 ,1,3,4,7, 杨泉女3, 郑洪建4, 许彦芬2, 桑志勤5, 郭子锋1, 彭海6, 张丛2, 蓝昊发2, 王蕴波3, 吴坤生2, 陶家军2, 张嘉楠,2
,1,3,4,7, 杨泉女3, 郑洪建4, 许彦芬2, 桑志勤5, 郭子锋1, 彭海6, 张丛2, 蓝昊发2, 王蕴波3, 吴坤生2, 陶家军2, 张嘉楠,2Genotyping by Target Sequencing (GBTS) and Its Applications
XU Yunbi,1,3,4,7, YANG QuanNü3, ZHENG HongJian4, XU YanFen2, SANG ZhiQin5, GUO ZiFeng1, PENG Hai6, ZHANG Cong2, LAN HaoFa2, WANG YunBo3, WU KunSheng2, TAO JiaJun2, ZHANG JiaNan,2通讯作者:
责任编辑: 李莉
收稿日期:2020-05-6接受日期:2020-06-16网络出版日期:2020-08-01
| 基金资助: |
Received:2020-05-6Accepted:2020-06-16Online:2020-08-01

摘要
关键词:
Abstract
Keywords:
PDF (2275KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
徐云碧, 杨泉女, 郑洪建, 许彦芬, 桑志勤, 郭子锋, 彭海, 张丛, 蓝昊发, 王蕴波, 吴坤生, 陶家军, 张嘉楠. 靶向测序基因型检测(GBTS)技术及其应用[J]. 中国农业科学, 2020, 53(15): 2983-3004 doi:10.3864/j.issn.0578-1752.2020.15.001
XU Yunbi, YANG QuanNü, ZHENG HongJian, XU YanFen, SANG ZhiQin, GUO ZiFeng, PENG Hai, ZHANG Cong, LAN HaoFa, WANG YunBo, WU KunSheng, TAO JiaJun, ZHANG JiaNan.
在分子水平进行遗传变异的检测,简称分子检测,是生物遗传变异分析的重要手段。理想的分子检测,是在个体和群体水平上对生物进行完整检测,例如全基因组测序。尽管随着分子检测技术的发展,单位时间所能获得的信息呈指数增加,而且检测成本呈指数降低,但生物体的全信息分子检测在信息处理和分析上仍然面临重大挑战。因此,以点带面、基于分子标记的遗传变异检测技术仍然是一种简便、高效、低成本的分子检测手段[1]。分子标记技术就是采用分布在生物基因组上的每个标记作为靶标,测定特定基因组位点和区域的相关基因型变异,简称基因型检测,从而在整体上代表全基因组水平的遗传变异。从20世纪以蛋白质(主要是同工酶)作为分子标记开始,分子标记技术在数量、种类、通量、分析成本等方面均发生了一系列革命性的变化,并在应用生物学的许多领域得到日益广泛的应用[2,3]。在动植物的遗传改良中,分子标记辅助选择与转基因和基因编辑技术相结合,成为支撑分子育种的三大核心技术。
分子标记辅助选择所主导的分子育种,在国际跨国种业公司早已成为与常规育种技术密切整合的重要育种手段[4,5]。大型跨国种业公司凭借其完整的产业链、巨大的体量、高昂的研发投入和较高的市场占有率,发展了可以高效利用分子标记检测系统支撑其育种流程的平台、技术和方法。一套完整分子育种技术和平台可以为其在世界各地的育种中心提供统一的支撑,在极大地降低成本的同时,也提高了平台的运转和使用效率[6,7,8]。然而,在中小育种公司和发展中国家,因为小而分散的育种活动,无法建立起高效低成本的分子标记辅助育种体系,成为制约分子育种的重要瓶颈因素[5]。
四十年来,分子检测技术日新月异,各种分子标记技术和检测设备不停地更新换代,经历了从凝胶电泳、荧光检测、固相芯片到液相芯片的4G发展过程(图1),但更新所需的巨大投入使共享技术和平台难以与最新的技术保持同步发展。纵观分子标记技术的革新,以DNA序列变异为基础的单核苷酸多态性(SNP),已经接近在分子水平进行变异检测的终极标准。预期今后若干年分子检测技术和平台的发展大致是在此基础上进行提升和改进。因此,高效、低成本的SNP基因型检测技术成为发展共享技术和平台的最佳选择。
图1
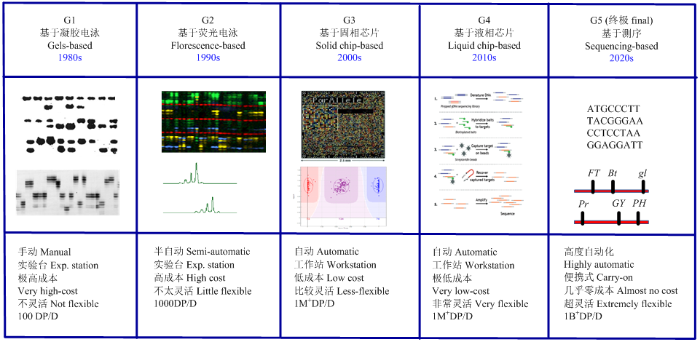 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1基因型检测平台从1G到5G的变革
Fig. 1Evolution of genotyping platforms from G1 to G5
本文将综合讨论近年发展起来的基于SNP的靶向测序基因型检测(genotyping by target sequencing,GBTS)技术[9,10,11]。该技术具有检测效率高、成本低、适应性广、应用灵活等特点,适合于动物、植物和微生物等所有生物遗传变异和基因型的检测,有望成为各种生物可以共享的技术和平台,并广泛应用于种质资源评价、遗传图谱构建、基因定位和克隆、分子标记辅助选择、品种权保护、种子质量监控、生物检测和安全评价等领域。本文将讨论GBTS技术的基本原理、平台、优势、应用与展望。
1 靶向测序基因型检测(GBTS)技术的基本原理
分子或基因型检测的终极目标是在DNA水平上进行全基因组测序。这类技术被称为测序式基因型检测(genotyping by sequencing,GBS)。以人类基因组为例,全基因组DNA测序的成本已经从早期第一个人类基因组测序时的27亿美元,降到现在的500美元。但要针对育种计划相关的成千上万的样本进行高效快速的全基因组测序,依然面临成本高昂和海量数据处理的巨大挑战。为了降低成本和数据量,对全基因组DNA进行限制性酶切,可以仅挑选一部分片段进行随机测序。然后经过大规模的生物信息分析,获得代表全基因组简化序列的分子标记(图2)[1,12-13]。在高质量参考基因组或单倍型图谱的辅助下,借助于大规模累计的简化测序数据,通过海量数据的对比分析和缺失替补(imputation)[14],可以较小的测序量获得覆盖全基因组的高密度分子标记。这类GBS平台的构建和应用在玉米中已经十分成功[15]。但这样的大规模数据积累和强大的生物信息支撑,在很多作物中,特别是小宗作物中是难以实现的。另外,不同材料、实验室和平台之间所获得的GBS标记数据很难进行比对和累加,导致难以进行GBS数据的长期保存和综合利用。图2
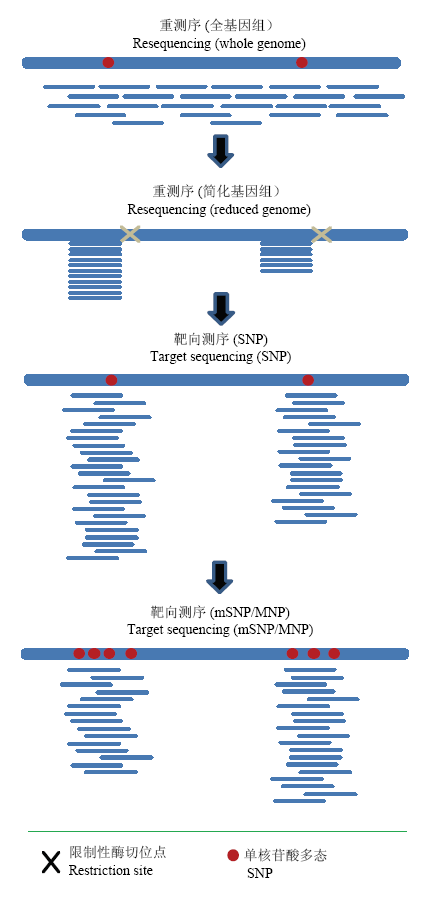 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2基因型检测从随机重测序(全基因组)到简化基因组测序、靶向测序(SNP和mSNP)的发展
Fig. 2Evolution of genotyping platforms from random whole genome resequencing to reduced genome resequencing and target sequencing (SNP and mSNP)
鉴于随机简化基因组测序存在的种种限制,理想的GBS策略就是从基因组上定点进行测序并获得物理位置相对固定的标记。GBTS技术就是要从浩瀚的基因组DNA中,挑选特定的靶向位点,进行测序和基因型检测(图2)[16,17]。由于GBTS技术能够通过不同平台在不同实验室稳定地获得相同的SNP标记,为检测数据的累积、共享、比较和整合提供了简单可靠的技术平台。这种靶向或固定的简化基因组测序大大减少了DNA测序量,简化了生物信息分析和数据处理,提高了对各种基因型检测平台的适应性。因此,推测GBTS将成为未来很长一段时间基因型检测的首选。
目前,中国已经开发出具有自主知识产权、不受其他专利技术制约、不依赖于特定检测设备的GBTS技术体系,并广泛应用于不同物种的分子和基因型检测[11]。根据所涉及的标记数量,GBTS由2个独特但又相互交叉的技术体系组成:GenoPlexs和GenoBaits。前者基于多重PCR,而后者基于液相探针杂交。2种技术均可实现对基因组任意位置、任意长度的非高度重复区的精准捕获,可同时检测SSR、SNP、InDel等多种类型的基因型变异。GBTS是通过测序获取的不同等位变异的Reads来判断纯合和杂合。在人类检测中,由于人都是基于单个个体的分析,所以会将0.05(即reads数占比少的等位基因类型超过5%)作为阈值进行杂合体判断。但是在植物检测中通常采用多植株混合取样,为避免混杂的干扰,将阈值调整为0.2,即覆盖目标位点的100条reads中,占比较少的等位基因类型的reads数大于20条就认为是杂合,否则就是纯合。
GBTS技术的基本流程包括:通过甲基化敏感内切酶介导的简化基因组技术或重测序技术鉴定出多态SNP位点;利用标记技术,开发40—50K SNP的原始GBTS标记或基于mSNP的240—270K SNP标记;经过优化和精选,最后形成高效、低成本的GenoBaits或GenoPlexs标记。
1.1 基于多重PCR的靶向测序基因型检测(GBTS)技术:GenoPlexs
GenoPlexs的工作原理是依靠PCR对于靶向位点的定点扩增。对多个待测SNP位点设计特异扩增引物,在第一轮PCR中抑制引物干扰和非特异扩增,使数以千计的靶向引物能够在一管PCR反应中实现高度均一化的扩增,从而大量富集目标片段。随后,在第二轮PCR中,加上测序接头和文库条形码,最终获得测序所需的文库。最后通过大规模并行测序(massively parallel sequencing,MPS)揭示目标位点的标记基因型(图3)。目前,该技术已可以实现单管高达2 000对以上特定引物的混合扩增,如果考虑后面将要讨论的多聚SNP技术,同时检测的分子标记数量可高达10K(张嘉楠等,私人通讯)。图3
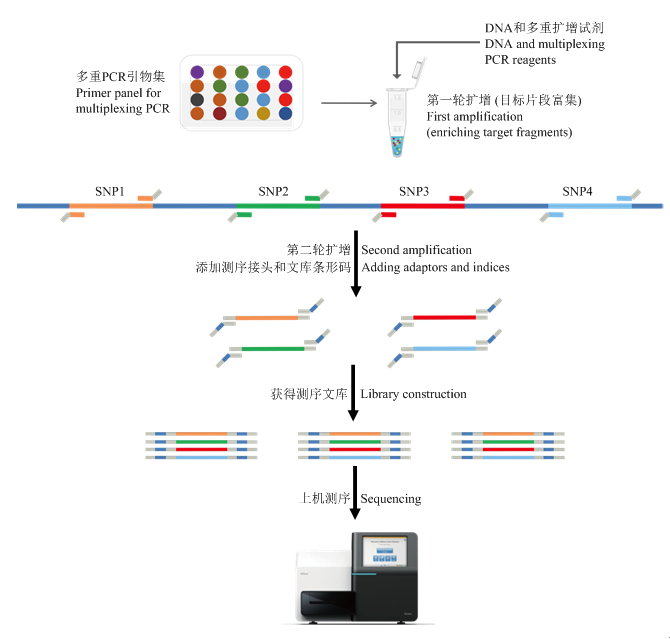 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3基于靶向测序基因型检测(GBTS)技术的GenoPlexs流程图
Fig. 3Flowchart for genotyping by target sequencing with GenoPlexs
1.2 基于液相探针杂交的靶向基因型检测(GBTS)技术:GenoBaits
GenoBaits工作原理是基于目标探针与靶向序列互补结合进行定点捕获(图4)。首先对要测试的材料进行gDNA文库构建。同时根据DNA互补原理,在每个待测位点设计覆盖目标SNP的探针,采用生物素(Biotin)标记对目标探针进行修饰。然后,在液态中利用生物素修饰的探针与基因组目标区域杂交形成双链。随后利用链霉亲和素包被的磁珠对携有生物素修饰的探针进行分子吸附,从而捕获与探针杂交的靶点。最后,对捕获的靶点序列进行洗脱、靶点扩增和测序,最终获得目标SNP的基因型。利用GenoBaits技术,目前可以对多达4.5万个目标位点进行单管同时检测(张嘉楠等,私人通讯)。图4
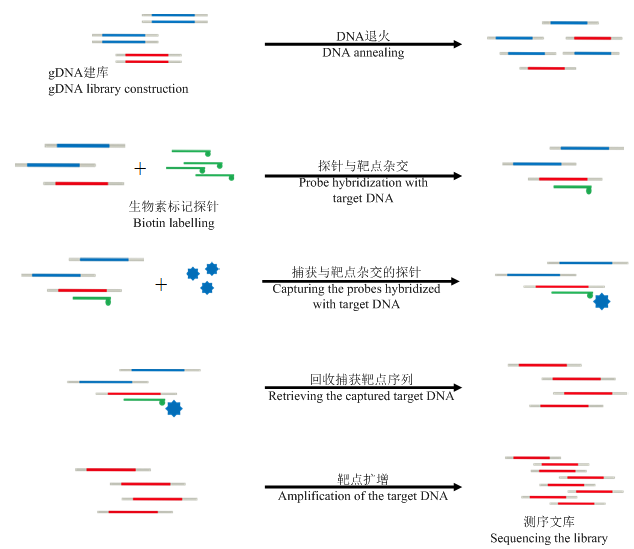 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4基于靶向测序基因型检测(GBTS)技术的GenoBaits流程图
Fig. 4Flowchart for genotyping by target sequencing with GenoBaits
1.3 多聚单核苷酸多态性(mSNP)检测技术
传统的SNP标记,一般是根据每一个SNP标记设计一对特定的扩增引物,在所获得的扩增子内产生一个SNP标记,即一个扩增子对应一个SNP标记。因此,总体来看,所检测到的SNP在基因组上形成单个的均匀分布。为了最大限度地利用每对引物扩增所获得的DNA片段的信息,发展了一种在单个扩增子内可以检测多个SNP,称之为多聚单核苷酸多态性(multiple single- nucleotide-polymorphism cluster,mSNP或multiple dispersed nucleotide polymorphism,MNP)的技术。生物基因组中存在大量的SNP变异。人类基因组含有300万个SNP,平均每1 000个碱基中就有1个SNP。根据SNP在基因中的位置,可分为基因编码区SNP(coding-region SNP)、基因周边SNP(perigenic SNP)和基因间SNP(intergenic SNP)。SNP的分布不均匀,非转录区要多于转录区,大多数位于蛋白的非编码区。SNP频率在5’端非编码区、3’端非编码区、内含子、沉默位点及编码区有显著差异。在外显子内,其变异率仅及周围序列的五分之一。玉米中,根据对55M SNP的分析,78.7%的SNP位于基因间,内含子中的SNP占10.5%[18]。玉米HapMap3包括了83M SNP,平均大约25 bp一个SNP[19]。生物基因组上存在大量由转座子等引起的变异热点,其SNP频率大大高于基因组的平均水平。
