a single cell without nucleic acid purification. Like standard TaqMan gene expression assays, TaqMan miRNA assays exhibit a dynamic range of seven orders of magnitude. Quantification of five miRNAs in seven mouse tissues showed variation from less than 10 to more than 30,000 copies per cell. This method enables fast, accurate and sensitive miRNA expression profiling and can identify and monitor potential biomarkers specific to tissues or diseases. Stem-loop RT-PCR can be used for the quantification of other small RNA molecules such as short interfering RNAs (siRNAs). Furthermore, the concept of stem-loop RT primer design could be applied in small RNA cloning and multiplex assays for better specificity and efficiency.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.funbio.2012.09.001URLPMID:23153806 [本文引用: 1]
Metarhizium anisopliae is one of the most common species of entopathogenic fungi. It has economic and social benefits in many countries where used in agriculture as an important biological control agent of insect pests. M. anisopliae can exist as multiple cell types, which suggests that this fungus has a complex way of gene regulation. MicroRNAs (miRNAs) are endogenous small noncoding RNAs. They play a crucial role in the regulation of gene expression and cell function in plants, animals, and in fungi where they were termed miRNA-like RNAs (milRNAs). In this study, we aimed to identify potential milRNAs in M. anisopliae that may regulate the processes of mycelium growth and conidiogenesis (CO). Two small RNA (sRNA) libraries were constructed and submitted to Solexa sequencing. Fifteen milRNAs were identified using deep-sequencing and computational analysis; most of these milRNAs originated from single genes. Database searches revealed that these novel milRNAs had no homologues in other organisms and were, therefore, M. anisopliae-specific. Many of the milRNAs had differential expression profiles for either mycelium growth or CO. The expression of the selected milRNAs was validated by quantitative reverse transcription polymerase chain reaction. Seventy-eight potential target mRNAs for 14 of the milRNAs were identified successfully by computational analysis. These milRNAs may play an important role in the regulation of mycelial growth and conidiation in M. anisopliae. To our knowledge, this study is the first report of milRNA profiles of organisms in the order Hypocreales. This information could be used to study the regulation of genes and their networks in M. anisopliae.
DOI:10.3389/fmicb.2019.00083URLPMID:30761116 [本文引用: 2]
Cordyceps militaris readily performs sexual reproduction, thus providing a remarkably rich model for understanding the processes involved in sexual development. It could regulate expression of human genes by diet-derived miRNA-like RNAs (milRNAs). However, the study of miRNAs in C. militaris has been limited. In the present study, genes encoding Dicers, Argonautes, and RNA-dependent RNA polymerases were identified. Illumina deep sequencing was performed to characterize the milRNAs in C. militaris at asexual and sexual development stages. Total 38 milRNAs were identified and five milRNAs were validated by northern blot and qRT-PCR, out of which, 19 were specific for sexual development. Importantly, the fungi could not form fruiting bodies after disruption of milR4, while the perithecium was formed in advance after over-expression of milR4. Abnormal pale yellow fruiting body primordium, covered with abnormal primordium, was formed in the strain with miR16 disruption. Although no milR4 or milR16 target genes were identified, differential expression of many different genes involved in mycelium growth and sexual development (mating process, mating signaling, and fruiting body development) among these mutants were found. Overall, milRNAs play vital roles in sexual development in C. militaris.
DOI:10.1371/journal.pone.0198234URLPMID:30231028 [本文引用: 2]
Coprinopsis cinerea is a model mushroom particularly suited for the study of fungal fruiting body development and the evolution of multicellularity in fungi. While microRNAs (miRNAs) have been extensively studied in animals and plants for their essential roles in post-transcriptional regulation of gene expression, miRNAs in fungi are less well characterized and their potential roles in controlling mushroom development remain unknown. To identify miRNA-like RNAs (milRNAs) in C. cinerea and explore their expression patterns during the early developmental transition of mushroom development, small RNA libraries of vegetative mycelium and primordium were generated and putative milRNA candidates were identified following the standards of miRNA prediction in animals and plants. Two out of 22 novel predicted milRNAs, cci-milR-12c and cci-milR-13e-5p, were validated by northern blot and stem-loop reverse transcription real-time PCR. Cci-milR-12c was differentially expressed whereas the expression levels of cci-milR-13e-5p were similar in the two developmental stages. Target prediction of the validated milRNAs resulted in genes associated with fruiting body development, including pheromone, hydrophobin, cytochrome P450, and protein kinase. Essential genes for miRNA biogenesis, including three coding for Dicer-like (DCL), one for Argonaute (AGO), one for AGO-like and one for quelling deficient-2 (QDE-2) proteins, were also identified in the C. cinerea genome. Phylogenetic analysis showed that the DCL and AGO proteins of C. cinerea were more closely related to those in other basidiomycetes and ascomycetes than to those in animals and plants. Taken together, our findings provided the first evidence for milRNAs in the model mushroom and their potential roles in regulating fruiting body development. New information on the evolutionary relationship of milRNA biogenesis proteins across kingdoms has also provided new insights for guiding further functional and evolutionary studies of miRNAs.