为了在每一靶向测序位点检测尽量多的SNP变异,设计了一套在单个扩增子内检测多个SNP的方法(图2)。与每个扩增位点只包括一个SNP的液相芯片相比,mSNP液相芯片具有四方面的改进。一是在每一个扩增位点可以产生多个SNP标记,即多个SNP的聚合体(mSNP),使可检测的SNP数目扩大到位点数的4倍以上。二是同一扩增子内的多个SNP标记之间可以构成单倍型,提高了变异的检测效率。三是可以从每个mSNP(扩增子)内挑选低频等位基因频率(MAF)最大的SNP组成核心标记。四是mSNP提供了更为精细的遗传变异检测,包括mSNP位点内和位点间的变异,而且可以采用单倍型和SNP 2种方式分别进行检测。mSNP技术不仅大大提高了标记的利用率,同时通过“一点多标”提升了标记鉴定的准确度和灵敏度。
mSNP开发共包括6个步骤。(1)获得该物种的参考基因组和重测序/变异组信息;(2)筛选出变异位点中MAF>0.05、缺失比例<20%、杂合比例<5%(自交物种)的位点作为候选位点集;(3)以100 bp为窗口,计算候选位点在窗口区段内的SNP数量,筛选SNP数量大于2、小于10的区段作为目标候选区段集;(4)依据PIC值,筛选PIC最高的十万个区段作为候选区段集,利用染色体均匀分布的原则筛选候选区段集,构成候选目标区段集;(5)利用其他区段和SNP信息对候选目标区段集中的染色体空洞部分(标记之间的距离≥基因组大小÷目标区段数量×10)进行补充,最终构成目标区段集;(6)利用GenoBaits探针设计软件对目标区段进行设计,并合成测试,最终完成标记位点组合的开发。同样地,mSNP检测技术也适用于基于多重PCR的GenoPlexs。
2 靶向测序基因型检测(GBTS)技术的平台与比较优势
2.1 平台广适性
GBTS标记的检测不需要借助于特定的昂贵设备,而可以配适多种测序平台,包括目前3种主流的测序平台Illumina、Ion Torrent和MGI。毫无疑问,GBTS标记检测也将适合于所有改进和新开发的任何测序技术和平台(表1)。Table 1
表1
表1靶向测序基因型检测(GBTS)技术的平台与比较优势
Table 1
| 优势 Advantages | 具体特征 Description of advantages | 展望 Prospects | |
|---|---|---|---|
| 平台 Platform | 广适性 Wide suitability | 适合所有二代和三代测序系统,包括Illumina、Ion Torrent和MGI Applicable to the second and third generations of sequencing facilities, including Illumina, Ion Torrent and MGI | 与下一代测序或其他检测设备兼容 Also applicable to the next generation of sequencing and other genotyping facilities |
| 标记 Markers | 灵活性 Flexibility | 适合各种标记类型(SNP、短SSR、长/短InDel、已知融合基因、甲基化位点);不同的标记密度;一款多用 Suitable for various marker types (SNPs, short SSRs, long/short InDels, known fusion genes and methylated loci), densities and applications | 广泛利用单倍型、LD区段和其他标记衍生物 Wide applications with haplotypes, LD blocks and other marker-derivatives |
| 检测 Genotyping | 高效性 High efficiency | 样本多重化、多重PCR;开发和升级简便;设计、测试和检测成本低 Sample- and PCR-multiplexing, readily development and upgrade of marker panels, and low cost in design, test and genotyping | 随检测技术进步推动检测的自动化、智能化、超高通量 More robotic, intelligent, and high-throughput with advanced genotyping facilities |
| 信息 Information | 可加性 Accumulativity | 数据重复率高,缺失数据少;不同时间、地点、项目间的数据可比和累加;整合度高 High duplication rate; less missing; accumulative across times, locations and projects; integrative | 可加性程度随技术进步而增强 Increasingly accumulative with technical advancement |
| 支撑 Support systems | 便捷性 Less demanding | 不依赖检测技术或专业化的生物信息团队;通用而简化的实验室信息管理系统;通用的信息整合、处理、分析流程 Independent of professional genotyping and informatics supports, manageable through regular LIMS and data integration, treatment and analytical protocols | 随技术进步而更加便捷、快速、智能化 Less demanding, much quicker but more intelligent, with technical development |
| 应用 Applications | 广谱性 Wide application | 广泛应用于动物、植物、微生物及其互作群体的进化、遗传、育种、知识产权保护等领域 Wide application in the fields of evolution, genetics, breeding and variety right protection in animals, plants and microorganisms | 随着海量信息的累计,将拓展在群体生物学、生态学等领域的应用 Applications extended to population biology and ecology as huge data accumulated |
新窗口打开|下载CSV
Illumina Hiseq/Miseq测序平台:Solexa高通量测序技术以单分子阵列技术为基础,是对合成测序技术的发展与延伸。Solexa是一种基于边合成边测序(sequencing-by-synthesis,SBS)的新型测序技术。通过单分子阵列实现在小型芯片(FlowCell)上进行桥式PCR。通过可逆阻断技术实现每次只合成一个碱基,再利用相应的激光激发荧光基团,捕获激发光,从而读取碱基信息。现有的Illumina测序平台包括Illumina HiSeq系列、Illumina NovaSeq系列、Illumina NextSeq系列以及Illumina MiSeq等。
Ion Torrent测序平台:其核心是使用半导体技术在化学和数字信息之间建立直接的联系。将DNA链固定在半导体芯片的微孔中,随后依次掺入ACGT。随着每个碱基的掺入,释放出氢离子H+,在它们穿过每个孔底部时进行检测。通过对H+的检测来实时判读碱基。Ion Torrent由于硬件设备无需光学检测和扫描系统,并且使用天然核苷酸和聚合酶、无需焦磷酸酶化学级联,无需标记荧光染料和化学发光的配套试剂,因此,测序速度快。其应用范围涵盖Sanger方法和已有的高通量测序技术,如基因组DNA序列测定、DNA扩增子测序等。现有的Ion Torrent测序平台包括Ion S5、Ion PGM、Ion Proton等。
MGI测序平台:这是一款华大智造的基因测序仪,其采用先进的DNBSEQTM测序核心技术,通过仪器气液系统先将DNA纳米球(DNA nanoball,DNB)泵入到规则阵列芯片(patterned array)并加以固定,然后泵入测序模板及测序试剂。测序模板与芯片上的DNB接头互补杂交,在DNA聚合酶的催化下,测序模板与测序试剂中的带荧光标记的探针相结合。然后由激光器激发荧光基团发光,不同荧光基团所发射的光信号被相机采集,经过处理后转换成数字信号,传输到计算机进行处理,从而获取待测样本的碱基序列信息。现有的MGI测序平台包括BGISEQ-500、BGISEQ-50、MGISEQ-200、MGISEQ- 2000以及DNBSEQ-T7等。
2.2 标记灵活性
采用GenoBaits和GenoPlexs 2种技术可以对基因组任意位置、任意长度区段,包括散点、外显子、基因组片段等进行精准捕获,可同时对SNP、短SSR、长/短InDel、已知/未知融合基因、甲基化位点等多种遗传变异进行单种检测和混合检测。理论上,GBTS所能检测的标记数量没有限制。GenoBaits和GenoPlexs 2种系统结合使用,可实现同时对5—260K标记检测。GBTS标记具有适用性广的特点,目标序列保守性要求低,目标位点侧翼序列可存在多个变异位点;目标序列GC含量容忍性高,GenoBaits可以对GC含量高达80%以上的区域进行捕获。同时,适合于分析SNP和mSNP、单个标记和单倍型。GBTS具有定制、使用和升级的灵活性(表1)。首先,没有起始样本量和标记位点数量的限制,片段测序与标记基因型检测可在同一管内完成。使用的灵活性在于没有单次检测样本量限制,定制和使用不需要凑足96或384样品或最小样本量;虽然样本量越大,单个样本的成本就越低。其次,升级的灵活性在于可向体系中随时加入新的引物或探针而不需对已有引物或探针进行调整。其三,一套多用:根据同一套高密度标记,可以通过调整测序深度来获得不同数量的标记。传统上,为了满足不同应用场景对于标记密度的需求,必须开发不同标记数量的成套标记。对于基于固相芯片的高密度标记组合,因为定制成本相对较高,很难照顾不同应用场景对标记数目的需求。因此,能够根据不同应用场景而灵活开发标记数量不同的成套组合,是高效分子检测系统的基本要求。满足这一要求的最佳方式就是利用同一套高密度分子标记,通过控制标记检测的某个环节来产生不同数量的标记,实现一款多用。
GBTS标记系统具备产生一款多用标记的潜力。一方面,因为每个目标标记区域的捕获效率存在差异,为了以99%以上的概率捕获特定的目标区域,必须在测序时保证一定的测序深度。另一方面,通过控制测序深度,就可以捕获到不同数量的标记。越容易捕获的标记,所需要的测序深度就越少。因此,如果标记捕获的难易程度在基因组上是随机分布的,在理论上就可以通过控制测序深度来捕获不同数目且在基因组上均匀分布的标记。
2.3 检测高效性
GBTS技术通过PCR多重化(multiplexing PCR)和样本多重化(multiplexing samples,SamPlexs)来大幅度提升分析的通量。与通用的KASP技术相比,GenoPlexs标记开发流程大大简化,不需要二轮标记测试。GenoPlexs可以在一管PCR反应体系中实现数千重高度均一的PCR反应。扩增子数量在5—2 000个范围时,可检测标记数量在5—10K范围内,单个标记的分析成本显著降低。这种高度多重的PCR,极大地提高了标记检测效率,降低了检测成本。SamPlexs技术在多重PCR的基础上实现了样品的混合测定,从而进一步降低了检测成本。此外,所有检测试剂基本实现了国产化或本地化,与其他检测技术相比,试剂成本大大降低。同时由于目前大量的测序设备闲置,以及测序技术的进步和测序设备的更新,与测序相关的成本逐渐降低。利用现有的测序设备可以大大降低设备的维护、管理和运营成本(表1)。与高密度分子标记技术——固相芯片相比,GenoBaits标记开发技术简便,开发、测试、验证的成本远低于传统的固相芯片。GBTS检测的最终成本将低于KASP、TaqMan、琼脂糖电泳、HRM、Agena等技术。例如,对于5—200个标记位点的单样品检测,费用明显低于KASP。在高密度标记情况下,固相芯片通过大规模定制样本,GBS通过大规模共享平台的支撑,可以获得比GenoBaits成本略低的微弱优势。但特定固相芯片需要达到行业垄断水平才能获得足够的定制规模,而GBS则需要在公共项目的无偿支持下建立共享平台并积累足够的数据量才能实现标记检测程序和质量的最优化。
2.4 信息可加性
目前所获得的GBTS基因型信息具有数据重复率高,缺失数据少的优点。GBTS采用靶向位点的深度测序,位点结果准确。而固相芯片检测采用荧光信号,结果受诸多因素影响。很多作物的55K的芯片,通常只能得到40K左右的可靠标记数据。GBTS标记的结果可靠性高,同批建库、同时测序的重复性,同批建库、不同时间上机的重复性,不同批次建库、同时测序的重复性均在99.9%以上。标记和基因型信息的高度重复性和可靠性,使特定目标区域标记信息的可加性成为可能(表1)。信息的可加性是检测最需要的特征,因为需要将不同时间、地点和试验所获得的信息累加起来,进行比较和综合分析。全基因组重测序和简化基因组测序只是对整个基因组的随机和部分测序,不同时间、地点和实验所获得的信息无法一一对应,很难进行数据的比对、累加和整合,必须借助于精细的参考基因组、泛基因组或单倍型图谱。但是,很多物种目前尚不具备这样精细可靠的比较标准。
在玉米中,基于对数万份玉米材料的GBS分析, 结合多个参考基因组,构建了完整的单倍型图谱[18,19]。据此,任何独立的GBS试验所获得的序列信息,通过与此单倍型图谱进行比对,不仅可以校正测序结果的偏差,同时也可以利用单倍型图谱进行缺失数据的替补(imputation),获得某个GBS试验中缺失、但存在于单倍型图谱的标记位点的等位形式。借助于这样的大数据平台和完整的单倍型图谱,可以大大降低GBS的测序量,同时提高标记检测的可靠性[15]。但高精度单倍型图谱的构建需要通过长期努力,检测大量的样本,累积大量的数据。相比而言,在信息的可加性方面,GBTS技术对高精度参考基因组和单倍型图谱依赖程度很低。
2.5 支撑便捷性
与固相芯片、全基因组重测序和随机的简化基因组测序等技术相比,GBTS技术对于平台和支撑的要求很低,不需要额外的检测技术团队或高度专业化的生物信息团队(表1)。由于整个检测流程的规范化,一般实验室和研究机构很容易建立起自己的GBTS检测系统,同时为其他需求者提供检测服务和技术支撑。GBTS技术因为其信息的高度可靠性和可加性,将大大简化实验室信息管理系统(LIMS)的构建、管理和运营(表1)。现有的LIMS系统不经任何升级或改造就可以直接用于管理GBTS信息。事实上,GBTS技术的支撑便捷性为大家提供了分子检测的一种近似傻瓜的操作模式和一站式数据分析管理系统。通过交互报告系统,可快速实现基因型和标记数据的文件格式转化和多样化数据分析。
2.6 应用广谱性
正如下一节要详细讨论的,基于GBTS的SNP检测,其应用领域非常广泛(表1)。首先,该技术适用于所有的生物物种,包括各种动物、植物和微生物。除了适用于大宗动植物以外,特别适合于研究基础较差、基因组学信息较为匮乏的物种。对于其他分子检测系统应用难度较大的物种,如多倍体物种,GenoPlexs的特异性好,可进行特异的基因组捕获。其次,该技术适合所有标记位点的检测,包括功能已知位点(如已克隆的基因)、功能未知位点(候选基因)以及中性位点。该检测系统标记数量的可塑性为各种应用提供了巨大弹性空间,基本上适合需要不同标记数量的全部场景,除了标记数量极少的应用场合以及超高密度的GWAS分析。
其三,GBTS检测技术的标记灵活性导致应用场景的多样化,包括后续将要讨论的全部应用场景,如标记辅助的主基因选择、回交育种及其背景选择、多基因聚合育种、种子纯度检测、转基因成份鉴定等(表2)。
Table 2
表2
表2靶向测序基因型检测(GBTS)的应用
Table 2
| 应用领域 Applications | 40K+ | 30K | 20K | 10K | 5K | 1K | <200 |
|---|---|---|---|---|---|---|---|
| 生物进化Biological evolution | +++ | +++ | ++ | ++ | + | ||
| 种质资源评价Germplasm evaluation | +++ | +++ | ++ | ++ | + | + | |
| 分类Classification | +++ | +++ | +++ | +++ | ++ | ++ | |
| 图谱构建Linkage map construction | +++ | +++ | +++ | ++ | + | + | |
| 基因定位和克隆Gene mapping/cloning | +++ | +++ | +++ | +++ | ++ | + | |
| 标记-性状关联Marker-trait association | +++ | ++ | ++ | + | |||
| 后裔测验Progeny testing | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| 基因渐渗Gene introgression | +++ | +++ | +++ | +++ | +++ | ++ | + |
| 基因累加Gene pyramiding | +++ | +++ | +++ | +++ | ++ | + | + |
| 品种权保护Variety right protection | +++ | +++ | +++ | +++ | ++ | ++ | + |
| 质量控制Quality control | +++ | +++ | +++ | +++ | ++ | ++ | + |
| 生物检测Bioassay | +++ | +++ | +++ | +++ | ++ | + | + |
新窗口打开|下载CSV
3 靶向测序基因型检测(GBTS)技术的潜在应用
3.1 物种进化分析
评估物种内种群结构和种间进化问题往往受可使用标记数据的质量和范围限制,采用更多位点的信息一般可以获得更准确的结果。利用捕获测序的方法,针对物种间相对保守的基因,设计捕获探针,利用探针的高容错性捕获的特点,可以实现对目标序列的跨物种捕获,从而获得可用于系统进化分析的高可比性跨物种序列信息[10]。采用GenoBaits技术,设计单拷贝保守基因探针,可在序列相似度较低时实现跨物种的目标序列捕获,从而获得不同物种的单拷贝保守基因序列并加以比较分析,进而推测物种间的进化关系。可以利用现有的进化分析检测标记直接对感兴趣的样本进行检测,也可以根据特定需求开发新的保守基因捕获标记开展进化研究。GenoBaits可以对相似度90%以上的序列实现正常捕获,因而可用于跨物种保守基因的捕获。由于仅用于分析保守基因,而不需要对整个基因组进行测序,因此可以大大降低进化研究所需的检测成本。单样本平均测序深度控制在100×以上,一般可捕获1—3 000范围内任意数量的基因外显子序列,可以用于种间系统发生关系和种内亚种间系统发生关系的研究。
JOHNSON等[10]从600种被子植物中筛选出353个单拷贝保守基因作为目标基因。为了获得较好的序列代表性,利用K-mers算法对不同物种的353基因序列进行聚类,挑选出可以代表所有序列的4 781条核心序列作为捕获目标序列,用于液相探针的设计。对42个被子植物物种进行了序列捕获,结果显示平均每个物种可以捕获到283个目标基因(120—344),其中21个样本每个都可以检测到300个以上的目标基因。对于部分检出基因较少、捕获效率低的样本,可以通过增加测序量来提高检出率。通过外显子序列捕获,获得了平均长度为216 kb的非编码序列。这些序列可以用来构建物种间的系统进化树。
3.2 种质资源评价与DNA指纹鉴定
种质资源的搜集、保存、评估、挖掘和利用是应用生物学研究的重要内容。应用基因组学和分子标记对各类种质资源进行系统分析,可以揭示种质资源的驯化进程,评估种质间的遗传多样性,构建核心种质,了解群体结构,进行系谱鉴定、聚类分析、品种杂合度分析和杂种优势群划分等[20],对于指导种质资源的创新和利用具有很重要意义。GBTS技术提供的中密度分子标记分析平台,特别适合于种质资源评价(表2)。因研究目标和物种的不同,可以采用3种不同标记密度的标记:(1)1—2K+ GenoPlexs(低密度),适合对分辨率要求较低的小宗作物。(2)10—20K GenoBaits(中密度),适合于主要农作物和动物物种。(3)20—40K GenoBaits(高密度),适合于动植物核心种质资源。以玉米为例,通过20K SNP标记的GBTS分析,将自交系划分为瑞德、兰卡、黄早四、唐四平头、旅大红骨、Iodent等多个杂种优势群[11]。据此,育种家就可以根据育种目标,进行群内改良或者群间杂交组配,达到事半功倍的选择效果。包括中草药在内的特色物种种质资源长期处于保护缺失的状态,许多特色地方品种已经濒临灭绝或者与其他外来品种杂交,严重威胁到特色地方品种的物种多样性。物种特色资源的鉴定和保护是种质资源评价的重要内容之一,目前,主要依赖于分子标记基因型检测所获得的DNA指纹图谱。通过采集每个个体的DNA指纹数据以及“户籍”信息(包括品种名称、出生地、出生日期),就可以建立DNA指纹-个体“户籍”两者互相关联的数据库。通过远程访问该数据库,任何人都可通过DNA指纹信息确认其所查询的特色地方品种的相关信息。DNA指纹数据库及其在线溯源系统的建设,可以为品种及其制品生产和销售的企业或个人提供精确到个体的溯源系统,从而为产品追踪、质量监控、打假维权等提供最科学最权威的证据支持。
对于GenoPlexs指纹鉴定标记的开发,可以根据几种容易混淆的近似物种基因组基因注释和变异信息,挑选100个左右的mSNP位点作为区分这些近似物种的“DNA指纹”,并开发形成GenoPlexs检测标记,用来对市场上假冒或近似的品种进行检测。GenoBaits指纹鉴定标记的开发一般涉及以下步骤:根据转录组表达筛选、基因注释筛选、同源基因筛选等多种方式确定品种特定的候选基因;设计基于长片段多重PCR技术的候选基因全长引物(或基于探针杂交技术的候选基因外显子探针),对已有自然群体材料进行检测,发掘候选基因位点的遗传变异;对自然群体进行表现型鉴定,结合已经获取的基因型信息进行候选基因关联分析,筛选与表现型明确关联的遗传变异;根据现有重测序数据筛选缺失率低、多态性高、分布均匀的背景标记,优先考虑位于基因外显子区、基因启动子区的标记;将基因注释中明确的重要功能基因位点作为背景位点的补充;加入候选基因功能标记,利用GenoBaits技术开发特定品种的40K检测标记,并通过对部分材料的测试,筛选出对应的20K和10K等位点组合,实现一款多用。为筛选特色地方品种的DNA指纹标记,可根据已有的重测序数据,挑选50个优良且能够满足各方面要求的mSNP标记,进行引物设计和合成。根据验证结果从中精选出20—30个mSNP作为检测标记。利用中选标记对若干样品进行测试以验证开发标记的检测效果,开发特色地方品种GBTS检测标记。
3.3 分子遗传图谱构建、基因/QTL定位和基因克隆
遗传图谱是遗传研究最基础也是最重要的工具。高密度分子遗传图谱可以用于定位表型性状的基因,确定基因与标记间、标记与标记间的相互关系,进而开展重要基因的图位克隆(表2)。在植物上,分子连锁图谱的构建通常采用双亲群体如F2、DH、RIL等。采用GBTS基因型检测技术,能够快速、低成本地构建连锁图谱。主基因的定位可以采用标记连锁定位相同的原理。而数量性状基因座位(QTL)的定位,通常涉及存在于不同染色体上的多个基因。通常采用永久性分离群体(DH和RIL等)或以家系为单位的分离群体(F3等)进行QTL定位,因为表型的测定可以基于多环境或多个重复下的平均值,以尽量减少或消除环境误差。同时,同步检测多个QTL的分离和重组需要采用远远大于主基因定位所需的群体,且必须采用高密度的分子标记以区分不同的QTL重组体。高效、低成本、高密度的GBTS技术成为QTL定位的最适合技术,因为基于GenoBaits的GBTS技术,标记密度可以根据定位的需要进行任意调整。