DOI:10.3389/fmicb.2018.00818URLPMID:29755439 [本文引用: 1]
Ascospores act as the primary inoculum of Fusarium graminearum, which causes the destructive disease Fusarium head blight (FHB), or scab. MicroRNAs (miRNAs) have been reported in the F. graminearum vegetative stage, and Fgdcl2 is involved in microRNA-like RNA (milRNA) biogenesis but has no major impact on vegetative growth, abiotic stress or pathogenesis. In the present study, we found that ascospore discharge was decreased in the Fgdcl1 deletion mutant, and completely blocked in the double-deletion mutant of Fgdcl1 and Fgdcl2. Besides, more immature asci were observed in the double-deletion mutant. Interestingly, the up-regulated differentially expressed genes (DEGs) common to DeltaFgdcl1 and DeltaFgdcl1/2 were related to ion transmembrane transporter and membrane components. The combination of small RNA and transcriptome sequencing with bioinformatics analysis predicted 143 novel milRNAs in wild-type perithecia, and 138 of these milRNAs partly or absolutely depended on Fgdcl1, while only 5 novel milRNAs were still obtained in the Fgdcl1 and Fgdcl2 double-deletion mutant. Furthermore, 117 potential target genes were predicted. Overall, Fgdcl1 and Fgdcl2 genes were partly functionally redundant in ascospore discharge and perithecium-specific milRNA generation in F. graminearum, and these perithecium-specific milRNAs play potential roles in sexual development.
DOI:10.1007/s00438-015-1128-1URLPMID:26481645 [本文引用: 1]
Deep sequencing of small RNAs is a useful tool to identify novel small RNAs that may be involved in fungal growth and pathogenesis. In this study, we used HiSeq deep sequencing to identify 747,487 unique small RNAs from Curvularia lunata. Among these small RNAs were 1012 microRNA-like RNAs (milRNAs), which are similar to other known microRNAs, and 48 potential novel milRNAs without homologs in other organisms have been identified using the miRBase(c) database. We used quantitative PCR to analyze the expression of four of these milRNAs from C. lunata at different developmental stages. The analysis revealed several changes associated with germinating conidia and mycelial growth, suggesting that these milRNAs may play a role in pathogen infection and mycelial growth. A total of 8334 target mRNAs for the 1012 milRNAs that were identified, and 256 target mRNAs for the 48 novel milRNAs were predicted by computational analysis. These target mRNAs of milRNAs were also performed by gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway analysis. To our knowledge, this study is the first report of C. lunata's milRNA profiles. This information will provide a better understanding of pathogen development and infection mechanism.