基因/QTL的定位除了可以采用上述基于双亲群体的策略之外,还可以利用自然群体进行关联分析[21,22]。与主基因定位对应的是基于候选基因的关联分析,即通过检测特定的候选基因在自然群体中的分离来验证候选基因与特定表型之间的关联,从而对候选基因进行确认和验证。候选基因一般是根据其他物种的已有研究结果,或通过特定结构域预测提示,可能与目标性状变异有关的某些或某类基因。GenoPlexs技术可用于富集感兴趣的某些或某类基因,通过深度测序,挖掘候选基因变异信息并进行候选基因关联分析,从而快速定位或克隆相关的候选基因。
与QTL定位相对应的关联分析是全基因组关联分析(genomewide association study,GWAS)[23,24]。GWAS利用覆盖全基因组的分子标记对自然群体中的所有个体进行检测,旨在寻找与某个表型性状相关联的所有遗传变异。一般而言,所用的标记数量越多,检测的群体越大,找到与目标性状相关联的遗传变异的概率也越高。因此,大群体和高密度分子标记是高标准GWAS分析的2个最基本要求。GenoBaits技术可以检测包含多达45K的目标位点(区段),每个区段长约150 bp。结合mSNP技术,可以检测多达260K以上的SNP标记。同时由于GenoBaits标记的共享性,可以对同一群体不同环境、不同群体的GWAS结果进行比较和综合。利用GBTS技术,可以富集感兴趣的某些或某类基因,降低单样本检测成本;可自由选择目标区域,可对基因全长加启动子进行分析,也可仅对任意数量的基因外显子区域进行分析,包括全外显子检测。
为了简化基因/QTL的连锁作图和候选基因鉴定,混样分析(BSA)方法得到越来越广泛的应用。早期的BSA方法是基于分离体的混合分析法(bulked segregant analysis)[25,26],就是从双亲分离群体中挑选具有两类极端表型的个体,将其DNA混合后构建两类DNA池,通过比较DNA池间的差异,找到影响目标性状的连锁标记甚至候选基因。近年,BSA方法扩展到了混样分析(bulked sample analysis),即从任意群体或群体的混合物选择两类极端表型个体,由此构成2个不同的样本DNA池[27]。
BSA的有效性取决于5个重要因素:(1)原始群体的大小。群体越大,两类极端个体之间的表型差异可能就越大,精细的基因定位需要数千或数万个个体。(2)选择率:入选率越低越好。一般不要超过5%,最好在1%以下。(3)极端个体数。极端个体数越少,由于机会造成的类群间差异的可能性就越大。要把假阳性的概率降低到1%以下,需要进行40 VS 40或更多极端个体间的比较。建议进行50 VS 50以上的比较。(4)标记密度。紧密连锁的标记之间同时表现为假阳性的概率是2个标记各自假阳性概率的乘积。采用高密度的分子标记,可以极大地降低假阳性的概率。(5)表型鉴定的准确性。由于极端个体的选择是基于表型的,任何程度的表型鉴定误差都会对BSA的结果产生重要的影响。严格控制上述5个条件,结合多池(群)比较[27]完全可以一步定位多个QTL,甚至确定影响目标性状的候选基因。灵活采用GenoBaits和GenoPlexs 2种方法,可以在全基因组范围筛选目标区域的同时,通过聚焦来锁定特定位点的候选基因。为了混池中不同个体DNA/RNA的均一性以及后续可能需要的个体基因型验证,最好按个体进行DNA/RNA提取后再混合,或保留个体的叶片等组织。
图位基因克隆是指从基因组位置入手,通过遗传和物理作图,最终找到控制某性状的基因。随着高质量参考基因组的完成,图位克隆流程现已变得更加简单。利用GBTS技术,可以利用目标性状上有分离的群体快速构建遗传图谱。对候选区段进行标记加密,根据参考基因组筛选和鉴定候选基因。亦可针对所有的候选基因设计标记,进行候选基因捕获。最后通过基因功能验证,最终确定影响性状变异的基因。
小麦秆锈病抗性基因的定位和克隆是一个颇具代表性的实例[28]。栽培小麦D基因组的二倍体祖先种节节麦(Aegilops tauschii)对小麦秆锈病病原菌具有较好的抗性,是抗性基因的宝贵来源。采用174份Ae. tauschii ssp. Strangulata亚型节节麦为材料,21份Ae. tauschii ssp. Tauschii作为外群材料,选择已克隆的R基因Sr33和Sr45作为阳性对照。针对节节麦的NLR基因组区段和在基因组上均匀分布的317个SNP位点设计了捕获探针,通过深度测序获得了249—336个全长以及1 312—2 170个非全长NLR基因,并鉴定了目标区域内的变异。结合151个样本的抗病性和候选基因关联分析,鉴定出了多个Sr基因,其中包括已克隆的Sr33、Sr45、Sr46和SrTA1662,研究表明,这四个基因是从节节麦亚群渗入到普通小麦中。
3.4 分子标记辅助选择:主基因累加和转移
与重要农艺性状高度连锁或位于基因内功能位点内的标记,或称诊断性标记(diagnostic marker),可以通过各种杂交育种,高效、快速地鉴定所需的性状,并能聚合有利主基因或在不加背景选择的情况下转移到新品种中去(表2)。这类借助于前景标记进行的分子标记辅助选择,具有以下5个特点[5,20]:不需要测交就能筛选雄性不育恢复性、广亲和性、配合力、恢复力等;不依赖于环境对各种胁迫性进行选择;不依赖于昂贵的仪器设备或者大量测试材料就能对品质性状、生物活性物质、生理生化性状等进行选择;可以在育种的早期阶段进行品质、杂种优势、产量潜力等进行选择和早代淘汰;一步完成在不同环境条件、不同育种阶段、或需要昂贵测试或测交才能完成的所有选择。最近十年来,随着许多物种高质量参考基因组的构建,控制农艺性状的许多主基因已经被图位克隆[29],很多基因已被跨国种业公司用于开发与性状关联的分子标记并应用于相关性状的基因累加和转移。因此,将重要农艺性状的克隆基因以及紧密连锁的分子标记,开发或转化成为可以进行高通量、快速和低成本检测的SNP功能标记,就可以采用GenoPlexs技术进行同步检测,从而推动分子标记辅助的主基因累加和转移在育种上的广泛应用。
3.5 分子标记辅助选择:目标基因和转基因回交育种
分子标记辅助的回交育种已广泛应用于重要农艺性状的基因转移或渐渗,且已有大量的研究和报道[30,31,32,33]。因此,这里仅讨论分子标记辅助的转基因回交育种。转基因就是将外源基因导入受体细胞并让其表达以获得新的遗传性状的生物技术。转基因技术具有针对性强、育种效率高、周期短的特点,已经成为植物分子育种的重要手段。考虑到中国转基因作物种植市场的开放是大势所趋,做好转基因产品的储备,等待时机,是每个育种单位和企业值得考虑的未雨绸缪之事。由于部分转基因产品专利保护期已过,而这些产品仍有很高的应用价值。通过转基因元件边缘探针可以直接调取跨元件序列,帮助锁定转基因事件。因此,可以通过分子标记辅助的转基因鉴定,将这些事件转入理想的品种中去,培育适合未来转基因种植市场、具有很强竞争优势的品种。在遗传转化中,不同基因型材料的转化效率和植株再生能力存在较大差异,导致只有少数材料适宜作为遗传转化的受体。以玉米为例,目前常用的受体材料多为A188、H99及杂交种HiII等,但这些材料往往不具备优良的综合性状,同时长时间组织培养过程中会发生大量基因位点突变,所以这些转化体不适宜直接作为生产材料应用。利用转化体和优良品系进行针对转化位点的回交转育成为转基因育种的主要手段(吴坤生,私人通讯)。
传统的回交育种往往需八代以上的回交才能高度恢复到轮回亲本的基因组。应用分子标记进行辅助回交,在每轮后代个体中筛选出背景恢复率最高的材料用于下一轮回交,可以大大提高回交效率,一般仅用3个世代就可恢复95%以上轮回亲本的基因组[31]。也就是通过2次回交选择得到理想的双重组个体AMB,即在BC1,选择aMB或者AMb单重组;在BC2中选择另一单重组,即在M与b或者在a与M之间重组。假设a和b位于与目标基因1 cM遗传距离,只要在BC1和BC2世代分别检测约185和370个单株,就有95%的概率获得到一个理想的单重组个体。
采用GenoPlexs技术,可以利用同一标记组合完成目标基因鉴定与轮回亲本的背景选择,使回交选择更加简单易行(表2)。同一套1—2K背景标记,可以保证在任何回交群体中都有足够数量的多态性标记来监测每个世代的轮回亲本基因组恢复情况,确保鉴定到既有目标性状且轮回亲本基因组比率较高的个体。当然也可以根据特殊育种需求来定制适用于特定育种目标的专用成套标记。高效低成本的标记检测技术, 辅之以从克隆基因所开发的功能标记,将推动分子标记辅助的回交育种的广泛应用。
3.6 分子标记辅助选择:轮回选择和全基因组选择
标记辅助的轮回选择(marker-assisted recurrent selection,MARS)[34,35,36]和全基因组选择(GS)[37,38,39]是分子标记辅助选择的延伸和扩展,适合于多基因控制的复杂性状(表1)。MARS是通过性状-标记的关联分析,挑选对表现型具有显著效应的标记来构建遗传模型,对个体的育种值进行预测。由于复杂性状一般涉及大量的基因,且每个基因的效应较小,常规的标记性状关联分析只能检测出效应较大的标记位点,而大量微效位点可能达不到统计学上的显著水平而被忽略。因此,常规的标记辅助选择或MARS,都不适合复杂性状的选择。GS同时考虑覆盖全基因组的所有标记对目标性状的效应,通过估算基因组育种值(genomic estimated breeding value,GEBV),根据育种值大小进行选择。具体做法是:首先对训练群体同时检测基因型和表现型,利用所有标记建立表型预测模型;然后对育种群体中待测个体进行SNP检测,利用标记效应的预测模型对待测个体进行表型预测,获得每个待测个体的GEBV。由于GS所需的分子标记数量多、育种群体大,导致单个样本的基因型检测成本较高。因此,长期以来,GS的大规模应用仅限于具有共享技术和平台的国际跨国种业公司内部以及单个样本经济价值较高的物种,如奶牛和种猪等。高效、低成本和共享技术平台是中小种业公司以及发展中国家开展大规模GS的前提[8]。GBTS作为高通量低成本的基因型检测技术,同一套标记可以通过控制测序深度,来满足不同群体GS对标记数量的需求(表2)。高密度的GenoBaits标记系统可以开发成为适合不同群体的通用系列标记,而中密度的GenoPlexs标记系统可以根据特定群体GS的需要进行特别定制,特别适合功能标记丰富、待选群体大、具有大数据和信息支撑的物种和群体。另外,GBTS技术的灵活性,可以将一部分标记固定作为背景位点而应用于所有的群体,而另一部分标记可以根据特定育种群体的需求进行灵活选择。GBTS标记的高度可累加性,使利用所有信息不断进行GS模型的优化成为可能。
3.7 知识产权保护和种业安全
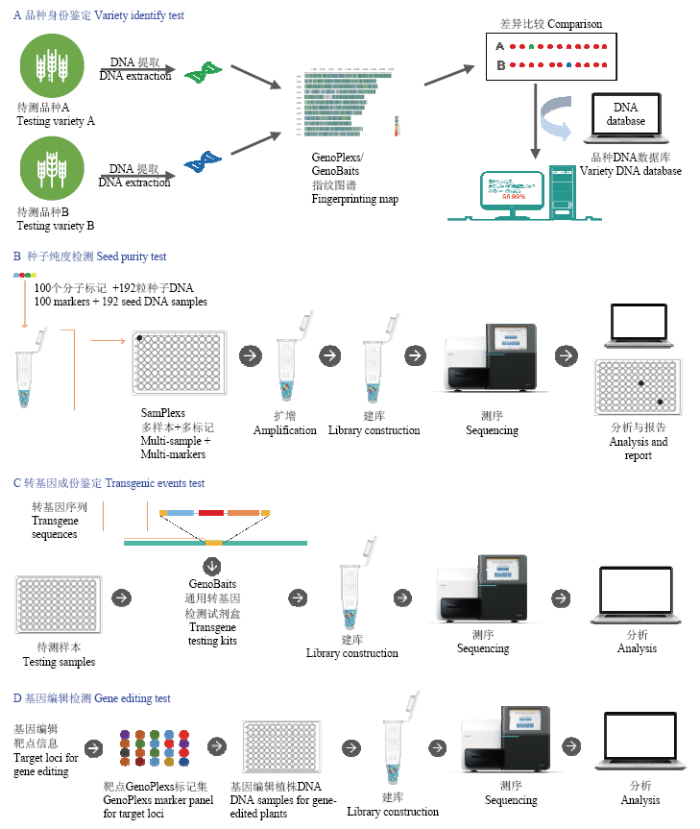
3.7.1 品种身份鉴定和品种权保护 随着育种过程及其产品的商业化以及分子生物学对育种技术进步的贡献日益增加,保护与育种技术和产品有关的知识产权,保护品种的经营和利益获取成为日益重要的问题[20,40-41]。育种技术和核心分子(基因)元件一般可以通过专利来保护。而品种权保护涉及到品种的身份鉴定和确权。传统的品种身份鉴定依靠表型特征来实现,国际上普遍接受的DUS方法,就是根据品种的特异性、一致性和稳定性来鉴定的。由于表型性状易受环境的影响且需要较长时间的培育和观察,实际操作难度较大,标准也难以规范和统一。因此,利用分子水平的基因型差异开展品种的身份鉴定成为当今和未来的重要技术选择[20]。分子标记作为可以检测分子水平变异的工具,在国内外已被广泛应用于品种身份鉴定(图5-A;表2)。中国当前品种权保护的标记技术多采用SSR标记法,所用标记位点数有限,一般不能鉴定实质性派生品种(essentially derived varieties,EDV),品种真实性鉴定结果也在众多判决案例中遭到质疑。SSR标记法应用于品种保护时,由于标准样品难以获得,跨实验室间的数据难以精准整合和精准比对,因此很难在不同机构间普遍推广与应用,只有屈指可数的几家单位拥有开展品种DNA指纹鉴定的条件与能力,使鉴定工作无法全面开展,无法有效遏制种业市场的品种权侵权问题。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5GBTS技术的4种检测应用(品种身份、种子纯度、转基因事件、基因编辑)
Fig. 5Four applications of GBTS in the tests of variety identity, seed purity, transgenic events and gene editing
近两年,国家有关部门正在探讨和制订更加可靠的分子检测方法,重点是采用统一的平台、标记和标准。以测序为基础的SNP检测方法、采用尽可能多且覆盖全基因组的分子标记是未来品种身份鉴别的发展方向。在国家标准制订过程中,认为基于GBTS技术的分子检测是品种身份鉴定的理想工具。通过GBTS检测,可以高效低成本地构建全基因组DNA指纹图谱,精确描述品种间的差异,重构品种间系谱。相关结果可以为品种身份鉴定、DUS测定和EDV分析等提供重要信息。长期积累的品种DNA指纹图谱和分子检测数据将为品种身份鉴定和品种权保护提供所需的全国性物种大数据。最近,基于GBTS技术的MNP(mSNP)标记法[42]发布成为品种鉴定的国家标准,有望以技术和标准为基础,整合全社会的力量有效解决当前品种权保护中碰到的诸多难题。
3.7.2 种子纯度鉴定 种子纯度由种子样本中所包含的特定基因型与非特定基因型之间的比例确定,是种子质量的一项重要指标。《中华人民共和国种子法》规定种子纯度必须达到规定指标才能在市场上销售。传统的纯度鉴定方法是通过田间种植,根据表现型来进行评价和估计。但是由于表型鉴定时间长、易受环境因素影响,现已逐渐被分子鉴定法所取代,并以DNA鉴定为主(图5-B;表2)。
常规品种的种子纯度鉴定相对比较简单,只需要鉴别出特定品种的目标基因型所占比例。而对于杂交种,不仅需要鉴定杂交种中所含自交种的比例,更希望能鉴定出异交种的比例。笔者已建立了一套基于mSNP的全新DNA纯度鉴定方法。采用近百个SNP标记对近二百粒种子进行单粒检测,能够检出杂交种子中混有的自交种和异交种,从而快速、精准地进行种子纯度鉴定。同时,由于采用比较多的分子标记,还能鉴定亲本纯度以及外来花粉导致的其他混杂基因型。
3.7.3 转基因成份检测 转基因农作物产品在过去二十几年累计种植面积超过25亿公顷,为全球粮食供应做出了巨大贡献[43]。然而,在中国主要粮食作物的转基因产品还未批准上市,种植转基因作物属于违法行为。另外,在批准商业化的物种中,也只能推广经过审批、含有特定转基因成分和事件的品种。因此,进行转基因成份和事件检测,可避免由于商业化含有未经审批的转基因事件的品种(转基因污染和违规)而造成的责任风险。
迄今为止,全球批准上市的农作物转基因事件已超过400个,更多正在开发中。如何保证育种材料不含非意愿生物性状不是一件容易的事。现有的国家转基因检测标准仅局限于特定转基因成份的鉴定。目前,采用的一般PCR方法,可因气溶胶实验室污染和烟草花叶病毒等植株污染导致假阳性。采用GenoBaits技术,可以构建包括所有转基因成分的分子标记检测系统,一次性检测多个元件,在检测出转基因共性成分的同时确定转基因具体事件(图5-C;表2)。此外,还可以屏蔽气溶胶和烟草花叶病毒等污染对结果的干扰。转基因检测可以基于单个样本,也可以采用多个样本的混合物以进一步提高检测效率。基于GBTS技术的目标区域测序法[44]已经于近期成为植物转基因鉴定的国家标准,可望在国家层面对已知与未知的转基因事件进行更加全面的监管,彻底避免由转基因检测假阳性而引发的大量纠纷与矛盾。
3.7.4 基因编辑后代的快速鉴定 基于CRISP/Cas9对特定DNA序列的敲除、插入和重组置换等改变的基因编辑技术被认为是21世纪生物领域的一项重大突破[45,46],近年来被广泛应用于动植物品种改良[47]。如何快速鉴定转化后代是否含有预期的变异并同时评估脱靶状况,现已成为基因编辑中急需解决的问题。基于超级多重PCR的GenoPlexs技术可以用于基因编辑后代的快速检测。利用带有特异性条码的引物进行扩增,将产物混合建库,经过高深度测序鉴定低频突变,就可以高效低成本地检测出基因编辑后代所携带的DNA序列变异(图5-D;表2)。GenoPlexs技术可以对来自同一载体相同靶点的后代、同一载体多个靶点的后代、混合载体多个靶点的后代进行快速鉴定。此外,基于GenoBaits的液相探针系统可以包含全基因组的高密度分子标记,可设计用于基因编辑中的脱靶分析。
在玉米中,利用CRISPR/Cas9高度特异和靶向性的特征,对1 000多个初定位的QTL和候选基因进行大规模基因编辑试验,通过调查QTL区间所有基因突变体的表型即可快速鉴定到功能基因,实现了高通量、低成本的基因鉴定[48]。面对大规模基因编辑后代的鉴定工作,采用GenoPlexs技术与不同编辑产物混合检测的方案相结合,大大提高了鉴定速度,降低了鉴定成本。
3.7.5 动植物病虫害鉴定 动植物病虫害的有效管理和防治需要对病虫害实施检测和监控,而防止检疫性病虫害的入侵和传播需要快速可靠的检测手段。过去主要在形态和生理水平上根据病虫害对生物的危害进行判断,涉及病虫害的发生、发展及其演变规律等。分子水平上的检测,有助于深刻理解病原菌和害虫不同生理种群或小种对生物的危害及其作用机制。理论上,可以针对不同病虫害的生物种群或小种开发特异性的分子标记,并整合成为成套的GBTS标记。采用一套2K的GenoPlexs标记,有可能从混合样本中检测同一病虫害的多个不同生理种群或小种,或者多种不同的病虫害类型(表2)。对不同环境下采集的病虫害样本进行全方位检测,将有助于了解病虫害的发生、发展、变异和流行规律,从而制定合理的病虫害管理和防治措施。
为了同步开展多种检疫性病虫害的监测,可利用针对检疫性动植物病虫害设计的分子标记,提高检测的灵敏性和可靠性。同时,采用高密度的GenoBaits液相探针系统,可以设计覆盖多个现存或检疫性病虫害生理种群或小种的全基因组分子标记,以检测其全基因组变异,实现对其种群或小种突变、新的病虫害生理种群或小种的检测,以便随时发现新的变异种群或小种。
对于食品安全造成巨大威胁的动物疫病,例如非洲猪瘟等,目前为止还没有切实有效的药物或者疫苗用于防范,捕杀还是唯一切实可行的处理措施。为尽可能控制捕杀的规模和数量,降低经济损失,就需要在最短时间内通过病原菌的分子标记检测,确认疑似病例的“户籍”信息。DNA指纹数据库及在线溯源系统的建立可以为动植物疫病防控部门提供全面而及时的疑似病例信息查询服务。
4 靶向测序基因型检测(GBTS)标记开发和应用实例
4.1 GenoPlexs标记的开发和应用
在植物上,利于GenoPlexs技术首次开发了玉米1K SNP标记集,并应用于杂种优势群的划分和转基因回交育种(袁隆平农业高科技股份有限公司卢东林、林海燕,石家庄博瑞迪生物技术公司张嘉楠等,私人通讯)。该套标记共包含1 085对引物,其中900个均匀分布在玉米基因组上的背景SNP位点、129个功能性SNP标记和4个功能性InDel标记、已经公开的34个转基因事件的检测标记。经过近2万份样本的测试,显示该套标记的检出率可达97%以上,均一性达到95%,捕获效率达97%。现已被大规模应用于杂种优势群划分、种质资源鉴定和转基因回交育种。在动物方面,开发了用于宠物猫和宠物狗的血统检测、遗传病评估的GenoPlexs标记集。在微生物方面,为更好地研究和检测新型冠状病毒(2019-nCoV),开发了针对305个2019-nCoV病毒已知变异位点的标记集,包括187对引物,其中新冠引物176对,人类内参基因引物11对(张嘉楠,私人通讯)。4.2 GenoBaits标记的开发和应用
GBTS技术最早在玉米上得到应用。目前已开发了两款基于GenoBaits的液相芯片,分别含有20K和40K SNP标记位点(扩增子)。20K SNP液相芯片采用了来自55K固相芯片[49]的SNP标记。首先从中挑选出在染色体上均匀分布的24 495个SNP标记。根据平均缺失率和测序深度以及染色体分布的均匀性,挑选出20K标记。随后,利用20K SNP标记集对483份国内外代表性玉米材料进行检测,以测试该液相芯片的有效性以及玉米材料的遗传多样性。最后,根据不同标记位点的捕获效率(同一条件下不同标记位点所获得的测序深度),从20K标记中挑选出捕获效率较高、染色体上均匀分布的10K SNP亚套。进一步从10K SNP中挑选出5K和1K SNP标记[11]。标记的捕获效率越高,越容易在较低的测序深度下捕获到,检测成本就越低。因此,同一套20K SNP标记,通过控制测序深度,可以获得20K、10K、5K、1K共4种不同标记数量的液相芯片组合,将缺失率控制在2%而所需的平均测序深度分别为50×、20×、7.5×和2.5×。检测结果表明,20K SNP液相芯片与55K固相芯片之间相同SNP标记的平均一致性高达95.3%。4个不同自交系,每个含2个生物学重复的检测表明,20K SNP液相芯片的基因型检测可重复性在98.0%—98.4%。针对483份材料所进行的多样性和聚类分析,将玉米分为热带和温带2个大群,而温带材料又可以进一步划分为7个主要的亚群。采用不同标记密度的4套液相芯片获得了非常类似的结果[11]。目前,这款玉米20K SNP液相芯片已被20多家大学、科研单位和种业公司所采用。第二款GenoBaits液相芯片是最新开发的40K mSNP标记集。这也是基于mSNP的第一款高密度SNP芯片。首先,根据MAF>0.05、缺失比例<20%、杂合比例<5%的条件对玉米HapMap3的位点进行筛选获得候选位点集合。其次,以100 bp为窗口,挑选出计算窗口内SNP数量大于2、小于15、单倍型总数大于30、频率5%以上的单倍型数大于4、频率大于1%以上的单倍型数为10的区段。最后,依据基因组均匀分布的原则构成40K位点的目标区段(徐云碧等,待发表)。
与每个扩增位点只包括一个SNP的20K液相芯片相比,这款40K mSNP液相芯片具有五个特点。一是在几乎同样的检测成本下,将标记位点的数目增加了一倍,即在玉米基因组中可以找到40K以上高变异的目标区域(平均100 bp)。二是在每一个扩增位点可以产生多个SNP标记,组成mSNP。三是同一扩增子内的多个SNP标记之间可以形成大量的单倍型,大大提高了变异的检测效率。四是可以从每个mSNP内挑选出MAF最大的SNP组成40K SNP核心标记。五是mSNP提供了更为精细的遗传变异检测,包括采用单倍型和SNP对mSNP位点内和位点间的变异进行检测。
随后,利用867份代表性玉米材料(包括621份国内外常规玉米和246份甜玉米)对40K mSNP标记集的有效性进行检测。尽管2套材料来源于不同的实验室,在多样性、杂合性、样本DNA质量等方面存在明显差异,仍然获得了高质量的mSNP、SNP和单倍型。采用621份常规玉米,获得了260K SNP和754K单倍型的可用数据(徐云碧等,待发表)。最后,根据不同标记的捕获效率,从同一套40K mSNP标记位点,通过控制测序深度,可以获得1—40K任意位点数量的液相芯片组合(徐云碧等,待发表)。采用11个不同自交系,每个含2个生物学重复的检测表明,40K mSNP液相芯片的基因型检测可重复性在97.8%—99.7%。针对867份材料所进行的多样性和聚类分析,获得了与20K SNP液相芯片类似的聚类结果,同时提供了甜玉米材料的相互关系图谱。借助于高密度的分子标记,可以对基因组不同区域(包括UTR5、CDS、UTR3、intronic和intergenic)的SNP及其单倍型进行比较分析,从而发现更深层次的遗传多样性和不同材料之间的精细差异。
除玉米外,依据同样的方案,现已开发出水稻和大豆的40K SNP标记集,并筛选出相应的20K、10K及1K标记位点,应用于遗传学和育种研究(中国农业科学院作物科学研究所徐建龙,中国科学院遗传与发育研究所田志喜,石家庄博瑞迪生物技术有限公司张嘉楠等,私人通讯)。其中水稻40K标记集中共包含了26 983个背景SNP位点,覆盖2 747个水稻已经克隆基因的14 226个位点,并针对56个水稻重点基因设置了共540个目标位点。目前,在20种主要大田和蔬菜植物中,开发了50余套标记集(表3)。在动物方面,目前正在开发奶牛和对虾的GenoBaits标记集。在微生物方面,开发了针对新型冠状病毒(2019-nCoV)序列变异的标记集,专注于新型冠状病毒完整序列的捕获,利用标记对序列变异的高容忍性来跟踪病毒序列的变异。该标记集共包含508个液相探针,覆盖2019-nCoV全长共29 914 bp,以及GAPDH、EIF3A等共10个人类常用内参基因的外显子区。经过检测,该探针集对目标全长序列的捕获覆盖率均在99.9%以上(张嘉楠,私人通讯)。
Table 3
表3
表3已经开发的动植物GenoPlexs和GenBaits标记集
Table 3
| 物种名称 Species names | GenoBaits 标记集GenoBaits panels | GenoPlexs 标记集GenoPlexs panels | ||||
|---|---|---|---|---|---|---|
| 40K | 20K | 10K | 500-1K | <100 | 功能标记 Functional markers | |
| 玉米 Zea mays L. | ● | ● | ● | ● | ○ | |
| 水稻Oryza sativa L. | ● | ● | ● | ● | ○ | |