DOI:10.1371/journal.pntd.0002398URLPMID:23991243 [本文引用: 1]
BACKGROUND: Penicillium marneffei is the most important thermal dimorphic fungus causing systemic mycosis in China and Southeast Asia. While miRNAs are increasingly recognized for their roles in post-transcriptional regulation of gene expression in animals and plants, miRNAs in fungi were less well studied and their potential roles in fungal dimorphism were largely unknown. Based on P. marneffei genome sequence, we hypothesize that miRNA-like RNAs (milRNAs) may be expressed in the dimorphic fungus. METHODOLOGY/PRINCIPAL FINDINGS: We attempted to identify milRNAs in P. marneffei in both mycelial and yeast phase using high-throughput sequencing technology. Small RNAs were more abundantly expressed in mycelial than yeast phase. Sequence analysis revealed 24 potential milRNA candidates, including 17 candidates in mycelial and seven in yeast phase. Two genes, dcl-1 and dcl-2, encoding putative Dicer-like proteins and the gene, qde-2, encoding Argonaute-like protein, were identified in P. marneffei. Phylogenetic analysis showed that dcl-2 of P. marneffei was more closely related to the homologues in other thermal dimorphic pathogenic fungi than to Penicillium chrysogenum and Aspergillus spp., suggesting the co-evolution of dcl-2 among the thermal dimorphic fungi. Moreover, dcl-2 demonstrated higher mRNA expression levels in mycelial than yeast phase by 7 folds (P<0.001). Northern blot analysis confirmed the expression of two milRNAs, PM-milR-M1 and PM-milR-M2, only in mycelial phase. Using dcl-1(KO), dcl-2(KO), dcl(DKO) and qde-2(KO) deletion mutants, we showed that the biogenesis of both milRNAs were dependent on dcl-2 but not dcl-1 or qde-2. The mRNA expression levels of three predicted targets of PM-milR-M1 were upregulated in knockdown strain PM-milR-M1 (KD), supporting regulatory function of milRNAs. CONCLUSIONS/SIGNIFICANCE: Our findings provided the first evidence for differential expression of milRNAs in different growth phases of thermal dimorphic fungi and shed light on the evolution of fungal proteins involved in milRNA biogenesis and possible role of post-transcriptional control in governing thermal dimorphism.
DOI:10.3390/ijms20215357URLPMID:31661835 [本文引用: 1]
Although the regulatory function of miRNAs and their targets have been characterized in model plants, a possible underlying role in the cotton response to herbivore infestation has not been determined. To investigate this, we performed small RNA and degradome sequencing between resistant and susceptible cotton cultivar following infestation with the generalist herbivore whitefly. In total, the 260 miRNA families and 241 targets were identified. Quantitative-PCR analysis revealed that several miRNAs and their corresponding targets exhibited dynamic spatio-temporal expression patterns. Moreover, 17 miRNA precursors were generated from 29 long intergenic non-coding RNA (lincRNA) transcripts. The genome-wide analysis also led to the identification of 85 phased small interfering RNA (phasiRNA) loci. Among these, nine PHAS genes were triggered by miR167, miR390, miR482a, and two novel miRNAs, including those encoding a leucine-rich repeat (LRR) disease resistance protein, an auxin response factor (ARF) and MYB transcription factors. Through combined modeling and experimental data, we explored and expanded the miR390-tasiARF cascade during the cotton response to whitefly. Virus-induced gene silencing (VIGS) of ARF8 from miR390 target in whitefly-resistant cotton plants increased auxin and jasmonic acid (JA) accumulation, resulting in increased tolerance to whitefly infestation. These results highlight the provides a useful transcriptomic resource for plant-herbivore interaction.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1128/mmbr.66.3.447-459.2002URLPMID:12208999 [本文引用: 3]
Filamentous fungi are unique organisms-rivaled only by actinomycetes and plants-in producing a wide range of natural products called secondary metabolites. These compounds are very diverse in structure and perform functions that are not always known. However, most secondary metabolites are produced after the fungus has completed its initial growth phase and is beginning a stage of development represented by the formation of spores. In this review, we describe secondary metabolites produced by fungi that act as sporogenic factors to influence fungal development, are required for spore viability, or are produced at a time in the life cycle that coincides with development. We describe environmental and genetic factors that can influence the production of secondary metabolites. In the case of the filamentous fungus Aspergillus nidulans, we review the only described work that genetically links the sporulation of this fungus to the production of the mycotoxin sterigmatocystin through a shared G-protein signaling pathway.