| 棉花Gossypium spp | ● | ○ | ||||
| 大豆Glycine max (Linn.) Merr. | ● | ● | ● | ● | ||
| 花生 Arachis hypogaea L. | ● | ● | ● | ● | ||
| 小麦Triticum aestivum L. | ○ | ○ | ● | ● | ||
| 谷子 Setaria italica | ● | |||||
| 大麦 Hordeum vulgare L. | ● | ● | ● | |||
| 番茄Solanum lycopersicum | ● | ● | ○ | |||
| 黄瓜 Cucumis sativus L. | ● | ● | ○ | |||
| 辣椒 Capsicum annuum L. | ● | ● | ○ | |||
| 西瓜Citrullus lanatus (Thunb.) Matsum. et Nakai | ● | ● | ||||
| 白菜Brassica pekinensis (Lour.) Rupr. | ● | ● | ||||
| 荔枝Litchi chinensis Sonn. | ● | |||||
| 猕猴桃Actinidia spp. | ● | |||||
| 甘蓝Brassica oleracea L. | ● | |||||
| 胡萝卜Daucus carota L. var. sativa Hoffm. | ● | |||||
| 西葫芦Cucurbita pepo L. | ● | |||||
| 白萝卜 Raphanus sativus | ● | |||||
| 苹果Malus domestica | ● | |||||
| 牛Bos holsatiae | ● | |||||
新窗口打开|下载CSV
5 靶向基因型检测(GBTS)技术应用展望
5.1 便携式、自动化、高通量、智能化检测平台
为推动基因型检测技术的广泛应用,需要发展便携式、自动化、高通量检测平台。一方面,测序技术的进步可能推动便携式、小型化测序仪和检测系统的发展。另一方面,应用场景的多样化需求也会反过来催生灵活多样性检测系统的开发。检测系统或系统的一部分,比如取样和DNA提取,甚至PCR,均可能实现小型化,方便携带至生物采样和分析现场,实现现场分析直至进行选择决策。大规模的样本检测可能需要高度自动化的高通量检测设备,包括DNA提取和DNA文库构建。笔者曾设想未来的基因型检测系统可能是手机一样大小的掌上机,与人工智能和机器人相结合,可以在现场采集样本,立刻完成并显示基因型检测结果,对待选样本进行实时处理,或选择保留,或现场淘汰清理。相关基因型信息和检测结果可以通过智能化系统,进入数据库并发送至用户终端。5.2 可变密度、多功能分子检测系统
如果DNA测序的成本大幅度降低以至测序量不再是成本的主要决定因素,同时信息支撑系统的进步导致海量数据的处理和分析成为常态,则可以从低成本产生的海量检测信号中根据需要提取不同密度(数量)的信号(标记),实现同一检测系统的多种应用。另一种可变密度的分子检测系统就是本文介绍的GenoBaits技术,同一套SNP液相芯片,可以调整测序深度,以最佳的成本获得所需的标记数目。GBTS技术的灵活性为基因型检测的多功能应用奠定了基础。同一套标记,通过控制测序深度,几乎可以用于表2所列的所有十二类标记应用场景。这几乎涉及到动物、植物、微生物科学研究和应用的全产业链。此外,还有3个应用领域值得特别提及。一是基因组特定区域的饱和检测,可以通过靶向捕获特定的基因组区域,进行深度测序,获取不同生物材料的全序列,进行特定区域遗传变异的深度挖掘、比较和分析,以获取有用的信息。二是全基因组候选基因的检测。玉米中,采用GenoBaits技术,开发出覆盖全基因组的40K mSNP位点,每个位点平均包含6.2个SNP(徐云碧等,待发表)。如果每个位点对应于一个候选基因,这样的位点数目足够覆盖一个物种的所有基因。因此可以开发代表全基因组候选基因的标记系统,开展基于所有候选基因的GWAS分析。三是部分候选基因的检测和验证。目前许多研究涉及大量候选基因及其等位基因变异和单倍型变异。GBTS技术提供了测试和验证大量候选基因的快速简便途径。
在最近关于高通量靶向基因编辑加速玉米功能基因挖掘的研究中,利用近期CUBIC群体所获大量初定位QTL及其他重要候选基因,进行了一千多个候选基因的大规模检测[48]。为了实现目标基因的高效捕获,将不同编辑靶点的样本进行等比混合,利用基于超多重PCR的GenoPlexs技术一次性对所有靶点进行多重PCR扩增,然后对富集的目标位点进行深度测序,鉴定出混合样本中每个位点的低频变异及其序列,根据不同样本的编辑位点情况,即可直接锁定混合样本中单个样本是否编辑成功以及编辑后的序列信息。因此,GenoPlexs技术实现了高效、精准、低成本的大规模、多靶点的基因编辑检测和目标基因捕获。
5.3 与其他技术的整合
基于GenoBaits和GenPlexs两类技术,可以覆盖5—40 000个mSNP位点(或200+ K SNP标记)的检测分析,理论上包括了从低到高的所有标记密度范围。但不能满足进行低成本单标记检测的要求,因为在某些情况下,主基因性状或个别转基因/基因编辑事件的检测,可能只是需要采用一个到少数几个标记。KASP标记检测系统目前仍然最适合单标记的检测,在系统和平台已经建立的条件下,具有成本和价格优势,但需要针对目标位点进行标记的设计和优化。由于很难在每个实验室都建立起检测系统并进行目标标记的开发,KASP系统还是应该包括在公共的设施平台中,与其他标记检测系统一起进行运营、维护和管理。分子检测的另一个极端就是超高密度的标记系统,目前,Affymetrix系统最高可以达到660K[50]。这类超高密度芯片主要用于高精度的GWAS分析,需要与大群体和高精度的表型鉴定相结合,以确定复杂性状的候选基因。由于单个材料的检测成本、单个项目的整体费用高昂,这类超高密度芯片只适合少数科研单位使用。同时这类芯片需要特别定制,标记的检测需要借助于专用的昂贵设备和试剂,系统的运转和维护需要专人负责。因此,超高密度的标记检测适合由国际性、区域性、共享性平台来进行。GBTS技术与KASP和超高密度的固相芯片相结合,提供了可以检测所有标记的完整方案;而GBTS技术为绝对部分广谱的分子检测应用提供了更为高效、节省、灵活的解决方案。未来随着测序和信息处理技术的革命,基于全基因组测序的基因型检测技术将有可能极大地降低个体的测序成本,迎来基因型检测技术的5G时代(图1)。
GBTS技术与BSA相结合,可以进一步提升标记检测的通量并降低单个样本的分析成本。实践上共有三类BSA。第一类是将多个样本分别采用不同的探针进行标志,混合起来进行PCR和其他分析,主要目标是降低成本。第二类是将多个样本直接混合进行PCR和其他分析,通过高敏感性检测所发现的微量检出物进行判断。第三类是典型的BSA,从混合的样本中提取不同个体共有的标记或信号,与另一个混合样本进行比较,获得2个混样之间的差异信息,而假定其他的背景信息在2个混样之间保持相同。GBTS与BSA相结合,在大幅度降低检测成本的同时,进一步提高了检测效率。BSA已广泛应用于基于DNA、RNA和蛋白质水平上的基因组学、遗传学和育种学的研究,主要包括基因定位、特定成分鉴定、种子质量和纯度检测等[27]。近期进行的千万武汉人大规模新冠病毒核酸筛查就采用了BSA方法,使检测效率成倍提高。
5.4 资源共享与开源育种
GBTS技术采用固定的靶向检测获得高度可重复的基因型信息,使不同实验室、不同项目、不同样本的信息比较、累加、整合和共享成为可能[11]。对于基因组学研究比较深入、具有高质量参考基因组和泛基因组的物种,通过大规模重测序或简化基因组测序,可以构建覆盖全基因组的高精度单倍型图谱。因此,不同技术平台所产生的基因型数据可以通过与单倍型图谱进行比对、定位和整合,同时根据LD对区间标记位点进行缺失数据替补(imputation),推测出标记等位形式。借助于GBTS技术系统,不需要强大的数据和信息系统的支撑,甚至在没有完整参考基因组的情况下,就可以依靠自身的特定标记组合进行信息的比较和整合,因而大大简化了数据的分析、处理以及实验室的信息管理系统(LIMS),也同时降低了中小企业或研究机构等对于强大生物信息技术支撑团队的需求[8]。GBTS技术在信息比较、累加、整合等方面的优势,为基因型信息的共享创造了条件,同时推动其他资源的共享,这将大大提高平台的使用效率,降低单位时间的运营成本,同时避免区域范围内平台的重复和过度建设。由于GBTS标记可以采用多种测序仪进行基因型检测,增加了平台共享的机会和可能性。此外,无论动物和植物,均能采用同类的GBTS标记和相同的平台进行检测。因此,平台的共享可以横跨动物、植物和微生物。共享的平台、共性的检测技术和可标准化的信息系统为跨物种跨行业共享分子检测平台和支撑系统奠定了基础[11]。在康奈尔大学和包括国际玉米小麦改良中心在内的几家国际农业研究机构的努力下,建立了基因组开源育种信息项目(genomic open-source breeding informatics initiative,GOBii,
GBTS技术由于其高度可重复性以及低成本,可以为大规模的育种项目和几乎所有育种材料提供完整可靠的精准基因型指纹图谱,并追踪基因和染色体片段在育种过程中的变异、选择和传递。因此,不同的育种单位或公司可以建立育种联盟或联合体,在高精度分子检测平台的支持下,对不同育种材料进行身份鉴定,为广泛的材料交流、资源互通、利益共享、风险同担提供证据和支撑。由于育成品种或品系的遗传成分、亲本贡献、后代选择等都可以通过精细的指纹图谱来确定,合作育成品种的利益也可以借助于分子检测所确定的遗传贡献等来定量分配。在这一设想的推动下,国内已经组织了几个信息和资源共享的大规模玉米开源育种项目[8],包括通州国际种业组织的GS项目、中国农业科学院作物科学研究所组织的“1(所)+8(公司)”育种联合体、多家南繁单位参与创建的九所育种联盟等,其主要的开源育种活动是复杂性状的GS。大量GS模型和选择结果所积累的大数据,共享性育种技术、平台和信息,日益改进的预测技术,将不断优化GS的选择模型,提高其选择效果。这类开源育种项目可望在共享育种平台和支撑系统的支持下,实现扬长避短、优势互补、资源共享,以推动育种事业,特别是分子育种的跨越式发展。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/nrg3012URLPMID:21681211 [本文引用: 2]

The advent of next-generation sequencing (NGS) has revolutionized genomic and transcriptomic approaches to biology. These new sequencing tools are also valuable for the discovery, validation and assessment of genetic markers in populations. Here we review and discuss best practices for several NGS methods for genome-wide genetic marker development and genotyping that use restriction enzyme digestion of target genomes to reduce the complexity of the target. These new methods -- which include reduced-representation sequencing using reduced-representation libraries (RRLs) or complexity reduction of polymorphic sequences (CRoPS), restriction-site-associated DNA sequencing (RAD-seq) and low coverage genotyping -- are applicable to both model organisms with high-quality reference genome sequences and, excitingly, to non-model species with no existing genomic data.
[本文引用: 1]
//
[本文引用: 1]
//
[本文引用: 1]
DOI:10.2135/cropsci2007.04.0191URL [本文引用: 3]
DOI:10.1093/jxb/erx135URLPMID:28830098 [本文引用: 2]
As one of the important concepts in conventional quantitative genetics and breeding, genetic gain can be defined as the amount of increase in performance that is achieved annually through artificial selection. To develop pro ducts that meet the increasing demand of mankind, especially for food and feed, in addition to various industrial uses, breeders are challenged to enhance the potential of genetic gain continuously, at ever higher rates, while they close the gaps that remain between the yield potential in breeders' demonstration trials and the actual yield in farmers' fields. Factors affecting genetic gain include genetic variation available in breeding materials, heritability for traits of interest, selection intensity, and the time required to complete a breeding cycle. Genetic gain can be improved through enhancing the potential and closing the gaps, which has been evolving and complemented with modern breeding techniques and platforms, mainly driven by molecular and genomic tools, combined with improved agronomic practice. Several key strategies are reviewed in this article. Favorable genetic variation can be unlocked and created through molecular and genomic approaches including mutation, gene mapping and discovery, and transgene and genome editing. Estimation of heritability can be improved by refining field experiments through well-controlled and precisely assayed environmental factors or envirotyping, particularly for understanding and controlling spatial heterogeneity at the field level. Selection intensity can be significantly heightened through improvements in the scale and precision of genotyping and phenotyping. The breeding cycle time can be shortened by accelerating breeding procedures through integrated breeding approaches such as marker-assisted selection and doubled haploid development. All the strategies can be integrated with other widely used conventional approaches in breeding programs to enhance genetic gain. More transdisciplinary approaches, team breeding, will be required to address the challenge of maintaining a plentiful and safe food supply for future generations. New opportunities for enhancing genetic gain, a high efficiency breeding pipeline, and broad-sense genetic gain are also discussed prospectively.
URLPMID:30569365 [本文引用: 1]
DOI:10.1016/j.xplc.2019.100005URL [本文引用: 5]
DOI:10.1111/pbi.12834URLPMID:28913866 [本文引用: 1]
Wheat breeders and academics alike use single nucleotide polymorphisms (SNPs) as molecular markers to characterize regions of interest within the hexaploid wheat genome. A number of SNP-based genotyping platforms are available, and their utility depends upon factors such as the available technologies, number of data points required, budgets and the technical expertise required. Unfortunately, markers can rarely be exchanged between existing and newly developed platforms, meaning that previously generated data cannot be compared, or combined, with more recently generated data sets. We predict that genotyping by sequencing will become the predominant genotyping technology within the next 5-10 years. With this in mind, to ensure that data generated from current genotyping platforms continues to be of use, we have designed and utilized SNP-based capture probes from several thousand existing and publicly available probes from Axiom(R) and KASP genotyping platforms. We have validated our capture probes in a targeted genotyping by sequencing protocol using 31 previously genotyped UK elite hexaploid wheat accessions. Data comparisons between targeted genotyping by sequencing, Axiom(R) array genotyping and KASP genotyping assays, identified a set of 3256 probes which reliably bring together targeted genotyping by sequencing data with the previously available marker data set. As such, these probes are likely to be of considerable value to the wheat community. The probe details, full probe sequences and a custom built analysis pipeline may be freely downloaded from the CerealsDB website (http://www.cerealsdb.uk.net/cerealgenomics/CerealsDB/sequence_capture.php).
DOI:10.1093/sysbio/syy086URLPMID:30535394 [本文引用: 3]
Sequencing of target-enriched libraries is an efficient and cost-effective method for obtaining DNA sequence data from hundreds of nuclear loci for phylogeny reconstruction. Much of the cost of developing targeted sequencing approaches is associated with the generation of preliminary data needed for the identification of orthologous loci for probe design. In plants, identifying orthologous loci has proven difficult due to a large number of whole-genome duplication events, especially in the angiosperms (flowering plants). We used multiple sequence alignments from over 600 angiosperms for 353 putatively single-copy protein-coding genes identified by the One Thousand Plant Transcriptomes Initiative to design a set of targeted sequencing probes for phylogenetic studies of any angiosperm group. To maximize the phylogenetic potential of the probes, while minimizing the cost of production, we introduce a k-medoids clustering approach to identify the minimum number of sequences necessary to represent each coding sequence in the final probe set. Using this method, 5-15 representative sequences were selected per orthologous locus, representing the sequence diversity of angiosperms more efficiently than if probes were designed using available sequenced genomes alone. To test our approximately 80,000 probes, we hybridized libraries from 42 species spanning all higher-order groups of angiosperms, with a focus on taxa not present in the sequence alignments used to design the probes. Out of a possible 353 coding sequences, we recovered an average of 283 per species and at least 100 in all species. Differences among taxa in sequence recovery could not be explained by relatedness to the representative taxa selected for probe design, suggesting that there is no phylogenetic bias in the probe set. Our probe set, which targeted 260 kbp of coding sequence, achieved a median recovery of 137 kbp per taxon in coding regions, a maximum recovery of 250 kbp, and an additional median of 212 kbp per taxon in flanking non-coding regions across all species. These results suggest that the Angiosperms353 probe set described here is effective for any group of flowering plants and would be useful for phylogenetic studies from the species level to higher-order groups, including the entire angiosperm clade itself.
DOI:10.1007/s11032-019-0940-4URL [本文引用: 9]
[本文引用: 1]
DOI:10.1371/journal.pone.0019379URLPMID:21573248 [本文引用: 1]
Advances in next generation technologies have driven the costs of DNA sequencing down to the point that genotyping-by-sequencing (GBS) is now feasible for high diversity, large genome species. Here, we report a procedure for constructing GBS libraries based on reducing genome complexity with restriction enzymes (REs). This approach is simple, quick, extremely specific, highly reproducible, and may reach important regions of the genome that are inaccessible to sequence capture approaches. By using methylation-sensitive REs, repetitive regions of genomes can be avoided and lower copy regions targeted with two to three fold higher efficiency. This tremendously simplifies computationally challenging alignment problems in species with high levels of genetic diversity. The GBS procedure is demonstrated with maize (IBM) and barley (Oregon Wolfe Barley) recombinant inbred populations where roughly 200,000 and 25,000 sequence tags were mapped, respectively. An advantage in species like barley that lack a complete genome sequence is that a reference map need only be developed around the restriction sites, and this can be done in the process of sample genotyping. In such cases, the consensus of the read clusters across the sequence tagged sites becomes the reference. Alternatively, for kinship analyses in the absence of a reference genome, the sequence tags can simply be treated as dominant markers. Future application of GBS to breeding, conservation, and global species and population surveys may allow plant breeders to conduct genomic selection on a novel germplasm or species without first having to develop any prior molecular tools, or conservation biologists to determine population structure without prior knowledge of the genome or diversity in the species.
URLPMID:20517342 [本文引用: 1]
DOI:10.1371/journal.pone.0090346URLPMID:24587335 [本文引用: 2]
Genotyping by sequencing (GBS) is a next generation sequencing based method that takes advantage of reduced representation to enable high throughput genotyping of large numbers of individuals at a large number of SNP markers. The relatively straightforward, robust, and cost-effective GBS protocol is currently being applied in numerous species by a large number of researchers. Herein we describe a bioinformatics pipeline, TASSEL-GBS, designed for the efficient processing of raw GBS sequence data into SNP genotypes. The TASSEL-GBS pipeline successfully fulfills the following key design criteria: (1) Ability to run on the modest computing resources that are typically available to small breeding or ecological research programs, including desktop or laptop machines with only 8-16 GB of RAM, (2) Scalability from small to extremely large studies, where hundreds of thousands or even millions of SNPs can be scored in up to 100,000 individuals (e.g., for large breeding programs or genetic surveys), and (3) Applicability in an accelerated breeding context, requiring rapid turnover from tissue collection to genotypes. Although a reference genome is required, the pipeline can also be run with an unfinished
URLPMID:20111037 [本文引用: 1]
URLPMID:25528188 [本文引用: 1]
DOI:10.1038/ng.2313URL [本文引用: 2]
Whereas breeders have exploited diversity in maize for yield improvements, there has been limited progress in using beneficial alleles in undomesticated varieties. Characterizing standing variation in this complex genome has been challenging, with only a small fraction of it described to date. Using a population genetics scoring model, we identified 55 million SNPs in 103 lines across pre-domestication and domesticated Zea mays varieties, including a representative from the sister genus Tripsacum. We find that structural variations are pervasive in the Z. mays genome and are enriched at loci associated with important traits. By investigating the drivers of genome size variation, we find that the larger Tripsacum genome can be explained by transposable element abundance rather than an allopolyploid origin. In contrast, intraspecies genome size variation seems to be controlled by chromosomal knob content. There is tremendous overlap in key gene content in maize and Tripsacum, suggesting that adaptations from Tripsacum (for example, perennialism and frost and drought tolerance) can likely be integrated into maize.
URLPMID:29342277 [本文引用: 2]
[本文引用: 4]
DOI:10.1016/j.copbio.2006.02.003URL [本文引用: 1]
Association mapping, a high-resolution method for mapping quantitative trait loci based on linkage disequilibrium, holds great promise for the dissection of complex genetic traits. The recent assembly and characterization of maize association mapping panels, development of improved statistical methods, and successful association of candidate genes have begun to realize the power of candidate-gene association mapping. Although the complexity of the maize genome poses several significant challenges to the application of association mapping, the ongoing genome sequencing project will ultimately allow for a thorough genome-wide examination of nucleotide polymorphism-trait association.