DOI:10.1186/1471-2164-13-285URLPMID:22747707 [本文引用: 2]
BACKGROUND: We present a comprehensive transcriptome analysis of the fungus Ascosphaera apis, an economically important pathogen of the Western honey bee (Apis mellifera) that causes chalkbrood disease. Our goals were to further annotate the A. apis reference genome and to identify genes that are candidates for being differentially expressed during host infection versus axenic culture. RESULTS: We compared A. apis transcriptome sequence from mycelia grown on liquid or solid media with that dissected from host-infected tissue. 454 pyrosequencing provided 252 Mb of filtered sequence reads from both culture types that were assembled into 10,087 contigs. Transcript contigs, protein sequences from multiple fungal species, and ab initio gene predictions were included as evidence sources in the Maker gene prediction pipeline, resulting in 6,992 consensus gene models. A phylogeny based on 12 of these protein-coding loci further supported the taxonomic placement of Ascosphaera as sister to the core Onygenales. Several common protein domains were less abundant in A. apis compared with related ascomycete genomes, particularly cytochrome p450 and protein kinase domains. A novel gene family was identified that has expanded in some ascomycete lineages, but not others. We manually annotated genes with homologs in other fungal genomes that have known relevance to fungal virulence and life history. Functional categories of interest included genes involved in mating-type specification, intracellular signal transduction, and stress response. Computational and manual annotations have been made publicly available on the Bee Pests and Pathogens website. CONCLUSIONS: This comprehensive transcriptome analysis substantially enhances our understanding of the A. apis genome and its expression during infection of honey bee larvae. It also provides resources for future molecular studies of chalkbrood disease and ultimately improved disease management.
DOI:10.1139/m73-117URLPMID:4712507 [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/j.1574-6968.2010.02192.xURLPMID:21204928 [本文引用: 1]
Autophagy is a degradation system in which cellular components are digested via vacuoles/lysosomes, and involved in differentiation in addition to helping cells to survive starvation. The autophagic process is composed of several steps: induction of autophagy, formation of autophagosomes, transportation to vacuoles, and degradation of autophagic bodies. To further understand autophagy in the filamentous fungus Aspergillus oryzae, we first constructed A. oryzae mutants defective for the Aoatg13, Aoatg4, and Aoatg15 genes and examined the resulting phenotypes. The DeltaAoatg13 mutant developed conidiophores and conidia, although the number of conidia was decreased compared with the wild-type strain, while conidiation in the DeltaAoatg4 and DeltaAoatg15 mutants was not detected. The DeltaAoatg15 mutants displayed a marked reduction of development of aerial hyphae. Moreover, autophagy in these mutants was examined by observation of the behavior of enhanced green fluorescent protein (EGFP)-AoAtg8. In the DeltaAoatg13 mutant, the slight accumulation of EGFP-AoAtg8 in vacuoles, preautophagosomal structures (PAS), and autophagosomes was observed, whereas only PAS-like structures were detected in the DeltaAoatg4 mutant. In the DeltaAoatg15 mutant, autophagic bodies accumulated in vacuoles, suggesting that the uptake process proceeded. We therefore propose that the level of autophagy is closely correlated with the degree of differentiation in A. oryzae.
DOI:10.1128/EC.00224-07URLPMID:17921348 [本文引用: 1]
Autophagy is the major cellular pathway for bulk degradation of cytosolic material and is required to maintain viability under starvation conditions. To determine the contribution of autophagy to starvation stress responses in the filamentous fungus Aspergillus fumigatus, we disrupted the A. fumigatus atg1 gene, encoding a serine/threonine kinase required for autophagy. The DeltaAfatg1 mutant showed abnormal conidiophore development and reduced conidiation, but the defect could be bypassed by increasing the nitrogen content of the medium. When transferred to starvation medium, wild-type hyphae were able to undergo a limited amount of growth, resulting in radial expansion of the colony. In contrast, the DeltaAfatg1 mutant was unable to grow under these conditions. However, supplementation of the medium with metal ions rescued the ability of the DeltaAfatg1 mutant to grow in the absence of a carbon or nitrogen source. Depleting the medium of cations by using EDTA was sufficient to induce autophagy in wild-type A. fumigatus, even in the presence of abundant carbon and nitrogen, and the DeltaAfatg1 mutant was severely growth impaired under these conditions. These findings establish a role for autophagy in the recycling of internal nitrogen sources to support conidiophore development and suggest that autophagy also contributes to the recycling of essential metal ions to sustain hyphal growth when exogenous nutrients are scarce.