DOI:10.1016/j.pbi.2009.12.004URL [本文引用: 1]
Increased availability of high throughput genotyping technology together with advances in DNA sequencing and in the development of statistical methodology appropriate for genome-wide association scan mapping in presence of considerable population structure contributed to the increased interest association mapping in plants. While most published studies in crop species are candidate gene-based, genome-wide studies are on the increase. New types of populations providing for increased resolution and power of detection of modest-size effects and for the analysis of epistatic interactions have been developed. Classical biparental mapping remains the method of choice for mapping the effects of alleles rare in germplasm collections, such as some disease resistance genes or alleles introgressed from exotic germplasm.
URLPMID:19185530 [本文引用: 1]
[本文引用: 1]
DOI:10.1093/nar/19.23.6553URLPMID:1684420 [本文引用: 1]
We present a general method for isolating molecular markers specific to any region of a chromosome using existing mapping populations. Two pools of DNA from individuals homozygous for opposing alleles for a targeted chromosomal interval, defined by two or more linked RFLP markers, are constructed from members of an existing mapping population. The DNA pools are then screened for polymorphism using random oligonucleotide primers and PCR (1). Polymorphic DNA bands should represent DNA sequences within or adjacent to the selected interval. We tested this method in tomato using two genomic intervals containing genes responsible for regulating pedicle abscission (jointless) and fruit ripening (non-ripening). DNA pools containing 7 to 14 F2 individuals for each interval were screened with 200 random primers. Three polymorphic markers were thus identified, two of which were subsequently shown to be tightly linked to the selected intervals. The third marker mapped to the same chromosome (11) but 45 cM away from the selected interval. A particularly attractive attribute of this method is that a single mapping population can be used to target any interval in the genome. Although this method has been demonstrated in tomato, it should be applicable to any sexually reproducing organism for which segregating populations are being used to construct genetic linkage maps.
DOI:10.1073/pnas.88.21.9828URLPMID:1682921 [本文引用: 1]
We developed bulked segregant analysis as a method for rapidly identifying markers linked to any specific gene or genomic region. Two bulked DNA samples are generated from a segregating population from a single cross. Each pool, or bulk, contains individuals that are identical for a particular trait or genomic region but arbitrary at all unlinked regions. The two bulks are therefore genetically dissimilar in the selected region but seemingly heterozygous at all other regions. The two bulks can be made for any genomic region and from any segregating population. The bulks are screened for differences using restriction fragment length polymorphism probes or random amplified polymorphic DNA primers. We have used bulked segregant analysis to identify three random amplified polymorphic DNA markers in lettuce linked to a gene for resistance to downy mildew. We showed that markers can be reliably identified in a 25-centimorgan window on either side of the targeted locus. Bulked segregant analysis has several advantages over the use of near-isogenic lines to identify markers in specific regions of the genome. Genetic walking will be possible by multiple rounds of bulked segregation analysis; each new pair of bulks will differ at a locus identified in the previous round of analysis. This approach will have widespread application both in those species where selfing is possible and in those that are obligatorily outbreeding.
DOI:10.1111/pbi.12559URLPMID:26990124 [本文引用: 3]
Biological assay has been based on analysis of all individuals collected from sample populations. Bulked sample analysis (BSA), which works with selected and pooled individuals, has been extensively used in gene mapping through bulked segregant analysis with biparental populations, mapping by sequencing with major gene mutants and pooled genomewide association study using extreme variants. Compared to conventional entire population analysis, BSA significantly reduces the scale and cost by simplifying the procedure. The bulks can be built by selection of extremes or representative samples from any populations and all types of segregants and variants that represent wide ranges of phenotypic variation for the target trait. Methods and procedures for sampling, bulking and multiplexing are described. The samples can be analysed using individual markers, microarrays and high-throughput sequencing at all levels of DNA, RNA and protein. The power of BSA is affected by population size, selection of extreme individuals, sequencing strategies, genetic architecture of the trait and marker density. BSA will facilitate plant breeding through development of diagnostic and constitutive markers, agronomic genomics, marker-assisted selection and selective phenotyping. Applications of BSA in genetics, genomics and crop improvement are discussed with their future perspectives.
DOI:10.1038/s41587-018-0007-9URLPMID:30718880 [本文引用: 1]
Disease resistance (R) genes from wild relatives could be used to engineer broad-spectrum resistance in domesticated crops. We combined association genetics with R gene enrichment sequencing (AgRenSeq) to exploit pan-genome variation in wild diploid wheat and rapidly clone four stem rust resistance genes. AgRenSeq enables R gene cloning in any crop that has a diverse germplasm panel.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
//
[本文引用: 1]
//
[本文引用: 1]
//
[本文引用: 1]
[本文引用: 1]
//
[本文引用: 1]
URLPMID:11290733 [本文引用: 1]
Recent advances in molecular genetic techniques will make dense marker maps available and genotyping many individuals for these markers feasible. Here we attempted to estimate the effects of approximately 50,000 marker haplotypes simultaneously from a limited number of phenotypic records. A genome of 1000 cM was simulated with a marker spacing of 1 cM. The markers surrounding every 1-cM region were combined into marker haplotypes. Due to finite population size N(e) = 100, the marker haplotypes were in linkage disequilibrium with the QTL located between the markers. Using least squares, all haplotype effects could not be estimated simultaneously. When only the biggest effects were included, they were overestimated and the accuracy of predicting genetic values of the offspring of the recorded animals was only 0.32. Best linear unbiased prediction of haplotype effects assumed equal variances associated to each 1-cM chromosomal segment, which yielded an accuracy of 0.73, although this assumption was far from true. Bayesian methods that assumed a prior distribution of the variance associated with each chromosome segment increased this accuracy to 0.85, even when the prior was not correct. It was concluded that selection on genetic values predicted from markers could substantially increase the rate of genetic gain in animals and plants, especially if combined with reproductive techniques to shorten the generation interval.
DOI:10.2135/cropsci2009.11.0662URL [本文引用: 1]
DOI:10.3389/fgene.2015.00049URLPMID:25750652 [本文引用: 1]
Genomic selection is a promising development in agriculture, aiming improved production by exploiting molecular genetic markers to design novel breeding programs and to develop new markers-based models for genetic evaluation. It opens opportunities for research, as novel algorithms and lab methodologies are developed. Genomic selection can be applied in many breeds and species. Further research on the implementation of genomic selection (GS) in breeding programs is highly desirable not only for the common good, but also the private sector (breeding companies). It has been projected that this approach will improve selection routines, especially in species with long reproduction cycles, late or sex-limited or expensive trait recording and for complex traits. The task of integrating GS into existing breeding programs is, however, not straightforward. Despite successful integration into breeding programs for dairy cattle, it has yet to be shown how much emphasis can be given to the genomic information and how much additional phenotypic information is needed from new selection candidates. Genomic selection is already part of future planning in many breeding companies of pigs and beef cattle among others, but further research is needed to fully estimate how effective the use of genomic information will be for the prediction of the performance of future breeding stock. Genomic prediction of production in crossbreeding and across-breed schemes, costs and choice of individuals for genotyping are reasons for a reluctance to fully rely on genomic information for selection decisions. Breeding objectives are highly dependent on the industry and the additional gain when using genomic information has to be considered carefully. This review synthesizes some of the suggested approaches in selected livestock species including cattle, pig, chicken, and fish. It outlines tasks to help understanding possible consequences when applying genomic information in breeding scenarios.
// Available at:
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
http://www.isaaa.org/resources/publications/briefs/54/default.asp,
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1126/science.1229223URL [本文引用: 1]
Allostery is well documented for proteins but less recognized for DNA-protein interactions. Here, we report that specific binding of a protein on DNA is substantially stabilized or destabilized by another protein bound nearby. The ternary complex's free energy oscillates as a function of the separation between the two proteins with a periodicity of similar to 10 base pairs, the helical pitch of B-form DNA, and a decay length of similar to 15 base pairs. The binding affinity of a protein near a DNA hairpin is similarly dependent on their separation, which-together with molecular dynamics simulations-suggests that deformation of the double-helical structure is the origin of DNA allostery. The physiological relevance of this phenomenon is illustrated by its effect on gene expression in live bacteria and on a transcription factor's affinity near nucleosomes.
URLPMID:24906146 [本文引用: 1]
DOI:10.1038/s43016-020-0051-8URL [本文引用: 1]
DOI:10.1186/s13059-020-1930-xURLPMID:31980033 [本文引用: 2]
BACKGROUND: Identifying genotype-phenotype links and causative genes from quantitative trait loci (QTL) is challenging for complex agronomically important traits. To accelerate maize gene discovery and breeding, we present the Complete-diallel design plus Unbalanced Breeding-like Inter-Cross (CUBIC) population, consisting of 1404 individuals created by extensively inter-crossing 24 widely used Chinese maize founders. RESULTS: Hundreds of QTL for 23 agronomic traits are uncovered with 14 million high-quality SNPs and a high-resolution identity-by-descent map, which account for an average of 75% of the heritability for each trait. We find epistasis contributes to phenotypic variance widely. Integrative cross-population analysis and cross-omics mapping allow effective and rapid discovery of underlying genes, validated here with a case study on leaf width. CONCLUSIONS: Through the integration of experimental genetics and genomics, our study provides useful resources and gene mining strategies to explore complex quantitative traits.
DOI:10.1007/s11032-017-0622-zURLPMID:28255264 [本文引用: 1]
With the decrease of cost in genotyping, single nucleotide polymorphisms (SNPs) have gained wide acceptance because of their abundance, even distribution throughout the maize (Zea mays L.) genome, and suitability for high-throughput analysis. In this study, a maize 55 K SNP array with improved genome coverage for molecular breeding was developed on an Affymetrix(R) Axiom(R) platform with 55,229 SNPs evenly distributed across the genome, including 22,278 exonic and 19,425 intronic SNPs. This array contains 451 markers that are associated with 368 known genes and two traits of agronomic importance (drought tolerance and kernel oil biosynthesis), 4067 markers that are not covered by the current reference genome, 734 markers that are differentiated significantly between heterotic groups, and 132 markers that are tags for important transgenic events. To evaluate the performance of 55 K array, we genotyped 593 inbred lines with diverse genetic backgrounds. Compared with the widely-used Illumina(R) MaizeSNP50 BeadChip, our 55 K array has lower missing and heterozygous rates and more SNPs with lower minor allele frequency (MAF) in tropical maize, facilitating in-depth dissection of rare but possibly valuable variation in tropical germplasm resources. Population structure and genetic diversity analysis revealed that this 55 K array is also quite efficient in resolving heterotic groups and performing fine fingerprinting of germplasm. Therefore, this maize 55 K SNP array is a potentially powerful tool for germplasm evaluation (including germplasm fingerprinting, genetic diversity analysis, and heterotic grouping), marker-assisted breeding, and primary quantitative trait loci (QTL) mapping and genome-wide association study (GWAS) for both tropical and temperate maize.
DOI:10.1111/pbi.13361URLPMID:32065714 [本文引用: 1]
The rapid development and application of molecular marker assays have facilitated genomic selection and genome-wide linkage and association studies in wheat breeding. Although PCR-based markers (e.g. simple sequence repeats and functional markers) and genotyping by sequencing have contributed greatly to gene discovery and marker-assisted selection, the release of a more accurate and complete bread wheat reference genome has resulted in the design of single-nucleotide polymorphism (SNP) arrays based on different densities or application targets. Here, we evaluated seven types of wheat SNP arrays in terms of their SNP number, distribution, density, associated genes, heterozygosity and application. The results suggested that the Wheat 660K SNP array contained the highest percentage (99.05%) of genome-specific SNPs with reliable physical positions. SNP density analysis indicated that the SNPs were almost evenly distributed across the whole genome. In addition, 229 266 SNPs in the Wheat 660K SNP array were located in 66 834 annotated gene or promoter intervals. The annotated genes revealed by the Wheat 660K SNP array almost covered all genes revealed by the Wheat 35K (97.44%), 55K (99.73%), 90K (86.9%) and 820K (85.3%) SNP arrays. Therefore, the Wheat 660K SNP array could act as a substitute for other 6 arrays and shows promise for a wide range of possible applications. In summary, the Wheat 660K SNP array is reliable and cost-effective and may be the best choice for targeted genotyping and marker-assisted selection in wheat genetic improvement.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}