DOI:10.1128/EC.00216-07URLPMID:17715363 [本文引用: 1]
URLPMID:18473669 [本文引用: 1]
DOI:10.1111/1462-2920.13198URLPMID:26714892 [本文引用: 1]
Metarhizium robertsii has been used as a model to study fungal pathogenesis in insects, and its pathogenicity has many parallels with plant and mammal pathogenic fungi. MAPK (Mitogen-activated protein kinase) cascades play pivotal roles in cellular regulation in fungi, but their functions have not been characterized in M. robertsii. In this study, we identified the full complement of MAPK cascade components in M. robertsii and dissected their regulatory roles in pathogenesis, conidiation and stress tolerance. The nine components of the Fus3, Hog1 and Slt2-MAPK cascades are all involved in conidiation. The Fus3- and Hog1-MAPK cascades are necessary for tolerance to hyperosmotic stress, and the Slt2- and Fus3-MAPK cascades both mediate cell wall integrity. The Hog1 and Slt2-MAPK cascades contribute to pathogenicity; the Fus3-MAPK cascade is indispensable for fungal pathogenesis. During its life cycle, M. robertsii experiences multiple microenvironments as it transverses the cuticle into the haemocoel. RNA-seq analysis revealed that MAPK cascades collectively play a major role in regulating the adaptation of M. robertsii to the microenvironmental change from the cuticle to the haemolymph. The three MAPKs each regulate their own distinctive subset of genes during penetration of the cuticle and haemocoel colonization, but they function redundantly to regulate adaptation to microenvironmental change.
Agrobacterium-mediated insertional mutagenesis (AIM) of the entomopathogenic fungus Beauveria bassiana
DOI:10.1007/s00294-003-0468-2URLPMID:14634789 [本文引用: 1]
Agrobacterium tumefaciens was used to stably transform the entomopathogenic deuteromycete Beauveria bassiana to hygromycin B resistance by integration of the hph gene of Escherichia coli into the fungal genome. The transformation protocol was optimized to generate a library of insertion mutants of Beauveria. Transformation frequencies around 10(-4) and suppression of background growth were achieved. Over 90% of the AIM mutants investigated contained single-copy T-DNA integrations at different chromosomal locations. Integrated T-DNAs were re-isolated from ten transformants by a marker rescue approach. When the sequences flanking these T-DNAs were compared with the corresponding locations of the wild-type genome, truncations of T-DNA borders were found to be common, while none of the sites of integration had suffered deletion or rearrangement. Thus, AIM can be considered a promising tool for insertional mutagenesis studies of entomopathogenic filamentous fungi.
DOI:10.1139/w06-014URLPMID:16917517 [本文引用: 1]
A simple, highly efficient, and reliable Agrobacterium tumefaciens-mediated transformation method was developed for the insect pathogenic fungus Metarhizium anisopliae. Expression of the green fluorescent protein gene, egfp, and the benomyl resistance gene, benA3, were used as markers in transformed M. anisopliae. Transformation efficiencies were dependent on the strain of A. tumefaciens used. With strain AGL-1, 17.0 +/- 1.4 transformants per plate could be obtained using conidial concentrations of 10(6) conidia/mL and a 2 day co-cultivation in the presence of 200 micromol/L acetosyringone. On the other hand, transformations using strain LBA4404 were unsuccessful. Ten transformants were tested by Southern analysis and found to contain a single copy T-DNA. Twenty transformants were subcultured for five generations on nonselective media, and 95% of the transformants were mitotically stable. Agrobacterium tumefaciens-mediated transformation of M. anisopliae can serve as a useful tool to investigate genes involved in insect pathogenicity.
DOI:10.1016/j.mimet.2014.07.033URLPMID:25107375 [本文引用: 1]
Lecanicillium lecanii has been used in the biological control of several insects in agricultural practice. Since the gene manipulation tools for this entomopathogenic fungus have not been sufficiently developed, Agrobacterium tumefaciens-mediated transformation (ATMT) in L. lecanii was investigated in this study, using the wild-type isolate FZ9906 as a progenitor strain and the hygromycin B resistance (hph) gene as a selection marker. Furthermore, a field carbendazim-resistant (mrt) gene from Botrytis cinerea was expressed in L. lecanii FZ9906 via the ATMT system. The results revealed that the frequency of transformation surpassed 25transformants/10(6) conidia, most of the putative transformants contained a single copy of T-DNA, and the T-DNA inserts were stably inherited after five generations. All putative transformants had indistinguishable biological characteristics relative to the wild-type strain, excepting two transformants with altered growth habits or virulence. Moreover, the resistance of the putative transformants to carbendazim (MBC) was improved, and the highest one was 380-fold higher than the wild-type strain. In conclusion, ATMT is an effective and suitable system for L. lecanii transformation, and will be a useful tool for the basic and application research of gene functions and gene modifications of this strain.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}